

Summary
Regulatory T (Treg) cells induce an immunosuppressive microenvironment that is a major obstacle for successful tumor immunotherapy. Dissecting the regulatory mechanisms between energy metabolism and functionality in Treg cells will provide insight toward developing novel immunotherapies against cancer. Here we report that human, naturally occurring and tumor-associated Treg cells exhibit distinct metabolic profiles with selectivity for glucose metabolism compared with effector T cells. Treg-mediated accelerated glucose consumption induces cellular senescence and suppression of responder T cells through cross-talk. TLR8 signaling selectively inhibits glucose uptake and glycolysis in human Treg cells, resulting in reversal of Treg suppression. Importantly, TLR8 signaling-mediated reprogramming of glucose metabolism and function in human Treg cells can enhance anti-tumor immunity in vivo in a melanoma adoptive transfer T cell therapy model. Our studies identify mechanistic links between innate signaling and metabolic regulation of human Treg suppression, which may be used as a strategy to advance tumor immunotherapy.
Keywords: Treg cells, T cell subsets, metabolism, glycolysis, Toll-like receptor, tumor immunotherapy
In Brief
Treg-mediated immunosuppression is a significant obstacle for tumor immunotherapy. Li et al. show that human Treg cells heighten glucose consumption and subsequently trigger cellular senescence and suppression of effector T cells. With TLR8 signaling, they selectively inhibit glucose uptake and glycolysis in human Treg cells, resulting in Treg function reversal and enhanced anti-tumor immunity.
Graphical Abstract
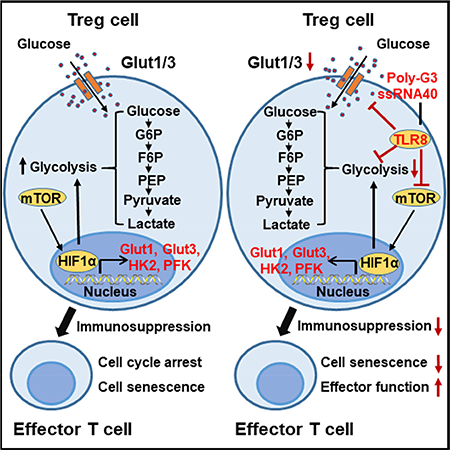
Introduction
Different T cell subsets have distinct respective effector functions in response to physiological and pathological conditions. Increasing evidence indicates that cellular energy metabolism directs T cell survival, proliferation and the ability to perform its specific and functional immune responses (Fox et al., 2005; Jacobs et al., 2008; Macintyre et al., 2014). In response to environmental cues, there are specific drivers of cellular metabolism that regulate the expression of enzymes crucial for various metabolic processes (Fox et al., 2005; Gerriets and Rathmell, 2012; MacIver et al., 2013; Newton et al., 2016; Pearce, 2010; Zeng and Chi, 2015). Aerobic glycolysis is the main metabolic pathway and is specifically required for effector function in T cells upon T cell activation (Macintyre et al., 2014; MacIver et al., 2013). Increasing glycolysis in T cells results in enhanced effector functions such as IFN-γ production (Chang et al., 2013; Chang et al., 2015; Ho et al., 2015; Michalek et al., 2011; Sukumar et al., 2013); while decreased glycolysis has been shown to inhibit both IFN-γ and IL-17 production (Cham et al., 2008; Cham and Gajewski, 2005; Chang et al., 2013). In contrast, lipid oxidation has been identified as a primary metabolic pathway for induced Treg cells and memory CD8+ T cells (Buck et al., 2016; Michalek et al., 2011; Pearce et al., 2009; Shi et al., 2011). Furthermore, the endogenous fatty acid synthesis pathway and the glycolytic-lipogenic axis are crucial for the metabolic programming of Th17 cell development but not for Treg cells (Berod et al., 2014). A better understanding of the molecular processes and metabolic profiles of different T cell subsets will facilitate the development of novel strategies via metabolic reprogramming of T cell fate and function for immunotherapy.
It has become clear that tumor-infiltrating Treg cells induce an immunosuppressive microenvironment that is a major obstacle for successful tumor immunotherapy (Curiel, 2008; Wang et al., 2004; Zou, 2006). A challenge in developing novel immunotherapies against cancer is to develop effective strategies for breaking immune tolerance induced by Treg cells (Curiel, 2008; Zou, 2006). Given the importance of metabolism in directing T cell fate and functions, defining the metabolic processes of Treg cells should provide alternative novel strategies and more specific checkpoint targets for controlling Treg-induced suppression. Recent studies suggest that metabolic regulations of Treg differentiation, Foxp3 expression and Treg stability and homeostasis involves both glycolysis and lipid metabolism (Dang et al., 2011; De Rosa et al., 2015; Michalek et al., 2011; Newton et al., 2016; Procaccini et al., 2016; Shi et al., 2011; Zeng et al., 2013). Several molecular signaling pathways and/or molecules have been identified, which are critical and required for Treg metabolic programming and development, including Akt-mTOR signaling, Toll-like receptor (TLR) signaling, autophagy, as well as transcription factors HIF1α, cMyc, and FoxP3 (De Rosa et al., 2015; Gerriets et al., 2016; Maj et al., 2017; Newton et al., 2016; Shi et al., 2011; Shrestha et al., 2015; Wang et al., 2011; Wei et al., 2016; Zeng et al., 2013). Furthermore, both glycolysis and lipid metabolism are important for Treg suppressive functions (Procaccini et al., 2016). Although these more recent studies have substantially increased our understanding of Treg metabolism, the active metabolic pathways and regulations in human Treg cells are still unclear. Furthermore, whether the metabolic profiles of tumor-derived Treg cells are different or similar from that of naturally occurring Treg cells and/or other T cell subsets is unknown (Biswas, 2015; Chang et al., 2015). In addition, the metabolic regulation of established Treg cell function, including tumor-associated Treg cells has not yet been fully explored (De Rosa et al., 2015; Newton et al., 2016; Procaccini et al., 2016; Zeng et al., 2013). We have recently identified that senescence induction in responder T cells is a novel suppressive mechanism mediated by human Treg cells (Liu et al., 2018; Ye et al., 2012; Ye et al., 2013). However, how the metabolic activity of Treg cells influence the fate in responder T cells during their cross-talk and interactions is also important and urgent to be investigated. Precisely dissecting these challenging issues will enhance the development of novel strategies to specifically reprogram Treg metabolism for immunotherapy against cancer and other diseases.
TLRs are critical components of the innate immune system acting as a link between innate and adaptive immunity. TLRs are also very important for regulating Treg cell function (Caramalho et al., 2003; Kiniwa et al., 2007; Peng et al., 2005; Peng et al., 2007; Sutmuller et al., 2006; Wang et al., 2008). TLR signaling in dendritic cells or Treg cells can reverse mouse Treg suppression (Pasare and Medzhitov, 2003; Sutmuller et al., 2006). Recent studies suggest that TLR signaling also directly regulates energy metabolism in immune cells regulating saturated fatty acids and proinflammatory signaling (Huang et al., 2012; Lee et al., 2001; Lee et al., 2004; Shi et al., 2006), and driving early glycolytic reprogramming of DCs for their activation and function (Everts et al., 2014). In addition, TLR1 and TLR2 signaling activation in mouse Treg cells increases Treg glycolysis and proliferation and reduces their suppressive capacity (Gerriets et al., 2016). We have demonstrated that TLR8 signaling reverses the suppressive functions of human tumor-derived CD4+, CD8+ and γδ Treg cells resulting in enhanced anti-tumor immunity (Kiniwa et al., 2007; Peng et al., 2005; Peng et al., 2007; Ye et al., 2012; Ye et al., 2013). Our more recent studies have shown that TLR8 signaling activation in human Treg cells and tumor cells can prevent their induction of senescence in responder T cells and DCs (Ye et al., 2012; Ye et al., 2014; Ye et al., 2013). However, whether TLR8 signaling can also regulate energy metabolism in human Treg cells is still unknown. In addition, the unique signaling pathway(s) regulated by TLR8 signaling, leading to reversal of human Treg suppression and selective effects on Treg function are unknown. A better understanding of the molecular mechanisms and unique signaling pathways involved in the effects of how TLR8 signaling regulates Treg functions will be critical for development of therapeutic interventions.
In our efforts to control Treg suppression for tumor immunotherapy, we explored the cell metabolic profiles of human Treg and effector T cells. We found that human naturally occurring Treg and tumorassociated Treg cells display predominant dependence on glucose metabolism compared with effector T cells. Treg-mediated heightened glucose consumption triggers cell senescence and suppression in responder T cells during their cross-talk. Furthermore, we discovered that TLR8 signaling significantly inhibits glucose uptake and suppresses metabolic processes of glycolysis in human Treg cells, resulting in reversal of Treg suppression. Our in vivo studies further confirmed our hypothesis and concept that TLR8 signaling-mediated reprogramming of glucose metabolism and function in human Treg cells can enhance anti-tumor immunity and tumor immunotherapy in a melanoma therapy model.
Results
Human Treg cells exhibit a distinct metabolic profile compared with effector T cells
The distinct functional requirement of CD4+ Th cells suggests that each subset may require specific metabolic programs to meet their differing energetic and biosynthetic demands. However, the active metabolic profiles and regulations in human Treg cells, especially tumor-derived Treg cells are unclear. To precisely understand the metabolic regulations in human CD4+ T cell subsets, we obtained naïve CD4+ T cells purified from healthy donors, and Th1, Th2 and Th17 cells differentiated under the different polarized cytokine conditions. Furthermore, CD4+CD25+ naturally occurring Treg cells (nTreg) were freshly purified form PBMCs using FACS sorting or microbeads (Ye et al., 2012). We then characterized the key metabolic gene profiles involving both glucose and lipid metabolism in different CD4+ T cell subsets using real-time quantitative PCR analyses. Those molecules include glucose transporters Glut1 and Glut3, as well as glycolysis-related enzymes hexokinase 2 (HK2), glucose-6-phosphate isomerase (GPI), phosphofructokinase 1 (PFK1), triosephosphate isomerase 1 (TPI1), enolase 1 (ENO1), pyruvate kinase muscle 2 (PKM2) and lactate dehydrogenase A (LDHα). Furthermore, the genes of key enzymes related to both cholesterol synthesis, and fatty acid oxidation and synthesis were also included, such as 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR), 3-hydroxy-3-methylglutaryl-CoA synthase 1 (HMGCS1), squalene monooxygenase (SQLE), isopentenyl-diphosphate delta isomerase 1 (IDI1), carnitine palmitoyltransferase I (CPT-1), fatty acid synthase (FASN) and acetyl-CoA carboxylases 1 (ACC1). Our studies clearly suggested that human nTreg cells have more active glucose and lipid metabolism than that of effector T cell subsets, showing elevated expression levels of those metabolic genes (Figure 1A & 1B). We also compared the metabolic gene expression levels in freshly purified nTreg cells with that of in vitro expanded nTreg cells using anti-CD3 and IL-2 stimulation. Fresh nTreg cells and expanded nTreg cells have similar expression levels of those metabolic molecules (Supplemental Figure 1A & 1B). Notably, all the effector T cell subsets expressed high levels of glucose transporter Glut1. Furthermore, Th17 cells also had increased levels of fatty acid metabolic enzymes CPT-1 and ACC1 compared with Th1 and Th2 cells. Given that Treg cells play a critical role in maintaining tumor suppressive microenvironments, we explored metabolic profiles of tumor-derived Treg cells established from cancer patient TILs, including melanoma-specific CD4+ Treg cells (CD4 TregE1) and breast cancer-associated γδ Treg cells (γδ Treg31 and γδ Treg76) (Peng et al., 2007; Wang et al., 2004; Ye et al., 2013). Consistent with the expression profiles of nTreg cells, the majority of metabolic gene levels in tumor-derived Treg cell lines were also much higher than those in normal Th1 cells (Figure 1C & 1D). In addition to the characterization of gene expression of key metabolic enzymes, we further performed glucose metabolomic analyses to determine the metabolites related to glycolysis and tricarboxylic acid cycle (TCA) pathways in different T cell subsets using a LC-triple quadruple mass spectrometry (Supplemental Figure 1C). Metabolomic analysis revealed that nTreg cells produced higher amounts of key metabolites compared with effector T cell subsets, including the glycolytic metabolites Fructose-1,6-biphosphate (F-1,6-P) and lactate, as well as the TCA metabolites citrate, isocitrate, α-ketoglutatrate, succinate, malate, and ADP (Figure 1E). Notably, some key metabolites were significantly increased in the cell lysates in nTreg cells but were undetectable in effector T cells, such as 1, 3-bisphosphoglycerate (1, 3-BPG), phosphoenolpyuvate (PEP) and pyruvate (Data not shown). These results collectively suggest that both nTreg cells and tumor-derived Treg cells have heightened glucose and lipid metabolism distinct from effector T cells.
Figure1. Human nTreg and tumor-derived Treg cells have heightened glucose and lipid metabolism distinct from effector T cells.

(A) Gene expression levels of glucose transporters (Glut1 and Glut3) and the key enzymes in glycolysis (HK2, GPI, PFK1, TPI, ENO1, PKM2 and LDHα) in different T cell subsets. Th1, Th2 and Th17 cells were polarized from naïve T cells purified from healthy donors in the presence of related polarization cytokine conditions. nTreg cells were directly purified from PBMCs of healthy donors. Total RNA was isolated from each cell type and analyzed by real-time PCR. Expression levels of each gene were normalized to β-actin expression level and adjusted to the levels in naïve CD4+ T cells (served as 1). Data shown are mean ± SD from four independent donors. (B) Gene expression levels of key enzymes in cholesterol synthesis (HMGCR, HMGCS1, SQLE, and IDI1), as well as fatty acid oxidation (CPT-1) and synthesis (ACC1 and FASN) in different T cell subsets. Cell preparations and assays were identical to (A). (C) and (D) Tumor-derived CD4+ Treg and γδ Treg cells had higher gene expression levels of glucose transporters and the key enzymes in glycolysis (in C) and lipid metabolism (in D) than those of naïve CD4+ and Th1 cells. Tumor-derived Treg cells: CD4 TregE1 is a melanoma-specific Treg cells and γδ Treg31 & 76 cells are derived from TILs of breast cancer patients. Relative mRNA expression level of each gene was determined by real-time PCR, normalized to β-actin expression and then adjusted to the level in naïve CD4+ T cells. (E) nTreg cells produced higher amounts of the key metabolites involved in the glycolysis and tricarboxylic acid cycle than other T cell subsets. Th1, Th2, Th17 and nTreg cells were prepared as (A). The cell lysates from different T cell subsets were extracted and analyzed using a LCtriple quadruple mass spectrometry for determination of cellular glucose metabolites. Metabolite levels are normalized to naïve CD4 cell group. Relative levels of intermediate metabolites in the glycolysis and TCA-cycle pathways are shown as mean ± SD from representative of three independent T cell subsets with similar results. (F) and (G) Inhibition of glycolysis and lipid metabolism dramatically blocked Treg cell suppressive capacity on T cell proliferation (in F) and prevented Treg-induced responder T cell senescence (in G). nTreg cells were pretreated with pharmacological glucose transporter, glycolysis and lipid metabolism inhibitors for 48 hours, including phloretin (2 μM), 2-DG (1 mM), LND (125 μM), and 3BrPA (30 μM), etomoxir (100 μM), C75 (5 μM), orlistat (10 μM), 25-HC (0.25 μg/ml), simvastatin (2 μM), respectively. Naïve CD4+ T cells were then co-cultured with inhibitor-pretreated or untreated Treg cells for 3 days. Proliferation of co-cultured naïve T cells stimulated by anti-CD3 antibody was determined by [3H]-thymidine incorporation assays, and SA-β-Gal expression in treated T cells was also determined. Data shown are mean ± SD from representative of three independent experiments with similar results.* p<0.05 and ** p<0.01, compared with the medium only group.
Activated nTreg and tumor-associated Treg cells uniquely depend on glucose metabolism for their suppressive activity
More recent studies from mouse Treg cells suggest that metabolic regulation of Treg differentiation, Foxp3 expression, Treg cell lineage stability and homeostasis involves both glycolysis and lipid metabolism (Dang et al., 2011; De Rosa et al., 2015; Michalek et al., 2011; Newton et al., 2016; Procaccini et al., 2016; Shi et al., 2011; Zeng et al., 2013). To determine whether metabolic processes are critical for regulatory function of human Treg cells, we determined Treg suppressive capacities on responder T cell proliferation and senescence induction in the presence of a panel of potent pharmaceutical inhibitors targeting glucose (glucose transport and glycolysis) and lipid (cholesterol and fatty acids) metabolic pathways (Supplemental Figure 2A, 2B & 2C)(Peng et al., 2005; Ye et al., 2012; Ye et al., 2013). We found that blockage of both glucose transporter and glycolysis with inhibitors phloretin, 2-deoxy-D-glucose (2-DG), lonidamine (LND) and 3-bromopyruvate (3-BrPA), can significantly block Treg suppressive effect on the proliferation of responder T cells (Figure 1F). Furthermore, general lipid synthesis inhibitor 25-hydroxycholesterol (25-HC), and cholesterol and isoprenoid lipid synthesis inhibitor simvastatin, also impaired Treg suppression on responder T cell proliferation (Figure 1F). In addition, we further tested whether those metabolic inhibitors could prevent Treg-induced responder T cell senescence (Peng et al., 2005; Ye et al., 2012; Ye et al., 2013). In consistent with our previous report, we found a significant increase of senescent populations (SA-β-Gal+) in responder CD4+ T cells after co-culture with nTreg cells. However, pretreatment of nTreg cells with glucose transporter inhibitor (phloretin), glycolysis inhibitors (LND, 2-DG, 3-BrPA), or lipid biosynthesis inhibitors (C75, 25-HC and simvastatin) markedly decreased responder T cell senescence induced by nTreg cells (Figure 1G and Supplemental Figure 2D). Our results clearly indicate that both glucose and lipid metabolism are required for Treg biological functions.
T cell subsets, including Treg cells require activation and TCR engagement before performing their biological functions. We next investigated whether activated human Treg cells have different metabolic features with naïve Treg cells. We determined the metabolic gene expressions in different CD4+ T cell subsets after TCR activation with plated-bounded anti-CD3 antibody. Consistent with the metabolic profiles in naïve Treg cells, anti-CD3-activated Treg cells also had increased gene expression levels of those key molecules/enzymes involving both glucose and lipid metabolism compared with activated Th1, Th2 and Th17 effector T cell subsets (Figure 2A & Supplemental Figure 3A). Notably, after TCR activation, all the T cell subsets exhibited significantly increased gene expression of glucose transporters and enzymes in glycolysis, but not in enzymes of lipid metabolism (Figure 2B & Supplemental Figure 3B). However, the fold increases in those glycolytic genes in activated Treg cells were much higher than that of other activated CD4+ T cell subsets (Figure 2B). To further confirm that Treg cells have higher glucose metabolism than other T cell subsets, we determined the glucose uptake ability of different subsets of T cells with or without anti-CD3 activation, using a fluorescent glucose analog 2-NBDG labeling assay. Our results clearly demonstrated that nTreg had a higher glucose uptake than effector T cells regardless of activation status (Figure 2C). In addition, both unactivated and activated Treg cells produced more L-lactate than other T cell subsets (Figure 2D). Importantly, anti-CD3-activated Treg cells had much higher glucose uptake activity and lactate production compared with unactivated Treg cells, suggesting a heightened glycolytic activity in activated Treg cells. In addition, the majority of metabolic gene levels in anti-CD3-activated tumor-derived CD4+ Treg and γδ Treg cells were much higher than those in activated Th1 cells (Figure 2E & Supplemental Figure 3C). These strongly suggest that Treg cells may need more glucose metabolism to obtain energy after TCR activation to execute their biological functions.
Figure 2. Activated human Treg cells display high levels of glucose metabolism than that of effector T cells.

(A) Gene expression levels of glucose transporters and the key enzymes in glycolysis in different T cell subsets after stimulation with anti-CD3 antibody. Cell subset preparations and assays were identical to Figure 1. Total RNA was isolated from each cell type and analyzed by real-time PCR. Expression levels of each gene were normalized to β-actin expression levels and adjusted to the levels in naïve CD4 cells (served as 1). Data shown are mean ± SD from four independent healthy donors. (B) Comparisons of gene expression levels of key enzymes involved in glucose metabolism in different T cell subsets before and after anti-CD3 stimulations. Anti-CD3 activated Treg cells displayed strong desire for glucose metabolism compared with effector T cells. T cells were stimulated with or without anti-CD3 for 8 hours and total RNA was isolated from each cell type and analyzed by real-time PCR. Expression levels of each gene were normalized to β-actin expression levels and adjusted to the levels in naïve CD4+ T cells (served as 1). Data shown are mean ± SD from four independent healthy donors. *p<0.05 and **p<0.01, compared with the Treg results before anti-CD3 activation. #p<0.01, compared with the other T cell subsets with anti-CD3 activation. (C) Treg cells had higher glucose uptake than other T cell subsets no matter activation status. Cell subset preparations were identical to Figure 1. T cell subsets were stimulated with/without anti-CD3 antibody for 24 hours, and glucose uptake was determined by the flow cytometry after addition of 2-NBDG for 15 min. Results shown are a representative from three independent experiments. (D) Treg cells produced more L-lactate than other T cell subsets. Cell subset preparations and stimulations were identical to Figure 1. The L-lactate levels in the culture supernatants were determined by the Glycolysis cell-based assay kit. Results shown are mean ± SD from the summary of three independent experiments. **p<0.01, compared with the levels in naïve CD4 T cells. #p<0.01, compared with the other T cell subsets. (E) Activated tumor-derived CD4 Treg and γδ Treg cells also have high gene expression levels of glucose transporters and the key enzymes in glycolysis compared with those of activated Th1 cells. Cell preparations and assays are identical to (B). Data shown are mean ± SD from three independent experiments with similar results.
TLR8 signaling inhibits glucose uptake and metabolism in human Treg cells
Recent studies suggest that tumor cells and TILs compete for glucose within the tumor suppressive microenvironment, which is a driver for tumor suppression and progression (Chang et al., 2015; Sukumar et al., 2015). Furthermore, we have more recently identified that the suppression on responder T cells mediated by Treg cells is due to the induction of T cell senescence (Ye et al., 2012; Ye et al., 2013). We therefore determined the possibility that lack of enough glucose highly consumed by Treg cells may result in responder T cell senescence. We found that low concentrations of glucose can significantly induce both CD4+ and CD8+ T cell senescence (Figure 3A & Supplemental Figure 4A). However, normal (11 mM) and higher (25 mM) concentrations of glucose did not induce T cell senescence. We have more recently identified that both nTreg cells and tumor-derived Treg cells have a high glucose uptake capacity, and Treg-mediated acceleration of glucose consumption triggers cell senescence and DNA damage in responder T cells during their cross-talk (Liu et al., 2018). In addition, addition of high concentration of glucose (25 mM) dramatically prevented responder T cell senescence mediated by nTreg cells and tumor-derived Treg cells (Figure 3B). Collectively, these results clearly indicate that glucose competition between Treg and responder effector T cells triggers cell senescence and suppression in responder T cells during their interactions.
Figure 3. TLR8 signaling suppresses glucose uptake and metabolism in human Treg cells.

(A) Significantly increased SA-β-Gal+ T cell populations were induced in anti-CD3-activated naïve CD4+ and CD8+ T cells cultured in the medium with different concentrations of glucose for 3 days. Data shown are mean ± SD of T cells from three individual healthy donors. Normal medium with 11 mM glucose served as a control. (B) Addition of high concentration of glucose markedly rescued responder T cell senescence induced by nTreg cells and tumor-derived Treg cells. Anti-CD3 activated CD4+ T cells were co-cultured with Treg cells for 3 days with different concentrations of glucose. SA-β-Gal expression in responder CD4+ T cells was determined. Data shown are mean ± SD from three independent experiments. **p<0.01, compared with the naïve CD4 only group. #p<0.01, compared with the Tregtreated with normal concentration of glucose (11mM) group. (C) and (D) TLR8 ligand Poly-G3 treatment significantly reversed Treg suppressive capacity on T cell proliferation (in C) and prevented Treg-induced responder T cell senescence (in D). Different types of Treg cells were co-cultured with naïve CD4+ T cells in the presence or absence of Poly-G3 (3 μg/ml) or Poly-T3 (control) for 3 days. Proliferation of cocultured naïve T cells stimulated by anti-CD3 antibody was determined by [3H]-thymidine incorporation assays (in C), and SA-β-Gal expression in treated naïve T cells was determined (in D). Data shown are mean ± SD from representative of three independent experiments with similar results. **p<0.01, compared with the respective medium only and Poly-T3 treatment groups. (E) Alterations of genes involved in glucose metabolism were identified and ranked in nTreg cells after treatment with Poly-G3 at 24 hours. Gene alterations were normalized to log2 expression level. Human nTreg cells were isolated from PBMCs of two healthy donors and treated with Poly-G3 for different time points. Total RNA was purified and pooled, and transcriptome analyses of Treg cells were performed using the Illumina wholegenome Human HT-12 BeadChips. (F) Poly-G3 treatment increased glucose levels in the culture medium of both nTreg and tumor-derived Treg cells. Different types of Treg cells and naïve CD4+ T cells were cultured in the presence or absence of Poly-G3 or Poly-T3 (Control) for 3 days, and glucose levels in the culture medium were determined. **p<0.01, compared with the None and Poly-T3 treatment groups. (G) and (H) Poly-G3 treatment significantly decreased glucose uptake by nTreg and tumorderived Treg cells. Glucose uptake was determined by the flow cytometry with addition of 2-NBDG for 15 min after 3 day culture. Results shown in histogram (H) are mean ± SD from four independent experiments. *p<0.05 and **p<0.01, compared with the respective medium only and control Poly-T3 treatment groups.
We have demonstrated that human TLR8 signaling reverses the suppressive functions of nTreg cells and tumor-derived CD4+, CD8+ and γδ Treg cells on responder T cell proliferation and senescence development, resulting in enhanced anti-tumor immunity (Figure 3C & 3D). (Kiniwa et al., 2007; Peng et al., 2005; Peng et al., 2007; Ye et al., 2012; Ye et al., 2013). To further dissect how TLR8 signaling molecularly affects Treg functions, we conducted transcriptome analyses of human nTreg cells after TLR8 ligand Poly-G3 treatment using Illumina whole-genome Human HT-12 BeadChips. We identified genes that were significantly altered in Treg cells after Poly-G3 treatment for 24 hours, which are involved in cytokines, cytokine receptors, metabolic signaling, Treg associated markers and transcription factors (Supplemental Table 2).
Since we have shown above that glucose metabolism is critical for Treg suppressive activities and functions, we hypothesized that TLR8-mediated reversal of Treg suppression may be through the regulation of Treg glucose metabolism. As expected, our transcriptome analyses demonstrated that TLR8 ligand Poly-G3 treatment significantly inhibited the gene expression levels of enzymes involved in glucose metabolism, especially in glycolysis in Treg cells (Figure 3E). In support of the results shown in metabolic gene expression profiles and transcriptome analyses of Treg cells, we further observed that both nTreg and tumor-derived Treg cells (CD4 TregE1 and γδ Treg 31) highly consumed glucose in the medium during 3 days of culture compared with the control CD4+ T cells, indicating a heightened metabolic activity. However, TLR8 ligand Poly-G3 treatment dramatically prevented glucose consumption by these Treg cells, suggesting that Poly-G3 may inhibit glucose metabolism in both human nTreg and tumor-derived Treg cells (Figure 3F). To further test this possibility, we then determined the glucose uptake ability of Treg cells in the presence and absence of TLR8 ligand, using a fluorescent glucose analog 2-NBDG labeling assay. Our results clearly showed that nTreg and tumor-derived Treg cells have a significantly higher glucose uptake than that of control CD4+ effector T cells (Figure 3G & 3H). However, Poly-G3 treatment, but not control Poly-T3, markedly decreased glucose uptake in Treg cells (Figure 3G & 3H). Notably, TLR8 ligand PolyG3 treatment did not suppress glucose consumption and uptake of normal CD4+ T cells, which is consistent with the results showing that Treg cells have more desire for glucose (Figures 1 & 2). These data indicate that reversal of human Treg cell suppression mediated by TLR8 signaling results from the inhibition of glucose metabolism in Treg cells.
TLR8 signaling down-regulates the expression and function of glucose transporters Glut1 and Glut3 in Treg cells
Given that the movement of glucose in and out of cells is controlled by facilitative glucose transporters (Gluts), we next determined whether the changes in glucose consumption in Treg cells mediated by TLR8 activation are due to alterations of Gluts (Jacobs et al., 2008; Wofford et al., 2008). Our transcriptome analyses suggested the changes of Glut genes in human nTreg cells after TLR8 activation (Supplemental Figure 4B), but we only detected significant gene expression levels of Glut1 and Glut3, but not other Glut family members in Treg cells using real-time PCR. Furthermore, Poly-G3 treatment significantly down-regulated gene expression levels of Glut1 and Glut3 in both nTreg and tumor-derived CD4 TregE1 and γδ Treg31 cells (Figure 4A). In contrast, Poly-G3 treatment did not affect Glut1 and Glut3 expression in control effector CD4+ T cells. We also confirmed that Poly-G3 treatment dramatically inhibited protein expression levels of Glut1 and Glut3 in nTreg cells (Figure 4B).
Figure 4. TLR8 signaling down-regulates the expression levels and membrane translocation of glucose transporters Glut1 and Glut3 in Treg cells.

(A) and (B) Poly-G3 treatment down-regulated gene (in A) and protein (in B) expressions of Glut1 and Glut3 in human Treg cells. Treg and control CD4+CD25− effector cells were treated with Poly-G3 (3 μg/ml) for 48 hours. Total RNA was isolated from the T cells and analyzed by real-time PCR. The expression levels of each gene were normalized to β-actin expression levels and adjusted to the levels in untreated T cells (in A). Treated nTreg cells were also determined for Glut1 and Glut3 protein expression using the flow cytometry analysis (in B). Data shown in histograms are representative of average of three independent experiments ± SD. *p<0.05 and **p<0.01, compared with the medium only group. (C) Decreased Glut1 and Glut3 protein expression was induced by Poly-G3 treatment in nTreg cells but not in control CD4+ T cells after 3-day culture. Glut1 and Glut3 (green) expression was determined by an indirect immunofluorescence assay with a confocal microscopy. Scale bar, 50 μm. Results shown in the right histograms are mean ± SD of fluorescence intensity (MFI) quantifications of glucose transporters from three independent experiments. *p<0.05 and **p<0.01, compared with the medium only group. (D) Poly-G3 treatment down-regulated Glut1 and Glut3 membrane expression and promoted its intracellular translocation in nTreg cells. Cell treatment and procedure were identical to (C). Percentages of glucose transporter expression in cell membrane or intracellular were counted and shown in the right histograms. Scale bar, 25 μm. Results are mean ± SD of positive cells from three independent experiments. **p<0.01, compared with the medium only group. (E) and (F) Inhibition of glucose transport significantly promoted the Poly-G3-mediated reversal of Treg suppression on responder T cell proliferation (in E) and induction of cell senescence (in F). nTreg cells were pretreated with or without glucose transporter inhibitor phloretin (2 μM) for 2 days, and then co-cultured with naive CD4+ T cells in the presence or absence of Poly-G3 (3 μg/ml) for 3 days. Proliferation of co-cultured naïve T cells stimulated with anti-CD3 antibody was determined by [3H]-thymidine incorporation assays (in E), and SA-β-Gal expression in treated T cells was determined (in F). Data shown are mean ± SD from three independent experiments with similar results. **p<0.01 between the comparison groups.
In addition to expression levels, the localization, intracellular trafficking and translocation of these Gluts are critical for cell glucose uptake (Jacobs et al., 2008; Wofford et al., 2008). We then investigated whether TLR8 signaling affects Glut protein localization, trafficking, and translocation in Treg cells using the immunofluorescence analyses with a confocal microscopy. Consistent with above results, we observed that nTreg cells exhibited high fluorescence intensities in cellular membranes of Glut1 and Glut3 compared with control effector T cells, suggesting high membrane expression of Glut1 and Glut3 in nTreg cells (Figure 4C). However, the fluorescence intensities of Glut1 and Glut3 expression in cellular membranes were obviously decreased in nTreg cells after Poly-G3 treatment. Besides the downregulation of membrane expression of Glut1 and Glut3, Poly-G3 treatment dramatically promoted the translocation of Glut1 and Glut3 from cellular membranes to intracellular storage sites (Figure 4D). In contrast, control Poly-T3 treatment did not change the membrane expression and translocation of Glut1 and Glut3 in Treg cells. Furthermore, Poly-G3 treatment did not affect the expression levels and intracellular translocation of Glut1 and Glut3 in control effector CD4+ T cells (Figure 4C and Supplemental Figure 4C). To further investigate the importance of Glut on biological functions of Treg cells, we utilized the pharmacological Glut inhibitor phloretin and tested whether blockage of Glut function can reverse Treg suppressive activities. We found that treatment of Treg cells with phloretin significantly reversed Treg suppression on responder T cell proliferation and prevented the induction of senescence in responder T cells (Figure 4E and 4F). In addition, combined treatments of Treg cells with phloretin and Poly-G3 markedly increased the effects of Poly-G3-mediated reversal of Treg cell suppression on responder T-cell proliferation and decrease of senescence in responder T cells (Figure 4E and 4F). Collectively, these results indicate that effect on human Treg suppression and glucose metabolism medicated by TLR8 signaling is partially due to the alterations of Glut expression and function within human Treg cells.
TLR8 signaling suppresses metabolic processes of glycolysis in Treg cells
Given that activated human Treg cells have high expression levels of glycolysis-related enzymes (Figures 1 & 2), we reasoned that TLR8 signaling may also inhibit glycolysis in Treg cells, resulting in the reversal of their function. We first determined the potential suppression of key enzymes encompassing the entire spectrum of the glycolytic pathway, including HK2, PFK1, GPI, TPI, ENO1, PKM, and LDHα in human Treg cells during TLR8-mediated their function reversal (Shi et al., 2011). We found that TLR8 ligand Poly-G3, but not control Poly-T3 treatment markedly inhibited the expression of major glycolytic enzymes in both human nTreg and tumor-derived Treg cells (Figure 5A). We also determined whether TLR8 signaling changes the enzymes involving in the cholesterol and fatty acid synthesis and oxidation, since lipid metabolism is also important for Treg suppression. However, we did not observe significant effects mediated by Poly-G3 on lipid metabolism-related enzyme expression in Treg cells, except partial inhibition of ACC1 in nTreg and CD4 TregE1 but not in γδ Treg31 cells (Supplemental Figure 5A). We next determined the synergetic effects of glycolysis inhibitors and PolyG3 on the reversal of Treg functions. Consistent with the previous results shown in Figure 1F and 1G, blockage of glycolysis in Treg cells using specific pharmacological inhibitors 2-DG, LND and 3-BrPA can significantly block Treg suppressive effects on cell proliferation and senescence induction in responder T cells (Figure 5B and 5C). Furthermore, combined use of these inhibitors and Poly-G3 dramatically increased the effects of Poly-G3-mediated reversal of Treg suppression on responder T-cell proliferation and senescence induction (Figure 5B & 5C, and Supplemental Figure 5B). However, combination with lipid metabolism-related inhibitors including etomoxir, C75, orlistat, 25-HC simvastatin had no synergetic effects on Poly-G3-mediated reversal of Treg inhibitory functions (Supplemental Figure 5B–5D). These results clearly indicated that TLR8 signaling inhibits glycolysis of human Treg cells, resulting in their function reversal.
Figure 5. TLR8 signaling inhibits molecular processes of glycolysis in Treg cells.

(A) Poly-G3 treatment significantly down-regulated gene expression levels of key glycolytic enzymes in both nTreg and tumor-derived Treg cells. Different types of human Treg cells and control effector CD4+ T cells were treated with or without Poly-G3 or Poly-T3 for 48 hours. Total RNA was isolated from the T cells and analyzed by real-time PCR. The expression levels of each gene were normalized to β-actin expression levels and adjusted to the levels in untreated T cells (medium). Data shown in nTreg and control CD4+ T cells are mean ± SD from four independent donors. Data for CD4 TregE1 and γδ Treg31 are averages of three independent experiments. *p<0.05 and **p<0.01, compared with the medium only group. (B) and (C) Blockage of glycolysis in nTreg cells using specific pharmacological inhibitors dramatically enhanced the effects of Poly-G3-mediated reversal of Treg suppression on responder T cell proliferation (in B) and induction of cell senescence (in C). nTreg cells were pretreated with glycolysis inhibitors, including 2-DG (1 mM), LND (125 μM), and 3-BrPA (30 μM), respectively for 48 hours. Naïve CD4+ T cells were then co-cultured with inhibitor-pretreated or untreated Treg cells for 3 days in the presence or absence of Poly-G3. Proliferation of co-cultured naïve T cells stimulated with anti-CD3 antibody was determined by [3H]-thymidine incorporation assays (in B), and SA-β-Gal expression in treated T cells was determined (in C). Data shown are mean ± SD from representative of three independent experiments with similar results. **p<0.01 and #p<0.01, compared with the respective medium only group. (D) TLR8 signaling activation decreased the key metabolites involved in glycolysis and TCA in nTreg cells. nTreg cells were cultured in T cell medium in the presence of Poly-G3 or Poly-T3 for 72 hours. Glucose metabolites from the nTreg cell lysates were analyzed using a LC-triple quadruple mass spectrometry, and metabolite levels are normalized to medium group. Relative levels of intermediate metabolites in the glycolysis and TCA-cycle pathways are shown. Data shown are mean ± SD from representative of three independent nTreg cells with similar results. *p<0.05 and **p<0.01, compared with the medium only group.
Hypoxia-derived metabolites, such as adenosine and lactate are potent immune suppressors that can protect tumor cells from T cell mediated anti-tumor immune responses (Ohta et al., 2006; Sitkovsky et al., 2008; Xia et al., 2017). We have demonstrated that tumor-derived cAMP is responsible for tumor cell-induced T cell senescence (Ye et al., 2014). Therefore, we determined whether TLR8-mediated reversal of Treg suppression is involved in the changes of glucose metabolites from Treg cells. We performed glucose metabolomic analyses to determine the metabolites related to glycolysis and TCA pathways in human Treg cells treated with or without TLR8 signaling using a LC-triple quadruple mass spectrometry as described in Supplemental Figure 1C. Consistent with the results shown in Figure 1E, some key metabolites significantly increased in the cell lysates in nTreg cells (Figure 5D). However, similar changes as shown in metabolic enzymes mediated by TLR8 signaling, glycolytic metabolites including 1, 3-bisphosphoglycerate (1, 3-BPG), phosphoenolpyuvate (PEP), pyruvate and lactate were dramatically decreased after TLR8 ligand Poly-G3 treatment. In addition, some down-stream metabolites related to the TCA cycle, including citrate, α-ketoglutatrate and succinate were also down-regulated in Treg cells mediated by Poly-G3 treatment (Figure 5D). These suggest that TLR8 signaling not only inhibits glycolysis but also decreases glucose metabolites of human Treg cells that results in the reversal of Treg suppressive functions.
TLR8 activation down-regulates mTORC1-HIF1α signaling in Treg cells that controls molecular processes of Treg glucose metabolism and suppressive functions
Recent studies have demonstrated that mTOR kinase signaling regulates Treg cell lineage commitment and differentiation (Delgoffe et al., 2009; Shi et al., 2011; Zeiser et al., 2008; Zeng et al., 2013). Furthermore, mTOR signaling plays a critical role in regulation of glucose uptake and energy balance, and subsequently directs cell growth and proliferation (Dukhande et al., 2011; Mori et al., 2009). We therefore dissected the potential mechanistic crosstalk between the TLR8 and mTOR signaling in Treg cells. Transcriptome analyses of human nTreg cells after TLR8 ligand Poly-G3 treatment showed that Poly-G3 treatment induced significant alterations in 24 genes/adaptors involved in the upstream or downstream of mTOR signaling (Figure 6A). To further confirm whether mTOR signaling is involved in TLR8-mediated reversal of Treg cell suppression, we analyzed the phosphorylation and activation of mTOR and its downstream substrates p70S6K and 4E-BP1 in Treg cells activated with or without TLR8 signaling. We found that nTreg cells have higher phosphorylated mTOR and p70S6K cell populations than that of control effector T cells, indicating elevated steady-state mTORC1 activity in Treg cells. In addition, treatment with Poly-G3 significantly suppressed the phosphorylation of mTOR and its downstream substrates p70S6K and 4E-BP1 in Treg cells, but not in effector CD4+ T cells, further confirming the inhibition of mTOR signaling in Treg cells after TLR8 activation (Figure 6B). We then determined whether TLR8 signaling in Treg cells directly regulates the mTOR signaling pathway. We have previously identified p38α MAPK as a key molecule in TLR8 signaling pathway (TLR8-MyD88IRAK4-TRAF6-p38 signaling) controlling the reversal of Treg suppression (Peng et al., 2005; Peng et al., 2007; Ye et al., 2014). We therefore used a specific pharmacological inhibitor SB203580 against p38 to test the involvement of p38 in regulating mTOR signaling in Treg cells (Peng et al., 2005; Peng et al., 2007). We observed that blockage of p38 signaling in nTreg cells completely reversed the downregulated phosphorylation of mTOR signaling mediated by Poly-G3, suggesting that p38 MAPK controls mTOR signaling (Figure 6C). We then confirmed the functional importance of mTOR signaling in regulating reversal of Treg activities induced by TLR8 signaling. We showed that the mTOR inhibitor rapamycin markedly abolished Treg activities of suppression on T cell proliferation and promotion of senescence induction in responder T cells (Figure 6D & 6E). Furthermore, combined treatments with rapamycin and Poly-G3 significantly increased the effect of Poly-G3-mediated reversal of Treg cell suppression. In addition, we performed a functional rescue experiment with over-expression of the mTOR upstream activator RHEB gene in Treg cells (retrovirus-based RHEB linked to GFP)(Peng et al., 2005; Peng et al., 2007; Yang et al., 2017). Activation of mTOR signaling with Retro-RHEB transfection significantly increased gene expression levels of glucose transporters and glycolytic enzymes in Treg cells, and promoted Treg suppression of responder T cell proliferation and senescence induction (Figure 6F & 6G and Supplemental Figure 6A). However, mTOR signaling activation markedly prevented the Poly-G3-mediated reversal of Treg suppressive activities on responder T cell proliferation and generation of cell senescence, as well as on TLR8 signaling-mediated inhibition of glycolysis (Figure 6F & 6G and Supplemental Figure 6A). These results indicate that mTOR signaling pathway is critical and involved in regulating TLR8-mediated reversal of Treg suppression.
Figure 6. TLR8 activation down-regulates mTORC1-HIFα signaling in Treg cells that controls the molecular processes of Treg glucose metabolism and suppressive functions.

(A) Significant alterations in 24 genes involved in the mTOR signaling pathway were identified and ranked in nTreg cells after treatment with or without TLR8 ligand Poly-G3 at 24 hours. Gene alterations were normalized to log2 expression level. Human nTreg cells were isolated from PBMCs of two healthy donors and treated with Poly-G3 for different time points. Total RNA was purified and transcriptome analyses of Treg cells were performed using the Illumina whole-genome Human HT-12 BeadChips. (B) Suppression of phosphorylation and subsequent activation of mTOR signaling in Treg cells treated with Poly-G3. Treg cells were treated with or without Poly-G3 (3 μg/ml) for 30 minutes and then phosphorylated mTOR, p70S6K, and 4E-BP1 in Treg cells were determined by the flow cytometry. Protein levels shown in the right histograms are summarized as the mean ± SD from three independent experiments. **p<0.01 compared with the medium only group. (C) Blockage of p38 signaling prevented TLR8-mediated inhibition of mTOR signaling. nTreg cells were treated with p38 inhibitor SB203580 (10 μM) in the presence or absence of Poly-G3 (3 μg/ml) for 30 minutes and then phosphorylated mTOR and p70S6K in Treg cells were determined by the flow cytometry. Results shown in the right histogram are summarized as the mean ± SD from three independent experiments. **p<0.01 compared with the other treatment groups. (D) and (E) Blockage of mTOR signaling with mTOR inhibitor rapamycin partially reversed Treg suppression and promoted the Poly-G3-mediated reversal of Treg suppressive activities on responder T cell proliferation (in D) and induction of cell senescence (in E). nTreg cells were pretreated with or without rapamycin (300 nM) for 1 day, and then co-cultured with naive CD4+ T cells in the presence or absence of Poly-G3 (3 μg/ml) for 3 days. Proliferation of co-cultured naïve T cells stimulated by anti-CD3 antibody was determined by [3H]-thymidine incorporation assays (in D), and SAβ-Gal expression in treated T cells was determined (in E). Data shown are mean ± SD from representative of three independent experiments. **p<0.01 between the comparison groups. (F) and (G) Activation of mTOR signaling with Retro-RHEB transfection promoted Treg suppression and prevented the Poly-G3-mediated reversal of Treg suppressive activities on responder T cell proliferation (in F) and induction of cell senescence (in G). Activated nTreg cells were infected with retrovirus carrying RHEB gene or control vector for 48 hours. Infected Treg cells were then co-cultured with naive CD4+ T cells in the presence or absence of Poly-G3 (3 μg/ml) for 3 days. Proliferation and SA-β-Gal expression of cocultured naïve T cells were determined as above. Data shown are mean ± SD from three independent experiments with similar results. *p<0.05 and **p<0.01 between the comparison groups. (H) Relative expression levels of HIF1α in different T cell subsets before and after anti-CD3 stimulations. TCR-activated Treg cells showed significantly elevated HIF1α expression compared with that of effector T cells. T cells were stimulated with or without anti-CD3 for 8 hours and total RNA was isolated from each cell type and analyzed by real-time PCR. Expression level of HIF1α was normalized to β-actin expression and adjusted to the level in naïve CD4+ T cells (served as 1). Data shown are mean ± SD from four independent healthy donors. (I) Activated tumor-derived CD4+ Treg and γδ Treg cells also had higher HIF1α gene expression than activated Th1 cells. Cell treatment and assays were the same as in (H). Data shown are mean ± SD from three independent experiments with similar results. (J) and (K) Poly-G3 treatment down-regulated HIF1α mRNA expression in human Treg cells. nTreg and tumorderived Treg cells, and control CD4+ T cells were treated with Poly-G3 (3 μg/ml) or Poly-T3 for 48 hours. Total RNA was isolated from the T cells and analyzed by real-time PCR. The gene expression levels of HIF1α were normalized to β-actin expression levels and adjusted to the levels in untreated T cells. Data shown in histograms are representative of mean ± SD from three independent experiments. *p<0.05 and **p<0.01, compared with the medium only group. (L) and (M) Inhibition of HIF1α signaling alleviated Treg suppression and significantly promoted the Poly-G3-mediated reversal of Treg suppressive activities on responder T cell proliferation (in L) and induction of cell senescence (in M). nTreg cells were pretreated with or without HIF1α inhibitors YC-1 (5 μM) or 2-ME (10 μM) for 1 day, and then co-cultured with naive CD4+ T cells in the presence or absence of Poly-G3 (3 μg/ml) for 3 days. Proliferation and SA-β-Gal expression of co-cultured naïve T cells were determined as above. Data shown are mean ± SD from three independent experiments with similar results. *p<0.05 and #p<0.01, compared with the respective medium only group. (N) and (O) Activation of HIF1α signaling in nTreg cells dramatically augmented Treg suppression and blocked the effects of Poly-G3-mediated reversal of Treg suppression on responder T cell proliferation (in N) and induction of cell senescence (in O). nTreg cells were pretreated with or without HIF1α activator DMOG (0.1mM) for 1 day and then co-cultured with naive CD4+ T cells in the presence or absence of Poly-G3 (3 μg/ml) for 3 days. Proliferation and SA-β-Gal expression of cocultured naïve T cells were determined. Data shown are mean ± SD from representative of three independent experiments with similar results. *p<0.05 and **p<0.01, between the comparison groups.
HIF1α serves as a key transcription factor that performs important functions in regulation of cellular metabolism. The mTORC1–HIF1α pathway also promotes glucose metabolism and glycolysis in Th17 cells and effector CD8+ T cells. We compared mRNA levels of HIF1α in different effector T cell subsets, nTreg and tumor-derived Treg cells. HIF1α expression levels in nTreg cells were much higher than that of effector T cell subsets. Furthermore, activated nTreg cells with anti-CD3 stimulation significantly increased HIF1α expression (Supplemental Figure 6B & 6C, and Figure 6H). Freshly-isolated nTreg cells and expanded Treg cells have similar HIF1α expression levels (Supplemental Figure 6D). In addition, tumor-derived CD4+ Treg and γδ Treg cells were highly expressed HIF1α independent of cell activation status (Figure 6I). To test the hypothesis that TLR8-mediated reversal of Treg suppression involves the mTORC1–HIF1α pathway regulation, we compared gene expression profiles of HIF1α pathway in nTreg cells treated with or without poly-G3 using transcriptome analyses. We showed that most HIF1α-targeted genes, related to molecules in metabolism, cell growth and angiogenesis, were significantly down-regulated in nTreg cells after Poly-G3 treatment, suggesting down-regulation of HIF1α pathway in Treg cells by TLR8 signaling (Supplementary Figure 6E). Furthermore, Poly-G3 treatment markedly decreased the HIF1α mRNA expression in both nTreg and tumor-derived Treg cells (Figure 6J & 6K). In addition, we confirmed that Poly-G3 inhibited HIF1α in nTreg cells but not in effector CD4+ T cells in protein levels (Supplemental Figure 6F). To further determine the functional regulation of Treg activity by HIF1α, we blocked HIF1α expression and function with specific pharmacological inhibitors YC1 and 2-methoxyestradiol (2-ME) and then evaluated Treg suppressive activities on T cell proliferation and senescence induction (Mabjeesh et al., 2003; Sun et al., 2007). Treatments with HIF1α inhibitors YC-1 and 2-ME significantly alleviated the suppression on T cell proliferation and prevented senescence induction of responder T cells induced by Treg cells (Figure 6L & 6M). Furthermore, combined treatments with HIF1α inhibitors and Poly-G3 synergistically increased the Poly-G3 effects on the reversal of Treg cell suppressive activities on T cell proliferation and senescence induction (Figure 6L & 6M). In addition, we activated HIF1α function with the specific pharmacological activator dimethyloxalylglycine (DMOG) and then evaluated Treg suppressive activities in the presence of TLR8 signaling (Cosin-Roger et al., 2017; Zhdanov et al., 2015). Activation of HIF1α signaling with DMOG in nTreg cells dramatically augmented Treg suppression on responder T cell proliferation and induction of cell senescence (Figure 6N & 6O). However, DMOG treatment significantly blocked the effect of PolyG3-mediated reversal of Treg suppressive activities (Figure 6N & 6O). These results collectively suggest that TLR8 signaling modulates the mTOR-HIF1α axis and its downstream glycolytic program in human Treg cells, resulting in the reversal of Treg inhibitory functions.
TLR8 signaling-mediated reprogramming of glucose metabolism and function in Treg cells enhances anti-tumor immunity and tumor immunotherapy in vivo
We have demonstrated that human TLR8 signaling can reverse the suppressive functions of nTreg and tumor-derived γδ Treg cells in vivo in the established adoptive transfer models (Kiniwa et al., 2007; Peng et al., 2005; Peng et al., 2007; Ye et al., 2012; Ye et al., 2013). We next explored whether TLR8mediated reversal of Treg suppression in vivo is due to the inhibition of Treg glucose metabolism in the same adoptive transfer models as we previously described (Ye et al., 2012; Ye et al., 2013). Anti-CD3pre-activated naïve CD8+ T cells were co-transferred with expanded nTreg cells into NSG mice. In parallel, Treg cells were pretreated with TLR8 ligand Poly-G3, glycolytic metabolism inhibitor 2-DG, or HIF1α inhibitor 2-ME for 24 hours prior to adoptively transfer into the mice, and then Poly-G3 and inhibitors were continued to be intraperitoneally injected into mice for a total of 3 doses with 3-day intervals. Consistent with our previous findings, we showed that Treg cells can significantly suppress responder T cells in vivo, converting them into senescent T cells (SA-β-gal+) (Figure 7A). However, administration of Poly-G3 significantly blocked the induction of senescence in transferred CD8+ T cells (Figure 7B). Importantly, treatment with glycolytic metabolism inhibitor 2-DG, or HIF1α inhibitor 2-ME had similar effects as Poly-G3 on responder T cells that significantly blocked induction of cell senescence in transferred pre-activated CD8+ T cells co-injected with Treg cells in vivo (Figure 7B).
Figure 7. Reprogramming of glucose metabolism in Treg cells via TLR8 signaling activation or specific metabolic inhibition enhances anti-tumor immunity and tumor immunotherapy in vivo.

(A) Increased senescent cell populations were markedly induced in pre-activated CD8+ T cells after cotransfer with nTreg cells but not with control effector CD4+ T cells. Naïve CD8+ T cells (5 × 106/mouse), expanded nTreg (3 × 106/mouse) and CD4+CD25− T cells (3 × 106/mouse) were pre-activated with antiCD3 antibody and adoptively co-transferred into NSG mice. Blood and Spleens were harvested at 12 days post-injection. The transferred human CD8+ T cells were isolated for subsequent SA-β-Gal staining. **p<0.01, compared with the groups co-transferred with CD4+CD25− T cells or CD8+ T cells alone. (B) Treatment with Poly-G3, 2-DG, or 2-ME significantly prevented induction of senescence in transferred CD8+ T cells. nTreg cells were pretreated with Poly-G3 (3 μg/mL), glycolytic metabolism inhibitor 2-DG (1 mM) or HIF1α inhibitor 2-ME (10 μM) for 24 hours prior to adoptively transfer into the mice. After adoptive transfer of T cells into the mice, Poly-G3 (50 μg/mouse), 2-DG (10 mg /mouse), or 2-ME (0.3 mg /mouse) were intraperitoneally injected into mice for a total of 3 doses with 3-day intervals. Cell preparation and injection procedures were the same as in (A). The transferred human CD8+ T cells in different organs were isolated at 12 days post-injection for subsequent SA-β-Gal staining. **p<0.01, compared with the medium group. (C) and (D) Real-time PCR quantification of expression changes of glucose transporters and glycolytic enzymes in purified nTreg cells from blood and spleens of NSG mice treated with Ploy-G3 and different inhibitors. Cell treatment and adoptive transfer procedure were identical to (B). The transferred nTreg cells were isolated for RT-PCR analyses.*p<0.05 and **p<0.01, compared with the co-transferred group without inhibitor treatment. (E) to (G) Treatments with Poly-G3 and inhibitors prevented tumor-specific T cell senescence and enhanced antitumor immunity in NSG mice. Human 586mel tumor cells (5 × 106/mouse) were subcutaneously injected into NSG mice. Tumorspecific CD8+ TIL586 cells (5 × 106/mouse) were i.v. injected on day 5 with or without nTreg cells (3 × 106/mouse). In addition, nTreg cells were pretreated with TLR8 ligand Poly-G3, glycolytic metabolism inhibitor 2-DG and HIF1α inhibitor 2-ME for 24 hours prior to adoptively transfer into the mice, and mice were then intraperitoneally injected with Poly-G3, 2-DG or 2-ME for a total of 3 doses with 3-day intervals after T cell transfer. The treatment procedures and doses were identical to the experiments in (B). Tumor volumes were measured and presented as mean ± SD (in E) (n=5 mice per group). Blood, spleens, and tumors were harvested at day 33 post injection. The transferred human TIL586 T cells in different organs were isolated for SA-β-Gal staining. *p<0.05 and **p<0.01, compared between the comparison groups (in E), or compared with the groups of TIL586 alone (in F) or co-transferred group without inhibitor treatment (in G), respectively. (H) to (J) Real-time PCR quantification of glucose transporter and glycolytic enzyme expression changes in purified Treg cells from TILs, blood and spleens of tumor-bearing NSG mice treated with Ploy-G3 and different inhibitors. Cell treatment and adoptive transfer procedure were identical to (E). The transferred nTreg cells were isolated from different organs and tumor tissues for RT-PCR analyses.*p<0.05 and **p<0.01, compared with the co-transferred group without inhibitor treatment.
To further determine whether the reversal of Treg suppressive activity mediated by TLR8 signaling in vivo is also due to the inhibition of Treg metabolism as shown in vitro studies, we harvested the adoptively transferred Treg cells from blood and spleens in NSG mice and glucose metabolic gene expression profiles determined. Consistent with the results shown in in vitro studies, we observed that administration of TLR8 ligand Poly-G3 strongly inhibited gene expression of Glut1, Glut3, GPI, PFK1 and ENO1 in recovered Treg cells in vivo (Figure 7C & 7D). We obtained very similar metabolic gene expression profiles as shown in Poly-G3 treatment that treatments with 2-DG and 2-ME also inhibited Treg glucose metabolism in vivo (Figure 7C and 7D). We also explored whether TLR8 ligand Poly-G3 treatment could suppress the metabolism of effector responder T cells in vivo. We first tested this possibility in vitro in co-cultures and found that responder CD8+ T cells co-cultured with nTreg cells had significantly increased gene expression levels of Glut transporters and glycolytic enzymes, further suggesting that senescent T cells have an active glucose metabolism (Supplemental Figure 7A). Furthermore, Poly-G3 treatment did not induce suppression of metabolic gene expression in responder CD8+ T cells co-cultured with nTreg cells. Interesting, even treatment with glycolytic metabolism inhibitor 2-DG, or HIF1α inhibitor 2-ME, the co-cultured responder CD8+ T cells still had higher gene expression levels of the key metabolic molecules than those of normal CD8+ T cells (Supplemental Figure 7A). These in vitro in co-culture results were further confirmed in vivo in adoptive transferred CD8+ T cells in mice. In contrast to the transferred nTreg cells, administrations with Poly-G3, 2-DG, or 2-ME did not induce significant suppression on gene expression of metabolic molecules in CD8+ T cells co-transferred with Treg cells in vivo (Supplemental Figure 7B). Collectively, these results indicate that human Treg cells can convert responder T cells into senescent T cells both in vitro and in vivo, and that TLR8 signaling can specifically inhibit Treg but not responder T cell glucose metabolism, resulting in prevention of Treg-mediated induction of T-cell senescence and subsequent immune suppression.
We next investigated whether reprogramming of Treg metabolism via TLR8 activation or glucose metabolism blockade can enhance anti-tumor immunity in the humanized NSG model. We used our established adoptive T cell transfer immunotherapy melanoma model to test our hypothesis (Peng et al., 2005; Peng et al., 2007; Ye et al., 2014). Human 586mel tumor cells were subcutaneously injected into NSG mice on day 0. Tumor-specific CD8+ TIL586 T cells (which recognize and kill autologous 586mel tumor cells) were adoptively co-transferred through i.v. injection on day 5 with or without Treg, followed by intrapeaneral injection of Poly-G3, 2-DG or 2-ME. We found that 586 melanoma cells grew progressively in NSG mice. While adoptive transfer of tumor-specific CD8+ TIL586 T cells, tumor growth was significantly inhibited. However, co-transfer of CD8+ TIL586 T cells and expanded nTreg cells, tumor growth was markedly promoted, suggesting that Treg cells inhibit anti-tumor immunity by tumor-specific T cells (Figure 7E). Importantly, Poly-G3 treatment dramatically promoted the inhibition of tumor growth mediated by tumor-specific TIL586 T cells, suggesting that Poly-G3 treatment enhances the anti-tumor ability mediated by TIL586. In addition, administrations of 2-DG and 2-ME also enhanced the anti-tumor ability mediated by TIL586 T cells, markedly suppressing tumor growth (Figure 7E). Notably, 2-DG treatment has a very similar effect on tumor growth inhibition as Poly-G3. To confirm the molecular processes responsible for anti-tumor effects mediated by TLR8 signaling, we isolated both Treg cells and tumor-specific TIL586 T cells from blood, spleens and tumor tissues, and their phenotypes and functions were determined. We found that high percentages of senescent T cells were also induced in transferred CD8+ TIL586 cells recovered from different organs and tumor tissues in the tumor-bearing NSG mice, suggesting that tumor cells also induce T cell senescence (Figure 7F). Furthermore, co-transfer with Treg cells significantly increased the senescent T populations in transferred CD8+ TIL586 cells. However, intraperitoneal administrations with Poly-G3, 2-DG or 2-ME dramatically decreased senescence in transferred CD8+ TIL586 cells in the 586mel-bearing NSG mice (Figure 7G). We also confirmed gene expression profiles of Treg glucose metabolism and demonstrated that treatments with Poly-G3, 2-DG or 2-ME significantly suppressed glucose transporters and glycolytic enzymes in Treg cells recovered from tumor, blood and spleens in the tumor-bearing mice (Figure 7H, 7I & 7J). In addition, treatments with Poly-G3, 2-DG or 2-ME markedly reversed Treg suppressive activities (Supplemental Figure 7C & 7D). These results further indicate that reprogramming of glucose metabolism and function in Treg cells via TLR8 signaling is an effective strategy that reverses Treg-mediated immune suppression and enhances anti-tumor immunity and tumor immunotherapy.
Discussion
It has become clear that tumor-infiltrating Treg cells induce an immunosuppressive microenvironment that is a major obstacle for successful tumor immunotherapy (Curiel, 2008; Wang et al., 2004; Zou, 2006). Given the importance of metabolism in directing T cell fate and functions, a better understanding of Treg metabolism, especially tumor-associated Treg cells, should provide alternative novel strategies for tumor immunotherapy. In the current study, we demonstrated that human naturally occurring Treg and tumorassociated Treg cells display heightened glucose consumption compared with effector T cells that molecularly triggers cell senescence and suppression in responder T cells during their cross-talk. Our previous studies have shown that human TLR8 signaling functionally inactivates Treg cells without changing the Treg repertoire or inhibiting effector T cell functions (Kiniwa et al., 2007; Peng et al., 2005; Peng et al., 2007). Here we further discovered that TLR8 signaling-mediated reversal of human Treg suppression is due to selective inhibition of glucose uptake and metabolic processes of glycolysis in human Treg cells. Importantly, using the melanoma adoptive transfer immunotherapy model, we further established our novel concept that reprogramming of Treg metabolism and function via manipulating TLR8 signaling and/or glucose metabolic processes is a novel immunotherapeutic strategy for human cancer treatment.
Increasing evidence suggests that each T cell subset requires specific metabolic programs to meet their distinct developmental and functional processes. Recent studies have shown that metabolic regulation of Treg differentiation, Foxp3 expression, lineage stability and homeostasis involves both glycolysis and lipid metabolism (Dang et al., 2011; De Rosa et al., 2015; Michalek et al., 2011; Newton et al., 2016; Procaccini et al., 2016; Shi et al., 2011; Zeng et al., 2013). However, the active metabolic profiles and regulations in human Treg cells are still unclear. Furthermore, metabolic regulation of human Treg functionality, especially in tumor-associated Treg cells is unknown, which should be more critical for the development of effective immunotherapeutic strategies against cancer (Biswas, 2015; Chang et al., 2015). To address these challenging issues, our current studies clearly demonstrate that both naturally occurring Treg and tumor-associate Treg cells display more active glycolytic and lipid metabolism compared with other T cell subsets. Functional assays further suggest that both glucose and lipid metabolism are critical for human Treg cells performing their suppressive activities. However, after TCR activation, human Treg cells exhibit more glycolysis but not lipid metabolism compared with any other Th subsets, although activated Treg cells still have a high level of lipid metabolism. The metabolic profiles of human Treg cells are distinct from murine Treg cells shown to use lipid oxidation as a primary metabolic pathway (Buck et al., 2016; Michalek et al., 2011; Pearce et al., 2009; Shi et al., 2011). Moreover, mTORC1 was found to be essential for mouse Treg-cell homeostasis and suppressive function via promotion of lipid and cholesterol biosynthesis (Zeng et al., 2013). Furthermore, mitochondrial respiratory and oxidative phosphorylation (OXPHOS) are also critical for mouse Treg cell proliferation and suppressive function (Beier et al., 2015). Our current human Treg studies clearly indicate that metabolic control of human Treg cell suppression, especially glycolysis inhibition, is a potential novel concept for anti-tumor immunotherapy in the future.
We have recently identified that senescence induction in responder T cells is a novel suppressive mechanism mediated by human Treg cells (Ye et al., 2012; Ye et al., 2013). Importantly, these senescent T cells have potent suppressive activity on other immune cells amplifying immune suppression in the tumor microenvironment (Ye et al., 2012; Ye et al., 2014; Ye and Peng, 2015). Therefore, dissecting the molecular mechanism utilized by Treg cells to induce senescence and dysfunction in T cells should provide insights for developing novel immunotherapies against cancer. Recent studies suggest that tumor cells and TILs compete for glucose within the tumor suppressive microenvironment, which is a driver for tumor suppression and progression (Chang et al., 2015; Sukumar et al., 2015). Given that heightened glycolysis of Treg cells similar to tumor cells, we therefore presumed that the lack of enough glucose highly consumed by Treg cells may result in responder T cell senescence. As expected, our studies clearly demonstrated that shortage of glucose significantly promotes cell cycle regulatory gene expression and ATM-associated DNA damage response in T cells, which is the key molecular process of cellular senescence (Liu et al., 2018; Rodier et al., 2009; Van Nguyen et al., 2007). Furthermore, our studies also confirmed that human Treg cells can trigger the DNA damage response and senescence in responder T cells through glucose competition (Liu et al., 2018). In addition, the addition of glucose can rescue and prevent Treg-mediated senescence induction in responder T cells. Collectively, our current studies identify the causative link between Treg metabolism and the molecular process on responder T cell fate and function mediated by glucose competition/deprivation during their cross-talk and interactions.
In our efforts to develop strategies to reverse Treg suppression for tumor immunity, we have discovered that human TLR8 signaling, but not other TLRs, can reverse the suppressive functions of tumor-derived CD4+, CD8+ and γδ Treg cells, resulting in enhanced anti-tumor immunity (Kiniwa et al., 2007; Peng et al., 2005; Peng et al., 2007; Ye et al., 2012; Ye et al., 2013). Our more recent studies have shown that TLR8 signaling activation in human Treg cells and tumor cells can prevent their induction of senescence in responder T cells and DCs (Ye et al., 2012; Ye et al., 2014; Ye et al., 2013). Therefore, defining the molecular mechanisms and unique signaling pathways involved in TLR8 signaling regulation of Treg functions will be critical for development of therapeutic interventions. More recent studies suggest that TLR1 and TLR2 signaling activation in mouse Treg cells increases Treg glycolysis and proliferation and reduces their suppressive capacity (Gerriets et al., 2016). Given that TLR8 is nonfunctional in mice and that TLR1 and TLR2 signaling activation could not reverse the suppressive function of human Treg cells, these studies further suggest the distinct regulations between mouse and human Treg cells. Based on the transcriptome analysis and metabolic profiles of human Treg cells, we hypothesized that TLR8-mediated reversal of Treg suppression is due to the regulation of energy metabolism in human Treg cells. In this study, we clearly showed that TLR8-mediated Treg functional reversal occurs through the comprehensive inhibition of glucose metabolism in Treg cells in the following three metabolic processes. First, TLR8 signaling significantly inhibits glucose uptake in human Treg cells. This effect is molecularly due to the down-regulation of the membrane expression of glucose transporters Glut1 and Glut3 in Treg cells induced by TLR8 activation. Besides inhibition of Glut1/3 expression, TLR8 signaling dramatically promotes the translocation of Glut1 and Glut3 from cellular membranes into intracellular storage sites, resulting in decreased Glut biological function for glucose transport into Treg cells (Jacobs et al., 2008; Wofford et al., 2008). Secondly, TLR8 signaling activation markedly suppresses glycolysis in Treg cells via down-regulating the key enzymes encompassing the glycolytic pathway, including HK2, PFK1, GPI, TPI, ENO1, PKM2, and LDHα in human Treg cells. Third, our glucose metabolomic analyses further suggested that TLR8 signaling decreases glucose metabolites related to glycolysis and TCA pathways of human Treg cells. All of this evidence strongly supports our prediction and hypothesis that TLR8-mediated reversal of Treg suppression is mechanistically due to inhibition of glucose metabolism in human Treg cells.
In addition, our current study answers another challenging question of what unique signaling pathway(s) are regulated by TLR8 signaling, leading to the reversal of human Treg suppression and selective inhibition on Treg metabolism and function. mTOR kinase signaling regulates decisions between effector and Treg cell lineage commitment (Dang et al., 2011; Delgoffe et al., 2009; Shi et al., 2011; Zeiser et al., 2008; Zeng et al., 2013). Furthermore, mTOR signaling pathway plays a critical role in regulating glucose uptake and energy balance, and subsequently directs cell growth and proliferation (Buller et al., 2008; Dukhande et al., 2011; Macintyre et al., 2014; Mori et al., 2009; Shi et al., 2011). Glycolysis is also regulated by HIF1α (Semenza, 2011). The activity of mTORC1 enhances HIF1α expression at both the transcriptional and translational levels, and thereby stimulates glycolysis and glucose transport, responsible for the glycolytic response downstream of mTOR. mTORC1–HIF1α pathway upregulates glucose metabolism, and differentiation and function in Th17 cells (Dang et al., 2011; Shi et al., 2011). However, limited information has been generated regarding the potential links between TLR and mTOR-HIFα signaling pathways (Cao et al., 2008; Geng et al., 2010). As expected, we found that TLR8 signaling could significantly decrease the phosphorylation and activation of mTOR signaling, as well as down-regulate HIF1α expression in Treg cells. Our results collectively indicate that TLR8 signaling molecularly modulates the mTOR-HIF1α axis and its downstream glycolytic program in human Treg cells, resulting in the reversal of Treg inhibitory functions. These novel studies identify the mechanistic links between TLR signaling, T-cell energy metabolism, mTOR signaling, and Treg cell suppression, which should potentially lead to novel strategies capable of augmenting immune responses directed against cancer.
Our previous and current studies strongly support the concept that reprogramming/rebalancing energy metabolism in Treg cells within tumor microenvironments is potentially a novel strategy for tumor treatment. Our current studies have molecularly addressed the previous concern of why human TLR8 signaling uniquely inactivates Treg cells without changing the Treg repertoire or inhibiting effector T cell functions (Kiniwa et al., 2007; Peng et al., 2005; Peng et al., 2007). Our in vitro studies identify that TLR8-mediated reversal of Treg suppression is mechanistically dependent on inhibition of Treg glucose metabolism. Based on the metabolic profiles of different types of human T cells in this study, it is easy to understand that TLR8 signaling selectively inhibits Treg cells with heightened glucose metabolism. However, a significant concern remains that broad inhibition of glucose metabolism in effector T cells could be induced by pharmaceutical inhibitors including TLR8 ligands. Interestingly, in our efforts exploring whether TLR8 ligand Poly-G3 treatment could suppress the metabolism of effector responder T cells, we unexpectedly discovered that responder CD8+ T cells co-cultured with Treg cells had significantly increased gene expression levels of Glut transporters and glycolytic enzymes, further suggesting that senescent T cells have an active glucose metabolism. This result is also supported by our recent studies showing that senescent T cells induced by Treg cells are functionally active, secreting proinflammatory and suppressive cytokines that are distinct from exhausted or anergic T cells(Liu et al., 2018). Importantly, our in vivo studies clearly demonstrated that reprogramming of Treg metabolism via TLR8 activation or glucose metabolism blockade (2-DG and 2-ME treatments) can enhance anti-tumor immunity in the humanized NSG model. Our in vivo analyses of metabolic profiles of Treg cells and tumor-specific T cells further confirmed that treatments with Poly-G3, 2-DG, or 2-ME only suppressed glucose transporters and glycolytic enzymes in Treg cells but not in tumor-specific T cells in tumorbearing mice. These studies strongly indicate the feasibility of development of targeted Treg metabolism and function for anti-tumor immunotherapy.
In summary, we identified the metabolic profiles of different human T cell subsets and further demonstrated that glycolysis is more critical for Treg suppressive function. We further dissected the unique molecular mechanism of the TLR8-mediated reversal of Treg cell suppression involved in inhibition of glucose metabolism and mTOR-HIF1α signaling in human Treg cells. Importantly, our studies established the proof-of-concept that reprogramming human Treg cell metabolism by manipulating TLR8 signaling and glucose metabolic processes represents a novel immunotherapeutic strategy to treat human cancers.
Limitations of the Study
While our studies provide compelling evidence that TLR8 signaling selectively inhibits glucose uptake and glycolysis in human Treg cells, resulting in reversal of Treg suppression, this novel concept has not been validated in a real tumor suppressive microenvironment. Because TLR8 is nonfunctional in mice, a suitable human TLR8 transgenic mouse model is essential for future studies to explore the translational potential of this novel immunotherapeutic strategy. Furthermore, the limited cell numbers of human Treg cells has restricted the scope of our work, especially in the tumor-associated Treg cells established from the TILs of cancer patients. In vitro expanded human Treg cells have been utilized in some experiments in this study, which may have metabolic differences compared with primary Treg cells in the tumor microenvironment. In addition to cell numbers, the limited availability of fresh human tissue samples used to purify tumor-derived Treg cells is another restriction for our studies. Our current studies investigated several human tumor-derived Treg cell lines from melanoma and breast cancer patients regulated by TLR8 signaling. However, more studies with human Treg cells directly purified from fresh tumor tissues from different types of cancer patients should be included in future studies to explore the generality and applicability of this novel mechanism and therapeutic strategy.
STAR ★ METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE Mouse Anti-Human CD25 (Clone: M-A251) | BD Biosciences | Cat# 555432; RRID:AB_395826 |
| Anti-human CD3 (Clone OKT-3) | Bio X Cell | Cat# BE0001–2; RRID:AB_1107632 |
| Anti-human CD28 (Clone 9.3) | Bio X Cell | Cat # BE0248; RRID:AB_2687729 |
| Anti-IL-4 neutralizing antibody (Clone MP4–25D2) | Bio X Cell | Catalog # BE0240; RRID:AB_2687722 |
| Anti-IFN−γ neutralizing antibody (Clone 133.5) | Bio X Cell | Cat # BE0235; RRID:AB_2687717 |
| Phospho-mTOR (Ser2448) Antibody | Cell Signaling Technology | Cat# 2971; RRID:AB_330970 |
| Phospho-p70 S6 Kinase (Thr389) (108D2) Rabbit mAb | Cell Signaling Technology | Cat# 9234; RRID:AB_2269803 |
| Phospho-4E-BP1 (Thr37/46) (236B4) Rabbit mAb | Cell Signaling Technology | Cat# 2855; RRID:AB_560835 |
| Glut1 (H-43) antibody | Santa Cruz Biotechnology | Cat# sc-7903; RRID:AB_2190936 |
| Glut3 (H-50) antibody | Santa Cruz Biotechnology | Cat# sc-30107; RRID:AB_2254979 |
| HIF1α antibody | Thermo Fisher Scientific | Cat# PA1–16601; RRID:AB_2117128 |
| GAPDH (14C10) Rabbit mAb | Cell Signaling Technology | Cat# 2118; RRID:AB_561053 |
| Anti-Rabbit IgG (H+L), Alexa Fluor®488 Conjugate | Cell Signaling Technology | Cat# 4412; RRID:AB_1904025) |
| Biological Samples | ||
| Human buffy coats | Gulf Coast Regional Blood Center, Houston | N/A |
| Human melanoma TILs | Derived from patient tissues | N/A |
| Human breast cancer tumor TILs | Derived from patient tissues | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human IL-1β | R & D | Cat# 201-LB |
| Recombinant Human IL-4 | R & D | Cat# 204-IL |
| Recombinant Human IL-6 | R & D | Cat# 206-IL |
| Recombinant human IL-12 | R & D | Cat# 219-IL |
| Recombinant Human IL-23 | R & D | Cat# 1290-IL |
| Recombinant Human TGF-β1 | R & D | Cat# 240-B |
| Trizol reagent | Invitrogen | Cat# 15596026 |
| SuperScript II Reverse Transcriptase | Invitrogen | Cat# 18064014 |
| Phloretin | Cayman Chemical | Cat# 14452; CAS:60–82-2 |
| LND | Cayman Chemical | Cat# 14640; CAS:50264–69-2 |
| 2-DG | Cayman Chemical | Cat# 14325; CAS:154–17-6 |
| 3-BrPA | Cayman Chemical | Cat# 19068; CAS:1113–59-3 |
| Etomoxir | Cayman Chemical | Cat# 11969; CAS:828934–41-4 |
| C75 | Cayman Chemical | Cat# 10005270; CAS:191282–48-1 |
| Orlistat | Cayman Chemical | Cat# 10005426; CAS:96829–58-2 |
| 25-HC | Cayman Chemical | Cat# 11097; CAS:2140–46-7 |
| Simvastatin | Cayman Chemical | Cat# 10010344 CAS:79902–63-9 |
| Rapamycin | Sigma Aldrich | Cat# R0395; CAS: 53123–88-9 |
| 2-methoxy Estradiol | Cayman Chemical | Cat# 13021; CAS: 362–07-2 |
| YC-1 | Cayman Chemical | Cat# 81560; CAS:170632–47-0 |
| SB203580 | AdipoGen | Cat# AG-CR1–0030; CAS: 152121–47-6 |
| 2-NBDG | Cayman Chemical | Cat# 11046; CAS:186689–07-6 |
| DMOG | Sigma Aldrich | Cat# D3695; CAS:89464–63-1 |
| DAPI | Invitrogen | Cat# D1306; CAS: 28718–90-3 |
| Polybrene | Sigma Aldrich | Cat# H9268; CAS: 28728–55-4 |
| RNeasy Kit | Qiagen | Cat# 74106 |
| RPMI-1640 (no Glucose) | Thermo Fisher Scientific | Cat# 11879–020 |
| 5-Bromo-4-chloro-3-indolyl β-D-galactopyranoside (XGal) | Sigma Aldrich | Cat# B4252 CAS:7240–90-6 |
| Ficoll-Paque | Thermo Fisher Scientific | Cat# 45001750 |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo Fisher Scientific | Cat# 34080 |
| Critical Commercial Assays | ||
| EasySep Human CD8 Positive Selection Kit | StemCell Technologies | Cat# 18053 |
| EasySep Human CD4 Positive Selection Kit | StemCell Technologies | Cat# 18052 |
| Human CD4+CD127lowCD49d- Treg cell enrichment kit | StemCell Technologies | Cat# 19232 |
| Glucose (GO) Assay Kit | Sigma | Cat# GAGO-20 |
| Glycolysis cell-based assay kit | Cayman Chemical | Cat# 600450 |
| Deposited Data | ||
| Transcriptome analyses dataset/Microarray data | This paper | GEO: GSE118311 |
| Experimental Models: Cell Lines | ||
| Human 586mel tumor cells | National Cancer Institute | N/A |
| Tumor-specific CD8+ TIL586 cells | National Cancer Institute | N/A |
| Experimental Models: Organisms/Strains | ||
| NOD-scid IL2Rγnull mice (NSG) | Jackson Laboratory | Stock# 005557 |
| Oligonucleotides | ||
| Primers for qPCR | This paper | See Table S1 |
| Poly-G3 | IDT Inc | Custom |
| Poly-T3 | IDT Inc | Custom |
| Recombinant DNA | ||
| Phoenix cell packaging system | Gift from Dr. Thirugnana | N/A |
| Software and Algorithms | ||
| FlowJo 7.6.2 | FlowJ, LLC | https://www.flowjo.com/ |
| Prism 5 | GraphPad Software | https://www.graphpad.com/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Guangyong Peng (guangyong.peng@health.slu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Blood Samples and T cell Lines
Buffy coats from healthy donors of both adult men and women were obtained from the Gulf Coast Regional Blood Center at Houston. Peripheral blood mononuclear cells (PBMCs) were purified from buffy coats using Ficoll-Paque. Human naïve CD4+ and CD8+ T cells were purified from PBMCs by EasySep enrichment kits (StemCell Technologies). Naturally occurring human CD4+CD25hi Treg cells (nTreg) were purified from CD4+ T cells by FACS sorting after staining with anti-CD25-PE (BD Biosciences), or isolated from PBMCs by negative selection with the human CD4+CD127lowCD49d− Treg cell enrichment kit (StemCell Technologies). Tumor-associated Treg cell lines were generated from the primary melanoma or breast cancer tumor tissues and maintained in T cell medium of RPMI-1640 containing 10% human AB serum supplemented with L-glutamine, 2-mercaptethanol, and 50 U/mL of IL-2, as we previously described (Ma et al., 2012; Peng et al., 2007; Su et al., 2010; Wang et al., 2004). All human subject studies have been approved by the Saint Louis University Institutional Review Board (IRB# 15283), and the informed consent was obtained from all subjects. In some experiments, human Treg cells were performed in vitro expansion with anit-CD3/CD28 and allogeneic PBMC stimulations.
In vivo Mouse Studies
NOD-scid IL2Rγnull mice (NSG, Stock No. 005557, strain NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) were purchased from The Jackson Laboratory and maintained in the institutional animal BSL2 facility. The facility is fully accredited by Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International and treated in accordance with principles outlined in the Guide for the Care and Use of Laboratory Animals. Animals were grouped housed (5 per cage or less) in specific pathogen free conditions on a 12-h light/12-h dark cycle with free access to autoclave water and standard rodent diet. The caging and bedding are sterilized by autoclave and food is irradiated. Mice were kept in a temperature (72 0F) and humidity (30–70%) controlled environment with air changes (10–13 per hour). Both male and female NSG mice with 6–8 weeks were used for the studies. All animal studies have been approved by the Institutional Animal Care Committee at Saint Louis University (Protocol No. 2411).
Naïve CD8+ T cells (5 × 106/mouse), expanded CD4+CD25hi Treg (nTreg, 3 × 106/mouse) and CD4+CD25− effector T cells (3 × 106/mouse) were pre-activated with anti-CD3 (2 μg/ml, Bio × Cell) and adoptively co-transferred into NSG mice through intravenous injection into the following groups: naïve CD8+ T cells alone, naïve CD8+ T cells plus Treg cells, or naïve CD4+ T cells plus CD4+CD25− effector T cells. In a parallel experiment, Treg cells were pretreated with TLR8 ligand Poly-G3 (3 μg/mL, synthesized by Integrated DNA Technologies Inc), glycolytic metabolism inhibitor 2-DG (1 mM, Cayman Chemical) and HIF1α inhibitor 2-ME (10 μM, Cayman Chemical) for 24 hours prior to adoptively transfer into the mice. After adoptive transfer of T cells into the mice, Poly-G3 (50 μg/mouse), 2-DG (10 mg /mouse), 2-ME (0.3 mg /mouse) were intraperitoneally injected into mice for a total of 3 doses with 3-day intervals. Five to ten mice were included in each group. Blood and spleens (SP) were harvested at 12 days post-injection. The transferred human CD4+ and CD8+ T cells were isolated by antibody-coated microbeads (StemCell Technologies) for subsequent phenotypic and functional analyses in vitro. SA-βGal staining, flow cytometry analyses, and 3H-thymidine incorporation assays, as well as real-time quantitative RT- PCR analysis were performed as described in the following studies.
For tumor growth and anti-tumor immunity studies, human 586mel tumor cells (5 × 106/mouse) in 100 μl of buffered saline were subcutaneously injected into NSG mice. Tumor-specific CD8+ TIL586 cells (5 × 106/mouse) were i.v. injected on day 5 with or without expanded nTreg cells. In a parallel experiment, Treg cells were pretreated with TLR8 ligand Poly-G3, glycolytic metabolism inhibitor 2-DG and HIF1α inhibitor 2-ME for 24 hours prior to adoptively transfer into the mice, and then Poly-G3 and inhibitors were intraperitoneally injected after T cell transfer. The treatment procedures and doses were identical to the above experiments. Tumor size was measured with calipers every 3 days. Tumor volume was calculated on the basis of two-dimension measurements. Blood, spleens, and tumors were harvested at 33 days post injection. The transferred human CD4+ and CD8+ T cells were isolated by antibody-coated microbeads (StemCell Technologies) for subsequent phenotypic and functional analyses in vitro, as described below.
METHOD DETAILS
T Cell Subset Generation
Human CD4+ T cell subsets in vitro differentiation was performed as previously described (Su et al., 2010; Ye et al., 2011). Briefly, naïve CD4+ T cells were purified from PBMCs of healthy donors with CD4+ T-cell enrichment kit (Stemcell Technologies) and then cultured with plate-bound anti-CD3 (2 μg/ml) and anti-CD28 (2 μg/ml) (Bio X Cell) plus polarization condition medium of RPMI-1640 containing 10% human AB serum supplemented with the following cytokines/antibodies at 37°C for 6 days. For Th1 differentiation, naïve T cells were cultured in the presence of anti-IL-4 neutralizing antibody (10 μg/ml, Bio X Cell), and recombinant human IL-12 (5 ng/ml, R & D). For Th2 differentiation, naïve T cells were cultured in the presence of anti-IFN-γ neutralizing antibody (10 μg/ml, Bio X Cell) and rhIL-4 (4 ng/ml, R & D). For Th17 differentiation, naïve T cells were cultured in the presence of IL-1β (20 ng/ml), IL-6 (20 ng/ml), rhTGF-β (3 ng/ml) and IL-23 (10 ng/ml) (R & D) for 6 days.
Naturally occurring human nTreg cells were purified from CD4+ T cells by FACS sorting or isolated from PBMCs by negative selection with the human CD4+CD127lowCD49d− Treg cell enrichment kit as described above. The purified nTreg cells were further performed phenotypic analysis of FoxP3 expression by flow cytometry and FoxP3 demethylation status by Real-time PCR, as well as suppressive activity on other T cell proliferation using a [3H]-thymidine incorporation assay, as we previously described (Liu et al., 2018; Ye et al., 2012). Tumor-associated Treg cell lines were generated from the primary melanoma or breast cancer tumor tissues (Ma et al., 2012; Peng et al., 2005; Peng et al., 2007; Su et al., 2010; Wang et al., 2004; Wang et al., 2005). Briefly, tumor tissues were minced into small pieces followed by digestion with collagenase type IV, hyaluronidase, and deoxyribonuclease. After digestion, cells were washed in RPMI-1640 and cultured in RPMI-1640 containing 10% human AB serum supplemented with L-glutamine, 2-mercaptethanol, and 50 U/mL of IL-2 for the generation of tumorinfiltrating T cells (TILs). Tumor-associated Treg cells were further characterized based on the TCR specificity, tumor recognition, expression of CD25, CTLA-4 and FoxP3, as well as suppressive activity, as described in our published reports (Peng et al., 2005; Peng et al., 2007; Wang et al., 2004; Wang et al., 2005) .
RT-qPCR Analysis
Total RNA was extracted from T cells using Trizol reagent (Invitrogen), and cDNA was transcribed using a SuperScript II RT kit (Invitrogen), both according to manufacturers’ instructions. Expression levels of each gene were determined by reverse-transcription PCR using specific primers, and mRNA levels in each sample were normalized to the relative quantity of β-actin gene expression. All experiments were performed in triplicate. The specific primers used for T cells are listed in Supplemental Table 1.
SA-β-Gal Staining
Senescence associated β-Galactosidase (SA-β-Gal) activity in senescent T cells was detected as we previously described (Ye et al., 2012; Ye et al., 2013). Naïve CD4+ T cells or CD8+ T cells were labeled with Carboxyfluorescein succinimidyl ester (CFSE) (4.5 μM), and co-cultured with Treg or control T cells at different ratios of 10:1 to 1:1 in anti-CD3-coated 24-well plates for 3 or 5 days. Co-cultured naïve T cells were then separated from co-cultures using FACS sorting gated on CFSE positive populations, and then stained with SA-β-Gal staining reagent.
For some experiments, Treg cells were pretreated with the following inhibitors and then SA-β-Gal expression in the co-cultured naïve T cells determined. For metabolic inhibitor blockage experiments, Treg cells were pretreated with phloretin (2 μM), LND (125 μM), 2-DG (1 mM), 3-BrPA (30 μM), etomoxir (100 μM), C75 (5 μM), orlistat (10 μM), 25-HC (0.25 μg/ml), simvastatin (2 μM) (Cayman Chemical) for 48 hours and then co-cultured with naïve CD4+ T cells in the presence or absence Poly-G3 for another 3 days. For mTOR and HIF1α signaling blockage or activation experiments, Treg cells were pretreated with rapamycin (300 nM, Sigma), YC-1 (5 μM, Cayman Chemical ), 2-methoxy Estradiol (2-ME) (10 μM, Cayman Chemical), or DMOG (0.1mM, Sigma) for 24 h, then co-cultured with naïve CD4+ T cells for 3 days. For glucose usage experiment, naïve CD4+ T cells were co-cultured with Treg cells in the medium with different glucose concentrations for 3 days, and then SA-β-Gal expression determined.
Flow Cytometry Analysis
The expression markers on T cells were determined by flow cytometry analysis after staining with antihuman specific antibodies, including anti–phospho-mTOR, anti–phospho-p70S6K, anti–phospho-4E-BP1 (Cell Signaling Technology), anti-Glut1 and anti-Glut3 (Santa Cruz Biotechnology). The second antibody was anti-rabbit IgG (H+L) conjugated with Alexa Flour488 (Cell Signaling Technology). All stained cells were analyzed on a Guava EasyCyte Plus Flow Cytometer (Millipore) and data analyzed with FlowJo software (Tree Star).
Indirect Immunofluorescence Staining
Treg or control CD4+ T cells treated with Poly-G3 (3 μg/ml) or Poly-T3 (3 μg/ml) (synthesized by Integrated DNA Technologies Inc) for 3 days. The cells were fixed in 4% paraformaldehyde, permeabilized in 0.5% Triton X-100, and stained with the anti-human Glut1 and anti-human Glut3 antibodies (Santa Cruz Biotechnology) followed by a secondary antibody Alexa Fluor488–conjugated anti-rabbit (Cell Signaling Technology), and then were further counterstained with 40, 6-diamidino-2phenylindole (DAPI, Invitrogen). The expression and localization of Glut1 and Glut3 in Treg cells were observed with a confocal microscopy.
Western-blotting Analysis
Treg or control CD4+ T cells treated with Poly-G3 (3 μg/ml) or Poly-T3 (3 μg/ml) for various time points. Whole cell lysates of the T cells were prepared for western blot analyses. Western blots were developed with Chemiluminescent Substrate (Thermo Fisher Scientific). The rabbit polyclonal antibodies used in western blotting are anti-HIF1α (Thermo Fisher Scientific) and anti-GAPDH (Cell Signaling Technology).
Functional Proliferation Assay
Proliferation assays were performed using a [3H]-thymidine incorporation assay, as we previously described (Peng et al., 2005; Peng et al., 2007; Ye et al., 2012; Ye et al., 2014). Naïve CD4+ T cells (1 × 105/well) purified from healthy donors were co-cultured with Treg cells with different treatments at a ratio of 10:1 in 200 μl of T cell assay medium containing 2% human AB. After 56 hours of culture, [3H]thymidine was added at a final concentration of 1 μCi/well, followed by an additional 16 hours of culture. The incorporation of [3H]-thymidine was measured with a liquid scintillation counter.
Glucose and L-lactate Assays
Glucose concentrations in cell culture supernatants were measured by Glucose Assay Kits (Sigma). nTreg cells and CD4+ T cells were treated with poly-G3 and Poly-T3 for 3 days. Furthermore, L-lactate concentrations in cell culture supernatants from different T cell subsets were determined using the Glycolysis cell-based assay kit (Cayman Chemical). The culture supernatant was collected and glucose and L-lactate concentrations determined according to the manufacturer’s protocols.
Glucose Uptake Assay
Glucose uptake was measured following 15 min incubation of T cells with a fluorescent D-glucose analog 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-D-glucose (2-NBDG) (Cayman Chemical), as previously described (Wofford et al., 2008). Different T cell subsets were stimulated with/without the antiCD3 antibody for 24 hours, and then glucose uptake was determined after addition of 2-NBDG for 15 min. In addition, nTreg cells and CD4+ T cells were treated with Poly-G3 or Poly-T3 for 3 days, and then cultured in glucose-free RPMI 1640 medium (Gibco) with 2% AB serum for another 30 min, and followed addition of 2-NBDG (100 μM) for 15 min. The T cells were collected and analyzed on a Guava EasyCyte Plus Flow Cytometer (Millipore) and data analyzed with FlowJo software (Tree Star).
Glucose Metabolite Profiling Analysis
Glucose metabolomic analyses of T cells were performed as described (Gerriets et al., 2015; Wang et al., 2011). Different T cell subsets after polarization from naïve T cells for 6 days, and naturally occurring human Treg cells purified from PBMCs and treated with Poly-G3 (3 μg/ml) or Poly-T3 (3 μg/ml) for 24 or 72 hours, were collected to snap freeze in ethanol/dry ice. Lysate was sonicated, spun to remove cell debris, dried and kept at −80°C until time of use. Metabolite separation and detection were performed using an Agilent 1260 LC pump with a RPLC Atlantis T3 column (4.6 mm x 150 mm, 3 μm particles) coupled with a TSQ Quantum Access Max triple quadrupole mass spectrometer fitted with an ESI source. Stable isotope amino acids were used to account for differences in sample preparation. For each analysis, the average ratio for each isotope organic acid was used as a multiplication factor for the signal intensities for each of the endogenous organic acids.
Retrovirus Generation and T Cell Transduction
Generation of recombinant retrovirus carrying GFP and RHEB gene were performed using the Phoenix cell packaging system (kindly provided by Dr. Thirugnana Subramanian in the Institution of Molecular Virology at Saint Louis University). For virus transduction, Treg cells (2 × 106) were activated by platebound anti-CD3 (2 μg/ml) and mixed with the concentrated retrovirus supernatant with a multiplicity of infection (MOI) of 10–15 in a total volume of 0.5 ml T cell medium containing 8 μg/ml polybrene (Sigma), and then spun at 1000 x g for 1 h at room temperature. The infected cells were sorted based on GFP expression with a FACS ARIA sorter at 2 days post-transduction, and were then used to determine suppressive function and T cell senescence induction in the presence or absence of Poly-G3.
QUANTIFICATION AND STATISTICAL ANALYSIS
Transcriptome Analyses of Treg Cells
Transcriptome analyses of naturally occurring human Treg cells were performed using the Illumina whole-genome HumanHT-12 BeadChips, as we previously described (Ye et al., 2012). The normalized log2 expression level of each gene was calculated. nTreg cells were purified from CD4+ T cells by FACS sorting after staining with anti-CD25-PE (BD Biosciences) from healthy donors, and were treated with or without Poly-G3 for different time points, including 0 h, 12 h and 24 h. Total RNA was purified from the Treg cells treated and untreated with Poly-G3 using RNeasy Kit (Qiagen). Samples with OD 260/280 ratios of 1.8 or greater will be used for transcriptome analyses. Integrity of RNA was confirmed in the Agilent Technologies 2100 Bioanalyzer Lab-on-a-chip system before use (Agilent Technologies). At least two biological replicates were generated using total RNA from the pool of 5 donors for each experimental condition. Transcriptional profiling of nTreg cells was performed using illumina’s whole-genome HumanHT-12 BeadChips (Genome Center at Washington University in St. Louis). The labeled RNA was hybridized to the array and later stained with fluorescently labeled anti-biotin antibody. After staining, the BeadChip was scanned on the Illumina BeadArray Reader. Normalization was accomplished in six steps: self-normalization uses information contained within the array itself (normalization ID) followed by additional outlier removal, background estimation, rotational estimation, shear estimation, and scaling estimation. The normalized data was used for all further analyses.
To identify genes differentially expressed in Poly-G3 treated and untreated nTreg cells, a t-test was performed for each gene at the 2 fixed time points separately, and an F-test based on ANOVA was performed as an overall test for the combined data from 2 time points. P-values were adjusted using Benjamini and Hochberg’s FDR method (Benjamini Y & Hochberg Y. 1995), and an adjusted p<0.01 was used as a cutoff to define the significance. Hierarchical clustering was utilized to present the selected significant down-regulated and up-regulated genes. In addition, Gene Ontology (GO) terms associated with each gene were used to characterize the functionally-related genes and identify processes associated with networks of differentially expressed genes. Enrichment test of GO categories was conducted and associated p-values were calculated based on hypergeometric distribution.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism5 software. Unless indicated otherwise, data are expressed as mean ± standard deviation (SD). D’Agostino and Pearson test was used to test whether the data come from a Gaussian distribution. For multiple group comparison in vivo studies, the one-way analysis of variance (ANOVA) was used, followed by the Dunnett’s test for comparing experimental groups against a single control. For single comparison between two groups, paired Student’s t test was used. Nonparametric t-test was chosen if the sample size was too small and not fit Gaussian distribution. The statistical parameters can be found in the figure legends.
Supplementary Material
Highlights.
Human Tregs exhibit distinct metabolic profiles compared with effector T cells
Activated Tregs uniquely accelerate glucose consumption for their suppression
TLR8 signaling selectively inhibits glucose uptake and glycolysis in human Tregs
TLR8-mediated reprogramming of Treg glucose metabolism enhances anti-tumor immunity
ACKNOWLEDGMENTS
The authors would like to thank Joy Eslick and Sherri Koehm for FACS sorting and analyses. We thank Drs. Seth Crosby and Qunyuan Zhang at Washington University in St. Louis for performing microarray analyses. This work was partially supported by grants from the American Cancer Society (RSG-10–16001-LIB, to G. P), Melanoma Research Alliance (to G. P), and the NIH (AI097852 and CA184379 to G. P).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
Microarray data that support the findings of this study have been deposited in Gene Expression Omibus with the primary accession code GSE118311.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two tables and can be found with this article online.
References
- Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R, et al. (2015). Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 29, 2315–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, et al. (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nature medicine 20, 1327–1333. [DOI] [PubMed] [Google Scholar]
- Biswas SK (2015). Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 43, 435–449. [DOI] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, et al. (2016). Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 166, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller CL, Loberg RD, Fan MH, Zhu Q, Park JL, Vesely E, Inoki K, Guan KL, and Brosius FC 3rd (2008). A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol 295, C836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Manicassamy S, Tang H, Kasturi SP, Pirani A, Murthy N, and Pulendran B (2008). Tolllike receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol 9, 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, and Demengeot J (2003). Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. The Journal of experimental medicine 197, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cham CM, Driessens G, O’Keefe JP, and Gajewski TF (2008). Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol 38, 24382450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cham CM, and Gajewski TF (2005). Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol 174, 4670–4677. [DOI] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosin-Roger J, Simmen S, Melhem H, Atrott K, Frey-Wagner I, Hausmann M, de Valliere C, Spalinger MR, Spielmann P, Wenger RH, et al. (2017). Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nature communications 8, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ (2008). Regulatory T cells and treatment of cancer. Curr Opin Immunol 20, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al. (2011). Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146, 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, Romano A, De Simone S, Procaccini C, La Rocca C, et al. (2015). Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol 16, 1174–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, and Powell JD (2009). The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukhande VV, Sharma GC, Lai JC, and Farahani R (2011). Chronic hypoxia-induced alterations of key enzymes of glucose oxidative metabolism in developing mouse liver are mTOR dependent. Mol Cell Biochem. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, et al. (2014). TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nature immunology 15, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CJ, Hammerman PS, and Thompson CB (2005). Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol 5, 844–852. [DOI] [PubMed] [Google Scholar]
- Geng D, Zheng L, Srivastava R, Asprodites N, Velasco-Gonzalez C, and Davila E (2010). When Toll-like receptor and T-cell receptor signals collide: a mechanism for enhanced CD8 T-cell effector function. Blood 116, 3494–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, Warmoes MO, de Cubas AA, MacIver NJ, Locasale JW, et al. (2016). Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nature immunology 17, 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. (2015). Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest 125, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, and Rathmell JC (2012). Metabolic pathways in T cell fate and function. Trends in immunology 33, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Rutkowsky JM, Snodgrass RG, Ono-Moore KD, Schneider DA, Newman JW, Adams SH, and Hwang DH (2012). Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. Journal of lipid research 53, 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, and Rathmell JC (2008). Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol 180, 4476–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM, Thompson TC, Old LJ, and Wang RF (2007). CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 13, 69476958. [DOI] [PubMed] [Google Scholar]
- Lee JY, Sohn KH, Rhee SH, and Hwang D (2001). Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem 276, 16683–16689. [DOI] [PubMed] [Google Scholar]
- Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, and Hwang DH (2004). Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem 279, 16971–16979. [DOI] [PubMed] [Google Scholar]
- Liu X, Mo W, Ye J, Li L, Zhang Y, Hsueh EC, Hoft DF, and Peng G (2018). Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nature communications 9, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Zhang Q, Ye J, Wang F, Zhang Y, Wevers E, Schwartz T, Hunborg P, Varvares MA, Hoft DF, et al. (2012). Tumor-infiltrating gammadelta T lymphocytes predict clinical outcome in human breast cancer. J Immunol 189, 5029–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW, and Giannakakou P (2003). 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer cell 3, 363–375. [DOI] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, et al. (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell metabolism 20, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, and Rathmell JC (2013). Metabolic regulation of T lymphocytes. Annual review of immunology 31, 259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, Zhao L, Vatan L, Shao I, Szeliga W, et al. (2017). Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1blockade resistance in tumor. Nat Immunol 18, 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, and Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186, 3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Inoki K, Munzberg H, Opland D, Faouzi M, Villanueva EC, Ikenoue T, Kwiatkowski D, MacDougald OA, Myers MG Jr., et al. (2009). Critical role for hypothalamic mTOR activity in energy balance. Cell Metab 9, 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R, Priyadharshini B, and Turka LA (2016). Immunometabolism of regulatory T cells. Nature immunology 17, 618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, et al. (2006). A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A 103, 13132–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasare C, and Medzhitov R (2003). Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036. [DOI] [PubMed] [Google Scholar]
- Pearce EL (2010). Metabolism in T cell activation and differentiation. Current opinion in immunology 22, 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, and Choi Y (2009). Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, Wang DY, Li Y, Wang HY, and Wang RF (2005). Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 309, 13801384. [DOI] [PubMed] [Google Scholar]
- Peng G, Wang HY, Peng W, Kiniwa Y, Seo KH, and Wang RF (2007). Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity 27, 334–348. [DOI] [PubMed] [Google Scholar]
- Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, Faicchia D, Marone G, Tramontano D, Corona M, et al. (2016). The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity 44, 406–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, and Campisi J (2009). Persistent DNA damage signalling triggers senescenceassociated inflammatory cytokine secretion. Nature cell biology 11, 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2011). Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harbor symposia on quantitative biology 76, 347–353. [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, and Flier JS (2006). TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116, 3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, and Chi H (2011). HIF1alphadependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. The Journal of experimental medicine 208, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha S, Yang K, Guy C, Vogel P, Neale G, and Chi H (2015). Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nature immunology 16, 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitkovsky MV, Kjaergaard J, Lukashev D, and Ohta A (2008). Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res 14, 5947–5952. [DOI] [PubMed] [Google Scholar]
- Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, and Peng G (2010). Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol 184, 1630–1641. [DOI] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. (2013). Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. The Journal of clinical investigation 123, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Roychoudhuri R, and Restifo NP (2015). Nutrient Competition: A New Axis of Tumor Immunosuppression. Cell 162, 1206–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HL, Liu YN, Huang YT, Pan SL, Huang DY, Guh JH, Lee FY, Kuo SC, and Teng CM (2007). YC-1 inhibits HIF-1 expression in prostate cancer cells: contribution of Akt/NF-kappaB signaling to HIF-1alpha accumulation during hypoxia. Oncogene 26, 3941–3951. [DOI] [PubMed] [Google Scholar]
- Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, and Adema GJ (2006). Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest 116, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nguyen T, Puebla-Osorio N, Pang H, Dujka ME, and Zhu C (2007). DNA damage-induced cellular senescence is sufficient to suppress tumorigenesis: a mouse model. J Exp Med 204, 1453–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lee DA, Peng G, Guo Z, Li Y, Kiniwa Y, Shevach EM, and Wang RF (2004). Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity 20, 107–118. [DOI] [PubMed] [Google Scholar]
- Wang HY, Peng G, Guo Z, Shevach EM, and Wang RF (2005). Recognition of a new ARTC1 peptide ligand uniquely expressed in tumor cells by antigen-specific CD4+ regulatory T cells. J Immunol 174, 2661–2670. [DOI] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RF, Miyahara Y, and Wang HY (2008). Toll-like receptors and immune regulation: implications for cancer therapy. Oncogene 27, 181–189. [DOI] [PubMed] [Google Scholar]
- Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, Wu C, Vogel P, Neale G, Green DR, et al. (2016). Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nature immunology 17, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wofford JA, Wieman HL, Jacobs SR, Zhao Y, and Rathmell JC (2008). IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 111, 2101–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Wang W, Crespo J, Kryczek I, Li W, Wei S, Bian Z, Maj T, He M, Liu RJ, et al. (2017). Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Science immunology 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, Dhar A, and Pavletich NP (2017). Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Huang X, Hsueh EC, Zhang Q, Ma C, Zhang Y, Varvares MA, Hoft DF, and Peng G (2012). Human regulatory T cells induce T-lymphocyte senescence. Blood 120, 2021–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ma C, Hsueh EC, Dou J, Mo W, Liu S, Han B, Huang Y, Zhang Y, Varvares MA, et al. (2014). TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO molecular medicine 6, 1294–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ma C, Hsueh EC, Eickhoff CS, Zhang Y, Varvares MA, Hoft DF, and Peng G (2013). Tumor-derived gammadelta regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J Immunol 190, 2403–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, and Peng G (2015). Controlling T cell senescence in the tumor microenvironment for tumor immunotherapy. Oncoimmunology 4, e994398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Su X, Hsueh EC, Zhang Y, Koenig JM, Hoft DF, and Peng G (2011). Human tumorinfiltrating Th17 cells have the capacity to differentiate into IFN-gamma+ and FOXP3+ T cells with potent suppressive function. Eur J Immunol 41, 936–951. [DOI] [PubMed] [Google Scholar]
- Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, Hou JZ, and Negrin RS (2008). Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood 111, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, and Chi H (2015). Metabolic control of regulatory T cell development and function. Trends in immunology 36, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Yang K, Cloer C, Neale G, Vogel P, and Chi H (2013). mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhdanov AV, Okkelman IA, Collins FW, Melgar S, and Papkovsky DB (2015). A novel effect of DMOG on cell metabolism: direct inhibition of mitochondrial function precedes HIF target gene expression. Biochimica et biophysica acta 1847, 1254–1266. [DOI] [PubMed] [Google Scholar]
- Zou W (2006). Regulatory T cells, tumour immunity and immunotherapy. Nature reviews. Immunology 6, 295–307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.