

Abstract
Hybrid molecules have been developed which are comprised of a tyrosine kinase-targeted, quinazoline-based scaffold and a flexibly linked dia(m)minechloridoPt(II) moiety. The target compounds maintain high affinity and selectivity for ErbB family kinase proteins and one of the derivatives induces platinum adducts with a pharmacologically important cysteine residue.
Activating mutations within the protein kinase complement of the human genome (kinome) are responsible for a large number of diseases, including cancer.1–3 Because of their favorable toxicity profiles and cancer tissue-specific mechanism of action, protein kinase inhibitors have become a major class of personalized oncology drugs.4 Tyrosine kinases belonging to the epidermal growth factor receptor family (ErbB, e.g. EGFR and Her2) are a major target for treating patients diagnosed with tumors harboring mutationally activated or overexpressed oncogenes.5 One promising oncology drug, gefitinib (Fig. 1a), a 4-(3’-chloro-4’-fluoroanilino)-quinazoline derivative, is a molecularly targeted therapy indicated for the management of EGFR-positive lung adenocarcinomas and other solid tumors.6
Fig. 1.
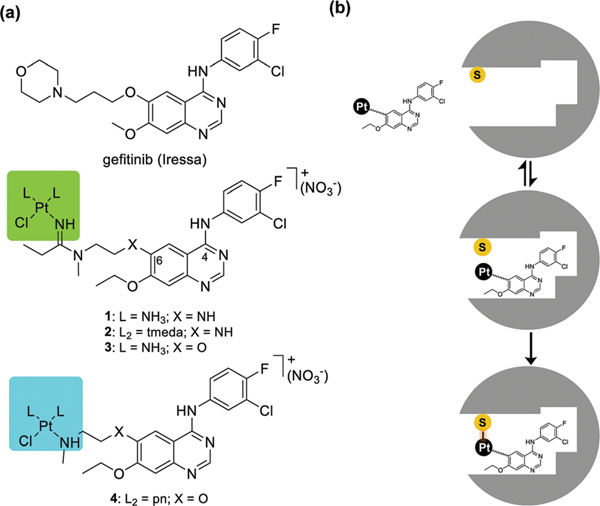
(a) Structures of gefitinib and the newly designed platinum–TKI hybrid agents 1–4 (tmeda stands for N,N,N’,N’-tetramethylethylenediamine), and (b) proposed mechanism of the hybrid agents involving reversible, high-affinity recognition of the ATP-binding pocket and subsequent irreversible cysteine modification.
Tyrosine kinase inhibitors (TKIs) such as gefitinib have been designed to outcompete adenosine triphosphate (ATP), the enzymes’ natural substrate, for active site binding, thereby suppressing phosphorylation signals that lead to uncontrolled cell proliferation and cancer progression.7,8 Unfortunately, secondary active site mutations often diminish the therapeutic efficacy of gefitinib and other reversible kinase inhibitors.9 This obstacle has recently been addressed by designing TKIs into irreversible binders to block ATP binding more effectively. These derivatives most commonly employ covalent bond formation via Michael addition chemistry between a suitably modified group on the inhibitor and a noncatalytic cysteine residue near the ATP binding pocket.10,11 This strategy has proven successful in cancers that have not acquired resistance due to EGFR-independent bypass signaling pathways.12
We have recently begun to explore the use of precious metals as electrophilic “warheads” in molecularly targeted therapies.13,14 In the present study, we wanted to answer the question whether a monofunctional Pt(II) moiety could be introduced into a TKI as athiol-reactive metalloelectrophile and if such a compound would be capable of inducing permanent adducts within the enzyme’s active site. Here, we demonstrate that the modification of a TKI with platinum is compatible with EGFR kinase–inhibitor recognition and provide mass spectrometric evidence for adduct formation with cysteine in a model peptide and in EGFR kinase.
The quinazoline-derived pharmacophore tolerates a wide range of structural modifications of the side chain at C6 of the quinazoline C6 ring, which protrudes out of the ATP binding pocket of the EGFR kinase domain.13,15 Installation of a platinum(II) moiety in that location positions the metal in proximity to EGFR–C797 in the hinge region of the kinase domain, in complete analogy with the design of Michael acceptor-modified TKIs.11 In the monofunctional [PtCl(diam(m)ine)]+ moieties introduced in this study, the chlorido ligand serves as a leaving group to promote ligand exchange with cysteine sulfur. Based on previous docking experiments,13 this design should favor formation of a platinum-mediated TKI–enzyme cross-link (Fig. 1b). The target compounds in this study were generated by reacting a terminal secondary amino group installed on the C6 side chain of a quinazoline derivative13 with a platinum–nitrile complex to produce an amidine linkage16 (Fig. 1a, compounds 1–3). Another derivative was generated via direct replacement of a chlorido ligand with amine nitrogen in a silver ion-activated cis-platinum precursor (Fig. 1a, compound 4) (see the ESI† for synthetic schemes and procedures). In prototypes 1 and 2, the side chain on C6 is connected to the quinazoline ring via an amino group. In compounds 3 and 4 the NH-group was replaced with an ether linkage, which improved the stability of the hybrids. Monodentate NH3 and bidentate amine ligands were introduced as nonleaving groups to study the effects of ligand sterics on receptor–ligand interactions.
Because the new hybrid agents were derived from gefitinib’s core scaffold, the design promises strong binding to EGFR kinase. An ATP-independent competitive binding assay17 was performed with compounds 1 and 2 to determine if the newly introduced cationic Pt(II) moieties affect kinase binding affinity and selectivity (reversible binding step in Fig. 1b). Compounds 1 and 2 showed dissociation constants (Kd) in wild-type EGFR of 1.0 nM and 3.4 nM, respectively (see the ESI† for dose–responsecurves and details of the assay). For comparison, the Kd reported for gefitinib determined by the same assay is 0.9 nM.17 These results suggest that installation of platinum does not compromise the nanomolar binding affinity of the quinazoline pharmacophore. Replacement of NH3 (in 1) with the sterically more hindered bidentate tmeda ligand (in 2) leads only to a minor decrease in binding affinity, demonstrating that the binding site tolerates major structural variations within the platinum coordination sphere.
Using the same assay, compound 1 was also screened against a panel of 145 kinase domains representing the human protein kinase “cysteinome”, a group of approximately 200 kinases containing targetable active-site cysteines.18,19 These include the ErbB family kinases EGFR (wild-type enzyme and 11 oncogenic mutants), ErbB2 (Her2), ErbB3, and ErbB4. Compound 1 showed a high binding affinity for 17 of the 145 tested kinases, based on its ability to displace more than 65% of kinase from an immobilized substrate surrogate (reported as the percentage of kinase that remains bound).17 High-affinity targets are highlighted as red circles in a phylogenetic tree in Fig. 2 (see the caption and the ESI† for details). The highest affinities among the top 17 kinases were observed for ErbB family kinases, except for ErbB3, which show relatively weak binding (81% of control). Wild-type EGFR kinase and several of its mutated forms show complete displacement of surrogate (0% of control), which corresponds to low-nanomolar Kd values.17 The only non-ErbB targets mapped in this assay are FMS-related tyrosine kinase (FLT3) and lymphocyte-specific protein tyrosine kinase (LCK). LCK has previously also been identified as a target of gefitinib.17 Overall, the selectivity profile established for compound 1 closely mimics that reported for gefitinib.17 These findings indicate that modification of the quinazoline scaffold at the C6 position with a monofunctional Pt(II) moiety via a flexible linker (X–(CH2)2–N, X = NH or O) does not affect the reversible binding step of the quinazoline ligand with the ATP-binding pocket. This outcome is consistent with solid state structures of EGFR kinase–inhibitor complexes15 and our previous findings using protein–ligand modeling.13 These studies demonstrated that the extended side chain at quinazoline-C6 is located at the entrance of the ATP-binding pocket and is not involved in specific interactions with the active site that might be disrupted by platinum and compromise TKI–kinase recognition.
Fig. 2.
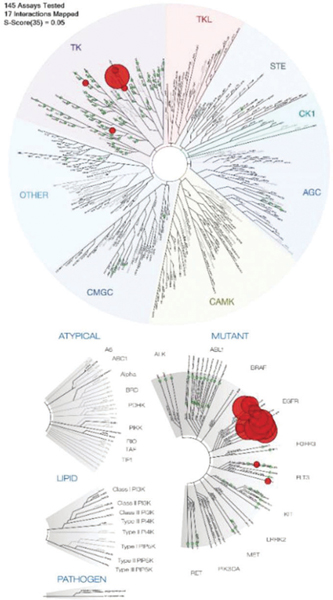
Competitive kinase binding assay for compound 1 in 145 protein kinases of the human cysteinome, including several of their mutated forms. Relative binding affinities are represented by red and green circles in a phylogenetic kinome tree for wild-type enzymes (top) and mutant variants (bottom). A larger red circle indicates a higher binding affinity. A total of 17 interactions with binding levels of less than 35% of control (red circles) at a fixed concentration of test compound of 1 μM were mapped. Selectivity factor, S35 = 0.052. See the ESI† for assay details and complete binding data (Table S3).
The results from the kinome screening experiments unequivocally demonstrate that the platinum moieties in the respective hybrid agents have no major effect on the binding affinity and selectivity of the quinazoline scaffold. Likewise, cell-free kinase assays performed in the presence of ATP to study substrate phosphorylation demonstrated that our hybrids are as efficient at inhibiting kinase function as gefitinib (see the ESI†). The above assays are designed to measure equilibrium binding and do not provide insight into the proposed reactivity of platinum in the hybrids with kinase cysteine (Fig. 1b, irreversible binding step). To detect and structurally characterize potential cysteine modifications, reactions were monitored by in-line high performance liquid chromatography–high resolution mass spectrometry (LC–HRMS) in conjunction with tandem mass spectrometry (MS/MS). Prior to experiments with human recombinant EGFR kinase, the reactivity of two representative compounds, 3 and 4, with a synthetic octapeptide, QLMPFGCL, was studied. This sequence represents amino acid residues 791–798 in the hinge region of the EGFR kinase domain, including the proposed target, C797.
LC–MS analysis of the mixtures resulting from incubations of the octapeptide with compounds 3 and 4 indicate that platinum in both cases reacts with C797 in two distinct ways. Furthermore, tandem mass spectroscopy was used to confirm the amino acid and sequence selectivity of the adducts formed. For both compounds, the data is consistent with a mechanism by which cysteine sulfur replaces the chloride leaving group. This results in monofunctional adducts in which platinum produces a cross-link between the quinazoline ligands and the peptide (adducts A1 and A3, Fig. 3A and B; for LC–MS traces and product analysis, see the ESI†). In addition, a second ligand exchange occurs for both hybrid agents. In compound 3, one of the two ammine (NH3) ligands is lost, resulting in a bifunctional S,N-chelate involving cysteine sulfur and deprotonated amide nitrogen of the peptide backbone, a common motif previously observed in cis-platinum–peptide adducts (adduct A2, Fig. 3A).20–22 A similar situation is observed for compound 4. In this case the reaction results in the substitution of the quinazoline ligand (T2), which becomes a leaving group, while the propane-1,3diamine (pn) chelate remains intact. The result of this reactivity is the transfer of the [Pt(pn)]2+ moiety from the quinazoline ligand to C797, yielding adduct A4 (Fig. 3B).
Fig. 3.
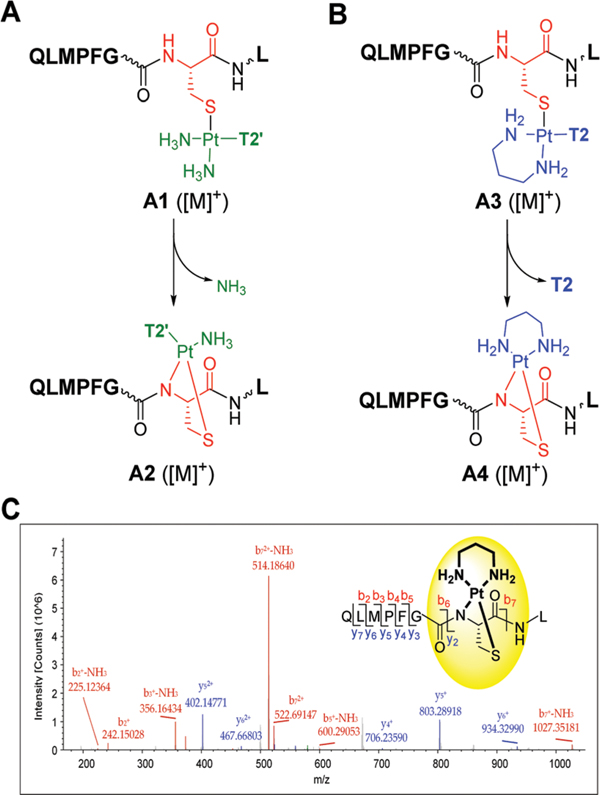
Reaction of compounds 3 (A) and 4 (B) with the target peptide QLMPFGCL based on product analysis by LC–MS. T2’ and T2 stand for the amidine-linked and amine-linked quinazoline ligand (X=O), respectively. (C) Characterization of adduct A4 by MS/MS, confirming the structure and the sequence specificity of a [Pt(pn)]2+ chelate (inset). Selected fragment ions are labeled; see the ESI† for a complete list of b and y ions.
When tandem MS was performed on A1, A2, and A3 to confirm the structures and sequence specificity of the adducts, fragmentation of the modified sequence by higher-energy collisional dissociation (HCD) also led to the dissociation of platinum from the peptide. By contrast, the bis-chelated platinum adduct A4 remained intact, and several relevant platinum-containing b and y ions (see the ESI†) were observed (Fig. 3C). The results unequivocally demonstrate that platinum selectively targets cysteine. Adduct formation with methionine sulfur (M793) in this sequence, which is also known to bind platinum(II) with high affinity,20 is not observed. Finally, based on the pattern of platinum-modified b and y ions, the chelate forms with sulfur and amide nitrogen of cysteine and bidentate coordination involving a non-cysteine nucleophile is unlikely.
To determine if any of the adducts identified in the synthetic octapeptide (A1–A4) also form in the intact enzyme, the reactions of compounds 3 and 4 with the 88 kDa truncated intracellular kinase domain of human recombinant EGFR were investigated. Tryptic digests were analyzed by HR-MS and tandem MS to sequence the peptides and locate the sites of modification. (To eliminate the possibility of nonspecific reactions with the digested peptides and ensure that only adducts formed in the intact protein are detected, excess hybrid agent was removed prior to proteolytic digestion.) Masses (m/z) representing both the monofunctional adducts and chelates in A1–A4 were included as possible modifications when searching for platinum-containing peptides.
The data generated for compound 4 confirms that the [Pt(pn)]2+ chelate observed in model adduct A4 also forms within the native enzyme. By contrast, no monofunctional adducts were detected in which the quinazoline moiety remained cross-linked to the protein (see adduct A3). Three peptides were identified in which platinum chelated one of the following cysteine residues: C797, C939, and C781 (Fig. 4; see ESI† for MS/MS data). Platination of C797, the pharmacologically relevant of the six cysteine residues in EGFR kinase, supports the proposed mechanism of targeted delivery of platinum into the ATP-binding hinge region of the enzyme. Because incubations were performed with excess hybrid agent, formation of additional (nonspecific) adducts is also observed with C939 in the C-terminal lobe and C781 in the N-terminal lobe of the kinase domain. These residues are located at the protein surface but exhibit more limited solvent accessibility than C797 (Fig. 4). No adducts were detected for the deeply buried cysteine residues. Surprisingly, none of the adducts formed by compound 3 in the model peptide were observed in EGFR kinase. One reason for this outcome may be that platinum in 3 is positioned within the binding pocket in a way that disfavors binding to C797. In addition to undetectable levels of modification, it is also possible that any adducts formed by this agent may be too labile and susceptible to collision-induced dissociation, as demonstrated for this analogue in the model octapeptide.
Fig. 4.
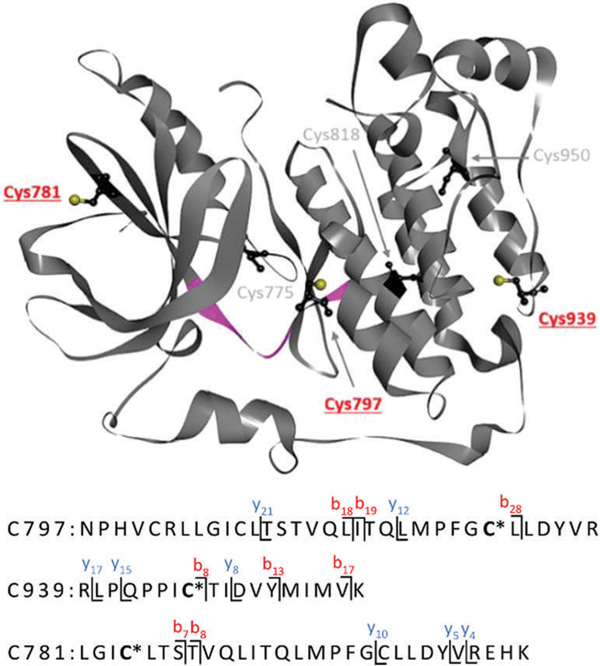
Summary of cysteine adducts identified in EGFR tyrosine kinase based on tandem mass spectrometry analysis of tryptic digests of enzyme treated with compound 4. Pt-Modified cysteine residues are highlighted in red. [Pt(pn)]2+ is represented by an asterisk. The model octapeptide sequence, Q791–L798, in the hinge region between the N- and C-lobes, is highlighted in magenta. Structure adopted from PDB ID: 3W2S.
In this study we have demonstrated using competitive kinase binding and inhibition assays that it is possible to equip a classical TKI structure with an electrophilic platinum(II) moiety that is compatible with kinase active site recognition. In addition, we demonstrated for one of the new hybrid agents a unique mechanism that leads to the formation of a platinum(II) chelate at C797. This form of TKI-mediated delivery of a metallo-electrophile has potential applications in modulating ATP-driven kinase signaling, which contrasts other metal-based hybrid designs.14,23 Kinase cysteine is not only a pharmacologically relevant target for irreversible inhibitors. Cysteine modifications under oxidative stress conditions, for instance, have also been implicated in kinase regulation.24,25 Similarly, platination of this site by our hybrid agents may also lead to conformational changes affecting protein phosphorylation and signal transduction. In conclusion, the unique reactivity and cysteine modifications observed for the platinum-modified TKIs suggest a potential new role of platinum, the classical anticancer metal, as an electrophile in molecularly targeted drugs. The results of this study warrant future experiments to delineate structure–function relationships and the molecular mechanism of these hybrid agents in cancer cells harboring mutated EGFR kinase. This work was funded by the Wake Forest Innovation and Commercialization Services and the Wake Forest University Comprehensive Cancer Center (NCI CCSG P30CA012197).
Supplementary Material
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures, compound characterization, enzyme assays, and mass spectrometry data. See DOI: 10.1039/c8cc04251a
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Olow A, Chen Z, Niedner RH, Wolf DM, Yau C, Pankov A, Lee EP, Brown-Swigart L, van ‘t Veer LJ and Coppe JP, Cancer Res, 2016, 76, 1733–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M and Varmus H, Proc. Natl. Acad. Sci. U. S. A, 2004, 101, 13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma SV, Bell DW, Settleman J and Haber DA, Nat. Rev. Cancer, 2007, 7, 169–181. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Yang PL and Gray NS, Nat. Rev. Cancer, 2009, 9, 28–39. [DOI] [PubMed] [Google Scholar]
- 5.Roskoski R Jr., Pharmacol. Res, 2014, 79, 34–74 [DOI] [PubMed] [Google Scholar]
- 6.Herbst RS, Morgensztern D and Boshoff C, Nature, 2018, 553, 446–454. [DOI] [PubMed] [Google Scholar]
- 7.Kim Y, Li ZM, Apetri M, Luo BB, Settleman JE and Anderson KS, Biochemistry, 2012, 51, 5212–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gajiwala KS, Feng J, Ferre R, Ryan K, Brodsky O, Weinrich S, Kath JC and Stewart A, Structure, 2013, 21, 209–219. [DOI] [PubMed] [Google Scholar]
- 9.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE and Meyerson M, Science, 2004, 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Z and Bourne PE, Drug Discovery Today, 2018, 23, 727–735. [DOI] [PubMed] [Google Scholar]
- 11.Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH and Gray NS, Chem. Biol, 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuwano M, Sonoda K, Murakami Y, Watari K and Ono M, Pharmacol. Ther, 2016, 161, 97–110. [DOI] [PubMed] [Google Scholar]
- 13.Yang M, Pickard AJ, Qiao X, Gueble MJ, Day CS, Kucera GL and Bierbach U, Inorg. Chem, 2015, 54, 3316–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang M and Bierbach U, Eur. J. Inorg. Chem, 2017, 1561–1572. [Google Scholar]
- 15.Park JH, Liu Y, Lemmon MA and Radhakrishnan R, Biochem. J, 2012, 448, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Z, Rao L and Bierbach U, J. Med. Chem, 2009, 52, 3424–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabian MA, Biggs WH 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP and Lockhart DJ, Nat. Biotechnol, 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- 18.Chaikuad A, Koch P, Laufer SA and Knapp S, Angew. Chem., Int. Ed, 2018, 57, 4372–4385. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Zhang D, Tian H, Jiao Y, Shi Z, Ran T, Liu H, Lu S, Xu A, Qiao X, Pan J, Yin L, Zhou W, Lu T and Chen Y, Mol. Pharmaceutics, 2016, 13, 3106–3118. [DOI] [PubMed] [Google Scholar]
- 20.Messori L and Merlino A, Coord. Chem. Rev, 2016, 315, 67–89. [Google Scholar]
- 21.Boal AK and Rosenzweig AC, J. Am. Chem. Soc, 2009, 131, 14196–14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi DH, Hambley TW and Freeman HC, J. Inorg. Biochem, 1999, 73, 173–186. [Google Scholar]
- 23.Wei Y, Poon DC, Fei R, Lam AS, Au-Yeung SC and To KK, Sci. Rep, 2016, 6, 25363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wani R, Nagata A and Murray BW, Front. Pharmacol, 2014, 5, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Truong TH, Ung PMU, Palde PB, Paulsen CE, Schlessinger A and Carroll KS, Cell Chem. Biol, 2016, 23, 837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.