

In this study, Vizioli et al. investigated the upstream signaling events that promote cytoplasmic formation of chromatin fragments (CCFs), which are extruded from the nucleus of senescent cells and trigger the senescence-associated secretory phenotype (SASP). They show that dysfunctional mitochondria, linked to down-regulation of nuclear-encoded mitochondrial oxidative phosphorylation genes, trigger a ROS–JNK retrograde signaling pathway that drives CCF formation and hence the SASP.
Keywords: cytoplasmic chromatin, inflammation, mitochondria, senescence
Abstract
Cellular senescence is a potent tumor suppressor mechanism but also contributes to aging and aging-related diseases. Senescence is characterized by a stable cell cycle arrest and a complex proinflammatory secretome, termed the senescence-associated secretory phenotype (SASP). We recently discovered that cytoplasmic chromatin fragments (CCFs), extruded from the nucleus of senescent cells, trigger the SASP through activation of the innate immunity cytosolic DNA sensing cGAS–STING pathway. However, the upstream signaling events that instigate CCF formation remain unknown. Here, we show that dysfunctional mitochondria, linked to down-regulation of nuclear-encoded mitochondrial oxidative phosphorylation genes, trigger a ROS–JNK retrograde signaling pathway that drives CCF formation and hence the SASP. JNK links to 53BP1, a nuclear protein that negatively regulates DNA double-strand break (DSB) end resection and CCF formation. Importantly, we show that low-dose HDAC inhibitors restore expression of most nuclear-encoded mitochondrial oxidative phosphorylation genes, improve mitochondrial function, and suppress CCFs and the SASP in senescent cells. In mouse models, HDAC inhibitors also suppress oxidative stress, CCF, inflammation, and tissue damage caused by senescence-inducing irradiation and/or acetaminophen-induced mitochondria dysfunction. Overall, our findings outline an extended mitochondria-to-nucleus retrograde signaling pathway that initiates formation of CCF during senescence and is a potential target for drug-based interventions to inhibit the proaging SASP.
Cellular senescence is a stable cell cycle arrest that can be induced by replicative exhaustion and other stresses, such as DNA damage and aberrant oncogenic activation. By preventing the growth of oncogene-expressing, damaged, or stressed cells, senescence is a potent barrier to tumorigenesis (Herranz and Gil 2018).
Senescent cells display important and unique properties, including changes in morphology and metabolism, subcellular organelles, and profound chromatin remodeling. In addition, senescent cells secrete a complex combination of proinflammatory cytokines, chemokines, growth factors, and proteases, termed the senescence-associated secretory phenotype (SASP) (Coppé et al. 2008; Rodier and Campisi 2011). The SASP acts in a cell-autonomous manner to reinforce the senescent state (Acosta et al. 2008; Kuilman et al. 2008) but also communicates with and alters the microenvironment (Nelson et al. 2012; Acosta et al. 2013). SASP factors can contribute to tumor suppression by promoting immune-mediated clearance of damaged or premalignant cells (Kang et al. 2011). However, the SASP can also be responsible for persistent chronic inflammation and tissue damage. Chronic inflammation can cause or greatly accelerate degenerative and neoplastic diseases associated with aging, including cancer (Baker et al. 2011; Yoshimoto et al. 2013; van Deursen 2014; Milanovic et al. 2018). Consequently, there is growing interest in developing therapeutic agents to suppress the deleterious effects of senescent cells. There is currently much effort devoted to developing compounds (termed senolytics) that selectively kill senescent cells (Kirkland et al. 2017). Given the role of the SASP in mediating the detrimental effects of senescent cells, SASP inhibition (by so-called senostatics or senomorphics) can be an alternative to senolytics to promote healthy aging (Niedernhofer and Robbins 2018; Short et al. 2019). Hence, it is important to better understand how the SASP is regulated because its modulation may be a potential avenue to combat the proaging effects of senescent cells.
We previously showed that senescent cells shed fragments of nuclear chromatin into the cytoplasm, termed cytoplasmic chromatin fragments (CCFs), via a nucleus-to-cytoplasmic blebbing process (Ivanov et al. 2013). CCFs are positive for the DNA damage marker γH2AX but lack its usual partner, 53BP1. They are enriched in heterochromatin markers including H3K9me3 and H3K27me3 but devoid of certain euchromatin markers, such as H3K9ac (Ivanov et al. 2013). Importantly, we discovered that CCF formation depends on a nuclear lamin B1-LC3 interaction (Dou et al. 2015) and that these fragments initiate the SASP program during senescence (Dou et al. 2017; Glück et al. 2017; Yang et al. 2017). Specifically, CCFs are recognized by the cytosolic DNA sensor cGAS that produces the second messenger cyclic GMP-AMP (cGAMP), which binds and activates the adaptor protein STING. The cGAS–STING pathway leads to activation of NFκB signaling that turns on transcription of proinflammatory genes (Dou et al. 2017; Glück et al. 2017; Yang et al. 2017). However, the initiating signals of CCF formation are unknown.
Senescent cells are metabolically active but accumulate dysfunctional mitochondria resulting in increased mitochondrial mass, increased oxygen consumption, decreased efficiency of oxidative phosphorylation, decreased membrane potential, and increased levels of reactive oxygen species (ROS) (Passos et al. 2007; Moiseeva et al. 2009; Kaplon et al. 2013; Korolchuk et al. 2017). Mitochondria have been reported to contribute to SASP induction in senescence (Correia-Melo et al. 2016). However, the underlying mechanism and whether dysfunctional mitochondria are functionally connected to the SASP through CCFs is unclear. Here, we examined the role of mitochondria in mediating CCF formation in senescence and discovered a mitochondria-to-nucleus retrograde signaling event that triggers CCFs and the SASP. We show that this pathway is a druggable target to dampen expression of proaging inflammatory genes and ameliorate features of inflammatory disease.
Results
Mitochondria are required for CCF formation in senescent cells
We initiated our study by investigating the potential connection of mitochondria and CCFs. As the first step, we analyzed the RNA-seq data from IMR90 primary human fibroblasts induced to senesce by ionizing irradiation (IR). This analysis showed that expression of nuclear-encoded mitochondrial oxidative phosphorylation genes belonging to the complexes I, II, III, IV, and V are generally reduced (Fig. 1A). Gene set enrichment analysis (GSEA) confirmed significant down-regulation of nuclear-encoded mitochondrial genes in senescent (IR) cells (Supplemental Fig. S1A). This was also confirmed by qPCR of gene transcripts, showing that the steady-state mRNA levels of NDUFA10, NDUFA12, NDUFC2, and NDUFS8 were significantly reduced in irradiated senescent cells (Supplemental Fig. S1B). In line with this, senescent cells accumulated mitochondria that showed decreased mitochondrial membrane potential and increased mitochondrial ROS and increased levels of mitochondrial SOD2 mRNA, product of a ROS-responsive gene (Fig. 1B–D; Kops et al. 2002). Together, these data show that senescent cells down-regulate expression of nuclear-encoded oxidative phosphorylation genes, accompanied by marked mitochondria dysfunction and ROS production.
Figure 1.

Mitochondria are required for formation of CCFs. (A) Heat map of RNA-seq analysis of nuclear-encoded mitochondrial genes belonging to complexes I, II (CII), III (CIII), IV (CIV), and V (CV) in proliferating (Prolif) and senescent (IR) IMR90 cells 10 d after IR, from n = 3 independent experiments. The color intensity represents the log2 fold change, where red indicates highly and blue lowly expressed. (B) Ten days after IR, proliferating (Prolif) and senescent (IR) IMR90 cells were incubated with TMRE (100 nM) and Mitotracker Green (MTG; 100 nM) followed by flow cytometric analysis. The ratio of TMRE and MTG fluorescence intensity was used to represent mitochondrial membrane potential (MMP). (C) Mitochondrial reactive oxygen species (mROS) were measured by flow cytometry using MitoSOX (5 μM) in proliferating (Prolif) and senescent (IR) IMR90 cells 10 d after IR. For raw data, see Supplemental Figures S6 and S7. (D) Expression of anti-ROS gene SOD2 was determined by RT-qPCR in proliferating (Prolif) and senescent (IR) IMR90 cells 10 d after IR. Data shown in B–D are mean ± SEM of n = 3 independent experiments. P-value was calculated by unpaired two-tailed Student's t-test. (***) P < 0.001; (*) P < 0.05. (E) Scheme summarizing the experimental design of Parkin-directed mitophagy experiments in F–K. (F) The expression of mitochondrial proteins from the different mitochondrial complexes NDUFB8 (complex I), SDHA (complex II), and UQCR2 (complex III) and for the mitochondrial outer membrane marker Tom20 were analyzed by immunoblotting in senescent (IR) control (C) and Parkin (P)-expressing IMR90 cells treated with or without CCCP. (G) Representative fluorescent images of Tom20 staining in senescent (IR) Parkin-expressing cells treated with or without CCCP. Scale bar, 20 μm. (H) Representative confocal immunofluorescence images of proliferating (Prolif) and senescent (IR) Parkin-expressing cells stained for DAPI (blue), anti-γH2AX (green). CCFs are indicated by arrows and quantified. Scale bar, 5 μm. Graph shows mean ± SEM of n = 3 independent experiments. P-value was calculated by unpaired two-tailed Student's t-test. (****) P < 0.0001. (I) The expression of the indicated SASP components was monitored by RT-qPCR in proliferating (Prolif) and senescent (IR) Parkin-expressing cells. Data are mean ± SEM of n = 3 independent experiments. P-value was calculated by unpaired two-tailed Student's t-test. (***) P < 0.001; (**) P < 0.01. (J) Heat map of RNA-seq analysis of SASP genes in the depicted IMR90 cells from n = 3 independent experiments. The color intensity represents the Z-score of the mean FPKM expression, where red indicates highly and blue lowly expressed. (K) Lysates from proliferating (Prolif) and senescent (IR) of control (C) and Parkin-expressing (P) cells were probed for the expression of IL8 by immunoblotting.
We then tested whether dysfunctional mitochondria are connected to CCFs, because mitochondria and CCFs have both been reported to drive the SASP (Correia-Melo et al. 2016; Dou et al. 2017; Glück et al. 2017; Yang et al. 2017). We used a Parkin-mediated mitophagy model to remove mitochondria in senescent cells (Correia-Melo et al. 2016). Briefly, IMR90 primary human fibroblasts were stably transduced with control or a Parkin-YFP fusion protein (hereafter referred to as Parkin) followed by induction of senescence by IR. Two days after irradiation, cells were treated with the mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP) to induce mitochondrial depolarization and Parkin-mediated mitophagy. Cells were harvested 10 d after irradiation (Fig. 1E). We confirmed that expression of mitochondrial proteins was dramatically reduced in Parkin CCCP-treated senescent cells but not in control CCCP-treated senescent cells, as assessed by immunoblotting (Fig. 1F). Clearance of mitochondria was also confirmed by staining for mitochondrial Tom20 (Fig. 1G). Absence of mitochondria did not impair the senescence growth arrest, as shown by loss of phosphorylated pRB (ph-Rb) and cyclin A and induction of p21 (Supplemental Fig. S1C). Consistent with our previous studies (Ivanov et al. 2013; Dou et al. 2017), CCFs were detected in ∼30%–50% of control senescent cells by staining with DAPI and γH2AX (Fig. 1H). Strikingly, senescent cells lacking mitochondria showed strong suppression of CCFs (Fig. 1H; Supplemental Fig. S1D), suggesting that mitochondria are necessary for CCF formation.
Because CCF generation is a pivotal event triggering the SASP (Dou et al. 2017; Glück et al. 2017; Yang et al. 2017), we next examined the effects of mitochondria elimination on SASP gene expression: The induction of IL1α, IL1β, IL6, and IL8 mRNAs was drastically impaired in mitochondria-depleted senescent cells, determined by qPCR (Fig. 1I; Supplemental Fig. S1E). We thus performed RNA-seq on control and Parkin-expressing proliferating cells and control and Parkin-expressing senescent cells treated with or without CCCP. Principal component analysis (PCA) showed clear separation of the mitochondria-depleted senescent cells (Parkin IR CCCP) from the other five treatment groups, along the first principal component (Supplemental Fig. 1F). Differential gene expression analysis showed that senescence-associated expression changes of 3466 genes were abrogated in senescent cells depleted of mitochondria (Parkin IR CCCP) compared with control senescent cells (Parkin IR). Specifically, 1505 genes were up-regulated in senescent cells (Parkin IR–Parkin Prolif) and down-regulated upon mitochondria removal (Parkin IR CCCP–Parkin IR), whereas 1961 genes were down-regulated in senescent cells (Parkin IR–Parkin Prolif) and up-regulated upon mitochondria clearance (Parkin IR CCCP–Parkin IR) (Supplemental Fig. S1G). By analyzing the SASP components and genes associated with cell cycle progression, we confirmed that Parkin-mediated mitophagy strongly attenuated the SASP expression (Fig. 1J) without reversing the senescent growth arrest (Supplemental Fig. S1H). As an additional confirmation of SASP reduction at the mRNA level in senescent cells depleted of mitochondria, we performed gene expression analysis using a custom NanoString panel targeting 31 SASP mRNAs (Coppé et al. 2010). Most SASP mRNAs that were significantly up-regulated in senescent cells (Parkin IR) were down-regulated upon mitochondria clearance (Parkin IR CCCP) (Supplemental Fig. S1I). Analysis at the protein level by IL8 immunoblotting also showed the ability of mitochondria clearance to dampen the SASP (Fig. 1K).
To confirm that these results were not unique to the IR-induced senescence model, we removed mitochondria in cells induced to enter senescence by the DNA damaging agent etoposide. Specifically, control and Parkin-expressing cells were exposed to etoposide for 24 h, and after 2 d cells were treated with or without CCCP. Parkin-mediated mitophagy led to elimination of mitochondria in senescent cells (Supplemental Fig. S1J), again without reversing the senescence growth arrest (Supplemental Fig. S1K). Similar to the IR model, clearance of mitochondria prevented formation of CCFs (Supplemental Fig. S1L) and abrogated expression of the SASP (Supplemental Fig. S1M). Taken together, our results indicate that mitochondria are required for the formation of CCFs and hence the SASP program.
Mitochondria–ROS–JNK signaling pathway drives CCFs and the SASP
We next sought to define the mechanisms by which mitochondria trigger formation of CCFs. Because dysfunctional mitochondria increase ROS production (Fig. 1C; McCubrey et al. 2006; Schieber and Chandel 2014) and it has been shown that ROS control the SASP (Nelson et al. 2018), we investigated whether ROS promote CCF formation. Mitochondria-deficient senescent cells showed significantly reduced mitochondrial ROS production when compared with control senescent cells treated with CCCP (Fig. 2A), consistent with the idea that mitochondria signal to CCFs via ROS. To directly test this, we elevated or reduced cellular ROS levels and examined the corresponding effects on CCFs. First, we nonspecifically elevated cellular ROS levels in proliferating IMR90 cells by hydrogen peroxide (H2O2) (Fig. 2B). This procedure triggered cell cycle arrest, as judged by loss of ph-Rb and cyclin A and induction of p21 (Fig. 2C), as well as CCFs and the SASP (Fig. 2D,E). Second, we specifically increased mitochondrial ROS by MitoParaquat (Fig. 2F; Robb et al. 2015), which again triggered growth arrest (Fig. 2G) and induced formation of CCFs (Fig. 2H) and expression of the SASP genes (Fig. 2I). Third, conversely, we suppressed mitochondrial ROS with mitochondrial antioxidant MitoQ (Fig. 2J). This strongly decreased both CCFs and the SASP in irradiated senescent cells (Fig. 2K,L) without reverting senescence-associated cell cycle arrest, assessed by EdU incorporation, proliferation markers including ph-Rb and cyclin A, and growth arrest marker p21 (Fig. 2M,N). Taken together, these results indicate that elevated mitochondrial ROS induce formation of CCFs.
Figure 2.

ROS promote formation of CCFs and expression of the SASP. (A) Mitochondrial reactive oxygen species (mROS) were measured by flow cytometry using MitoSOX (5 μM) in proliferating and senescent (IR) of control and Parkin-expressing cells with or without CCCP treatment 10 d after IR. For raw data, see Supplemental Figure S7. (B) Proliferating IMR90 cells were exposed to exogenous ROS (H2O2, 600 μM) for 2 h. Quantification of mitochondrial reactive oxygen species (mROS) was assessed by flow cytometry using MitoSOX (5 μM) at day 4. For raw data, see Supplemental Figure S7. (C–E) Proliferating IMR90 cells exposed to exogenous ROS (H2O2, 600 μM) and harvested at the indicated times were analyzed for the depicted proteins by immunoblotting (C), quantified for CCFs (D), and analyzed for the expression of SASP components by RT-qPCR (E). Data shown in A, B, D, and E are mean ± SEM of n = 3 independent experiments. P-value was calculated by unpaired two-tailed Student's t-test or one-way ANOVA coupled with Dunnett's test. (****) P < 0.0001; (***) P < 0.001; (**) P < 0.01; (*) P < 0.05; (NS) not significant. (F–I) Proliferating IMR90 cells treated with MitoPQ (40 μM) for 8 d were analyzed for mitochondrial superoxide production by flow cytometry using MitoSOX (5 μM) (for raw data, see Supplemental Figure S7) (F), probed for the indicated proteins by immunoblotting (G), quantified for CCFs (H), and assessed for expression of the indicated SASP genes by RT-qPCR (I). For F, H, and I, data are mean ± SEM of n = 3 independent experiments. P-value was calculated by unpaired two-tailed Student's t-test. (***) P < 0.001; (**) P < 0.01; (*) P < 0.05. (J–N) IMR90 cells were treated with 100 nM MitoQ and then irradiated to initiate senescence. Cells were harvested after 10 d of irradiation. (J) Quantification of mitochondrial reactive oxygen species (mROS) by flow cytometry using MitoSOX (5 μM) in proliferating and senescent (IR) with or without MitoQ. For raw data, see Supplemental Figure S7. (K) Formation of CCFs was monitored by immunofluorescence, imaged, and quantified in proliferating and senescent (IR) IMR90 cells treated with or without MitoQ. Representative fluorescent images are shown. CCFs are indicated by arrows. Scale bar, 8 μm. (L) Expression of the selected SASP genes was measured by RT-qPCR in proliferating and senescent (IR) IMR90 cells treated with or without MitoQ. Mean ± SEM of n = 3 independent experiments are shown in J–L. P-value was calculated by unpaired two-tailed Student's t-test. (****) P < 0.001; (***) P < 0.001; (**) P < 0.01; (*) P < 0.05. (M) The effect of MitoQ on cell proliferation was assessed by EdU incorporation. Representative images and quantification from two independent experiments are shown. Scale bar, 15 μm. (N) Lysates from proliferating and senescent (IR) IMR90 cells with or without MitoQ were analyzed for the indicated proteins by immunoblotting.
We next investigated the mechanism by which ROS induce CCF formation. ROS can promote the activation of the stress-activated JNK kinase (McCubrey et al. 2006; Schieber and Chandel 2014; Shi et al. 2014). Therefore, we set out to test whether ROS signal to CCFs via JNK. In line with this idea, JNK was activated in senescent cells (Fig. 3A) and suppressed in mitochondria-depleted senescent cells as well as in MitoQ-treated senescent cells (Supplemental Fig. S2A,B), supporting the mitochondria–ROS–JNK signaling pathway. To directly test whether JNK activity is required for CCF formation and induction of the SASP, we treated IMR90 cells with JNK inhibitor SP600125 and then induced cells to senesce by IR. Like parkin-directed mitophagy, JNK inhibition did not affect several senescence markers, including loss of ph-Rb and cyclin A and induction of p21 (Fig. 3B), but it did compromise accumulation of CCFs and induction of representative SASP genes assessed by qPCR (Fig. 3C,D). As an additional confirmation of SASP reduction upon JNK inhibitor treatment, we performed targeted gene expression profiling of proliferating and senescent cells, with or without JNK inhibitor, via a NanoString panel probing 249 inflammatory genes. PCA analysis showed that JNK inhibitor-treated senescent cells are more similar to proliferating cells (Fig. 3E). Differential expression analysis showed that 84 of the 249 genes are up-regulated in senescence and 64 of these were down-regulated in senescent cells treated with JNK inhibitor. Suppressed genes included all SASP genes (Freund et al. 2010) on the panel (Fig. 3F) and a slew of other senescence-up-regulated inflammation genes (Supplemental Fig. S2D). JNK inhibitor treatment also diminished the SASP at the protein level in senescent cells, as shown by IL8 immunoblotting (Fig. 3G). Importantly, inhibition of JNK did not suppress mitochondrial ROS (Fig. 3H), in line with the idea that ROS act upstream of JNK activation. The effects of the JNK inhibitor were validated using shRNAs targeting JNK1 and JNK2. Knockdown of JNK1/2 strongly reduced CCF accumulation and the expression of SASP genes in senescent cells (Fig. 3I–K). Finally, inhibition of JNK in already senescent cells (9 d after IR) (Supplemental Fig. S2F) also resulted in reduction of CCFs and down-regulation of the SASP (Supplemental Fig. S2G,H) but did not rescue cell cycle arrest (Supplemental Fig. S2I), corroborating our hypothesis that JNK mediates CCFs and the SASP. Together, these data show that a mitochondria–ROS–JNK signaling pathway drives CCF formation and hence the SASP.
Figure 3.

JNK mediates formation of CCFs and expression of the SASP. (A) Proliferating (Prolif) and senescent (IR) IMR90 cells harvested at the indicated times were analyzed for ph-JNK and JNK by immunoblotting. (B–H) IMR90 cells were treated with JNK inhibitor (JNKi, SP600125, 20 μM) and then irradiated to induce senescence. Cells were harvested 10 d after IR. (B) Immunoblot analysis of the indicated proteins in proliferating (Prolif) and senescent (IR) IMR90 cells with or without JNKi. (C) Proliferating (Prolif) and senescent cells (IR) with or without JNKi were imaged and quantified for CCF formation. CCFs are indicated by arrows. Scale bar, 8 μm. (D) Expression of the SASP measured by RT-qPCR in the depicted IMR90 cells. (E) Principal component analysis scatter plot showing the global gene expression of the cohorts used in the NanoString analysis. (F) Heat map of SASP genes included in the NanoString analysis in proliferating (Prolif) and senescent (IR) with or without JNKi. Mean of three independent replicates is shown. The color intensity represents the Z-score of the mean FPKM expression. (G) Immunoblot analysis of IL8 in proliferating and senescent (IR) IMR90 cells in the absence or presence of JNKi. (H) Determination of mitochondrial reactive oxygen species (mROS) by flow cytometry using MitoSOX (5 μM) in proliferating (Prolif) and senescent (IR) IMR90 cells, in the absence or presence of JNKi. The percentage of positive cells is shown. For raw data, see Supplemental Figure S7. Data shown in C,D, and H are mean ± SEM of three independent experiments. P-value was calculated by unpaired two-tailed Student's t-test. (***) P < 0.001; (**) P < 0.01; (*) P < 0.05. (I–K) Proliferating IMR90 cells were infected with a nontargeting control shRNA (sh-NTC) or with shRNA against JNK1/2 (sh-JNK1/2), followed by mock (Prolif) or ionizing irradiation (IR). Ten days after IR, cells were assessed for expression of JNK by immunoblotting (I) or percentage of CCF-positive cells by immunofluorescence (J) or monitored for the expression of the indicated SASP genes by RT-qPCR (K). Data shown in J and K are mean ± SEM of three independent experiments. Statistical significance was calculated using unpaired two-tailed Student's t-test. (**) P < 0.01; (*) P < 0.05.
53BP1 controls formation of CCF in the nucleus
We next sought to investigate the nuclear events that generate CCFs and the connection to JNK. Currently, the mechanisms that initiate CCFs in the nucleus are not well understood. A clue is provided from our previous study investigating the molecular markers of CCFs, where we found that CCFs stain negative for 53BP1 (Fig. 4A; Ivanov et al. 2013). This observation is unusual, as CCFs are enriched in γH2AX, and γH2AX and 53BP1 typically colocalize (Kleiner et al. 2015). We therefore suspected that 53BP1 might be a regulated inhibitor of CCF formation. We speculated that JNK signals to 53BP1 and promotes CCF formation. Concordant with this idea, coimmunoprecipitation analysis showed that a 53BP1-JNK interaction occurs between the endogenous proteins in senescent cells but not in proliferating cells (Fig. 4B). To directly test whether 53BP1 negatively regulates CCF formation, we stably suppressed 53BP1 by shRNA and then induced senescence by IR (Fig. 4C). Knockdown of 53BP1 led to elevated CCFs and SASP (Fig. 4D,E) without affecting the senescence-associated cell cycle arrest (Supplemental Fig. S3A). We then stably overexpressed 53BP1 in IMR90 cells and induced senescence by IR. While overexpression of 53BP1 did not impair the senescence growth arrest, reflected in loss of ph-Rb and abundance of p21 (Fig. 4F), it markedly and significantly suppressed induction of CCFs and the SASP (Fig. 4G–I). Taken together, these results, notably 53BP1's exclusion from CCFs, its senescence-specific interaction with JNK, and its ability to increase or decrease CCFs when ectopically expressed or knocked down, suggest that 53BP1 is a JNK-regulated inhibitor of CCF formation.
Figure 4.

53BP1 controls formation of CCFs in the nucleus. (A) Confocal fluorescent images of senescent (IR) IMR90 cells stained for DAPI (blue), γH2AX (green), and 53BP1(red) 10 d after IR. CCFs are indicated with arrows. Scale bar, 10 μm. (B) Immunoblot analysis for 53BP1 and JNK after immunoprecipitation of 53BP1 from proliferating (Prolif) and senescent (IR) IMR90 cells 10 d after IR. One representative experiment out of four experiments is shown. (C–E) Proliferating IMR90 cells were infected with a nontargeting control shRNA (sh-NTC) or with shRNA against 53BP1 (sh-53BP1), followed by mock (Prolif) or ionizing irradiation (IR). Ten days after IR, cells were analyzed for expression of 53BP1 by immunoblotting (C), stained and quantified for formation of CCF (indicated by arrows) (D), or examined for the expression of SASP components by RT-qPCR (E). Scale bar, 10 μm. Mean ± SEM of three independent experiments is shown in D and E. Statistical significance was calculated using unpaired two-tailed Student's t-test. (*) P < 0.05; (NS) not significant. (F–I) IMR90 cells were infected with lentivirus directing expression of 53BP1 or control, induced to senesce by irradiation (IR) or control (Prolif). Ten days after IR, cells were analyzed for the depicted proteins by immunoblotting (F), stained and quantified for CCFs (indicated by arrows) (G), assessed for the indicated SASP genes by RT-qPCR (H), or probed for the expression of IL8 by immunoblotting (I). Scale bar, 6 μm. Data shown in G and H represent mean ± SEM of three independent experiments. P-values were calculated by unpaired two-tailed Student's t-test. (****) P < 0.0001; (***) P < 0.001; (**) P < 0.01.
At the molecular level, 53BP1 inhibits double-strand break (DSB) end resection (Zimmermann and de Lange 2014), suggesting that DSB end resection is required for CCF formation. To test this idea, we directly inhibited DSB end resection by Mirin, an inhibitor of MRE11-mediated end resection (Dupré et al. 2008). Upon IR-induced senescence, as expected, Mirin suppressed γH2AX (Dupré et al. 2008), as shown by immunoblotting (Supplemental Fig. S3B). Strikingly, Mirin inhibited formation of CCFs and the SASP (Supplemental Fig. S3C,D), supporting the idea that 53BP1 suppresses CCFs by restraining DSB end resection. Future studies are needed to decipher the specific regulation of 53BP1 by JNK and the role of end resection in CCF formation.
HDAC inhibitors improve mitochondria function and suppress formation of CCFs and expression of the SASP
Given the important role of mitochondria as drivers of CCFs and the SASP, we considered the possibility that drugs could be used to restore mitochondria function and hence suppress CCFs and the SASP in senescent cells. Already we showed that JNK and MRE11 inhibitors block mito–nuclear signaling to suppress CCFs and the SASP (Fig. 3; Supplemental Figs. S2, S3). We showed above that nuclear-encoded mitochondrial oxidative phosphorylation genes were down-regulated in senescent cells (Fig. 1A; Supplemental Fig. S1A), and this is linked to mitochondria dysfunction (Fig. 1B–D). We speculated that this decrease in expression might be rescued by treatment with histone deacetylase inhibitors (HDACi), agents that often increase expression of genes by increasing histone acetylation (Haberland et al. 2009). We therefore examined whether HDACi can antagonize repression of nuclear-encoded mitochondrial oxidative phosphorylation genes and improve mitochondria function in senescence.
As expected, in senescent IMR90 cells, Trichostatin A (TSA) treatment increased histone acetylation in a dose-dependent manner, as shown by immunoblotting for H4K16ac (Fig. 5A,B; Sharma et al. 2010). For subsequent experiments, we used 100 nM TSA, a dose substantially below that typically found to be cytotoxic to cancer cells (Venkataramani et al. 2010; Khan and La Thangue 2012). This low dose of TSA was nontoxic, in contrast to higher doses, which were preferentially toxic to senescent cells compared with proliferating cells (i.e., senolytic, as noted previously for HDACi) (Supplemental Fig. S4A,B; Di Micco et al. 2011). Low-dose TSA did not revert the senescence-associated growth arrest, reflected in EdU incorporation and analysis of proliferation markers, including ph-Rb, cyclin A, and p21 (Fig. 5C,D). To verify this result, we performed RNA-seq on proliferating and senescent cells, pretreated with or without TSA. PCA showed separation of the four treatment groups (Supplemental Fig. S4C). However, analysis of expression of cell cycle promoting genes in RNA-seq data confirmed that TSA treatment of senescent cells did not rescue the proliferation arrest (Supplemental Fig. S4D). These results indicate that TSA does not affect repression of cell cycle genes nor the cell cycle arrest characteristic of senescence.
Figure 5.

HDAC inhibitors improve mitochondria function and suppress formation of CCFs and expression of the SASP. (A) Scheme of experimental design in B–M. (B) Proliferating or senescent (IR) IMR90 cells treated with TSA at indicated dose range (20–50–100 nM) were analyzed for H4K16ac and H4 by immunoblotting. (C) The effect of TSA (100 nM) on cell proliferation was assessed by EdU incorporation. Representative fluorescent images and quantification are shown. Scale bar, 10 μm. (D) Lysates from proliferating (Prolif) and senescent (IR) IMR90 cells with or without TSA (100 nM) were analyzed for the indicated proteins by immunoblotting. (E) Relative fold change in nuclear-encoded oxidative phosphorylation genes (KEGG hsa00190; subunits of complexes I–V) was represented in a histogram with bin size of 0.1 log2 fold change units. Counts in each bin were normalized to total number of nuclear-encoded mitochondrial genes detected. (F) The expression of the indicated nuclear-encoded mitochondrial genes was analyzed by RT-qPCR in senescent cells (IR) treated with or without TSA (100 nM). (G) Proliferating (Prolif) and senescent (IR) cells treated with or without TSA (100 nM) were analyzed for the indicated proteins by immunoblotting. (H) Proliferating (Prolif) and senescent (IR) cells treated with or without TSA were incubated with TMRE (100 nM) and Mitotracker Green (MTG; 100 nM) and analyzed by flow cytometry. The ratio of TMRE and MTG fluorescence intensity was used to represent mitochondrial membrane potential (MMP). (I) Senescent (IR) IMR90 cells with or without TSA (100 nM) were incubated with MitoSOX (5 μM) and analyzed by flow cytometry for mitochondrial reactive oxygen species. Percentage of positive cells is shown. For raw data, see Supplemental Figure S8. (J) Expression of ROS-responsive gene SOD2 was determined by RT-qPCR in proliferating (Prolif) and senescent (IR) IMR90 cells with or without TSA (100 nM). (K,L) Proliferating (Prolif) and senescent (IR) cells treated with or without TSA (100 nM) were imaged by confocal microscope and quantified for formation of γH2AX-positive CCFs (indicated by arrows) (K) or monitored for the expression of indicated SASP genes by RT-qPCR (L). Scale bar, 8 μm. Data shown in C, F, and H–L are mean ± SEM of three independent experiments. Statistical analysis was performed with unpaired two-tailed Student's t-test. (****) P < 0.0001; (***) P < 0.001; (**) P < 0.01; (*) P < 0.05. (M) Heat map of RNA-seq analysis of SASP genes in the indicated IMR90 cells from n = 3 independent experiments. Color intensity represents the Z-score of mean FPKM expression.
In contrast, examination of expression of nuclear-encoded mitochondrial oxidative phosphorylation genes demonstrated a general increase in their expression in TSA-treated senescent cells, shifting the expression profile in TSA-treated senescent cells toward that of proliferating cells (Fig. 5E). GSEA also revealed significant up-regulation of these genes in TSA-treated senescent (IR) cells (Supplemental Fig. S4E). We confirmed this observation by qPCR of mRNA transcripts. Nuclear-encoded oxidative phosphorylation mRNAs (NDUFA10, NDUFA12, NDUFC2, and NDUFS8) were higher in TSA-treated senescent cells relative to the control senescent cells (Fig. 5F). Similarly, protein levels of nuclear-encoded NDUFB8 and SDHA genes were increased by TSA treatment (Fig. 5G). Concomitant with this, TSA significantly increased mitochondria membrane potential (Fig. 5H), decreased mitochondrial ROS (Fig. 5I), and lowered expression of ROS-responsive gene SOD2 (Fig. 5J). NFE2L2 (NRF2) is a master regulator of cellular redox homeostasis (Ma 2013). A subset of NRF2 target genes was up-regulated in senescence, and some of these were down-regulated by TSA (Supplemental Fig. S4F), also consistent with a model whereby TSA suppresses mitochondria dysfunction.
We next asked whether TSA suppressed CCFs and the SASP in senescent cells. TSA treatment significantly decreased accumulation of CCFs in senescent cells (Fig. 5K). Consistent with this, TSA also strongly reduced the expression of key SASP genes, including IL1α, IL1β, IL6, and IL8 (Fig. 5L). Suppression of the SASP by HDACi was confirmed by RNA-seq. Overall, TSA antagonized the effect of senescence on expression of 606 genes (471 genes up-regulated in senescence (IR) and down-regulated by TSA (IR TSA); 135 down-regulated in senescence (IR) and up-regulated by TSA (IR TSA) (Supplemental Fig. S4G). Gene Ontology (GO) analysis of the 471 senescence-induced genes down-regulated by TSA identified several categories associated with the SASP, including immune response, response to wounding, inflammatory response, and cytokine activity (Supplemental Fig. S4H). In fact, specific analysis of SASP factors confirmed that TSA blunted the expression of most of them (Fig. 5M). The ability of TSA to impair the SASP was also observed at the secreted protein level in the conditioned medium by immunoblotting for IL8 (Supplemental Fig. S4I). We also tested a second HDAC inhibitor, Vorinostat (SAHA), already approved for use in humans, for example, against cutaneous T-cell lymphoma (Mann et al. 2007). IMR90 cells were treated with low doses of SAHA (as for TSA, lower than doses typically investigated as cytotoxic to cancer cells (Shi et al. [2017]) and induced to senesce by IR. Treatment with SAHA reduced formation of CCFs (Supplemental Fig. S4J) and attenuated induction of IL8, without rescuing the senescence growth arrest (Supplemental Fig. S4K). We also treated IMR90 senescent cells with TSA only after senescence was established (Supplemental Fig. S4L). In this setting, TSA also reduced formation of CCFs and expression of the SASP without affecting cell cycle arrest (Supplemental Fig. S4M–O). In addition, based on gene expression profiling by RNA-seq, there was a marked overlap (3.7 times larger than predicted from random; P < 0.001) between the effects of Parkin-mediated mitophagy and TSA on the senescence-associated gene expression program. Most notably, ∼50% of the senescence-regulated genes antagonized by TSA were also antagonized by Parkin-mediated mitophagy (Supplemental Fig. S4P). This overlap is consistent with the view that both mitophagy and TSA exert their effects, in part, by eradication of dysfunctional mitochondria, the former by their autophagic clearance and the latter by improving their function. In sum, HDACi improve mitochondria function and suppress CCF accumulation and the SASP program.
Despite these data, it is well-documented that HDACi can exert wide-ranging effects on cells (Xu et al. 2007). Therefore, we considered the possibility that HDACi might exert their effects on CCFs and the SASP by other mechanisms, aside from improved mitochondria function. HDACi did not affect proinflammatory signaling by CCFs, once present in the cytoplasm. As a CCF mimetic, transfection of cells with DNA or chromatin activates cytoplasmic cGAS and the SASP (Dou et al. 2017). Secretion of IL8 induced by transfection of chromatin fragments into proliferating cells was not inhibited in the presence of TSA (Fig. 6A), suggesting that TSA did not desensitize CCF sensing and the cGAS/STING signaling pathway. Aside from the CCF–cGAS–STING pathway, various other effectors have been implicated in SASP activation in senescent cells, including DNA damage, p38MAPK and mTOR as upstream drivers, and NFκB and C/EBPβ as proximal transcription factors (Acosta et al. 2008; Kuilman et al. 2008; Orjalo et al. 2009; Freund et al. 2011; Laberge et al. 2015; Herranz and Gil 2018). To gauge the specificity of HDACi for inhibition of the SASP by targeting of dysfunctional mitochondria, we assessed these other known SASP inducers. We found that TSA did not significantly alter DNA damage signaling, at least as reflected in the number of intranuclear γH2AX foci in senescent cells (Fig. 6B). Likewise, TSA did not inhibit p38MAPK and the mTOR pathway, as judged by phosphorylation of p38MAPK and S6, respectively (Fig. 6C,D). We next tested the possibility that TSA interfered with the NFκB signaling axis, thereby preventing SASP activation. In senescent cells, IL1α binds its receptor (IL1R1) and activates a signaling cascade that leads to degradation of IRAK1 and nuclear translocation of NFκB (Orjalo et al. 2009). We examined IRAK1 protein levels in cells treated with or without TSA and induced to senesce by IR (Fig. 6E). In the absence of TSA, IRAK1 was reduced and IL8 increased in response to irradiation, indicating activated IL1R1 and NFκB signaling, while in the presence of TSA the levels of IL8 decreased and IRAK1 increased, suggesting suppression of IL1R1 and NFκB signaling (Fig. 6E,F). However, when irradiated senescent cells were exposed to recombinant IL1α, TSA did not prevent the further down-regulation of IRAK1 (Fig. 6E) as well as the secretion of IL8 (Fig. 6F), suggesting that TSA did not suppress the SASP by making the NFκB pathway refractory to activation. Taken together, these results demonstrate a new class of agents able to target the senescence-associated inflammation, namely HDAC inhibitors. HDAC inhibitors preferentially improve mitochondria function and accordingly are potent suppressors of both CCF and SASP.
Figure 6.
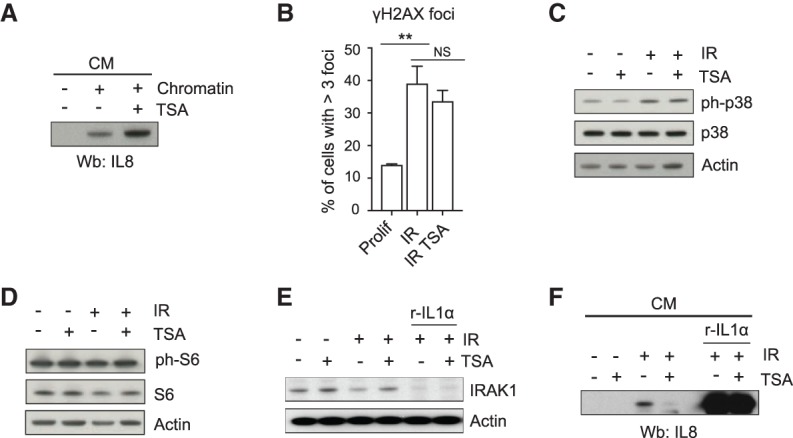
Effects of HDAC inhibitors on other pathways. (A) Proliferating IMR90 cells were transfected with chromatin fragments and treated with TSA as indicated. On day 4, conditioned medium (CM) was collected and analyzed for IL8 by immunoblotting. (B) Proliferating (Prolif) and senescent (IR) cells with or without TSA as in Figure 5A were immunostained for γH2AX. Shown is the percentage of cells with more than three intranuclear γH2AX foci. Data are mean ± SEM of three independent experiments. Statistical analysis was performed with unpaired two-tailed Student's t-test. (**) P < 0.01; (NS) not significant. (C) Proliferating and senescent (IR) IMR90 cells treated as in Figure 5A were analyzed for phospho-p38 (p-p38) and p38 by immunoblotting. (D) Immunoblot for phospho-S6 (p-S6) and S6 in proliferating and senescent (IR) cells 10 d after IR. (E,F) IMR90 cells were treated with TSA, irradiated to induce senescence, and incubated with recombinant IL1α (r-IL1α) as indicated. (E) Cell lysates were analyzed for the indicated proteins by immunoblotting 10 d after IR. (F) Conditioned medium (CM) was collected and probed for IL8 by immunoblotting 10 d after IR.
HDAC inhibitors suppress inflammation in vivo
Next we investigated the ability of HDAC inhibitors to suppress the SASP and inflammation associated with senescence and mitochondria dysfunction in vivo. We and others have shown that sublethal IR induces senescence, CCF formation, and the SASP in mouse liver (Dou et al. 2017; Glück et al. 2017). To test the ability of HDACi to suppress the SASP in this in vivo model, young adult (9-wk-old) C57BL/6 mice were administered vehicle (HPO-β-CD) or SAHA in drinking water for 1 d before IR and 7 d after IR (Fig. 7A). Liver samples were harvested at day 8 and analyzed by immunoblotting and immunohistochemistry. SAHA-treated mice showed increased histone acetylation (Fig. 7B,C). SAHA did not significantly attenuate the DNA damage marker γH2AX (Fig. 7D), similar to our in vitro results (Fig. 6B), and as expected since DNA damage is proximal to IR in this model. In contrast, SAHA strongly suppressed the induction of IL1α compared with vehicle-treated mice (Fig. 7D).
Figure 7.

HDAC inhibitors suppress inflammation in vivo. (A) Scheme of experimental design in B–D. C57BL/6 mice were pretreated with vehicle (HPO-β-CD) or SAHA (0.1 g/L) in drinking water, subjected to irradiation (IR, 4Gy), and administrated vehicle (HPO-β-CD) or SAHA in drinking water for seven more days. Seven days after IR, livers were harvested. (B,C) Livers were lysed and analyzed for H4K16ac, H4, H3K9ac, and H3 by immunoblotting. (D) Livers were stained for γH2AX and IL1α and quantified. Representative immunohistochemistry images are shown. N = 5 mice. Data are the mean ± SEM. Statistical significance was calculated using unpaired two-tailed Student's t-test. (*) P < 0.05; (NS) not significant. Scale bar, 20 μm. (E) Livers from mice treated as indicated were harvested 48 h after APAP administration, stained for γH2AX and IL1α. CCFs are indicated by arrows. The percentage of cells positive for CCFs and IL1α is shown. Representative immunohistochemistry images are shown. N = 5 mice for DMSO; n = 8 for APAP DMSO and APAP TSA. Scale bar, 20 μm. Data represent the mean ± SEM. Statistical analysis was performed with unpaired two-tailed Student's t-test. (****) P < 0.0001; (*) P < 0.05. (F) Livers from mice treated as indicated were harvested 48 h after APAP administration and stained for γH2AX and quantified. Representative immunohistochemistry images are shown. N = 5 mice for DMSO; n = 8 for APAP DMSO and APAP TSA. Scale bar, 20 μm. Data represent the mean ± SEM. Statistical analysis was performed with unpaired two-tailed Student's t-test. (****) P < 0.0001. (G,H) Livers were harvested 14 h after APAP injection, lysed, and subjected to RNA-seq analysis. (G) Heat map of RNA-seq analysis showing all genes that are significantly up-regulated in APAP DMSO versus untreated mice and significantly down-regulated in APAP TSA versus APAP DMSO. N = 4 mice per group. (H) Heat map of RNA-seq analysis of NRF2 target genes in mice treated as indicated, 14 h after APAP administration. N = 4 mice for cohort. Color intensities are Z-scores of the mean FPKM expression. (I) RT-qPCR analysis of NRF2 target genes in the liver of mice treated as indicated, 14 h after APAP administration. N = 10 mice for group. Data represent the mean ± SEM. Statistical analysis was performed with unpaired two-tailed Student's t-test. (**) P < 0.01; (*) P < 0.05.
To test the protective effect of HDACi in a model of mitochondria dysfunction, we took advantage of an established mouse model of mitochondria redox stress-induced liver injury, caused by acetaminophen (APAP) overdose. The toxicity of APAP is initiated by production of reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI), which depletes glutathione and covalently modifies mitochondrial proteins. This, in turn, causes mitochondrial oxidative stress and activation of JNK (Ramachandran and Jaeschke 2017), events that we have shown trigger CCF formation and the SASP in senescent cells in vitro. Indeed, it has been recently shown that APAP-induced liver injury is also associated with the induction of senescent-like cells, reflected by SA-β-gal and γH2AX, and increased expression of p21 and p16, as well as growth arrest and expression of the SASP (Bird et al. 2018). To probe the effects of HDACi on responses to APAP, we injected young (9-wk-old) C57BL/6 mice with TSA or DMSO 1 d before and simultaneously with APAP administration (here, we used TSA to complement the previous in vivo experiments with SAHA). TSA prevented CCF and SASP induction, assessed 48 h after APAP administration by staining for γH2AX and IL1α, respectively (Fig. 7E; Supplemental Fig. S5A). Consistent with a propagating role for SASP-mediated secondary senescence (Bird et al. 2018), TSA also markedly reduced p21 and intranuclear γH2AX levels induced by APAP exposure (Fig. 7F; Supplemental Fig. S5B).
We next harvested liver tissue after APAP dosing and performed RNA-seq, using four mice per cohort. PCA showed separation of the four cohorts based on gene expression (Supplemental Fig. S5C). Consistent with our in vitro data (Fig. 5E–G), RNA-seq analysis of nuclear-encoded oxidative phosphorylation genes revealed that TSA antagonized their global down-regulation in liver of APAP-treated mice after drug administration (Supplemental Fig. S5D,E). Analysis of all genes that are differentially expressed showed that 1825 genes were affected upon APAP treatment (1201 genes up-regulated and 624 genes down-regulated) (Supplemental Fig. S5F). Interestingly, 445 genes whose expression was increased by APAP were suppressed in APAP TSA-treated mice (Fig. 7G; Supplemental Fig. S5F). IPA analysis of these genes showed NRF2 as one of the top upstream regulators that is activated in APAP-treated mice and down-regulated by TSA (Supplemental Fig. S5G). Upon oxidative stress, NRF2 accumulates in the nucleus, promoting the expression of target genes, including Heme oxygenase 1 (Hmox1), Sulfiredoxin 1 (Srxn1), NAD(P)H dehydrogenase (quinone) 1 (Nqo1), and Glutathione S-transferase 1 (GSTA1) (Walsh et al. 2014; Forootan et al. 2017). Specifically, ∼20 out of 76 documented NRF2 target genes (Hayes and Dinkova-Kostova 2014) showed a trend to up-regulation in APAP-treated mice (Fig. 7H), and TSA suppressed the majority of these, including Cbr1, Ptgr1, Nqo1, Gsta1, Gpx4, Srxn1, and Hmox1. These results were confirmed by qPCR for selected NRF2 targets, Gsta1, Hmox1, Nqo1, and Srnx1 (Fig. 7I). To more directly ask whether TSA affects APAP-induced oxidative stress, we stained liver tissues from APAP mice treated with or without TSA with the lipid peroxidation product 4-hydroxy-2-nonenal (4-HNE). We found that TSA reduced the levels of 4-HNE after APAP administration (Supplemental Fig. S5H). The observation that TSA suppresses down-regulation of nuclear-encoded mitochondrial oxidative phosphorylation genes, up-regulation of NRF2 target genes, oxidative damage, CCFs, inflammation, and secondary senescence is consistent with the idea that TSA exerts its effects by suppression of ROS and DNA damage at the source—in other words, by countering mitochondria dysfunction in this in vivo model.
Discussion
This work addresses a major challenge of research into the biology of aging, specifically to define the molecular relationships between the so-called hallmarks of aging (López-Otín et al. 2013; Kennedy et al. 2014). To this end, here, we charted a molecular pathway that links dysfunctional mitochondria to inflammatory signaling of senescent cells. Specifically, we defined a novel mitochondria–nuclear retrograde signaling pathway that triggers formation of CCFs, a trigger of the SASP. This pathway is initiated by dysfunctional mitochondria producing ROS, an activator of JNK kinase. JNK kinase's initiation of CCF formation in senescent cells is linked to its interaction with 53BP1, a negative regulator of DSB end resection and CCF formation. Importantly, this pathway is targetable by drug interventions, some of which show promise as anti-inflammatory/prohealthy aging interventions. As proof of this concept, we show here that MRE11, JNK, and HDAC inhibitors suppress CCF formation and the SASP in senescent cells. Aside from their anticancer activities (Falkenberg and Johnstone 2014), HDACi have been previously reported to suppress inflammation and extend the health span and life span in multiple animal models (Hockly et al. 2003; Tao et al. 2004; Gao et al. 2009; Guan et al. 2009; Krishnan et al. 2011; Hull et al. 2016). However, the underlying mechanisms are unknown.
Since we observe decreased nuclear-encoded mitochondrial oxidative phosphorylation gene expression in senescent cells, our data is consistent with increased mitochondrial mass due to decreased mitophagy in senescent cells, as established previously in the literature (Dalle Pezze et al. 2014; Korolchuk et al. 2017). Decreased mitophagy is typically associated with mitochondrial dysfunction (Korolchuk et al. 2017; Pickles et al. 2018), hence the decreased mitochondria membrane potential and ROS production in senescent cells. However, since mitochondria ROS production results from electron leakage from the electron transport change during oxidative phosphorylation (Brand and Nicholls 2011), it is also tempting to speculate that down-regulation of nuclear-encoded oxidative phosphorylation genes is directly related to increased ROS production in senescent cells.
By several measures, HDACi improve mitochondria function, thereby suppressing oxidative stress, CCFs, and the SASP, both in vitro and in vivo. Improved mitochondria function correlates with specific up-regulation of nuclear-encoded mitochondrial oxidative phosphorylation genes, compared with cell cycle genes whose senescence-associated repression is unaffected by HDACi in the in vitro model. HDACi's ability to increase expression of oxidative phosphorylation genes may contribute to their beneficial effects on mitochondria function. By perhaps the simplest scenario, HDACi might act by increasing histone acetylation at the oxidative phosphorylation genes themselves (Haberland et al. 2009), thereby promoting their expression. However, HDACi have many nonhistone substrates and pleiotropic effects on cells (Xu et al. 2007), so alternative more indirect mechanisms are possible. For example, HDACi may also act to enhance mitophagy, thereby also improving mitochondrial function. The relationship between expression of nuclear-encoded oxidative phosphorylation genes and mitophagy in senescence is unknown. Regardless of the specific mechanism, the demonstration that HDACi can improve mitochondria function is important because it may contribute to the documented, but poorly understood, anti-inflammatory and prolongevity effects of these drugs (Hockly et al. 2003; Tao et al. 2004; Gao et al. 2009; Guan et al. 2009; Krishnan et al. 2011; Hull et al. 2016). Future studies are required to address outstanding questions.
In apparent contrast to the anti-SASP activity shown here, TSA and other HDACi have been previously reported to induce senescence or to kill senescent human cells. Importantly, previous prosenescence or senolytic effects of HDACi in human cells have been observed with substantially higher doses of HDACi than we found to inhibit the SASP. Previous studies have employed >1 µM TSA, whereas here we report anti-SASP activity at 0.05–0.5 µM TSA. We confirmed that higher doses of TSA are toxic to senescent cells (Supplemental Fig. S4A,B). In sum, there is a clear dose dependence, with anti-SASP activity at low doses and senolytic activity at higher doses.
We acknowledge that unresolved issues remain; for example, the precise regulatory interaction between JNK, 53BP1, and DSB end resection in CCF formation is unknown at this stage, as is their relationship to other regulators of CCFs, such as autophagy factor LC3 and nuclear scaffold component lamin B (Dou et al. 2015). Regarding the relationship between JNK and 53BP1, we speculate that JNK kinase phosphorylates 53BP1 to modulate its control of end resection and hence CCF formation. If so, JNK kinase may also control 53BP1's role in DNA repair in senescent cells. Alternatively, the regulation of CCFs by JNK-53BP1 may be separable from 53BP1's role in DNA repair. Ongoing and future studies will address this.
To conclude, this study assimilates previously disparate facets of the biology of senescence, defines a novel mitochondria-to-nucleus signaling pathway in senescent cells that initiates formation of CCFs and the SASP, and demonstrates the potential utility of this pathway as a target for suppression of inflammatory responses.
Materials and methods
Cell culture
Human primary IMR90 fibroblasts were grown in Dulbecco's modified Eagle's medium (DMEM) (Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM glutamine at 37°C in a humidified atmosphere with 5% CO2 and 3% oxygen. Human embryonic kidney HEK293 and Phoenix amphotropic cells were grown in DMEM (Thermo Fisher Scientific), supplemented with 10% foetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM glutamine at 37°C in a humidified atmosphere with 5% CO2 and 20% oxygen. Cells were regularly checked to confirm absence of mycoplasma contamination.
Induction of senescence
Proliferating IMR90 cells at ∼40%–50% confluency were induced to senesce by X-ray irradiation with 20 Gy and allowed to recover overnight. The media were then replaced, and cells were trypsinized and split 1:4 2 d later; cells were analyzed at day 10. Alternatively, IMR90 cells at ∼60%–70% confluency were induced to senesce by incubation with 600 μM H2O2 (Sigma-Aldrich) in serum-free media for 2 h or with 40 μM MitoParaquat (MitoPQ, Abcam) for 9 d. For etoposide-induced senescence, IMR90 cells at ∼60%–70% confluency were treated with 50 μM for 24 h. The media were then refreshed, and cells were harvested at day 8.
Retroviral and lentiviral infection
Retroviral LZRS or LZRS-YFP_Parkin (a kind gift from Dr. Stephen Tait, University of Glasgow, UK) and pLPC-N Myc (Addgene 12540) or N-Myc-53BP1 WT pLPC (Addgene 19836) constructs were used to transfect a Phoenix packaging cell line. Lentiviral pLKO constructs along with packaging plasmids were used to transfect HEK293T cells. Phoenix and HEK293T cells were seeded at 70% confluency and transfected using Lipofectamine 2000 (Thermo Fisher Scientific). Six hours later, the transfection medium was replaced by new culture medium. At 24 h after transfection, viral supernatant was filtered through a 0.45-μm-pore-size PVDF filter (Millipore), mixed with new culture medium in the proportion 1:1, supplemented with 8 μg/mL polybrene and added to recipient cells for 20 h. The infected cells were selected with either 250 μM zeocin (Thermo Fisher Scientific; for LZRS and LZRS-YFP_Parkin) or 1 μg/mL puromycin (Thermo Fisher Scientific; for PLPC-N Myc, N-Myc-53BP1 WT pLPC and pLKO constructs). pLKO-based shRNA was from Dharmacon. The following shRNAs were used: MAPK8 (JNK1, TRCN0000001055), MAPK9 (JNK2, TRCN000000943), and TP53BP1 (TRCN0000018865).
Treatments
For mitochondrial depolarization, LZRS and YFP-Parkin cells were treated for 48 h with 12.5 μM CCCP (Sigma-Aldrich), 2 d after irradiation or etoposide treatment.
Antioxidant treatment was performed daily in irradiated cells treated 8 h before irradiation with 100 nM MitoQ (MedKoo Bioscience) for 10 d. Unless stated otherwise, for JNK inhibition cells were treated daily for 8 h before the irradiation with 20 μM SP600125 (Selleck Chemicals) for 10 d. For MRE11 inhibition, cells were treated with 100 μM Mirin (Selleck Chemicals) immediately after the irradiation for 10 d. The following HDAC inhibitors were used: 100 nM Trichostatin A (TSA) (Sigma- Aldrich) and Vorinostat (SAHA) (Selleck Chemicals) at the indicated doses were added 8 h before the irradiation for 10 d, unless stated otherwise. Drugs were replaced daily. Recombinant human IL1α protein (R&D Systems) was added to irradiated cells at 20 ng/mL for 24 h in serum-free medium.
Conditioned medium
Cells were cultured in serum-free medium for 24 h. The cultured media were then collected, and the cell numbers were counted for normalization. The media were filtered with 0.45-μm PVDF filter (Millipore) to remove cells and debris. The resulting supernatant was concentrated with a 5000 MWCO spin concentrator (Agilent Technologies) and analyzed by immunoblotting.
Antibodies
The following antibodies were used: NDUFB8 (Abcam ab110242), SDHA (Cell Signaling Technology 11998), UQCR2 (Abcam ab14745), Tom20 (Santa Cruz Biotechnology sc-11415), p-Rb ser807/811 (Cell Signaling Technology 9308), Cyclin A (Santa Cruz Biotechnology sc-751), p21 (Santa Cruz Biotechnology sc-471 and HUGO 291 Abcam ab107099), IL8 (Abcam ab18672), p-JNK Thr183/Tyr188 (Cell Signaling Technology 9251), JNK (Cell Signaling Technology 9252 and Santa Cruz Biotechnology sc-7345), γH2AX ser139 (Millipore-Merck 05-636 and Cell Signaling Technology 9718), 53BP1 (Abcam 21083 and Cell Signaling Technology 4937), H4K16ac (Millipore-Merck 07-329), H4 (Active Motif 39163), H3K9ac (Active Motif 61252), H3 (Active Motif 39763), cleaved caspase 3 (Cell Signaling Technology 9661), p-38 MAPK Thr180/Tyr182 (Cell Signaling Technology 9211), p38 MAPK (Cell Signaling Technology 9212), p-S6 ser235/236 (Cell Signaling Technology 2211), pS6 (Cell Signaling Technology 2317), IRAK1 (Santa Cruz Biotechnology 5288), Actin (Sigma-Aldrich A1978), GAPDH (Cell Signaling Technology 5174), IL1α (R&D Systems AF-400), and 4-HNE (JaICA).
Immunoblotting
Cells were lysed in SDS sample buffer (62.5 mM Tris at pH 6.8, 0.01% bromophenol blue, 10% glycerol, 2% SDS) or in EBC buffer (50 mM Tris-HCl at pH 8.0, 120 mM NaCl, 0.5% NP40). Tissues were removed and homogenized in SDS sample buffer. Proteins were separated on a 4%–12% Bis-Tris gel (NuPAGE, Thermo Fisher Scientific) by electrophoresis and transferred to PVDF membrane. The membranes were blocked in 4% BSA in Tris-buffered saline (TBS) for 1 h at room temperature and then incubated with the primary antibodies overnight at 4°C. Primary antibodies were diluted in 4% BSA in TBS supplemented with 0.1% Tween 20 (TBST). Membranes were washed three times with TBST, followed by incubation with horseradish-peroxidase-conjugated secondary antibodies at room temperature for 1 h in 4% BSA/TBS. The membranes were washed again three times. Antibody binding was visualized using a Pierce ECL Western blotting substrate (Pierce 32106).
Immunoprecipitation
Cells were lysed in EBC buffer containing 50 mM Tris-HCl pH 8.0, 120 mM NaCl, 0.5% NP40 supplemented with protease inhibitor cocktail (Sigma-Aldrich). The lysate was rotated for 30 min at 4°C followed by centrifugation at 13000g for 10 min at 4°C. The supernatant was incubated with antibody-conjugated Dynabeads (Thermo Fisher Scientific) and rotated overnight at 4°C. The IP was washed and collected by magnet five times with buffer containing 20 mM Tris (pH 8), 1 mM EDTA, 0.5% NP40, and 100 mM NaCl. After washing, the IP was boiled with SDS sample buffer. Samples were analyzed by immunoblotting.
Immunofluorescence
Cells were seeded onto coverslips and fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. Cells were washed twice with PBS and permeabilized with 0.2% Triton X-100 in PBS for 5 min. After washing twice, cells were blocked in 3% BSA in PBS supplemented with 1% goat serum for 1 h at room temperature. Cells were then incubated with primary antibodies in 3% BSA in PBS supplemented with 1% goat serum for 1 h at room temperature. Next, cells were washed three times with PBS supplemented with 0.1% Tween 20 (PBST) for 5 min each followed by incubation with Alexa Fluor-conjugated secondary antibody (Thermo Fisher Scientific) in 3% BSA in PBS supplemented with 1% goat serum for 1 h at room temperature. The cells were then washed three times with PBS and incubated with 1 μg/mL DAPI for 5 min. The cells were then washed twice with PBS and mounted with ProLong Gold (Thermo Fisher Scientific) and imaged with a Leica or NikonA1R laser scanning confocal microscope. Quantification of the percentage of positive cells for γH2AX was done manually by using ImageJ software. Over 200 cells from 10 randomly selected fields were analyzed.
Quantitative real-time PCR
Total RNA from cells was extracted using the RNeasy Mini kit (Qiagen), with a DNase I (Qiagen) digestion to minimize genomic DNA contamination. Complementary DNAs were generated using SuperScript III reverse transcriptase (Thermo Fisher Scientific), dNTPs, and random hexamers. Quantitative real-time PCR was carried out in a C1000TM Thermal Cycler (Bio-Rad) using TaqMan Universal PCR Master Mix (Bio-Rad). The average of the ΔΔC(t) of triplicate samples was calculated using GAPDH as a housekeeping gene. The following TaqMan probes (Thermo Fisher Scientific) were used for RT-qPCR of human cells: IL1α (Hs00174092_m1), IL1β (Hs01555410_m1), CXCL8 (Hs01553824_g1), IL6 (Hs00174131_m1), SOD2 (Hs00167309_m1), and GAPDH (Hs02786624_g1). Mouse liver tissue samples were weighed and total RNA was extracted from 30 mg using the RNeasy Mini kit (Qiagen). Reverse transcription was performed as previously described. Quantitative real-time PCR was carried out in a C1000TM Thermal Cycler (Bio-Rad) using DyNAmo SYBR Green Master Mix (Thermo Fisher Scientific). The average of the ΔΔC(t) of triplicate samples was calculated using GAPDH as a housekeeping gene. The following primers were used for RT-qPCR of mouse tissues: Hmox1 (GTCAAGCACAGGGTGACAGA and ATCACCTGCAGCTCCTCAAA), Gsta1 (CAGCCTGGCAGCCAGAGA and TCTGTGGCTCCATCAATGCA), Nqo1 (TTTAGGGTCGTCTTGGCAAC and GTCTTCTCTGAATGGGCCAG), Srxn1 (AAAGTGCAGAGCCTGGTG and CTTTGATCCAGAGGACGTCG), and Gapdh (AACTTTGGCATTGTGGAAGG and ACACATTGGGGGTAGGAACA).
RNA sequencing
Total RNA was extracted from cells or 30 mg of mouse tissue using the RNeasy minikit (Qiagen). Purified RNA was tested on an Agilent 2200 TapeStation using RNA screentape. mRNA-seq libraries were prepared using the TruSeq Stranded LT (Illumina kit). Samples were run on the Illumina NextSeq 500 using a High Output 75 cycle kit (twice for 36 cycles, paired-end reads, single index). Sample data were demultiplexed using the bcl2fastq tool (Illumina) before read quality was assessed using the FastQC algorithm, and then reads were trimmed appropriately using the TrimGalore! tool (Babraham Bioinformatics). Data were then aligned to an hg19 or mm9 reference genome (Ensembl), as appropriate, using the Tophat2 algorithm (Trapnell et al. 2010; Kim et al. 2013). FPKM determination and differential expression analysis were performed using the Cufflink and Cuffdiff tools, respectively, from the Cufflinks suite. PCA analysis was performed using custom R scripts, then plotted using the Rgl package in R. Heat maps showing FPKM values of named gene subsets were produced using custom R scripts, with cohort means of Z-scores plotted.
RNA sequencing data were uploaded to GEO under accession number GSE132371.
Nanostring
Total RNA from cells was extracted using an RNeasy minikit (Qiagen). Purified RNA was tested on an Agilent 2200 TapeStation using RNA screentape. Samples were run on an nCounter platform using the nCounter custom human SASP panel containing 31 canonical SASP genes (Coppé et al. 2010) and three internal reference genes or nCounter human inflammation panel containing 249 inflammation-related genes and six internal reference genes. Hybridization reactions were performed according the manufacturer's instructions (NanoString Technologies). To assess data quality, the field of view counts and binding density measurements were manually extracted from the data and verified as acceptable according to the manufacturer's protocol. Positive control linearity and limit of detection were then verified using custom R scripts. Counts were normalized using the NanoStringNorm R package, using the “mean.3ds” and “geo.mean” parameters to return high confidence values. Differential expression analysis was performed using nSolver or custom R scripts to determine log2 fold changes and P-values (obtained using Student's t-test). P-values of <0.05 were considered statistically significant. PCA analysis was performed using custom R scripts, then plotted using the Rgl package in R. Heat maps depicting the normalized counts of named gene subsets were produced using GraphPad Prism v8 or custom R scripts, with cohort means of Z-scores plotted.
Click-iT EdU incorporation assay
Cells were seeded onto coverslips and incubated with 10 μM EdU (Thermo Fisher Scientific) in complete medium for 2 h. Cells were then washed twice with PBS and fixed with 3.7% formaldehyde in PBS for 15 min at room temperature, followed by permeabilization with 0.5% Triton X-100 in PBS for 20 min at room temperature. The EdU detection was performed following the instructions of the Click-iT imaging kit (Thermo Fisher Scientific). Ten images per condition were acquired with a 40× magnification objective. The number of EdU-positive cells and DAPI-positive cells was determined using ImageJ.
Measurement of mitochondrial ROS
Mitochondrial ROS were determined by harvesting and incubating ∼ 5 × 105 live cells with 5 µM MitoSOX Red (Molecular Probes, Thermo Fisher Scientific) in serum-free DMEM for 10 min at 37°C in the dark. Cells were centrifuged at 1200 rpm for 2 min, the supernatant was discarded, and the cell pellet was resuspended in PBS for flow cytometry analysis. For raw data, see Supplemental Figures S6–S8.
Measurement of mitochondrial membrane potential
The indicated cells (5 × 105) were seeded and incubated with 100 nM tetramethylrhodamine ethyl ester (TMRE) (Thermo Fisher Scientific) and 100 nM Mitotracker Green FM (MTG, Thermo Fisher Scientific) in complete medium for 30 min at 37°C in the dark. Cells were trypsinized, and the cell pellet was washed with PBS and analyzed by flow cytometry. The ratio of TMRE to MTG fluorescence intensity was used to represent the mitochondrial membrane potential per unit of mitochondrial mass.
Chromatin fragment extraction and transfection
Extraction of chromatin fragments was performed as described previously (Dou et al. 2017). Briefly, proliferating IMR90 cells were scraped with buffer containing 10 mM Tris (pH 7.5), 30 mM NaCl, 3 mM MgCl2, and 0.1% NP40, supplemented with protease inhibitor cocktail (Sigma-Aldrich). Following 10 min of incubation on ice, the suspension was centrifuged at 300g for 3 min at 4°C. The supernatant was removed, and the pellet was incubated with benzonase buffer containing 50 mM Tris (pH 7.5), 300 mM NaCl, 0.5% NP40, and 2.5 mM MgCl2, with protease inhibitor cocktail (Sigma-Aldrich) supplemented with 10 U of benzonase (Sigma-Aldrich). The lysate was incubated for 30 min on ice. The product was centrifuged at 300g for 5 min at 4°C. The supernatant was diluted twice with buffer containing 50 mM Tris (pH 7.5), 300 mM NaCl, 0.5% NP40, and 15 mM EDTA. Chromatin fragments were transfected to proliferating IMR90 cells using Lipofectamine 2000 (Thermo Fisher Scientific). Transfected cells were harvested 4 d after transfection.
Mouse experiments
Male C57BL/6 mice (6–8 wk old) were purchased from Charles River Laboratories. Mice were maintained under a specific pathogen-free environment and kept under standard conditions with a 12-h light/dark cycle and fed ad libitum on a regular diet. All animal experiments were carried out under procedural guidelines and severity protocols with ethical permission form the Animal Welfare and the Ethical Review Body and Home Office (UK).
For ionizing radiation, mice were treated with 300 mg/L Vorinostat (SAHA) (Selleck Chemicals) or the equivalent concentration of vehicle 2-hydroxypropyl-β-cyclodextrin (HOP-β-CD) (Sigma-Aldrich) in drinking water 1 d prior to irradiation for 8 d. SAHA and HOP-β-CD were prepared as described previously (Hockly et al. 2003). Briefly, 0.3 g of SAHA was added to a solution of 9 g of HOP-β-CD in 1 L of water and heated until fully dissolved. An equivalent concentration of HOP-β-CD without SAHA was given to the vehicle-treated mice. Mice were then exposed to a sublethal dose of 4 Gy irradiation and were harvested 7 d after the procedure.
For acetaminophen (APAP) studies, 9-wk-old male C57BL/6 mice were administered 0.5 mg/kg Trichostatin A (TSA) (Sigma-Aldrich) or vehicle 10% DMSO via intraperitoneal (i.p.) injection 1 d prior to APAP administration. APAP was delivered in PBS at 350 mg/kg by single i.p. injection of 20 μL/g after a 10-h fast.
Mice were sacrificed by CO2 inhalation. Liver tissue was harvested and fixed in formalin for histological analysis. Part of the tissue was also frozen and stored at −80°C for biochemical analysis.
Immunohistochemistry
After formalin fixation for 24 h, tissues were embedded in paraffin wax. Three-micrometer-thick sections were subjected to immunohistochemical staining using standard procedures at the institutional histology core facilities. Briefly, sections were dewaxed, dehydrated, and subjected to antigen retrieval (25-min incubation at 98°C in citrate buffer at pH 6). After incubation with the primary antibodies for 35 min at room temperature and blocking of endogenous peroxidase (Dako) for 5 min at room temperature, sections were incubated for 30 min at room temperature with the detection system (Envision system, Dako), followed by incubation with 3,3′-diaminobenzidine tetrahydrochloride (Dako) and counterstaining with hematoxylin (Cell Path). 4-HNE staining was performed using the Vectastain ABC system (Vector Laboratories) with the avidin/biotin peroxidase complex (ABC) method and M.O.M. (mouse on mouse) immunodetection kit. Sections were incubated in NovaRed (Vector Laboratories) solution and counterstained in hematoxylin (Sigma Aldrich). Quantification of positively stained hepatocytes is the mean value of four randomly imaged fields per mouse.
Statistical analysis
Statistical significance was calculated by unpaired two-tailed Student's t-test and one-way ANOVA coupled with Dunnett's test as described in the figure legends. Prism 6 software was used to generate graphs and to perform statistical analysis. P-values of statistical significance are represented as P < 0.0001 (****), P < 0.001 (***), P < 0.01 (**), and P < 0.05 (*). Observed/expected overlaps analysis was performed using permutation analysis to simulate >1000 random overlaps of appropriate size. Gene set enrichment analysis (http://software.broadinstitute.org/gsea/index.jsp) was conducted using a custom gene set of 85 nuclear-encoded oxidative phosphorylation genes and was run with default parameters using 1000 gene set permutations
Supplementary Material
Acknowledgments
We thank all members of the Adams laboratory for critical discussion. We thank Dr. Stephan Tait for generous sharing of reagents, and Dr. Karthic Swaminathan for his help with confocal microscopy. We acknowledge the Core Services and Advanced Technologies at the Cancer Research UK Beatson Institute (C596/A17196), with particular thanks to the Animal Core Facility and Biological Services Unit and Histology. Special thanks to Karen Blyth for advice on animal experiments. The work in the laboratory of P.D.A. and S.L.B. was supported by AG031862. K.N.M. and N.D. were supported by Glenn Foundation for Medical Research Postdoctoral Fellowships (PD18082 and PD19131, respectively). K.N.M. was also supported by F32 AG066459. A.B.L. and J.F.P. were supported by the Ted Nash Foundation. C.K. was supported by Cancer Research UK Beatson Institute Core funding (A171196). T.G.B. was funded by the Wellcome Trust (WT107492Z). Z.D. was supported by National Institutes of Health K99AG053406. S.L.B. and P.D.A. were supported by AG031862.
Author contributions: M.G.V., Z.D., and P.D.A. conceived the project. M.G.V. carried out the bulk of the experiments. T.L., K.N.M., A.B.L., A.P.-G., C.K., N.D, P.J.K, D.J., C.N., and W.C. carried out further experiments. K.N.M., N.A.R., K.G., and X.L. carried out the bioinformatics analysis. T.G.B., J.F.P., and S.L.B. provided substantial and critical intellectual input. M.G.V. and P.D.A. wrote the manuscript. All authors discussed the results and revised the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.331272.119.
References
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. 2008. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018. 10.1016/j.cell.2008.03.038 [DOI] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang T-W, Lasitschka F, Andrulis M, et al. 2013. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15: 978–990. 10.1038/ncb2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232–236. 10.1038/nature10600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird TG, Müller M, Boulter L, Vincent DF, Ridgway RA, Lopez-Guadamillas E, Lu W-Y, Jamieson T, Govaere O, Campbell AD, et al. 2018. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci Transl Med 10: eaan1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. 2011. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312. 10.1042/BJ20110162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868. 10.1371/journal.pbio.0060301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118. 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia-Melo C, Marques FDM, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, et al. 2016. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35: 724–742. 10.15252/embj.201592862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle Pezze P, Nelson G, Otten EG, Korolchuk VI, Kirkwood TBL, von Zglinicki T, Shanley DP. 2014. Dynamic modelling of pathways to cellular senescence reveals strategies for targeted interventions. PLoS Comput Biol 10: e1003728 10.1371/journal.pcbi.1003728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, dal Zuffo R, Matti V, d'Ario G, Montani E, et al. 2011. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol 13: 292–302. 10.1038/ncb2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan J-A, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. 2015. Autophagy mediates degradation of nuclear lamina. Nature 527: 105–109. 10.1038/nature15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. 2017. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550: 402–406. 10.1038/nature24050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupré A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee J-H, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, et al. 2008. A forward chemical genetic screen reveals an inhibitor of the Mre11–Rad50–Nbs1 complex. Nat Chem Biol 4: 119–125. 10.1038/nchembio.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg KJ, Johnstone RW. 2014. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov 13: 673–691. 10.1038/nrd4360 [DOI] [PubMed] [Google Scholar]
- Forootan SS, Mutter FE, Kipar A, Iwawaki T, Francis B, Goldring CE, Park BK, Copple IM. 2017. Real-time in vivo imaging reveals localised Nrf2 stress responses associated with direct and metabolism-dependent drug toxicity. Sci Rep 7: 16084 10.1038/s41598-017-16491-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Orjalo AV, Desprez P-Y, Campisi J. 2010. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16: 238–246. 10.1016/j.molmed.2010.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Patil CK, Campisi J. 2011. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 30: 1536–1548. 10.1038/emboj.2011.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J. 2009. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58: 1509–1517. 10.2337/db08-1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. 2017. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19: 1061–1070. 10.1038/ncb3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J-S, Haggarty SJ, Giacometti E, Dannenberg J-H, Joseph N, Gao J, Nieland TJF, Zhou Y, Wang X, Mazitschek R, et al. 2009. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459: 55–60. 10.1038/nature07925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. 2009. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10: 32–42. 10.1038/nrg2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Dinkova-Kostova AT. 2014. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39: 199–218. 10.1016/j.tibs.2014.02.002 [DOI] [PubMed] [Google Scholar]
- Herranz N, Gil J. 2018. Mechanisms and functions of cellular senescence. J Clin Invest 128: 1238–1246. 10.1172/JCI95148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PAS, et al. 2003. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci 100: 2041–2046. 10.1073/pnas.0437870100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull EE, Montgomery MR, Leyva KJ. 2016. HDAC inhibitors as epigenetic regulators of the immune system: impacts on cancer therapy and inflammatory diseases. Biomed Res Int 2016: 8797206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. 2013. Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202: 129–143. 10.1083/jcb.201212110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. 2011. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: 547–551. 10.1038/nature10599 [DOI] [PubMed] [Google Scholar]
- Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EME, Cascante M, Shlomi T, et al. 2013. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498: 109–112. 10.1038/nature12154 [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, et al. 2014. Geroscience: linking aging to chronic disease. Cell 159: 709–713. 10.1016/j.cell.2014.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, La Thangue NB. 2012. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 90: 85–94. 10.1038/icb.2011.100 [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. 2017. The clinical potential of senolytic drugs. J Am Geriatr Soc 65: 2297–2301. 10.1111/jgs.14969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner RE, Verma P, Molloy KR, Chait BT, Kapoor TM. 2015. Chemical proteomics reveals a γH2AX-53BP1 interaction in the DNA damage response. Nat Chem Biol 11: 807–814. 10.1038/nchembio.1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJPL, Dansen TB, Polderman PE, Saarloos I, Wirtz KWA, Coffer PJ, Huang T-T, Bos JL, Medema RH, Burgering BMT. 2002. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419: 316–321. 10.1038/nature01036 [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. 2017. Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine 21: 7–13. 10.1016/j.ebiom.2017.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Chow MZY, Wang Z, Zhang L, Liu B, Liu X, Zhou Z. 2011. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci 108: 12325–12330. 10.1073/pnas.1102789108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133: 1019–1031. 10.1016/j.cell.2008.03.039 [DOI] [PubMed] [Google Scholar]
- Laberge R-M, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et al. 2015. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17: 1049–1061. 10.1038/ncb3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153: 1194–1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q. 2013. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53: 401–426. 10.1146/annurev-pharmtox-011112-140320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann BS, Johnson JR, He K, Sridhara R, Abraham S, Booth BP, Verbois L, Morse DE, Jee JM, Pope S, et al. 2007. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin Cancer Res 13: 2318–2322. 10.1158/1078-0432.CCR-06-2672 [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Lahair MM, Franklin RA. 2006. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal 8: 1775–1789. 10.1089/ars.2006.8.1775 [DOI] [PubMed] [Google Scholar]
- Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, et al. 2018. Senescence-associated reprogramming promotes cancer stemness. Nature 553: 96–100. 10.1038/nature25167 [DOI] [PubMed] [Google Scholar]
- Moiseeva O, Bourdeau V, Roux A, Deschênes-Simard X, Ferbeyre G. 2009. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol 29: 4495–4507. 10.1128/MCB.01868-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. 2012. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11: 345–349. 10.1111/j.1474-9726.2012.00795.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. 2018. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech Ageing Dev 170: 30–36. 10.1016/j.mad.2017.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Robbins PD. 2018. Senotherapeutics for healthy ageing. Nat Rev Drug Discov 17: 377 10.1038/nrd.2018.44 [DOI] [PubMed] [Google Scholar]
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. 2009. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci 106: 17031–17036. 10.1073/pnas.0905299106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, et al. 2007. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol 5: e110 10.1371/journal.pbio.0050110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Vigié P, Youle RJ. 2018. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28: R170–R185. 10.1016/j.cub.2018.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Jaeschke H. 2017. Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J Clin Transl Res 3: 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb EL, Gawel JM, Aksentijević D, Cochemé HM, Stewart TS, Shchepinova MM, Qiang H, Prime TA, Bright TP, James AM, et al. 2015. Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free Radic Biol Med 89: 883–894. 10.1016/j.freeradbiomed.2015.08.021 [DOI] [PubMed] [Google Scholar]
- Rodier F, Campisi J. 2011. Four faces of cellular senescence. J Cell Biol 192: 547–556. 10.1083/jcb.201009094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber M, Chandel NS. 2014. ROS function in redox signaling and oxidative stress. Curr Biol 24: R453–R462. 10.1016/j.cub.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, Bhadra U, Pandita RK, Porteus MH, Chen DJ, et al. 2010. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol Cell Biol 30: 3582–3595. 10.1128/MCB.01476-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Nikulenkov F, Zawacka-Pankau J, Li H, Gabdoulline R, Xu J, Eriksson S, Hedström E, Issaeva N, Kel A, et al. 2014. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ 21: 612–623. 10.1038/cdd.2013.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X-Y, Ding W, Li T-Q, Zhang Y-X, Zhao S-C. 2017. Histone deacetylase (HDAC) inhibitor, suberoylanilide hydroxamic acid (SAHA), induces apoptosis in prostate cancer cell lines via the Akt/FOXO3a signaling pathway. Med Sci Monit 23: 5793–5802. 10.12659/MSM.904597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short S, Fielder E, Miwa S, von Zglinicki T. 2019. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 41: 683–692. 10.1016/j.ebiom.2019.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao D, Lu J, Sun H, Zhao Y-M, Yuan Z-G, Li X-X, Huang B-Q. 2004. Trichostatin A extends the lifespan of Drosophila melanogaster by elevating hsp22 expression. Acta Biochim Biophys Sin (Shanghai) 36: 618–622. 10.1093/abbs/36.9.618 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM. 2014. The role of senescent cells in ageing. Nature 509: 439–446. 10.1038/nature13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataramani V, Rossner C, Iffland L, Schweyer S, Tamboli IY, Walter J, Wirths O, Bayer TA. 2010. Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the Alzheimer amyloid precursor protein. J Biol Chem 285: 10678–10689. 10.1074/jbc.M109.057836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh J, Jenkins RE, Wong M, Olayanju A, Powell H, Copple I, O'Neill PM, Goldring CEP, Kitteringham NR, Park BK. 2014. Identification and quantification of the basal and inducible Nrf2-dependent proteomes in mouse liver: biochemical, pharmacological and toxicological implications. J Proteomics 108: 171–187. 10.1016/j.jprot.2014.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WS, Parmigiani RB, Marks PA. 2007. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26: 5541–5552. 10.1038/sj.onc.1210620 [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ. 2017. cGAS is essential for cellular senescence. Proc Natl Acad Sci 114: E4612–E4620. 10.1073/pnas.1705499114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. 2013. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101. 10.1038/nature12347 [DOI] [PubMed] [Google Scholar]
- Zimmermann M, de Lange T. 2014. 53BP1: pro choice in DNA repair. Trends Cell Biol 24: 108–117. 10.1016/j.tcb.2013.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.