

In this review, Slade provides an overview of the molecular mechanisms and cellular consequences of PARP and PARG inhibition. The author also highlights the clinical performance of four PARP inhibitors used in cancer therapy (olaparib, rucaparib, niraparib, and talazoparib) and discusses the predictive biomarkers of inhibitor sensitivity and mechanisms of resistance as well as the means of overcoming them through combination therapy.
Keywords: poly(ADP-ribose) polymerases, poly(ADP-ribose) glycohydrolase, PARP inhibitor, PARG inhibitor, cancer therapy
Abstract
Oxidative and replication stress underlie genomic instability of cancer cells. Amplifying genomic instability through radiotherapy and chemotherapy has been a powerful but nonselective means of killing cancer cells. Precision medicine has revolutionized cancer therapy by putting forth the concept of selective targeting of cancer cells. Poly(ADP-ribose) polymerase (PARP) inhibitors represent a successful example of precision medicine as the first drugs targeting DNA damage response to have entered the clinic. PARP inhibitors act through synthetic lethality with mutations in DNA repair genes and were approved for the treatment of BRCA mutated ovarian and breast cancer. PARP inhibitors destabilize replication forks through PARP DNA entrapment and induce cell death through replication stress-induced mitotic catastrophe. Inhibitors of poly(ADP-ribose) glycohydrolase (PARG) exploit and exacerbate replication deficiencies of cancer cells and may complement PARP inhibitors in targeting a broad range of cancer types with different sources of genomic instability. Here I provide an overview of the molecular mechanisms and cellular consequences of PARP and PARG inhibition. I highlight clinical performance of four PARP inhibitors used in cancer therapy (olaparib, rucaparib, niraparib, and talazoparib) and discuss the predictive biomarkers of inhibitor sensitivity, mechanisms of resistance as well as the means of overcoming them through combination therapy.
Cancer is one of the most devastating diseases of our time. Uncontrolled and abnormal growth of cancer cells relies on a panel of acquired functions referred to as cancer hallmarks: sustaining proliferative signaling, enabling replicative immortality, evading growth suppressors, resisting cell death, inducing angiogenesis, activating invasion and metastasis, reprogramming energy metabolism, and evading immune destruction (Hanahan and Weinberg 2011). Acquisition of these cancer traits is facilitated by two “enabling characteristics”: genomic instability and inflammation (Hanahan and Weinberg 2011). The common denominator of genomic instability and inflammation is oxidative stress. Cancer cells experience high levels of oxidative stress. Oncogenes such as MYC and RAS induce the production of reactive oxygen species (ROS) and replication stress (Vafa et al. 2002; Maya-Mendoza et al. 2015). Inflammatory cells such as macrophages and neutrophils can induce oxidative stress themselves by releasing ROS (Grivennikov et al. 2010; Forrester et al. 2018). ROS induce DNA damage and mutations, resulting in genomic instability (Tubbs and Nussenzweig 2017). ROS also activate proinflammatory transcription factors that induce expression of inflammatory molecules (Grivennikov et al. 2010; Forrester et al. 2018). Anticancer drugs have been designed to target the whole panel of cancer traits. Arguably, targeting genomic instability and inflammation and amplifying these “enabling characteristics” to turn them into “disabling factors” is a promising way to eradicate cancer.
Over the past decade poly(ADP-ribose) polymerases (PARPs) have emerged as a new target in cancer therapy (Mateo et al. 2019). PARP inhibitors capitalize on genomic instability caused by oxidative and replication stress, as well as deficiencies in DNA repair pathways. Four PARP inhibitors, olaparib, rucaparib, niraparib, and talazoparib, have been approved by the U.S. Food and Drug Administration (FDA) and by the European Medicines Agency (EMA). In 2014, olaparib was approved as maintenance therapy for platinum-sensitive advanced ovarian cancer with germline mutations in DNA repair genes BRCA1/2 that are required for the homologous recombination (HR) pathway of double-strand break (DSB) repair. In 2016, rucaparib was approved for advanced ovarian cancer with both germline and somatic BRCA1/2 mutations. In 2017 and 2018, olaparib, rucaparib, and niraparib were approved for the maintenance treatment of recurrent, epithelial ovarian, fallopian tube, or primary peritoneal cancer irrespective of the BRCA status. Last, in 2018, olaparib and talazoparib were approved for human epidermal growth factor receptor type 2 (HER2)-negative locally advanced or metastatic breast cancer with germline BRCA1/2 mutations. Multiple clinical trials carried out since 2009 have demonstrated PARP inhibitor efficacy in BRCA mutated ovarian and breast cancer, but also prostate, pancreatic cancer, and small cell lung carcinoma (SCLC), irrespective of the BRCA status (Weaver and Yang 2013; Sonnenblick et al. 2015; Mirza et al. 2018; Franzese et al. 2019; Keung et al. 2019; Mateo et al. 2019; Pant et al. 2019; Pilie et al. 2019a). Inhibitors of poly(ADP-ribose) glycohydrolase (PARG) joined the stage once structures of the PARG catalytic site became available (Slade et al. 2011; Dunstan et al. 2012; Kim et al. 2012; Barkauskaite et al. 2013). Rather than synergizing with deficiencies in DNA repair pathways, PARG inhibitors seem to exploit deficiencies in replication machinery and higher levels of replication stress in cancer cells (Pillay et al. 2019).
In general, cancers with high levels of replication stress and genomic instability due to DNA repair deficiency and/or oncogene-induced increase in replication origin firing are particularly responsive to PARP and PARG inhibition. PARP and PARG inhibitors exploit and exacerbate these tumor vulnerabilities by inducing further DNA damage, preventing DNA repair and amassing unresolved replication intermediates that instigate replication and mitotic catastrophe.
Molecular mechanisms of PARP and PARG inhibitors
PARPs synthesize poly(ADP-ribose) (PAR) from NAD, releasing nicotinamide as the reaction product (Okayama et al. 1977). PARP1, as the major producer of cellular PAR, is activated by binding DNA lesions (Benjamin and Gill 1980a,b). Catalytic activation of PARP1 is a multistep process of binding to DNA through N-terminal zinc fingers (ZnF), unfolding of the helical domain (HD), binding of NAD to the catalytic pocket, and PAR catalysis (Langelier et al. 2012; Eustermann et al. 2015). The first PARP1 inhibitor was nicotinamide itself (Clark et al. 1971), followed by 3-aminobenzamide (3-AB) (Purnell and Whish 1980). All subsequently developed PARP1 inhibitors contain nicotinamide/benzamide pharmacophores and compete with NAD for the catalytic pocket of PARPs (Fig. 1; Ferraris 2010; Steffen et al. 2013). PARP1 inhibitors dock into the catalytic site by forming hydrogen bonds with Gly, Ser, and Glu as well as hydrophobic stacking interactions with two Tyr residues within the nicotinamide-binding pocket (Fig. 1; Ferraris 2010). Given the high degree of conservation of the catalytic pocket among different PARPs, additional interactions are required for selective inhibition (Steffen et al. 2013). A screen for more potent and selective inhibitors identified different scaffolds from which new-generation PARP1 inhibitors evolved; phthalazinone and tetrahydropyridophthalazinone served as a scaffold for olaparib and talazoparib, benzimidazole and indazole carboxamide for veliparib and niraparib, tricyclicindole lactam for rucaparib (Banasik et al. 1992; White et al. 2000; Canan Koch et al. 2002). Olaparib was the first PARP inhibitor that entered clinical trials due to its selectivity for inhibiting PARP1/2 as well as its potency, oral availability, and favorable pharmacokinetic and pharmacodynamic properties (Menear et al. 2008; Fong et al. 2009). All clinically relevant PARP1/2 inhibitors have high catalytic activity with IC50 in the low nanomolar range and inhibit PARP1 and PARP2 with similar efficiency (Fig. 1; Menear et al. 2008; Jones et al. 2009; Shen et al. 2013, 2015; Wang et al. 2016a).
Figure 1.

Structures of PARP and PARG inhibitors. (A,C) Chemical structures. IC50 denotes half-maximal inhibitory concentration based on measurements of PARP/PARG activity in vitro. Cellular PAR synthesis EC50 denotes half-maximal effective concentration determined by measuring PAR levels in cellular extracts treated with inhibitors. Cytotoxicity CC50 denotes half-maximal cytotoxic concentration determined by measuring cell viability after PARP/PARG inhibitor treatment. (B,D) X-ray structures. (B) Veliparib bound to PARP1 active site (PDB: 2RD6). (D) JA2131 bound to PARG active site (PDB: 6OA3). Inhibitors are labeled in green, PARP1/PARG residues in the binding pocket are labeled in blue, water molecules are shown as red dots, and hydrogen bonds are represented by yellow dashes.
Despite improved selectivity, many PARP1/2 inhibitors are not highly selective over other family members (Wahlberg et al. 2012; Papeo et al. 2014; Thorsell et al. 2017). Among clinically relevant inhibitors, veliparib is the most selective PARP1/2 inhibitor, followed by niraparib (Thorsell et al. 2017). Their selectivity is based on formation of a PARP1/2-unique water-mediated hydrogen bond interaction with a regulatory subdomain residue (D766 in PARP1), which is conserved in PARP1/2 but not in other PARPs (Fig. 1). Compared with veliparib, which exhibits >100-fold higher selectivity for PARP1/2 compared with other family members, olaparib and talazoparib show only 15-fold to 20-fold higher selectivity (Thorsell et al. 2017). Rucaparib is the least selective clinical PARP1 inhibitor, which inhibits different PARPs (PARP1, PARP2, PARP5A, and PARP5B) as well as mono(ADP-ribosyl) transferases PARP3, PARP4, PARP10, PARP15, and PARP16 (Thomas et al. 2007; Wahlberg et al. 2012). Moreover, some PARP inhibitors such as rucaparib and niraparib also inhibit non-PARP targets, albeit with lower efficiency; rucaparib inhibits hexose-6-phosphate dehydrogenase (H6PD), while niraparib inhibits deoxycytidine kinase (DCK) (Knezevic et al. 2016). Such cross-inhibition may potentiate cancer cell death, as in the case of rucaparib and PARP/H6PD inhibition, but may also be detrimental for combination therapy with niraparib and nucleoside analogs such as gemcitabine due to cross-inhibition of DCK required for their activation (Knezevic et al. 2016).
In addition to inhibiting PARP catalytic activity, PARP inhibitors also trap PARP1 and PARP2 on DNA (Murai et al. 2012, 2014a). PARP1 is the dominant target for DNA trapping by PARP inhibitors, as depletion of PARP1—but not PARP2—reduces sensitivity to PARP inhibitors (Murai et al. 2012). PARP1 being the relevant target is consonant with its high nuclear abundance and its requirement for synthetic lethality with HR deficiency (Amé et al. 1999; Ronson et al. 2018; Murai and Pommier 2019). Entrapment of PARP1 on DNA can be determined based on the shift in distribution of PARP1 from nuclear-soluble to chromatin-bound fraction (Murai et al. 2012). PARP entrapment can occur on DNA-strand breaks as well as topoisomerase I (TOP1)-processed ribonucleotides and unligated Okazaki-fragment intermediates of DNA replication (Fig. 2; Strom et al. 2011; Hanzlikova et al. 2018; Zimmermann et al. 2018). Once trapped, PARP1 cannot dissociate from DNA due to inhibition of its catalytic activity, which is required for repulsion between auto-PARylated PARP1 and DNA (Pommier et al. 2016). Catalytic inhibition of PARP1 auto-PARylation is thus a prerequisite for PARP1-DNA trapping (Hopkins et al. 2015).
Figure 2.

DNA lesions recognized by PARP1 as potential PARP-trapping sites. Unligated Okazaki fragments are DNA replication intermediates. Single-strand DNA breaks (SSBs) are a frequent form of endogenous DNA damage and are particularly hazardous for replication forks. Ribonucleotides incorporated into DNA need to be removed by RNase H2-mediated ribonucleotide excision repair. In RNase H2-deficient cells these ribonucleotides are removed by topoisomerase I (TOP1)-mediated excision. TOP1 cleavage results in nicks, covalent TOP1–DNA adducts, and single-strand DNA gaps that can engage PARP1.
The catalytic inhibitory effects of the clinically relevant PARP inhibitors olaparib, rucaparib, niraparib, and talazoparib are comparable; however, their potency in trapping PARP–DNA complexes varies considerably, which is why PARP1 trapping was proposed to rely on allosteric changes in the PARP1 DNA-binding domain induced by the PARP inhibitor binding to the D-loop at the outer border of the NAD site (Murai et al. 2012, 2014a). Talazoparib exhibits the highest trapping efficiency (talazoparib >> niraparib > olaparib = rucaparib >> veliparib) and has the most rigid structure (Murai et al. 2014a). Veliparib is one of the weakest PARP1/2 inhibitors with low PARP trapping efficiency (Murai et al. 2012).
PARG hydrolyzes ribose–ribose bonds within PAR with high specific activity and processivity, particularly after DNA damage (Wielckens et al. 1982; Hatakeyama et al. 1986; Alvarez-Gonzalez and Althaus 1989). PARG has a macro domain that binds ADP-ribose moiety and a PARG-specific loop with conserved glutamates that cleave ribose–ribose bonds in an exoglycohydrolase mode (Slade et al. 2011; Dunstan et al. 2012; Kim et al. 2012; Barkauskaite et al. 2013). The first PARG inhibitors gallotannin and GPI-16552 showed low activity in vitro and off-target effects in cells (Falsig et al. 2004; Erdèlyi et al. 2005). ADP-HPD and rhodanine-based PARG inhibitors (RBPIs) are potent and specific inhibitors, but lack cell permeability (Slama et al. 1995; Finch et al. 2012). The quinazolinedione-type PARG inhibitor PDD00017273 inhibits PARG selectively and with high efficiency, is cell-permeable and cell-active, but has limited bioavailability, which makes it unsuitable for clinical application (James et al. 2016). The naphthalen-type PARG inhibitor COH34 is a potent, specific, and cell-permeable inhibitor with a terminal half-life of 3.9 h, and may thus prove a good candidate for clinical studies (Chen and Yu 2019). Chemical library screening identified thioxanthine/methylxanthine derivatives JA2–4 and JA2131 as potent, specific, cell-permeable, and cell-active PARG inhibitors, which are also likely to show good bioavailability given their structural similarity with caffeine (Houl et al. 2019). PARG inhibitors compete with PAR for the PARG active site by occupying the subsite normally occupied by the adenine moiety of ADP-ribose (Fig. 1; James et al. 2016; Chen and Yu 2019; Houl et al. 2019).
Cellular mechanisms of PARP and PARG inhibitors
Functions of PARP1 and PARG in DNA repair and replication fork protection
Nuclear functions of PARP1 and PARG in DNA repair, replication fork protection, and transcription regulation are critical for understanding the mechanism of action of PARP inhibitors. PARP1 is involved in different pathways of DNA repair, including single-strand DNA break (SSB) repair, nucleotide excision repair (NER), alternative nonhomologous end-joining (alt-NHEJ), and homologous recombination (HR) (Ray Chaudhuri and Nussenzweig 2017). PARP1 and PARG are also critical for preserving the integrity of replication forks under conditions that induce replication stress (Hanzlikova and Caldecott 2019). PARP1 or PARG depletion or inhibition exert the most profound effects on SSB repair and replication fork stability.
In SSB repair, PARP1 activity is important for the recruitment of the scaffold protein XRCC1 to the sites of DNA damage, while PARG regulates XRCC1 dissociation (Fig. 3; El-Khamisy et al. 2003; Okano et al. 2003; Fisher et al. 2007; Chen and Yu 2019). PARP1/XRCC1-dependent SSB repair was implicated as an alternative pathway of Okazaki fragment processing (Fig. 2; Hanzlikova et al. 2018).
Figure 3.

Single-strand break (SSB) repair and replication fork protection by PARP1. PARP1 acts as a sensor of SSBs and recruits XRCC1. XRCC1 is a scaffold for the recruitment of proteins that process damaged termini, DNA polymerase β that fills the gap, and DNA ligase III that seals the nick. PARP1 rescues damaged replication forks through fork reversal or homologous recombination (HR). SSBs on the leading strand trigger fork reversal by fork remodeling proteins. PARP1 promotes fork reversal by inhibiting the RECQ1 helicase involved in fork restart. PARP1 stabilizes RAD51 filaments on reversed forks and together with BRCA1 and BRCA2 protects forks from degradation by the MRE11 nuclease. If forks collapse when encountering an SSB on the lagging strand, PARP1 promotes HR-mediated fork repair and restart by recruiting MRE11, EXO1, and BRCA1-CtIP for end resection, and BRCA2 for RAD51 filament formation.
PARP1 interacts with DNA replication machinery and is active during S phase and in response to replication stress (Jump et al. 1979; Anachkova et al. 1989; Dantzer et al. 1998; Simbulan-Rosenthal et al. 1998; Bryant et al. 2009). Replication stress leads to uncoupling between DNA polymerase and helicase activities, which generates single-stranded DNA (ssDNA). RPA binds ssDNA and recruits the S/G2 checkpoint kinase ATR. ATR suppresses silent origin firing and activates the checkpoint kinase CHK1 to induce cell cycle arrest (Zeman and Cimprich 2014). By preventing unscheduled origin firing, replication checkpoints prevent accumulation of ssDNA and exhaustion of RPA, and thereby safeguard against fork breakage (Toledo et al. 2013). Replication stress generates SSBs and exposes unligated Okazaki fragments as DNA substrates for PARP1 binding (Hanzlikova and Caldecott 2019). In response to replication stress, PARP1 slows down replication forks to promote fork reversal by antagonizing the RECQ1 helicase (Yang et al. 2004; Sugimura et al. 2008; Bryant et al. 2009; Ray Chaudhuri et al. 2012; Berti et al. 2013), protects replication forks from degradation by the MRE11 nuclease (Ying et al. 2012), stabilizes RAD51 nucleofilaments at stalled forks together with PARP2 (Ronson et al. 2018), and activates the S-phase checkpoint kinase CHK1 (Fig. 3; Min et al. 2013). PARG localizes at replication forks by binding PCNA and promotes recovery from prolonged replication stress (Mortusewicz et al. 2011; Illuzzi et al. 2014; Kaufmann et al. 2017). PARP1 also regulates replication and DNA repair at the transcription level by stimulating activity of the transcription factor E2F1, which regulates the expression of replication and HR genes (Simbulan-Rosenthal et al. 1998, 2003).
Replication forks are prone to breakage if they encounter an SSB, which is why homologous recombination is a critical pathway for repairing replication forks to prevent fork collapse (Ait Saada et al. 2018). PARP1 contributes to the homologous recombination pathway of DSB repair by promoting rapid recruitment of MRE11, EXO1, BRCA1, and BRCA2 to DNA damage sites (Fig. 3; Haince et al. 2008; Li and Yu 2013; Zhang et al. 2015a,b). The MRE11 nuclease is responsible for the early processing of DNA lesions, while EXO1 and BRCA1-CtIP contribute to extensive end resection. BRCA2 is required for the loading of RAD51 filaments onto ssDNA generated by end resection. PARP1 also counteracts nonhomologous end-joining (NHEJ) as the alternative pathway of DSB repair by preventing the binding of the NHEJ protein Ku to DNA ends (Hochegger et al. 2006; Wang et al. 2006; Patel et al. 2011; Yang et al. 2018).
In addition to PARP1 and PARG, other members of the PARP family have also been implicated in DSB repair and replication fork stability, most notably PARP2, PARP3, PARP10, and PARP14 (Martin-Hernandez et al. 2017), and may contribute to cellular phenotypes of PARP1/2 inhibitors given that some of them exhibit weaker target specificity.
Synthetic lethality between PARP or PARG inhibitors and genomic instability in cancer cells
Genomic instability underlies the ability of cancer cells to acquire different tumorigenic properties. Genomic instability entails chemical alterations in DNA known as mutations as well as changes in the chromosome number or structure defined as chromosomal instability. Genomic instability arises due to high levels of DNA damage caused by oxidative or replication stress, defects in DNA repair pathways, and/or dysfunctional surveillance mechanisms that fail to trigger cellular senescence or apoptosis (Tubbs and Nussenzweig 2017). Cancer cells experience high levels of oxidative and replication stress, resulting in high mutational rates (Bartkova et al. 2005; Gorgoulis et al. 2005; Dobbelstein and Sørensen 2015; Macheret and Halazonetis 2015; Kotsantis et al. 2018). Activation of oncogenes such as MYC, RAS, and cyclin E1 (CCNE1) induces replication stress by promoting premature entry into S phase, increasing replication origin firing, changing replication fork rates, causing nucleotide depletion, and inducing replication–transcription conflicts (Bester et al. 2011; Kotsantis et al. 2016; Macheret and Halazonetis 2018). Replication stress leads to accumulation of replication errors and DNA lesions that compromise fork stability and require DNA repair pathways to restore fork progression (Dobbelstein and Sørensen 2015; Macheret and Halazonetis 2015; Kotsantis et al. 2018). Many cancers have germline or somatic mutations in DNA repair genes. Mutations in tumor suppressor genes such as the cell cycle checkpoint gene TP53 are common across different cancer types and allow cancer cells to escape senescence or apoptosis and continue proliferating in the presence of DNA damage (The Cancer Genome Atlas Research Network 2011; Kandoth et al. 2013; Nik-Zainal et al. 2016; Robinson et al. 2017; Hafner et al. 2019).
Genotoxic agents have been used routinely in cancer therapy in order to induce high levels of DNA damage that render cancer cells particularly vulnerable due to their high proliferation rates. These include ionizing radiation and chemotherapeutic drugs that damage DNA by inducing DSBs (e.g., bleomycin, doxorubicin, topoisomerase inhibitors), intrastrand or interstrand DNA cross-links (platinum compounds; e.g., cisplatin, carboplatin, and oxaliplatin), DNA base alkylation (e.g., temozolomide), or that interfere with DNA replication such as nucleoside and base analogs (e.g., gemcitabine and 5-fluorouracil). Mitotic drugs that inhibit cell division such as taxanes (e.g., docetaxel and paclitaxel) are also used in chemotherapy. Chemotherapy is often combined with radiotherapy. Synergistic effects are additionally achieved in patients with genetic deficiencies in DNA repair pathways. For example, platinum drugs (carboplatin) improved response rate in BRCA mutated advanced triple-negative breast cancer (TNBC) patients and are more effective than taxanes (Telli et al. 2016; Tutt et al. 2018). However, genotoxic agents also affect normal cells and have severe side effects such as myelosuppression.
Precision medicine has revolutionized cancer therapy by putting forth the concept of selective targeting of cancer cells. PARP inhibitors represent a successful example of precision medicine applied in the clinic. PARP inhibitors act through synthetic lethality, whereby genetic DNA repair defects are enhanced by drug-induced defects in a compensatory pathway (Lord and Ashworth 2017). Two seminal studies showed how PARP inhibitors specifically kill HR-deficient cells mutated in BRCA1/2 (Fig. 4; Bryant et al. 2005; Farmer et al. 2005). Carriers of heterozygous BRCA1/2 mutations are sensitive to PARP inhibitor treatment as they lose the wild-type allele during tumorigenesis and thereby become BRCA1/2-null. Since this first example of synthetic lethality between genetic defects and PARP inhibitors, it has become clear that oxidative stress and genomic instability, manifested not just through mutations in DNA repair proteins but also replication stress, sensitize cells to PARP and PARG inhibitors (Bryant et al. 2005; Farmer et al. 2005; McCabe et al. 2006; Bunting et al. 2010; Lord and Ashworth 2012, 2017; McLellan et al. 2012; Murai et al. 2012; Dréan et al. 2016; Gravells et al. 2017; Zimmermann et al. 2018; Chen and Yu 2019; Giovannini et al. 2019; Pillay et al. 2019). In addition to BRCA1/2, mutations in DNA damage response genes such as ATM, PRKDC, ATR, RPA1, DSS1, NBN, RAD51, RAD54, CHEK1, CHEK2, FANC genes, ERCC1, POLB, FEN1, and CDK12 have shown synthetic lethality in combination with PARP inhibitors (Bryant and Helleday 2006; McCabe et al. 2006; Murai et al. 2012; Postel-Vinay et al. 2013; Bajrami et al. 2014). Synthetic lethality between mutations in HR-related genes and PARP inhibition was confirmed by CRISPR screens, which enable high-throughput investigation of synthetic lethal interactions (Zimmermann et al. 2018).
Figure 4.
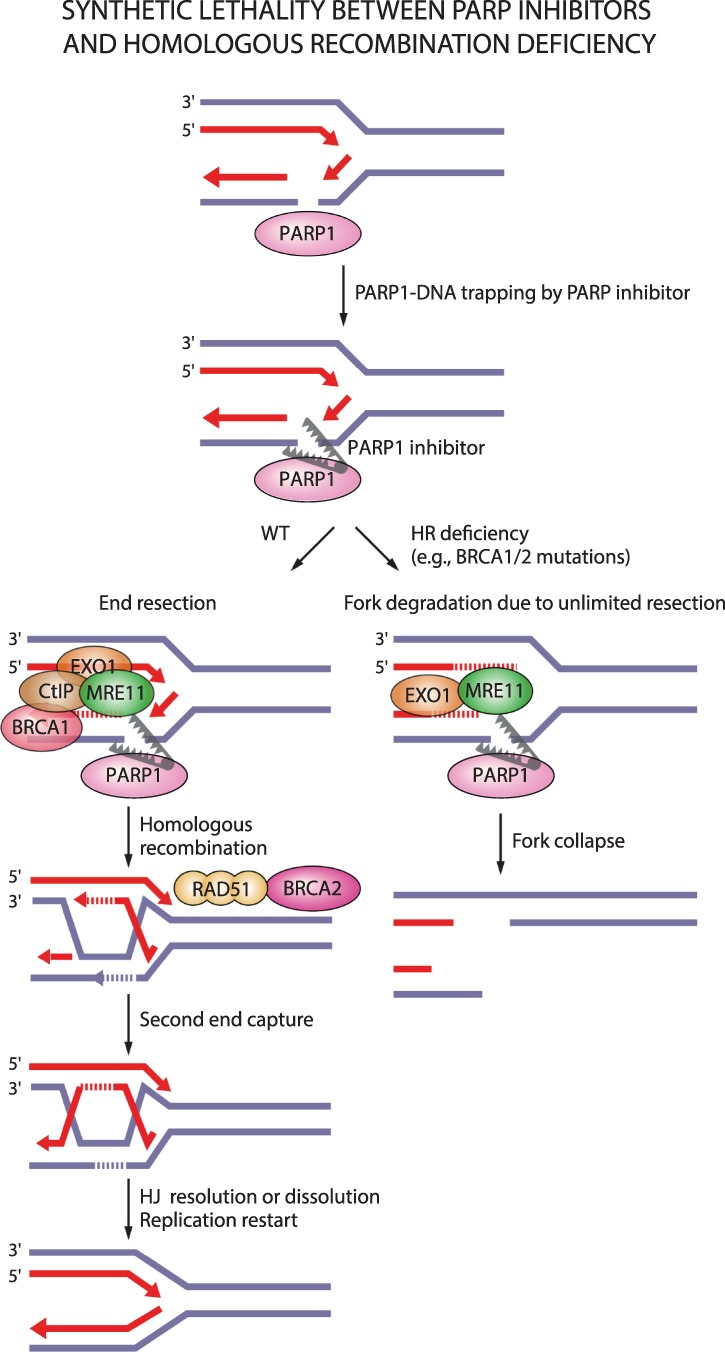
Synthetic lethality between PARP inhibitors and homologous recombination deficiency. PARP entrapment on DNA lesions blocks replication machinery and loss of PARP activity prevents fork protection, fork reversal, and fork restart. This results in DSBs that need to be repaired by homologous recombination. In the case of homologous recombination deficiency due to, for example, mutations in BRCA1/2, PARP1-trapping lesions elicit excessive fork degradation by the MRE11 nuclease, the activity of which is unrestrained in the absence of BRCA1/2 and PARP1. This results in fork collapse.
In contrast to PARP inhibitors, a clear correlation between HR deficiency and synthetic lethality with PARG inhibitors is lacking. Depletion of the HR proteins BRCA1/2, PALB2, ABRAXAS, and BARD1 in MCF7 breast cancer cells was shown to elicit synthetic lethal interactions with PARG depletion or PARG inhibition (with gallotannin or PDD00017273) (Fathers et al. 2012; Gravells et al. 2017). The PARG inhibitor COH34 efficiently kills BRCA mutated or olaparib-resistant ovarian and breast cancer cells (Chen and Yu 2019). However, PARG depletion did not show synthetic lethality with BRCA1 mutations in different cancer cell lines (Noll et al. 2016), the PARG inhibitor JA2131 efficiently killed BRCA-proficient cancer cells (Houl et al. 2019), and only one out of six tested ovarian cancer cells with BRCA1/2 mutations showed sensitivity to PARG inhibition with PDD00017273 (Pillay et al. 2019). Instead, synthetic lethal interactions with the PARG inhibitor PDD00017273 involve replication-associated genes such as TIMELESS, HUS1, and RFC2 (Fig. 5; Pillay et al. 2019).
Figure 5.
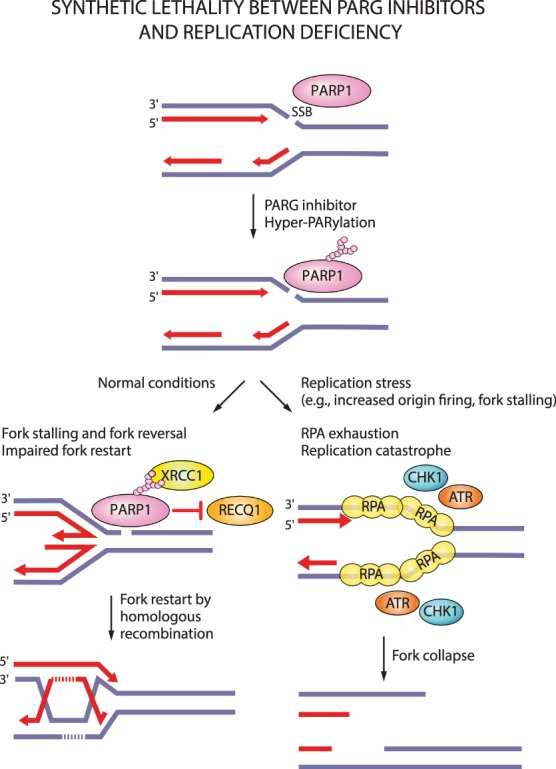
Synthetic lethality between PARG inhibitors and replication stress. PARG inhibition increases PARylation levels and may prevent dissociation of PARP1 and PAR-binding repair proteins (e.g., XRCC1) from DNA damage sites. Loss of PARG activity causes fork stalling and impairs restart of reversed forks. Forks can presumably restart by homologous recombination. Under replication stress conditions, increased origin firing and prolonged fork stalling generates excessive ssDNA and causes RPA exhaustion. Such replication catastrophe results in fork collapse.
Amplifying genomic instability with PARP and PARG inhibitors
PARP1 depletion or inhibition increases replication fork speed, impairs replication fork reversal, and causes untimely fork restart following replication stress (Fig. 6). This leads to an accumulation of DNA damage in S-phase cells, S-phase stalling, and G2 delay (Sugimura et al. 2008; Bryant et al. 2009; Ray Chaudhuri et al. 2012; Berti et al. 2013; Dale Rein et al. 2015; Farrés et al. 2015; Maya-Mendoza et al. 2018; Michelena et al. 2018; Ronson et al. 2018). In PARP1-depleted or inhibited cells, DSBs arise due to deprotection of stalled replication forks and their degradation by the MRE11 nuclease, due to impaired fork reversal, or from unligated Okazaki fragments encountered by replication forks (Lonn and Lonn 1985; Ray Chaudhuri et al. 2012; Ying et al. 2012; Hanzlikova et al. 2018).
Figure 6.

Cellular consequences of PARP and PARG inhibition on replication and mitosis. PARP inhibitors increase replication fork rate, reduce fork reversal, and cause premature fork restart. In HR-deficient cells, PARP inhibitors cause fork collapse and DSBs. PARG inhibitors reduce fork rate, increase fork reversal, and impair fork restart. In the presence of replication stress, PARG inhibitors cause replication catastrophe. Destabilization of replication forks causes mitotic defects and death by mitotic catastrophe. PARG inhibitors cause mitotic defects in combination with DNA-damaging agents.
In line with the opposing catalytic activities of PARP1 and PARG, PARG depletion or inhibition show opposite effects. PARG depletion or inhibition slows down replication forks, causes accumulation of reversed forks and ssDNA gaps, and prevents fork restart through suppression of the RECQ1 helicase (Fig. 6; Illuzzi et al. 2014; Ray Chaudhuri et al. 2015; Gravells et al. 2017; Houl et al. 2019; Pillay et al. 2019). This causes cell cycle stalling in the S/G2 phase, accumulation of ssDNA as judged by RPA foci, and accumulation of DNA damage, as indicated by pan-nuclear γH2AX staining (Illuzzi et al. 2014; Pillay et al. 2019). Prolonged exposure of PARG-depleted cells to replication stress results in the loss of RPA foci, indicating RPA exhaustion due to replication stress-induced formation of ssDNA—a concept known as replication catastrophe (Illuzzi et al. 2014; Toledo et al. 2017). PARP1 or PARG depletion or inhibition sensitize cells to DNA-damaging agents and cause increased DNA damage levels due to decreased HR efficiency (Rottenberg et al. 2008; Ame et al. 2009; Min et al. 2010; Shirai et al. 2013; Murai et al. 2014b; Dréan et al. 2016; James et al. 2016; Gravells et al. 2018; Houl et al. 2019). Collectively, loss or inhibition of PARP1 or PARG destabilize replication forks and cause fork breakage, particularly under conditions of HR deficiency, oxidative and replication stress, or exogenous DNA damage.
Impaired fork progression and accumulation of DNA damage are more pronounced in PARP inhibitor-treated cells compared with PARP-depleted cells, which suggests that PARP inhibition does more than just inhibit PARP1 catalytic activity. For example, PARP inhibition increases fork rate and reduces fork reversal following camptothecin exposure more than PARP depletion (Sugimura et al. 2008; Ray Chaudhuri et al. 2012; Maya-Mendoza et al. 2018). Furthermore, PARP inhibition results in more strand breaks (SSBs and DSBs), as judged by γH2AX foci (Murai et al. 2012) and delayed SSB repair after ionizing radiation or alkylation damage (Godon et al. 2008; Strom et al. 2011). Consequently, PARP inhibition exhibits stronger synthetic lethality with HR deficiency compared with PARP depletion (Bryant et al. 2005; Ronson et al. 2018).
In fact, PARP inhibitor cytotoxicity was found to correlate with the strength of PARP–DNA entrapment rather than a reduction in PARP1 catalytic activity (Murai et al. 2012; Pettitt et al. 2013). A combination of the chemical inhibition of PARP1 catalytic activity and the physical obstruction caused by PARP–DNA entrapment seems to be responsible for the greater cytotoxicity of PARP inhibitors compared with PARP depletion. The physical obstruction caused by PARP–DNA entrapment exacerbates replication problems caused by loss of PARP activity and induces mitotic phenotypes of premature loss of cohesion or anaphase DNA bridges, which are not observed upon PARP depletion (Murai et al. 2012; Shen et al. 2015; Kukolj et al. 2017; Schoonen et al. 2017). Therefore, the cytotoxicity of PARP inhibitors seems to arise from an accumulation of replication problems, which are carried over into mitosis resulting in death by mitotic catastrophe (Slade 2019).
PARP and PARG inhibitors activate the S/G2 checkpoint kinases ATR and CHK1 (Ray Chaudhuri et al. 2015; Colicchia et al. 2017; Kim et al. 2017; Maya-Mendoza et al. 2018; Pillay et al. 2019), which generally halt the cell cycle to allow DNA repair and completion of DNA replication before mitotic entry (Lecona and Fernandez-Capetillo 2018). In accordance with the activation of the ATR checkpoint, PARG inhibitor PDD00017273-treated cells stall in the S/G2 phase without progressing into mitosis and assume a “fried egg” morphology (Pillay et al. 2019). Conversely, despite the activation of ATR and CHK1, cells treated with PARP inhibitors progress into mitosis and exhibit different mitotic defects, which arise from problems during S phase (Fig. 6; Colicchia et al. 2017; Kim et al. 2017; Maya-Mendoza et al. 2018).
As explained above, fork destabilization caused by PARP inhibition results in DSBs. DSBs cannot be repaired by the HR pathway in BRCA-deficient cells, which forms the basis for synthetic lethality approaches with PARP inhibitors. Moreover, PARP inhibition itself induces HR deficiency by reducing the expression of the E2F1 target genes involved in DNA replication and cell cycle regulation (e.g., PCNA, MCM7, and CCNA2) and HR factors such as BRCA1/2 and RAD51, as shown in prostate and small cell lung cancer (Byers et al. 2012; Schiewer et al. 2018). A contingency pathway for DSB repair, NHEJ, which is functional throughout the cell cycle, is thought to compensate for inactive HR in PARP inhibitor-treated cells. NHEJ is an error-prone pathway that can lead to small mutations as well as chromatid fusions (Bunting et al. 2010). Fusion of two broken sister chromatids, chromosomes, or telomeres during interphase can generate dicentric chromosomes visible as radial fusions in metaphase and as anaphase DNA bridges (Ganem and Pellman 2012). Chromatid fusions also give rise to acentric chromosomes, which cannot attach to the mitotic spindle and cannot segregate accurately during anaphase, thus appearing as lagging chromosomes. During telophase and cytokinesis, acentric or lagging chromosomes obtain their own nuclear envelope and form micronuclei (Fenech et al. 2011). Chromosomes within micronuclei are prone to chromothripsis, whereby reduced and asynchronous DNA replication results in DNA damage and chromosome fragmentation (Crasta et al. 2012). DNA released from micronuclei triggers cGAS accumulation and activation of proinflammatory response (Harding et al. 2017; Mackenzie et al. 2017). Following chromothripsis, fragmented chromosomes can assemble randomly, resulting in chromosome rearrangements (Luijten et al. 2018). Chromatid breaks, radial chromosomes, anaphase DNA bridges, lagging chromosomes, and micronuclei are all common in BRCA1/2-deficient cells treated with PARP inhibitors (Fig. 6; Bunting et al. 2010; Schoonen et al. 2017).
Genomic regions called common fragile sites (CFSs) are particularly sensitive to impaired fork progression. CFS are found within long genes and are prone to form abnormal replication intermediates due to transcription–replication conflicts (Helmrich et al. 2011). CFSs are late-replicating and remain underreplicated at the G2/M transition (Le Beau et al. 1998). Underreplicated CFSs remain connected through thin threads of DNA in mitosis known as ultrafine anaphase DNA bridges (Le Beau et al. 1998). Replication intermediates that remain unresolved during mitosis are marked by 53BP1 in G1 cells (Lukas et al. 2011). Replication stress induced by PARP inhibitors gives rise to ultrafine anaphase DNA bridges in mitotic cells and 53BP1-positive nuclear bodies in G1 cells (Gemble et al. 2015; Michelena et al. 2018). Furthermore, PARP inhibition during S phase causes weakening of sister chromatid cohesion, resulting in premature loss of cohesion (“cohesion fatigue”) and chromosome alignment problems in metaphase (Fig. 6; Kukolj et al. 2017).
It is clear that mitotic defects in PARP-inhibitor treated cells arise from destabilization of replication forks and DNA damage acquired during S phase. Replication stress-induced mitotic defects result in death by mitotic catastrophe (Dale Rein et al. 2015; Gemble et al. 2015; Majuelos-Melguizo et al. 2015; Colicchia et al. 2017; Kukolj et al. 2017; Schoonen et al. 2017; Maya-Mendoza et al. 2018; Michelena et al. 2018). Mitotic catastrophe is a special type of cell death whereby cells die by apoptosis or slip out of mitosis through multinucleation or macronucleation due to chromosome missegregation, as well as micronucleation that results from lagging or acentric chromosomes (Galluzzi et al. 2018).
While HR-deficient cancer cells were shown to respond better to PARP inhibitors, PARP inhibitors are also effective in HR-proficient cells that experience high levels of oxidative and replication stress. Indeed, anaphase DNA bridges, lagging chromosomes, micronuclei, ultrafine anaphase DNA bridges, and premature loss of cohesion occur in HR-proficient cells exposed to PARP inhibitors (Majuelos-Melguizo et al. 2015; Kukolj et al. 2017; Schoonen et al. 2017; Michelena et al. 2018). Moreover, PARP inhibitor efficacy was shown to correlate with basal levels of replication stress in cancer cells (Kukolj et al. 2017).
Unlike PARP inhibitors, the PARG inhibitor PDD00017273 exhibits cytostatic rather than cytotoxic effects by causing a replication catastrophe that is not transferred into mitosis but remains contained in interphase (Pillay et al. 2019). PARP trapping is most likely the reason why PARP inhibitors lead to mitotic catastrophe and are more potent in killing cells compared with PARG inhibitors. However, the combination of PARG inhibitors with cell cycle checkpoint inhibitors such as CHK1 or combination of PARG inhibition/depletion with DNA-damaging agents allows cells to progress into mitosis where they experience various mitotic abnormalities (Koh et al. 2004; Ame et al. 2009; Min et al. 2010; Gravells et al. 2018; Pillay et al. 2019; Slade 2019). For example, PARG-depleted cells or PARG hypomorphic cells lacking nuclear and cytoplasmic PARG isoforms show centrosome amplification, centrosome fragmentation, multipolar spindles, chromosome misalignment, and missegregation, which are more pronounced after exposure to ionizing radiation (Ame et al. 2009; Min et al. 2010). PARG inhibition coupled with ionizing radiation also yields aberrant spindle formation and metaphase arrest (Fig. 6; Gravells et al. 2018).
In sum, cytotoxicity of PARP inhibitors is a multistage process of the destabilization of replication forks through PARP entrapment and loss of PARP activity, the generation of unresolved replication intermediates and DSBs, their transmission into mitosis, and the induction of mitotic defects (premature loss of cohesion, misalignment, missegregation) that ultimately result in mitotic catastrophe. The cytostatic effects of PARG inhibitors involve fork stalling, which results in replication catastrophe and cell cycle arrest in S/G2.
Targeting transcription, RNA metabolism, and ribosome biogenesis through PAPR inhibition
In addition to DNA repair and replication fork stability, PARP1 is also implicated in gene expression regulation, RNA processing, and ribosome biogenesis, which may contribute to cellular effects of PARP inhibition (Weaver and Yang 2013; Feng et al. 2015) and give rise to synthetic lethal interactions with transcription and splicing factors identified in CRISPR screens (Zimmermann et al. 2018). PARP1 modulates the activity of different transcription regulators implicated in cancer (e.g., p53, nuclear receptors) or inflammation (e.g., NF-κB) (Schiewer and Knudsen 2014; Bai 2015). Hence, transcriptional deregulation may sensitize cancer cells to PARP inhibitors, as shown for DNA-repair proficient HER2-positive breast cancer cells whereby NF-κB overactivation is attenuated through PARP inhibition (Nowsheen et al. 2012). PARP inhibitors are also effective in Ewing's sarcomas by blocking, on the one hand, PARP1-dependent transcriptional activation effects of ETS gene fusions such as EWS-FLI-1, and by exacerbating DNA damage on the other (Brenner et al. 2012). Furthermore, PARP inhibitors reduce rDNA transcription and ribosome biogenesis in BRCA1/2-proficient cancer cells by preventing DDX21 ADP-ribosylation, and thereby reduce breast cancer growth (Kim et al. 2019).
Determinants of PARP inhibitor sensitivity in cancer cells
Since the first example of synthetic lethality between PARP inhibitors and BRCA1/2 mutations, it has become clear that any form of HR deficiency in tumors that phenocopies BRCA1/2 mutations, often referred to as BRCAness, may sensitize cells to PARP inhibitors (Lord and Ashworth 2016). Different patient biomarkers have been used to assess HR deficiency as a measure of sensitivity to PARP inhibitor treatment, such as mutations in DNA repair genes, their expression levels, as well as mutational and genomic signatures of HR deficiency. Replication stress markers and transcriptome profiles are complementary means of evaluating PARP inhibitor response given the importance of PARP1 for replication fork stability and gene expression regulation.
Homologous recombination (HR) deficiency
Germline or somatic mutations in DNA repair genes as well as their transcriptional down-regulation are frequently used as biomarkers of PARP inhibitor response. Ten percent to 15% of breast and ovarian cancer patients carry germline mutations in HR genes BRCA1 and BRCA2 (Gallagher et al. 2011; Mavaddat et al. 2012; Nik-Zainal et al. 2016). Strikingly, 75% of germline mutations in metastatic cancers affect DNA repair genes such as MUTYH, BRCA2, CHEK2, and BRCA1 (Robinson et al. 2017). Moreover, BRCA1/2, ATM, and CHEK2 are the most frequently mutated DNA repair genes in somatic cancer cells (Heeke et al. 2018). In addition to being directly inactivated by mutation, BRCA1 and RAD51C were also found to be down-regulated through promoter hypermethylation in breast and ovarian cancer (Chiang et al. 2006; The Cancer Genome Atlas Research Network 2011; Lips et al. 2013; Timms et al. 2014; Polak et al. 2017; Bernards et al. 2018; Castroviejo-Bermejo et al. 2018; Kondrashova et al. 2018). BRCA1 promoter methylation confers the same degree of sensitivity to PARP inhibitors as BRCA1 mutations (Veeck et al. 2010). Furthermore, the expression level of BRCA1 was shown to be reduced due to depletion of the CDK12 kinase, which sensitizes breast and ovarian cancer cells to PARP inhibition (Bajrami et al. 2014). CDK12 regulates transcription of HR genes by suppressing intronic polyadenylation (Dubbury et al. 2018). CDK12 is often mutated in ovarian and prostate cancer and CDK12 deficiency may thus prove useful as a biomarker of PARP inhibitor response (The Cancer Genome Atlas Research Network 2011; Bajrami et al. 2014; Wu et al. 2018). A recent CRISPR screen identified TP53-induced glycolysis and apoptosis regulator (TIGAR) as another modulator of expression of HR genes (Fang et al. 2019a). TIGAR is amplified in different cancer types and its down-regulation sensitizes cancer cells to PARP inhibitors through inhibition of the pentose phosphate pathway, increase in ROS and DNA damage, down-regulation of BRCA1/2 and RAD51, and induction of cellular senescence (Fang et al. 2019a).
HR deficiency can also be scored based on different mutational and genomic signatures. A mutational signature of HR deficiency in BRCA mutated breast, ovarian, and pancreatic cancers are large indels (3- to 50-bp insertions and deletions) with overlapping microhomology at breakpoint junctions that result from NHEJ as the alternative pathway of DSB repair (Alexandrov et al. 2013). NHEJ joins two broken DNA ends, which may lead to small insertions or deletions (indels) (Chang et al. 2017). In addition to microhomology-mediated indels as the main signature of BRCA1/2 deficiency, base substitutions and rearrangements also reflect an abrogation of DSB repair pathways (Nik-Zainal et al. 2016). Mutational signatures identified from whole-genome sequencing of breast, ovarian, and pancreatic cancers were used to generate an HRDetect tool that can predict HR deficiencies (Davies et al. 2017). However, mutational signatures are not prognostic of PARP inhibitor sensitivity in the case of tumors with restored HR, which harbor mutational signatures but are resistant to PARP inhibitors due to restoration of the HR pathway (see “Mechanisms of Resistance to PARP Inhibitors”; Mateo et al. 2019).
Genomic signatures of HR deficiency comprise loss of heterozygosity (LOH), telomeric allelic imbalances (TAIs), and large-scale state transitions (LSTs). LOH results in irreversible loss of one of the parental alleles in regions >15 Mb (Abkevich et al. 2012). TAI refers to unequal contribution of maternal and paternal telomeric DNA sequences (Birkbak et al. 2012). LSTs are defined as chromosomal breaks between adjacent regions of at least 10 Mb (Popova et al. 2012). LOH, TAIs, and LSTs were shown to correlate well with mutations in HR genes BRCA1/2 in breast and ovarian cancer (Abkevich et al. 2012; Birkbak et al. 2012; Popova et al. 2012). All these genomic signatures of HR deficiency have been combined in a “homologous recombination deficiency” (HRD) score as a measure of genomic instability (Timms et al. 2014; Telli et al. 2016). HRDetect and HRD scores are both used to predict PARP inhibitor sensitivity in clinical settings.
Examining transcriptional signatures (or RNA) of cancer cells rather than their mutational signatures (or DNA) emerged as another means of predicting PARP inhibitor sensitivity. Gene expression profile derived from BRCA mutated ovarian cancers, termed “the BRCAness profile,” was found to correlate with platinum and PARP inhibitor sensitivity and was efficient in predicting platinum sensitivity of non-BRCA mutated ovarian cancers (Konstantinopoulos et al. 2010). Furthermore, gene expression profiles from cell lines depleted in different HR proteins revealed an HRD transcriptome signature that can predict HR deficiency and PARP inhibitor sensitivity (Peng et al. 2014).
Last, the cytological signature of HR deficiency is given by the number of RAD51 foci. Reduced RAD51 foci formation indicates HR deficiency and correlates with PARP inhibitor sensitivity, as shown in BRCA mutated breast tumor samples 2 h after 5 Gy of IR (Naipal et al. 2014), ovarian cancer cell lines 8 h after 4 Gy of IR (Shah et al. 2014), and breast cancer patient-derived xenografts (PDXs) (Castroviejo-Bermejo et al. 2018; Cruz et al. 2018). Unlike RAD51, γH2AX foci are not a reliable predictor of sensitivity to PARP inhibition as γH2AX foci may correlate positively or negatively with HR deficiency and PARP inhibitor sensitivity (Fong et al. 2009; Mukhopadhyay et al. 2010; Dale Rein et al. 2015; Michelena et al. 2018).
Replication stress
High levels of replication stress and depletion of replication-associated genes may render cancer cells sensitive to PARP inhibitors even in the absence of HR deficiency. For example, loss of TP53 and RB1 coupled with amplification of MYC generate replication stress and sensitize small cell lung cancer cells (SCLC) to PARP inhibitors (George et al. 2015; Sen et al. 2018). Schlafen 11 (SLFN11) is a recently identified biomarker of PARP inhibitor response in SCLC. SLFN11 binds to RPA in response to replication stress and blocks replication fork progression by changing chromatin structure (Murai et al. 2018). SLFN11 overexpression correlates with PARP inhibitor sensitivity, as shown in SCLC PDXs treated with olaparib or talazoparib, as well as in SCLC patients treated with veliparib in combination with temozolomide (Allison Stewart et al. 2017; Lok et al. 2017).
Overexpression of the cytidine deaminase APOBEC3, which causes an increase in abasic sites at replication forks, is frequently encountered in cancer and was shown to sensitize cells to PARP inhibitors (Burns et al. 2013; Roberts et al. 2013; Nikkilä et al. 2017). Mutations in genes required for Okazaki fragment processing such as FEN1 also sensitize cells to PARP inhibition, but FEN1 is rarely mutated in cancer (Murai et al. 2012). Depletion of replication-associated proteins such as cohesin (SMC1, SMC3, and RAD21), cohesin-associated factors (ESCO1 and ESCO2), core replication machinery (MCM2/3/6 helicases), and topoisomerases (TOP2B and TOP3A) increases sensitivity to PARP inhibition, as shown in colon and breast cancer cell lines (McLellan et al. 2012; Bajrami et al. 2014).
A recent CRISPR screen revealed synthetic lethality between mutations in RNASEH2 and PARP inhibition due to increased levels of replication-dependent DNA damage (Zimmermann et al. 2018). RNase H2 deficiency results in impaired ribonucleotide excision repair and accumulation of ribonucleotides that are cleaved by TOP1 (Zimmermann et al. 2018). Cleavage of these ribonucleotides produces nicks, covalent TOP1–DNA adducts, and ssDNA gaps that can act as PARP-trapping lesions, thus contributing to PARP inhibitor efficacy (Zimmermann et al. 2018). RNASEH2B deletions are frequently found in chronic lymphocytic leukemia and metastatic prostate cancer, which renders them more sensitive to PARP inhibition (Zimmermann et al. 2018).
Transcriptome profiles, PARP1 expression levels, and PARP1 activity
Given the important roles of PARP1 in transcription regulation (Kraus and Hottiger 2013), gene expression profiles from ovarian and breast cancer cell lines with known sensitivity to olaparib and rucaparib were used to derive a transcriptional algorithm that can predict sensitivity to PARP inhibitors (McGrail et al. 2017). The expression levels of PARP1 itself may also determine PARP inhibitor response. PARP1 expression is increased in different cancer types, particularly at advanced stages (Ossovskaya et al. 2010; Domagala et al. 2011; Mascolo et al. 2012; Bi et al. 2013; Bieche et al. 2013; Gan et al. 2013; Salemi et al. 2013; Dziaman et al. 2014; Park et al. 2015; Zhai et al. 2015; Li et al. 2016; Hou et al. 2018), and in some cases correlates positively with the cytotoxic effects of PARP inhibition (Byers et al. 2012; Kukolj et al. 2017).
PARP1 catalytic activity is enhanced by the receptor tyrosine kinase c-Met-mediated phosphorylation on Y907, which in turn reduces PARP inhibitor binding. Blocking PARP1 phosphorylation with a c-Met inhibitor can increase the efficacy of PARP inhibitors (Du et al. 2016). Endogenous inhibition of PARP activity through increased levels of NADP+ was shown to render ovarian cancer cells hypersensitive to PARP inhibitors irrespective of the BRCA status, suggesting that NADP+ levels could also be used as a biomarker of PARP inhibitor sensitivity (Bian et al. 2019).
Clinical studies with PARP inhibitors
The very first clinical trials demonstrated the efficacy of the PARP inhibitor olaparib in breast and ovarian cancer patients carrying germline mutations in BRCA1/2, thus supporting the rationale for synthetic lethality (Fong et al. 2009; Audeh et al. 2010; Tutt et al. 2010). The phase 1 trial showed the antitumor activity of olaparib at 400 mg twice daily and acceptable adverse effects (nausea, fatigue, vomiting, taste alteration, anorexia) (Table 1; Fong et al. 2009). The PARP inhibitor niraparib showed antitumor activity at 300 mg daily with more pronounced hematologic adverse effects compared with olaparib (anemia, thrombocytopenia, and neutropenia) (Sandhu et al. 2013). Rucaparib showed partial or complete response in ovarian, breast, and pancreatic cancer patients given 600 mg twice daily, with fatigue, nausea, anemia, and vomiting as the most common adverse effects (Kristeleit et al. 2017). Talazoparib administered at the recommended dose of 1 mg/d demonstrated high antitumor activity in BRCA mutated breast and ovarian cancer patients with fatigue, anemia, and thrombocytopenia as the most pronounced adverse effects (de Bono et al. 2017). Clinically recommended doses and the severity of side effects correlate with the PARP inhibitor trapping potency; talazoparib as the strongest PARP trapper has the lowest recommended dose and shows the highest occurrence of anemia (de Bono et al. 2017; Litton et al. 2018; Pilié et al. 2019a).
Table 1.
Phase 1 clinical trials with PARP inhibitors

Olaparib treatment in advanced breast or ovarian cancer patients with BRCA1/2 germline mutations showed a 41% and a 33% objective response rate defined as the proportion of patients with tumor size reduction of a predefined amount and for a minimum time period (Table 2; Audeh et al. 2010; Tutt et al. 2010). Olaparib administered in high-grade serous and/or undifferentiated ovarian cancer patients showed a 41% and 24% objective response rate with or without BRCA1/2 mutations; however, there was no response in TNBC patients (Gelmon et al. 2011). A study comparing olaparib with placebo in platinum-sensitive, relapsed, high-grade serous ovarian cancer patients who had received two or more platinum-based regimens showed longer median progression-free survival from 4.3 to 11.2 mo for BRCA mutated cancer and from 5.5 to 7.4 mo for wild-type (WT) BRCA (Ledermann et al. 2012, 2014). However, the overall survival of olaparib-treated versus placebo patients was not significantly different after 5 yr (Ledermann et al. 2016). Long-term responders to olaparib with progression-free survival >2 yr had a prevalence of BRCA2 mutations and a high HRD score, confirming that mutations in HR genes such as BRCA2 and HRD score can be used as predictive biomarkers for PARP inhibitor response (Lheureux et al. 2017b). A study with platinum-resistant ovarian and breast cancer patients with three or more chemotherapy regimens for metastatic disease, all carrying BRCA1/2 mutations, showed a 31.1% and 12.9% tumor response rate to olaparib, indicating that olaparib may indeed be more effective in ovarian than breast cancer (Kaufman et al. 2015). Based on these studies, olaparib was approved in 2014 by EMA for the treatment of germline BRCA mutated ovarian cancer after three or more lines of chemotherapy, and by the FDA for the maintenance treatment of BRCA mutated ovarian cancer patients who have responded to platinum-based chemotherapy. The currently approved dose is 300 mg twice daily.
Table 2.
Phase 2 and phase 3 clinical trials with PARP inhibitors

In 2017, olaparib was approved by the FDA as maintenance therapy in platinum-sensitive high-grade ovarian cancer patients irrespective of the BRCA status, based on phase 3 clinical trials that showed longer median progression-free survival compared with placebo, from 5.5 mo to 19.1 mo (Pujade-Lauraine et al. 2017) or from 13.8 to 49.9 mo in patients with germline BRCA1/2 mutations (Table 2; Moore et al. 2018). In a phase-3 trial focusing on BRCA mutated and HER2-negative metastatic breast cancer patients, olaparib was compared with standard single-agent therapy (e.g., microtubule inhibitors, nucleoside, or fluoropyrimidine analogs) and showed a longer median progression-free survival and 59.9% objective response rate compared with 28.8% for patients receiving standard therapy (Robson et al. 2017). As a result, olaparib was approved by the FDA for germline BRCA mutated metastatic breast cancer in 2018. It remains to be clarified whether olaparib also confers an advantage over standard chemotherapy in ovarian cancer, given that one clinical trial in advanced ovarian cancer patients with BRCA1/2 mutations whose disease had progressed or recurred after the use of platinum-based chemotherapy showed that the effect of olaparib was not superior to pegylated liposomal doxorubicin (PLD) (Kaye et al. 2012). Nevertheless, chemotherapy is invariably associated with side effects on normal cells, which makes PARP inhibitors the therapy of choice even without appreciable differences in overall survival.
Rucaparib administered at 600 mg twice daily in platinum-sensitive, high-grade recurrent ovarian carcinoma patients showed longer median progression-free survival in BRCA mutated cancer compared with WT (12.8 mo vs. ∼5 mo) and a 53.8% objective response rate in BRCA mutated patients (Table 2; Oza et al. 2017; Swisher et al. 2017). As a result, rucaparib was approved in 2016 for advanced ovarian cancer with germline and somatic BRCA1/2 mutations. Phase 3 trials showed that rucaparib (600 mg twice daily) and niraparib (300 mg once daily) increased the median duration of progression-free survival compared with placebo, from ∼5 mo for placebo to 16.6 or 21 mo for rucaparib and niraparib (Mirza et al. 2016; Coleman et al. 2017). Niraparib was effective not only in the treatment of recurrent ovarian cancer but also newly diagnosed advanced ovarian cancer (Mirza et al. 2016; González-Martin et al. 2019). While the effect was more pronounced in BRCA mutated cancers, the patients without BRCA mutations also showed a good response to niraparib and rucaparib, indicating that PARP inhibitors can be effective irrespective of the BRCA status (Mirza et al. 2016; Coleman et al. 2017; González-Martin et al. 2019). Niraparib and rucaparib were approved in 2017 and 2018 by the FDA and the EMA as a maintenance treatment of recurrent, epithelial ovarian, fallopian tube, or primary peritoneal cancer irrespective of the BRCA status.
Talazoparib was approved in 2018 for BRCA mutated and HER2-negative breast cancer. Talazoparib administered at 1 mg daily showed a 21%–37% objective response rate in BRCA mutated advanced breast cancer patients (Table 2; Turner et al. 2019). Compared with standard single-agent therapy in BRCA mutated advanced breast cancer patients, talazoparib showed longer median progression-free survival and a 62.6% objective response rate compared with 27.2% for patients receiving standard therapy (Litton et al. 2018).
Veliparib applied as monotherapy did not improve clinical outcomes in ovarian and pancreatic cancer patients (Coleman et al. 2015; Lowery et al. 2018) or in combination with carboplatin/paclitaxel in TNBC (Table 2; Loibl et al. 2018). However, the most recent phase 3 study in high-grade serous ovarian carcinoma showed increased progression-free survival for veliparib in combination with carboplatin and paclitaxel followed by veliparib maintenance therapy compared with carboplatin and paclitaxel alone (Coleman et al. 2019). Veliparib is so far the only clinically relevant PARP inhibitor that is tolerated in combination with standard doses of chemotherapy.
PARP inhibitors have also shown good response in prostate and pancreatic patients. Germline BRCA2 mutations in prostate cancer patients are associated with worse clinical outcomes (Castro et al. 2013; Taylor et al. 2019). Olaparib administered in BRCA mutated pancreatic and prostate cancer patients elicited a 21.7% and 50% tumor response rate (Kaufman et al. 2015). Another study showed a 33% response rate to olaparib in prostate cancer patients, where most of the responders had BRCA2 or ATM mutations (Mateo et al. 2015). A phase 3 trial in metastatic pancreatic cancer patients showed longer median progression-free survival for olaparib versus placebo (7.4 vs. 3.8 mo) (Golan et al. 2019). Rucaparib showed a 15.8% response rate in BRCA mutated pancreatic cancer patients (Shroff et al. 2018). To date, PARP inhibitors have not yielded promising results in the treatment of metastatic gastric cancer, according to a phase 3 study where olaparib in combination with paclitaxel (taxane drug) did not improve overall survival (Bang et al. 2017). However, PARP inhibitors have shown encouraging results in small cell lung cancer (SCLC). Veliparib combined with temozolomide showed improved objective response rate in SCLC patients (Pietanza et al. 2018), while talazoparib alone or in combination with temozolomide yielded promising results in PDX models of SCLC (Lok et al. 2017; Laird et al. 2018).
PARG inhibitors have not yet reached clinical trials due to low metabolic stability. However, newly identified PARG inhibitors COH34 and JA2131 exhibit favorable pharmacokinetic properties and may enter clinical studies (Chen and Yu 2019; Houl et al. 2019).
Mechanisms of resistance to PARP inhibitors
Restoration of the HR pathway
Resistance to chemotherapy is a frequent problem in clinical practice that also affects PARP inhibitors (Gogola et al. 2019; Noordermeer and van Attikum 2019). The most common avenue of PARP inhibitor resistance is restoration of the HR pathway. The first example of PARP inhibitor resistance was identified in Capan-1 cells derived from a pancreatic epithelial tumour, which had an intragenic deletion in BRCA2 of 458 bp resulting in removal of the inactivating frameshift mutation and expression of an almost full-length BRCA2 protein lacking 153 amino acids (Edwards et al. 2008). Since then, many examples of secondary mutations in BRCA1/2, as well as RAD51C, RAD51D, and PALB2, that genetically revert the mutation and restore functional full-length protein have been reported in breast, ovarian, pancreatic, and prostate carcinoma (Sakai et al. 2008; Norquist et al. 2011; Barber et al. 2013; Patch et al. 2015; Christie et al. 2017; Goodall et al. 2017; Kondrashova et al. 2017; Lheureux et al. 2017a; Pishvaian et al. 2017; Quigley et al. 2017; Weigelt et al. 2017; Lin et al. 2019). Platinum and cisplatin chemotherapy seem to select for these secondary mutation events (Sakai et al. 2008; Norquist et al. 2011).
Secondary mutations in BRCA1 may result in the expression of functional hypomorphic variants in PARP inhibitor-resistant patients (Drost et al. 2016; Wang et al. 2016c). Such mutant BRCA1 protein may be stabilized by the heat shock chaperone HSP90 as shown in MDA-MB-436 breast cancer cells (Johnson et al. 2013).
Increased levels of RAD51 as the core component of the HR pathway were also shown to contribute to PARP inhibitor resistance in TNBC (Liu et al. 2017). In TNBC cell lines, down-regulation of the early mitotic inhibitor 1 (EMI1), which assembles a ubiquitin ligase complex to degrade RAD51, induces PARP inhibitor resistance (Marzio et al. 2019).
Finally, inactivation of different NHEJ-promoting factors that inhibit DNA end resection, such as 53BP1, RIF1, PTIP, Artemis, REV7 (MAD2L2), the Shieldin complex (SHLD1–3 and REV7), and the CTC1–STN1–TEN1 (CST) complex, can also lead to a partial restoration of HR in BRCA1-deficient cells and mammary tumors (Bunting et al. 2010; Callen et al. 2013; Chapman et al. 2013; Escribano-Diaz et al. 2013; Jaspers et al. 2013; Zimmermann et al. 2013; Wang et al. 2014; Xu et al. 2015; Barazas et al. 2018; Dev et al. 2018; Ghezraoui et al. 2018; Gupta et al. 2018; Noordermeer et al. 2018). Furthermore, inactivation of dynein DYNLL1 and the HELB helicase can also promote end resection and restore the HR pathway independent of 53BP1, resulting in PARP inhibitor resistance in BRCA1-deficient cells (Tkáč et al. 2016; Dev et al. 2018; He et al. 2018). Of note, while BRCA1 mutations are compatible with HR restoration upon inactivation of NHEJ-promoting factors, restoration of the HR pathway fully depends on functional BRCA2 (Bouwman et al. 2010).
Stabilization of replication forks
In BRCA1/2-deficient cells, stabilization of replication forks independent of HR restoration and DSB repair emerged as an alternative mechanism of PARP inhibitor resistance (Ray Chaudhuri et al. 2016). PARP1, BRCA1, and BRCA2 protect stalled forks from nucleolytic degradation and enable their restart after DNA damage; their loss therefore leads to fork degradation (Lomonosov et al. 2003; Bryant et al. 2009; Ray Chaudhuri et al. 2012; Schlacher et al. 2012; Ying et al. 2012; Berti et al. 2013; Pathania et al. 2014; Ronson et al. 2018). Compensatory mechanisms of restoring fork protection in the absence of BRCA1/2 or PARP1 may be at play in PARP inhibitor-resistant cancers. For example, loss or inhibition of PTIP, a downstream effector of 53BP1, protects forks from degradation in BRCA2-deficient cells by inhibiting the recruitment of the MRE11 nuclease to stalled forks, resulting in PARP inhibitor resistance (Ray Chaudhuri et al. 2016). Furthermore, reduced expression of the Polycomb protein EZH2 in BRCA2 mutated cancer results in fork stabilization due to reduced H3K27 methylation and reduced recruitment of the MUS81 nuclease as a result (Rondinelli et al. 2017). Depletion of fork remodeling factors such as SMARCAL1, ZRANB3, and HLTF, which promote MRE11-dependent fork degradation, also reduces sensitivity to olaparib (Taglialatela et al. 2017). Increased expression of miR-493-5p was shown to induce resistance to PARP inhibitors in BRCA2 mutated carcinomas by down-regulating MRE11 and EXO1 and thereby stabilizing replication forks (Meghani et al. 2018). While SLFN11 overexpression is used as a biomarker of PARP inhibitor sensitivity (Allison Stewart et al. 2017; Lok et al. 2017), SLFN11 is inactivated in 50% of cancers and frequently silenced due to promoter methylation (Nogales et al. 2016; Murai et al. 2018). SLFN11 repression occurs frequently in talazoparib-resistant cancer cell lines and was linked with impaired S-phase arrest and G2 progression, which may also be due to fork stabilization (Murai et al. 2016, 2018).
PARP1 and PARG mutations
PARP1 mutations within and outside the DNA-binding domain were found to confer resistance to PARP inhibitors by hindering PARP trapping (Pettitt et al. 2013, 2018). A clinically relevant PARP mutation found in an olaparib-resistant ovarian cancer patient occurs within the WGR domain (R591C) (Pettitt et al. 2018). The mutated residue is critical for interdomain communication between the WGR and the DNA-binding domain; while being proficient in DNA binding, the PARP1 R591C mutant dissociates rapidly from DNA damage sites, indicating inefficient PARP DNA trapping (Pettitt et al. 2018). PARP1 mutations can only confer PARP inhibitor resistance in HR-proficient cells or BRCA1 mutated cells that have residual BRCA1 activity, as PARP1 and BRCA1 mutations are synthetic lethal (Pettitt et al. 2018).
PARP1 depletion was shown to reduce sensitivity to PARP inhibitors in DT40 cells (Murai et al. 2012). BRCA1 mutated breast cancer PDX models resistant to PARP inhibitors showed reduced PARP1 expression levels (Dev et al. 2018). However, PARP inhibitor resistance due to reduced PARP1 expression levels might not be very common in vivo due to the high cellular abundance of PARP1 (Thomas et al. 2018).
PARG mutations were identified in TNBC and high-grade serous ovarian cancers eligible for PARP inhibitor treatment (Gogola et al. 2018). On the one hand, PARG depletion was shown to reduce sensitivity of HeLa cells to PARP inhibitors combined with DNA-damaging agents or mouse Brca2 mutated cells to PARP inhibitors alone (Feng and Koh 2013; Gogola et al. 2018). PARG deficiency may contribute to PARP inhibitor resistance by increasing PARP1 auto-PARylation that allows PARP1 dissociation from DNA (i.e., reduction in PARP trapping) and restoration of PARP signaling (Gogola et al. 2018). On the other hand, down-regulation of PARG due to loss of the RNA stabilizing protein HuR (ELAV-L1) was shown to enhance sensitivity to PARP inhibitors in pancreatic ductal carcinoma (Chand et al. 2017). Increased PARylation may sensitize cells to PARP inhibitors by enhancing PARP1 trapping and promoting DNA damage accumulation (Chand et al. 2017). The variable effects of PARG depletion on reduction or increase in PARP trapping may be due to the nature of stabilized PAR polymers or the site of their attachment.
Drug efflux
Overexpression of ATP-binding cassette (ABC) drug transporters is often associated with drug resistance (Patch et al. 2015). Olaparib and rucaparib, but not veliparib, are substrates of the ATP-dependent drug efflux P-glycoprotein (P-gp) pump (also known as MDR1, encoded by ABCB1) (Lawlor et al. 2014; Henneman et al. 2015; Parrish et al. 2015). Long-term treatment with PARP inhibitors leads to the up-regulation of P-gp, thereby reducing the intracellular concentrations of PARP inhibitors (Rottenberg et al. 2008). Coadministration of the pump inhibitor verapamil, elacridar, or tariquidar can resensitize cancer cells to PARP inhibitors (Rottenberg et al. 2008; Vaidyanathan et al. 2016).
PARP inhibitor combination therapies
Frequently acquired resistance to PARP inhibitors has spurred efforts to combine PARP inhibitors with other agents (Dréan et al. 2016; Pilié et al. 2019b). Phase 1 clinical trials with PARP inhibitors in combination with chemotherapeutic agents such as temozolomide, cisplatin, carboplatin, paclitaxel, gemcitabine, or the topoisomerase inhibitor I topotecan showed severe myelosuppression in the form of neutropenia and thrombocytopenia as a side effect (Matulonis and Monk 2017; Yap et al. 2019). Veliparib is the only clinically relevant PARP inhibitor that can be combined with chemotherapy (see “Clinical Studies with PARP Inhibitors”). Administration of PARP inhibitors following chemotherapy has proven to be a better strategy (Ledermann et al. 2012; Coleman et al. 2015, 2017; Mirza et al. 2016; Oza et al. 2017; Pujade-Lauraine et al. 2017; Robson et al. 2017; Swisher et al. 2017; Moore et al. 2018). Moreover, mutations that cause resistance to PARP inhibitors, such as loss of NHEJ factors, PARP1, or PARG, were shown to sensitize cells to different DNA-damaging agents such as radiotherapy, topoisomerase I inhibitors and temozolomide (Murai et al. 2014b; Gogola et al. 2018; Barazas et al. 2019).
Cell cycle checkpoint inhibitors
Combination of PARP inhibitors with cell cycle checkpoint inhibitors is a promising strategy in overcoming PARP inhibitor resistance caused by replication fork stabilization (Kim et al. 2017; Yazinski et al. 2017; Haynes et al. 2018). The S/G2 checkpoint is essential to allow repair of DSBs induced by PARP inhibitors and to prevent premature mitotic entry and mitotic catastrophe. Inhibition of the S/G2 checkpoint kinases ATR and CHK1 results in unscheduled replication origin firing, exhausts nuclear RPA pools due to excess ssDNA, depletes dNTPs, and allows mitotic entry in the presence of underreplicated DNA and unrepaired DNA damage from interphase (Toledo et al. 2017; Lecona and Fernandez-Capetillo 2018). The combination of PARP inhibitors and ATR or CHK1 inhibitors in HR-deficient cells causes the release of the G2/M arrested cells, accumulation of chromosomal breaks, and aberrations in mitosis followed by cell death (Kim et al. 2017; Fang et al. 2019b). Chromosomal breaks in mitosis, indicative of unrepaired DSBs, are more pronounced for the PARP–ATR inhibitor combination, which is also more effective in inducing tumor regression in ovarian cancer PDX models (Kim et al. 2017). The ATR inhibitor overcomes fork stabilization in BRCA-deficient cells by preventing RAD51 loading onto stalled forks and triggering MRE11-mediated fork degradation, as shown in PARP-resistant ovarian cancer cells and PDXs (Yazinski et al. 2017). The ATR inhibitor can also overcome resistance to talazoparib due to SLFN11 repression (Murai et al. 2016). Although PARG inhibitors have not yet reached clinical trials, they have shown synergistic effects with CHK1 inhibitors in ovarian cancer cell lines and ovarian PDX models (Pillay et al. 2019).
Furthermore, PARP inhibitors were successfully combined with WEE1 kinase inhibitors (Lallo et al. 2018; Parsels et al. 2018; Fang et al. 2019b). WEE1 kinase regulates S phase and G2/M progression by inhibiting cell cycle-dependent kinases CDK1 and CDK2. The combination of PARP and WEE1 inhibitors abrogates G2 arrest and induces mitotic catastrophe, yielding promising results in small cell lung cancer (Lallo et al. 2018), KRAS mutated nonsmall cell lung cancers (Parsels et al. 2018), gastric cancer (Lin et al. 2018), and TP53 mutated cancer (Meng et al. 2018). Sequential rather than concurrent inhibition of PARP and ATR or WEE1 is preferable in a clinical setting due to lower toxicity while preserving efficacy (Fang et al. 2019b).
PARP inhibitors also synergize with the pan-CDK inhibitor dinaciclib in reducing MYC expression in TNBC and other cancer types, resulting in down-regulation of HR genes and induction of DNA damage (Carey et al. 2018).
Inhibitors of transcription regulators and epigenetic modifiers
Inhibition of BRD4 as the global transcription regulator is synthetic lethal with PARP inhibitors (Karakashev et al. 2017; Yang et al. 2017; Sun et al. 2018). BRD4 inhibition induces HR deficiency independent of the BRCA status by impairing CTIP, BRCA1, and RAD51 expression, inducing DNA damage and eventually resulting in mitotic catastrophe in various cancer cell lines and PDX models (Karakashev et al. 2017; Yang et al. 2017; Sun et al. 2018). Inhibition of the Polycomb protein EZH2 synergizes with PARP inhibitors in BRCA mutated breast and ovarian cancer cells (Yamaguchi et al. 2018). Histone deacetylase (HDAC) inhibitors synergize with PARP inhibitors through the down-regulation of HR genes and an increase in DNA damage in PTEN-positive TNBC and prostate cancer (Chao and Goodman 2014; Min et al. 2015). EZH2 and HDAC inhibitors may restore PARP inhibitor sensitivity by restoring SLFN11 expression in SCLC (Gardner et al. 2017; Tang et al. 2018). The combination with inhibitors of DNA methyltransferase 1 (DNMT1) was shown to enhance the PARP inhibitor effect in TNBC through an increase in DSBs due to PARP retention at DNA damage sites (Muvarak et al. 2016).
Agents that pharmacologically induce HR deficiency: androgen receptor inhibitors, PI3K–AKT–mTOR inhibitors, and antiangiogenic agents
PARP inhibitors have been successfully combined with agents that pharmacologically recreate HR deficiency and BRCAness. For example, the combination of olaparib with the androgen receptor (AR) inhibitors enzalutamide or bicalutamid is effective in HR-proficient prostate cancer due to suppression of HR gene expression (Asim et al. 2017; Li et al. 2017). Olaparib in combination with the AR antagonist abiraterone in metastatic prostate cancer patients extended median progression-free survival from 8.2 mo (abiraterone alone) to 13.8 mo regardless of the HR mutation status (Table 3; Clarke et al. 2018).
Table 3.
Clinical trials with PARP inhibitor combination treatments

PARP inhibitors further synergize with inhibitors of the PI3K pathway, as shown in BRCA-proficient TNBC, BRCA1 mutated breast cancer mouse models, PIK3CA mutated ovarian cancer cells, and PTEN mutated endometrial cancer cells (Ibrahim et al. 2012; Juvekar et al. 2012; Wang et al. 2016b; Philip et al. 2017). A phase I clinical trial revealed synergistic effects between olaparib and the PI3K inhibitor alpelisib in epithelial ovarian cancer, which are most likely based on HR suppression through reduced RAD51 protein levels and RAD51 foci formation (Konstantinopoulos et al. 2019). The combination of talazoparib and the mTOR inhibitor everolimus synergistically kills BRCA-proficient TNBC by inducing HR deficiency through repression of the histone methyltransferase SUV39H1 (Mo et al. 2016).
Combination with antiangiogenic agents may sensitize tumors by inducing hypoxia and thereby reducing the expression of HR genes (Bindra et al. 2005). Hypoxia induces reduction in binding of the activating transcription factor E2F1 to the BRCA1 promoter, resulting in reduced BRCA1 expression (Bindra et al. 2005). PARP inhibition also reduces E2F1 genomic binding and expression of the E2F1 target genes (Byers et al. 2012; Schiewer et al. 2018) and may thereby synergize with hypoxic effects induced by antiangiogenic agents. A successful example of such a synergy is given by the combination of olaparib with the pan-VEGF inhibitor cediranib, which increased median progression-free survival from 9 to 17.7 mo compared with olaparib alone in platinum-sensitive ovarian cancer (Liu et al. 2014). Another VEGF inhibitor bevacizumab has also shown promising results in combination with niraparib in platinum-sensitive ovarian cancer patients (Mirza et al. 2019).
Immune checkpoint inhibitors
Replication stress induced by PARP inhibitors was recently shown to stimulate the expression of type I interferons by activating the cGAS–STING pathway (Shen et al. 2019). DNA damage or DNA repair defects caused by BRCA or ERCC1 mutations can also activate this pathway and thereby potentiate PARP inhibitor effects in ERCC1-deficient nonsmall cell lung cancer, BRCA1-deficient TNBC, and ovarian cancer (Ding et al. 2018; Chabanon et al. 2019). The cGAS–STING pathway is activated by cytosolic DNA fragments coming from micronuclei or destabilized replication forks bearing ssDNA and unresolved DNA lesions (Harding et al. 2017; Mackenzie et al. 2017; Parkes et al. 2017). Nucleases such as MRE11, EXO1, and CtIP are required for production of DNA fragments and activation of the cGAS–STING pathway (Shen et al. 2019). Stimulation of interferon production by the cGAS–STING pathway promotes antitumor immunity in immunogenic tumors but also triggers the expression of immune checkpoints such as PD-L1 (Deng et al. 2014; Parkes et al. 2017). PD-L1 is a ligand that binds to the PD-1 receptor on T cells and suppresses T-cell proliferation and cytokine release. Immune checkpoint inhibitors are antibodies that block PD-L1 to restore T-cell function. Immunotherapy with immune checkpoint inhibitors relies on the activation of the cGAS–STING pathway (Harding et al. 2017). Therefore, combining PARP inhibitors that activate the cGAS–STING pathway with immune checkpoint inhibitors has proven effective as an antitumor therapy. PARP inhibitors combined with antibodies against PD-L1 show synergistic effects in breast cancer cells and PDXs (Jiao et al. 2017) as well as ovarian and colon-cancer mouse models (Ding et al. 2018; Shen et al. 2019). Phase II trials of the PD-L1 antibody durvalumab or pembrolizumab in combination with olaparib or niraparib showed clinical response in patients with advanced SCLC, germline BRCA1/2 mutated breast and platinum-sensitive ovarian cancer, but also in prostate cancer and platinum-resistant ovarian cancer patients irrespective of BRCA mutations (Pilié et al. 2019a).
Concluding remarks
PARP and PARG are critical regulators of DNA damage response and replication fork stability. Cancer cells experience high levels of replication stress and often harbor germline or somatic mutations in DNA damage response genes. Replication stress compromises replication fork stability, whereas mutations in homologous recombination genes prevent the restoration of fork stability. PARP and PARG inhibitors exploit and exacerbate these vulnerabilities of cancer cells by destabilizing replication forks and inducing DNA damage. The original concept of synthetic lethality between PARP inhibitors and breast and ovarian cancer mutations in BRCA1/2 has now expanded to include a range of cancer types exhibiting homologous recombination deficiency and replication stress. PARP inhibitors have demonstrated the success of the “DNA damage response synthetic lethality” paradigm in various clinical trials by markedly extending disease progression-free survival while being well-tolerated. As clinical trials exploring further indications and combination treatments with PARP inhibitors are ongoing and PARG inhibitor trials still pending, the PARP community should invest further efforts into understanding on a molecular and cellular level how PARP and PARG maintain replication fork integrity and how replication stress and genomic instability resulting from their inhibition instigate mitotic defects and cell death by replication and mitotic catastrophe. This would advance new biomarkers of PARP inhibitor sensitivity and would guide new combination strategies.
Acknowledgments
I thank Rupert Thorough for editing English, and Ortrun Mittelsten Scheid and Hedwig Sutterlüty-Fall for their kind help with paper access. This work was supported by the Austrian Science Fund Grant (P 31112-B28).
Footnotes
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.334516.119.
Freely available online through the Genes & Development Open Access option.
References
- Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, Smith-McCune K, Broaddus R, Lu KH, Chen J, et al. 2012. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer 107: 1776–1782. 10.1038/bjc.2012.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait Saada A, Lambert SAE, Carr AM. 2018. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 71: 135–147. 10.1016/j.dnarep.2018.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison Stewart C, Tong P, Cardnell RJ, Sen T, Li L, Gay CM, Masrorpour F, Fan Y, Bara RO, Feng Y, et al. 2017. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 8: 28575–28587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Gonzalez R, Althaus FR. 1989. Poly(ADP-ribose) catabolism in mammalian cells exposed to DNA-damaging agents. Mutat Res 218: 67–74. 10.1016/0921-8777(89)90012-8 [DOI] [PubMed] [Google Scholar]
- Amé JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Höger T, Menissier-de Murcia J, de Murcia G. 1999. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 274: 17860–17868. 10.1074/jbc.274.25.17860 [DOI] [PubMed] [Google Scholar]
- Ame JC, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, Schreiber V. 2009. Radiation-induced mitotic catastrophe in PARG-deficient cells. J Cell Sci 122: 1990–2002. 10.1242/jcs.039115 [DOI] [PubMed] [Google Scholar]
- Anachkova B, Russev G, Poirier GG. 1989. DNA replication and poly(ADP-ribosyl)ation of chromatin. Cytobios 58: 19–28. [PubMed] [Google Scholar]
- Asim M, Tarish F, Zecchini HI, Sanjiv K, Gelali E, Massie CE, Baridi A, Warren AY, Zhao W, Ogris C, et al. 2017. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun 8: 374 10.1038/s41467-017-00393-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, et al. 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376: 245–251. 10.1016/S0140-6736(10)60893-8 [DOI] [PubMed] [Google Scholar]
- Bai P. 2015. Biology of poly(ADP-ribose) polymerases: the factotums of cell maintenance. Mol Cell 58: 947–958. 10.1016/j.molcel.2015.01.034 [DOI] [PubMed] [Google Scholar]
- Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, Campbell J, Sims D, Rafiq R, Hooper S, et al. 2014. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res 74: 287–297. 10.1158/0008-5472.CAN-13-2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasik M, Komura H, Shimoyama M, Ueda K. 1992. Specific inhibitors of poly(ADP-ribose) synthetase and mono(ADP-ribosyl)transferase. J Biol Chem 267: 1569–1575. [PubMed] [Google Scholar]
- Bang YJ, Xu RH, Chin K, Lee KW, Park SH, Rha SY, Shen L, Qin S, Xu N, Im SA, et al. 2017. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 18: 1637–1651. 10.1016/S1470-2045(17)30682-4 [DOI] [PubMed] [Google Scholar]
- Barazas M, Annunziato S, Pettitt SJ, de Krijger I, Ghezraoui H, Roobol SJ, Lutz C, Frankum J, Song FF, Brough R, et al. 2018. The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-deficient cells. Cell Rep 23: 2107–2118. 10.1016/j.celrep.2018.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barazas M, Gasparini A, Huang Y, Küçükosmanoğlu A, Annunziato S, Bouwman P, Sol W, Kersbergen A, Proost N, de Korte-Grimmerink R, et al. 2019. Radiosensitivity is an acquired vulnerability of PARPi-resistant BRCA1-deficient tumors. Cancer Res 79: 452–460. 10.1158/0008-5472.CAN-18-2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, Assiotis I, Rodrigues DN, Reis Filho JS, Moreno V, et al. 2013. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol 229: 422–429. 10.1002/path.4140 [DOI] [PubMed] [Google Scholar]
- Barkauskaite E, Brassington A, Tan ES, Warwicker J, Dunstan MS, Banos B, Lafite P, Ahel M, Mitchison TJ, Ahel I, et al. 2013. Visualization of poly(ADP-ribose) bound to PARG reveals inherent balance between exo- and endo-glycohydrolase activities. Nat Commun 4: 2164 10.1038/ncomms3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Hořejší Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434: 864–870. 10.1038/nature03482 [DOI] [PubMed] [Google Scholar]
- Benjamin RC, Gill DM. 1980a. ADP-ribosylation in mammalian cell ghosts. Dependence of poly(ADP-ribose) synthesis on strand breakage in DNA. J Biol Chemistry 255: 10493–10501. [PubMed] [Google Scholar]
- Benjamin RC, Gill DM. 1980b. Poly(ADP-ribose) synthesis in vitro programmed by damaged DNA. A comparison of DNA molecules containing different types of strand breaks. J Biol Chem 255: 10502–10508. [PubMed] [Google Scholar]
- Bernards SS, Pennington KP, Harrell MI, Agnew KJ, Garcia RL, Norquist BM, Swisher EM. 2018. Clinical characteristics and outcomes of patients with BRCA1 or RAD51C methylated versus mutated ovarian carcinoma. Gynecol Oncol 148: 281–285. 10.1016/j.ygyno.2017.12.004 [DOI] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. 2013. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20: 347–354. 10.1038/nsmb.2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. 2011. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145: 435–446. 10.1016/j.cell.2011.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi FF, Li D, Yang Q. 2013. Hypomethylation of ETS transcription factor binding sites and upregulation of PARP1 expression in endometrial cancer. BioMed Res Int 2013: 946268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian C, Zhang C, Luo T, Vyas A, Chen SH, Liu C, Kassab MA, Yang Y, Kong M, Yu X. 2019. NADP+ is an endogenous PARP inhibitor in DNA damage response and tumor suppression. Nat Commun 10: 693 10.1038/s41467-019-08530-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieche I, Pennaneach V, Driouch K, Vacher S, Zaremba T, Susini A, Lidereau R, Hall J. 2013. Variations in the mRNA expression of poly(ADP-ribose) polymerases, poly(ADP-ribose) glycohydrolase and ADP-ribosylhydrolase 3 in breast tumors and impact on clinical outcome. Int J Cancer 133: 2791–2800. [DOI] [PubMed] [Google Scholar]
- Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, Bristow RG, Classon MK, Glazer PM. 2005. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res 65: 11597–11604. 10.1158/0008-5472.CAN-05-2119 [DOI] [PubMed] [Google Scholar]
- Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, Bowman-Colin C, Li Y, Greene-Colozzi A, Iglehart JD, et al. 2012. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov 2: 366–375. 10.1158/2159-8290.CD-11-0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 17: 688–695. 10.1038/nsmb.1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner JC, Feng FY, Han S, Patel S, Goyal SV, Bou-Maroun LM, Liu M, Lonigro R, Prensner JR, Tomlins SA, et al. 2012. PARP-1 inhibition as a targeted strategy to treat Ewing's sarcoma. Cancer Res 72: 1608–1613. 10.1158/0008-5472.CAN-11-3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Helleday T. 2006. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res 34: 1685–1691. 10.1093/nar/gkl108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917. 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. 2009. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 28: 2601–2615. 10.1038/emboj.2009.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141: 243–254. 10.1016/j.cell.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Temiz NA, Harris RS. 2013. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet 45: 977–983. 10.1038/ng.2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, Giri U, Peyton M, Fan YH, Diao L, et al. 2012. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov 2: 798–811. 10.1158/2159-8290.CD-12-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. 2013. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153: 1266–1280. 10.1016/j.cell.2013.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canan Koch SS, Thoresen LH, Tikhe JG, Maegley KA, Almassy RJ, Li J, Yu XH, Zook SE, Kumpf RA, Zhang C, et al. 2002. Novel tricyclic poly(ADP-ribose) polymerase-1 inhibitors with potent anticancer chemopotentiating activity: design, synthesis, and X-ray cocrystal structure. J Med Chem 45: 4961–4974. 10.1021/jm020259n [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615. 10.1038/nature10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey JPW, Karakas C, Bui T, Chen X, Vijayaraghavan S, Zhao Y, Wang J, Mikule K, Litton JK, Hunt KK, et al. 2018. Synthetic lethality of PARP inhibitors in combination with MYC blockade is independent of BRCA status in triple-negative breast cancer. Cancer Res 78: 742–757. 10.1158/0008-5472.CAN-17-1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami K, Guy M, et al. 2013. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol 31: 1748–1757. 10.1200/JCO.2012.43.1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castroviejo-Bermejo M, Cruz C, Llop-Guevara A, Gutiérrez-Enríquez S, Ducy M, Ibrahim YH, Gris-Oliver A, Pellegrino B, Bruna A, Guzmán M, et al. 2018. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol Med 10 10.15252/emmm.201809172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, Lamb A, Hénon C, Dorvault N, Rouanne M, et al. 2019. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest 129: 1211–1228. 10.1172/JCI123319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chand SN, Zarei M, Schiewer MJ, Kamath AR, Romeo C, Lal S, Cozzitorto JA, Nevler A, Scolaro L, Londin E, et al. 2017. Posttranscriptional regulation of PARG mRNA by HuR facilitates DNA repair and resistance to PARP inhibitors. Cancer Res 77: 5011–5025. 10.1158/0008-5472.CAN-16-2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR. 2017. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18: 495–506. 10.1038/nrm.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao OS, Goodman OB Jr. 2014. Synergistic loss of prostate cancer cell viability by coinhibition of HDAC and PARP. Mol Cancer Res 12: 1755–1766. 10.1158/1541-7786.MCR-14-0173 [DOI] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. 2013. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 49: 858–871. 10.1016/j.molcel.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Yu X. 2019. Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies. Sci Adv 5: eaav4340 10.1126/sciadv.aav4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JW, Karlan BY, Cass L, Baldwin RL. 2006. BRCA1 promoter methylation predicts adverse ovarian cancer prognosis. Gynecol Oncol 101: 403–410. 10.1016/j.ygyno.2005.10.034 [DOI] [PubMed] [Google Scholar]
- Christie EL, Fereday S, Doig K, Pattnaik S, Dawson SJ, Bowtell DDL. 2017. Reversion of BRCA1/2 germline mutations detected in circulating tumor DNA from patients with high-grade serous ovarian cancer. J Clin Oncol 35: 1274–1280. 10.1200/JCO.2016.70.4627 [DOI] [PubMed] [Google Scholar]
- Clark JB, Ferris GM, Pinder S. 1971. Inhibition of nuclear NAD nucleosidase and poly ADP-ribose polymerase activity from rat liver by nicotinamide and 5′-methyl nicotinamide. Biochim Biophys Acta 238: 82–85. 10.1016/0005-2787(71)90012-8 [DOI] [PubMed] [Google Scholar]
- Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, Chiuri VE, Jassem J, Fléchon A, Redfern C, et al. 2018. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 19: 975–986. 10.1016/S1470-2045(18)30365-6 [DOI] [PubMed] [Google Scholar]
- Coleman RL, Sill MW, Bell-McGuinn K, Aghajanian C, Gray HJ, Tewari KS, Rubin SC, Rutherford TJ, Chan JK, Chen A, et al. 2015. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation—an NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol 137: 386–391. 10.1016/j.ygyno.2015.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G, et al. 2017. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390: 1949–1961. 10.1016/S0140-6736(17)32440-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, Okamoto A, Moore KN, Efrat Ben-Baruch N, Werner TL, et al. 2019. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med 381: 2403–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicchia V, Petroni M, Guarguaglini G, Sardina F, Sahún-Roncero M, Carbonari M, Ricci B, Heil C, Capalbo C, Belardinilli F, et al. 2017. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene 36: 4682–4691. [DOI] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. 2012. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482: 53–58. 10.1038/nature10802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz C, Castroviejo-Bermejo M, Gutiérrez-Enríquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, Bonache S, Morancho B, Bruna A, Rueda OM, et al. 2018. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann Oncol 29: 1203–1210. 10.1093/annonc/mdy099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale Rein I, Solberg Landsverk K, Micci F, Patzke S, Stokke T. 2015. Replication-induced DNA damage after PARP inhibition causes G2 delay, and cell line-dependent apoptosis, necrosis and multinucleation. Cell Cycle 14: 3248–3260. 10.1080/15384101.2015.1085137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer F, Nasheuer HP, Vonesch JL, de Murcia G, Ménissier-de Murcia J. 1998. Functional association of poly(ADP-ribose) polymerase with DNA polymerase α-primase complex: a link between DNA strand break detection and DNA replication. Nucleic Acids Res 26: 1891–1898. 10.1093/nar/26.8.1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, et al. 2017. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23: 517–525. 10.1038/nm.4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, Kaye S, Sachdev J, Heymach J, Smith DC, et al. 2017. Phase I, dose-Escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov 7: 620–629. 10.1158/2159-8290.CD-16-1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. 2014. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41: 843–852. 10.1016/j.immuni.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W, Ostermaier M, et al. 2018. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 20: 954–965. 10.1038/s41556-018-0140-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. 2018. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep 25: 2972–2980.e5. 10.1016/j.celrep.2018.11.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelstein M, Sørensen CS. 2015. Exploiting replicative stress to treat cancer. Nat Rev Drug Discov 14: 405–423. 10.1038/nrd4553 [DOI] [PubMed] [Google Scholar]
- Domagala P, Huzarski T, Lubinski J, Gugala K, Domagala W. 2011. PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: possible implications for PARP-1 inhibitor therapy. Breast Cancer Res Treat 127: 861–869. 10.1007/s10549-011-1441-2 [DOI] [PubMed] [Google Scholar]
- Dréan A, Lord CJ, Ashworth A. 2016. PARP inhibitor combination therapy. Crit Rev Oncol Hematol 108: 73–85. 10.1016/j.critrevonc.2016.10.010 [DOI] [PubMed] [Google Scholar]
- Drew Y, Ledermann J, Hall G, Rea D, Glasspool R, Highley M, Jayson G, Sludden J, Murray J, Jamieson D, et al. 2016. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer 114: 723–730. 10.1038/bjc.2016.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost R, Dhillon KK, van der Gulden H, van der Heijden I, Brandsma I, Cruz C, Chondronasiou D, Castroviejo-Bermejo M, Boon U, Schut E, et al. 2016. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J Clin Invest 126: 2903–2918. 10.1172/JCI70196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee G, et al. 2016. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med 22: 194–201. 10.1038/nm.4032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbury SJ, Boutz PL, Sharp PA. 2018. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 564: 141–145. 10.1038/s41586-018-0758-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunstan MS, Barkauskaite E, Lafite P, Knezevic CE, Brassington A, Ahel M, Hergenrother PJ, Leys D, Ahel I. 2012. Structure and mechanism of a canonical poly(ADP-ribose) glycohydrolase. Nat Commun 3: 878 10.1038/ncomms1889 [DOI] [PubMed] [Google Scholar]
- Dziaman T, Ludwiczak H, Ciesla JM, Banaszkiewicz Z, Winczura A, Chmielarczyk M, Wisniewska E, Marszalek A, Tudek B, Olinski R. 2014. PARP-1 expression is increased in colon adenoma and carcinoma and correlates with OGG1. PLoS One 9: e115558 10.1371/journal.pone.0115558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. 2008. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451: 1111–1115. 10.1038/nature06548 [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. 2003. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res 31: 5526–5533. 10.1093/nar/gkg761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdèlyi K, Kiss A, Bakondi E, Bai P, Szabó C, Gergely P, Erdödi F, Virág L. 2005. Gallotannin inhibits the expression of chemokines and inflammatory cytokines in A549 cells. Mol Pharmacol 68: 895–904. 10.1124/mol.105.012518 [DOI] [PubMed] [Google Scholar]
- Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. 2013. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 49: 872–883. 10.1016/j.molcel.2013.01.001 [DOI] [PubMed] [Google Scholar]
- Eustermann S, Wu WF, Langelier MF, Yang JC, Easton LE, Riccio AA, Pascal JM, Neuhaus D. 2015. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol Cell 60: 742–754. 10.1016/j.molcel.2015.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsig J, Christiansen SH, Feuerhahn S, Bürkle A, Oei SL, Keil C, Leist M. 2004. Poly(ADP-ribose) glycohydrolase as a target for neuroprotective intervention: assessment of currently available pharmacological tools. Eur J Pharmacol 497: 7–16. 10.1016/j.ejphar.2004.06.042 [DOI] [PubMed] [Google Scholar]
- Fang P, De Souza C, Minn K, Chien J. 2019a. Genome-scale CRISPR knockout screen identifies TIGAR as a modifier of PARP inhibitor sensitivity. Commun Biol 2: 335 10.1038/s42003-019-0580-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, et al. 2019b. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell 35: 851–867.e7. 10.1016/j.ccell.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- Farrés J, Llacuna L, Martin-Caballero J, Martínez C, Lozano JJ, Ampurdanés C, López-Contreras AJ, Florensa L, Navarro J, Ottina E, et al. 2015. PARP-2 sustains erythropoiesis in mice by limiting replicative stress in erythroid progenitors. Cell Death Differ 22: 1144–1157. 10.1038/cdd.2014.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathers C, Drayton RM, Solovieva S, Bryant HE. 2012. Inhibition of poly(ADP-ribose) glycohydrolase (PARG) specifically kills BRCA2-deficient tumor cells. Cell Cycle 11: 990–997. 10.4161/cc.11.5.19482 [DOI] [PubMed] [Google Scholar]
- Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD, Thomas P. 2011. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 26: 125–132. 10.1093/mutage/geq052 [DOI] [PubMed] [Google Scholar]
- Feng X, Koh DW. 2013. Inhibition of poly(ADP-ribose) polymerase-1 or poly(ADPribose) glycohydrolase individually, but not in combination, leads to improved chemotherapeutic efficacy in HeLa cells. Int J Oncol 42: 749–756. 10.3892/ijo.2012.1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng FY, de Bono JS, Rubin MA, Knudsen KE. 2015. Chromatin to clinic: the molecular rationale for PARP1 inhibitor function. Mol Cell 58: 925–934. 10.1016/j.molcel.2015.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris DV. 2010. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J Med Chem 53: 4561–4584. 10.1021/jm100012m [DOI] [PubMed] [Google Scholar]
- Finch KE, Knezevic CE, Nottbohm AC, Partlow KC, Hergenrother PJ. 2012. Selective small molecule inhibition of poly(ADP-ribose) glycohydrolase (PARG). ACS Chem Biol 7: 563–570. 10.1021/cb200506t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher AE, Hochegger H, Takeda S, Caldecott KW. 2007. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol 27: 5597–5605. 10.1128/MCB.02248-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. 2009. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361: 123–134. 10.1056/NEJMoa0900212 [DOI] [PubMed] [Google Scholar]
- Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. 2018. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res 122: 877–902. 10.1161/CIRCRESAHA.117.311401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzese E, Centonze S, Diana A, Carlino F, Guerrera LP, Di Napoli M, De Vita F, Pignata S, Ciardiello F, Orditura M. 2019. PARP inhibitors in ovarian cancer. Cancer Treat Rev 73: 1–9. 10.1016/j.ctrv.2018.12.002 [DOI] [PubMed] [Google Scholar]
- Gallagher DJ, Konner JA, Bell-McGuinn KM, Bhatia J, Sabbatini P, Aghajanian CA, Offit K, Barakat RR, Spriggs DR, Kauff ND. 2011. Survival in epithelial ovarian cancer: a multivariate analysis incorporating BRCA mutation status and platinum sensitivity. Ann Oncol 22: 1127–1132. 10.1093/annonc/mdq577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. 2018. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 25: 486–541. 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan A, Green AR, Nolan CC, Martin S, Deen S. 2013. Poly(adenosine diphosphate-ribose) polymerase expression in BRCA-proficient ovarian high-grade serous carcinoma; association with patient survival. Hum Pathol 44: 1638–1647. 10.1016/j.humpath.2013.01.015 [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D. 2012. Linking abnormal mitosis to the acquisition of DNA damage. J Cell Biol 199: 871–881. 10.1083/jcb.201210040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, Ni A, Khodos I, de Stanchina E, Nguyen T, et al. 2017. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2–SLFN11 axis. Cancer Cell 31: 286–299. 10.1016/j.ccell.2017.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, et al. 2011. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 12: 852–861. 10.1016/S1470-2045(11)70214-5 [DOI] [PubMed] [Google Scholar]
- Gemble S, Ahuja A, Buhagiar-Labarchède G, Onclercq-Delic R, Dairou J, Biard DS, Lambert S, Lopes M, Amor-Guéret M. 2015. Pyrimidine pool disequilibrium induced by a cytidine deaminase deficiency inhibits PARP-1 activity, leading to the under replication of DNA. PLoS Genet 11: e1005384 10.1371/journal.pgen.1005384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, et al. 2015. Comprehensive genomic profiles of small cell lung cancer. Nature 524: 47–53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar-Lele M, Sanchiz-Calvo M, Fueyo-Marcos E, et al. 2018. 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 560: 122–127. 10.1038/s41586-018-0362-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini S, Weller MC, Repmann S, Moch H, Jiricny J. 2019. Synthetic lethality between BRCA1 deficiency and poly(ADP-ribose) polymerase inhibition is modulated by processing of endogenous oxidative DNA damage. Nucleic Acids Res 47: 9132–9143. 10.1093/nar/gkz624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godon C, Cordelieres FP, Biard D, Giocanti N, Megnin-Chanet F, Hall J, Favaudon V. 2008. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res 36: 4454–4464. 10.1093/nar/gkn403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, James DI, Guerrero Llobet S, Vis DJ, Annunziato S, et al. 2018. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell 33: 1078–1093.e12. 10.1016/j.ccell.2018.05.008 [DOI] [PubMed] [Google Scholar]
- Gogola E, Rottenberg S, Jonkers J. 2019. Resistance to PARP inhibitors: lessons from preclinical models of BRCA-associated cancer. Ann Rev Cancer Biol 3: 235–254. 10.1146/annurev-cancerbio-030617-050232 [DOI] [Google Scholar]
- Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, et al. 2019. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med 381: 317–327. 10.1056/NEJMoa1903387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G, et al. 2019. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 381: 2391–2402. 10.1056/NEJMoa1910962 [DOI] [PubMed] [Google Scholar]
- Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, Perez-Lopez R, Dolling D, Robinson DR, Sandhu S, et al. 2017. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov 7: 1006–1017. 10.1158/2159-8290.CD-17-0261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, DiTullio RA Jr, Kastrinakis NG, Levy B, et al. 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434: 907–913. 10.1038/nature03485 [DOI] [PubMed] [Google Scholar]
- Gravells P, Grant E, Smith KM, James DI, Bryant HE. 2017. Specific killing of DNA damage-response deficient cells with inhibitors of poly(ADP-ribose) glycohydrolase. DNA Repair 52: 81–91. 10.1016/j.dnarep.2017.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravells P, Neale J, Grant E, Nathubhai A, Smith KM, James DI, Bryant HE. 2018. Radiosensitization with an inhibitor of poly(ADP-ribose) glycohydrolase: a comparison with the PARP1/2/3 inhibitor olaparib. DNA Repair 61: 25–36. 10.1016/j.dnarep.2017.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. 2010. Immunity, inflammation, and cancer. Cell 140: 883–899. 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A, et al. 2018. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173: 972–988.e23. 10.1016/j.cell.2018.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A, Bulyk ML, Jambhekar A, Lahav G. 2019. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol 20: 199–210. 10.1038/s41580-019-0110-x [DOI] [PubMed] [Google Scholar]
- Haince J-F, McDonald D, Rodrigue A, Déry U, Masson J-Y, Hendzel MJ, Poirier GG. 2008. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 283: 1197–1208. 10.1074/jbc.M706734200 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hanzlikova H, Caldecott KW. 2019. Perspectives on PARPs in S phase. Trends Genet 35: 412–422. 10.1016/j.tig.2019.03.008 [DOI] [PubMed] [Google Scholar]
- Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, Caldecott KW. 2018. The importance of poly(ADP-ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol Cell 71: 319–331.e3. 10.1016/j.molcel.2018.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. 2017. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548: 466–470. 10.1038/nature23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama K, Nemoto Y, Ueda K, Hayaishi O. 1986. Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose). J Biol Chem 261: 14902–14911. [PubMed] [Google Scholar]
- Haynes B, Murai J, Lee JM. 2018. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat Rev 71: 1–7. 10.1016/j.ctrv.2018.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YJ, Meghani K, Caron MC, Yang C, Ronato DA, Bian J, Sharma A, Moore J, Niraj J, Detappe A, et al. 2018. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 563: 522–526. 10.1038/s41586-018-0670-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen WJ, Baker TM, Marshall JL, Isaacs C. 2018. Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis Oncol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Tora L. 2011. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 44: 966–977. 10.1016/j.molcel.2011.10.013 [DOI] [PubMed] [Google Scholar]
- Henneman L, van Miltenburg MH, Michalak EM, Braumuller TM, Jaspers JE, Drenth AP, de Korte-Grimmerink R, Gogola E, Szuhai K, Schlicker A, et al. 2015. Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proc Natl Acad Sci 112: 8409–8414. 10.1073/pnas.1500223112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, Zhao GY, Saberi A, Masutani M, Adachi N, et al. 2006. Parp-1 protects homologous recombination from interference by Ku and ligase IV in vertebrate cells. EMBO J 25: 1305–1314. 10.1038/sj.emboj.7601015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, DiGiammarino EL, Panchal SC, Wilsbacher JL, Gao W, Olson AM, et al. 2015. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol Cancer Res 13: 1465–1477. 10.1158/1541-7786.MCR-15-0191-T [DOI] [PubMed] [Google Scholar]
- Hou D, Liu Z, Xu X, Liu Q, Zhang X, Kong B, Wei JJ, Gong Y, Shao C. 2018. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol 17: 99–111. 10.1016/j.redox.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houl JH, Ye Z, Brosey CA, Balapiti-Modarage LPF, Namjoshi S, Bacolla A, Laverty D, Walker BL, Pourfarjam Y, Warden LS, et al. 2019. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat Commun 10: 5654 10.1038/s41467-019-13508-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J, et al. 2012. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov 2: 1036–1047. 10.1158/2159-8290.CD-11-0348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illuzzi G, Fouquerel E, Amé JC, Noll A, Rehmet K, Nasheuer HP, Dantzer F, Schreiber V. 2014. PARG is dispensable for recovery from transient replicative stress but required to prevent detrimental accumulation of poly(ADP-ribose) upon prolonged replicative stress. Nucleic Acids Res 42: 7776–7792. 10.1093/nar/gku505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James DI, Smith KM, Jordan AM, Fairweather EE, Griffiths LA, Hamilton NS, Hitchin JR, Hutton CP, Jones S, Kelly P, et al. 2016. First-in-class chemical probes against poly(ADP-ribose) glycohydrolase (PARG) inhibit DNA repair with differential pharmacology to olaparib. ACS Chem Biol 11: 3179–3190. 10.1021/acschembio.6b00609 [DOI] [PubMed] [Google Scholar]
- Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, et al. 2013. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov 3: 68–81. 10.1158/2159-8290.CD-12-0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, et al. 2017. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res 23: 3711–3720. 10.1158/1078-0432.CCR-16-3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, et al. 2013. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci 110: 17041–17046. 10.1073/pnas.1305170110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C, Lamartina S, Monteagudo E, Ontoria JM, Orsale MV, et al. 2009. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J Med Chem 52: 7170–7185. 10.1021/jm901188v [DOI] [PubMed] [Google Scholar]
- Jump DB, Butt TR, Smulson M. 1979. Nuclear protein modification and chromatin substructure. 3. Relationship between poly(adenosine diphosphate) ribosylation and different functional forms of chromatin. Biochemistry 18: 983–990. 10.1021/bi00573a008 [DOI] [PubMed] [Google Scholar]
- Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, et al. 2012. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov 2: 1048–1063. 10.1158/2159-8290.CD-11-0336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339. 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakashev S, Zhu H, Yokoyama Y, Zhao B, Fatkhutdinov N, Kossenkov AV, Wilson AJ, Simpkins F, Speicher D, Khabele D, et al. 2017. BET bromodomain inhibition synergizes with PARP inhibitor in epithelial ovarian cancer. Cell Rep 21: 3398–3405. 10.1016/j.celrep.2017.11.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. 2015. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 33: 244–250. 10.1200/JCO.2014.56.2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann T, Grishkovskaya I, Polyansky AA, Kostrhon S, Kukolj E, Olek KM, Herbert S, Beltzung E, Mechtler K, Peterbauer T, et al. 2017. A novel non-canonical PIP-box mediates PARG interaction with PCNA. Nucleic Acids Res 45: 9741–9759. 10.1093/nar/gkx604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, Amnon A, Bell-McGuinn KM, Chen LM, Friedlander M, et al. 2012. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol 30: 372–379. 10.1200/JCO.2011.36.9215 [DOI] [PubMed] [Google Scholar]
- Keung MYT, Wu Y, Vadgama JV. 2019. PARP inhibitors as a therapeutic agent for homologous recombination deficiency in breast cancers. J Clin Med 8: E435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IK, Kiefer JR, Ho CM, Stegeman RA, Classen S, Tainer JA, Ellenberger T. 2012. Structure of mammalian poly(ADP-ribose) glycohydrolase reveals a flexible tyrosine clasp as a substrate-binding element. Nat Struct Mol Biol 19: 653–656. 10.1038/nsmb.2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. 2017. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin Cancer Res 23: 3097–3108. 10.1158/1078-0432.CCR-16-2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, Kraus WL. 2019. Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol Cell 75: 1270–1285.e14. 10.1016/j.molcel.2019.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic CE, Wright G, Remsing Rix LL, Kim W, Kuenzi BM, Luo Y, Watters JM, Koomen JM, Haura EB, Monteiro AN, et al. 2016. Proteome-wide profiling of clinical PARP inhibitors reveals compound-specific secondary targets. Cell Chem Biol 23: 1490–1503. 10.1016/j.chembiol.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. 2004. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci 101: 17699–17704. 10.1073/pnas.0406182101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, Kuiper MJ, Ho GY, Barker H, Jasin M, et al. 2017. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov 7: 984–998. 10.1158/2159-8290.CD-17-0419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, Zapparoli GV, Hadley A, Holian R, Boehm E, et al. 2018. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun 9: 3970 10.1038/s41467-018-05564-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, Levine DA, Cannistra SA. 2010. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol 28: 3555–3561. 10.1200/JCO.2009.27.5719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Barry WT, Birrer M, Westin SN, Cadoo KA, Shapiro GI, Mayer EL, O'Cearbhaill RE, Coleman RL, Kochupurakkal B, et al. 2019. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol 20: 570–580. 10.1016/S1470-2045(18)30905-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, Petermann E. 2016. Increased global transcription activity as a mechanism of replication stress in cancer. Nat Commun 7: 13087 10.1038/ncomms13087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Petermann E, Boulton SJ. 2018. Mechanisms of oncogene-induced replication stress: jigsaw falling into place. Cancer Discov 8: 537–555. 10.1158/2159-8290.CD-17-1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WL, Hottiger MO. 2013. PARP-1 and gene regulation: progress and puzzles. Mol Aspects Med 34: 1109–1123. 10.1016/j.mam.2013.01.005 [DOI] [PubMed] [Google Scholar]
- Kristeleit R, Shapiro GI, Burris HA, Oza AM, LoRusso P, Patel MR, Domchek SM, Balmaña J, Drew Y, Chen LM, et al. 2017. A phase I–II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin Cancer Res 23: 4095–4106. 10.1158/1078-0432.CCR-16-2796 [DOI] [PubMed] [Google Scholar]
- Kukolj E, Kaufmann T, Dick AE, Zeillinger R, Gerlich DW, Slade D. 2017. PARP inhibition causes premature loss of cohesion in cancer cells. Oncotarget 8: 103931–103951. 10.18632/oncotarget.21879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird JH, Lok BH, Ma J, Bell A, de Stanchina E, Poirier JT, Rudin CM. 2018. Talazoparib Is a potent radiosensitizer in small cell lung cancer cell lines and xenografts. Clin Cancer Res 24: 5143–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallo A, Frese KK, Morrow CJ, Sloane R, Gulati S, Schenk MW, Trapani F, Simms N, Galvin M, Brown S, et al. 2018. The combination of the PARP inhibitor olaparib and the WEE1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res 24: 5153–5164. [DOI] [PubMed] [Google Scholar]
- Langelier MF, Planck JL, Roy S, Pascal JM. 2012. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 336: 728–732. 10.1126/science.1216338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor D, Martin P, Busschots S, Thery J, O'Leary JJ, Hennessy BT, Stordal B. 2014. PARP inhibitors as P-glyoprotein substrates. J Pharm Sci 103: 1913–1920. 10.1002/jps.23952 [DOI] [PubMed] [Google Scholar]
- Le Beau MM, Rassool FV, Neilly ME, Espinosa R 3rd, Glover TW, Smith DI, McKeithan TW. 1998. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Hum Mol Genet 7: 755–761. 10.1093/hmg/7.4.755 [DOI] [PubMed] [Google Scholar]
- Lecona E, Fernandez-Capetillo O. 2018. Targeting ATR in cancer. Nat Rev Cancer 18: 586–595. 10.1038/s41568-018-0034-3 [DOI] [PubMed] [Google Scholar]
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, et al. 2012. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 366: 1382–1392. 10.1056/NEJMoa1105535 [DOI] [PubMed] [Google Scholar]
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, et al. 2014. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15: 852–861. 10.1016/S1470-2045(14)70228-1 [DOI] [PubMed] [Google Scholar]
- Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, et al. 2016. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol 17: 1579–1589. 10.1016/S1470-2045(16)30376-X [DOI] [PubMed] [Google Scholar]
- Lheureux S, Bruce JP, Burnier JV, Karakasis K, Shaw PA, Clarke BA, Yang SY, Quevedo R, Li T, Dowar M, et al. 2017a. Somatic BRCA1/2 recovery as a resistance mechanism after exceptional response to poly (ADP-ribose) polymerase inhibition. J Clin Oncol 35: 1240–1249. 10.1200/JCO.2016.71.3677 [DOI] [PubMed] [Google Scholar]
- Lheureux S, Lai Z, Dougherty BA, Runswick S, Hodgson DR, Timms KM, Lanchbury JS, Kaye S, Gourley C, Bowtell D, et al. 2017b. Long-term responders on olaparib maintenance in high-grade serous ovarian cancer: clinical and molecular characterization. Clin Cancer Res 23: 4086–4094. 10.1158/1078-0432.CCR-16-2615 [DOI] [PubMed] [Google Scholar]
- Li M, Yu X. 2013. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 23: 693–704. 10.1016/j.ccr.2013.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Yan X, Sun Y, Yang X. 2016. Expression of ADP-ribosyltransferase 1 is associated with poor prognosis of glioma patients. Tohoku J Exp Med 239: 269–278. 10.1620/tjem.239.269 [DOI] [PubMed] [Google Scholar]
- Li L, Karanika S, Yang G, Wang J, Park S, Broom BM, Manyam GC, Wu W, Luo Y, Basourakos S, et al. 2017. Androgen receptor inhibitor-induced ‘BRCAness’ and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Chen D, Zhang C, Zhang X, Li Z, Dong B, Gao J, Shen L. 2018. Augmented antitumor activity by olaparib plus AZD1775 in gastric cancer through disrupting DNA damage repair pathways and DNA damage checkpoint. J Exp Clin Cancer Res 37: 129 10.1186/s13046-018-0790-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, Helman E, Radke MR, Say C, Vo LT, et al. 2019. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov 9: 210–219. 10.1158/2159-8290.CD-18-0715 [DOI] [PubMed] [Google Scholar]
- Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FB, Imholz AL, Wesseling J, Rodenhuis S, Nederlof PM. 2013. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer 108: 2172–2177. 10.1038/bjc.2013.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, et al. 2018. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med 379: 753–763. 10.1056/NEJMoa1802905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, Rimel B, Buss MK, Nattam S, Hurteau J, et al. 2014. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol 15: 1207–1214. 10.1016/S1470-2045(14)70391-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Burness ML, Martin-Trevino R, Guy J, Bai S, Harouaka R, Brooks MD, Shang L, Fox A, Luther TK, et al. 2017. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triple-negative breast cancer. Clin Cancer Res 23: 514–522. 10.1158/1078-0432.CCR-15-1348 [DOI] [PubMed] [Google Scholar]
- Loibl S, O'Shaughnessy J, Untch M, Sikov WM, Rugo HS, McKee MD, Huober J, Golshan M, von Minckwitz G, Maag D, et al. 2018. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol 19: 497–509. 10.1016/S1470-2045(18)30111-6 [DOI] [PubMed] [Google Scholar]
- Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, de Stanchina E, Teicher BA, Riaz N, Powell SN, et al. 2017. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res 23: 523–535. 10.1158/1078-0432.CCR-16-1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR. 2003. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev 17: 3017–3022. 10.1101/gad.279003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonn U, Lonn S. 1985. Accumulation of 10-kilobase DNA replication intermediates in cells treated with 3-aminobenzamide. Proc Natl Acad Sci 82: 104–108. 10.1073/pnas.82.1.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. 2012. The DNA damage response and cancer therapy. Nature 481: 287–294. 10.1038/nature10760 [DOI] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. 2016. BRCAness revisited. Nat Rev Cancer 16: 110–120. 10.1038/nrc.2015.21 [DOI] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. 2017. PARP inhibitors: synthetic lethality in the clinic. Science 355: 1152–1158. 10.1126/science.aam7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery MA, Kelsen DP, Capanu M, Smith SC, Lee JW, Stadler ZK, Moore MJ, Kindler HL, Golan T, Segal A, et al. 2018. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur J Cancer 89: 19–26. 10.1016/j.ejca.2017.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijten MNH, Lee JXT, Crasta KC. 2018. Mutational game changer: chromothripsis and its emerging relevance to cancer. Mutat Res 777: 29–51. 10.1016/j.mrrev.2018.06.004 [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grøfte M, Chan KL, Hickson ID, Bartek J, et al. 2011. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13: 243–253. 10.1038/ncb2201 [DOI] [PubMed] [Google Scholar]
- Macheret M, Halazonetis TD. 2015. DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10: 425–448. 10.1146/annurev-pathol-012414-040424 [DOI] [PubMed] [Google Scholar]
- Macheret M, Halazonetis TD. 2018. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555: 112–116. 10.1038/nature25507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. 2017. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548: 461–465. 10.1038/nature23449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majuelos-Melguizo J, Rodríguez MI, López-Jiménez L, Rodríguez-Vargas JM, Martí Martín-Consuegra JM, Serrano-Sáenz S, Gavard J, de Almodóvar JM, Oliver FJ. 2015. PARP targeting counteracts gliomagenesis through induction of mitotic catastrophe and aggravation of deficiency in homologous recombination in PTEN-mutant glioma. Oncotarget 6: 4790–4803. 10.18632/oncotarget.2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Hernandez K, Rodriguez-Vargas JM, Schreiber V, Dantzer F. 2017. Expanding functions of ADP-ribosylation in the maintenance of genome integrity. Semin Cell Dev Biol 63: 92–101. 10.1016/j.semcdb.2016.09.009 [DOI] [PubMed] [Google Scholar]
- Marzio A, Puccini J, Kwon Y, Maverakis NK, Arbini A, Sung P, Bar-Sagi D, Pagano M. 2019. The F-box domain-dependent activity of EMI1 regulates PARPi sensitivity in triple-negative breast cancers. Mol Cell 73: 224–237.e6. 10.1016/j.molcel.2018.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascolo M, Ilardi G, Romano MF, Celetti A, Siano M, Romano S, Luise C, Merolla F, Rocco A, Vecchione ML, et al. 2012. Overexpression of chromatin assembly factor-1 p60, poly(ADP-ribose) polymerase 1 and nestin predicts metastasizing behaviour of oral cancer. Histopathology 61: 1089–1105. 10.1111/j.1365-2559.2012.04313.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. 2015. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373: 1697–1708. 10.1056/NEJMoa1506859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo J, Lord CJ, Serra V, Tutt A, Balmaña J, Castroviejo-Bermejo M, Cruz C, Oaknin A, Kaye SB, de Bono JS. 2019. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol 30: 1437–1447. 10.1093/annonc/mdz192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulonis UA, Monk BJ. 2017. PARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: does a development pathway forward exist? Ann Oncol 28: 443–447. 10.1093/annonc/mdw697 [DOI] [PubMed] [Google Scholar]
- Mavaddat N, Barrowdale D, Andrulis IL, Domchek SM, Eccles D, Nevanlinna H, Ramus SJ, Spurdle A, Robson M, Sherman M, et al. 2012. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol Biomarkers Prev 21: 134–147. 10.1158/1055-9965.EPI-11-0775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Mendoza A, Ostrakova J, Kosar M, Hall A, Duskova P, Mistrik M, Merchut-Maya JM, Hodny Z, Bartkova J, Christensen C, et al. 2015. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol Oncol 9: 601–616. 10.1016/j.molonc.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. 2018. High speed of fork progression induces DNA replication stress and genomic instability. Nature 559: 279–284. 10.1038/s41586-018-0261-5 [DOI] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Białkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ, et al. 2006. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66: 8109–8115. 10.1158/0008-5472.CAN-06-0140 [DOI] [PubMed] [Google Scholar]
- McGrail DJ, Lin CC, Garnett J, Liu Q, Mo W, Dai H, Lu Y, Yu Q, Ju Z, Yin J, et al. 2017. Improved prediction of PARP inhibitor response and identification of synergizing agents through use of a novel gene expression signature generation algorithm. NPJ Syst Biol Appl 3: 8 10.1038/s41540-017-0011-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan JL, O'Neil NJ, Barrett I, Ferree E, van Pel DM, Ushey K, Sipahimalani P, Bryan J, Rose AM, Hieter P. 2012. Synthetic lethality of cohesins with PARPs and replication fork mediators. PLoS Genet 8: e1002574 10.1371/journal.pgen.1002574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meghani K, Fuchs W, Detappe A, Drané P, Gogola E, Rottenberg S, Jonkers J, Matulonis U, Swisher EM, Konstantinopoulos PA, et al. 2018. Multifaceted impact of microRNA 493-5p on genome-stabilizing pathways induces platinum and PARP inhibitor resistance in BRCA2-mutated carcinomas. Cell Rep 23: 100–111. 10.1016/j.celrep.2018.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S, et al. 2008. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem 51: 6581–6591. 10.1021/jm8001263 [DOI] [PubMed] [Google Scholar]
- Meng X, Bi J, Li Y, Yang S, Zhang Y, Li M, Liu H, Li Y, McDonald ME, Thiel KW, et al. 2018. AZD1775 increases sensitivity to olaparib and gemcitabine in cancer cells with p53 mutations. Cancers (Basel) 10: 149 10.3390/cancers10050149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelena J, Lezaja A, Teloni F, Schmid T, Imhof R, Altmeyer M. 2018. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat Commun 9: 2678 10.1038/s41467-018-05031-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W, Cortes U, Herceg Z, Tong WM, Wang ZQ. 2010. Deletion of the nuclear isoform of poly(ADP-ribose) glycohydrolase (PARG) reveals its function in DNA repair, genomic stability and tumorigenesis. Carcinogenesis 31: 2058–2065. 10.1093/carcin/bgq205 [DOI] [PubMed] [Google Scholar]
- Min W, Bruhn C, Grigaravicius P, Zhou ZW, Li F, Krüger A, Siddeek B, Greulich KO, Popp O, Meisezahl C, et al. 2013. Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat Commun 4: 2993 10.1038/ncomms3993 [DOI] [PubMed] [Google Scholar]
- Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee KH, Kim TY, Han SW, Oh DY, Kim TY, et al. 2015. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res 17: 33 10.1186/s13058-015-0534-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, et al. 2016. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med 375: 2154–2164. 10.1056/NEJMoa1611310 [DOI] [PubMed] [Google Scholar]
- Mirza MR, Pignata S, Ledermann JA. 2018. Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann Oncol 29: 1366–1376. 10.1093/annonc/mdy174 [DOI] [PubMed] [Google Scholar]
- Mirza MR, Åvall Lundqvist E, Birrer MJ, dePont Christensen R, Nyvang GB, Malander S, Anttila M, Werner TL, Lund B, Lindahl G, et al. 2019. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol 20: 1409–1419. 10.1016/S1470-2045(19)30515-7 [DOI] [PubMed] [Google Scholar]
- Mo W, Liu Q, Lin CC, Dai H, Peng Y, Liang Y, Peng G, Meric-Bernstam F, Mills GB, Li K, et al. 2016. mTOR inhibitors suppress homologous recombination repair and synergize with PARP inhibitors via regulating SUV39H1 in BRCA-proficient triple-negative breast cancer. Clin Cancer Res 22: 1699–1712. 10.1158/1078-0432.CCR-15-1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, Lisyanskaya A, Floquet A, Leary A, Sonke GS, et al. 2018. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 379: 2495–2505. 10.1056/NEJMoa1810858 [DOI] [PubMed] [Google Scholar]
- Moore KN, Secord AA, Geller MA, Miller DS, Cloven N, Fleming GF, Wahner Hendrickson AE, Azodi M, DiSilvestro P, Oza AM, et al. 2019. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol 20: 636–648. 10.1016/S1470-2045(19)30029-4 [DOI] [PubMed] [Google Scholar]
- Mortusewicz O, Fouquerel E, Amé JC, Leonhardt H, Schreiber V. 2011. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res 39: 5045–5056. 10.1093/nar/gkr099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Elattar A, Cerbinskaite A, Wilkinson SJ, Drew Y, Kyle S, Los G, Hostomsky Z, Edmondson RJ, Curtin NJ. 2010. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin Cancer Res 16: 2344–2351. 10.1158/1078-0432.CCR-09-2758 [DOI] [PubMed] [Google Scholar]
- Murai J, Pommier Y. 2019. PARP trapping beyond homologous recombination and platinum sensitivity in cancers. Ann Rev Cancer Biol 3: 131–150. 10.1146/annurev-cancerbio-030518-055914 [DOI] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. 2012. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 72: 5588–5599. 10.1158/0008-5472.CAN-12-2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, Pommier Y. 2014a. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther 13: 433–443. 10.1158/1535-7163.MCT-13-0803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, Pommier Y. 2014b. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther 349: 408–416. 10.1124/jpet.113.210146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, Pommier Y. 2016. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 7: 76534–76550. 10.18632/oncotarget.12266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Tang SW, Leo E, Baechler SA, Redon CE, Zhang H, Al Abo M, Rajapakse VN, Nakamura E, Jenkins LMM, et al. 2018. SLFN11 blocks stressed replication forks independently of ATR. Mol Cell 69: 371–384.e6. 10.1016/j.molcel.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y, Bellani M, Zou Y, Singh ZN, Duong VH, et al. 2016. Enhancing the cytotoxic effects of PARP inhibitors with DNA demethylating agents - a potential therapy for cancer. Cancer Cell 30: 637–650. 10.1016/j.ccell.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naipal KA, Verkaik NS, Ameziane N, van Deurzen CH, Ter Brugge P, Meijers M, Sieuwerts AM, Martens JW, O'Connor MJ, Vrieling H, et al. 2014. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin Cancer Res 20: 4816–4826. 10.1158/1078-0432.CCR-14-0571 [DOI] [PubMed] [Google Scholar]
- Nikkilä J, Kumar R, Campbell J, Brandsma I, Pemberton HN, Wallberg F, Nagy K, Scheer I, Vertessy BG, Serebrenik AA, et al. 2017. Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br J Cancer 117: 113–123. 10.1038/bjc.2017.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534: 47–54. 10.1038/nature17676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales V, Reinhold WC, Varma S, Martinez-Cardus A, Moutinho C, Moran S, Heyn H, Sebio A, Barnadas A, Pommier Y, et al. 2016. Epigenetic inactivation of the putative DNA/RNA helicase SLFN11 in human cancer confers resistance to platinum drugs. Oncotarget 7: 3084–3097. 10.18632/oncotarget.6413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll A, Illuzzi G, Amé JC, Dantzer F, Schreiber V. 2016. PARG deficiency is neither synthetic lethal with BRCA1 nor PTEN deficiency. Cancer Cell Int 16: 53 10.1186/s12935-016-0333-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer SM, van Attikum H. 2019. PARP inhibitor resistance: a tug-of-war in BRCA-mutated cells. Trends Cell Biol 29: 820–834. 10.1016/j.tcb.2019.07.008 [DOI] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Álvarez-Quilón A, Moatti N, Zimmermann M, et al. 2018. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560: 117–121. 10.1038/s41586-018-0340-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM. 2011. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol 29: 3008–3015. 10.1200/JCO.2010.34.2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowsheen S, Cooper T, Bonner JA, LoBuglio AF, Yang ES. 2012. HER2 overexpression renders human breast cancers sensitive to PARP inhibition independently of any defect in homologous recombination DNA repair. Cancer Res 72: 4796–4806. 10.1158/0008-5472.CAN-12-1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano S, Lan L, Caldecott KW, Mori T, Yasui A. 2003. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol Cell Biol 23: 3974–3981. 10.1128/MCB.23.11.3974-3981.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayama H, Edson CM, Fukushima M, Ueda K, Hayaishi O. 1977. Purification and properties of poly(adenosine diphosphate ribose) synthetase. J Biol Chem 252: 7000–7005. [PubMed] [Google Scholar]
- Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. 2010. Upregulation of poly (ADP-ribose) polymerase-1 (PARP1) in triple-negative breast cancer and other primary human tumor types. Genes Cancer 1: 812–821. 10.1177/1947601910383418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza AM, Tinker AV, Oaknin A, Shapira-Frommer R, McNeish IA, Swisher EM, Ray-Coquard I, Bell-McGuinn K, Coleman RL, O'Malley DM, et al. 2017. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: integrated analysis of data from study 10 and ARIEL2. Gynecol Oncol 147: 267–275. 10.1016/j.ygyno.2017.08.022 [DOI] [PubMed] [Google Scholar]
- Pant S, Maitra A, Yap TA. 2019. PARP inhibition—opportunities in pancreatic cancer. Nat Rev Clin Oncol 16: 595–596. 10.1038/s41571-019-0257-6 [DOI] [PubMed] [Google Scholar]
- Papeo G, Avanzi N, Bettoni S, Leone A, Paolucci M, Perego R, Quartieri F, Riccardi-Sirtori F, Thieffine S, Montagnoli A, et al. 2014. Insights into PARP inhibitors’ selectivity using fluorescence polarization and surface plasmon resonance binding assays. J Biomol Screen 19: 1212–1219. 10.1177/1087057114538319 [DOI] [PubMed] [Google Scholar]
- Park SH, Jang KY, Kim MJ, Yoon S, Jo Y, Kwon SM, Kim KM, Kwon KS, Kim CY, Woo HG. 2015. Tumor suppressive effect of PARP1 and FOXO3A in gastric cancers and its clinical implications. Oncotarget 6: 44819–44831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, McCloskey KD, Buckley NE, Savage KI, Salto-Tellez M, et al. 2017. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst 109: djw199 10.1093/jnci/djw199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish KE, Cen L, Murray J, Calligaris D, Kizilbash S, Mittapalli RK, Carlson BL, Schroeder MA, Sludden J, Boddy AV, et al. 2015. Efficacy of PARP inhibitor rucaparib in prthotopic glioblastoma xenografts is limited by ineffective drug penetration into the central nervous system. Mol Cancer Ther 14: 2735–2743. 10.1158/1535-7163.MCT-15-0553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsels LA, Karnak D, Parsels JD, Zhang Q, Vélez-Padilla J, Reichert ZR, Wahl DR, Maybaum J, O'Connor MJ, Lawrence TS, et al. 2018. PARP1 trapping and DNA replication stress enhance radiosensitization with combined WEE1 and PARP inhibitors. Mol Cancer Res 16: 222–232. 10.1158/1541-7786.MCR-17-0455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, et al. 2015. Whole-genome characterization of chemoresistant ovarian cancer. Nature 521: 489–494. 10.1038/nature14410 [DOI] [PubMed] [Google Scholar]
- Patel AG, Sarkaria JN, Kaufmann SH. 2011. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci 108: 3406–3411. 10.1073/pnas.1013715108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, et al. 2014. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat Commun 5: 5496 10.1038/ncomms6496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM, Peng Y, Mo Q, Siwko S, Hu R, et al. 2014. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun 5: 3361 10.1038/ncomms4361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt SJ, Rehman FL, Bajrami I, Brough R, Wallberg F, Kozarewa I, Fenwick K, Assiotis I, Chen L, Campbell J, et al. 2013. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS One 8: e61520 10.1371/journal.pone.0061520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, et al. 2018. Genome-wide and high-density CRISPR–Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun 9: 1849 10.1038/s41467-018-03917-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip CA, Laskov I, Beauchamp MC, Marques M, Amin O, Bitharas J, Kessous R, Kogan L, Baloch T, Gotlieb WH, et al. 2017. Inhibition of PI3K–AKT–mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer 17: 638 10.1186/s12885-017-3639-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, Owonikoko TK, Woo KM, Cardnell RJ, Fujimoto J, et al. 2018. Randomized, double-blind, phase II study of temozolomide in combination with either veliparib or placebo in patients with relapsed-sensitive or refractory small-cell lung cancer. J Clin Oncol 36: 2386–2394. 10.1200/JCO.2018.77.7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilié PG, Gay CM, Byers LA, O'Connor MJ, Yap TA. 2019a. PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res 25: 3759–3771. 10.1158/1078-0432.CCR-18-0968 [DOI] [PubMed] [Google Scholar]
- Pilié PG, Tang C, Mills GB, Yap TA. 2019b. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 16: 81–104. 10.1038/s41571-018-0114-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay N, Tighe A, Nelson L, Littler S, Coulson-Gilmer C, Bah N, Golder A, Bakker B, Spierings DCJ, James DI, et al. 2019. DNA replication vulnerabilities render ovarian cancer cells sensitive to poly(ADP-ribose) glycohydrolase inhibitors. Cancer Cell 35: 519–533.e8. 10.1016/j.ccell.2019.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishvaian MJ, Biankin AV, Bailey P, Chang DK, Laheru D, Wolfgang CL, Brody JR. 2017. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br J Cancer 116: 1021–1026. 10.1038/bjc.2017.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kübler K, Mouw KW, et al. 2017. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 49: 1476–1486. 10.1038/ng.3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, O'Connor MJ, de Bono J. 2016. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 8: 362ps17 10.1126/scitranslmed.aaf9246 [DOI] [PubMed] [Google Scholar]
- Popova T, Manie E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal-Zafrani B, Bollet M, Longy M, et al. 2012. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res 72: 5454–5462. 10.1158/0008-5472.CAN-12-1470 [DOI] [PubMed] [Google Scholar]
- Postel-Vinay S, Bajrami I, Friboulet L, Elliott R, Fontebasso Y, Dorvault N, Olaussen KA, André F, Soria JC, Lord CJ, et al. 2013. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene 32: 5377–5387. 10.1038/onc.2013.311 [DOI] [PubMed] [Google Scholar]
- Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, Korach J, Huzarski T, Poveda A, Pignata S, et al. 2017. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 18: 1274–1284. 10.1016/S1470-2045(17)30469-2 [DOI] [PubMed] [Google Scholar]
- Purnell MR, Whish WJ. 1980. Novel inhibitors of poly(ADP-ribose) synthetase. Biochem J 185: 775–777. 10.1042/bj1850775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, Aggarwal R, Kim W, Lu E, Schwartzman J, et al. 2017. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov 7: 999–1005. 10.1158/2159-8290.CD-17-0146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Nussenzweig A. 2017. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 18: 610–621. 10.1038/nrm.2017.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. 2012. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19: 417–423. 10.1038/nsmb.2258 [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Ahuja AK, Herrador R, Lopes M. 2015. Poly(ADP-ribosyl) glycohydrolase prevents the accumulation of unusual replication structures during unperturbed S phase. Mol Cell Biol 35: 856–865. 10.1128/MCB.01077-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. 2016. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535: 382–387. 10.1038/nature18325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. 2013. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 45: 970–976. 10.1038/ng.2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar-Sinha C, Raymond V, et al. 2017. Integrative clinical genomics of metastatic cancer. Nature 548: 297–303. 10.1038/nature23306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, et al. 2017. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 377: 523–533. 10.1056/NEJMoa1706450 [DOI] [PubMed] [Google Scholar]
- Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, et al. 2017. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol 19: 1371–1378. 10.1038/ncb3626 [DOI] [PubMed] [Google Scholar]
- Ronson GE, Piberger AL, Higgs MR, Olsen AL, Stewart GS, McHugh PJ, Petermann E, Lakin ND. 2018. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat Commun 9: 746 10.1038/s41467-018-03159-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. 2008. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci 105: 17079–17084. 10.1073/pnas.0806092105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. 2008. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451: 1116–1120. 10.1038/nature06633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi M, Galia A, Fraggetta F, La Corte C, Pepe P, La Vignera S, Improta G, Bosco P, Calogero AE. 2013. Poly (ADP-ribose) polymerase 1 protein expression in normal and neoplastic prostatic tissue. Eur J Histochem 57: e13 10.4081/ejh.2013.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, et al. 2013. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol 14: 882–892. 10.1016/S1470-2045(13)70240-7 [DOI] [PubMed] [Google Scholar]
- Schiewer MJ, Knudsen KE. 2014. Transcriptional roles of PARP1 in cancer. Mol Cancer Res 12: 1069–1080. 10.1158/1541-7786.MCR-13-0672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiewer MJ, Mandigo AC, Gordon N, Huang F, Gaur S, de Leeuw R, Zhao SG, Evans J, Han S, Parsons T, et al. 2018. PARP-1 regulates DNA repair factor availability. EMBO Mol Med 10 10.15252/emmm.201708816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51–BRCA1/2. Cancer Cell 22: 106–116. 10.1016/j.ccr.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonen PM, Talens F, Stok C, Gogola E, Heijink AM, Bouwman P, Foijer F, Tarsounas M, Blatter S, Jonkers J, et al. 2017. Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells. Nat Commun 8: 15981 10.1038/ncomms15981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Gay CM, Byers LA. 2018. Targeting DNA damage repair in small cell lung cancer and the biomarker landscape. Transl Lung Cancer Res 7: 50–68. 10.21037/tlcr.2018.02.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Dobbin ZC, Nowsheen S, Wielgos M, Katre AA, Alvarez RD, Konstantinopoulos PA, Yang ES, Landen CN. 2014. An ex vivo assay of XRT-induced Rad51 foci formation predicts response to PARP-inhibition in ovarian cancer. Gynecol Oncol 134: 331–337. 10.1016/j.ygyno.2014.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, Wang B, Lord CJ, Post LE, Ashworth A. 2013. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res 19: 5003–5015. 10.1158/1078-0432.CCR-13-1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Aoyagi-Scharber M, Wang B. 2015. Trapping poly(ADP-ribose) polymerase. J Pharmacol Exp Ther 353: 446–457. 10.1124/jpet.114.222448 [DOI] [PubMed] [Google Scholar]
- Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G. 2019. PARPi Triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res 79: 311–319. 10.1158/0008-5472.CAN-18-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai H, Poetsch AR, Gunji A, Maeda D, Fujimori H, Fujihara H, Yoshida T, Ogino H, Masutani M. 2013. PARG dysfunction enhances DNA double strand break formation in S-phase after alkylation DNA damage and augments different cell death pathways. Cell Death Dis 4: e656 10.1038/cddis.2013.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, Goble S, Lin KK, Biankin AV, Giordano H, et al. 2018. Rucaparib monotherapy in patients with pancreatic cancer and a known deleterious BRCA mutation. JCO Precis Oncol 10.1200/PO.17.00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simbulan-Rosenthal CM, Rosenthal DS, Boulares AH, Hickey RJ, Malkas LH, Coll JM, Smulson ME. 1998. Regulation of the expression or recruitment of components of the DNA synthesome by poly(ADP-ribose) polymerase. Biochemistry 37: 9363–9370. 10.1021/bi9731089 [DOI] [PubMed] [Google Scholar]
- Simbulan-Rosenthal CM, Rosenthal DS, Luo R, Samara R, Espinoza LA, Hassa PO, Hottiger MO, Smulson ME. 2003. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene 22: 8460–8471. 10.1038/sj.onc.1206897 [DOI] [PubMed] [Google Scholar]
- Slade D. 2019. Mitotic functions of poly(ADP-ribose) polymerases. Biochem Pharmacol 167: 33–43. 10.1016/j.bcp.2019.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, Ahel I. 2011. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 477: 616–620. 10.1038/nature10404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slama JT, Aboul-Ela N, Goli DM, Cheesman BV, Simmons AM, Jacobson MK. 1995. Specific inhibition of poly(ADP-ribose) glycohydrolase by adenosine diphosphate (hydroxymethyl)pyrrolidinediol. J Med Chem 38: 389–393. 10.1021/jm00002a021 [DOI] [PubMed] [Google Scholar]
- Sonnenblick A, de Azambuja E, Azim HA Jr, Piccart M. 2015. An update on PARP inhibitors–moving to the adjuvant setting. Nat Rev Clin Oncol 12: 27–41. 10.1038/nrclinonc.2014.163 [DOI] [PubMed] [Google Scholar]
- Steffen JD, Brody JR, Armen RS, Pascal JM. 2013. Structural implications for selective targeting of PARPs. Front Oncol 3: 301 10.3389/fonc.2013.00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Helleday T. 2011. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res 39: 3166–3175. 10.1093/nar/gkq1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura K, Takebayashi S, Taguchi H, Takeda S, Okumura K. 2008. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J Cell Biol 183: 1203–1212. 10.1083/jcb.200806068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, et al. 2018. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell 33: 401–416.e8. 10.1016/j.ccell.2018.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O'Malley DM, et al. 2017. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol 18: 75–87. 10.1016/S1470-2045(16)30559-9 [DOI] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. 2017. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol Cell 68: 414–430.e8. 10.1016/j.molcel.2017.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SW, Thomas A, Murai J, Trepel JB, Bates SE, Rajapakse VN, Pommier Y. 2018. Overcoming resistance to DNA-targeted agents by epigenetic activation of Schlafen 11 (SLFN11) expression with class I histone deacetylase inhibitors. Clin Cancer Res 24: 1944–1953. 10.1158/1078-0432.CCR-17-0443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RA, Fraser M, Rebello RJ, Boutros PC, Murphy DG, Bristow RG, Risbridger GP. 2019. The influence of BRCA2 mutation on localized prostate cancer. Nat Rev Urol 16: 281–290. 10.1038/s41585-019-0164-8 [DOI] [PubMed] [Google Scholar]
- Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, et al. 2016. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res 22: 3764–3773. 10.1158/1078-0432.CCR-15-2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, Maegley KA, Newell DR, Skalitzky D, Wang LZ, et al. 2007. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther 6: 945–956. 10.1158/1535-7163.MCT-06-0552 [DOI] [PubMed] [Google Scholar]
- Thomas A, Murai J, Pommier Y. 2018. The evolving landscape of predictive biomarkers of response to PARP inhibitors. J Clin Invest 128: 1727–1730. 10.1172/JCI120388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell AG, Ekblad T, Karlberg T, Löw M, Pinto AF, Trésaugues L, Moche M, Cohen MS, Schüler H. 2017. Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J Med Chem 60: 1262–1271. 10.1021/acs.jmedchem.6b00990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, Kalva S, Potter J, Tran TV, Chen J, et al. 2014. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res 16: 475 10.1186/s13058-014-0475-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkáč J, Xu G, Adhikary H, Young JTF, Gallo D, Escribano-Díaz C, Krietsch J, Orthwein A, Munro M, Sol W, et al. 2016. HELB is a feedback inhibitor of DNA end resection. Mol Cell 61: 405–418. 10.1016/j.molcel.2015.12.013 [DOI] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. 2013. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103. 10.1016/j.cell.2013.10.043 [DOI] [PubMed] [Google Scholar]
- Toledo L, Neelsen KJ, Lukas J. 2017. Replication catastrophe: when a checkpoint fails because of exhaustion. Mol Cell 66: 735–749. 10.1016/j.molcel.2017.05.001 [DOI] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A. 2017. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168: 644–656. 10.1016/j.cell.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Telli ML, Rugo HS, Mailliez A, Ettl J, Grischke EM, Mina LA, Balmaña J, Fasching PA, Hurvitz SA, et al. 2019. A phase II study of talazoparib after platinum or cytotoxic nonplatinum regimens in patients with advanced breast cancer and germline BRCA1/2 mutations (ABRAZO). Clin Cancer Res 25: 2717–2724. 10.1158/1078-0432.CCR-18-1891 [DOI] [PubMed] [Google Scholar]
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, et al. 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376: 235–244. 10.1016/S0140-6736(10)60892-6 [DOI] [PubMed] [Google Scholar]
- Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, Owen J, Abraham J, Barrett S, Barrett-Lee P, et al. 2018. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med 24: 628–637. 10.1038/s41591-018-0009-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. 2002. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell 9: 1031–1044. 10.1016/S1097-2765(02)00520-8 [DOI] [PubMed] [Google Scholar]
- Vaidyanathan A, Sawers L, Gannon AL, Chakravarty P, Scott AL, Bray SE, Ferguson MJ, Smith G. 2016. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer 115: 431–441. 10.1038/bjc.2016.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeck J, Ropero S, Setien F, Gonzalez-Suarez E, Osorio A, Benitez J, Herman JG, Esteller M. 2010. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J Clin Oncol 28: e563–e564. 10.1200/JCO.2010.30.1010 [DOI] [PubMed] [Google Scholar]
- Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell AG, Pol E, Frostell A, Ekblad T, Öncü D, et al. 2012. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol 30: 283–288. 10.1038/nbt.2121 [DOI] [PubMed] [Google Scholar]
- Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. 2006. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res 34: 6170–6182. 10.1093/nar/gkl840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Aroumougame A, Lobrich M, Li Y, Chen D, Chen J, Gong Z. 2014. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev 28: 2693–2698. 10.1101/gad.252478.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Chu D, Feng Y, Shen Y, Aoyagi-Scharber M, Post LE. 2016a. Discovery and characterization of (8S,9R)-5-fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-te trahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (BMN 673, talazoparib), a novel, highly potent, and orally efficacious poly(ADP-ribose) polymerase-1/2 inhibitor, as an anticancer agent. J Med Chem 59: 335–357. 10.1021/acs.jmedchem.5b01498 [DOI] [PubMed] [Google Scholar]
- Wang D, Li C, Zhang Y, Wang M, Jiang N, Xiang L, Li T, Roberts TM, Zhao JJ, Cheng H, et al. 2016b. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol Oncol 142: 548–556. 10.1016/j.ygyno.2016.07.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O'Brien SW, et al. 2016c. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res 76: 2778–2790. 10.1158/0008-5472.CAN-16-0186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver AN, Yang ES. 2013. Beyond DNA repair: additional functions of PARP-1 in cancer. Front Oncol 3: 290 10.3389/fonc.2013.00290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Comino-Méndez I, de Bruijn I, Tian L, Meisel JL, García-Murillas I, Fribbens C, Cutts R, Martelotto LG, Ng CKY, et al. 2017. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin Cancer Res 23: 6708–6720. 10.1158/1078-0432.CCR-17-0544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AW, Almassy R, Calvert AH, Curtin NJ, Griffin RJ, Hostomsky Z, Maegley K, Newell DR, Srinivasan S, Golding BT. 2000. Resistance-modifying agents. 9. Synthesis and biological properties of benzimidazole inhibitors of the DNA repair enzyme poly(ADP-ribose) polymerase. J Med Chem 43: 4084–4097. 10.1021/jm000950v [DOI] [PubMed] [Google Scholar]
- Wielckens K, Schmidt A, George E, Bredehorst R, Hilz H. 1982. DNA fragmentation and NAD depletion. Their relation to the turnover of endogenous mono(ADP-ribosyl) and poly(ADP-ribosyl) proteins. J Biol Chem 257: 12872–12877. [PubMed] [Google Scholar]
- Wu YM, Cieślik M, Lonigro RJ, Vats P, Reimers MA, Cao X, Ning Y, Wang L, Kunju LP, de Sarkar N, et al. 2018. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 173: 1770–1782.e14. 10.1016/j.cell.2018.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. 2015. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521: 541–544. 10.1038/nature14328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Du Y, Nakai K, Ding M, Chang SS, Hsu JL, Yao J, Wei Y, Nie L, Jiao S, et al. 2018. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene 37: 208–217. 10.1038/onc.2017.311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ. 2004. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 23: 3872–3882. 10.1038/sj.onc.1207491 [DOI] [PubMed] [Google Scholar]
- Yang L, Zhang Y, Shan W, Hu Z, Yuan J, Pi J, Wang Y, Fan L, Tang Z, Li C, et al. 2017. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci Transl Med 9: eaal1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Liu C, Chen SH, Kassab MA, Hoff JD, Walter NG, Yu X. 2018. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res 46: 3446–3457. 10.1093/nar/gky088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TA, Plummer R, Azad NS, Helleday T. 2019. The DNA damaging revolution: PARP inhibitors and beyond. Am Soc Clin Oncol Educ Book 39: 185–195. 10.1200/EDBK_238473 [DOI] [PubMed] [Google Scholar]
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, et al. 2017. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 31: 318–332. 10.1101/gad.290957.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T. 2012. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72: 2814–2821. 10.1158/0008-5472.CAN-11-3417 [DOI] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol 16: 2–9. 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai L, Li S, Li H, Zheng Y, Lang R, Fan Y, Gu F, Guo X, Zhang X, Fu L. 2015. Polymorphisms in poly (ADP-ribose) polymerase-1 (PARP1) promoter and 3′ untranslated region and their association with PARP1 expression in breast cancer patients. Int J Clin Exp Pathol 8: 7059–7071. [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Shi J, Bian C, Yu X. 2015a. Poly(ADP-ribose) mediates the BRCA2-dependent early DNA damage response. Cell Rep 13: 678–689. 10.1016/j.celrep.2015.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Shi J, Chen S-H, Bian C, Yu X. 2015b. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Res 43: 10782–10794. 10.1093/nar/gkv939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 2013. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339: 700–704. 10.1126/science.1231573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaitė Z, Muir M, Fluteau A, Aregger M, McEwan A, et al. 2018. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559: 285–289. 10.1038/s41586-018-0291-z [DOI] [PMC free article] [PubMed] [Google Scholar]