

Abstract
Massively parallel sequencing of the polyadenylated RNAs has played a key role in delineating transcriptome complexity, including alternative use of an exon, promoter, 5′ or 3′ splice site or polyadenylation site, and RNA modification. However, reads derived from the current RNA-seq technologies are usually short and deprived of information on modification, compromising their potential in defining transcriptome complexity. Here, we applied a direct RNA sequencing method with ultralong reads using Oxford Nanopore Technologies to study the transcriptome complexity in Caenorhabditis elegans. We generated approximately six million reads using native poly(A)-tailed mRNAs from three developmental stages, with average read lengths ranging from 900 to 1100 nt. Around half of the reads represent full-length transcripts. To utilize the full-length transcripts in defining transcriptome complexity, we devised a method to classify the long reads as the same as existing transcripts or as a novel transcript using sequence mapping tracks rather than existing intron/exon structures, which allowed us to identify roughly 57,000 novel isoforms and recover at least 26,000 out of the 33,500 existing isoforms. The sets of genes with differential expression versus differential isoform usage over development are largely different, implying a fine-tuned regulation at isoform level. We also observed an unexpected increase in putative RNA modification in all bases in the coding region relative to the UTR, suggesting their possible roles in translation. The RNA reads and the method for read classification are expected to deliver new insights into RNA processing and modification and their underlying biology in the future.
Alternative splicing is a hallmark of eukaryotic transcriptomes. Over 90% of human genes show evidence of alternative splicing (Pan et al. 2008; Wang et al. 2018). It plays a key role not only in cell fate specification (Gerstein et al. 2010; Linker et al. 2019) but also in the development of higher organisms with sophisticated tissues, organs, and developmental processes by expanding the complexity of the transcriptome and thus of the proteome (Mudge et al. 2011; Ragle et al. 2015; Angiolini et al. 2019). Aberrant splicing has been frequently linked to various diseases, including cancer (Zhang et al. 2019), aging (Adusumalli et al. 2019), diabetes (Eizirik et al. 2012), abnormal nutritional response (Maxwell et al. 2012), and neuronal disorders (Lee et al. 2016). In addition, the transcriptome is further subjected to various base modifications with different biological implications (Yang et al. 2018). Systematic detection of such modifications and understanding of their roles in vivo remains a significant challenge.
Identification of all types of transcripts produced by a genome is crucial for understanding the functional complexity of normal development and disease progression but remains a challenging task even in an organism with a relatively small genome. For example, to facilitate annotation of the transcriptome of Caenorhabditis elegans or C. briggsae with a genome size of ∼100 Mb (The C. elegans Sequencing Consortium 1998; Stein et al. 2003; Ross et al. 2011; Li et al. 2016; Ren et al. 2018), various data sets have been used, including ESTs, full-length cDNAs, and RNA sequencing (RNA-seq) of cDNA fragments using massively parallel sequencing (Reboul et al. 2003; Ramani et al. 2011; Uyar et al. 2012; Grün et al. 2014; Boeck et al. 2016; Tourasse et al. 2017). Short RNA-seq reads, typically shorter than 200 nt, have played a leading role in transcriptome annotation during the past decade. However, it is difficult to reconstruct and quantify alternative transcripts using short reads, which is further complicated by a requirement of an amplification step (Steijger et al. 2013). Clearly, the ability to produce longer reads using the native RNA molecule without amplification would minimize perturbation of transcript integrity, permitting capturing of full-length RNA molecules, which would be ideal for elucidating transcriptome complexity, including alternative splicing, alternative transcriptional start and ending, as well as the underlying biology. To this end, synthetic long-read RNA sequencing has been introduced (Tilgner et al. 2015), which relies on subpooling of full-length cDNAs followed by sequence amplification, fragmentation, and assembly to produce a long read. The method has been shown to be able to recover many novel isoforms in humans and mice. However, the amplification and reverse transcription steps make it problematic for quantification and detection of native modifications. The current method of choice for profiling RNA methylation is RNA immunoprecipitation using modification-specific antibodies followed by reverse transcription and massively parallel sequencing (Helm and Motorin 2017; Yang et al. 2018). However, it provides poor resolution in terms of modification site. Third generation sequencing technology, for example, the Pacific Biosciences (PacBio) RSII platform, is able to produce long reads and detect DNA methylation based on polymerase kinetics during DNA synthesis, but a reverse transcription step is required for sequencing of the RNA molecule indirectly (Flusberg et al. 2010). Therefore, direct sequencing of native RNA molecules is still not feasible.
Recently, Oxford Nanopore Technologies (ONT) has developed a direct sequencing method for both DNA and RNA based on changes in the ion current profile when a nucleotide passes through a nanopore (Loman and Watson 2015). Due to its ultralong read length, it has been adopted for many applications, including resolving repeats within human Y Chromosome centromeres (Jain et al. 2018), improving the existing genome assembly (Ren et al. 2018), the rapid on-site sequencing of pathogens (Jain et al. 2016), and detecting 5-methylcytosine (5mC) in the genomes of humans and yeast. Direct sequencing of single-molecule native RNA is expected to benefit transcript integrity by getting rid of the steps for reverse transcription and amplification. The DNA modifications detected with ONT are highly correlated with those from the bisulfite sequencing-based method (Rand et al. 2017; Simpson et al. 2017; Jain et al. 2018). Because ONT relies on the change of profile in electric current to differentiate nucleotide bases, with appropriate positive and negative training data sets, the platform may be able to detect known or unknown modifications in native RNA molecules without any pretreatment step (Garalde et al. 2018).
Given a relatively high error rate of the long reads, using them to define transcriptome complexity is not trivial. Several methods have been developed to call transcript isoforms with a reference genome using long reads, including ToFU (Gordon et al. 2015) and SQANTI (Tardaguila et al. 2018), which were designed for PacBio cDNA reads. These methods depend heavily on existing splicing junctions to classify the reads into representative isoforms, which may compromise the potential of the long read in defining novel splicing junction. Therefore, they demand precise junctions for each individual read track. To satisfy this requirement, the junctions must be precorrected for each read using existing junctions or massively parallel sequencing reads (referred to as short reads hereafter). A method for calling transcript isoforms without a reference genome has also been developed (Marchet et al. 2019). However, the method suffers from a higher false-positive rate and is problematic in handling close paralogs, which are often associated with short reads (Grabherr et al. 2011). With the decreasing costs of third generation sequencing, it has become increasingly desirable to define the transcriptome complexity of an existing genome using long reads only. However, a method capable of meeting this challenge is still lacking.
RNA modifications are emerging as a significant player not only in the regulation of rRNAs and tRNAs but also in post-transcriptional regulation of mRNAs. More than 150 RNA modifications are known (Helm and Motorin 2017), but the true potential of only a few of these has recently been revealed at the transcriptome scale, which is mainly due to the rapid development in detection technology based on high-throughput sequencing (Dominissini et al. 2012). For example, transcriptome-wide RNA modification is mainly achieved by coupling antibody immunoprecipitation (Meyer et al. 2012) or chemical treatments (Schaefer et al. 2008) to massively parallel sequencing. However, these techniques suffer from low resolution or limited capacity for generalization. A more straightforward method for detection of transcriptome-wide modification is necessary.
The C. elegans genome is one of the best characterized metazoan genomes due to its homozygosity and lack of gaps (The C. elegans Sequencing Consortium 1998). The 5′ end of most of its mRNAs carries a unique SL that is derived from independent loci (Lasda and Blumenthal 2011), making it straightforward to evaluate the completeness of mRNA transcripts purified using oligo(dT) magnetic beads. To examine the potential of ONT RNA sequencing in defining transcriptome complexity, we first performed direct sequencing of poly(A)-tailed RNAs from different developmental stages of C. elegans. We next devised a novel method for de novo discovery of alternative splicing events by using the mapping tracks of full-length RNA transcripts, which allowed us to identify 57,000 novel isoforms that are absent in the current annotation. We detected putative stage-specific expression of isoforms that was independent of the stage-specific expression of genes. Finally, we observed coding sequence–specific candidate RNA modification in all types of nucleotides.
Results
Statistics of read length and mappability
To evaluate the potential of direct RNA sequencing in identifying novel splicing isoforms, we first purified poly(A)-tailed RNAs. Most of these RNAs were expected to be mRNAs encoding proteins. We then performed direct RNA sequencing using portable MinION devices and generated a total of approximately six million long reads from three developmental stages, that is, embryo (EMB), L1 larva (L1), and young adult (YA). For each stage, we produced at least 1.6 million reads with an average read length of 1118, 908, and 925 nt for EMB, L1, and YA, respectively (Table 1). These reads are substantially longer than the short reads produced by massively parallel sequencing, which are typically <200 nt in length. We refer to the direct RNA sequencing reads as long reads hereafter. We expect that a subset of these reads represents full-length transcripts derived from the C. elegans genome.
Table 1.
Read statistics

We mapped the reads against the C. elegans genome (WormBase release 260) (Lee et al. 2018) using minimap2 through “split-read” alignment, which implements “concave gap cost” for long insertions and deletions to accommodate intron skipping (Li 2018). Taking into account mismatches and small insertions and deletions (indels) against the C. elegans genome, the overall read accuracy is roughly 85% (Table 1). Despite this relatively low read accuracy, the percentage of long reads that can be mapped back to the C. elegans genome, referred to as mappability hereafter, is 99.7%, indicating the high specificity of the long reads, consistent with previous mapping results using other types of long reads, including PacBio cDNA reads (Pan et al. 2008; Wang et al. 2018). Despite a relatively low read quality score (average Q score = 11) of the long read compared with the short reads, this mappability is significantly higher than the short reads that are routinely used in RNA-seq, the mappability of which is around 80% (Derrien et al. 2012). The substantially elevated mappability over an extended genomic interval provides an advantage in discovery of novel splicing isoforms. Consistent with this, when we mapped the long reads against existing annotated spliced exons and UTRs and noncoding transcripts in the WormBase WS260, the overall mappability dropped to 76.7%. The nearly perfect mappability against the C. elegans genome contrasts sharply with a much lower mappability against its annotated transcripts, suggesting a possibility of novel transcripts that are currently missing in the WormBase, highlighting the value of the long reads in defining novel splicing isoforms.
To examine to what extent the long reads represent a full-length transcript, we classified them into two categories, that is, full-length and partial-length reads. Given that the mRNAs were purified using oligo(dT) beads, most of them had intact 3′ ends. Therefore, we defined the full-length reads as those that span at least 95% of the length of their best hit of an existing transcript as described previously (Jenjaroenpun et al. 2018) or those that carry a splicing leader (SL) at 5′ end. The remaining reads were defined as partial-length reads (Fig. 1A). Over 65% of the SL sites (34,639 out of 52,846) identified using the long reads were also identified by meta-analysis of RNA-seq data (Tourasse et al. 2017) or were currently annotated in WormBase (Supplemental Fig. S1; Supplemental Table S1). Approximately half of the long reads were defined as full-length ones with these criteria. YA showed a slightly higher percentage of full-length reads than L1 and EMB (Fig. 1B). Of the long reads, 23% carried an SL (Fig. 1C). A previous analysis showed that ∼80% of C. elegans protein-coding transcripts carried a SL at the 5′ end (Tourasse et al. 2017). Judged by that benchmark, the percentage of SL-containing long reads in this study was lower than the expected 80% of the full-length reads, which would have corresponded to roughly 40% of the total long reads with the assumption that 3′ ends are intact. Two factors may have contributed to the underrepresentation of SL-containing reads. First, the mRNAs were purified from their 3′ end, which was expected to preferentially enrich reads that were intact at that end. Consistent with this, the 5′ but not 3′ end of the long reads seemed to undergo degradation (Supplemental Fig. S2). Second, the technical issues associated with the ONT platform could have contributed to this phenomenon. The platform has been demonstrated to be problematic in resolving the very end of the last few nucleotides (Byrne et al. 2017; Garalde et al. 2018). This was further complicated with a relatively higher rate of read error by the Nanopore platform than by the massively parallel sequencing platform (Table 1).
Figure 1.

Fraction of full-length RNA reads (defined as reads with an SL signal or covering ≥95% length [indicated by dashed line] of an annotated transcript) out of all RNA reads. (A) Count distribution of reads over the fraction of the mapped length out of the length of existing transcripts (WS260). (B) Proportion of reads over the fraction of their mapped lengths against those of the annotated transcripts derived from embryos (EMB), larvae (L1), and young adults (YA). (C) Distributions of read count over read length. The reads with and without SL and the simulated reads are colored in brown, gray, and blue, respectively. The annotation simulation curve was generated by replacing the actual length of long reads with the length of an annotated transcript to which it shows the highest similarity. (D) Distribution of full-length reads derived from the mitochondrial genome. Top: Read coverages of mitochondrial genes color-coded based on the respiratory chain complex. Bottom: Fractions of full-length reads for individual genes. Read counts are shown proportional to the circle area.
To independently evaluate the capability of Nanopore long reads in recovering full-length transcript, we calculated the percentage of full-length transcripts encoded by mitochondrial genes, which also carry a poly(A) tail but no intron in C. elegans (Li et al. 2018), for which we expect few complications associated with splicing. Nearly 80% of the mitochondrial transcripts were full-length, although for nduo-5, the total was <70% (Fig. 1D), indicating that the long reads were able to recover the majority of full-length transcripts that undergo no alternative splicing. The higher percentage of full-length transcripts from mitochondrial than from nuclear genes was probably due to their smaller size or lack of introns. Although the shorter genes tend to have a higher coverage of full-length reads, the long reads were still able to cover 80% length of the transcripts for both mitochondrial and nuclear genes whose transcripts were up to 3000 nt in length (Supplemental Fig. S3). Only 5% of existing nuclear genes were shown to produce transcript over 3000 nt in length. The results highlight the value of these reads in resolving transcript complexity.
A pipeline for reference genome–based identification of alternative splicing events
Current methods for identifying alternative splicing events using long reads mainly depend on predefined exon-intron junctions, leading to a high false-positive rate in calling novel isoforms (Tardaguila et al. 2018). For example, if an existing junction is inaccurately predicted, it will overwrite any isoforms defined by the long read mapping. In addition, existing methodologies for using long reads to identify isoforms are not designed for quantifying expression levels (Tardaguila et al. 2018; Marchet et al. 2019). Given the extremely high mappability of our long reads against the high-quality C. elegans genome, these full-length reads hold promise for de novo identification of intron-exon junctions, alternative promoter and polyadenylation sites, as well as variation in the UTR, which were collectively referred to as transcriptome complexity.
To take advantage of the long reads in defining transcriptome complexity, we devised a new method, called TrackCluster, which took full advantage of the mapping tracks of the full-length long reads to de novo construct a transcript isoform and determine its expression level. Using a customized classifier, TrackCluster either de novo identifies an isoform using a full-length transcript or groups the transcript with an existing isoform by their similarity score (Fig. 2; Supplemental Fig. S4). To increase the confidence of calling of an isoform, we demanded that the calling of an isoform must be simultaneously supported by at least five independent full-length long reads. Specifically, the mapping tracks of full-length reads were subjected to two rounds of clustering through calculating their intersection/overlapping scores (see Methods). The first round clustered all of the full-length reads with similar mapping tracks (exon/intron combinations) into distinct groups (Fig. 2B,C), whereas the second round merged the partial-length reads with an established group defined in the first round so as to quantify the expression level of the isoform (Fig. 2D,E). All of the transcript isoforms annotated in the WormBase WS260 were also included as a single track and clustered along with the long-read tracks. Therefore, TrackCluster not only outputs the isoforms that are consistent with the existing isoforms but also holds promise to identify novel isoforms that could be missing from the existing annotations. As a result, TrackCluster outputs 12 categories of novel splicing isoforms relative to the existing isoforms to which they bear the highest similarity (Fig. 2). Four of these categories involve the alternative use of promoter or polyadenylation sites, that is, bearing extra or missing exon(s) at the 5′ or 3′ end. Another four categories involve UTR extensions or truncations at the 5′ or 3′ end, in which all of the newly identified intron-exon boundaries match with those of an existing isoform except the first or the last exon. To satisfy those latter four categories, the difference must be at least 5% of the summed length of all exons from the existing isoform. Two categories involve new combinations of exons within the gene body, including extra or missing exon(s). One category involves intron retention, and the last category involves fusion of two separate isoforms from adjacent genes into a single isoform (Fig. 2). Many of those with a retained intron contained a premature codon. Some of them did have an intact open reading frame. However, we did not know the role of these isoforms from our data only. Alternatively, TrackCluster is also able to de novo identify an isoform independent of a reference isoform, making it more useful for annotation of any newly sequenced genome.
Figure 2.

Flowchart of new isoform calling by TrackCluster. (A) Assignment of sequence tracks (shown as gray bar with different numbers) to a given locus based on read mapping against the C. elegans genome. Existing isoforms are also included as individual tracks (black bar). Two reads that show few overlaps with any existing exons in or are antisense to a given locus are excluded from subsequent analysis. (B) First round of clustering of tracks based on their distance scores (see Methods). (C) Read tracks (excluding existing transcripts) are merged if their distance scores satisfy our cutoff. Only the one with the biggest size of summed exons is retained (indicated with “√”) from each group along with the existing one (indicated with “#”) for subsequent isoform calling. The remaining tracks (indicated with “×”) including existing transcripts are assigned as “subreads” and used only for expression quantification and boundary correction. Note, during track merging, a minor shift (indicated with “*”, within 5% change in “score 1” defined in Methods) in exon-intron boundary caused by read error is permitted to avoid overcalling of novel isoform. (D) The retained tracks from C are subjected to a 2nd round of track clustering based on mutual distance scores (see “score 2” in Methods). (E) The tracks (including existing transcripts) are merged if their distances satisfy our cutoff to avoid calling a novel isoform from a possible partially degraded read retained in C except for those starting with an SL. (F) Existing annotated (black) and novel isoforms (gray) after junction correction (see Supplemental Fig. S4). The retained track is called a novel isoform due to its distance score with any existing transcript satisfying our cutoff. (G–I) Schematic representation of each category of the newly identified isoform. Novel isoforms involving newly defined 5′ and/or 3′ end. “5′ and/or 3′ extra or missing” are/is defined as novel isoform with an extra or missing exon at both or either end(s) of a novel isoform relative to an existing transcript. “UTR extensions” or “UTR truncations” is defined as a novel isoform involving changes only in the UTR relative to an existing transcript. (J–M) Novel isoform involving the exon change within the gene body. Note that straightforward assignment of an exon combination constitutes the main advantage of the long reads (J).
To demonstrate the performance of TrackCluster, we generated simulation data sets for the gene unc-52, for which 17 isoforms are currently annotated in the WormBase. For each isoform, we randomly generated 10–300 long reads. To mimic the characteristics of our actual reads, we generated the reads with around 85% accuracy (3.9% mismatch, 6.1% deletion, and 5% insertion), around 65% of which were expected to be full-length based on the statistics of our Nanopore reads. In addition, 23% of the full-length reads were marked with an SL signal. In 100 simulations, we achieved a FDR smaller than 5% in terms of novel isoform calling and a variation smaller than 10% in terms of isoform quantification.
An underestimation of transcriptome complexity in C. elegans
With approximately three million full-length long reads and approximately another three million partial-length long reads, we identified 169,804 splicing junctions. Out of those junctions, 150,591 (88.7%) were identical to those annotated in the WormBase (WS260), And 4537 (23.6%) of the remaining junctions (19,213) were also identified by meta-analysis of RNA-seq data (Supplemental Fig. S1B; Supplemental Table S3; Tourasse et al. 2017). Consequently, we recovered approximately 25,000 (75%) out of the existing 33,500 isoforms and identified a total of about 57,000 novel isoforms, which significantly expanded the complexity of existing isoforms annotated in the WormBase WS260 (Fig. 3A; Supplemental Fig. S5). Given our relatively low read coverage compared with the aggregated coverage of short RNA-seq reads used for exon annotation, it was not surprising that we missed approximately 7000 existing isoforms (Fig. 3A). The novel isoforms we identified involved 11,921 genes (Supplemental Table S2). Given that the summed exonic regions currently annotated in the WormBase are around 31.5 million bps, about 4.99 million (15.8%) of these are missed by our long reads. Most of the novel isoforms were contributed by variations in the 5′ and/or 3′ end of the existing isoforms, that is, an alternative promoter or alternative polyadenylation site (Fig. 2G,H). For example, it was frequently observed that an exon was missing at either end of a novel isoform relative to an existing transcript. It was also common that a UTR was expanded or truncated relative to an existing UTR. “5′ or 3′ extra or missing” was defined as a novel isoform with an extra or missing exon at the 5′ or the 3′ end of a novel isoform relative to an existing transcript, respectively. Such variations in the 5′ and 3′ end were referred to as an alternative promoter and alternative polyadenylation site, respectively (Zahler 2012). “UTR extensions or truncations” was defined as a novel isoform involving an extra or missing part only in the UTR relative to an existing transcript, respectively. We identified a total of 17,646 5′ UTRs (Supplemental Fig. S1D). About half of them are annotated in WormBase WS260. Roughly one-third of the 5′ UTRs identified by Nanopore reads were also identified with Cap-seq analyses (Gu et al. 2012; Chen et al. 2013; Kruesi et al. 2013; Saito et al. 2013), whereas the majority of the UTRs identified by the Cap-seq analyses were missed by Nanopore reads, which could be contributed by artifacts, very low-frequency events, or unannotated trans-spliced sites.
Figure 3.

Statistics of the newly called isoforms with long reads. (A) Summary of the isoforms called using long reads. Left: Bar plots of the number of existing isoforms that are recovered (defined as coverage of over 95% length of an existing transcript by at least a single long read, colored in black) or uncovered (the remaining isoforms, colored in gray). Right: Bar plots of the number of novel isoforms called by TrackCluster. Breakdown of the novel isoforms is color-coded as in Figure 2 and is also shown at the bottom. (B,C) Abundance (B) or length distribution (C) of the novel isoforms of various categories as defined in Figure 2. (D) Count of the isoforms output by TrackCluster (gray), existing isoforms (yellow), and their ratio (blue) over read length, in nt. (E) An example of TrackCluster predicted isoforms. Shaded in blue is the accumulative long read coverage of unc-52 in all developmental stages. Shaded in gray are three novel isoforms (colored in red or green as in B) supported by the highest read coverage along with the existing isoforms (black) to which they bear the highest similarity. Genomic coordinates are shown at the top, in kb.
It is worth noting that nearly half of the members of the category of “fusion gene” (defined as a fusion between two existing separate transcripts) belong to an operon (Supplemental Fig. S6), which serves as a nice validation of the identified isoform. These transcripts were likely captured before processing into two separate ones. Another half may contain some operonic transcripts that are currently unidentified. Part of the fusion transcripts could be derived from unidentified operonic transcripts. There were some reads that spanned two independent loci located even on different chromosomes. We manually removed these reads during calling of fusion isoform, resulting in a much smaller size of the category (Figs. 2A–C, 3A; Supplemental Fig. S6). The category of “missing or extra exon” (defined as an isoform output by TrackCluster that shows a different exon combination relative to that of any existing transcript) within a gene body contributes a relative small fraction of the novel isoforms, indicating that massively parallel sequencing-based RNA-seq analyses have been effective in recovering exons within the gene body. However, the ability of straightforward assignment of exon combinations highlights the main advantage of the long reads over the short reads. This is particularly true for genes with numerous exons and complicated splicing patterns. There are still 71,668 reads (around 1.2% of all our reads) that do not show obvious overlap with any existing isoforms. However, most of these reads are short, with a size smaller than 100 (Supplemental Table S4). We did not include these reads in our analysis of isoforms.
To examine whether different categories of novel isoforms demonstrated differential expression levels, we plotted their accumulated expression level from the three developmental stages. The category involving UTR extensions demonstrated a higher expression level than the remaining categories (Fig. 3B), indicating that many isoforms differentiate themselves from others by changing their UTR. To evaluate whether the sizes of novel isoforms varied across categories, we plotted the length for all categories of novel isoforms. The results showed that the isoforms associated with exon change within the gene body usually involved genes with a relatively large size (Fig. 3C). The one category that showed a significantly smaller size involved missing sequences in UTRs. The overall size of the novel isoforms identified by TrackCluster was comparable to that of the existing ones (Fig. 3D).
To help illustrate the use of long read coverage in identifying novel isoforms, we depicted three novel isoforms of unc-52 that were identified by TrackCluster with the highest coverage of long reads. Also shown were the three existing isoforms that have the highest level of similarity to the newly identified isoforms (Fig. 3E). One of the three novel isoforms was produced by skipping of multiple exons at its 3′ end relative to the existing isoform to which it has the highest level of similarity, that is, the category of “3′ missing” (Fig. 2B). A careful examination of the accumulated coverage of the long reads showed a sharp drop in one relevant exon compared with its neighbouring tracks. This exon was part of only one of the three novel isoforms (Fig. 3E). The remaining two isoforms skipped the exon relative to its closest reference isoform; that is, they belonged to the category of “missing exon.”
In a few cases, the long reads were able to recover missing sequence within the C. elegans genome. For example, the long read derived from the tsr-1 locus indicated absence of an exon along with its flanking sequences (Supplemental Fig. S7). Examination of the Illumina synthetic long reads we produced previously showed that the exon along with its flanking intronic area was also missing in the current genome (Li et al. 2015). By parsing the results from the read-to-genome alignments, we were able to obtain an additional 730 loci that possibly carry a missing sequence longer than 50 nt. All of these missing sequences were supported by at least five long reads. These loci involved 437 protein-coding genes in total (Supplemental Table S5). Consistent with this, recent publication of the genome of an N2-derived strain VC2010 suggested that roughly 2% of the C. elegans encoded genes were affected by the deficiencies in the existing N2 reference genome (Yoshimura et al. 2019). It remains a possibility that part of the missing sequences were produced by genetic variations unique to the strain we used for sequencing.
The polyadenylation sites from long reads form an independent resource for identification of its motif, that is, polyadenylation signal (PAS). We determined the PAS as described (Mangone et al. 2010) for both canonical (defined as those annotated in WormBase WS260 and with hits by long read in the locus) and noncanonical isoforms (defined as isoforms whose polyadenylation site is at least 15 nt away from canonical isoforms based on long reads) (Supplemental Fig. S8A,B). The occurrences of a canonical PAS (AATAAA) consist of 25% and 20% in the canonical and noncanonical isoforms, respectively (Supplemental Fig. S8C). However, the polyadenylation sites only show a moderate overlap with those of previous studies (Supplemental Fig. S1A; Supplemental Table S6; Mangone et al. 2010; Tourasse et al. 2017). This could be due to the difficulty of Nanopore reads in resolving the ion current signal of poly(A) sequence, which may lead to a slight shift of the boundary of the poly(A) tail.
The sets of genes with differential expression versus differential isoform usage are largely different
The capability to unambiguously assign isoforms using the long reads permitted quantification of stage-specific expression not only at the gene level but also at the isoform level. To evaluate whether stage-specific expression at the gene level is contributed by differential expression of their isoforms, we quantified the isoforms from three developmental stages, that is, EMB, L1, and YA, using TrackCluster (Supplemental Tables S3, S7, S8). Despite a relatively high correlation of expression between gene levels measured with RNA-seq and the long reads (Fig. 4A), we observed a moderate overlap between the genes with stage-specific expression at the gene and isoform levels (Fig. 4C; Supplemental Fig. S9; Supplemental Tables S7, S8). In addition, most of the stage-specific genes were shared among the three stages, but fractions of the shared isoforms were much reduced at the isoform level (Fig. 4D). Gene Ontology analysis of stage-specific expression at the gene and isoform levels also demonstrated little overlap with each other (Supplemental Fig. S10), indicating that stage-specific expression at the gene and isoform level is largely uncorrelated from each other.
Figure 4.

Relationship between expression at gene level and isoform level during development. (A) Correlations of expression at gene levels in three developmental stages determined by the long reads in this study and by the existing RNA-seq reads (WormBase release WS260). (B) An example of stage-specific abundance of TrackCluster predicted isoforms. Shaded in light blue is the coverage of long reads for gene efhd-1 over development. Existing (black) and TrackCluster predicted novel isoforms are shaded in gray. Novel isoforms are labeled as “1” to “5” and are colored as in Figure 2 and existing isoforms are labeled as “a”, “b,” or “c”. The existing isoform without any supporting long read is shown at the bottom. Note that the exon (indicated with arrowhead) with an elevated usage in young adult (55%) is highlighted in the black box. The elevation is produced by stage-specific expression of the isoform “1.” The abundance of the isoforms in EMB, L1, and YA is indicated (5%, 9%, and 55%, respectively). (C) Intersection of differentially expressed genes (DEG) and isoforms (DEI) between L1 and embryonic stages (L1_vs_EMB). Left: Venn diagram between DEG and DEI. Number of unique and shared genes are indicated. Right: Simulation of intersection between two sets of genes randomly chosen from the expressed genes either in L1 or embryos based on long reads. Number of genes sampled in each set is the same as that in DEG or DEI. (D) Venn diagrams showing the intersection of DEG (left) or DEI (right) among three stages.
To illustrate the power of the long reads in delineating stage-specific expression of the isoform, we plotted stage-specific read tracks along with the novel isoforms identified by the long reads as well as the existing isoforms for gene efhd-1 (Fig. 4B). TrackCluster identified a total of five isoforms supported by at least five long reads, while there were a total of four existing isoforms annotated in WormBase. Our isoform “1” showed the highest level of similarity to the existing isoform, efhd-16b. Approximately 55% of the long reads derived from YA were contributed by expression of the isoform “1,” whereas roughly 5% and 9% of the long reads from the EMB and L1 stages, respectively, were contributed by expression of that isoform.
Embryonic transcripts are longer than postembryonic transcripts
One unexpected observation was that the long reads derived from EMB were significantly longer in size than those of the remaining stages for both raw reads and mapped reads (Mann–Whitney U test, P < 10−15) (Fig. 5A,B). The poly(A) tails derived from EMB were also significantly longer than those of the remaining stages (Mann–Whitney U test, P < 10−15) (Fig. 5C). The sizes of both long reads and poly(A) tails were comparable between the L1 and YA stage. The size difference between the embryonic and postembryonic transcripts was not unique to the isoforms newly identified by the long reads (Fig. 5D). Existing transcripts annotated in the WormBase WS260 also showed a similar trend. The functional implications of the elevated size in embryonic transcripts remain to be determined.
Figure 5.

The long reads derived from embryos are significantly longer than those from L1 larvae or young adults. (A) Violin plots showing the length of raw reads derived from embryos (EMB), L1 larvae (L1), and young adults (YA). (B) Violin plots showing the length of mapped reads from EMB, L1, and YA stages. (C) Violin plots showing the length of poly(A) tails derived from EMB, L1, and YA stages. (D) Genes with a higher expression level (High) in embryo have an overall longer isoform. Box plots showing the length of isoforms from three categories of genes, that is, those that are expressed at a high (High), low (Low), or moderate (Mid) level in EMB compared to both L1 and YA stages (see Methods). Note that the genes with elevated expression in the embryo tend to have longer transcripts. The results from existing WormBase annotation (Release 260) and TrackCluster output are differentially color-coded in green and yellow boxes, respectively.
Elevated putative RNA modifications within coding regions
To explore the capability of direct RNA-seq to identify modifications in RNA molecules, we first identified all of the possible modified bases via the deviation of their ion current profile from that of known unmodified nucleotides using Tombo (Stoiber et al. 2017). It worked by computing the possibility of a modification on each site for every read and outputting the fraction of a possible modification on the site out of all input reads. The modification was detected by reproducible deviations of the ion current profile of a base in question from that of an unmodified base without knowledge of the exact chemical identity of the modification (Supplemental Figs. S11, S12; Supplemental Table S9). This was achieved with the assumption that the deviation in the ion current profile of read error occurs randomly. We next normalized the ratio of the modified bases against their relative position within the gene body, including UTRs. We then plotted the normalized ratio of each base along the gene body, including UTRs. A previous study in C. elegans predominantly identified modified adenosines in the noncoding regions of DNA transposons (Zhao et al. 2015). We observed an apparent increase in the modification of all four types of bases within the coding region relative to the UTR (Fig. 6).
Figure 6.
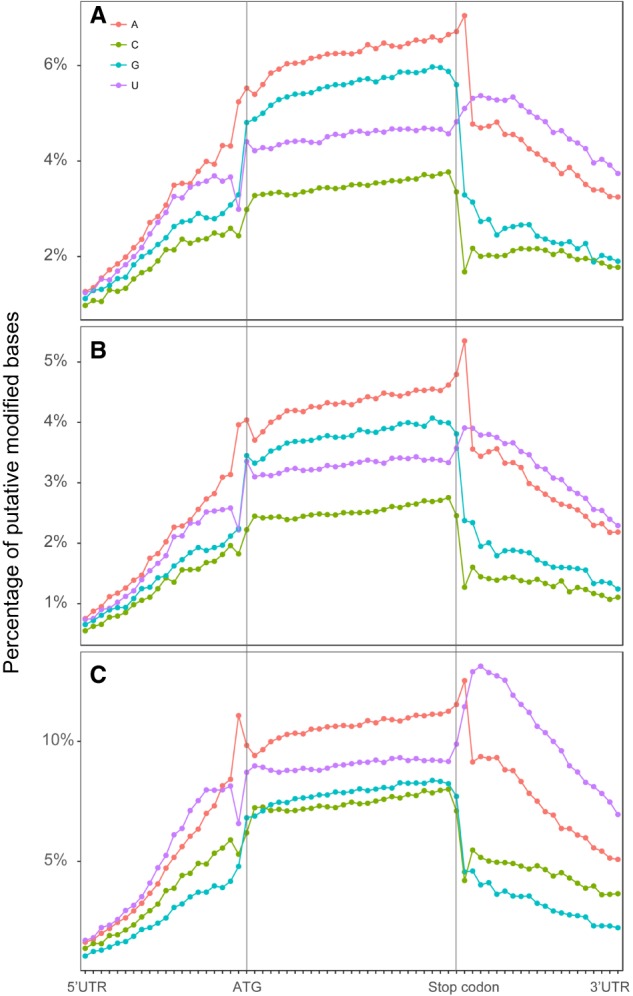
Percentage of putative modified bases along the gene body in three developmental stages: (A) embryos; (B) L1 larvae; (C) young adults. Putative base modifications are predicted with Tombo (version 1.4) and are differentially colored as indicated. The “de novo” model is used to identify all potential modifications on each base. The fraction of modification at each site indicates the normalized percentage of a putative modification out of all available reads at this site.
To investigate whether the modification in cytosine was contributed by 5mC only, we detected 5mC and quantified its ratio in RNA using a well-established method for 5mC detection (Stoiber et al. 2017). The pattern of 5mC is similar to that of total cytosine modifications but at a much smaller scale (Fig. 6; Supplemental Fig. S11), suggesting that 5mC contributed to a fraction of the observed ratio of modification in cytosine. The putative modifications in the bases A and U demonstrated opposite patterns from each other immediately before the start codon, whereas the modifications in all four bases except for U showed a sharp decrease immediately after the stop codon, suggesting that RNA modifications play an important role in protein translation and that U may have a unique role in termination of translation. In addition, the relative modification in U was higher in the YA stage than the remaining two stages, suggesting its stage-specific modification. It is worth noting that de novo detection of RNA modifications could be error-prone and should be treated with caution. It remains possible that the detected modification may be a sequence context-dependent artifact without independent validation. Independent validation is required before working on the roles of the modified bases.
Discussion
The capability of direct sequencing of full-length transcripts provides a key advantage over massively parallel sequencing-based RNA-seq analysis in several ways. First, direct sequencing of a full-length RNA transcript makes it straightforward to identify a transcript isoform with numerous exons. Second, it enables profiling of developmental stage-specific or cell-specific expression of isoforms, which is problematic using RNA-seq, which may provide important insights into developmental processes. Finally, it holds promise to define RNA modifications de novo without an extra treatment step, for example, by measuring the deviation in the ion current profile from that of wild-type RNA. Here, we performed direct RNA sequencing of C. elegans poly(A)-tailed RNAs derived from three developmental stages using ONT technology and devised a pipeline for identifying isoform variants based on read track, allowing straightforward characterization of transcriptome complexity in a stage-specific way.
The actual number of novel isoforms could be even higher
We recovered ∼75% out of the existing 33,500 isoforms and identified another 57,000 novel isoforms with approximately 6 million long reads. It is likely that the actual number of novel isoforms may be substantially higher due to the following reasons. First, our sampling depth was not as high as those of analyses using RNA-seq both spatially and temporally. For example, we only sampled three developmental stages, whereas RNA-seq has been performed for tens of developmental stages in different cell or tissue types (Gerstein et al. 2010). Second, we demanded simultaneous support by at least five full-length reads to define a novel isoform. If we reduced this requirement to two full-length long reads, approximately 20,000 more candidate novel isoforms could be identified (Supplemental Data Set). Third, the ratio of full-length reads may also be underestimated. This was because our definition of full-length was based on alignment against existing transcripts. Many transcript isoforms may not exist in the WormBase that is currently annotated. For example, the full-length ratio of mitochondrial reads that carry no intron was 78%, whereas the ratio of nuclear mRNAs was only 49% (Fig. 1D). Although the proportion of full-length reads decreases slightly over transcript length (Supplemental Fig. S3), this cannot account for the sharp decrease of the full-length proportion between the mitochondrial and nuclear transcripts. It also suggests that a portion of the partial-length reads may be bona fide full-length ones but are absent in the current WormBase. The main purpose of this study was not to capture as many isoforms as possible but to demonstrate the capacity of the direct RNA sequencing technology in characterizing transcriptome complexity and in identifying novel RNA modifications. Future work should focus on systematic identification of alternative splicing events at different developmental stages and tissue/cell types. This work also provides an entry point for biochemical and functional characterization of various RNA modifications observed in vivo.
Intron-retaining isoforms seem to be nonfunctional
One category of novel isoform output by TrackCluster is “intron retention,” defined as a novel transcript that carries a well-established intron supported by various RNA-seq data (Fig. 2). We initially speculated that the retention of introns could be due to incomplete processing of pre-mRNAs, which were expected to retain multiple introns and to be enriched at the 3′ end, that is, the end at which their processing terminated. However, most (89.9%) of the isoforms with intron retention only retained one intron and are shared between developmental stages, and location of the retained introns was enriched in the middle of the gene body rather than at the 3′ end (Supplemental Fig. S13), suggesting that these errors could be a product of incorrect rather than incomplete processing of pre-mRNA. Most of these intron-retaining isoforms carried a premature stop codon, suggesting that they were not functional in coding a protein.
Complications associated with Nanopore direct RNA sequencing
It is possible that at least a fraction of the long reads could be artifacts. This is because these reads contain sequences derived from different parts of a single chromosome or from different chromosomes. However, the chromosome assembly in the relevant regions seems to be intact, as judged by a lack of repetitive sequences or gaps in these regions. We speculate that these reads could be chimeric ones created during adaptor ligation; that is, two separate reads were ligated together. We estimate that about 0.5% of the long reads were likely to be the results of such artifacts.
Despite the ultralong read length offered by Nanopore direct RNA-seq, a few notable caveats might limit its application in the following respects. First, its sequence throughput is substantially lower than conventional RNA-seq, translating into a much higher sequencing cost per nucleotide. Currently, only one to two Gb of data of RNA sequences can be generated per flow cell, whereas over 10 Gb of data of DNA sequences can be produced using the same flow cell. This low throughput significantly inhibits its application in research areas that are heavily dependent on gene expression profiling, which demand an especially high coverage for these low-abundance transcripts. Second, the relatively high error rate in read sequences is problematic during alignment in some cases. A customized alignment algorithm must be used to accommodate these errors. Third, Nanopore direct RNA-seq is known to be deficient in calling the very last bases that it sequences. This could have contributed to our lower than expected percentage of calling of SL-containing transcripts. Given that Nanopore direct RNA-seq produces sequence from the 3′ to the 5′ end, we speculate that the underrepresentation of SL signals was partially due to incomplete sequence at the 5′ end of the long reads, which inhibited reliable calling of SL signals, typically only 22 nt in length (Lasda and Blumenthal 2011). Fourth, methodology for detecting RNA bases is in its infancy and under active development. A more robust method is needed to reliably detect the RNA base modification and its chemical identity. Any putative RNA base modifications reported in this study could be an artifact resulting from various noises. Functional characterization of these modifications is not warranted until they are independently validated. Finally, Nanopore direct RNA-seq demands a large amount of starting RNAs in the magnitude of ∼100 µg. This limits its use in single-cell analysis. Future development should focus on adaptation of Nanopore direct RNA-seq to small amounts of starting materials, which would maximize its potential in identifying novel isoforms.
Although numerous potential modifications were named in this study, the software we used (Stoiber et al. 2017) is likely to be error-prone. For example, we do not have a way to prove to what extent the observed modification is dependent on sequence context. The validity of the modification signal should be confirmed using an independent method. Methodology for RNA modification detection from a single molecule is under active development. A more robust method with thorough negative and positive controls in various sequence contexts will definitely improve the accuracy of modification detection.
Taken together, with our newly devised classifying method, the long reads generated by ONT greatly facilitate the unambiguous resolution of alternative splicing events. The reads also hold great potential in de novo identification of RNA modifications, which is expected to catalyze the functional characterization of the new isoforms and modifications. Given the evidence of conserved splicing events between nematode and mammals (Barberan-Soler and Zahler 2008; Irimia et al. 2008), some of the splicing events were expected to be conserved across species.
Methods
Purification and sequencing of mRNAs with MinION
Synchronized embryos, L1, and young adult N2 animals were collected, and total RNAs were extracted using TRIzol (Invitrogen). Approximately 900 ng of poly(A)-tailed mRNAs was purified using a Dynabeads mRNA Purification kit (Invitrogen), immediately followed by library construction using a Direct RNA sequencing kit (cat# SQK-RNA001), which was sequenced on MinION (ONT).
Mapping of mRNA reads
Sequences were separately mapped against the C. elegans transcriptome with parameters “-ax map-ont” and against the C. elegans genome with parameters “-ax splice” using minimap2 (Li 2018).
The resulting SAM files were sorted and indexed with SAMtools (v2.1) (Li et al. 2009) by sequence coordinate. For visualization on a genome browser, they were converted to bigGenePred format (https://genome.ucsc.edu/goldenpath/help/examples/bigGenePred.as) using customized script in the TrackCluster package. The coverage track was generated by using BEDTools (2.24) (Quinlan and Hall 2010).
Defining novel isoforms with TrackCluster using the long reads
The details of TrackCluster design are described in Supplemental Methods.
Identification of spliced leaders
A previous study showed that Nanopore direct RNA reads were truncated by a few nucleotides in the 5′ end (Garalde et al. 2018), which made the determination of the SL problematic. To maximize the possibility to recover an SL, a customized script was written as part of TrackCluster, which used the Smith-Waterman (SW) alignment algorithm to detect a putative SL signal by aligning the very first 22 nt of the long reads against seven SL sequences. Reads with SW scores over 11 were treated as SL-containing reads. Simulation suggested that the FDR was lower than 20% using these parameters and cutoff.
Identification of PAS motif
A PAS motif was identified as described (Mangone et al. 2010). A 50-nt region immediately upstream of poly(A) sites was scanned for all possible hexamer sequences to identify the top 50 overrepresented motifs. The overrepresented motifs were then scanned against the sequences of 14–24 nt (19±5 nt) upstream of a PAS to obtain occurrence of the motifs within these regions. The count of motifs with the same composition of nucleotides, for example, AATAAA, AAATAA, and ATAAAA, were not merged as described (Mangone et al. 2010).
Modification identification
Modifications of the RNA sequences were identified with Tombo package version 1.4 (Stoiber et al. 2017). The models of “5mC” and “de novo” were implemented separately to detect possible modification in each read. The score on each site indicated the fraction of a possible modification on a given site. For plotting the modification coverage along the gene body, the modification coverage was normalized for each isoform using a “w0” method with a bin size of 5 nt with EnrichedHeatmap (Gu et al. 2018). Only the isoforms with both 5′ and 3′ UTRs longer than 50 nt were used in the calculation.
Characterization of poly(A) tail length
The poly(A) lengths of each read were calculated using Nanopolish (Loman et al. 2015). The raw current signal from the 3′ unaligned ends of reads was extracted to estimate the length of poly(A) tail, which was deduced by the duration of the signal.
Analysis of differential expression
An isoform was defined as differentially expressed between stages when the change of its relative abundance (percentage of read count) out of all the transcripts derived from the same locus was >20% across stages. A gene was defined as differentially expressed between stages when the fold-change of its abundance of combined transcripts (read count per million) derived from its locus is greater than four across stages. Only genes supported by at least five long reads were used for the subsequent statistical analyses. Differences in the lengths of long reads between different developmental stages were calculated using a Mann–Whitney U test implemented in R 3.4.4 (R Core Team 2018).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE130044. The source code for TrackCluster has been deposited in GitHub (https://github.com/Runsheng/trackcluster) and is also available as Supplemental Code. All the isoforms are also included in Supplemental Data Set and Supplemental Table S3.
Competing interest statement
A U.S. patent was filed for de novo identification of transcript isoforms using TrackCluster.
Supplementary Material
Acknowledgments
We thank Mr. Chung Wai Shing and Dr. Cindy T. Tan for the logistic support and the members of Zhao's lab for helpful discussion and comments. This work was supported by Hong Kong Baptist University (HKBU) General Research Funds (HKBU12100917, HKBU12123716, HKBU201/18, HKBU12100118) from the Hong Kong Research Grant Council and HKBU Research Committee and Interdisciplinary Research Clusters Matching Scheme 2019/20 for 2017/18 to Z.Z. and was supported by the Hong Kong Branch of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (SMSEGL20SC02) to R.L.
Author contributions: Z.Z. and R.L. conceived and designed the study. X.R., Q.D., Y.B., and D.X. performed the experiments. R.L. conducted the computational analyses. Z.Z. provided the resources for experimentation and computation. Z.Z. and R.L. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.251512.119.
References
- Adusumalli S, Ngian Z-K, Lin W-Q, Benoukraf T, Ong C-T. 2019. Increased intron retention is a post-transcriptional signature associated with progressive aging and Alzheimer's disease. Aging Cell 18: e12928 10.1111/acel.12928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angiolini F, Belloni E, Giordano M, Campioni M, Forneris F, Paronetto MP, Lupia M, Brandas C, Pradella D, Di Matteo A, et al. 2019. A novel L1CAM isoform with angiogenic activity generated by NOVA2-mediated alternative splicing. eLife 8: e44305 10.7554/eLife.44305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan-Soler S, Zahler AM. 2008. Alternative splicing and the steady-state ratios of mRNA isoforms generated by it are under strong stabilizing selection in Caenorhabditis elegans. Mol Biol Evol 25: 2431–2437. 10.1093/molbev/msn181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeck ME, Huynh C, Gevirtzman L, Thompson OA, Wang G, Kasper DM, Reinke V, Hillier LW, Waterston RH. 2016. The time-resolved transcriptome of C. elegans. Genome Res 26: 1441–1450. 10.1101/gr.202663.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne A, Beaudin AE, Olsen HE, Jain M, Cole C, Palmer T, DuBois RM, Forsberg EC, Akeson M, Vollmers C. 2017. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat Commun 8: 16027 10.1038/ncomms16027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282: 2012–2018. 10.1126/science.282.5396.2012 [DOI] [PubMed] [Google Scholar]
- Chen RA-J, Down TA, Stempor P, Chen QB, Egelhofer TA, Hillier LW, Jeffers TE, Ahringer J. 2013. The landscape of RNA polymerase II transcription initiation in C. elegans reveals promoter and enhancer architectures. Genome Res 23: 1339–1347. 10.1101/gr.153668.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Estellé J, Marco Sola S, Knowles DG, Raineri E, Guigó R, Ribeca P. 2012. Fast computation and applications of genome mappability. PLoS One 7: e30377 10.1371/journal.pone.0030377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. 10.1038/nature11112 [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, Ortis F, Santin I, Colli ML, Barthson J, et al. 2012. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet 8: e1002552 10.1371/journal.pgen.1002552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7: 461–465. 10.1038/nmeth.1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, Pantic N, Admassu T, James P, Warland A, et al. 2018. Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods 15: 201–206. 10.1038/nmeth.4577 [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787. 10.1126/science.1196914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SP, Tseng E, Salamov A, Zhang J, Meng X, Zhao Z, Kang D, Underwood J, Grigoriev I V, Figueroa M, et al. 2015. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS One 10: e0132628 10.1371/journal.pone.0132628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün D, Kirchner M, Thierfelder N, Stoeckius M, Selbach M, Rajewsky N. 2014. Conservation of mRNA and protein expression during development of C. elegans. Cell Rep 6: 565–577. 10.1016/j.celrep.2014.01.001 [DOI] [PubMed] [Google Scholar]
- Gu W, Lee HC, Chaves D, Youngman EM, Pazour GJ, Conte D, Mello CC. 2012. CapSeq and CIP-TAP identify Pol II start sites and reveal capped small RNAs as C. elegans piRNA precursors. Cell 151: 1488–1500. 10.1016/j.cell.2012.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M, Ishaque N. 2018. EnrichedHeatmap: an R/Bioconductor package for comprehensive visualization of genomic signal associations. BMC Genomics 19: 234 10.1186/s12864-018-4625-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M, Motorin Y. 2017. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet 18: 275–291. 10.1038/nrg.2016.169 [DOI] [PubMed] [Google Scholar]
- Irimia M, Rukov JL, Penny D, Garcia-Fernandez J, Vinther J, Roy SW. 2008. Widespread evolutionary conservation of alternatively spliced exons in Caenorhabditis. Mol Biol Evol 25: 375–382. 10.1093/molbev/msm262 [DOI] [PubMed] [Google Scholar]
- Jain M, Olsen HE, Paten B, Akeson M. 2016. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol 17: 239 10.1186/s13059-016-1103-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al. 2018. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36: 338–345. 10.1038/nbt.4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenjaroenpun P, Wongsurawat T, Pereira R, Patumcharoenpol P, Ussery DW, Nielsen J, Nookaew I. 2018. Complete genomic and transcriptional landscape analysis using third-generation sequencing: a case study of Saccharomyces cerevisiae CEN.PK113-7D. Nucleic Acids Res 46: e38 10.1093/nar/gky014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruesi WS, Core LJ, Waters CT, Lis JT, Meyer BJ. 2013. Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. eLife 2: e00808 10.7554/eLife.00808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasda EL, Blumenthal T. 2011. Trans-splicing. Wiley Interdiscip Rev RNA 2: 417–434. 10.1002/wrna.71 [DOI] [PubMed] [Google Scholar]
- Lee J-A, Damianov A, Lin C-H, Fontes M, Parikshak NN, Anderson ES, Geschwind DH, Black DL, Martin KC. 2016. Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron 89: 113–128. 10.1016/j.neuron.2015.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RYN, Howe KL, Harris TW, Arnaboldi V, Cain S, Chan J, Chen WJ, Davis P, Gao S, Grove C, et al. 2018. WormBase 2017: molting into a new stage. Nucleic Acids Res 46: D869–D874. 10.1093/nar/gkx998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Hsieh C-L, Young A, Zhang Z, Ren X, Zhao Z. 2015. Illumina Synthetic Long Read Sequencing allows recovery of missing sequences even in the “finished” C. elegans genome. Sci Rep 5: 10814 10.1038/srep10814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Ren X, Bi Y, Ho VWS, Hsieh C, Young A, Zhang Z, Lin T, Zhao Y, Miao L, et al. 2016. Specific down-regulation of spermatogenesis genes targeted by 22G RNAs in hybrid sterile males associated with an X-Chromosome introgression. Genome Res 26: 1219–1232. 10.1101/gr.204479.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Ren X, Bi Y, Ding Q, Ho VWS, Zhao Z. 2018. Comparative mitochondrial genomics reveals a possible role of a recent duplication of NADH dehydrogenase subunit 5 in gene regulation. DNA Res 25: 577–586. 10.1093/dnares/dsy026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linker SM, Urban L, Clark SJ, Chhatriwala M, Amatya S, McCarthy DJ, Ebersberger I, Vallier L, Reik W, Stegle O, et al. 2019. Combined single-cell profiling of expression and DNA methylation reveals splicing regulation and heterogeneity. Genome Biol 20: 30 10.1186/s13059-019-1644-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Watson M. 2015. Successful test launch for nanopore sequencing. Nat Methods 12: 303–304. 10.1038/nmeth.3327 [DOI] [PubMed] [Google Scholar]
- Loman NJ, Quick J, Simpson JT. 2015. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12: 733–735. 10.1038/nmeth.3444 [DOI] [PubMed] [Google Scholar]
- Mangone M, Manoharan AP, Thierry-Mieg D, Thierry-Mieg J, Han T, Mackowiak SD, Mis E, Zegar C, Gutwein MR, Khivansara V, et al. 2010. The landscape of C. elegans 3′UTRs. Science 329: 432–435. 10.1126/science.1191244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchet C, Lecompte L, Da Silva C, Cruaud C, Aury J-M, Nicolas J, Peterlongo P. 2019. De novo clustering of long reads by gene from transcriptomics data. Nucleic Acids Res 47: e2 10.1093/nar/gky834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell CS, Antoshechkin I, Kurhanewicz N, Belsky JA, Baugh LR. 2012. Nutritional control of mRNA isoform expression during developmental arrest and recovery in C. elegans. Genome Res 22: 1920–1929. 10.1101/gr.133587.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. 10.1016/j.cell.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudge JM, Frankish A, Fernandez-Banet J, Alioto T, Derrien T, Howald C, Reymond A, Guigó R, Hubbard T, Harrow J. 2011. The origins, evolution, and functional potential of alternative splicing in vertebrates. Mol Biol Evol 28: 2949–2959. 10.1093/molbev/msr127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragle JM, Katzman S, Akers TF, Barberan-Soler S, Zahler AM. 2015. Coordinated tissue-specific regulation of adjacent alternative 3′ splice sites in C. elegans. Genome Res 25: 982–994. 10.1101/gr.186783.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Calarco JA, Pan Q, Mavandadi S, Wang Y, Nelson AC, Lee LJ, Morris Q, Blencowe BJ, Zhen M, et al. 2011. Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res 21: 342–348. 10.1101/gr.114645.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, Paten B. 2017. Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods 14: 411–413. 10.1038/nmeth.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2018. R: a language and environment for statistical computing. R; Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Reboul J, Vaglio P, Rual JF, Lamesch P, Martinez M, Armstrong CM, Li S, Jacotot L, Bertin N, Janky R, et al. 2003. C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat Genet 34: 35–41. 10.1038/ng1140 [DOI] [PubMed] [Google Scholar]
- Ren X, Li R, Wei X, Bi Y, Ho VWS, Ding Q, Xu Z, Zhang Z, Hsieh C-L, Young A, et al. 2018. Genomic basis of recombination suppression in the hybrid between Caenorhabditis briggsae and C. nigoni. Nucleic Acids Res 46: 1295–1307. 10.1093/nar/gkx1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JA, Koboldt DC, Staisch JE, Chamberlin HM, Gupta BP, Miller RD, Baird SE, Haag ES. 2011. Caenorhabditis briggsae recombinant inbred line genotypes reveal inter-strain incompatibility and the evolution of recombination. PLoS Genet 7: e1002174 10.1371/journal.pgen.1002174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito TL, Hashimoto S-i, Gu SG, Morton JJ, Stadler M, Blumenthal T, Fire A, Morishita S. 2013. The transcription start site landscape of C. elegans. Genome Res 23: 1348–1361. 10.1101/gr.151571.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M, Pollex T, Hanna K, Lyko F. 2008. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res 37: e12 10.1093/nar/gkn954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W. 2017. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods 14: 407–410. 10.1038/nmeth.4184 [DOI] [PubMed] [Google Scholar]
- Steijger T, Abril JF, Engström PG, Kokocinski F, The RGASP Consortium, Hubbard TJ, Guigó R, Harrow J, Bertone P, Behr J, et al. 2013. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods 10: 1177–1184. 10.1038/nmeth.2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, et al. 2003. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol 1: E45 10.1371/journal.pbio.0000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoiber M, Quick J, Egan R, Lee JE, Celniker S, Neely RK, Loman N, Pennacchio LA, Brown J. 2017. De novo identification of DNA modifications enabled by genome-guided nanopore signal processing. bioRxiv 10.1101/094672 [DOI]
- Tardaguila M, de la Fuente L, Marti C, Pereira C, Pardo-Palacios FJ, del Risco H, Ferrell M, Mellado M, Macchietto M, Verheggen K, et al. 2018. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res 28: 396–411. 10.1101/gr.222976.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilgner H, Jahanbani F, Blauwkamp T, Moshrefi A, Jaeger E, Chen F, Harel I, Bustamante CD, Rasmussen M, Snyder MP. 2015. Comprehensive transcriptome analysis using synthetic long-read sequencing reveals molecular co-association of distant splicing events. Nat Biotechnol 33: 736–742. 10.1038/nbt.3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourasse NJ, Millet JRM, Dupuy D. 2017. Quantitative RNA-seq meta-analysis of alternative exon usage in C. elegans. Genome Res 27: 2120–2128. 10.1101/gr.224626.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyar B, Chu JS, Vergara IA, Chua SY, Jones MR, Wong T, Baillie DL, Chen N. 2012. RNA-seq analysis of the C. briggsae transcriptome. Genome Res 22: 1567–1580. 10.1101/gr.134601.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Regulski M, Tseng E, Olson A, Goodwin S, McCombie WR, Ware D. 2018. A comparative transcriptional landscape of maize and sorghum obtained by single-molecule sequencing. Genome Res 28: 921–932. 10.1101/gr.227462.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hsu PJ, Chen Y-S, Yang Y-G. 2018. Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res 28: 616–624. 10.1038/s41422-018-0040-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura J, Ichikawa K, Shoura MJ, Artiles KL, Gabdank I, Wahba L, Smith CL, Edgley ML, Rougvie AE, Fire AZ, et al. 2019. Recompleting the Caenorhabditis elegans genome. Genome Res 29: 1009–1022. 10.1101/gr.244830.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler AM. 2012. Pre-mRNA splicing and its regulation in Caenorhabditis elegans. WormBook 1–21. 10.1895/wormbook.1.31.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Brown RL, Wei Y, Zhao P, Liu S, Liu X, Deng Y, Hu X, Zhang J, Gao XD, et al. 2019. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev 33: 166–179. 10.1101/gad.319889.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H-Q, Zhang P, Gao H, He X, Dou Y, Huang AY, Liu X-M, Ye AY, Dong M-Q, Wei L. 2015. Profiling the RNA editomes of wild-type C. elegans and ADAR mutants. Genome Res 25: 66–75. 10.1101/gr.176107.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.