

Abstract
Connections between the microbiome and health are rapidly emerging in a wide range of diseases. However, a detailed mechanistic understanding of how different microbial communities are influencing their hosts is often lacking. One method researchers have used to understand these effects are germ-free (GF) mouse models. Differences found within the organ systems of these model organisms may highlight generalizable mechanisms that microbiome dysbioses have throughout the host. Here, we applied multiplexed, quantitative proteomics on the brains, spleens, hearts, small intestines, and colons of conventionally raised and GF mice, identifying associations to colonization state in over 7000 proteins. Highly ranked associations were constructed into protein–protein interaction networks and visualized onto an interactive 3D mouse model for user-guided exploration. These results act as a resource for microbiome researchers hoping to identify host effects of microbiome colonization on a given organ of interest. Our results include validation of previously reported effects in xenobiotic metabolism, the innate immune system, and glutamate-associated proteins while simultaneously providing organism-wide context. We highlight organism-wide differences in mitochondrial proteins including consistent increases in NNT, a mitochondrial protein with essential roles in influencing levels of NADH and NADPH, in all analyzed organs of conventional mice. Our networks also reveal new associations for further exploration, including protease responses in the spleen, high-density lipoproteins in the heart, and glutamatergic signaling in the brain. In total, our study provides a resource for microbiome researchers through detailed tables and visualization of the protein-level effects of microbial colonization on several organ systems.
The gut microbiome is emerging as a critical component of human health. It has been shown that the microbial communities colonizing our bodies play important roles in the immune development of infants (Milani et al. 2017) and the regulation of the innate immune system (Thaiss et al. 2016). Further, a dysbiosis of the gut microbiome has been correlated with many diseases including inflammatory bowel disease (IBD) (Sartor and Wu 2017), diabetes (Tilg and Moschen 2014), obesity (Bouter et al. 2017), cardiovascular disease (Ahmadmehrabi and Tang 2017), and mental health disorders (Nguyen et al. 2018). Microbial production or modification of metabolites such as bile acids, choline derivatives, vitamins, and lipids provide some insight into the underlying host-microbe interactions in these diseases (Nicholson et al. 2012). However, many mechanisms mediating these disease states remain unknown.
Germ-free (GF) mouse models, wherein a mouse is raised without any exposure to microbes, have been an invaluable tool for assessing causal effects in microbiome research (Bhattarai and Kashyap 2016). GF models also provide an opportunity to understand the fundamental effects of microbial colonization at an organismal scale. Systems level analyses of the tissues of GF mice have been performed, but these studies have generally highlighted a select few organ tissues. Protein-level studies have shown varying responses to colonization along different regions of the gastrointestinal (GI) tract (Lichtman et al. 2016), changes in drug metabolizing proteins in livers and kidneys (Kuno et al. 2016), and differences in circulating fatty acids from an analysis of serum and livers (Kindt et al. 2018). They have also shown that microbial colonization alters posttranslational modifications, including histone acetylation and methylation in liver, colon, and adipose tissue (Krautkramer et al. 2016), as well as lysine acetylation in the gut and liver (Simon et al. 2012). An important transcriptomic study revealed a strong connection between colonization and increased Nnt, a mitochondrial protein that has functions in redox homeostasis and biosynthetic pathways through the generation of NADH and NADP+ (Mardinoglu et al. 2015). The authors found Nnt transcripts increased in several conventional mouse tissues, including sections of the small intestine, colon, and liver, which correlated with significant alterations in host amino acid levels and glutathione metabolism (Mardinoglu et al. 2015). Other related studies found transcript differences in the brain (Diaz Heijtz et al. 2011) and further highlighted the GI-dependent transcript effects of microbial colonization in Myd88-deficient mice (Larsson et al. 2012).
Here, we sought to further detail the protein effects of microbiota colonization occurring both inside and outside of the GI tract as a reference for microbiome researchers interested in a given host protein, organ system, or protein network. Associations of highly ranked proteins were constructed into protein–protein interaction networks for the brain, spleen, heart, small intestine, and colon, as well as a global network. While the brains and gastrointestinal tract of GF mice have been characterized in several studies, other organs such as the heart and spleen may be of interest given the emerging roles of the microbiota in atherosclerosis (Karlsson et al. 2012) and immune development (Chung et al. 2012). We hypothesized that applying methods for improved accuracy in quantitative proteomics (Ting et al. 2011) would further define the influence of the microbiota in each of these organs. With this, we hope to reveal microbiota-induced changes that could underlie disease states. Our results validate a body of literature in the field, provide visualization tools for contextualizing the organism-wide effects of microbial colonization, and identify several new host-microbiota associations for further investigation.
Results
Construction of protein–protein interaction networks
To determine the protein-level consequences of microbial colonization, three biological replicates of five different tissues (brain, small intestine, colon, spleen, heart) were analyzed from conventionally raised or GF mice. Multiplexed proteomic analysis of tissue homogenates resulted in the quantification of 7752 proteins overall, of which 4663 were quantified across all samples. These 4663 proteins were used for downstream analysis. A separate pilot study of the brain tissue resulted in the quantification of 6203 proteins.
On a per-organ basis, we contrasted the protein abundances of tissue collected from conventional mice against tissue from GF mice. We ranked the association of each protein to colonization state by accounting for both significance level and fold-change (Supplemental Table S1). Interaction networks were built in order to identify groups of proteins whose abundance was modulated by microbial colonization. We first constructed organ-specific protein interaction networks containing all the proteins with a highly ranked (|π| > 1) association with the colonization state of a given organ (Fig. 1). Functional enrichments within these organ networks were then assessed by gene-set enrichment analysis (Fig. 2; Supplemental Table S2). To identify overlapping mechanisms occurring throughout all organs, we compiled all highly ranked proteins within each organ (|π| > 1) into a single protein network (Fig. 3).
Figure 1.

Organ-specific protein networks modulated by microbial colonization. Proteins significantly increased in either GF or conventional animals were analyzed for interactions through STRINGdb. Edges in each node represent the combined score accounting for all interaction sources. The edges are sized by the combined score, with the minimum threshold being 0.4 (of a maximum confidence 1). Nodes represent gene names of significant proteins with a minimum statistical cutoff of |π| > 1. Red indicates a significantly higher presence in conventional mice and gray indicates the opposite. The nodes are sized by the level of significance as assessed by π-score.
Figure 2.

Functional enrichments associated with microbial colonization within each organ system. Proteins significantly increased in either GF or conventional animals within each organ were analyzed for functional enrichments using DAVID. All proteins identified within the experiment were used as a background. Displayed are bar plots showing the −Log10(adj. P-values) associated with selected functional groupings. Benjamini–Hochberg correction was applied to account for multiple hypothesis testing. The bars are plotted in red if they are associated with the proteins enriched among conventional mice and gray if they are associated with GF mice.
Figure 3.

Combined organ protein networks modulated by microbial colonization. Proteins significantly increased in either GF or conventional animals were analyzed for interactions through STRINGdb. Edges in each node represent the combined score accounting for all interaction sources. The edges are sized by the combined score, with the minimum threshold being 0.8 (of a maximum confidence 1). Nodes represent gene names of proteins with a highly ranked association (a minimum statistical cutoff of |π| > 1) within at least one organ. Nodes are sized by the number of organs with which the protein had a strong association. The level of association of each node to a particular organ is colored according to the fraction of the total |π|-score each organ contributes. Below each node is a bar plot of the π-scores for each organ within the node. Putative functional groupings within the network are highlighted. Select sections are highlighted in colored boxes and shown in 2× zoom.
Colonization state of the mouse appeared to have larger impacts on organs in direct contact with the gut microbiota, namely, the small intestine and colon. The GI organs analyzed had an average of 210 proteins associated with colonization state while the three organs outside of the GI tract averaged 52. The brain yielded the lowest number of associated proteins with only 22. GI tract organs also displayed a higher percentage of interconnectivity, with an average of 71% of associated proteins within GI organs having a moderate-confidence connection to another associated protein within the organ, while organs outside the GI tract averaged 39% (Fig. 1). We hypothesize that the interconnectivity and number of associations with colonization is related to the direct contact of GI organs with microbiota. However, it is possible that including a higher portion of intestinal tissue may have influenced these results.
Validation of protein networks through previously reported associations
Our networks provided support for previously reported broad-scale effects on GI organs, as well as abundance shifts from specific transcripts or proteins. As shown in a previous proteomic study (Lichtman et al. 2016), the small intestine and colon displayed distinct changes as a result of microbial colonization. One example of this was the larger portion of proteins associated with GF status within the small intestine (66%) than in the colon (42%) (Fig. 1).
Other studies identified similar results at a pathway and individual protein level. After a literature search for protein or transcript differences within GF models, we identified seven publications with related findings (Supplemental Table S3). In brief, changes in xenobiotic metabolism were reported in the GI, liver, and kidney, both at the transcriptional level (Fu et al. 2017) and the protein level (Kuno et al. 2016). Our networks also highlighted changes in glutamate-related proteins which were previously reported at the transcriptional level (El Aidy et al. 2013). We also report the regulation of innate immune proteins including the antimicrobial peptide REG3G (Larsson et al. 2012) and the regulation of NNT (Mardinoglu et al. 2015), which will be discussed further below.
Organ-specific network results
Functional enrichment analysis of GI tract organs resulted in stronger and more diverse associations with colonization status than organs outside the GI tract. Several enrichments emerged with potential links to redox shifts in the small intestine. These included disulfide bonds, oxidoreductase activity, NADP, and xenobiotic metabolism through cytochrome P450s (Figs. 2, 3). Functional differences in the colon highlighted pancreatic secretion, immunoglobulins, proteins of the heat shock protein 70 family, digestion, and stress as the functions increased in conventional mice (Figs. 2, 3). GF colons had enrichments for transporter activity, Calycin, fatty-acid binding, metalloproteases, and peroxisome proliferator-activated receptor (PPAR) signaling (Fig. 2). Together, these results may indicate stress response as an important factor mediating host-microbiota interactions in the colon and redox states influencing interactions of the small intestine.
Organs outside of the GI tract have been less studied in regard to their regulatory responses to the microbiome. Within the spleen, proteins increased in conventional mice were found to be part of a highly connected functional protein network consisting primarily of pancreatic digestive enzymes (CELA2A, CELA3B, CELA1, CPA1, CPA2, CTRB1, CTRL, CTSE, TRY5, etc.), iron-binding proteins (FST1, TFRC, STEAP3, FECH, LTF, and HP) and innate immune mediators (LCN2, S100A9, NGP, ITGAM, and CHIL3) (Figs. 1, 3). Many of the digestive enzymes have roles in the GI tract and were similarly associated to colonization state within the small intestine and colon (Fig. 3). Iron-binding and immune proteins were similarly regulated in other organs (Fig. 3), but the differential regulation of LTF (Lactotransferrin) and FSLT1 (Follistatin-related protein 1) were primarily restricted to the spleen. Given the biological roles of the spleen, the influence of these proteins in mediating immune processes may be of significant interest.
The networks associated with the brain and the heart displayed fewer interconnected proteins. Only 34% and 27% of proteins had a connection within the heart network and brain network, respectively. However, a group of primarily GF-associated proteins that included APOA1 and APOE was found among the heart proteins (Fig. 1). These results may suggest changes in lipid profiles in the heart: increased high-density lipoproteins (HDL) and chylomicrons in GF compared to conventional mice. The brain showed limited functional enrichments (Supplemental Table S2). However, both RASGRF1 and RASGRF2 were increased among conventional mice. These proteins may be of interest given implications in glutamatergic excitatory synaptic signaling and in facilitating long-term potentiation leading to enhanced memory, learning, and synaptic plasticity (Drake et al. 2011; Schwechter et al. 2013).
Organism-wide network results
One prominent finding from our generalized network was a significant increase in NNT in all conventional tissues. This protein is a key regulator of generalized biosynthetic processes and is related to glutamate synthesis (Mardinoglu et al. 2015). Statistically, NNT was among the strongest relationships found within all organs analyzed (π = 3.8, 4, 2.3, 7, and 3.7 for small intestine, colon, spleen, heart, and brain, respectively). We also identified subnetworks related to mitochondrial glutamate metabolism and mitochondrial respiratory chain NADH dehydrogenase, with most of the related proteins down-regulated in the small intestine of conventional mice (Fig. 3). In addition, proteins relating to the mitochondrial reduction of glutathione, GLUD1, and GLS had confirmed associations with previous transcriptomic analysis (Supplemental Table S3). While this subnetwork was largely derived from small intestine proteins, there was evidence that this system may also be affected in the brain through AMT, which is involved in the mitochondrial metabolism of glycine.
The protein networks identified generalized differences among all organs related to the innate and adaptive immune systems. Proteins related to innate immunity tended to be increased in conventional mice, while proteins associated with neutrophil degranulation were associated with GF mice (Fig. 3). REG3G, a protein associated with toll-like receptor (TLR) signaling subsequent to pathogen-associated molecular pattern (PAMP) activation in Paneth cells, was increased among conventional mice. Additionally, proteins related to antigen processing were moderately enriched in GF mice.
We next evaluated protein relationships to GF status at an organismal level by applying a compositionally aware multinomial regression technique. This technique accounts for organ type to assess organism-wide protein associations through ranks. Our top-ranked protein associated with conventional status was NNT, while the protein with the strongest association with GF status was IGKV5-39, a protein involved in immune response and immunoglobulin production (Fig. 4A). As observed from the traditional statistical approach, NNT was significantly higher in conventional mice within all organs included in the regression (Fig. 4B). IGKV5-39 was more strongly associated with the spleen, colon, and heart than the small intestine (Fig. 4B).
Figure 4.

Organism-level protein networks modulated by the microbiome. A multinomial regression controlling for organ and microbial colonization state of the mice was used to assess proteins associated with colonization status. (A) Proteins ranked by regression coefficient; proteins with coefficients of the greatest magnitude are most associated with colonization status. Proteins with positive coefficients are more abundant in conventional mice, while proteins with negative coefficients are more abundant in GF mice. (B) Log abundance of NNT or IGKV5-39 over the entire proteome in each organ. (C) Protein–protein interaction networks from the top-ranked proteins from the multinomial regression associated with both conventional and GF status when controlling for organ and mouse. The top 150 proteins associated with both GF and conventional status were analyzed (300 proteins total), and proteins with high confidence interactions (0.8) are shown. Nodes are sized by the absolute value of the regression coefficient and colored by association with GF (gray) or conventional (red) status. Putative functional groupings are indicated.
Next, we assessed the 150 top- and bottom-ranked proteins associated with GF status from the multinomial regression and created a protein–protein interaction network (Fig. 4C). Of interest was a cluster of proteins related to redox states in mitochondria. Several proteins including ECSIT, NDUFS4, NUBPL, NDUFA11, NDUFB6, NDUFA8, and ATPAF1 are all related to mitochondrial complex 1 of the electron transport chain. Other evidence of the organism-wide impact of microbial colonization on mitochondria included shifts in mitochondrial ribosomal proteins (MRPS23, MRPL50, MRPS17, MRPL49, and MRPL17) and proteins related to mitochondrial heat shock response (HSPD1, HSPE1, SOD2, LONP1). The data presented here suggest that microbial colonization may have organismal-level impacts on mitochondrial function.
Interactive 3D visualization of associations with colonization status
To encourage user interaction with our data, we created a web-based display of our results projected onto a 3D model of a mouse. This interactive display allows for user-guided exploration of the protein and pathway-level associations with colonization status. To access the model, users should access the ‘ili web server (https://ili.embl.de/), then drag-and-drop the provided texturization file (Supplemental File S1) and a supplemental table for either the proteins (Supplemental Table S4) or pathways (Supplemental Table S5) identified in this study. With this tool, users can search for proteins and pathways of interest and project the association scores onto all the organs analyzed in this study.
For the protein-level visualization, each protein is listed by the protein name with any Gene Ontology (GO) molecular function terms associated with the protein. With this, users are able to search for a protein of interest or identify proteins of interest by molecular functions. The ‘ili-compatible table of association scores (Supplemental Table S4) uses the π-statistic for conventional/GF status. These scores range from 6.98 (significantly increased in conventional mice) to −5.11 (significantly increased in GF mice). An example use case is displayed in Figure 5A, which depicts the strong association with conventional status for NNT within all organs.
Figure 5.
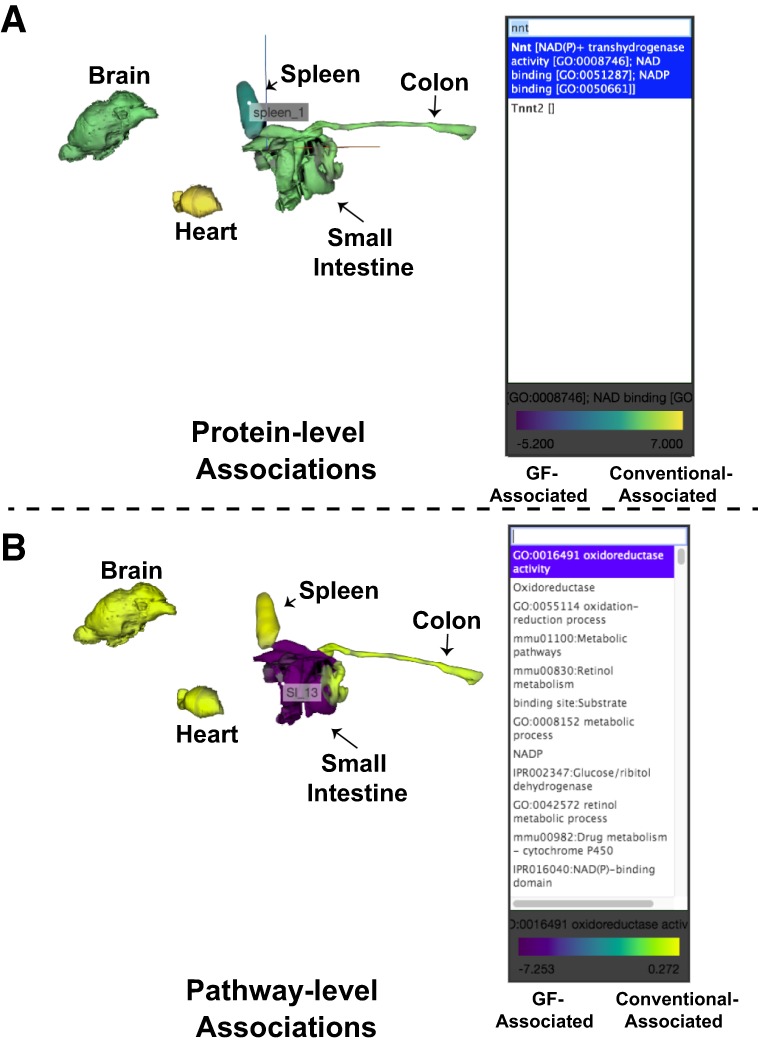
Interactive 3D visualization of associations with colonization status. A 3D mouse model was generated for use on the web-based ‘ili platform (https://ili.embl.de/). (A) An example use case for the protein-level association visualizations shown through plotting the π-score enrichment for conventional colonization status. (B) An example use case for the pathway level association visualizations shown through highlighting the enrichment scores for “Oxidoreductase.” Pathway association scores were generated through –Log10(Benjamini–Hochberg corrected P-values) of the conventional organs minus the GF organs (from Supplemental Table S2).
The pathway-level visualizations help to summarize and explore the gene set enrichment analyses. On a per-organ basis, an association with conventional or GF status for each functional category was calculated by comparing the statistical strength of gene set enrichments (Supplemental Table S2). These association scores have been summarized in an ‘ili-compatible file in Supplemental Table S5. Pathway association scores ranged from 12.90 (highly associated with conventional status) to −7.25 (highly associated with GF status). We demonstrate the use of this tool to visualize the associations with “Oxidoreductase” in Figure 5B.
Discussion
Our multiorgan analysis was utilized to generate protein interaction maps and 3D visualization tools that researchers can use to understand organ-specific and organism-wide changes that may underlie host-microbiome interactions. From our networks, we can identify common themes found from previous analyses of GF tissue. For example, we found changes in innate and adaptive immune responses to microbial colonization (Larsson et al. 2012), changes in the glutamine and glutamate pathway (El Aidy et al. 2013; Mardinoglu et al. 2015), and changes in xenobiotic degradation pathways (Kuno et al. 2016; Fu et al. 2017; Kindt et al. 2018). This study contributes to the field by moving toward an organism-wide understanding of host-microbiome interactions. Here, we incorporate new statistical and visualization tools for multiorgan analysis and include understudied organs from outside of the GI tract. As roles for the microbiome are expanding into immunity (Thaiss et al. 2016), cardiovascular disease (Ahmadmehrabi and Tang 2017), and mental health disorders (Nguyen et al. 2018), defining the influence of the microbiome on these tissues may be of importance. Indeed, our networks provided several putative organism-level roles for the microbiome.
Our protein networks highlight the potential of organism-wide redox state changes being linked to the microbiome. This is perhaps best highlighted in the distal increases of NNT associated with microbial colonization. NNT is a mitochondrial protein with well-described roles in glutathione redox reactions through the conversion of NADPH to NADP+ (Ronchi et al. 2013). Glutathione is interconverted with glutathione disulfide, collectively representing the most abundant redox pair in the body (Circu and Aw 2011). Abundances of this redox pair are often used as an indication of the general redox state (Circu and Aw 2011), which is involved in a large variety of biological processes (Circu and Aw 2011; Birben et al. 2012; Ray et al. 2012). Evidence of intestinal redox differences in GF animals has been known since the 1970s (Koopman et al. 1975; Celesk et al. 1976), and evidence of microbiota-dependent regulation of Nnt in the GI tract has been previously described (Mardinoglu et al. 2015). Here, we find evidence of increased NNT throughout the entire conventional mouse. Additionally, both the traditional and multivariate statistical methods used for the identification of organism-wide effects found networks of proteins related to mitochondria, including mitochondrial complex 1 proteins. Mitochondrial complex 1 activity might also be linked to redox states as complex 1 activity was shown to be dependent on glutathione transport into the mitochondria (Kamga et al. 2010). In summary, many of our protein networks may have been influenced by redox states, including shifts in mitochondrial proteins, the innate immune processes such as neutrophil degranulation, and degradation of xenobiotics through cytochrome P450s (Kramer and Darley-Usmar 2015).
Though our strongest relationships to microbial colonization were found within the GI tract, which was, unsurprisingly, the focus of most previous GF organ analyses (Larsson et al. 2012; El Aidy et al. 2013; Mardinoglu et al. 2015; Lichtman et al. 2016), the analyses of the brain, spleen, and heart did yield several interesting results. Within the heart, our analyses indicated a relationship to HDL and the GF state. Our results within the heart may be of importance given the increasing literature regarding microbiota regulation of lipids and lipoproteins including HDL (Nakaya and Ikewaki 2018), as well as the microbial links to metabolites leading to atherosclerosis (Wang et al. 2011b). Our findings may indicate a mechanism in which microbiota influence the makeup of the heart through changes in lipoproteins.
The data of the spleen proteomes revealed networks of proteases with similar increases within the small intestine and colon in the presence of the microbiota. These networks also suggested potential links between the gut microbiota and pancreatic secretion. Pancreatic secretion of enzymes is thought to be largely regulated through circulating hormones (Singh and Webster 1978). The gut microbiome has several potential links to pancreatitis and pancreatic cancer, which may be mediated through sensing of microbial compounds such as lipopolysaccharide through TLR4 (Leal-Lopes et al. 2015). Here, we have observed a link between microbial colonization and increased pancreatic secretion of digestive proteins, which may be of interest for further investigation.
The organism-wide effects of microbial colonization illustrated in our networks may have implications in several disease states. Dysregulation of complex 1 has been implicated in microbiome-related diseases, including ulcerative colitis (Haberman et al. 2019), and there is accumulating evidence of mitochondrial dysfunction in Crohn's disease (Mottawea et al. 2016). This association between the microbiome and mitochondria are thought to be mediated through three key microbiome metabolites: short-chain fatty acids, the urolithins, and lactate (Franco-Obregón and Gilbert 2017). While speculative, it is possible that the IBD microbiota may influence these interactions. Glutamatergic signaling has been suggested as a potential target for treating mood disorders (Zarate et al. 2010). Our identification of proteins in the brain related to glutamatergic signaling and glutamate (i.e., RASGRF proteins and NNT) may be of relevance to the discussions surrounding utilization of the microbiome to treat mood disorders (Mangiola et al. 2016).
There are likely many unknown mechanisms mediating host-microbiota interactions. Our detailed maps and visualization tools for understanding the organ-level impacts of microbial colonization give insight into the unique and common protein-level changes occurring throughout mice. These networks suggest changes throughout the mice related to mitochondrial dysfunction and redox states. Though fewer changes were found in organs apart from the GI tract, our protein networks of the spleen, brain, and heart may provide insight for researchers establishing connections between the microbiota and diseases related to these organ systems. We hope these networks and visualization tools may be useful in the microbiome research community to help dissect the specific effects that microbial communities have in a given organ system, protein, or protein network of interest. In total, we view our study as a step toward better understanding the role of the microbiota in health and disease.
Methods
Gnotobiotic mice
Three male GF C57BL/6 mice were kept under GF conditions in a Park Bioservices isolator in Georgia State University's (GSU's) GF facility, and three male conventional C57BL/6 were kept in regular housing at GSU's animal facility. At 5 mo of age, mice were euthanized and organs were collected followed by immediate snap-freezing. All mice were bred and housed at Georgia State University, Atlanta, Georgia, USA, under institutionally approved protocols (IACUC # A14033).
Protein digestion and TMT labeling
All organ proteome methods were performed as previously described (Lapek et al. 2018). Snap-frozen organs (stored at −80°C beforehand) were suspended in PBS and homogenized using a Mini BeadBeater (Biospec). Our final analysis contained three biological replicates of each colonization condition per organ, and no technical replicates were collected given strong correlation observed between biological replicates in our past work (Lapek et al. 2018). Organ homogenates were lysed in 1 mL of buffer composed of 75 mM NaCl (Sigma-Aldrich), 3% sodium dodecyl sulfate (SDS, Thermo Fisher Scientific), 1 mM NaF (Sigma-Aldrich), 1 mM beta-glycerophosphate (Sigma-Aldrich), 1 mM sodium orthovanadate (Sigma-Aldrich), 10 mM sodium pyrophosphate (Sigma-Aldrich), 1 mM phenylmethylsulfonyl fluoride (PMSF, Sigma-Aldrich), and Complete Mini EDTA-free protease inhibitors (1 tablet per 10 mL, Roche) in 50 mM HEPES (Sigma-Aldrich), pH 8.5 (Villén and Gygi 2008). To ensure full lysis, homogenates were passed through a 21-gauge syringe 20 times. Insoluble debris was then pelleted by centrifugation for 5 min at 14,000 rpm. Supernatants were transferred to new tubes, and an equal volume of 8 M urea in 50 mM HEPES, pH 8.5 was added to each sample. Samples were then vortexed and further lysed through two 10-sec intervals of probe sonication at 25% amplitude.
Proteins were reduced with dithiothreitol (DTT, Sigma-Aldrich) and alkylated with iodoacetamide (IAA, Sigma-Aldrich) (Haas et al. 2006). Proteins were next precipitated via methanol-chloroform precipitation (Wessel and Flügge 1984). Precipitated proteins were resolubilized in 300 µL of 1 M urea (Thermo Fisher Scientific) in 50 mM HEPES, pH 8.5. Proteins were digested in a two-step digestion process. First, 3 µg of LysC (Wako) was added to each sample, and samples were digested overnight at room temperature. Next, 3 µg of trypsin was added, and samples were digested for 6 h at 37°C. Digests were acidified with Pierce Trifluoroacetic Acid (TFA, Thermo Fisher Scientific) to quench the digestion reaction. Peptides were desalted with C18 Sep-Paks (Waters) as previously described (Tolonen and Haas 2014). Concentration of desalted peptides was determined using a Pierce Quantitative Colorimetric Peptide Assay (Thermo Fisher Scientific), and peptides were aliquoted into 50-µg portions. Aliquots were dried under vacuum and stored at −80°C until they were labeled with TMT reagents.
Peptides were labeled with 10-plex TMT reagents (Thermo Fisher Scientific) (Thompson et al. 2003; McAlister et al. 2014) as previously described (Wang et al. 2011a). TMT reagents were reconstituted in dry acetonitrile (Sigma-Aldrich) at 20 µg/µL. Dried peptides were resuspended in 30% dry acetonitrile in 200 mM HEPES, pH 8.5, and 7 µL of the appropriate TMT reagent was added to peptides. Reagents 126 and 131 (Thermo Fisher Scientific) were used to label peptide aliquots composed of an equal concentration of every sample within all mass spectrometry (MS) runs. These composite samples acted as a reference within all mass spectrometry runs for data normalization purposes described later. Remaining reagents were used to label samples in random order with no bias regarding animal of origin, organ, or colonization status. This randomization was performed to prevent known batch-effects in mass spectrometry experiments (Brenes et al. 2019). The brain samples were analyzed as a pilot experiment before the other organs. For brains, all procedures were performed as above, though the TMT experiment was separate from other organs. Within the brain TMT experiment, one channel, 129C, consisted of a 50-µg average of all peptides from brain samples. Labeling was carried out for 1 h at room temperature and was quenched by adding 8 µL of 5% hydroxylamine (Sigma-Aldrich). TMT-labeled peptides from each of the organ samples were acidified by adding 50 µL of 1% TFA and subsequently combined into a composite sample per TMT 10-plex experiment. During the pilot experiment with the brains, the pooled samples were desalted and fractionated using a Pierce High pH Reversed-Phase Peptide Fractionation kit (Thermo Fisher Scientific) per the manufacturer's instructions. For the larger study, samples were pooled per 10-plex experiment, desalted with C18 Sep-Paks, and further fractionated as described below.
Collection of LC-MS2/MS3 spectra for protein identification and quantification
Data acquisition methods were performed as previously described (Lapek et al. 2018). Sample fractionation, excluding the brain experiment, was performed by basic pH reverse-phase liquid chromatography with concatenated fractions as previously described (Wang et al. 2011a). Briefly, samples were resuspended in 5% formic acid/5% acetonitrile and separated over a 4.6 mm × 250 mm C18 column (Thermo Fisher Scientific) on an Ultimate 3000 HPLC fitted with a fraction collector, degasser, and variable wavelength detector. The separation was performed over a 22% to 35%, 60-min linear gradient of acetonitrile in 10 mM ammonium bicarbonate (Thermo Fisher Scientific) at 0.5 mL/min. The resulting 96 fractions were combined as previously described (Wang et al. 2011a). All fractions were dried under vacuum and resuspended in 5% formic acid/5% acetonitrile and analyzed by liquid chromatography (LC)-MS2/MS3 for identification and quantitation.
All LC-MS2/MS3 experiments were carried out on an Orbitrap Fusion (Thermo Fisher Scientific) with an in-line Easy-nLC 1000 (Thermo Fisher Scientific) and chilled autosampler. Home-pulled, home-packed columns (100 µm ID × 30 cm, 360 µm OD) were used for analysis. Analytical columns were triple-packed with 5 µm C4 resin, 3 µm C18 resin, and 1.8 µm C18 resin (Sepax) to lengths of 0.5, 0.5, and 30 cm, respectively. Peptides were loaded at 500 bar and eluted with a linear gradient of 11% to 30% acetonitrile in 0.125% formic acid over 165 min at a flow rate of 300 nL/min, with the column heated to 60°C. Nano-electrospray ionization was performed by applying 2000 V through a stainless-steel T-junction at the inlet of the microcapillary column.
The mass spectrometer was run in data-dependent mode, where a survey scan was performed over 500–1200 m/z at a resolution of 60,000 in the Orbitrap. Automatic gain control (AGC) was set to 2 × 105 for MS1 with a maximum ion injection time of 100 ms. The S-lens RF was set to 60, and centroided data was collected. Top-N mode was used to select the most abundant ions in the MS1 scan for MS2 and MS3 with N set to 10.
The decision tree option was used for MS2 analysis, using charge state and m/z range as qualifiers. Ions carrying two charges were analyzed from the m/z range of 600–1200, and ions carrying three and four charges were selected from the m/z range of 500–1200. An ion intensity threshold of 5 × 104 was used. MS2 spectra were obtained using quadrupole isolation at a 0.5 Th window and fragmented using Collision Induced Dissociation with a normalized collision energy of 30%. Fragment ions were detected and centroided data collected in the linear ion trap using rapid scan rate with an AGC target of 1 × 104 and maximum ion injection time of 35 ms.
MS3 analysis was performed using synchronous precursor selection (SPS) enabled to maximize TMT quantitation sensitivity (McAlister et al. 2014). A maximum of 10 MS2 precursors was specified for the SPS setting, which were simultaneously isolated and fragmented for MS3 analysis. Higher-Energy Collisional Dissociation fragmentation was used for MS3 analysis with a normalized collision energy of 55%. Resultant fragment ions (MS3) were detected in the Orbitrap at a resolution of 60,000 with a low mass cut-off of 110 m/z. AGC for MS3 spectra was set to 1 × 105 with a maximum ion injection time of 100 ms. Centroided data were collected, and MS2 ions between the range of 40 m/z below and 15 m/z above the precursor m/z were excluded by SPS.
Data processing and normalization
Data were processed using Proteome Discover 2.1 (Thermo Fisher Scientific). MS2 data were searched against UniProt mouse databases (downloaded 7/2/2018 and 5/11/2017 for the brain analysis and other organs, respectively) using the SEQUEST algorithm (Eng et al. 1994). A decoy search was also conducted with sequences in reverse order (Peng et al. 2003; Elias et al. 2005; Elias and Gygi 2007). A precursor mass tolerance of 50 ppm (Beausoleil et al. 2006; Huttlin et al. 2010) was specified and 0.6 Da tolerance for MS2 fragments. Static modification of TMT 10-plex tags on lysine and peptide n-termini (+229.162932 Da) and carbamidomethylation of cysteines (+57.02146 Da) were specified. Variable oxidation of methionine (+15.99492 Da) was also included in search parameters. Data were filtered to 1% peptide and protein level false discovery rates with the target-decoy strategy through Percolator (Käll et al. 2007; Spivak et al. 2009).
TMT reporter ion intensities were extracted from MS3 spectra for quantitative analysis, and signal-to-noise values were used for quantitation. Spectra were filtered and summed as previously described (Lapek et al. 2017b). Data were normalized in a multistep process, whereby they were first normalized to the pooled standards (TMT-126 and -131) for each protein and then to the median signal across the pooled standards from all experiments (Lapek et al. 2017a). An average of these normalizations was used for the next step. As the brain samples did not contain a composite bridge channel for normalization, raw signal/noise ratios were normalized by the average signal of the given protein divided by the median of all protein averages. To account for slight differences in amounts of protein labeled, these values were then normalized to the median of the entire data set and reported as final normalized summed signal-to-noise ratios per protein per sample.
Statistical analysis
Bioinformatic analysis was performed in Python (version 3.5.1), and records are available online in Jupyter Notebooks (https://github.com/rhmills/Germ-free-organ-proteomics). To prevent statistical artifacts generated due to the various methods of dealing with missing values, only proteins with quantification in all samples were used. A Student's t-test with unequal variance was performed through the package, SciPy (https://www.scipy.org). For ranking purposes, we evaluated associations with colonization state through π-score, which accounts for both fold-change and P-value (Xiao et al. 2014). A statistical cutoff of |π| > 1 was chosen based on previous work (Tran et al. 2019). This statistical measure corresponds to a significance level of α ∼ 0.05 and allowed for moderate stringency while including an adequate number of proteins for protein network construction and functional enrichment analysis.
Protein–protein interaction networks were created through STRINGdb (Szklarczyk et al. 2015). Associations between proteins were determined through default settings, accounting for text mining, experiments, databases, coexpression, neighborhood, gene fusion, and co-occurrence. Connections were restricted to interactions between proteins within the query list only. Networks were subsequently visualized through Cytoscape (version 3.5.1) (Shannon et al. 2003). Edges within protein networks were based on the combined evidence scores, with thicker edges indicating higher confidence. Per-organ networks were performed with a medium minimum confidence (0.4) to visualize connectivity through maximizing potential connections, while combined organ networks utilized a high minimum confidence (0.8) to identify putative functional groupings.
Functional enrichment analysis was performed through the DAVID server (Huang da et al. 2009a,b) to identify significant groups of proteins per organ, split between GF and colonized states. Parameters were set as previously described (Nicolay et al. 2015). Benjamini–Hochberg corrected P-values were reported for the most significant groupings. Bar plots were visualized through GraphPad Prism (version 7.0b).
Songbird (Morton et al. 2019) was used to implement the multinomial regression analysis with organ, mouse, and colonization status used in the regression formula. Model parameters were as follows: 10,000 epochs, batch size of 5, differential prior of 1.0, learning rate of 0.001, gradient clipping size of 10, and proteins with >5 counts were included. As the brain tissue was processed as an independent pilot study, we could not include this data as part of the multinomial regression analysis given the independent normalization used in each experiment.
3D mouse model
The 3D mouse model was generated as described previously (Quinn et al. 2019). In brief, MRI images were acquired at the UCSD Center for Functional MRI from a euthanized 8-wk-old female C57BL/6 mouse with a Bruker 7T/20 MRI scanner using a quadrature birdcage transceiver. MRI parameters were as follows: 3D FLASH protocol with TE/TR = 6 ms/15 ms and matrix size 128 × 64 × 156, field of view prescribed to match the body size. InVesalius (https://link.springer.com/chapter/10.1007/978-3-319-27857-5_5) was used to trace individual organs in each MRI slice to generate the 3D model. The model was then processed with Blender (https://www.blender.org/) for smoothing.
Interactive display of associations can be done through the following steps: (1) accessing the ‘ili-web browser (https://ili.embl.de) (Protsyuk et al. 2018); (2) uploading the 3D mouse model visualization file (Supplemental File S1); (3) uploading either the protein-based associations table (Supplemental Table S4) or the pathway-based associations table (Supplemental Table S5).
Pathway-level association scores were determined by comparing the −Log10(Benjamini–Hochberg corrected P-values) for each functional enrichment in Supplemental Table S2. The statistical strength of the functional enrichment in GF tissue was subtracted from the statistical strength of the functional enrichment in conventional tissue. The following formula summarizes the calculation:
Data access
The proteomic data generated in this study have been submitted to the Mass Spectrometry Interactive Virtual Environment (MassIVE) Repository (https://massive.ucsd.edu) under the study ID MSV000083874. Code is available through GitHub (https://github.com/rhmills/Germ-free-organ-proteomics) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
D.J.G. was supported by the Ray Thomas Edwards Foundation and the UC Office of the President. R.H.M. was supported by UCSD's Gastroenterology T32 Predoctoral fellowship. A.C. was supported by the UCSD Microbial Sciences Initiative Graduate Research Fellowship and by the UCSD Graduate Training Program in Cellular and Molecular Pharmacology through an institutional training grant from the National Institute of General Medical Sciences, T32 GM007752.
Author contributions: R.H.M., A.V., R.K., A.G., and D.J.G. conceived and designed the experiments. R.H.M., B.C., and A.V. performed the experiments. R.H.M., J.M.W., A.V., and A.C. analyzed the data. R.H.M., J.M.W., A.C., A.V., and D.J.G. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.256875.119.
References
- Ahmadmehrabi S, Tang WHW. 2017. Gut microbiome and its role in cardiovascular diseases. Curr Opin Cardiol 32: 761–766. 10.1097/HCO.0000000000000445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil SA, Villén J, Gerber SA, Rush J, Gygi SP. 2006. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24: 1285–1292. 10.1038/nbt1240 [DOI] [PubMed] [Google Scholar]
- Bhattarai Y, Kashyap PC. 2016. Germ-free mice model for studying host-microbial interactions. Methods Mol Biol 1438: 123–135. 10.1007/978-1-4939-3661-8_8 [DOI] [PubMed] [Google Scholar]
- Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. 2012. Oxidative stress and antioxidant defense. World Allergy Organ J 5: 9–19. 10.1097/WOX.0b013e3182439613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouter KE, van Raalte DH, Groen AK, Nieuwdorp M. 2017. Role of the gut microbiome in the pathogenesis of obesity and obesity-related metabolic dysfunction. Gastroenterology 152: 1671–1678. 10.1053/j.gastro.2016.12.048 [DOI] [PubMed] [Google Scholar]
- Brenes A, Hukelmann J, Bensaddek D, Lamond AI. 2019. Multibatch TMT reveals false positives, batch effects and missing values. Mol Cell Proteomics 18: 1967–1980. 10.1074/mcp.RA119.001472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celesk RA, Asano T, Wagner M. 1976. The size pH, and redox potential of the cecum in mice associated with various microbial floras. Proc Soc Exp Biol Med 151: 260–263. 10.3181/00379727-151-39187 [DOI] [PubMed] [Google Scholar]
- Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al. 2012. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149: 1578–1593. 10.1016/j.cell.2012.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Circu ML, Aw TY. 2011. Redox biology of the intestine. Free Radic Res 45: 1245–1266. 10.3109/10715762.2011.611509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S. 2011. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci 108: 3047–3052. 10.1073/pnas.1010529108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake NM, DeVito LM, Cleland TA, Soloway PD. 2011. Imprinted Rasgrf1 expression in neonatal mice affects olfactory learning and memory. Genes Brain Behav 10: 392–403. 10.1111/j.1601-183X.2011.00678.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Aidy S, Derrien M, Merrifield CA, Levenez F, Dore J, Boekschoten MV, Dekker J, Holmes E, Zoetendal EG, van Baarlen P, et al. 2013. Gut bacteria-host metabolic interplay during conventionalisation of the mouse germfree colon. ISME J 7: 743–755. 10.1038/ismej.2012.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4: 207–214. 10.1038/nmeth1019 [DOI] [PubMed] [Google Scholar]
- Elias JE, Haas W, Faherty BK, Gygi SP. 2005. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods 2: 667–675. 10.1038/nmeth785 [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5: 976–989. 10.1016/1044-0305(94)80016-2 [DOI] [PubMed] [Google Scholar]
- Franco-Obregón A, Gilbert JA. 2017. The microbiome-mitochondrion connection: common ancestries, common mechanisms, common goals. mSystems 2: e00018 10.1128/mSystems.00018-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu ZD, Selwyn FP, Cui JY, Klaassen CD. 2017. RNA-seq profiling of intestinal expression of xenobiotic processing genes in germ-free mice. Drug Metab Dispos 45: 1225–1238. 10.1124/dmd.117.077313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas W, Faherty BK, Gerber SA, Elias JE, Beausoleil SA, Bakalarski CE, Li X, Villén J, Gygi SP. 2006. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics 5: 1326–1337. 10.1074/mcp.M500339-MCP200 [DOI] [PubMed] [Google Scholar]
- Haberman Y, Karns R, Dexheimer PJ, Schirmer M, Somekh J, Jurickova I, Braun T, Novak E, Bauman L, Collins MH, et al. 2019. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun 10: 38 10.1038/s41467-018-07841-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. 2010. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143: 1174–1189. 10.1016/j.cell.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. 2007. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4: 923–925. 10.1038/nmeth1113 [DOI] [PubMed] [Google Scholar]
- Kamga CK, Zhang SX, Wang Y. 2010. Dicarboxylate carrier-mediated glutathione transport is essential for reactive oxygen species homeostasis and normal respiration in rat brain mitochondria. Am J Physiol Cell Physiol 299: C497–C505. 10.1152/ajpcell.00058.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. 2012. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun 3: 1245 10.1038/ncomms2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt A, Liebisch G, Clavel T, Haller D, Hörmannsperger G, Yoon H, Kolmeder D, Sigruener A, Krautbauer S, Seeliger C, et al. 2018. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nat Commun 9: 3760 10.1038/s41467-018-05767-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman JP, Janssen FG, van Druten JA. 1975. Oxidation-reduction potentials in the cecal contents of rats and mice. Proc Soc Exp Biol Med 149: 995–999. 10.3181/00379727-149-38942 [DOI] [PubMed] [Google Scholar]
- Kramer PA, Darley-Usmar VM. 2015. The emerging theme of redox bioenergetics in health and disease. Biomed J 38: 294–300. 10.4103/2319-4170.155591 [DOI] [PubMed] [Google Scholar]
- Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, Keller MP, Attie AD, Rey FE, Denu JM. 2016. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell 64: 982–992. 10.1016/j.molcel.2016.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno T, Hirayama-Kurogi M, Ito S, Ohtsuki S. 2016. Effect of intestinal flora on protein expression of drug-metabolizing enzymes and transporters in the liver and kidney of germ-free and antibiotics-treated mice. Mol Pharm 13: 2691–2701. 10.1021/acs.molpharmaceut.6b00259 [DOI] [PubMed] [Google Scholar]
- Lapek JD, Greninger P, Morris R, Amzallag A, Pruteanu-Malinici I, Benes CH, Haas W. 2017a. Detection of dysregulated protein association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat Biotechnol 35: 983–989. 10.1038/nbt.3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapek JD, Lewinski MK, Wozniak JM, Guatelli J, Gonzalez DJ. 2017b. Quantitative temporal viromics of an inducible HIV-1 model yields insight to global host targets and phospho-dynamics associated with protein Vpr. Mol Cell Proteomics 16: 1447–1461. 10.1074/mcp.M116.066019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapek JD, Mills RH, Wozniak JM, Campeau A, Fang RH, Wei X, van de Groep K, Perez-Lopez A, van Sorge NM, Raffatellu M, et al. 2018. Defining host responses during systemic bacterial infection through construction of a murine organ proteome atlas. Cell Syst 6: 579–592.e4. 10.1016/j.cels.2018.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson E, Tremaroli V, Lee YS, Koren O, Nookaew I, Fricker A, Nielsen J, Ley RE, Bäckhed F. 2012. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 61: 1124–1131. 10.1136/gutjnl-2011-301104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal-Lopes C, Velloso FJ, Campopiano JC, Sogayar MC, Correa RG. 2015. Roles of commensal microbiota in pancreas homeostasis and pancreatic pathologies. J Diabetes Res 2015: 284680 10.1155/2015/284680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman JS, Alsentzer E, Jaffe M, Sprockett D, Masutani E, Ikwa E, Fragiadakis GK, Clifford D, Huang BE, Sonnenburg JL, et al. 2016. The effect of microbial colonization on the host proteome varies by gastrointestinal location. ISME J 10: 1170–1181. 10.1038/ismej.2015.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiola F, Ianiro G, Franceschi F, Fagiuoli S, Gasbarrini G, Gasbarrini A. 2016. Gut microbiota in autism and mood disorders. World J Gastroenterol 22: 361–368. 10.3748/wjg.v22.i1.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinoglu A, Shoaie S, Bergentall M, Ghaffari P, Zhang C, Larsson E, Bäckhed F, Nielsen J. 2015. The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol Syst Biol 11: 834 10.15252/msb.20156487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. 2014. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86: 7150–7158. 10.1021/ac502040v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C, Duranti S, Bottacini F, Casey E, Turroni F, Mahony J, Belzer C, Delgado Palacio S, Arboleya Montes S, Mancabelli L, et al. 2017. The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol Rev 81 10.1128/MMBR.00036-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JT, Marotz C, Washburne A, Silverman J, Zaramela LS, Edlund A, Zengler K, Knight R. 2019. Establishing microbial composition measurement standards with reference frames. Nat Commun 10: 2719 10.1038/s41467-019-10656-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottawea W, Chiang CK, Mühlbauer M, Starr AE, Butcher J, Abujamel T, Deeke SA, Brandel A, Zhou H, Shokralla S, et al. 2016. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn's disease. Nat Commun 7: 13419 10.1038/ncomms13419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya K, Ikewaki K. 2018. Microbiota and HDL metabolism. Curr Opin Lipidol 29: 18–23. 10.1097/MOL.0000000000000472 [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Kosciolek T, Eyler LT, Knight R, Jeste DV. 2018. Overview and systematic review of studies of microbiome in schizophrenia and bipolar disorder. J Psychiatr Res 99: 50–61. 10.1016/j.jpsychires.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. 2012. Host-gut microbiota metabolic interactions. Science 336: 1262–1267. 10.1126/science.1223813 [DOI] [PubMed] [Google Scholar]
- Nicolay BN, Danielian PS, Kottakis F, Lapek JD, Sanidas I, Miles WO, Dehnad M, Tschöp K, Gierut JJ, Manning AL, et al. 2015. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev 29: 1875–1889. 10.1101/gad.264127.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. 2003. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res 2: 43–50. 10.1021/pr025556v [DOI] [PubMed] [Google Scholar]
- Protsyuk I, Melnik AV, Nothias LF, Rappez L, Phapale P, Aksenov AA, Bouslimani A, Ryazanov S, Dorrestein PC, Alexandrov T. 2018. 3D molecular cartography using LC–MS facilitated by Optimus and ‘ili software. Nat Protoc 13: 134–154. 10.1038/nprot.2017.122 [DOI] [PubMed] [Google Scholar]
- Quinn RA, Vrbanac A, Melnik AV, Patras KA, Christy M, Nelson AT, Aksenov A, Tripathi A, Humphrey G, da Silva R, et al. 2019. Chemical impacts of the microbiome across scales reveal novel conjugated bile acids. bioRxiv 10.1101/654756 [DOI]
- Ray PD, Huang BW, Tsuji Y. 2012. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24: 981–990. 10.1016/j.cellsig.2012.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi JA, Figueira TR, Ravagnani FG, Oliveira HC, Vercesi AE, Castilho RF. 2013. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic Biol Med 63: 446–456. 10.1016/j.freeradbiomed.2013.05.049 [DOI] [PubMed] [Google Scholar]
- Sartor RB, Wu GD. 2017. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 152: 327–339.e4. 10.1053/j.gastro.2016.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwechter B, Rosenmund C, Tolias KF. 2013. RasGRF2 Rac-GEF activity couples NMDA receptor calcium flux to enhanced synaptic transmission. Proc Natl Acad Sci 110: 14462–14467. 10.1073/pnas.1304340110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GM, Cheng J, Gordon JI. 2012. Quantitative assessment of the impact of the gut microbiota on lysine ε-acetylation of host proteins using gnotobiotic mice. Proc Natl Acad Sci 109: 11133–11138. 10.1073/pnas.1208669109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Webster PD. 1978. Neurohormonal control of pancreatic secretion. A review. Gastroenterology 74: 294–309. 10.1016/0016-5085(78)90812-0 [DOI] [PubMed] [Google Scholar]
- Spivak M, Weston J, Bottou L, Käll L, Noble WS. 2009. Improvements to the percolator algorithm for peptide identification from shotgun proteomics data sets. J Proteome Res 8: 3737–3745. 10.1021/pr801109k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. 2015. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452. 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaiss CA, Zmora N, Levy M, Elinav E. 2016. The microbiome and innate immunity. Nature 535: 65–74. 10.1038/nature18847 [DOI] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. 2003. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75: 1895–1904. 10.1021/ac0262560 [DOI] [PubMed] [Google Scholar]
- Tilg H, Moschen AR. 2014. Microbiota and diabetes: an evolving relationship. Gut 63: 1513–1521. 10.1136/gutjnl-2014-306928 [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. 2011. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8: 937–940. 10.1038/nmeth.1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolonen AC, Haas W. 2014. Quantitative proteomics using reductive dimethylation for stable isotope labeling. J Vis Exp 89: e51416. 10.3791/51416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HQ, Mills RH, Peters NV, Holder MK, de Vries GJ, Knight R, Chassaing B, Gonzalez DJ, Gewirtz AT. 2019. Associations of the fecal microbial proteome composition and proneness to diet-induced obesity. Mol Cell Proteomics 18: 1864–1879. 10.1074/mcp.RA119.001623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villén J, Gygi SP. 2008. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc 3: 1630–1638. 10.1038/nprot.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T, Shen Y, Monroe ME, Lopez-Ferrer D, Reno T, et al. 2011a. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11: 2019–2026. 10.1002/pmic.201000722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. 2011b. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472: 57–63. 10.1038/nature09922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel D, Flügge UI. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138: 141–143. 10.1016/0003-2697(84)90782-6 [DOI] [PubMed] [Google Scholar]
- Xiao Y, Hsiao TH, Suresh U, Chen HI, Wu X, Wolf SE, Chen Y. 2014. A novel significance score for gene selection and ranking. Bioinformatics 30: 801–807. 10.1093/bioinformatics/btr671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CJ, Machado-Vieira R, Henter I, Ibrahim L, Diazgranados N, Salvadore G. 2010. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry 18: 293–303. 10.3109/10673229.2010.511059 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.