

Abstract
Gametes constitute a critical stage of the plant life cycle during which the genome undergoes reprogramming in preparation for embryogenesis. Here, we examined genome-wide distributions of small RNAs in the sperm and egg cells of rice. We found that 24-nt siRNAs, which are a hallmark of RNA-directed DNA methylation (RdDM) in plants, were depleted from heterochromatin boundaries in both gametes relative to vegetative tissues, reminiscent of siRNA patterns in DDM1-type nucleosome remodeler mutants. In sperm cells, 24-nt siRNAs were spread across heterochromatic regions, while in egg cells, 24-nt siRNAs were concentrated at a smaller number of heterochromatic loci throughout the genome, especially at loci which also produced siRNAs in other tissues. In both gametes, patterns of CHH methylation, typically a strong indicator of RdDM, were similar to vegetative tissues, although lower in magnitude. These findings indicate that the small RNA transcriptome undergoes large-scale redistribution in both male and female gametes, which is not correlated with recruitment of DNA methyltransferases in gametes and suggestive of unexplored regulatory activities of gamete small RNAs.
During sexual reproduction in angiosperms, cells undergo a transition from vegetative to reproductive fates to produce spore mother cells. These spore mother cells then undergo meiosis to produce haploid spores that then undergo mitosis to produce gametes and other cells of the gametophyte. The development of reproductive cells is marked by multiple sex-specific changes in chromatin structure (for review, see Wang and Köhler 2017). For example, histone H1 and the centromere-specific histone variant CENH3 are depleted from the megaspore mother cell and the egg cell (Ingouff et al. 2010, 2017; She et al. 2013). Heterochromatin is decondensed in the central cell, which gives rise to the endosperm (Pillot et al. 2010; Yelagandula et al. 2014; Ingouff et al. 2017). A similar phenomenon occurs in the pollen vegetative nucleus (Schoft et al. 2009; Ingouff et al. 2010; Mérai et al. 2014; Hsieh et al. 2016). Most striking is the compaction of sperm cell chromatin, likely related to deposition of a set of histone variants including a male expressed H3 variant (for review, see Borg and Berger 2015). Specific activity of some types of euchromatic retrotransposons in the male germline in maize may also reflect male-specific chromatin changes (Dooner et al. 2019).
While DNA methylation patterns in general are transmitted stably across generations, there is also evidence for both loss and gain of methylation in specific sequence contexts and cell types in reproduction (for review, see Gehring 2019). RNA-directed DNA methylation (RdDM) is particularly interesting because of its potential to be erased and re-established de novo by siRNAs (for review, see Cuerda-Gil and Slotkin 2016). Methylation in the CHH context (mCHH), where H is A, C, or T, is a strong indicator of RdDM in rice, though not in Arabidopsis (Zemach et al. 2013; Stroud et al. 2014; Niederhuth et al. 2016; Tan et al. 2016). Live-cell analysis of transgene-driven expression of fluorescent proteins fused to methyl binding domains in Arabidopsis reveals reduced signal for mCHH in the megaspore mother cell, microspore, and in the sperm cell, and for mCG in the egg cell (Ingouff et al. 2017). Whole-genome bisulfite sequencing (WGBS) has been limited by the difficulty of obtaining sufficient quantities of pure reproductive cell types, but methylomes have been produced from sperm and pollen vegetative nuclei of both rice and Arabidopsis (Calarco et al. 2012; Ibarra et al. 2012; Hsieh et al. 2016; Kim et al. 2019) and from egg cells in rice (Park et al. 2016). Multiple differences are apparent between cell types by WGBS, including in mCHH. In Arabidopsis, the sperm cell has reduced mCHH content relative to the pollen vegetative cell (Calarco et al. 2012; Ibarra et al. 2012; Hsieh et al. 2016; Walker et al. 2018). The majority of mCHH in Arabidopsis pollen vegetative nuclei is primarily due to activity of the chromomethyltransferase CMT2 rather than RdDM (Hsieh et al. 2016; Borges et al. 2018). In both Arabidopsis and rice sperm, mCHH and mCHG levels at specific loci are correlated with reduced mCG in corresponding loci in the pollen vegetative nucleus, consistent with the hypothesis that chromatin changes in the pollen vegetative nucleus facilitate expression of siRNAs which are then transferred into the sperm cell to direct DNA methylation (Calarco et al. 2012; Ibarra et al. 2012; Kim et al. 2019).
Sequencing small RNAs from Arabidopsis whole pollen and isolated sperm indicates that 24-nt siRNAs typical of RNA polymerase IV (Pol IV) activity are the most abundant length, but 21-nt and 22-nt siRNAs increase in abundance in sperm cells relative to vegetative tissues (Slotkin et al. 2009; Borges et al. 2018). This increase in the ratio of 21- and 22-nt siRNAs to 24-nt siRNAs in Arabidopsis pollen is typical of siRNAs in vegetative tissues in mutants with compromised DNA methylation (Creasey et al. 2014; McCue et al. 2015; Corem et al. 2018; Fu et al. 2018; Long et al. 2018; Tan et al. 2018). The Arabidopsis pollen vegetative nucleus, however, does not have the reduced DNA methylation characteristic of ddm1 mutants, nor does the sperm, except in mCHH (Calarco et al. 2012; Ibarra et al. 2012; Hsieh et al. 2016; Walker et al. 2018).
Seeds produced from crosses with one parent defective in RdDM exhibit a variety of phenotypes, including the timing of zygotic genome activation, genomic imprinting, and genome dosage sensitivity (Autran et al. 2011; Erdmann et al. 2017; Borges et al. 2018; Grover et al. 2018; Martinez et al. 2018; Kirkbride et al. 2019; Satyaki and Gehring 2019). There are currently no available small RNA transcriptomes from sperm cells of flowering plants other than Arabidopsis and none from flowering plant egg cells. Due to the small size of the sperm cell relative to the egg cell (∼1/1000 the volume), the transcriptome of the newly fertilized egg cell is principally determined by the egg cell (Autran et al. 2011; Del Toro-De León et al. 2014; Anderson et al. 2017; Zhao et al. 2019). Given their likely importance both pre- and post-fertilization, we sequenced small RNAs from isolated sperm cells, egg cells, ovaries, and seedling shoots of rice, compared expression patterns of different classes of small RNAs, and investigated the relationship of 24-nt siRNAs to DNA methylation in the gametes.
Results
We prepared and sequenced small RNA Illumina libraries from seedling shoots, isolated sperm cells, isolated egg cells, and whole ovaries (after manual removal of egg cells) of rice variety Kitaake. For each cell or tissue type, we prepared at least three biological replicates. After removing PCR duplicates as described in Methods, we aligned all reads of 20 to 25 nt in length to the MSU7 reference genome assembly (Supplemental Table S1; Kawahara et al. 2013).
Distinct microRNA expression patterns in gametes
We examined microRNA (miRNA) expression across samples (values for each miRNA listed in Supplemental Data Set 1). Such miRNAs are predicted to be less mobile between cells than siRNAs (Grant-Downton et al. 2013), and since mRNA expression is globally different between egg, sperm, and seedling (Russell et al. 2012; Anderson et al. 2013, 2017), we also expected regulators of mRNA expression, such as miRNAs, to be differentially expressed across sample types. We performed principal component analysis (PCA) (Fig. 1A) to examine global differences of miRNA expression across samples. Each tissue formed a cluster on a miRNA PCA plot, with seedling leaf separated from reproductive tissues along the first PC axis, which explained ∼40% of variance; and male separated from female along the second PC axis, which explained ∼13% of the variance. To discover which individual miRNAs drove the variation between tissues, we performed hierarchical clustering and detected multiple miRNA clusters that distinguished seedling shoots from both gametes, as well as clusters that were enriched in one gamete but not the other (Supplemental Fig. S1A; Supplemental Table S3). Most clusters correlated with either one of the PCs (Supplemental Fig. S1B). Thus, the separation of samples on PCA plots is likely driven by miRNA clusters that have distinct expression patterns across tissues. miR159 family members were the most abundant miRNAs, especially in egg cell and ovary (Fig. 1B). In Arabidopsis, miR159 family members repress MYB transcription factors and are expressed in multiple tissues, including the sperm cell, and paternal miR159 regulates nuclear division in endosperm (Allen et al. 2010; Zhao et al. 2018). No maternal function for miR159 has been reported, but two members of the miR159 family—miR159a.1 and miR159b—account for greater than 60% of the total miRNAs in rice egg cell and ovary. In seedling shoot and sperm cell, miR159 accounted for greater than 20% of the total miRNAs.
Figure 1.

Small RNA composition of ovary, egg cell, sperm cell, and seedling shoot. (A) Principle component analysis (PCA) plot of tissues by miRNA expression pattern. (B) miRNA159 family expression. y-axis values are relative to the total number of miRNA reads in each tissue. Color code is the same as in A. (C) Small RNA compositions. y-axis values are relative to the total number of reads that mapped to the genome. (D) siRNA abundance by length and category. “siRNAs” refers to all 20- to 25-nt small RNAs that mapped to the genome but were not included in any category in C. “Uniquely mapped” refers to the subset that map with a MAPQ value of at least 20. “Gypsy” is the subset that overlaps with an annotated Gypsy element, and “TIR” is the subset that overlaps with an annotated DNA transposon of the Tc1/Mariner, PIF/Harbinger, Mutator, or hAT superfamilies. “CentO” is the subset that overlaps CentO tandem repeats. y-axis values are the number of siRNAs of each length normalized by the total number of siRNAs. In all panels, error bars are 95% confidence intervals based on biological replicates of each tissue.
The manual dissection of ovaries to isolate egg cells raises concern regarding the release of small RNA from the sporophytic ovary into the isolation medium and RNA being carried along with egg cell small RNAs into sequencing libraries. Three lines of evidence, however, indicated RNA contamination from the ovary in egg cells was minimal. First, our egg or zygote isolation method has previously been shown to have little mRNA contamination from the surrounding sporophytic ovule (Anderson et al. 2017; Li et al. 2019). Second, we performed differential expression analysis of the miRNAs and found that eight miRNAs had high levels in all three replicates of ovary but were nearly undetectable in all six replicates of egg cells (Supplemental Fig. S1C). Third, we did mock egg cell isolations in which we dissected ovaries using the same protocol, but we only collected cell-free solution rather than the egg cells. By qPCR quantification, library preparation from the mock samples yielded only about 5% of the number of library molecules as an actual egg cell small RNA library after 20 cycles of PCR amplification (Supplemental Fig. S2). Similarly, our sperm cell isolation method has previously been shown to have little mRNA contamination after isolation from surrounding floral tissues or pollen (Li et al. 2019).
Distinct small RNA composition across gametes and vegetative tissues
Small RNA compositions from gametes were distinct from general vegetative tissues, such as seedling shoot, most noticeably by the reduction of miRNAs relative to total small RNAs (Fig. 1C; Supplemental Fig. S3). In seedling shoots, 5.5% of the small RNA reads mapped to miRNA loci, as compared to 1.8% in egg cell and 0.7% in sperm cell. In addition to miRNAs, the relative abundance of phased secondary siRNAs (phasiRNAs), tRNAs, NOR RNAs, and 5S rRNAs varied across gametes and vegetative tissues. The relative abundance of 21-nt and 24-nt phasiRNAs clearly differentiated gametes. Twenty-four-nucleotide phasiRNAs are expressed in meiotic anthers in diverse angiosperms, including rice (Johnson et al. 2009; Zhai et al. 2015b; Fei et al. 2016; Xia et al. 2019). In female reproductive tissues, phasiRNAs are not well characterized, but the argonaute MEL1 binds 21-nt phasiRNAs and is required for both male and female meiosis in rice (Nonomura et al. 2007; Komiya et al. 2014). While both classes were rare in egg cell relative to seedling shoot, 21-nt phasiRNAs were even rarer in sperm cells (Fig. 1C). In contrast, 24-nt phasiRNAs were greater than sixfold more abundant in sperm cell as compared to ovary, egg cell, and seedling shoot. Comparison with published data (Li et al. 2017), however, revealed far higher relative abundance of both 21-nt and 24-nt phasiRNAs in anthers than in either egg or sperm cell (Supplemental Fig. S3).
The most abundant small RNAs in angiosperm vegetative tissues are 24-nt siRNAs that are involved in RNA-directed DNA methylation and, to a lesser extent, 21- and 22-nt siRNAs (for review, see Cuerda-Gil and Slotkin 2016). We grouped all 20- to 25-nt small RNAs that did not overlap at least 90% of their length with miRNA, phasiRNA, tRNA, NOR RNA, or 5S rRNA loci into a general siRNA category for further analysis. The length distribution of these siRNAs was similar between tissues, with a strong preference for 24-nt (Fig. 1D; Supplemental Fig. S4). Parsing out siRNAs that overlapped by at least 90% of their length with retrotransposons of the Gypsy superfamily and the centromeric tandem repeat CentO revealed strong siRNA enrichments for these elements in sperm cell. CentO tandem repeats are enriched in centromeres (Nagaki et al. 2003), and Gypsy retrotranposons are generally enriched in heterochromatic regions of the genome (Kawahara et al. 2013). siRNAs overlapping with terminal inverted repeat (TIR) DNA transposons of the PIF/Harbinger, Tc1/Mariner, Mutator, or hAT superfamilies were enriched in seedling shoot but were depleted in sperm cell, egg cell, and ovary. For brevity, we refer to siRNAs from these four superfamilies of DNA transposons as simply TIR siRNAs. These transposons are enriched near genes (Han et al. 2013).
Distinct genome-wide patterns of 24-nt siRNAs in gametes
Consistent with the high abundance of CentO and Gypsy siRNAs, a genome-wide view of 24-nt siRNA abundance revealed that 24-nt siRNAs in sperm cells had a complementary pattern to that of seedling shoot. Sperm cell 24-nt siRNAs were enriched in gene-poor, heterochromatic regions, whereas seedling shoot 24-nt siRNAs had the expected pattern for canonical RdDM in vegetative tissues, with enrichment in euchromatic regions that are gene-rich (Fig. 2A; Supplemental Figs. S5–S7). Egg cell 24-nt siRNAs showed a pattern distinct from both sperm cell and seedling shoot, with depletion across most of the genome except at a smaller number of loci with no clear relationship to gene density. The distribution of 21- and 22-nt siRNAs also did not have a clear relationship to gene density (Supplemental Fig. S5). Twenty-four-nucleotide siRNAs from ovary showed a nearly identical pattern with egg cell (Fig. 2A). Twenty-one- and 22-nt siRNAs were also similar between egg cell and ovary but less similar than 24-nt siRNAs (Supplemental Fig. S5). Comparison of 24-nt siRNAs from egg cell with ovaries in earlier stages of development (Li et al. 2017) indicated the egg/ovary pattern was established by the stage of megasporocyte formation (Supplemental Fig. S7). Consistent with the genome-wide analysis, metagene plots of the distribution of 24-nt siRNAs relative to genes revealed a depletion of 24-nt siRNAs both upstream of and downstream from genes in egg cell, sperm cell, and ovary (Fig. 2B), corresponding to where TIR transposons are enriched in the genome, with the exception of the CACTA superfamily (Han et al. 2013).
Figure 2.

siRNA distributions in ovary, egg cell, sperm cell, and seedling shoot. (A) Whole-genome 24-nt siRNA heat maps. Top track: gene density; second to fifth tracks: 24-nt siRNAs from seedling shoot, sperm cell, egg cell, and ovary. (B) Metagene coverage plot for 24-nt siRNAs. Coverage is measured over 100-bp intervals and normalized per 1000 total siRNAs. Vertical grid lines indicate 500-bp intervals. (TSS) Transcription start site, (poly[A]) polyadenylation site. (C) Venn diagram showing number and overlap of 24-nt siRNA loci. The genome was divided into 100-bp intervals and categorized as 24-nt siRNA loci based on coverage of 24-nt siRNAs from seedling shoot, sperm cell, and egg cell. (D) Number of 24-nt siRNA loci and abundance and enrichments of siRNAs from each tissue. Abundance (% 24-nt siRNAs in loci) is the number of 24-nt siRNAs that overlapped with the loci relative to the total number of 24-nt siRNAs from the tissue. Enrichment is the abundance normalized by the number of 24-nt loci. In locus types, “spec.” is short for “specific,” the set of loci only identified from a single tissue. (E) siRNA abundance at 24-nt siRNA loci. “Intersect.” refers to intersection loci, those identified as 24-nt siRNA in all three tissues. y-axis values are percent of siRNAs of each length relative to the total number of siRNAs that mapped to the genome. Error bars are 95% confidence intervals based on biological replicates of each tissue.
To identify representative sets of loci associated with siRNAs in each tissue, we divided the genome into nonoverlapping 100-bp intervals and counted the number of mapping siRNAs per two million total siRNAs from each tissue. Any 100-bp interval with at least three mapping 24-nt siRNAs that spanned at least one-third (34 bp) of the 100-bp sequence we categorized as a 24-nt siRNA locus. In seedling shoot, 192,428 loci met these criteria; in sperm, 102,325; and in egg, 54,476 (Fig. 2C). Consistent with the complementary patterns of 24-nt siRNA loci in sperm and seedling shoot in the whole-genome view of siRNA abundance (Fig. 2A), only 15% of the sperm loci were shared with seedling shoot loci (Fig. 2D). In contrast, 38% of the egg loci were shared with seedling shoot. The loci that were shared between all three tissues we called intersection 24-nt siRNA loci, whereas the loci that were unique to one tissue we called either seedling-specific, sperm-specific, or egg-specific. Twenty-four-nucleotide siRNAs from seedling shoot were the most sample-specific of the three in terms of total numbers of siRNAs overlapping seedling-specific loci (69%), whereas 24-nt siRNAs from egg cell were the least sample-specific (8% overlapping egg-specific loci). Instead, the majority of 24-nt siRNAs from egg cells overlapped 24-nt siRNA loci that were shared with one or both other tissues (Fig. 2D,E). Taking into account the number of loci in each category relative to the size of the genome revealed that intersection loci were more strongly enriched for 24-nt siRNAs than sample-specific loci in all three tissues but most strongly in egg cells (Fig. 2D). Twenty-four-nucleotide siRNAs from ovary had a similar pattern as egg cell, whereas other vegetative tissues were similar to seedling shoot (Fig. 2D,E; Supplemental Fig. S8). Counting overlaps between 24-nt siRNA loci and repetitive elements and measuring distances between 24-nt siRNA loci and nearest genes revealed expected trends of seedling-specific 24-nt loci being near genes and overlapping with TIR transposons, typical of canonical RdDM. (Supplemental Fig. S9). Gamete-specific loci were farther from genes and overlapped more with Gypsy retrotransposons, whereas intersection loci were similar to seedling-specific loci. Taken together, these data reveal that sperm and egg cells have distributions of siRNAs that are uncharacteristic of canonical RdDM in rice and that are distinct from each other. The sperm cell is complementary to vegetative tissues in its enrichment of siRNAs in large numbers of heterochromatic loci, whereas the egg cell and ovary have different patterns, with large numbers of siRNAs in a small number of loci that tend to be shared in sperm and vegetative tissues.
To test whether the pattern of 24-nt siRNAs in either sperm or egg cell could be explained by loss of 24-nt siRNAs from canonical RdDM loci, we removed all seedling shoot 24-nt siRNAs that overlapped with seedling-specific 24-nt siRNA loci and then examined the distribution of the remaining seedling 24-nt siRNAs in the genome. While this did result in fewer siRNAs near genes, it did not produce the enrichment in heterochromatic regions observed in sperm (Supplemental Fig. S6). Canonical RdDM is indicated by mCHH in addition to 24-nt siRNAs, so we also identified putative canonical RdDM loci independently, using mCHH levels in vegetative tissues from a previous study (Tan et al. 2016). We divided the genome into nonoverlapping 100-bp loci, then identified loci with an average mCHH of at least 5%. Eight percent of the genome, or 315,368 loci, met these criteria, and 47% of seedling shoot 24-nt siRNAs overlapped with these loci. After removing seedling 24-nt siRNAs from mCHH loci, the residual 24-nt siRNAs in seedling shoot were relatively depleted near genes, but again there was no specific enrichment in heterochromatic regions, as was the case with sperm, nor was there a pattern resembling that of egg cells (Supplemental Fig. S6).
The mRNA expression of sperm cell genes has an atypical distribution, with a smaller number of genes accounting for a larger proportion of mRNA in sperm cell than in egg cell (Supplemental Fig. S10A; Anderson et al. 2013). Since abundance of 24-nt siRNAs and mCHH near genes has been positively correlated with gene expression (Li et al. 2015), we wondered whether highly expressed sperm genes might be enriched for flanking 24-nt siRNAs. This was not the case, however. Genes with values of greater than 10 transcripts per million (TPM), which correspond to the top ∼25% of mRNA expression level in sperm cell, exhibited a similar lack of flanking siRNAs as the total set of genes (Supplemental Fig. S10B). We also asked whether differences in siRNAs could be explained by differences in mRNA levels of RdDM factors or DDM1 in egg cell and sperm cell. Analysis of published gamete transcriptomes did not reveal any obvious explanation for egg cell or sperm cell siRNA patterns, though they did show differences in mRNA levels for such RdDM factors as RDR2 (Supplemental Fig. S11; Anderson et al. 2013, 2017). At least one DDM1 homolog was strongly expressed in both gametes; however, in pollen vegetative cell, a single homolog was only moderately expressed. Analysis of 5′ nucleotide preferences of gametic and vegetative 24-nt siRNAs revealed a strong bias toward a 5′ A in seedling shoot and in sperm cell (Supplemental Fig. S12), consistent with known Pol IV activity (Blevins et al. 2015; Zhai et al. 2015a). The 5′ nucleotide biases were also present in ovary and egg cell, but less prominently than in seedling shoot or sperm cell, suggesting that maternal 24-nt siRNAs might also arise from different RNA polymerase activities or processing of siRNA precursors (both Pol II and Pol IV and multiple dicers and other factors can interact to produce diverse siRNAs [Cuerda-Gil and Slotkin 2016]). In sperm, 21- to 22-nt siRNAs also exhibited a preference for a 5′ A, though weaker than that of 24-nt siRNAs, consistent with reports that Pol IV can produce 21-nt siRNAs in Arabidopsis pollen (Borges et al. 2018; Martinez et al. 2018).
Transposon-related mRNAs made up a higher proportion of the total mRNA in sperm than in egg, zygote, or seedling (Supplemental Fig. S13; Anderson et al. 2013, 2017). This was true both for heterochromatic transposons (represented by LTR retrotransposons of the Gypsy superfamily) and euchromatic ones (represented by TIR transposons, except CACTA). Analysis of siRNA and mRNA expression of individual transposon copies is complicated by the limitation in mapping short reads to them uniquely. However, a strong trend was evident in that transposon copies with high mRNA expression in gametes tended to have fewer 24-nt siRNAs and vice versa (Supplemental Fig. S14). Both Gypsy and TIR transposons exhibited this relationship, suggesting that 24-nt siRNAs might be generally antagonistic to transposon mRNA accumulation.
Gametes have similar DNA methylation as vegetative tissues
Twenty-four-nucleotide siRNAs are key components of RdDM in vegetative tissues. To test whether the unusual genomic distributions of siRNAs in sperm and egg were associated with RdDM, we prepared and sequenced whole-genome bisulfite sequencing libraries using a post-bisulfite adapter tagging (PBAT) method from isolated egg cell and sperm cell (Supplemental Table S2). We also analyzed published PBAT libraries from egg cell, sperm cell, central cell, and pollen vegetative nuclei (Park et al. 2016; Kim et al. 2019). The DNA methylation in all of these resembled typical vegetative tissues in that mCHH was highest near genes (Fig. 3A; Supplemental Figs. S15, S16). Despite the genome-wide differences in distributions of 24-nt siRNAs in egg cell, sperm cell, and vegetative tissues, distributions of DNA methylation were similar between them, though the magnitude of methylation was variable between cell types and even between experiments. We also prepared libraries from whole ovary, bract (palea and lemma), embryo, and endosperm using a conventional WGBS method (Supplemental Table S2). All wild-type conventional libraries revealed the expected pattern of high mCHH flanking genes except mature endosperm (Fig. 3B). As with egg cell, the ovary retained robust mCHH at canonical RdDM loci, despite loss of 24-nt siRNAs. We also generated a knockout mutant for drm2 using CRISPR-Cas9 gene editing and prepared conventional WGBS libraries from mutant endosperm and embryo. DRM2 encodes a DNA methyltransferase of the Domains Rearranged Methyltransferase family, which function in RdDM. Near-gene mCHH was almost abolished in drm2 embryo and endosperm (Fig. 3B), demonstrating that the mCHH peaks near genes require functional DRM2 and consistent with prior work on methylation in drm2 mutant leaf (Tan et al. 2016).
Figure 3.

DNA methylation across tissues. (A) DNA methylation metagene plots for post-bisulfite adapter tagging (PBAT) libraries. Plots indicate average DNA methylation values over 100-bp intervals. Vertical grid lines indicate 500-bp intervals. DNA methylation is measured as the proportion of methylated cytosines relative to total cytosines in each sequence context. (TSS) Transcription start site, (poly[A]) polyadenylation site. Published data from Park et al. (2016) and Kim et al. (2019). (B) DNA methylation metagene plot for conventional WGBS libraries; x- and y-axes as in A. (C) DNA methylation of 24-nt siRNA loci for PBAT libraries. Center line is median; box spans interquartile range; whiskers span 2.5th to 97.5th percentiles. (D) DNA methylation of 24-nt siRNA loci for conventional WGBS libraries, as in C; color code as in C. (E) Abundance of hypomethylated CHH differential methylated regions (hypo-CHH DMRs) in drm2 mutant relative to wild-type mature embryo. Abundance is measured as the percent of hypomethylated regions relative to total number of regions with sufficient read coverage for statistical significance. Dotted line indicates genome-wide average percent hypo-CHH DMRs. Color code as in C.
To test whether 24-nt siRNAs directed DNA methylation in gametes, we examined gamete DNA methylation in the tissue-specific 24-nt siRNA loci that we identified (see above). All tissue-specific classes of 24-nt siRNA loci had high mCG and mCHG in all wild-type tissues, including gametes (Supplemental Fig. S17). Endosperm mCHG was an exception, being reduced by more than 50% relative to other vegetative tissues. mCHH was more variable across tissues, with endosperm having the lowest levels. Across all tissues, seedling-specific and intersection loci had the highest mCHH levels, independently of 24-nt siRNAs. In other words, despite the relative abundance of 24-nt siRNAs in egg-specific 24-nt siRNA loci, these loci had lower mCHH in egg than seedling-specific loci in egg. The same was true for sperm: Sperm-specific 24-nt siRNA loci had lower mCHH in sperm than seedling-specific loci in sperm (Fig. 3C). Intersection loci, like seedling-specific loci, had high mCHH across tissues, including pollen vegetative cell, central cell, and vegetative tissues (Supplemental Fig. S17). Although variation between tissues and even between experiments exists (Fig. 3A), within-library patterns are reproducible and confirm that redistribution of 24-nt siRNAs is not coupled to redistribution of DNA methylation in gametes (Fig. 3C). In all cases, differences between gamete-specific loci and seedling-specific loci were highly significant (P < 10−5, Tukey test using a generalized linear model with logit link). In addition, the drm2 mutation abolished mCHH in seedling-specific 24-nt loci and intersection 24-nt loci in mature embryo and leaf, further confirming that these are canonical RdDM loci (Figs. 3D, 4A). We also confirmed our inferences by counting the proportion of differentially methylated regions (DMRs) in the CHH context between mature WT and drm2 embryo across 24-nt siRNA loci categories (Fig. 3E). Genome-wide, 39% of eligible loci (loci that had sufficient coverage to be included in DMR analysis) were hypomethylated for mCHH in the drm2 embryo. Seedling-specific and intersection loci were enriched for hypo-mCHH DMRs (74% and 89%, respectively), whereas egg-specific and sperm-specific loci were depleted for hypo-mCHH DMRs (36% and 25%, respectively). Hypergeometric tests detected significant enrichment or depletion of DMRs in these sets of loci, respectively (P value = 0 for all four categories).
Figure 4.

siRNA and DNA methylation in ddm1 mutant leaf. (A) DNA methylation metagene plots. Plots indicate average DNA methylation values over 100-bp intervals in ddm1 (osddm1a osddm1b double mutant), drm2, and wild-type leaf. Vertical grid lines indicate 500-bp intervals. DNA methylation is measured as the proportion of methylated cytosines relative to total cytosines in each sequence context. (TSS) Transcription start site, (poly[A]) polyadenylation site. Data source: Tan et al. (2016). (B) DNA methylation of 24-nt siRNA loci. Center line is median; box spans interquartile range; whiskers span 2.5th to 97.5th percentile. (C) siRNA abundance at 24-nt siRNA loci in ddm1 and wild-type leaf. “Intersect.” refers to intersection loci, those identified as 24-nt siRNA in all three tissues. y-axis values are percent of siRNAs of each length relative to the total number of siRNAs that mapped to the genome. Error bars are 95% confidence intervals. Data source: Tan et al. (2018).
Gamete siRNAs mark loci dependent on DDM1, but not DRM2, for methylation in vegetative tissues
Loss of siRNAs from TIR DNA transposons and gain of siRNAs from LTR retrotransposons in gametes is reminiscent of ddm1 mutant phenotypes in vegetative tissues in diverse angiosperms (Creasey et al. 2014; McCue et al. 2015; Corem et al. 2018; Fu et al. 2018; Long et al. 2018; Tan et al. 2018). In mutants such as ddm1 that reduce heterochromatic DNA methylation, RNA polymerases may gain access to DNA from which they are normally restricted by inaccessible chromatin structure. The leaf phenotype of an osddm1a osddm1b double mutant in rice (which we will refer to simply as ddm1) includes substantial loss of mCHH and siRNAs flanking genes, coupled with increased DRM2 activity and siRNAs in heterochromatic regions (Tan et al. 2016, 2018). The reported differences in siRNAs between wild-type and ddm1 mutant leaf were not as severe as the differences we found between wild-type seedling shoot and wild-type gametes. Since the magnitude of such differences can be influenced by analysis methods, we reexamined the published ddm1 small RNA and methylation data in parallel with our gamete data. We also included published drm2 methylation data from the same study as the ddm1 mutant methylation data (Tan et al. 2016). Our analysis of methylation and siRNAs confirmed the prior study in that 24-nt siRNAs were reduced in gene flanks in the ddm1 mutant, but not nearly as severely as in gametes (Supplemental Fig. S18). In agreement with the previous study, the ddm1 mutant also had a strong reduction in methylation (∼50% lower in all contexts), in gene flanks in leaf (Fig. 4A). The drm2 mutant had a stronger reduction in mCHH but weaker reduction in mCG and mCHG near genes, as expected for a factor directly involved in RdDM (Fig. 4A).
To investigate whether gamete-specific 24-nt siRNA loci might be targets of DDM1 activity, we also evaluated DNA methylation in the mutants for each set of 24-nt siRNA loci. We found severe reductions in methylation—particularly in mCHG—in ddm1 mutants (P < 10−5, Tukey test using a generalized linear model with logit link), but not in drm2 mutants, at sperm-specific and egg-specific 24-nt siRNA loci (Fig. 4B). At these loci, spread of mCHH values was more variable in the ddm1 mutant than in wild type, and thus it was more difficult to confidently determine the overall trend. The seedling-specific and intersection 24-nt siRNA loci were strongly dependent on DRM2 for mCHH methylation (P < 10−5, Tukey test using a generalized linear model with logit link), consistent with canonical RdDM activity. The relative abundance of both 21-nt and 24-nt siRNAs increased in gamete-specific 24-nt siRNA loci, but 24-nt siRNAs decreased in seedling-specific and intersection loci in ddm1 leaf (Fig. 4C). Taken together, these analyses reveal that the ddm1 mutant causes leaf to resemble gametes in terms of siRNA expression and that loci that gain siRNA expression in either gamete are dependent on DDM1, but not DRM2, for methylation in vegetative tissues.
Discussion
The male and female gametes of flowering plants are highly dimorphic, which is reflected in their divergent transcriptomes (Anderson et al. 2013), as well as in their chromatin and histones (Ingouff et al. 2010), suggestive of major differences in reprogramming of their genomes prior to fertilization. The results from this study show that the small RNA transcriptomes of rice gametes differ significantly from each other, as well as from vegetative tissues. Whereas both sperm and egg cells had reduced abundance of miRNAs relative to siRNAs, each gamete expressed a diverse set of miRNAs (Supplemental Fig. S1). Similarities between miRNA expression patterns between egg and ovary might be indicative of cell-to-cell miRNA mobility, but any such miRNA mobility is unlikely to be general since ovary-specific miRNAs such as miR166 family members were not detectable or were detected at very low levels in the egg cell (Supplemental Fig. S1C). In Arabidopsis, expression of miR845 in pollen leads to production of 21- and 22-nt Gypsy and Copia siRNAs, termed epigenetically activated siRNAs, or easiRNAs, which are hypothesized to be transferred from the pollen vegetative cell into the sperm cell (Creasey et al. 2014; Borges et al. 2018; Martinez et al. 2018). However, we could not find any annotated miRNA or any genomic locus with significant homology to miR845 in rice, suggesting that Gypsy siRNAs in rice gametes are initiated differently. Likewise, 21- and 22-nt siRNAs were rare in sperm cell, including at gamete-specific 24-nt siRNA loci, as in other rice tissues (Fig. 2E; Supplemental Fig. S4).
The most notable feature of both gametes was the depletion of 24-nt siRNAs from heterochromatin boundaries (also called mCHH islands) (Fig. 5), where they are found in typical vegetative tissues (Gent et al. 2013; Li et al. 2015), constituting key components of canonical RdDM (for review, see Cuerda-Gil and Slotkin 2016). In spite of this loss of 24-nt siRNAs from canonical RdDM loci in both gametes, the abundance of 24-nt siRNAs relative to other siRNA lengths was unchanged. Each gamete, however, had a distinct genomic distribution of 24-nt siRNAs (Fig. 2). The redistribution of 24-nt siRNAs into heterochromatin in the sperm cell might be related to sperm-specific chromatin modifications, which could facilitate access of RNA Pol IV or another polymerase to repetitive loci that would normally be inaccessible. Sperm cells have been previously reported to have a suite of long noncoding RNAs (lncRNAs) distinct from vegetative cells but overlapping with lncRNAs in the emf2b polycomb group mutant (Johnson et al. 2018). Alternatively, sperm cell siRNAs could be inherited from progenitor cells or transferred from the pollen vegetative cell, which is known to lose heterochromatic modifications in Arabidopsis (Mérai et al. 2014). In sperm cells, egg cells, their companion gametophytic cells, or their progenitors, RdDM may not be fully functional and only methylate DNA at high-affinity loci, which are also shared with vegetative tissues. Gamete-specific 24-nt siRNAs could instead arise from a pathway that processes a Pol II primary transcript rather than a Pol IV primary transcript. Such an interpretation would be consistent with the lack of DNA methylation at gamete-specific sRNA loci.
Figure 5.
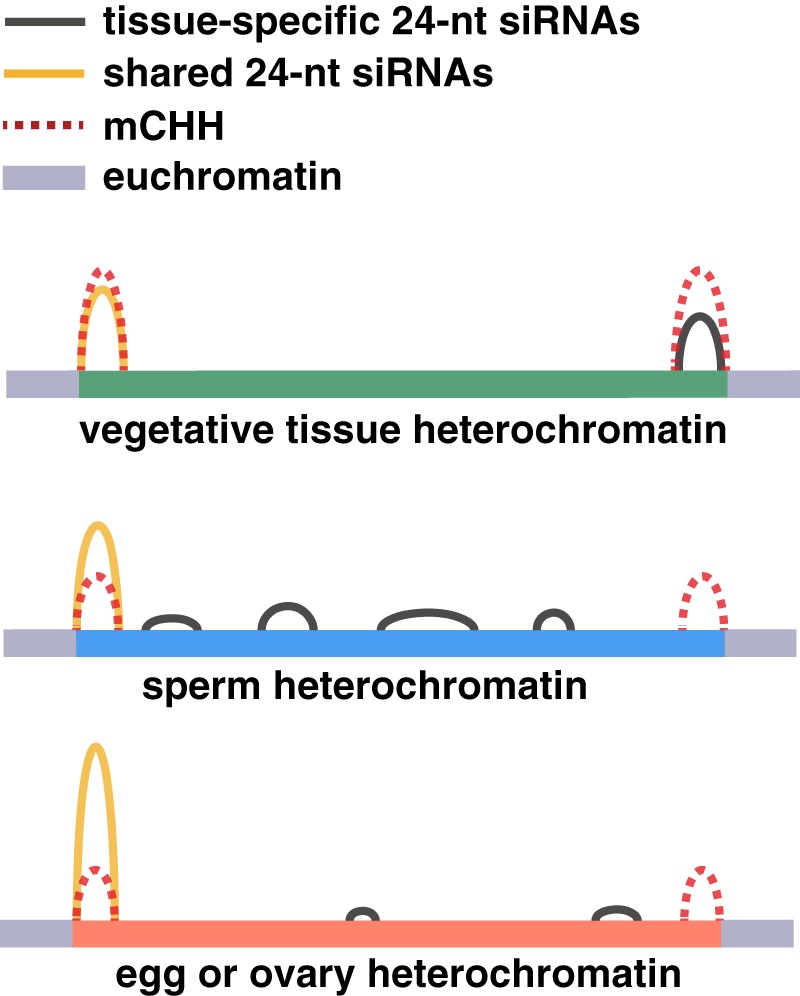
Schematic representation of 24-nt siRNAs and CHH methylation in different tissues. Gray segments represent euchromatic DNA and colored segments represent heterochromatic DNA, the same region in each tissue. Tissue-specific 24-nt siRNAs are abundant in either sperm cell, egg cell, or seedling leaf (tissue-specific loci), while shared 24-nt siRNAs are abundant in all three (intersection loci).
Changes in chromatin structure also occur in the production of female reproductive cells in plants, which may lead to corresponding changes in small RNA expression or activity (for review, see Wang and Köhler 2017). It is tempting to speculate that the absence of siRNAs from canonical RdDM loci in egg cell may be related to an absence of heterochromatin, and hence heterochromatin boundaries in egg cell, but this explanation would also require a similar absence of heterochromatin in ovaries to explain the similarities between egg cell and ovary 24-nt siRNAs. Alternatively, the chromatin status of egg may have more to do with chromatin in the ovary than egg cell. While it has been proposed that siRNAs are transferred from the central cell into the egg cell (Ibarra et al. 2012), the similarities between ovary and egg cell raise the possibility that 24-nt siRNAs in the egg cell might be acquired from the female reproductive tissues of the ovary.
A recent report on DNA methylation in rice pollen utilizing mutants in the DNA glycosylase ROS1 revealed a correlation between demethylation in the pollen vegetative nucleus and increased methylation in sperm (Ibarra et al. 2012; Kim et al. 2019), which is consistent with previous findings in Arabidopsis (Ibarra et al. 2012). In this study, comparison of rice sperm cell PBAT methylomes with a previously published rice egg cell PBAT methylome (Park et al. 2016) and methylomes of vegetative tissues by conventional WGBS methods detected different magnitudes of DNA methylation across these cell types, described as methylation reprogramming in the gametes (Park et al. 2016; Kim et al. 2019). We find from analysis of the published data from Kim et al. (2019) that they are consistent with our PBAT methylation libraries in this study but that the deduced differences in methylation in sperm and egg are subtle in comparison to the differences in 24-nt siRNAs (cf. Fig. 2 and Fig. 3). More specifically, our analysis also found magnitudes of DNA methylation to be different between the different cell types, but the overall genome-wide patterns remained largely unchanged in all wild-type tissues we examined except mature endosperm (Fig. 3; summarized in Fig. 5). We propose that distinct and major reprogramming of 24-nt siRNAs occurs both maternally and paternally in the germline, which are not correlated with a similar reprogramming of DNA methylation in the gametes. Since mCHH requires 24-nt siRNAs, one explanation for the presence of mCHH at heterochromatin boundaries without 24-nt siRNAs might be the removal of siRNAs after the deposition of DNA methylation. It is also possible that the observed mCHH pattern was inherited from progenitor cells, in which case immediate progenitor cells would have had double the levels of mCHH.
DNA methylation in drm2 mutants confirmed that mCHH and 24-nt siRNAs in vegetative tissues defined canonical RdDM loci, as they were strongly dependent upon DRM2 for mCHH. The siRNA expression patterns of neither sperm nor egg cell could be explained by simple loss of 24-nt siRNAs from canonical RdDM loci because 24-nt siRNAs remained abundant relative to other siRNA lengths and even increased in abundance relative to miRNAs. In addition, removal of 24-nt siRNAs from canonical RdDM loci in silico and analysis of the residual ones in seedling shoot yielded a genomic distribution unlike either gamete (Supplemental Fig. S6). Thus, the pattern in both egg and sperm cell requires both loss of 24-nt siRNAs from canonical RdDM loci and gain of siRNAs at novel loci that do not undergo RdDM. In this respect, gametes share similarities with ddm1 mutants in Arabidopsis, tomato, rice, and maize (Creasey et al. 2014; McCue et al. 2015; Corem et al. 2018; Long et al. 2018; Tan et al. 2018) and with chromomethylase mutants in maize (Fu et al. 2018), where loss of siRNAs at heterochromatin boundaries is accompanied by gain elsewhere. DDM1 facilitates methylation of heterochromatic DNA and has no known direct function in RdDM (Zemach et al. 2013; Lyons and Zilberman 2017). Our analysis of published ddm1 mutant rice data (Tan et al. 2016, 2018) revealed that the loci with novel 24-nt siRNA expression in either gamete tended to also gain siRNAs in ddm1 mutant leaves, not just 24-nt siRNAs but also 21-nt siRNAs (Fig. 4). Both mCHH and 24-nt siRNAs in ddm1 mutant leaves were reduced at canonical RdDM loci, as previously reported. The effect of ddm1 on siRNAs in leaves was not as strong as the difference between wild-type leaf and gametes, but the mutants clearly shifted leaf toward a more gamete-like profile. In maize ddm1 mutant embryos, the effect is much stronger than in ddm1 mutant rice leaf and more similar to rice gametes in that 24-nt siRNAs are nearly completely lost from canonical RdDM loci (Fu et al. 2018).
The finding that a transgenic DDM1::GFP construct under the native DDM1 promoter expresses in Arabidopsis sperm but not in pollen vegetative nucleus supports a model in which the absence of DDM1 in the pollen vegetative nucleus facilitates production of 21- and 22-nt siRNAs that are transferred into the sperm cell (Slotkin et al. 2009). While it is not clear how high levels of mCHG and mCG in pollen vegetative nucleus are produced in the absence of DDM1 (Calarco et al. 2012; Ibarra et al. 2012), it is clear that pollen vegetative nucleus undergoes a major loss of heterochromatin in Arabidopsis (Mérai et al. 2014). The mRNA expression data in rice (Anderson et al. 2013) do not clearly indicate the DDM1 status in the pollen vegetative nucleus, as it has reduced but not absent mRNA expression (Supplemental Fig. S11). Whether siRNAs of any length are transferred from the pollen vegetative nucleus into the sperm cell in rice is not known. Regardless of the origin of 24-nt siRNAs in rice gametes, their lack of correlation with mCHH in these cells raises two possibilities. The first is that they direct chromatin modifications other than DNA methylation in gametes. The second is that the gametic siRNAs are primed for RdDM in the early embryo. In this case, one or more RdDM factors missing in each gamete might become available upon fertilization and allow for immediate activity at loci not usually targeted in vegetative tissues.
Methods
Kitaake variety rice was used for all experiments. Gametes were isolated as described (Anderson et al. 2013; Li et al. 2019). Ovaries were dissected from pre-anthesis flowers. Seedling shoot segments were collected from 7-d-old water-germinated rice seeds. Total RNA was used as input for small RNA libraries: ∼30 ng for egg cells, ∼1 ng for sperm cells, ∼50 ng for ovaries, and ∼20 ng for seedling shoots. Small RNA libraries were made using the NEXTflex Small RNA-seq kit v3 (PerkinElmer NOVA-5132-05). Sperm samples and one of the four seedling replicates were amplified with 25 PCR cycles, all other samples with 15–20 cycles. After adapter trimming small RNA reads, PCR duplicates were removed using PRINSEQ “prinseq-lite.pl -fastq -out_format 3 -out_good -derep 1” (Schmieder and Edwards 2011). Reads were aligned to the genome with BWA-backtrack (version 0.7.15) (Li and Durbin 2009). Except where indicated otherwise, multimapping reads were included in all analyses. The complete set of read alignments was compared with miRNA, phasiRNA, tRNA, 5S rRNA, and NOR RNA loci in the genome (Supplemental Tables S4, S5). All other reads were categorized as siRNA reads and used for subsequent siRNA analyses. To evaluate reproducibility of low-input libraries, pairwise Pearson's correlation coefficients were calculated using the cor() function in R (R Core Team 2019). For this analysis, 24-nt siRNAs were first counted on 50-kb intervals (Supplemental Fig. S19A–D). Hierarchical clustering was performed using the hclust() function in R on these data and revealed the expected clustering by tissue (Supplemental Fig. S19E).
Whole-genome small RNA heat maps were made on 50-kb intervals using IGVtools (Thorvaldsdóttir et al. 2013). For better visualization of midrange values, heat map intensity was maxed out at 1.25× coverage (per 10 million siRNAs). To identify representative 24-nt siRNA loci, reads alignments were subsampled then combined from each sperm cell sample, from each egg cell sample, and from each seedling shoot sample to get as equal a representation as possible from each sample and a final combined number of two million in each. The genome was divided into nonoverlapping 100-bp loci, and siRNAs were counted for every locus. All loci that had at least three mapping 24-nt siRNAs that spanned at least one-third (34 bp) of the 100 bp were categorized as 24-nt siRNA loci; 5′ nucleotide frequencies were calculated with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
PBAT WGBS libraries were prepared using Pico Methyl-Seq Library Prep kits (Zymo, D5456). Conventional WGBS libraries from endosperm, embryo, and ovary were prepared using the methylC-seq method (Urich et al. 2015). MethylC-seq and PBAT reads were quality-filtered and trimmed of adapters using Cutadapt (Martin 2011), and reads were aligned to the genome with BS-Seeker2 (version 2.1.5) (Guo et al. 2013). For all reads except paired-end, previously published reads (Tan et al. 2016), PCR duplicates were removed prior to alignment with the BS-Seeker2 FilterReads.py module. DMRs were identified with CGmapTools, version 0.1.1 (Guo et al. 2018).
Additional details on tissue collection, CRISPR-Cas9 mutagenesis of drm2, small RNA library construction and sequencing analysis, mRNA sequencing analysis, mock egg isolations and qPCR quantification, genome annotations, and WGBS library construction and sequencing analysis are included in Supplemental Methods.
Data access
All raw sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA533115. All R scripts for statistical analyses and data visualization can be found at https://github.com/cxli233/gamete_smRNA_revision and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank Daniel Jones for assistance in sperm isolation; Zach Liechty, Christian Santos, and Joseph Edwards for assistance in R programming; and Alina Yalda and Jake Anichowski for greenhouse maintenance. The UC Davis Genome Center provided Illumina sequencing services. The UC Davis Plant Transformation Facility provided plant transformation services. This study was supported in part by resources and technical expertise from the Georgia Advanced Computing Resource Center, a partnership between the University of Georgia's Office of the Vice President for Research and Office of the Vice President for Information Technology. This research was funded by the National Science Foundation (IOS-1547760) and the U.S. Department of Agriculture (USDA) Agricultural Experiment Station (CA-D-XXX-6973-H).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.253674.119.
References
- Allen RS, Li J, Alonso-Peral MM, White RG, Gubler F, Millar AA. 2010. MicroR159 regulation of most conserved targets in Arabidopsis has negligible phenotypic effects. Silence 1: 18 10.1186/1758-907X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SN, Johnson CS, Jones DS, Conrad LJ, Gou X, Russell SD, Sundaresan V. 2013. Transcriptomes of isolated Oryza sativa gametes characterized by deep sequencing: evidence for distinct sex-dependent chromatin and epigenetic states before fertilization. Plant J 76: 729–741. 10.1111/tpj.12336 [DOI] [PubMed] [Google Scholar]
- Anderson SN, Johnson CS, Chesnut J, Jones DS, Khanday I, Woodhouse M, Li C, Conrad LJ, Russell SD, Sundaresan V. 2017. The zygotic transition is initiated in unicellular plant zygotes with asymmetric activation of parental genomes. Dev Cell 43: 349–358.e4. 10.1016/j.devcel.2017.10.005 [DOI] [PubMed] [Google Scholar]
- Autran D, Baroux C, Raissig MT, Lenormand T, Wittig M, Grob S, Steimer A, Barann M, Klostermeier UC, Leblanc O, et al. 2011. Maternal epigenetic pathways control parental contributions to Arabidopsis early embryogenesis. Cell 145: 707–719. 10.1016/j.cell.2011.04.014 [DOI] [PubMed] [Google Scholar]
- Blevins T, Podicheti R, Mishra V, Marasco M, Wang J, Rusch D, Tang H, Pikaard CS. 2015. Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. eLife 4: e09591 10.7554/eLife.09591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg M, Berger F. 2015. Chromatin remodelling during male gametophyte development. Plant J 83: 177–188. 10.1111/tpj.12856 [DOI] [PubMed] [Google Scholar]
- Borges F, Parent JS, van Ex F, Wolff P, Martínez G, Köhler C, Martienssen RA. 2018. Transposon-derived small RNAs triggered by miR845 mediate genome dosage response in Arabidopsis. Nat Genet 50: 186–192. 10.1038/s41588-017-0032-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calarco JP, Borges F, Donoghue MT, Van Ex F, Jullien PE, Lopes T, Gardner R, Berger F, Feijó JA, Becker JD, et al. 2012. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 151: 194–205. 10.1016/j.cell.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corem S, Doron-Faigenboim A, Jouffroy O, Maumus F, Arazi T, Bouché N. 2018. Redistribution of CHH methylation and small interfering RNAs across the genome of tomato ddm1 mutants. Plant Cell 30: 1628–1644. 10.1105/tpc.18.00167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasey KM, Zhai J, Borges F, Van Ex F, Regulski M, Meyers BC, Martienssen RA. 2014. miRNAs trigger widespread epigenetically activated siRNAs from transposons in Arabidopsis. Nature 508: 411–415. 10.1038/nature13069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuerda-Gil D, Slotkin RK. 2016. Non-canonical RNA-directed DNA methylation. Nat Plants 2: 16163 10.1038/nplants.2016.163 [DOI] [PubMed] [Google Scholar]
- Del Toro-De León G, García-Aguilar M, Gillmor CS. 2014. Non-equivalent contributions of maternal and paternal genomes to early plant embryogenesis. Nature 514: 624–627. 10.1038/nature13620 [DOI] [PubMed] [Google Scholar]
- Dooner HK, Wang Q, Huang JT, Li Y, He L, Xiong W, Du C. 2019. Spontaneous mutations in maize pollen are frequent in some lines and arise mainly from retrotranspositions and deletions. Proc Natl Acad Sci 116: 10734–10743. 10.1073/pnas.1903809116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann RM, Satyaki PRV, Klosinska M, Gehring M. 2017. A small RNA pathway mediates allelic dosage in endosperm. Cell Rep 21: 3364–3372. 10.1016/j.celrep.2017.11.078 [DOI] [PubMed] [Google Scholar]
- Fei Q, Yang L, Liang W, Zhang D, Meyers BC. 2016. Dynamic changes of small RNAs in rice spikelet development reveal specialized reproductive phasiRNA pathways. J Exp Bot 67: 6037–6049. 10.1093/jxb/erw361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu FF, Dawe RK, Gent JI. 2018. Loss of RNA-directed DNA methylation in maize chromomethylase and DDM1-type nucleosome remodeler mutants. Plant Cell 30: 1617–1627. 10.1105/tpc.18.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M. 2019. Epigenetic dynamics during flowering plant reproduction: evidence for reprogramming? New Phytol 224: 91–96. 10.1111/nph.15856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gent JI, Ellis NA, Guo L, Harkess AE, Yao Y, Zhang X, Dawe RK. 2013. CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res 23: 628–637. 10.1101/gr.146985.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant-Downton R, Kourmpetli S, Hafidh S, Khatab H, Le Trionnaire G, Dickinson H, Twell D. 2013. Artificial microRNAs reveal cell-specific differences in small RNA activity in pollen. Curr Biol 23: R599–R601. 10.1016/j.cub.2013.05.055 [DOI] [PubMed] [Google Scholar]
- Grover JW, Kendall T, Baten A, Burgess D, Freeling M, King GJ, Mosher RA. 2018. Maternal components of RNA-directed DNA methylation are required for seed development in Brassica rapa. Plant J 94: 575–582. 10.1111/tpj.13910 [DOI] [PubMed] [Google Scholar]
- Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen PY, Pellegrini M. 2013. BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics 14: 774 10.1186/1471-2164-14-774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Zhu P, Pellegrini M, Zhang MQ, Wang X, Ni Z. 2018. CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data. Bioinformatics 34: 381–387. 10.1093/bioinformatics/btx595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Qin S, Wessler SR. 2013. Comparison of class 2 transposable elements at superfamily resolution reveals conserved and distinct features in cereal grass genomes. BMC Genomics 14: 71 10.1186/1471-2164-14-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PH, He S, Buttress T, Gao H, Couchman M, Fischer RL, Zilberman D, Feng X. 2016. Arabidopsis male sexual lineage exhibits more robust maintenance of CG methylation than somatic tissues. Proc Natl Acad Sci 113: 15132–15137. 10.1073/pnas.1619074114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra CA, Feng X, Schoft VK, Hsieh TF, Uzawa R, Rodrigues JA, Zemach A, Chumak N, Machlicova A, Nishimura T, et al. 2012. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 337: 1360–1364. 10.1126/science.1224839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingouff M, Rademacher S, Holec S, Šoljić L, Xin N, Readshaw A, Foo SH, Lahouze B, Sprunck S, Berger F. 2010. Zygotic resetting of the HISTONE 3 variant repertoire participates in epigenetic reprogramming in Arabidopsis. Curr Biol 20: 2137–2143. 10.1016/j.cub.2010.11.012 [DOI] [PubMed] [Google Scholar]
- Ingouff M, Selles B, Michaud C, Vu TM, Berger F, Schorn AJ, Autran D, Van Durme M, Nowack MK, Martienssen RA, et al. 2017. Live-cell analysis of DNA methylation during sexual reproduction in Arabidopsis reveals context and sex-specific dynamics controlled by noncanonical RdDM. Genes Dev 31: 72–83. 10.1101/gad.289397.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, Kasprzewska A, Tennessen K, Fernandes J, Nan GL, Walbot V, Sundaresan V, Vance V, Bowman LH. 2009. Clusters and superclusters of phased small RNAs in the developing inflorescence of rice. Genome Res 19: 1429–1440. 10.1101/gr.089854.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, Conrad LJ, Patel R, Anderson S, Li C, Pereira A, Sundaresan V. 2018. Reproductive long intergenic noncoding RNAs exhibit male gamete specificity and Polycomb Repressive Complex 2-mediated repression. Plant Physiol 177: 1198–1217. 10.1104/pp.17.01269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, et al. 2013. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice (N Y) 6: 4 10.1186/1939-8433-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Ono A, Scholten S, Kinoshita T, Zilberman D, Okamoto T, Fischer RL. 2019. DNA demethylation by ROS1a in rice vegetative cells promotes methylation in sperm. Proc Natl Acad Sci 116: 9652–9657. 10.1073/pnas.1821435116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkbride RC, Lu J, Zhang C, Mosher RA, Baulcombe DC, Chen ZJ. 2019. Maternal small RNAs mediate spatial-temporal regulation of gene expression, imprinting, and seed development in Arabidopsis. Proc Natl Acad Sci 116: 2761–2766. 10.1073/pnas.1807621116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya R, Ohyanagi H, Niihama M, Watanabe T, Nakano M, Kurata N, Nonomura K. 2014. Rice germline-specific Argonaute MEL1 protein binds to phasiRNAs generated from more than 700 lincRNAs. Plant J 78: 385–397. 10.1111/tpj.12483 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Gent JI, Zynda G, Song J, Makarevitch I, Hirsch CD, Hirsch CN, Dawe RK, Madzima TF, McGinnis KM, et al. 2015. RNA-directed DNA methylation enforces boundaries between heterochromatin and euchromatin in the maize genome. Proc Natl Acad Sci 112: 14728–14733. 10.1073/pnas.1514680112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Shahid MQ, Xia J, Lu Z, Fang N, Wang L, Wu J, Chen Z, Liu X. 2017. Analysis of small RNAs revealed differential expressions during pollen and embryo sac development in autotetraploid rice. BMC Genomics 18: 129 10.1186/s12864-017-3526-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xu H, Russell SD, Sundaresan V. 2019. Step-by-step protocols for rice gamete isolation. Plant Reprod 32: 5–13. 10.1007/s00497-019-00363-y [DOI] [PubMed] [Google Scholar]
- Long JC, Xia AA, Liu JH, Jing JL, Wang YZ, Qi CY, He Y. 2018. Decrease in DNA methylation 1 (DDM1) is required for the formation of mCHH islands in maize. J Integr Plant Biol 61: 749–764. 10.1111/jipb.12733 [DOI] [PubMed] [Google Scholar]
- Lyons DB, Zilberman D. 2017. DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. eLife 6: e30674 10.7554/eLife.30674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17: 3 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Martinez G, Wolff P, Wang Z, Moreno-Romero J, Santos-González J, Conze LL, DeFraia C, Slotkin RK, Köhler C. 2018. Paternal easiRNAs regulate parental genome dosage in Arabidopsis. Nat Genet 50: 193–198. 10.1038/s41588-017-0033-4 [DOI] [PubMed] [Google Scholar]
- McCue AD, Panda K, Nuthikattu S, Choudury SG, Thomas EN, Slotkin RK. 2015. ARGONAUTE 6 bridges transposable element mRNA-derived siRNAs to the establishment of DNA methylation. EMBO J 34: 20–35. 10.15252/embj.201489499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mérai Z, Chumak N, García-Aguilar M, Hsieh TF, Nishimura T, Schoft VK, Bindics J, Slusarz L, Arnoux S, Opravil S, et al. 2014. The AAA-ATPase molecular chaperone Cdc48/p97 disassembles sumoylated centromeres, decondenses heterochromatin, and activates ribosomal RNA genes. Proc Natl Acad Sci 111: 16166–16171. 10.1073/pnas.1418564111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaki K, Talbert PB, Zhong CX, Dawe RK, Henikoff S, Jiang J. 2003. Chromatin immunoprecipitation reveals that the 180-bp satellite repeat is the key functional DNA element of Arabidopsis thaliana centromeres. Genetics 163: 1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederhuth CE, Bewick AJ, Ji L, Alabady MS, Kim KD, Li Q, Rohr NA, Rambani A, Burke JM, Udall JA, et al. 2016. Widespread natural variation of DNA methylation within angiosperms. Genome Biol 17: 194 10.1186/s13059-016-1059-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonomura K, Morohoshi A, Nakano M, Eiguchi M, Miyao A, Hirochika H, Kurata N. 2007. A germ cell–specific gene of the ARGONAUTE family is essential for the progression of premeiotic mitosis and meiosis during sporogenesis in rice. Plant Cell 19: 2583–2594. 10.1105/tpc.107.053199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Kim MY, Vickers M, Park JS, Hyun Y, Okamoto T, Zilberman D, Fischer RL, Feng X, Choi Y, et al. 2016. DNA demethylation is initiated in the central cells of Arabidopsis and rice. Proc Natl Acad Sci 113: 15138–15143. 10.1073/pnas.1619047114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillot M, Baroux C, Vazquez MA, Autran D, Leblanc O, Vielle-Calzada JP, Grossniklaus U, Grimanelli D. 2010. Embryo and endosperm inherit distinct chromatin and transcriptional states from the female gametes in Arabidopsis. Plant Cell 22: 307–320. 10.1105/tpc.109.071647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2019. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Russell SD, Gou X, Wong CE, Wang X, Yuan T, Wei X, Bhalla PL, Singh MB. 2012. Genomic profiling of rice sperm cell transcripts reveals conserved and distinct elements in the flowering plant male germ lineage. New Phytol 195: 560–573. 10.1111/j.1469-8137.2012.04199.x [DOI] [PubMed] [Google Scholar]
- Satyaki PRV, Gehring M. 2019. Paternally acting canonical RNA-directed DNA methylation pathway genes sensitize Arabidopsis endosperm to paternal genome dosage. Plant Cell 31: 1563–1578. 10.1105/tpc.19.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864. 10.1093/bioinformatics/btr026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoft VK, Chumak N, Mosiolek M, Slusarz L, Komnenovic V, Brownfield L, Twell D, Kakutani T, Tamaru H. 2009. Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep 10: 1015–1021. 10.1038/embor.2009.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- She W, Grimanelli D, Rutowicz K, Whitehead MW, Puzio M, Kotliński M, Jerzmanowski A, Baroux C. 2013. Chromatin reprogramming during the somatic-to-reproductive cell fate transition in plants. Development 140: 4008–4019. 10.1242/dev.095034 [DOI] [PubMed] [Google Scholar]
- Slotkin RK, Vaughn M, Borges F, Tanurdžić M, Becker JD, Feijó JA, Martienssen RA. 2009. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136: 461–472. 10.1016/j.cell.2008.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE. 2014. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol 21: 64–72. 10.1038/nsmb.2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan F, Zhou C, Zhou Q, Zhou S, Yang W, Zhao Y, Li G, Zhou DX. 2016. Analysis of chromatin regulators reveals specific features of rice DNA methylation pathways. Plant Physiol 171: 2041–2054. 10.1104/pp.16.00393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan F, Lu Y, Jiang W, Wu T, Zhang R, Zhao Y, Zhou DX. 2018. DDM1 represses noncoding RNA expression and RNA-directed DNA methylation in heterochromatin. Plant Physiol 177: 1187–1197. 10.1104/pp.18.00352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich MA, Nery JR, Lister R, Schmitz RJ, Ecker JR. 2015. MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat Protoc 10: 475–483. 10.1038/nprot.2014.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J, Gao H, Zhang J, Aldridge B, Vickers M, Higgins JD, Feng X. 2018. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat Genet 50: 130–137. 10.1038/s41588-017-0008-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Köhler C. 2017. Epigenetic processes in flowering plant reproduction. J Exp Bot 68: 797–807. 10.1093/jxb/erw486 [DOI] [PubMed] [Google Scholar]
- Xia R, Chen C, Pokhrel S, Ma W, Huang K, Patel P, Wang F, Xu J, Liu Z, Li J, et al. 2019. 24-nt reproductive phasiRNAs are broadly present in angiosperms. Nat Commun 10: 627 10.1038/s41467-019-08543-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelagandula R, Stroud H, Holec S, Zhou K, Feng S, Zhong X, Muthurajan UM, Nie X, Kawashima T, Groth M, et al. 2014. The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in Arabidopsis. Cell 158: 98–109. 10.1016/j.cell.2014.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D. 2013. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153: 193–205. 10.1016/j.cell.2013.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai J, Bischof S, Wang H, Feng S, Lee TF, Teng C, Chen X, Park SY, Liu L, Gallego-Bartolome J, et al. 2015a. A one precursor one siRNA model for Pol IV-dependent siRNA biogenesis. Cell 163: 445–455. 10.1016/j.cell.2015.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai J, Zhang H, Arikit S, Huang K, Nan GL, Walbot V, Meyers BC. 2015b. Spatiotemporally dynamic, cell-type–dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc Natl Acad Sci 112: 3146–3151. 10.1073/pnas.1418918112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang S, Wu W, Li L, Jiang T, Zheng B. 2018. Clearance of maternal barriers by paternal miR159 to initiate endosperm nuclear division in Arabidopsis. Nat Commun 9: 5011 10.1038/s41467-018-07429-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Zhou X, Shen K, Liu Z, Cheng T, Liu D, Cheng Y, Peng X, Sun MX. 2019. Two-step maternal-to-zygotic transition with two-phase parental genome contributions. Dev Cell 49: 882–893.e5. 10.1016/j.devcel.2019.04.016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.