

Abstract
In this research we describe the improvement of the water‐solubility of cyclic epitope mimics based on the HCV E2 glycoprotein by incorporation of suitable polar hinges. The poor solubility of epitope mimics based on peptide sequences in the envelope (E2) protein hampered their synthesis and purification and made it very difficult to prepare the molecular constructs for evaluation of their bioactivity. Since changes in the amino acid composition are hardly possible in these epitope mimics in order to increase water‐solubility, a polar cyclization hinge may offer a remedy leading to a significant increase of polarity and therefore water solubility. These polar hinges were applied in the synthesis of better water‐soluble HCV‐E2 epitopes. An azide functionality in the polar hinges allowed attachment of a tetraethylene glycol linker by Cu‐catalyzed azide‐alkyne cyclo‐addition (CuAAC) for a convenient conjugation to ELISA plates in order to evaluate the bio‐activity of the epitope mimics. The immunoassays showed that the use of more polar cyclization hinges still supported anti‐HCV antibody recognition and did not negatively influence their binding. This significantly increased solubility induced by polar hinges should therefore allow for the molecular construction and ultimate evaluation of synthetic vaccine molecules.
Keywords: cyclic peptides, cyclisation method, ELISA, epitope mimics, hepatitis C virus, polar hinge, synthetic vaccine
1. INTRODUCTION
The rapidly increasing therapeutic potential of cyclic peptides is due to multiple favourable molecular attributes.25 These attributes include the ability to access large and often flat surface areas within protein binding sites, thereby achieving high‐selectivity and binding‐affinity. Furthermore, peptide cyclization improves its proteolytic stability compared to the linear peptide. Earlier, we have shown that cyclic peptides therefore might be especially attractive in the molecular construction of epitope mimics.17,29 Moreover, it was recently found that cyclic peptides were better epitope mimics than the linear, non‐cyclized compounds.17,29 As part of our efforts directed towards synthetic vaccines we have been especially interested in HIV‐gp120 and the HCV‐E2.21, 29 Presently the focus is on HCV, which is a rapidly mutating and highly diverse virus. The natural immune response to this virus often falls short due to various immune evasive strategies and hypervariable immunogenic decoy domains of the virus that do not impede its viral efficiency.1, 18, 19, 22 In essence, the immune system of the host is exhausting itself by targeting ineffective viral epitopes. Despite enormous efforts, no vaccine is available as yet against HCV.20 An approach to remedy this might be selection of effective and conserved epitopes, which could be mimicked by cyclic peptides and thereby, circumvent problems with immunogenic decoys and inefficient epitopes.23
Multiple cyclic peptides were considered based on promising neutralizing epitopes located within the CD81 binding site of the HCV E2 glycoprotein. This binding site consists of 4 epitope domains (Epitopes I, II, III, and IV) to which various antibodies have been generated and isolated that bind these epitopes individually (continuous) or as parts of a larger domain (discontinuous).24 Their therapeutic potential is based on these antibodies being capable of neutralizing HCV infection by blocking the CD81 binding site of the E2 glycoprotein.2, 8, 26 Epitopes II and IV were found to bind the same class of HC84 antibodies.8, 24 Recently, we have shown that antibody HC84.1 binds our previously developed epitope II mimic without parts of epitope IV.17 Therefore, in this study we have focused on epitopes I, II, and III.
One of the most attractive cyclization methods, presently available is based on thioether‐formation with cysteine‐containing peptides. Electrophilic bromide (for example benzylic‐bromide) derivatives and Michael acceptors (Figure 1), among others, have been applied in the synthesis of many diverse (bi)cyclic peptides also towards synthetic vaccines.3, 4, 5, 9, 10, 11, 14, 15, 17, 27, 28, 29 However, depending on the sequence, the solubility of cyclized peptides onto a benzylic core (i.e. TBMB) or derivatives thereof can be very poor. Poor water‐solubility may hamper purification of cyclized peptides using reverse phase chromatography, resulting in very low yields thereby also limiting their therapeutic potential. In general, poor water‐solubility of peptides is often responsible for a large attrition rate similar to that observed in the small molecule drug discovery process.6
Figure 1.

Chemical structures of cyclization hinges for preparation of bicyclic peptides 1,3,5‐tris (bromomethyl)benzene (TBMB) (A); 1,3,5‐triacryloyl‐1,3,5‐triazinane (TATA), N,N′,N″‐(benzene‐1,3,5‐triyl)‐tris(2‐bromoacetamide) (TBAB) and N,N′,N″‐benzene‐1,3,5‐triyltrisprop‐2‐enamide (TAAB) described by Chen et al. 9 and Rim et al. 12 (B); and 2,4,6‐tris (bromomethyl)‐s‐triazine (TBMT) and 1,1′,1″‐(1,3,5‐triazinane‐1,3,5‐triyl)tris(2‐bromoethanone) (TATB) described by Van de Langemheen et al. 16 (C)
Several hinges with increasing polarity for cyclization of peptides have been described in the literature to improve the water solubility of resulting molecular constructs.3, 4, 5, 9, 10, 11, 12, 15, 16, 27 Examples of such polar hinges are shown in Figure 1 and include the 2,4,6‐1,3,5‐triacryloyl‐1,3,5‐triazinane (TATA), N,N′,N″‐(benzene‐1,3,5‐triyl)‐tris(2‐bromoacetamide) (TBAB) and N,N′,N″‐benzene‐1,3,5‐triyltrisprop‐2‐enamide (TAAB) tris (bromomethyl)‐s‐triazine (TBMT) and 1,1′,1″‐(1,3,5‐triazinane‐1,3,5‐triyl)tris(2‐bromoethanone) (TATB) cyclization hinges.9, 12, 16 Especially, the latter two, recently developed by us, have shown to be beneficial for the aqueous solubility of cyclic peptides.16
Recently, Heinis et al. have described a phage display protocol with respect to high‐throughput screening of bicyclic peptides, thereby opening up a wide range of potential therapeutics.4 Interestingly, they found that different hinge structures (i.e. TBMT, TATA, TBAB, and TAAB) incorporated into bicyclic peptides resulted in different conformations of the bicyclized peptide.9 Moreover, changing the cyclization hinge on a promising isolated sequence from the phage display library could reduce or even completely abolish (binding) activity.3, 9, 27 In addition, depending on the used cyclization hinge, different peptide consensus sequences were isolated from the phage display library.27 Taken together, this suggested that the core structure of the cyclization hinges does have a significant impact on the overall structure of the cyclized peptide.
The main goal of the work reported here focussed on the improvement of the water‐solubility of cyclic epitope mimics based on the HCV E2 glycoprotein by incorporation of suitable polar hinges. The poor solubility of the peptide sequences corresponding with these epitopes hampered the synthesis and purification of the epitope mimics, and made it very difficult to prepare the molecular construct for evaluation of their bioactivity. Evidently, possibilities to change amino acids in these epitope mimics in order to increase water‐solubility are very limited, as their peptide sequences have to resemble the mimicked peptide segments of the epitopes as closely as possible. Recently, we have addressed the issue of poor solubility of peptide loops for attachment to our molecular (TAC) scaffolds by development of polar hinges, which upon incorporation led to a significant increase of polarity and therefore their water solubility. Therefore, we wished to apply these polar hinges in the synthesis of better water‐soluble HCV‐E2 epitopes. Here, we describe the preparation of cyclized synthetic peptide sequences onto various cyclization (polar) hinges, equipped with a functional azide handle.16 In addition, an alkyne‐functionalized linker equipped with a trityl‐protected thiol moiety is described that allowed for conjugation of cyclic peptides with different core structures via by Cu‐catalyzed azide‐alkyne cyclo‐addition (CuAAC). Upon deprotection of the thiol‐moiety, the resulting epitope mimics were readily conjugated to a maleimide‐activated surface and their bio‐activity was evaluated by ELISA (Figure 2).
Figure 2.
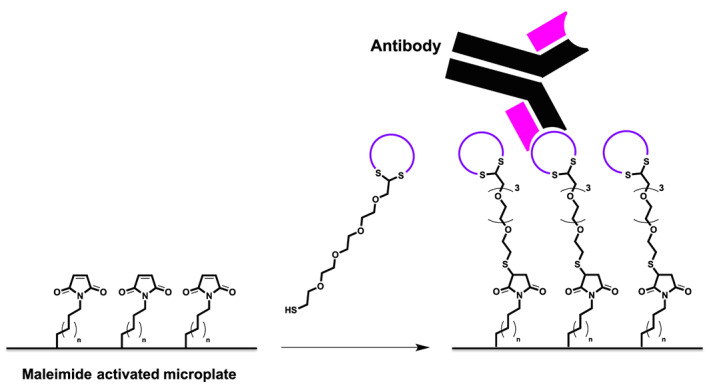
Thiol‐maleimide conjugation of epitope mimics forming a plate‐surface with uniformly oriented peptides used for ELISA to evaluate epitope mimicry
2. MATERIALS AND METHODS
2.1. General
All reagents and solvents were used as received. Fmoc‐amino acids were obtained from Activotec (Cambridge, United Kingdom) and N,N,N′,N′‐Tetramethyl‐O‐(6‐chloro‐1H‐benzotriazol‐1‐yl)uranium hexafluorophosphate (HCTU) was obtained from Matrix Innovation (Quebec, Canada). Tentagel S RAM resin (particle size 90 μm, capacity 0.25 mmol.g−1) was obtained from IRIS Biotech (Marktredwitz, Germany). Methyl tert‐butyl ether (MTBE), Hexane (HPLC grade) and TFA were obtained from Aldrich (Milwaukee, USA). DMF (Peptide grade) was obtained from VWR (Lutterworth, United Kingdom). Piperidine, DiPEA were obtained from AGTC Bioproducts (Hessle, United Kingdom) and 1,2‐ethanedithiol (EDT) was obtained from Merck (Darmstadt, Germany). HPLC grade CH2Cl2 and acetonitrile were obtained from Fischer Scientific (Loughborough, United Kingdom). Solid phase peptide synthesis was performed on a PTI Tribute‐UV peptide synthesizer. Lyophilizations were performed on a Christ Alpha 2–4 LDplus apparatus. Reactions were carried out at ambient temperature unless stated otherwise. Solvents were evaporated under reduced pressure at 40°C. Reactions in solution were monitored by TLC analysis and Rf‐values were determined on Merck pre‐coated silica gel 60 F‐254 (0.25 mm) plates. Spots were visualized by UV‐light and permanganate stain. Column chromatography was performed on Siliaflash P60 (40‐63 μm) from Silicycle (Canada) or on a Biotage Isolera One purification system using prepacked silica (KP‐SIL) Biotage SNAP cartridges. 1H NMR data was acquired on a Bruker 400 MHz spectrometer in CDCl3 as solvent. Chemical shifts (δ) are reported in parts per million (ppm) relative to trimethylsilane (TMS, 0.00 ppm). Analytical high‐pressure liquid chromatography (HPLC) was carried out on a Shimadzu instrument comprising a communication module (CBM‐20A), autosampler (SIL‐20HT), pump modules (LC‐20AT), UV/Vis detector (SPD‐20A) and system controller (Labsolutions V5.54 SP), with a Phenomenex Gemini C18 column (110 Å, 5 μm, 250 × 4.60 mm) or Dr. Maisch Reprosil Gold 200 C18 (5 μm, 250 × 4.60 mm). UV measurements were recorded at 214 and 254 nm, using a standard protocol: 100% buffer A (acetonitrile/H2O 5:95 with 0.1% TFA) for 2 min followed by a linear gradient of buffer B (acetonitrile/H2O 95:5 with 0.1% TFA) into buffer A (0–100% or 0–50%) over 30 min at a flow rate of 1.0 mL·min−1. Liquid chromatography mass spectrometry (LCMS) was carried out on a Thermo Scientific LCQ Fleet quadrupole mass spectrometer with a Dionex Ultimate 3000 LC using a Dr. Maisch Reprosil Gold 120 C18 column (110 Å, 3 μm, 150 × 4.0 mm) and the same linear gradients of buffer B into buffer A, flowrate and buffers as described for the analytical HPLC. Purification of the peptidic compounds was performed on an Agilent Technologies 1260 infinity preparative system using both UV and ELSD detectors with a Dr. Maisch Reprosil Gold 200 C18 (10 μm, 250 × 20 mm). Automatic collection of fractions was based on the UV measurements at 214 nm, using customized protocols using the same buffers as described for the analytical HPLC.
2.2. Hinge, linker and peptide synthesis
2.2.1. 2,4,6‐tris (bromomethyl)‐s‐triazine (TBMT) and 1,1′,1″‐(1,3,5‐triazinane‐1,3,5‐triyl)tris(2‐bromoethanone) (TATB)
These syntheses were carried out as described by van de Langemheen et al.16
2.2.2. 1‐(azidomethyl)‐3,5‐bis (bromomethyl)‐s‐triazine 1, azido triazinane‐tris(2‐bromoethanone) 2, and 1‐(azidomethyl)‐3,5‐bis (bromomethyl)benzene 3
These syntheses were carried out as described by van de Langemheen et al.16
2.2.3. Cyclization linker 4
These syntheses were carried out as described by van de Meuleman et al.17
2.2.4. Tetraethylene glycol mono‐trityl thioether 5
These syntheses were carried out as described earlier by Meuleman et al.17
2.2.5. Alkyne functionalized tetraethylene glycol monotrityl thioether 6
All steps were performed under N2 atmosphere. Tetraethylene glycol monotrityl thioether 5 (2.42 g, 5.3 mmol; 1.0 equiv.) was dissolved in dry THF (150 ml). The resulting solution was cooled to 0°C using an ice bath, followed by addition of sodium hydride (60% dispersion in mineral oil, 0.32 g, 8.0 mmol; 1.5 equiv.). The resulting reaction mixture was stirred for 1 hour at 0°C. After which, propargyl bromide (80 wt.% in toluene, 770 μl, 6.9 mmol; 1.3 equiv.) was added to the reaction mixture. The ice bath was removed and the reaction mixture was stirred for 1 day at RT, after which the reaction had not achieved complete conversion as yet according to TLC (EtOAc). Nevertheless, solvent was removed in vacuo and the residue was taken up in CH2Cl2 (100 ml). The resulting sodium bromide precipitate was filtered off over Celite and the filtrate was concentrated in vacuo. Purification by automated flash column chromatography using a linear gradient of EtOAc in petroleum ether 40–60°C (0–50% over 20 column volumes) afforded pure alkyne modified tetraethylene glycol monotrityl thioether 6 (1.50 g, 3.0 mmol; 58%). Rf = 0.86 (100% EtOAc); tR = 32.7 min; 1H‐NMR (400 MHz, CDCl3): δ = 7.41 (m, 6H, trityl o‐H), 7.27 (m, 6H, trityl m‐H), 7.21 (m, 3H, p‐H), 4.19 (d, 3JHH = 2.3 Hz, 2H, CH 2CCH), 3.67 (m, 4H, CH 2), 3.62 (m, 4H, CH 2), 3.56 (m, 2H, CH 2CH 2O‐Alkyne), 3.45 (m, 2H, CH 2CH 2O‐Alkyne), 3.30 (t, 3JHH = 6.9 Hz, 2H, SCH2CH 2), 2.43 (t, 3JHH = 6.9 Hz, 2H, SCH 2), 2.40 (t, 3JHH = 2.3 Hz, 1H, CCH); 13C‐NMR (101 MHz, CDCl3): δ = 144.8 (Ar‐C), 129.6 (trityl o‐C), 127.9 (trityl m‐C), 126.6 (trityl p‐C), 79.7 (CCH), 74.5 (CCH), 70.6 (CH2), 70.5 (CH2), 70.4 (CH2 CH2O‐Alkyne), 70.2 (CH2 CH2O‐Alkyne), 69.6 (SCH2 CH2), 69.1 (CH2), 66.6 (CS), 58.4 (CH2CCH), 31.7 (SCH2); HRMS: m/z calc. For C30H34O4S: 513.2076 [M + Na]+1; found: 513.2062.
2.2.6. General method for automated peptide synthesis
Peptides were synthesized on a PTI Tribute‐UV peptide synthesizer. Tentagel S RAM resin (1.0 g, 0.25 mmol, 1.0 equiv or 400 mg, 0.1 mmol, 1.0 equiv) was allowed to swell in DMF (3 x 10 min). De‐protection of the Fmoc group was achieved by treatment of the resin with 20% piperidine in DMF using the RV_top_UV_Xtend protocol from the Tribute‐UV peptide synthesizer followed by a DMF washing step (5 x 30 sec). Fmoc‐protected amino acids were coupled using HCTU (on the 0.1 mmol scale 5 equiv. were used and on the 0.25 mmol scale 4 equiv. were used) and DiPEA (on the 0.1 mmol scale 10 equiv. were used and on the 0.25 mmol scale 8 equiv. were used) in DMF, as a coupling reagent, with 2 min pre‐activation. The coupling time was 10 min when the peptide was synthesized on a 0.1 mmol scale and 20 min when the 0.25 mmol scale was conducted. After every coupling the resin was washed with DMF (6 x 30 sec), followed by a capping step for 10 minutes using capping solution (on both the 0.1 mmol scale and the 0.25 mmol scale 15 equiv. acetic anhydride and 3 equiv. DiPEA were used). After coupling of the last amino acid, the Fmoc group was cleaved using the standard de‐protection conditions (described above) and the resulting free N‐terminus was maintained (peptide 7) or acetylated (peptides 8–10) by treating the resin bounded peptide with capping solution (on both the 0.1 mmol scale and the 0.25 mmol scale 15 equiv. acetic anhydride and 3 equiv. DiPEA were used) for 10 minutes. After the last step the resin was washed with DMF (5 x 30 sec) and dried over a nitrogen flow for 10 min.
Cleavage and de‐protection was achieved by treatment of the resin with TFA/H2O/TIS/EDT (15 mL for the 0.25 mmol scale and 5 mL for the 0.1 mmol scale, 90:5:2.5:2.5, v/v/v/v) for 3 h at rt. The peptide was then precipitated in Et2O (90 mL for the 0.25 mmol scale and 45 mL for the 0.1 mmol scale), centrifuged (4500 rpm; 5 min), the supernatant decanted and the pellet washed 3 times with Et2O (90 mL for the 0.25 mmol scale and 45 mL for the 0.1 mmol scale). The resulting pellet was re‐dissolved in tBuOH:H2O (1:1, v/v) and lyophilized.
2.2.7. Peptide 7
This peptide was synthesized as described above by solid phase synthesis on a 0.25 mmol scale, which afforded crude peptide (384.0 mg). The crude peptide was used in the cyclization by alkylation step. t R = 16.5 min; LRMS: m/z calculated for C79H127N27O22S2: 935.96 ½[M + 2H]2+; found: 936.50.
2.2.8. Peptide 8
This peptide was synthesized as described above by solid phase synthesis on a 0.25 mmol scale, which afforded crude peptide (401.0 mg). The crude peptide was used in the cyclization by alkylation step. t R = 20.8 min; LRMS: m/z calculated for C89H117N21O19S2: 924.92 ½[M + 2H]2+; found: 925.42 (13C [+1]).
2.2.9. Peptide 9
This peptide was synthesized as described above by solid phase synthesis on a 0.25 mmol scale, which afforded crude peptide (354.2 mg). The crude peptide was used in the cyclization by alkylation step. t R = 18.8 min; LRMS: m/z calculated for C81H119N21O28S2: 949.91 ½[M + 2H]2+; found: 950.17 (13C [+1]).
2.2.10. Peptide 10
This peptide was synthesized as described above by solid phase synthesis on a 0.25 mmol scale, which afforded crude peptide (400.4 mg). The crude peptide was used in the cyclization by alkylation step. t R = 19.1 min; LRMS: m/z calculated for C90H119N21O19S2: 931.93 ½[M + 2H]2+; found: 932.33 (13C [+1]).
2.2.11. General method for peptide cyclization
Crude peptide (1.0 equiv.) was dissolved in DMF (30 ml), followed by addition of cyclization hinge (1.1–1.5 equiv.) in DMF (1 mL). Subsequently, an aqueous solution of NH4HCO3 (20 mM, pH 7.2<7.4; 10 mL) or H2O (10 mL) was added dropwise to the reaction mixture. The resulting reaction mixture consisted of <1mM peptide concentration was stirred for 15 minutes (NH4HCO3) or 1 hour (H2O) at room temperature, followed by removal of the solvents in vacuo (60°C). The residue was dissolved in tBuOH/H2O (1:1, v/v) aided by sonication. Lyophilization afforded the crude cyclized peptides.
2.2.12. Epitope mimics 34–37
Removal of the Trityl group was performed using TFA:H2O:TIS:EDT (90:5:2.5:2.5, v/v/v/v) (1 ml) for 15 minutes at room temperature, affording the free‐thiol moiety. Then, the product was precipitated in Et2O (15 ml) and collected by centrifugation (4500 rpm; 5 min). The collected precipitate was washed twice using Et2O (15 ml), followed by centrifugation (4500 rpm; 5 min). After which, the precipitate was dissolved in tBuOH:H2O (1:1, v/v) and lyophylized, which afforded the crude epitope mimics. The crude product was dissolved in HPLC buffer and the resulting solution was clarified by centrifugation (4500 rpm; 5 min), after which, the compound in the supernatant was purified by preparative HPLC.
2.2.13. Cyclic peptide 11
Peptide 7 (47.0 mg, 25.1 μmol; 1.0 equiv.) was treated with 1‐(azidomethyl)‐3,5‐bis (bromomethyl)‐s‐triazine 1 (12.1 mg, 37.6 μmol; 1.5 equiv.) in DMF/H2O as described above. The obtained crude product (54.5 mg) was dissolved in 6 mL buffer A and purified as two batches of 3 mL: 100% buffer A for 5 min followed by a linear gradient of buffer B into buffer A (0–40%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 11 (5.3 mg, 2.6 μmol; 9% overall yield). tR = 16.7 min; HRMS: calculated m/z for C85H131N33O22S2: 1015.9872 ½[M + 2H]+2; found 1015.9878; LRMS: calculated m/z for C85H131N33O22S2: 1015.99 ½[M + 2H]2+; found 1016.58 (13C [+1]).
2.2.14. Cyclic peptide 12
Peptide 7 (46.9 mg, 25.1 μmol; 1.0 equiv.) was treated with azido triazinane‐tris(2‐bromoethanone) 2 (15.8 mg, 38.4 μmol; 1.5 equiv.) in DMF/H2O as described above. The obtained crude product (66.5 mg) was dissolved in 6 mL buffer A and purified as two batches of 3 mL: 100% buffer A for 5 min followed by a linear gradient of buffer B into buffer A (0–40%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 12 (6.3 mg, 3.0 μmol; 10% overall yield). tR = 16.6 min; HRMS: calculated m/z for C88H137N33O25S2: 1061.0031 ½[M + 2H]2+; found 1061.0027; LRMS: calculated m/z for C88H137N33O25S2: 1061.00 ½[M + 2H]2+; found 1061.50 (13C [+1]).
2.2.15. Cyclic peptide 13
Peptide 7 (46.8 mg, 25.0 μmol; 1.0 equiv.) was treated with azido di (bromomethyl)benzene 3 (11.8 mg, 36.9 μmol; 1.5 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (60.6 mg) was dissolved in 6 mL buffer A and purified as two batches of 3 mL: 100% buffer A for 5 min followed by a linear gradient of buffer B into buffer A (0–40%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 13 (3.7 mg, 1.8 μmol; 6% overall yield). tR = 17.0 min; HRMS: calculated m/z for C88H134N30O22S2: 1014.4943 ½[M + 2H]2+; found 1014.4966; LRMS: calculated m/z for C88H134N30O22S2: 1014.49 ½[M + 2H]2+; found 1015.08 (13C [+1]).
2.2.16. Cyclic peptide 14
Peptide 8 (46.5 mg, 25.1 μmol; 1.0 equiv.) was treated with 1‐(azidomethyl)‐3,5‐bis (bromomethyl)‐s‐triazine 1 (9.9 mg, 30.7 μmol; 1.2 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (59.1 mg) was dissolved as batches of 10–15 mg in 1 mL buffer A:B (1:1, v/v) and purified: 100% buffer A for 5 min followed by a linear gradient of buffer B into buffer A (30–70%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 14 (11.6 mg, 5.8 μmol; 20% overall yield). tR = 21.0 min; HRMS: calculated m/z for C95H121N27O19S2: 1004.9465 ½[M + 2H]2+; found 1004.9470; LRMS: calculated m/z for C95H121N27O19S2: 1004.95 ½[M + 2H]2+; found 1005.50 (13C [+1]).
2.2.17. Cyclic peptide 15
Peptide 8 (46.5 mg, 25.1 μmol; 1.0 equiv.) was treated with azido triazinane‐tris(2‐bromoethanone) 2 (11.4 mg, 27.6 μmol; 1.1 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (63.2 mg) was dissolved in 3 mL buffer A:B (1:1, v/v) and purified: 20% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (20–60%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 15 (17.3 mg, 8.0 μmol; 28% overall yield). tR = 20.3 min; HRMS: calculated m/z for C98H127N27O22S2: 1049.9623 ½[M + 2H]2+; found 1049.9598; LRMS: calculated m/z for C98H127N27O22S2: 1049.96 ½[M + 2H]2+; found 1050.58 (13C [+1]).
2.2.18. Cyclic peptide 16
Peptide 8 (46.3 mg, 25.0 μmol; 1.0 equiv.) was treated with azido di (bromomethyl)benzene 3 (9.0 mg, 28.1 μmol; 1.1 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (57.6 mg) was dissolved as batches of 10–15 mg in 1 mL buffer A:B (1:1, v/v) and purified: 30% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (30–70%) over 80 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 16 (7.7 mg, 3.8 μmol; 13% overall yield). tR = 21.7 min; HRMS: calculated m/z for C98H124N24O19S2: 1003.4536 ½[M + 2H]2+; found 1003.4567; LRMS: calculated m/z for C98H124N24O19S2: 1003.45 ½[M + 2H]+2; found 1004.08 (13C [+1]).
2.2.19. Cyclic peptide 17
Peptide 9 (47.9 mg, 25.2 μmol; 1.0 equiv.) was treated with 1‐(azidomethyl)‐3,5‐bis (bromomethyl)‐s‐triazine 1 (10.7 mg, 33.3 μmol; 1.3 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (57.6 mg) was suspended in 3 mL buffer A:B (1:1, v/v) and centrifuged (4500 rpm; 5 min), which resulted in a pellet of un‐dissolved crude (29.8 mg; after lyophilization). The supernatant was purified: 20% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (20–60%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 17 (6.3 mg, 3.1 μmol; 18% overall yield). tR = 18.8 min; HRMS: calculated m/z for C87H123N27O28S2: 1029.9314 ½[M + 2H]2+; found 1029.9345; LRMS: calculated m/z for C87H123N27O28S2: 1029.93 ½[M + 2H]2+; found 1030.25 (13C [+1]).
2.2.20. Cyclic peptide 18
Peptide 9 (47.9 mg, 25.2 μmol; 1.0 equiv.) was treated with azido triazinane‐tris(2‐bromoethanone) 2 (11.3 mg, 27.5 μmol; 1.1 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (65.3 mg) was suspended in 3 mL buffer A:B (1:1, v/v) and centrifuged (4500 rpm; 5 min), which resulted in a pellet of un‐dissolved crude (16.8 mg; after lyophilization). The supernatant was purified: 20% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (20–60%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 18 (14.1 mg, 6.6 μmol; 26% overall yield). tR = 18.8 min; HRMS: calculated m/z for C90H129N27O31S2: 1074.9473 ½[M + 2H]2+; found 1074.9444; LRMS: calculated m/z for C90H129N27O31S2: 1074.95 ½[M + 2H]2+; found 1075.42 (13C [+1]).
2.2.21. Cyclic peptide 19
Peptide 9 (47.9 mg, 25.2 μmol; 1.0 equiv.) was treated with azido di (bromomethyl)benzene 3 (8.9 mg, 27.9 μmol; 1.1 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (56.3 mg) was suspended in 3 mL buffer A:B (1:1, v/v) and centrifuged (4500 rpm; 5 min), which resulted in a pellet of un‐dissolved crude (39.5 mg; after lyophilization). The supernatant was purified: 20% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (20–60%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 19 (1.4 mg, 0.7 μmol; 7% overall yield). tR = 19.6 min; HRMS: calculated m/z for C90H126N24O28S2: 1028.4386 ½[M + 2H]2+; found 1028.4374; LRMS: calculated m/z for C90H126N24O28S2: 1028.44 ½[M + 2H]2+; found 1028.92 (13C [+1]).
2.2.22. Cyclic peptide 20
Peptide 10 (46.6 mg, 25.0 μmol; 1.0 equiv.) was treated with 1‐(azidomethyl)‐3,5‐bis (bromomethyl)‐s‐triazine 1 (8.9 mg, 27.6 μmol; 1.1 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (59.7 mg) was dissolved in 3 mL buffer A:B (1:1, v/v) and purified: 20% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (20–60%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 20 (12.8 mg, 6.3 μmol; 22% overall yield). tR = 20.9 min; HRMS: calculated m/z for C96H123N27O19S2: 1011.9543 ½[M + 2H]2+; found 1011.9544; LRMS: calculated m/z for C96H123N27O19S2: 1011.95 ½[M + 2H]2+; found 1012.58 (13C [+1]).
2.2.23. Cyclic peptide 21
Peptide 10 (46.4 mg, 24.9 μmol; 1.0 equiv.) was treated with azido triazinane‐tris(2‐bromoethanone) 2 (11.5 mg, 27.9 μmol; 1.1 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (61.4 mg) was dissolved in 3 mL buffer A:B (1:1, v/v) and purified: 20% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (20–60%) over 40 min at a flow rate of 12.5 mL·min−1. Affording cyclic peptide 21 (15.0 mg, 7.1 μmol; 25% overall yield). tR = 20.5 min; HRMS: calculated m/z for C99H129N27O22S2: 1056.9702 ½[M + 2H]2+; found 1056.9688; LRMS: calculated m/z for C99H129N27O22S2: 1056.97 ½[M + 2H]2+; found 1057.58 (13C [+1]).
2.2.24. Cyclic peptide 22
Peptide 10 (46.5 mg, 25.0 μmol; 1.0 equiv.) was treated with azido di (bromomethyl)benzene 3 (8.8 mg, 27.6 μmol; 1.1 equiv.) DMF/NH4HCO3 (pH 7.2 < 7.4) as described above. The obtained crude product (59.2 mg) was dissolved in 3 mL buffer A:B (1:1, v/v) and purified: 20% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (20–60%) over 40 min at a flow rateof 12.5 mL·min−1. Affording cyclic peptide 22 (11.7 mg, 5.8 μmol; 20% overall yield). tR = 21.8 min; HRMS: calculated m/z for C99H126N24O19S2: 1010.4614 ½[M + 2H]2+; found 1010.4581; LRMS: calculated m/z for C99H126N24O19S2: 1010.46 ½[M + 2H]2+; found 1011.08 (13C [+1]).
2.2.25. General method for CuAAC of cyclized peptides to propargyl linker 6
Hinge cyclized peptide (1.0 equiv.), linker 3 (1.1 equiv.), and TBTA (0.5 equiv.) were dissolved separately in DMF (100 μl). The separate reagents were then combined. After which, CuSO4•5H2O (1.8 equiv.) was dissolved in H2O (100 μl), and sodium L‐ascorbate (10.0 equiv.) and aminoguanidine•HCl (11.0 equiv, addition of this was omitted in the preparation of epitope mimics 26–28, as no arginine residues are present in their peptide sequence) were each dissolved separately in H2O (50 μl). A premix was prepared by adding the sodium L‐ascorbate (50 μl) solution to the CuSO4•5H2O (100 μl) solution, followed by immediate addition of the aminoguanidine•HCl (50 μl) solution. Then, the premix (200 μl) was added to the reaction mixture, which was stirred for 15 minutes at room temperature under constant N2 flow. Analysis by analytical HPLC indicated completion of the reaction after 15 minutes, followed by addition of HPLC buffers A:B (1:1, v/v) (500 μl). The solution was centrifuged (12.000 rpm; 5 min) and the compound in the supernatant was purified by preparative HPLC: 50% buffer A for 5 min followed by linear gradient of buffer B into buffer A (50–100%) over 10 min at a flow rate of 12.5 mL·min−1 using the same buffers as described for the analytical HPLC. Fractions containing pure product were identified by analytical HPLC and were pooled and lyophilized, affording pure Trityl‐protected product as a fluffy white powder.
Removal of the Trityl group was performed using TFA:H2O:TIS:EDT (90:5:2.5:2.5, v/v/v/v) (1 ml) for 15 minutes at room temperature, affording the free‐thiol moiety. Then, the product was precipitated in Et2O (15 ml) and collected by centrifugation (4500 rpm; 5 min). The collected precipitate was washed twice using Et2O (15 ml), followed by centrifugation (4500 rpm; 5 min). After which, the precipitate was dissolved in tBuOH:H2O (1:1, v/v) and lyophylized, affording the pure product.
2.2.26. Epitope mimic 23
Cyclic peptide 11 (5.3 mg, 2.6 μmol) and linker 6 (5.0 mg, 10.2 μmol; dissolved in 355 μl DMF of which 100 μl was used) was subjected to the above described CuAAC with CuSO4•5H2O (1.9 mg, 7.6 μmol; dissolved in 162 μl H2O of which 100 μl was used), L‐ascorbate (16.0 mg, 80.8 μmol; dissolved in 162 μl H2O of which 50 μl was used), TBTA (1.1 mg, 2.1 μmol; dissolved in 179 μl DMF of which 100 μl was used), aminoguanidine•HCl (5.5 mg, 49.8 μmol; dissolved in 87 μl of which 50 μl was used). Purification and subsequent deprotection, afforded epitope mimic 23 (0.9 mg, 0.4 μmol, 15%). tR = 17.4 min; HRMS: calculated m/z for C96H151N33O26S3: 760.3635 1/3[M + 3H]+3; found 760.3664; LRMS: calculated m/z for C96H151N33O26S3: 1140.04 ½[M + 2H]2+/760.36 1/3[M + 3H]+3; found 1140.50/760.67 (13C [+1]).
2.2.27. Epitope mimic 24
Cyclic peptide 12 (3.7 mg, 1.7 μmol) and linker 6 (4.1 mg, 8.4 μmol; dissolved in 440 μl DMF of which 100 μl was used) was subjected to the above described CuAAC with CuSO4•5H2O (4.7 mg, 18.8 μmol; dissolved in 605 μl H2O of which 100 μl was used), L‐ascorbate (12.7 mg, 64.1 μmol; dissolved in 188 μl H2O of which 50 μl was used), TBTA (1.7 mg, 3.2 μmol; dissolved in 400 μl DMF of which 100 μl was used), aminoguanidine•HCl (6.5 mg, 58.8 μmol; dissolved in 157 μl of which 50 μl was used). Purification and subsequent deprotection, afforded epitope mimic 24 (1.3 mg, 0.6 μmol, 35%). tR = 17.3 min; HRMS: calculated m/z for C99H157N33O29S3: 790.3740 1/3[M + 3H]+3; found 790.3769; LRMS: calculated m/z for C99H157N33O29S3: 1185.06 ½[M + 2H]2+/790.37 1/3[M + 3H]+3; found 1185.50/790.67 (13C [+1]).
2.2.28. Epitope mimic 25
Cyclic peptide 13 (3.7 mg, 1.8 μmol) and linker 6 (4.6 mg, 9.4 μmol; dissolved in 475 μl DMF of which 100 μl was used) was subjected to the above described CuAAC with CuSO4•5H2O (3.3 mg, 13.2 μmol; dissolved in 405 μl H2O of which 100 μl was used), L‐ascorbate (9.9 mg, 50.0 μmol; dissolved in 139 μl H2O of which 50 μl was used), TBTA (0.9 mg, 1.7 μmol; dissolved in 210 μl DMF of which 100 μl was used), and aminoguanidine•HCl (6.8 mg, 61.5 μmol; dissolved in 155 μl of which 50 μl was used). Purification and subsequent deprotection, afforded only a trace of epitope mimic 25. tR = 17.5 min; HRMS: calculated m/z for C99H154N30O26S3: 759.3682 1/3[M + 3H]+3; found 759.3712; LRMS: calculated m/z for C99H154N30O26S3: 1138.55 ½[M + 2H]2+/759.37 1/3[M + 3H]3+; found 1139.08/759.67 (13C [+1]).
2.2.29. Epitope mimic 26
Cyclic peptide 14 (2.0 mg, 1.0 μmol) was dissolved in 200 μl of a linker 6 (10.9 mg, 22.2 μmol; dissolved in 4.0 ml DMF) solution. Next, 100 μl of a TBTA (0.3 mg, 0.6 μmol; dissolved in 125 μl DMF) solution was added. Then, 100 μl of a CuSO4•5H2O (1.6 mg, 6.4 μmol; dissolved in 355 μl H2O) solution and 100 μl of a L‐ascorbate (3.2 mg, 16.2 μmol; dissolved in 160 μl H2O) solution were premixed, of which 200 μl was added to the reaction mixture. After 15 minutes, the reaction was complete as gauged by analytical HPLC. Purification and subsequent deprotection, afforded epitope mimic 26 (0.4 mg, 0.2 μmol, 20%). tR = 21.2 min; HRMS: calculated m/z for C106H141N27O23S3: 1129.0006 ½[M + 2H]2+; found 1129.0049; LRMS: calculated m/z for C106H141N27O23S3: 1129.00 ½[M + 2H]2+; found 1129.33 (13C [+1]).
2.2.30. Epitope mimic 27
Cyclic peptide 15 (2.0 mg, 1.0 μmol) was dissolved in 200 μl DMF, followed by addition of 200 μl of a linker 6 (5.2 mg, 10.5 μmol; dissolved in 2.1 ml DMF) solution. Next, 200 μl of a TBTA (0.3 mg, 0.6 μmol; dissolved in 200 μl DMF) solution was added. Then, 400 μl of a CuSO4•5H2O (2.9 mg, 11.6 μmol; dissolved in 1.3 ml H2O) solution and 400 μl of a L‐ascorbate (6.8 mg, 34.3 μmol; dissolved in 690 μl H2O) solution were premixed, of which 400 μl was added to the reaction mixture. After 15 minutes, the reaction was complete as gauged by analytical HPLC. Purification and subsequent deprotection, afforded epitope mimic 27 (0.9 mg, 0.4 μmol, 40%). tR = 20.9 min; HRMS: calculated m/z for C109H147N27O26S3: 1174.0165 ½[M + 2H]2+; found 1174.0163; LRMS: calculated m/z for C109H147N27O26S3: 11174.02 ½[M + 2H]2+; found 1174.42 (13C [+1]).
2.2.31. Epitope mimic 28
Cyclic peptide 16 (2.0 mg, 1.0 μmol) was dissolved in 200 μl of a linker 6 (10.9 mg, 22.2 μmol; dissolved in 4.0 ml DMF) solution. Next, 100 μl of a TBTA (0.9 mg, 1.7 μmol; dissolved in 375 μl DMF) solution was added. Then, 400 μl of a CuSO4•5H2O (4.0 mg, 16.0 μmol; dissolved in 890 μl H2O) solution and 400 μl of a L‐ascorbate (11.7 mg, 59.0 μmol; dissolved in 590 μl H2O) solution were premixed, of which 200 μl was added to the reaction mixture. After 15 minutes, the reaction was complete by analytical HPLC. Purification and subsequent deprotection, afforded only a trace of epitope mimic 28. tR = 21.5 min; HRMS: calculated m/z for C109H144N24O23S3: 1127.5077 ½[M + 2H]2+; found 1127.5071; LRMS: calculated m/z for C109H144N24O23S3: 1127.51 ½[M + 2H]2+; found 1127.92 (13C [+1]).
2.2.32. Epitope mimic 29
Pure cyclic peptide 17 (3.3 mg, 1.6 μmol) and linker 6 (3.1 mg, 6.3 μmol; dissolved in 262 μl DMF of which 100 μl was used) was subjected to the above described CuAAC with CuSO4•5H2O (2.3 mg, 9.2 μmol; dissolved in 320 μl H2O of which 100 μl was used), L‐ascorbate (11.8 mg, 59.6 μmol; dissolved in 185 μl H2O of which 50 μl was used), TBTA (1.7 mg, 3.2 μmol; dissolved in 440 μl DMF of which 100 μl was used), aminoguanidine•HCl (9.0 mg, 81.4 μmol; dissolved in 230 μl of which 50 μl was used). Purification and subsequent deprotection, afforded only a trace of epitope mimic 29.
2.2.33. Epitope mimic 30
Cyclic peptide 18 (10.7 mg, 5.0 μmol) and linker 6 (4.1 mg, 8.4 μmol; dissolved in 112 μl DMF of which 100 μl was used) was subjected to the above described CuAAC with CuSO4•5H2O (10.2 mg, 40.9 μmol; dissolved in 455 μl H2O of which 100 μl was used), L‐ascorbate (17.7 mg, 89.3 μmol; dissolved in 89.3 μl H2O of which 50 μl was used), TBTA (4.9 mg, 9.2 μmol; dissolved in 405 μl DMF of which 100 μl was used), aminoguanidine•HCl (15.8 mg, 143.0 μmol; dissolved in 130 μl of which 50 μl was used). Purification and subsequent deprotection, afforded epitope mimic 30 (1.7 mg, 0.7 μmol, 14%). tR = 19.2 min; HRMS: calculated m/z for C101H149N27O35S3: 1220.9834 ½[M + 2Na]2+; found 1220.9838 LRMS: calculated m/z for C101H149N27O35S3: 1199.00 ½[M + 2H]2+; found 1199.42 (13C [+1]).
2.2.34. Scrambled mimic 31
Cyclic peptide 20 (6.0 mg, 3.0 μmol) and linker 6 (8.0 mg, 16.3 μmol; dissolved in 490 μl DMF of which 100 μl was used) was subjected to the above described CuAAC with CuSO4•5H2O (5.8 mg, 23.2 μmol; dissolved in 430 μl H2O of which 100 μl was used), L‐ascorbate (15.1 mg, 76.2 μmol; dissolved in 127 μl H2O of which 50 μl was used), TBTA (1.8 mg, 3.4 μmol; dissolved in 245 μl DMF of which 100 μl was used), aminoguanidine•HCl (7.4 mg, 66.9 μmol; dissolved in 101 μl of which 50 μl was used). Purification and subsequent deprotection, afforded epitope mimic 31 (1.0 mg, 0.4 μmol, 13%). tR = 21.0 min; HRMS: calculated m/z for C107H143N27O23S3: 1136.0084 ½[M + 2H]2+; found 1136.0112; LRMS: calculated m/z for C107H143N27O23S3: 1136.01 ½[M + 2H]2+/757.67 1/3[M + 3H]3+; found 1136.50/758.00 (13C [+1]).
2.2.35. Scrambled mimic 32
Cyclic peptide 21 (7.0 mg, 3.3 μmol; dissolved in 200 μl) and linker 6 (7.8 mg, 15.9 μmol; dissolved in 640 μl DMF of which 200 μl was used) was subjected to the above described CuAAC with CuSO4•5H2O (7.1 mg, 28.4 μmol; dissolved in 955 μl H2O of which 200 μl was used), L‐ascorbate (17.6 mg, 88.8 μmol; dissolved in 270 μl H2O of which 100 μl was used), TBTA (1.8 mg, 3.4 μmol; dissolved in 410 μl DMF of which 200 μl was used), aminoguanidine•HCl (10.9 mg, 98.6 μmol; dissolved in 150 μl of which 100 μl was used). Purification and subsequent deprotection, afforded epitope mimic 32 (3.1 mg, 1.3 μmol, 39%). tR = 20.9 min; HRMS: calculated m/z for C110H149N27O26S3: 1181.0243 ½[M + 2H]2+; found 1181.0253; LRMS: calculated m/z for C110H149N27O26S3: 1181.02 ½[M + 2H]2+/787.68 1/3[M + 3H]3+; found 1181.58/788.17 (13C [+1]).
2.2.36. Scrambled mimic 33
Cyclic peptide 22 (4.1 mg, 2.0 μmol) and linker 6 (7.4 mg, 15.1 μmol; dissolved in 685 μl DMF of which 100 μl was used) was subjected to the above described CuAAC with CuSO4•5H2O (3.3 mg, 13.2 μmol; dissolved in 365 μl H2O of which 100 μl was used), L‐ascorbate (11.6 mg, 58.6 μmol; dissolved in 147 μl H2O of which 50 μl was used), TBTA (1.3 mg, 2.4 μmol; dissolved in 265 μl DMF of which 100 μl was used), aminoguanidine•HCl (8.2 mg, 74.2 μmol; dissolved in 168 μl of which 50 μl was used). Purification and subsequent deprotection, afforded only a trace of epitope mimic 33. tR = 21.3 min; HRMS: calculated m/z for C110H146N24O23S3: 1134.5156 ½[M + 2H]2+; found 1134.5160; LRMS: calculated m/z for C110H146N24O23S3: 1134.52 ½[M + 2H]2+/756.68 1/3[M + 3H]3+; found 1135.00/756.92 (13C [+1]).
2.2.37. Epitope mimic 34
Crude peptide 7 (38.2 mg, 20.4 μmol, 1.1 equiv.) was treated with our previously developed cyclization linker 4 3 (22.3 mg, 30.6 μmol; 1.5 equiv.) as described above in the general method for peptide cyclization. The obtained crude product (34.0 mg) was suspended in 4 mL buffer A and centrifuged (4500 rpm; 5 min), which resulted in a pellet of un‐dissolved crude (9.5 mg; after lyophilization). The supernatant was purified in two batches of 2 mL: 0% buffer A for 5 min followed by a linear gradient of buffer B into buffer A (0–40%) over 40 min at a flow rate of 12.5 mL·min−1. Affording, epitope mimic 34 (1.7 mg, 0.8 μmol; 4% overall yield). tR = 17.5 min; HRMS: calculated m/z for C96H151N27O26S3: 1098.0321 ½[M + 2H]2+; found 1098.0355; LRMS: calculated m/z for C96H151N27O26S3: 1098.03 ½[M + 2H]2+/732.36 1/3[M + 3H]3+; found 1098.50/732.50 (13C [+1]).
2.2.38. Epitope mimic 35
Crude peptide 8 (46.2 mg, 25.0 μmol; 1.0 equiv.) was treated with our previously developed cyclization linker 4 3 (27.3 mg, 37.5 μmol; 1.5 equiv.) as described above in the general method for peptide cyclization. The obtained crude product (53.9 mg) was dissolved as three batches of 10–20 mg in 3 mL buffer A:B (1:1, v/v) and purified: 100% buffer A for 5 min followed by a linear gradient of buffer B into buffer A (0–50%) over 60 min at a flow rate of 12.5 mL·min−1. Affording epitope mimic 35 (9.7 mg, 4.5 μmol; 15% overall yield). tR = 22.3 min; HRMS: calculated m/z for C106H141N21O23S3: 1086.9914 ½[M + 2H]2+; found 1086.9949; LRMS: calculated m/z for C106H141N21O23S3: 1086.99 ½[M + 2H]2+; found 1087.25 (13C [+1]).
2.2.39. Epitope mimic 36
Crude peptide 9 (48.8 mg, 25.7 μmol; 1.0 equiv.) was treated with our previously developed cyclization linker 4 3 (28.0 mg, 38.4 μmol; 1.5 equiv.) as described above in the general method for peptide cyclization. The obtained crude product (56.6 mg) was suspended in 6 mL buffer A:B (1:1, v/v) and centrifuged (4500 rpm; 5 min), which resulted in a pellet of un‐dissolved crude (19.4 mg; after lyophilization). The supernatant was purified: 20% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (20–60%) over 40 min at a flow rate of 12.5 mL·min−1. Affording epitope mimic 36 (0.4 mg, 0.2 μmol; 1% overall yield). tR = 20.1 min; HRMS: calculated m/z for C98H143N21O32S3: 1109.9607 ½[M‐2H]−2; found 1109.9435; LRMS: calculated m/z for C98H143N21O32S3: 1111.98 ½[M + 2H]2+; found 1112.33 (13C [+1]).
2.2.40. Scrambled mimic 37
Crude cyclic precursor 10 (47.5 mg, 25.5 μmol; 1.0 equiv.) was treated with our previously developed cyclization linker 4 3 (20.9 mg, 28.7 μmol; 1.1 equiv.) as described above in the general method for peptide cyclization. The obtained crude product (52.4 mg) was dissolved in 3 mL buffer A:B (1:1, v/v) and purified: 20% buffer B in buffer A for 5 min followed by a linear gradient of buffer B into buffer A (20–60%) over 40 min at a flow rate of 12.5 mL·min−1. Affording epitope mimic 37 (10.1 mg, 4.6 μmol; 16% overall yield). tR = 22.0 min; HRMS: calculated m/z for C107H143N21O23S3: 1093.9992 ½[M + 2H]2+; found 1093.9997; LRMS: calculated m/z for C107H143N21O23S3: 1094.00 ½[M + 2H]2+; found 1094.58 (13C [+1]).
2.2.41. Monoclonal antibodies
The Mabs AP3313, HC84.18, and DAO531 used in this work were previously described.
2.2.42. Immobilisation method and ELISA
Immobilisation
Pierce® maleimide activated 96‐well plates were purchased from Thermo Scientific. The wells were washed three times with 200 μL wash buffer (0.1 M Na3PO4, 0.15 M NaCl, 0.05% Tween®‐20 detergent; pH 7.2). Then, the desired epitope mimic (100–500 μg) was suspended in 1 mL binding buffer (0.1 M Na3PO4, 0.15 M NaCl, 10mM EDTA; pH 7.2) and further diluted (10–50 fold) to a concentration of 10 μg/mL. To each well, 200 μL of the epitope mimic solution was added and incubated overnight at 4°C. After this, the wells were washed three times with 200 μL wash buffer. For capping unreacted maleimide groups, immediately before use, a solution of 10 μg/mL N‐acetylated cysteine was prepared and 200 μL was added to each of the wells, followed by incubation of 1–2 hours at room temperature. The wells were washed three times with 200 μL wash buffer and used or stored dry at −20°C.
ELISA
The wells were washed three times with 200 μL wash buffer. A three‐fold dilution series of primary anti‐HCV E2 antibodies (AP3313 [10.0–0.0 ng/ml]; HC84.18 [100.0–0.1 ng/ml]; DAO531 [15.0–0.0 ng/ml]) was prepared over 7 steps and 100 μL was transferred to each well, followed by incubation for 1–2 hours at room temperature. After this, the wells were washed three times with PBST, before supplying 100 μL 1:2000 secondary α‐mouse A4416 (Sigma) to each well. Incubation continued for 1–2 hours at room temperature, followed by washing the wells three times with PBST. The plates were developed using 100 μL 3, 3′, 5, 5′ tetramethylbenzidine (TMB) solution per well, obtained from Life Technologies, and incubating for 25 minutes at room temperature, after which, further development was stopped using 200 μL 0.5 M H2SO4 per well. Absorbance at 450 nm was measured on a Varioskan (Thermoscientific) or PHERAstar FS (BMG Labtech) instrument. Background signal (no Mab) was subtracted. The data are represented as a percentage relative to immobilized epitope mimic 34, 35, and 36 for antibodies AP33, HC84.1, and DAO5, respectively, with the absorbance of the highest Mab concentration being set to 100% per individual experiment. Data was collected from 3 to 6 independent experiments (AP33 [n = 5]; HC84.1 [n = 6]; DAO5 [n = 3]). All experiments were performed in duplicate. Error bars represent the standard deviation.
3. RESULTS AND DISCUSSION
Thus, in order to improve the water‐solubility of the recently developed cyclic and linear epitope HCV‐E2 mimics equipped with a TEG spacer for binding to a surface using maleimide‐thiol conjugation,17 we have incorporated our more polar cyclization hinges 1 and 2 (Figure 3)16 in addition to our previously used benzyl‐derivative 3.17 Instead of installing the different cyclization hinges on our previously developed TEG spacer directly, as was earlier described for the bis‐benzyl bromide hinge 4, we decided to opt for a more modular approach. The peptides were cyclized onto the hinges first, followed by CuAAC to install the S‐Trityl‐TEG linker used for conjugation to a maleimide plate. The required propargyl S‐trityl‐TEG linker 6 was easily accessible from the earlier synthesized S‐Trityl‐TEG 5 by alkylation (Scheme 1).17
Figure 3.

1‐(azidomethyl)‐3,5‐bis (bromomethyl)‐s‐triazine (DBMT‐N3) 1, azido triazinane‐tris(2‐bromoethanone) (TADB‐N3) 2, and 1‐(azidomethyl)‐3,5‐bis (bromomethyl)benzene (DBMB‐N3) 3 16,17
Scheme 1.

Synthesis of the propargyl moiety containing PEG‐based thiol linker for cu‐catalyzed azide‐alkyne cyclo‐addition of cyclic peptides
Next, the required dicysteine containing peptides were obtained by solid phase peptide synthesis (Table 1). These peptide sequences originated from promising antigenic regions located within the HCV‐E2 glycoprotein denoted as epitopes I (I411QLINTNGSWHINR424, 7), II (G436 WVAGLFYYHKFN448, 8), and III (T528WGENETDVFLLNN541, 9). Epitope I is a flexible broadly neutralizing immunogenic domain recognized by neutralizing antibody (Nab) AP33 (Table 1).7, 13, 30 Epitope II is a broadly neutralizing immunogenic domain recognized by Nab HC84.1 (Table 1).8 Particularly, broadly neutralizing antibodies AP3313 and HC84.18 are of special interest as they have been found to neutralize HCV pseudo‐particles (HCVpp) carrying E2 glycoprotein of various genotypes, indicating that these are highly conserved epitopes in an otherwise rapidly mutating virus. Epitope III is an immunogenic domain of interest, as together with epitopes I and II it is located in the CD81 binding site of the viral E2 glycoprotein.31 A sequence within epitope III is recognized by non‐neutralizing antibody DAO5 (Table 1),31 which despite being non‐neutralizing will still allow us to validate the conformation of our epitope mimics. In addition, a scrambled peptide sequence 10 was synthesized based on the epitope II region as a negative control.
Table 1.
Overview of synthetic peptide sequence corresponding to epitope I, II, and III of the HCV E2 glycoprotein. Including a scrambled negative control 10

Cyclization of dicysteine peptides with different hinges DBMT‐N3 1, TADB‐N3 2, and DBMB‐N3 3 (Figure 3) was carried out as described by Van de Langemheen et al (Scheme 2).16 Precursor peptide and the chosen cyclization hinge were dissolved in DMF, followed by dropwise addition of NH4HCO3 buffer (20mM; pH 7.2 < 7.4) or H2O. A lower pH was used when lysine‐amines and/or free N‐terminal amines were present in the peptide sequence to improve selectivity towards thioether formation and avoid alkylation of the amine. H2O was used for cyclic peptides 11 and 12 to prevent a suspension and potential polymerization, with the exception of cyclic peptide 13 that required NH4HCO3 buffer. The purification of peptides cyclized on polar hinges DBMT‐N3 1 and TADB‐N3 2 hinges was significantly improved, which resulted in higher yields compared to the peptides cyclized with the DBMB‐N3 3 hinge (Scheme 2). Furthermore, HPLC‐traces showed shorter retention times for peptides cyclized with the DBMT‐N3 1 and TADB‐N3 2 polar hinges, indicating a higher polarity compared to peptides cyclized with benzylic hinge 3 (Figure 4). Thus, the water solubility was indeed improved using the polar DBMT‐N3 1 and TADB‐N3 2 cyclization hinges. The effect of the latter was the most prominent.
Scheme 2.
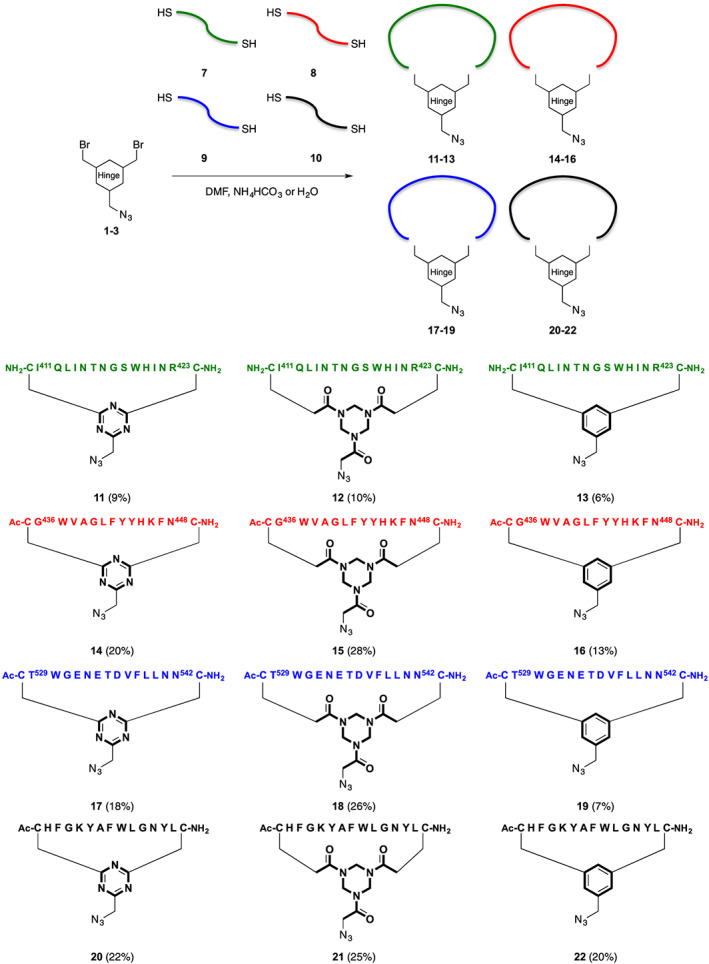
Cyclization of precursor peptides 7–10 with azido cyclization hinges DBMT‐N3 1, TADB‐N3 2, and DBMB‐N3 3. The reported yields are of purified products, after preparative reverse phase HPLC
Figure 4.

Analytical HPLC traces overlay of peptides 7–9 cyclized on DBMT‐N3 (1; purple), TADB‐N3 (2; pink), and DBMB‐N3 (3; black), cyclization hinges. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; gradient: 0 to 100% buffer B in 30 minutes
It was possible to gauge the improved aqueous solubility by assessing the concentration of the peptide cyclized using the different hinges DMBT‐N3 1, TADB‐N3 2, and DBMB‐N3 3. This was demonstrated for obtaining cyclic peptides 17, 18, and 19 from the same batch of (linear) peptide 9. Purification of these compounds resulted in a suspension that was clarified by centrifugation. The afforded pellet was resuspended and lyophilized to assess the amount of crude product that was present in the supernatant and subsequently purified. Crude cyclic peptide 18 based on TADB‐N3 2 afforded the highest concentration (16.2 mg/mL) in the supernatant, followed by the concentration (9.3 mg/mL) of cyclic peptide 17. As expected cyclic peptide 19 based on DBMB‐N3 3 resulted in the lowest concentration (5.6 mg/mL) in the supernatant. This showed that the aqueous solubility improved going from DBMB‐N3 3, DBMT‐N3 1 to TADB‐N3 2 used as linkers. Purification of crude cyclic peptides 11–13 was not investigated in this way, since the resulting suspension was too fine and could not be clarified by centrifugation or filtration. In addition, crude cyclic peptides 14–16 and 20–22 were readily soluble in the concentrations used for purification and yielded none to negligible insoluble pellets after centrifugation. Considering the low to modest yields obtained for the purified cyclic peptides, we did not perform a similar concentration study for the purified products.
It is important to note that the amino‐acid sequence does remain the more dominant factor with respect to overall aqueous solubility of the cyclic peptides. In addition, the therapeutic potential of the (cyclic) peptides is highly dependent on their amino‐acid composition and does not allow for a lot of changes. For example, peptide 7 exhibits a very poor water solubility despite leaving the N‐terminus non‐acetylated, as well as incorporation of an extra Arginine‐residue. This extra Arginine‐residue was only possible because it is present in roughly 50% of the known available sequences of the HCV E2 glycoprotein and was not part of the epitope sequence.24
CuAAC is a widely used and highly versatile conjugation method of an azide and alkyne moiety.32 Typically, the reaction can be conducted using moderate quantities of reagents being CuSO4•5H2O (0.3 equiv.), sodium‐L‐ascorbate (0.9 equiv.) and TBTA (0.15 equiv.) in a mixture of DMF and H2O (3:2, v/v).29 However, these conditions were found to be slow and unsuitable for using our newly developed alkyne linker 6. Possibly, this could be ascribed to an interaction of the thioether moieties with the formed Cu(I)‐catalyst. Optimization of the reaction conditions afforded our desired ‘clicked’ products 23, 24, 26, 27, 30, 31, and 32 within 15 minutes at room temperature using more equivalents of the reagents CuSO4•5H2O (1.8 equiv.), sodium‐L‐ascorbate (10 equiv.) and TBTA (0.5 equiv.) still in a mixture of DMF and H2O (3:2, v/v) (Scheme 3).
Scheme 3.

Cyclic epitope mimics with different hinges DBMT‐N3 (11, 14, 17, 20), TADB‐N3 (12, 15, 18, 21), and DBMB‐N3 (13, 16, 19, 22) based on loops I (green), II (red), III (blue), and a scrambled negative control (black). The red crosses indicate the mimics, which were only obtained in trace amounts. The green tick marks indicate the mimics, which were obtained in satisfactory amounts
Arginine‐dehydroascorbate adduct formation has been reported as a side‐reaction for CuAAC in the presence of arginine‐residues.33 Dehydroascorbate is a by‐product formed from ascorbate after reduction of Cu (II) to the catalytically active Cu(I)‐species. Similar dehydroascorbate adducts were formed here after prolonged reaction times with the DBMT‐N3 1 polar hinge structure. As was reported, formation of dehydroascorbate adducts could be prevented/decreased by supplementing aminoguanidine•HCl (11 equiv.) to the CuAAC reaction mixture.
After CuAAC, epitope mimics containing the trityl‐group protecting the thiol functionality, had to be purified. However, the presence of this trityl‐group decreased the water‐solubility of these epitope mimics considerably, resulting in benzylic‐hinge based mimics 25, 28, and 33 to be obtained only in trace amounts (indicated with red crosses in Scheme 3). Furthermore, the water‐solubility of peptide 9 was not sufficiently improved by the DBMT‐N3 1 polar hinge and also only trace amounts of the corresponding epitope mimic 29 (red cross in Scheme 3) were obtained (Scheme 3). It was therefore omitted from this study.
Recently Kale et al. reported on the influence of variation of the incorporated hinge on the structural diversity of macrocyclic peptides during screening.10 As a consequence we were interested in verifying the use of alternative azide‐functionalized cyclization hinges and study their impact on antibody binding. To investigate whether different cyclization hinges support the desired conformation of synthetic peptides based on promising HCV E2 epitopes for antibody binding we decided to employ our previously described ELISA screen.17 In order to compare the obtained results we wanted to include epitope mimics 34–37 (Figure 5) based on our previously reported cyclization linker 4 (Figure 3).17 Since these previously studied epitope mimics had not been obtained by CuAAC and therefore did not contain a triazole moiety, we had wished to include the corresponding epitope mimics 25, 28, and 33 containing a triazole moiety for comparison of their bio‐activity monitored by ELISA. Nevertheless, we did not expect a significant influence of the triazole moiety, because of its distance from the epitope sequence. Unfortunately, any effect of the triazole moiety could not be studied because epitope mimics 25, 28, and 33 were not obtained in sufficient yields (Scheme 3). The inability of obtaining these epitopes containing the hydrophobic TBMB hinge underlines the importance of having a hinge with improved solubility properties thereby providing better access to the said epitopes.
Figure 5.

Mimics of epitopes I (green), II (red), III (blue), and scrambled negative control (black) partly based on the previously reported cyclization linker. Epitope mimics 35 and 37 were described previously17 and epitope mimics 34 and 36 were included here for completeness of the study
Epitope mimics 23, 24, 26, 27, 30, 34, 35, and 36, as well as the scrambled negative controls 31, 32, and 37 (Scheme 3 and Figure 5) were covalently immobilized on Pierce® maleimide activated 96‐well plates by maleimide‐thiol conjugation. Subsequently, the resulting immobilized epitope mimics and scrambled negative controls were evaluated for antibody binding by ELISA.17
The immobilized epitope I mimics 23, 24, and 34 were evaluated in the ELISA against Nab AP33 (Figure 6). Immobilized epitope mimic 24 compared excellently to epitope mimic 34 towards binding of antibody Nab AP33. In addition, immobilized epitope mimic 23 showed excellent binding of Nab AP33. However, the immobilized scrambled loop 31, serving as a negative control, showed some unexpected binding of Nab AP33 that was not observed for immobilized scrambled negative controls 32 and 37 (Figure 6). This suggests that the DBMT‐N3 1 polar hinge causes indirect binding to Nab AP33 and would be unsuitable for developing an epitope I mimic.
Figure 6.
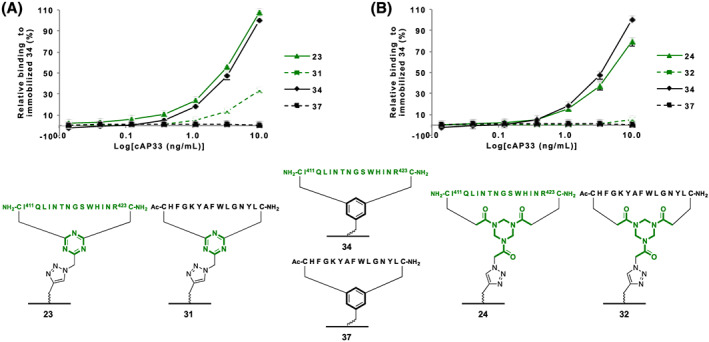
ELISA comparing immobilized epitope I mimics having (a): DBMT 23 and (B): TADB 24 hinges and immobilized scrambled negative controls (A): DBMT 31 and (B): TADB 32. Including immobilized epitope I mimic 34 and immobilized scrambled negative control 37 based on the previously published benzylic‐based linker 4 17
Immobilized loop II mimics 26, 27, and 35 were evaluated for binding of Nab HC84.1 (Figure 7). Both immobilized epitope mimics 26 and 27 showed similar if not slightly better binding towards Nab HC84.1 compared to immobilized epitope mimic 35. None of the immobilized scrambled negative controls 31, 32, and 37 showed binding of this antibody.
Figure 7.
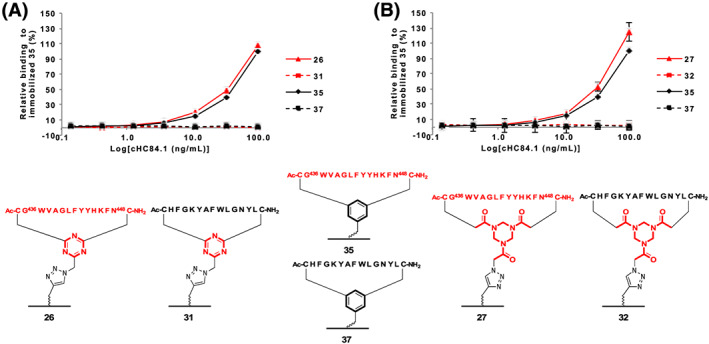
ELISA comparing immobilized epitope II mimics having (a): DBMT 26 and (B): TADB 27 hinges and immobilized scrambled negative controls (A): DBMT 31 and (B): TADB 32. Including immobilized epitope II mimic 35 and immobilized scrambled negative control 37 based on the previously published benzylic‐based linker 4 17
Finally, loop III immobilized mimics 30 and 36 were subjected to an ELISA against Mab DAO5 (Figure 8). No difference in binding of Mab DAO5 was observed between immobilized epitope mimics 30 and 36, and no binding of immobilized scrambled negative controls 32 and 37 was observed of this antibody.
Figure 8.

ELISA comparing immobilized epitope III mimics having TADB 30 hinge and immobilized scrambled negative controls 32. Including immobilized epitope III mimic 36 and immobilized scrambled negative control 37 based on the previously published benzylic‐based linker 4 17
4. CONCLUSIONS
In conclusion, utilizing more polar cyclization hinges did improve the water‐solubility of cyclic peptides. Higher yields were obtained for peptides cyclized on DBMT‐N3 1 and TADB‐N3 2 cyclization hinges compared to both DBMB‐N3 3 (Figure 3) and our previously reported hinge without the presence of a triazole ring (compound 4, Figure 3).17 As a consequence higher concentrations of compound could be dissolved in the buffer system for purification by preparative HPLC. In addition, analytical HPLC showed shorter retention times for DBMT‐N3 1 and TADB‐N3 2, indicating an increased polarity and likely an improved water‐solubility of the resulting cyclic peptides. The latter was confirmed by a solubility experiment. Heinis et al. showed that using different cyclization hinges have a significant influence on the conformation of the resulting (bi)cyclic peptides.3, 4, 9, 10, 27 However, the epitope mimics reported here did not show any difference with respect to antibody binding as indicated by ELISA for the different cyclization hinges used. This does not in anyway indicate that the different cyclization hinges do not induce conformational changes, but in view of the size of rings of the cyclic peptides, this may not be ‘detected’ by the antibody. It is known that the HCV E2 glycoprotein is a flexible protein, adopting multiple different conformations, and this may be reflected in the individual peptide epitopes. This dynamic nature of the viral protein may be one of the major hurdles that hinder the development of a therapeutic vaccine against HCV.7.30. 34 Nevertheless, our epitope mimics significantly benefit from the use of more polar cyclization hinges, with respect to water‐solubility, facilitating their purification and evaluation without compromising their ability for antibody binding.
ORCID
Rob Liskamp https://orcid.org/0000-0001-8897-8975
Arvind Patel http://orcid.org/0000-0003-4600-2047
Supporting information
Figure S1. 1H‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 1H‐NMR (400 MHz, CDCl3): δ = 7.41 (m, 6H, trityl o‐H 2–7), 7.27 (m, 6H, trityl m‐H 8–13), 7.21 (m, 3H, p‐H 14–16), 4.19 (d, 3JHH = 2.3 Hz, 2H, CH 2CCH17), 3.67 (m, 4H, CH 2 1, 18–20), 3.62 (m, 4H, CH 2 1, 18–20), 3.56 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.45 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.30 (t, 3JHH = 6.9 Hz, 2H, SCH2CH 2 23), 2.43 (t, 3JHH = 6.9 Hz, 2H, SCH 2 24), 2.40 (t, 3JHH = 2.3 Hz, 1H, CCH 25) ppm.
Figure S2. COSY‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 1H‐NMR (400 MHz, CDCl3): δ = 7.41 (m, 6H, trityl o‐H 2–7), 7.27 (m, 6H, trityl m‐H 8–13), 7.21 (m, 3H, p‐H 14–16), 4.19 (d, 3JHH = 2.3 Hz, 2H, CH 2CCH17), 3.67 (m, 4H, CH 2 1, 18–20), 3.62 (m, 4H, CH 2 1, 18–20), 3.56 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.45 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.30 (t, 3JHH = 6.9 Hz, 2H, SCH2CH 2 23), 2.43 (t, 3JHH = 6.9 Hz, 2H, SCH 2 24), 2.40 (t, 3JHH = 2.3 Hz, 1H, CCH 25) ppm.
Figure S3. 13C‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 13C‐NMR (101 MHz, CDCl3): δ = 144.8 (Ar‐C 26–28), 129.6 (trityl o‐C 2–7), 127.9 (trityl m‐C 8–13), 126.6 (trityl p‐C 14–16), 79.7 (CCH29), 74.5 (CCH25), 70.6 (CH2 1, 18–20), 70.5 (CH2 1, 18–20), 70.4 (CH2 CH2O‐Alkyne21, 22), 70.2 (CH2 CH2O‐Alkyne21, 22), 69.6 (SCH2 CH2 23), 69.1 (CH2 1, 18–20), 66.6 (CS30), 58.4 (CH2CCH17), 31.7 (SCH2 24) ppm.
Figure S4. 13C‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 13C‐NMR (101 MHz, CDCl3): δ = 144.8 (Ar‐C 26–28), 129.6 (trityl o‐C 2–7), 127.9 (trityl m‐C 8–13), 126.6 (trityl p‐C 14–16), 79.7 (CCH 29 ), 74.5 (CCH25), 70.6 (CH2 1, 18–20), 70.5 (CH2 1, 18–20), 70.4 (CH2 CH2O‐Alkyne21, 22), 70.2 (CH2 CH2O‐Alkyne21, 22), 69.6 (SCH2 CH2 23), 69.1 (CH2 1, 18–20), 66.6 (CS 30 ), 58.4 (CH2CCH17), 31.7 (SCH2 24) ppm.
Figure S5. HSQC‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 1H‐NMR (400 MHz, CDCl3): δ = 7.41 (m, 6H, trityl o‐H 2–7), 7.27 (m, 6H, trityl m‐H 8–13), 7.21 (m, 3H, p‐H 14–16), 4.19 (d, 3JHH = 2.3 Hz, 2H, CH 2CCH17), 3.67 (m, 4H, CH 2 1, 18–20), 3.62 (m, 4H, CH 2 1, 18–20), 3.56 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.45 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.30 (t, 3JHH = 6.9 Hz, 2H, SCH2CH 2 23), 2.43 (t, 3JHH = 6.9 Hz, 2H, SCH 2 24), 2.40 (t, 3JHH = 2.3 Hz, 1H, CCH 25) ppm. 13C‐NMR (101 MHz, CDCl3): δ = 144.8 (Ar‐C 26–28), 129.6 (trityl o‐C 2–7), 127.9 (trityl m‐C 8–13), 126.6 (trityl p‐C 14–16), 79.7 (CCH29), 74.5 (CCH25), 70.6 (CH2 1, 18–20), 70.5 (CH2 1, 18–20), 70.4 (CH2 CH2O‐Alkyne21, 22), 70.2 (CH2 CH2O‐Alkyne21, 22), 69.6 (SCH2 CH2 23), 69.1 (CH2 1, 18–20), 66.6 (CS30), 58.4 (CH2CCH17), 31.7 (SCH2 24) ppm.
Figure S6. Analytical HPLC analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S7. HRMS analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 Calculated m/z for C 30 H 34 O 4 S: 513.2076 [M + Na]+1; Found m/z 513.2062.
Figure S8. Analytical HPLC analysis of cyclic peptide 11. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S9. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 11, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S10. HRMS analysis of cyclic peptide 11. Calculated m/z for C 85 H 131 N 33 O 22 S 2: 1015.9872 ½[M + 2H+2; Found m/z 1015.9878.
Figure S11. LRMS analysis of cyclic peptide 11. Calculated m/z for C 85 H 131 N 33 O 22 S 2: 1015.99 ½[M + 2H]+2; Found m/z 1016.58.
Figure S12. Analytical HPLC analysis of cyclic peptide 12. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S13. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 12, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S14. HRMS analysis of cyclic peptide 12. Calculated m/z for C 88 H 137 N 33 O 25 S 2: 1061.0031 ½[M + 2H]+2; Found m/z 1061.0027.
Figure S15. LRMS analysis of cyclic peptide 12. Calculated m/z for C 88 H 137 N 33 O 25 S 2: 1061.00 ½[M + 2H]+2; Found m/z 1061.50.
Figure S16. Analytical HPLC analysis of cyclic peptide 13. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S17. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 13, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S18. HRMS analysis of cyclic peptide 13. Calculated m/z for C 88 H 134 N 30 O 22 S 2: 1014.4943 ½[M + 2H]2+; Found m/z 1014.4966.
Figure S19. LRMS analysis of cyclic peptide 13. Calculated m/z for C 88 H 134 N 30 O 22 S 2: 1014.49 ½[M + 2H]+2; Found m/z 1015.08.
Figure S20. Analytical HPLC analysis of cyclic peptide 14. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S21. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 14, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S22. HRMS analysis of cyclic peptide 14. Calculated m/z for C 95 H 121 N 27 O 19 S 2: 1004.9465 ½[M + 2H]+2; Found m/z 1004.9470.
Figure S23. LRMS analysis of cyclic peptide 14. Calculated m/z for C 95 H 121 N 27 O 19 S 2: 1004.95 ½[M + 2H]+2; Found m/z 1005.50.
Figure S24. Analytical HPLC analysis of cyclic peptide 15. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S25. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 15, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S26. HRMS analysis of cyclic peptide 15. Calculated m/z for C 98 H 127 N 27 O 22 S 2: 1049.9623 ½[M + 2H]+2; Found m/z 1049.9598.
Figure S27. LRMS analysis of cyclic peptide 15. Calculated m/z for C 98 H 127 N 27 O 22 S 2: 1049.96 ½[M + 2H]+2; Found m/z 1050.58.
Figure S28. Analytical HPLC analysis of cyclic peptide 16. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S29. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 16, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S30. HRMS analysis of cyclic peptide 16. Calculated m/z for C 98 H 124 N 24 O 19 S 2: 1003.4536 ½[M + 2H]+2; Found m/z 1003.4567.
Figure S31. LRMS analysis of cyclic peptide 16. Calculated m/z for C 98 H 124 N 24 O 19 S 2: 1003.45 ½[M + 2H]+2; Found m/z 1004.08.
Figure S32. Analytical HPLC analysis of cyclic peptide 17. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S33. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 17, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S34. HRMS analysis of cyclic peptide 17. Calculated m/z for C 87 H 123 N 27 O 28 S 2: 1029.9314 ½[M + 2H+2; Found m/z 1029.9345.
Figure S35. LRMS analysis of cyclic peptide 17. Calculated m/z for C 87 H 123 N 27 O 28 S 2: 1029.93 ½[M + 2H]+2; Found m/z 1030.25.
Figure S36. Analytical HPLC analysis of cyclic peptide 18. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S37. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 18, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S38. HRMS analysis of cyclic peptide 18. Calculated m/z for C 90 H 129 N 27 O 31 S 2: 1074.9473 ½[M + 2H+2; Found m/z 1074.9444.
Figure S39. LRMS analysis of cyclic peptide 18. Calculated m/z for C 90 H 129 N 27 O 31 S 2: 1074.95 ½[M + 2H]+2; Found m/z 1075.42.
Figure S40. Analytical HPLC analysis of cyclic peptide 19. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S41. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 19, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S42. HRMS analysis of cyclic peptide 19. Calculated m/z for C 90 H 126 N 24 O 28 S 2: 1028.4386 ½[M + 2H+2; Found m/z 1028.4374.
Figure S43. LRMS analysis of 19. Calculated m/z for C 90 H 126 N 24 O 28 S 2: 1028.44 ½[M + 2H+2; Found m/z 1028.92.
Figure S44. Analytical HPLC analysis of cyclic peptide 20. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S45. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 20, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S46. HRMS analysis of cyclic peptide 20. Calculated m/z for C 96 H 123 N 27 O 19 S 2: 1011.9543 ½[M + 2H+2; Found m/z 1011.9544.
Figure S47. LRMS analysis of cyclic peptide 20. Calculated m/z for C 96 H 123 N 27 O 19 S 2: 1011.95 ½[M + 2H+2; Found m/z 1012.58.
Figure S48. Analytical HPLC analysis of cyclic peptide 21. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S49. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 21, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S50. HRMS analysis of cyclic peptide 21. Calculated m/z for C 99 H 129 N 27 O 22 S 2: 1056.9702 ½[M + 2H]+2; Found m/z 1056.9688.
Figure S51. LRMS analysis of cyclic peptide 21. Calculated m/z for C 99 H 129 N 27 O 22 S 2: 1056.97 ½[M + 2H]+2; Found m/z 1057.58.
Figure S52. Analytical HPLC analysis of cyclic peptide 22. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S53. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 22, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S54. HRMS analysis of cyclic peptide 22. Calculated m/z for C 99 H 126 N 24 O 19 S 2: 1010.4614 ½[M + 2H+2; Found m/z 1010.4581.
Figure S55. LRMS analysis of cyclic peptide 22. Calculated m/z for C 99 H 126 N 24 O 19 S 2: 1010.46 ½[M + 2H+2; Found m/z 1011.08.
Figure S56. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 23, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S57. Analytical HPLC analysis of epitope mimic 23. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S58. HRMS analysis of epitope mimic 23. Calculated m/z for C 96 H 151 N 33 O 26 S 3: 760.3635 1/3[M + 3H]+3; Found m/z 760.3664.
Figure S59. LRMS analysis of epitope mimic 23. Calculated m/z for C 96 H 151 N 33 O 26 S 3: 1140.04 ½[M + 2H]2+ / 760.36 1/3[M + 3H]+3; Found m/z 1140.50 / 760.67.
Figure S60. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 24, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S61. Analytical HPLC analysis of epitope mimic 24. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S62. HRMS analysis of cyclic epitope mimic 24. Calculated m/z for C 99 H 157 N 33 O 29 S 3: 790.3740 1/3[M + 3H]+3; Found m/z 790.3769.
Figure S63. HRMS analysis of epitope mimic 24. Calculated m/z for C 99 H 157 N 33 O 29 S 3: 1185.06 ½[M + 2H]2+/ 790.37 1/3[M + 3H]+3; Found m/z 1185.50 / 790.67.
Figure S64. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 25, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S65. Analytical HPLC analysis of epitope mimic 25. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S66. HRMS analysis of epitope mimic 25. Calculated m/z for C 99 H 154 N 30 O 26 S 3: 759.3682 1/3[M + 3H+3; Found m/z 759.3712.
Figure S67. LRMS analysis of epitope mimic 25. Calculated m/z for C 99 H 154 N 30 O 26 S 3: 1138.55 ½[M + 2H]2+/ 759.37 1/3[M + 3H]+3; Found m/z 1139.08/ 759.67.
Figure S68. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 26, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S69. Analytical HPLC analysis of epitope mimic 26. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S70. HRMS analysis of epitope mimic 26. Calculated m/z for C 106 H 141 N 27 O 23 S 3: 1129.0006 ½[M + 2H]+2; Found m/z 1229.0049.
Figure S71. LRMS analysis of epitope mimic 26. Calculated m/z for C 106 H 141 N 27 O 23 S 3: 1129.00 ½[M + 2H]+2; Found m/z 1229.33.
Figure S72. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected cyclic epitope mimic 27, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S73. Analytical HPLC analysis of epitope mimic 27. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S74. HRMS analysis of epitope mimic 27. Calculated m/z for C 109 H 147 N 27 O 26 S 3: 1174.0165 ½[M + 2H]+2; Found m/z 1174.0163.
Figure S75. LRMS analysis of epitope mimic 27. Calculated m/z for C 109 H 147 N 27 O 26 S 3: 1174.02 ½[M + 2H]+2; Found m/z 1174.42.
Figure S76. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 28, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S77. Analytical HPLC analysis of epitope mimic 28. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S78. HRMS analysis of epitope mimic 28. Calculated m/z for C 109 H 144 N 24 O 23 S 3: 1127.5077 ½[M + 2H]+2; Found m/z 1127.5071.
Figure S79. LRMS analysis of epitope mimic 28. Calculated m/z for C 109 H 144 N 24 O 23 S 3: 1127.51 ½[M + 2H]+2; Found m/z 1127.92.
Figure S80. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 29, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S81. LRMS analysis of trityl‐protected epitope mimic 29. Calculated m/z for C 117 H 157 N 27 O 32 S 3: 1275.04 ½[M + 2H]+2; Found m/z 1275.08.
Figure S82. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 30, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S83. Analytical HPLC analysis of epitope mimic 30. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S84. HRMS analysis of epitope mimic 30. Calculated m/z for C 101 H 149 N 27 O 35 S 3: 1220.9834 ½[M + 2Na]+2; Found m/z 1220.9838.
Figure S85. LRMS analysis of epitope mimic 30. Calculated m/z for C 101 H 149 N 27 O 35 S 3: 1199.00 ½[M + 2H]+2; Found m/z 1199.42.
Figure S86. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected scrambled negative control 31, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S87. Analytical HPLC analysis of scrambled negative control 31. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S88. HRMS analysis of scrambled negative control 31. Calculated m/z for C 107 H 143 N 27 O 23 S 3: 1136.0084 ½[M + 2H]+2; Found m/z 1136.0112.
Figure S89. LRMS analysis of scrambled negative control 31. Calculated m/z for C 107 H 143 N 27 O 23 S 3: 1136.01 ½[M + 2H]+2 / 757.67 1/3[M + 3H]3+; Found m/z 1136.50 / 758.00.
Figure S90. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected scrambled negative control 32, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S91. Analytical HPLC analysis of scrambled negative control 32. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S92. HRMS analysis of scrambled negative control 32. Calculated m/z for C 110 H 149 N 27 O 26 S 3: 1181.0243 ½[M + 2H]+2; Found m/z 1181.0253.
Figure S93. LRMS analysis of scrambled negative control 32. Calculated m/z for C 110 H 149 N 27 O 26 S 3: 1181.02 ½[M + 2H]+2 / 787.68 1/3[M + 3H]3+; Found m/z 1181.58/ 788.17.
Figure S94. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected scrambled negative control 33, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S95. Analytical HPLC analysis of scrambled negative control 33. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S96. HRMS analysis of scrambled negative control 33. Calculated m/z for C 110 H 146 N 24 O 23 S 3: 1134.5156 ½[M + 2H]+2; Found m/z 1134.5160.
Figure S97. LRMS analysis of scrambled negative control 33. Calculated m/z for C 110 H 146 N 24 O 23 S 3: 1134.52 ½[M + 2H]+2/ 756.68 1/3[M + 3H]3+; Found m/z 1135.00/ 756.92.
Figure S98. Analytical HPLC analysis overlay of individual fraction(s) of cyclic epitope mimic 34, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S99. Analytical HPLC analysis of cyclic epitope mimic 34. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S100. HRMS analysis of cyclic epitope mimic 34. Calculated m/z for C 96 H 151 N 27 O 26 S 3: 1098.0321 ½[M + 2H]+2; Found m/z 1098.0355.
Figure S101. LRMS analysis of cyclic epitope mimic 34. Calculated m/z for C 96 H 151 N 27 O 26 S 3: 1098.03 ½[M + 2H]+2/732.36 1/3[M + 3H]3+; Found m/z 1098.50/732.50.
Figure S102. Analytical HPLC analysis overlay of individual fraction(s) of cyclic epitope mimic 35, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S103. Analytical HPLC analysis of cyclic epitope mimic 35. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S104. HRMS analysis of cyclic epitope mimic 35. Calculated m/z for C 106 H 141 N 21 O 23 S 3: 1086.9914 ½[M + 2H]+2; Found m/z 1086.9949.
Figure S105. LRMS analysis of cyclic epitope mimic 35. Calculated m/z for C 106 H 141 N 21 O 23 S 3: 1086.99 ½[M + 2H]+2; Found m/z 1087.25.
Figure S106. Analytical HPLC analysis overlay of individual fraction(s) of cyclic epitope mimic 36, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S107. Analytical HPLC analysis of cyclic epitope mimic 36. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S108. HRMS analysis of cyclic epitope mimic 36. Calculated m/z for C 98 H 143 N 21 O 32 S 3: 1109.9607 ½[M‐2H]−2; Found m/z 1109.9435.
Figure S109. LRMS analysis of cyclic epitope mimic 36. Calculated m/z for C 98 H 143 N 21 O 32 S 3: 1111.98 ½[M + 2H]+2; Found m/z 1112.33.
Figure S110. Analytical HPLC analysis overlay of individual fraction(s) of cyclic epitope mimic 37, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S111. Analytical HPLC analysis of cyclic epitope mimic 37. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S112. HRMS analysis of cyclic epitope mimic 37. Calculated m/z for C 107 H 143 N 21 O 23 S 3: 1093.9992 ½[M + 2H]+2; Found m/z 1093.9997.
Figure S113. LRMS analysis of cyclic epitope mimic 37. Calculated m/z for C 107 H 143 N 21 O 23 S 3: 1094.00 ½[M + 2H]+2; Found m/z 1094.58.
ACKNOWLEDGEMENTS
This research was funded by the University of Glasgow. Work in AHP's lab was supported by the Medical Research Council grants MC_UU12014/2.
Meuleman TJ, Cowton VM, Patel AH, Liskamp RMJ. Improving the aqueous solubility of HCV‐E2 glycoprotein epitope mimics by cyclization using POLAR hinges. J Pep Sci. 2020;26:e3222 10.1002/psc.3222
Contributor Information
Arvind H. Patel, Email: arvind.patel@glasgow.ac.uk.
Rob M.J. Liskamp, Email: robert.liskamp@glasgow.ac.uk.
REFERENCES
- 1. Lavie M, Hanoulle X, Dubuisson J. Glycan shielding and modulation of hepatitis C virus neutralizing antibodies. Front. Immunol. 2018;9(910):1‐9. 10.3389/fimmu.2018.00910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Owsianka A, Clayton RF, Loomis‐Price LD, McKeating JA, Patel AH. Functional analysis of hepatitis C virus E2 glycoproteins and virus‐like particles reveals structural dissimilarities between different forms of E2. J. Gen. Virol. 2001;82:1877‐1883. 10.1099/0022-1317-82-8-1877 [DOI] [PubMed] [Google Scholar]
- 3. Angelini A, Cendron L, Chen S, et al. Bicyclic peptide inhibitors reveals large contact interface with a protease target. ACS Chem. Biol. 2012;7(5):817‐821. 10.1021/cb200478t [DOI] [PubMed] [Google Scholar]
- 4. Deyle K, Kong XD, Heinis C. Phage selection of cyclic peptides for application in research and drug development. Acc. Chem. Res. 2017;50(8):1866‐1874. 10.1021/acs.accounts.7b00184 [DOI] [PubMed] [Google Scholar]
- 5. Streefkerk DE, Schmidt M, Ippel JH, et al. Synthesis of constrained tetracyclic peptides by consecutive CEPS, CLIPS, and oxime ligation. Org. Lett. 2019;21(7):2095‐2100. 10.1021/acs.orglett.9b00378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kalepu V, Nekkanti V. Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm. Sin. B. 2015;5(5):442‐453. 10.1016/j.apsb.2015.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meola A, Tarr AW, England P, et al. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J. Virol. 2015;89(4):2170‐2181. 10.1128/JVI.02190-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Keck ZY, Xia J, Wang Y, Krey T, Prentoe J. Carlsen, T, Li AY, Patel AH, lemon SM, Bukh J, Rey FA, Foung SK. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog. 2012;8(4):e1002653 10.1371/journal.ppat.1002653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen S, Morales‐Sanfrutos J, Angelini A, Cutting B, Heinis C. Structurally diverse cyclisation linkers impose different backbone conformations in bicyclic peptides. ChemBioChem. 2012;13(7):1032‐1038. DOI: 10.1002/cbic.201200049 [DOI] [PubMed] [Google Scholar]
- 10. Kale SS, Villequey C, Kong XD, Zorzi A, Deyle K, Heinis C. Cyclization of peptides with two chemical bridges affords large scaffold diversities. Nat. Chem. 2018;10(7):715‐723. 10.1038/s41557-018-0042-7 [DOI] [PubMed] [Google Scholar]
- 11. Richelle GJJ, Schmidt M, Ippel H, et al. A one pot “triple‐C” multicyclization methodology for the synthesis of highly constrained isomerically pure tetracyclic peptides. ChemBioChem. 2018;19(18):1934‐1938. 10.1002/cbic.201800346 [DOI] [PubMed] [Google Scholar]
- 12. Rim C, Lahey LJ, Patel VG, Zhang H, Son DY. Thiolene reactions of 1,3,5,triacryloylhexahydro‐1,3,5‐triazine (TAT): facile access to functional tripodal thioethers. Tetrahedron Lett. 2009;50(7):745‐747. 10.1016/j.tetlet.2008.11.094 [DOI] [Google Scholar]
- 13. Owsianka A, Tarr AW, Juttla VS, et al. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J. Virol. 2005;79(11):11095‐11104. 10.1128/JVI.79.17.11095-11104.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Werkhoven PR, Van de Langemheen H, van der Wal S, Kruijtzer JAW, Liskamp RMJ. Versatile convergent synthesis of a three peptide loop containing protein mimic of whooping cough pertactin by successive cu(I)‐catalyzed azide alkyne cycloaddition on an orthogonal alkyne functionalized TAC‐scaffold. J. Pept. Sci. 2014;20(4):235‐239. 10.1002/psc.2624 [DOI] [PubMed] [Google Scholar]
- 15. Richelle GJJ, Ori S, Hiemstra H, van Maarseveen JH, Timmerman P. general and facile route to isomerically pure tricyclic peptides based on template tandem CLIPS/CuAAC cyclizations. Angew. Chem. Int. Ed. Engl. 2018;57(2):501‐505. 10.1002/anie.201709127 [DOI] [PubMed] [Google Scholar]
- 16. Van de Langemheen H, Korotkovs V, Bijl J, et al. Polar hinges as functionalized conformational constraints in (bi)cyclic peptides. ChemBioChem. 2017;18(4):387‐395. 10.1002/cbic.201600612 [DOI] [PubMed] [Google Scholar]
- 17. Meuleman TJ, Dunlop JI, Owsianka AM, Van de Langemheen H, Patel AH, Liskamp RMJ. Immobilization by surface conjugation of cyclic peptides for effective mimicry of the HCV‐envelope E2 protein as a strategy toward synthetic vaccines. Bioconjug. Chem. 2018;29(4):1091‐1101. 10.1021/acs.bioconjchem.7b00755 [DOI] [PubMed] [Google Scholar]
- 18. Ray RB, Ray R. Hepatitis C Virus manipulates humans as its favourite host for a long‐term relationship. Hepatology. 2019;69(2):889‐900. 10.1002/hep.30214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwon YC, Ray R. Complement regulation and immune evasion by hepatitis C virus In: Law M. (eds) hepatitis C virus protocols. Methods in Molecular Biology, 2019;1911 Humana press, New York, NY: DOI: 10.1007/978-1-4939-8976-8_23 [DOI] [PubMed] [Google Scholar]
- 20. Walker CM. Designing an HCV vaccine: a unique convergence of prevention and therapy? Curr. Opin. Virol. 2017;23:113‐119. 10.1016/j.coviro.2017.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mulder GE, Quarles van Ufford HC, van Ameijde J, Brouwer AJ, Kruijtzer JAW, Liskamp RMJ. Scaffold optimization in discontinuous epitope containing protein mimics of gp120 using smart libraries. Org. Biomol. Chem. 2013;11(16):2676‐2684. 10.1039/c3ob27470e [DOI] [PubMed] [Google Scholar]
- 22. Dustin LB, Cashman SB, Laidlaw SM. Immune control and failure in HCV infection—tipping the balance. J. Leukoc. Biol. 2014;96(4):535‐548. DOI: 10.1189/jlb.4RI0214-126R [DOI] [PMC free article] [PubMed]
- 23. Tarr AW, Owsianka AM, Javaraj D, et al. Determination of the human antibody response to the epitope defined by the hepatitis C virus‐neutralizing monoclonal antibody AP33. J. Gen. Virol. 2007;88:2991‐3001. 10.1099/vir.0.83065-0 [DOI] [PubMed] [Google Scholar]
- 24. Cowton VM, Singer JB, Gifford RJ, Patel AH. Predicting the effectiveness of hepatitis C virus neutralizing antibodies by bioinformatics analysis of conserved epitope residues using public sequence data. Front Immunol. 2018;9:1470 10.3389/fimmu.2018.01470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zorzi A, Deyle K, Heinis C. Cyclic peptide therapeutics: past, present and future. Curr. Opin. Chem. Biol. 2017;(38):24‐29. 10.1016/j.cbpa.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 26. Sautto G, Tarr AW, Mancini N, Clementi M. Structural and antigenic definition of hepatitis C virus E2 glycoprotein epitopes targeted by monoclonal antibodies. Clin. Dev. Immunol. 2013;2013:450963 10.1155/2013/450963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen S, Bertoldo D, Angelini A, Pojer F, Heinis C. Peptide ligands stabilized by small molecules. Angew. Chem. Int. Ed. Engl. 2014;53(6):1602‐1606. 10.1002/anie.201309459 [DOI] [PubMed] [Google Scholar]
- 28. Longin O, Hezwani M, Van de Langemheen H, Liskamp RMJ. Synthetic antibody protein mimics of infliximab by molecular scaffolding on novel CycloTriVeratrilene (CTV) derivatives. Org. Biomol. Chem. 2018;16(29):5254‐5274. 10.1039/c8ob01104d [DOI] [PubMed] [Google Scholar]
- 29. Werkhoven PR, Elwakiel M, Meuleman TJ, Quarles van Ufford HC, Kruijtzer JAW, Liskamp RMJ. Molecular construction of HIV‐gp120 discontinuous epitope mimics by assembly of cyclic peptides on an orthogonal alkyne functionalized TAC‐scaffold. Org. Biomol. Chem. 2016;14(2):701‐710. DOI: 10.1039/c5ob02014j [DOI] [PubMed] [Google Scholar]
- 30. Kong L, Lee DE, Kadam RU, et al. Structural flexibility at a major conserved antibody target on hepatitis C virus E2 antigen. Proc. Natl. Acad. Sci. U. S. A. 2016;113(45):12768‐12773. 10.1073/pnas.1609780113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vasiliauskaite I, Owsianka A, England P, et al. Conformational flexibility in the immunoglobulin‐like domain of the hepatitis C virus glycoprotein E2. MBio. 2017;8(3):e00382‐17 10.1128/mBio.00382-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang L, Astruc D. The copper(I)‐catalyzed alkyne‐azide cycloaddition (CuAAC) “click” reaction and its applications. An Overview Coordination Chemistry Reviews. 2011;255:2933‐2945. 10.1016/j.ccr.2011.06.028 [DOI] [Google Scholar]
- 33. Conibear AC, Farbiarz K, Mayer RL, Matveenko M, Kählig H, Becker CF. arginine side‐chain modification that occurs during copper‐catalysed azide‐alkyne click reactions resembles an advanced glycation end product. Org. Biomol. Chem. 2016;14(16):6205‐6211. 10.1039/c6ob00932h [DOI] [PubMed] [Google Scholar]
- 34. Yost SA, Wang Y, Marcotrigiano J. Hepatitis c virus envelope glycoproteins: a balancing act of order and disorder. Front. Immunol. 2018;9:1917 10.3389/fimmu.2018.01917 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. 1H‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 1H‐NMR (400 MHz, CDCl3): δ = 7.41 (m, 6H, trityl o‐H 2–7), 7.27 (m, 6H, trityl m‐H 8–13), 7.21 (m, 3H, p‐H 14–16), 4.19 (d, 3JHH = 2.3 Hz, 2H, CH 2CCH17), 3.67 (m, 4H, CH 2 1, 18–20), 3.62 (m, 4H, CH 2 1, 18–20), 3.56 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.45 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.30 (t, 3JHH = 6.9 Hz, 2H, SCH2CH 2 23), 2.43 (t, 3JHH = 6.9 Hz, 2H, SCH 2 24), 2.40 (t, 3JHH = 2.3 Hz, 1H, CCH 25) ppm.
Figure S2. COSY‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 1H‐NMR (400 MHz, CDCl3): δ = 7.41 (m, 6H, trityl o‐H 2–7), 7.27 (m, 6H, trityl m‐H 8–13), 7.21 (m, 3H, p‐H 14–16), 4.19 (d, 3JHH = 2.3 Hz, 2H, CH 2CCH17), 3.67 (m, 4H, CH 2 1, 18–20), 3.62 (m, 4H, CH 2 1, 18–20), 3.56 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.45 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.30 (t, 3JHH = 6.9 Hz, 2H, SCH2CH 2 23), 2.43 (t, 3JHH = 6.9 Hz, 2H, SCH 2 24), 2.40 (t, 3JHH = 2.3 Hz, 1H, CCH 25) ppm.
Figure S3. 13C‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 13C‐NMR (101 MHz, CDCl3): δ = 144.8 (Ar‐C 26–28), 129.6 (trityl o‐C 2–7), 127.9 (trityl m‐C 8–13), 126.6 (trityl p‐C 14–16), 79.7 (CCH29), 74.5 (CCH25), 70.6 (CH2 1, 18–20), 70.5 (CH2 1, 18–20), 70.4 (CH2 CH2O‐Alkyne21, 22), 70.2 (CH2 CH2O‐Alkyne21, 22), 69.6 (SCH2 CH2 23), 69.1 (CH2 1, 18–20), 66.6 (CS30), 58.4 (CH2CCH17), 31.7 (SCH2 24) ppm.
Figure S4. 13C‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 13C‐NMR (101 MHz, CDCl3): δ = 144.8 (Ar‐C 26–28), 129.6 (trityl o‐C 2–7), 127.9 (trityl m‐C 8–13), 126.6 (trityl p‐C 14–16), 79.7 (CCH 29 ), 74.5 (CCH25), 70.6 (CH2 1, 18–20), 70.5 (CH2 1, 18–20), 70.4 (CH2 CH2O‐Alkyne21, 22), 70.2 (CH2 CH2O‐Alkyne21, 22), 69.6 (SCH2 CH2 23), 69.1 (CH2 1, 18–20), 66.6 (CS 30 ), 58.4 (CH2CCH17), 31.7 (SCH2 24) ppm.
Figure S5. HSQC‐NMR analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 1H‐NMR (400 MHz, CDCl3): δ = 7.41 (m, 6H, trityl o‐H 2–7), 7.27 (m, 6H, trityl m‐H 8–13), 7.21 (m, 3H, p‐H 14–16), 4.19 (d, 3JHH = 2.3 Hz, 2H, CH 2CCH17), 3.67 (m, 4H, CH 2 1, 18–20), 3.62 (m, 4H, CH 2 1, 18–20), 3.56 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.45 (m, 2H, CH 2CH 2O‐Alkyne21, 22), 3.30 (t, 3JHH = 6.9 Hz, 2H, SCH2CH 2 23), 2.43 (t, 3JHH = 6.9 Hz, 2H, SCH 2 24), 2.40 (t, 3JHH = 2.3 Hz, 1H, CCH 25) ppm. 13C‐NMR (101 MHz, CDCl3): δ = 144.8 (Ar‐C 26–28), 129.6 (trityl o‐C 2–7), 127.9 (trityl m‐C 8–13), 126.6 (trityl p‐C 14–16), 79.7 (CCH29), 74.5 (CCH25), 70.6 (CH2 1, 18–20), 70.5 (CH2 1, 18–20), 70.4 (CH2 CH2O‐Alkyne21, 22), 70.2 (CH2 CH2O‐Alkyne21, 22), 69.6 (SCH2 CH2 23), 69.1 (CH2 1, 18–20), 66.6 (CS30), 58.4 (CH2CCH17), 31.7 (SCH2 24) ppm.
Figure S6. Analytical HPLC analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S7. HRMS analysis of alkyne‐functionalized tetraethylene glycol monotrityl thioether.1 Calculated m/z for C 30 H 34 O 4 S: 513.2076 [M + Na]+1; Found m/z 513.2062.
Figure S8. Analytical HPLC analysis of cyclic peptide 11. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S9. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 11, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S10. HRMS analysis of cyclic peptide 11. Calculated m/z for C 85 H 131 N 33 O 22 S 2: 1015.9872 ½[M + 2H+2; Found m/z 1015.9878.
Figure S11. LRMS analysis of cyclic peptide 11. Calculated m/z for C 85 H 131 N 33 O 22 S 2: 1015.99 ½[M + 2H]+2; Found m/z 1016.58.
Figure S12. Analytical HPLC analysis of cyclic peptide 12. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S13. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 12, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S14. HRMS analysis of cyclic peptide 12. Calculated m/z for C 88 H 137 N 33 O 25 S 2: 1061.0031 ½[M + 2H]+2; Found m/z 1061.0027.
Figure S15. LRMS analysis of cyclic peptide 12. Calculated m/z for C 88 H 137 N 33 O 25 S 2: 1061.00 ½[M + 2H]+2; Found m/z 1061.50.
Figure S16. Analytical HPLC analysis of cyclic peptide 13. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S17. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 13, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S18. HRMS analysis of cyclic peptide 13. Calculated m/z for C 88 H 134 N 30 O 22 S 2: 1014.4943 ½[M + 2H]2+; Found m/z 1014.4966.
Figure S19. LRMS analysis of cyclic peptide 13. Calculated m/z for C 88 H 134 N 30 O 22 S 2: 1014.49 ½[M + 2H]+2; Found m/z 1015.08.
Figure S20. Analytical HPLC analysis of cyclic peptide 14. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S21. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 14, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S22. HRMS analysis of cyclic peptide 14. Calculated m/z for C 95 H 121 N 27 O 19 S 2: 1004.9465 ½[M + 2H]+2; Found m/z 1004.9470.
Figure S23. LRMS analysis of cyclic peptide 14. Calculated m/z for C 95 H 121 N 27 O 19 S 2: 1004.95 ½[M + 2H]+2; Found m/z 1005.50.
Figure S24. Analytical HPLC analysis of cyclic peptide 15. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S25. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 15, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S26. HRMS analysis of cyclic peptide 15. Calculated m/z for C 98 H 127 N 27 O 22 S 2: 1049.9623 ½[M + 2H]+2; Found m/z 1049.9598.
Figure S27. LRMS analysis of cyclic peptide 15. Calculated m/z for C 98 H 127 N 27 O 22 S 2: 1049.96 ½[M + 2H]+2; Found m/z 1050.58.
Figure S28. Analytical HPLC analysis of cyclic peptide 16. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S29. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 16, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S30. HRMS analysis of cyclic peptide 16. Calculated m/z for C 98 H 124 N 24 O 19 S 2: 1003.4536 ½[M + 2H]+2; Found m/z 1003.4567.
Figure S31. LRMS analysis of cyclic peptide 16. Calculated m/z for C 98 H 124 N 24 O 19 S 2: 1003.45 ½[M + 2H]+2; Found m/z 1004.08.
Figure S32. Analytical HPLC analysis of cyclic peptide 17. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S33. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 17, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S34. HRMS analysis of cyclic peptide 17. Calculated m/z for C 87 H 123 N 27 O 28 S 2: 1029.9314 ½[M + 2H+2; Found m/z 1029.9345.
Figure S35. LRMS analysis of cyclic peptide 17. Calculated m/z for C 87 H 123 N 27 O 28 S 2: 1029.93 ½[M + 2H]+2; Found m/z 1030.25.
Figure S36. Analytical HPLC analysis of cyclic peptide 18. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S37. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 18, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S38. HRMS analysis of cyclic peptide 18. Calculated m/z for C 90 H 129 N 27 O 31 S 2: 1074.9473 ½[M + 2H+2; Found m/z 1074.9444.
Figure S39. LRMS analysis of cyclic peptide 18. Calculated m/z for C 90 H 129 N 27 O 31 S 2: 1074.95 ½[M + 2H]+2; Found m/z 1075.42.
Figure S40. Analytical HPLC analysis of cyclic peptide 19. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S41. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 19, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S42. HRMS analysis of cyclic peptide 19. Calculated m/z for C 90 H 126 N 24 O 28 S 2: 1028.4386 ½[M + 2H+2; Found m/z 1028.4374.
Figure S43. LRMS analysis of 19. Calculated m/z for C 90 H 126 N 24 O 28 S 2: 1028.44 ½[M + 2H+2; Found m/z 1028.92.
Figure S44. Analytical HPLC analysis of cyclic peptide 20. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S45. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 20, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S46. HRMS analysis of cyclic peptide 20. Calculated m/z for C 96 H 123 N 27 O 19 S 2: 1011.9543 ½[M + 2H+2; Found m/z 1011.9544.
Figure S47. LRMS analysis of cyclic peptide 20. Calculated m/z for C 96 H 123 N 27 O 19 S 2: 1011.95 ½[M + 2H+2; Found m/z 1012.58.
Figure S48. Analytical HPLC analysis of cyclic peptide 21. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S49. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 21, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S50. HRMS analysis of cyclic peptide 21. Calculated m/z for C 99 H 129 N 27 O 22 S 2: 1056.9702 ½[M + 2H]+2; Found m/z 1056.9688.
Figure S51. LRMS analysis of cyclic peptide 21. Calculated m/z for C 99 H 129 N 27 O 22 S 2: 1056.97 ½[M + 2H]+2; Found m/z 1057.58.
Figure S52. Analytical HPLC analysis of cyclic peptide 22. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S53. Analytical HPLC analysis overlay of individual fraction(s) of cyclic peptide 22, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S54. HRMS analysis of cyclic peptide 22. Calculated m/z for C 99 H 126 N 24 O 19 S 2: 1010.4614 ½[M + 2H+2; Found m/z 1010.4581.
Figure S55. LRMS analysis of cyclic peptide 22. Calculated m/z for C 99 H 126 N 24 O 19 S 2: 1010.46 ½[M + 2H+2; Found m/z 1011.08.
Figure S56. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 23, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S57. Analytical HPLC analysis of epitope mimic 23. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S58. HRMS analysis of epitope mimic 23. Calculated m/z for C 96 H 151 N 33 O 26 S 3: 760.3635 1/3[M + 3H]+3; Found m/z 760.3664.
Figure S59. LRMS analysis of epitope mimic 23. Calculated m/z for C 96 H 151 N 33 O 26 S 3: 1140.04 ½[M + 2H]2+ / 760.36 1/3[M + 3H]+3; Found m/z 1140.50 / 760.67.
Figure S60. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 24, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S61. Analytical HPLC analysis of epitope mimic 24. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S62. HRMS analysis of cyclic epitope mimic 24. Calculated m/z for C 99 H 157 N 33 O 29 S 3: 790.3740 1/3[M + 3H]+3; Found m/z 790.3769.
Figure S63. HRMS analysis of epitope mimic 24. Calculated m/z for C 99 H 157 N 33 O 29 S 3: 1185.06 ½[M + 2H]2+/ 790.37 1/3[M + 3H]+3; Found m/z 1185.50 / 790.67.
Figure S64. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 25, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S65. Analytical HPLC analysis of epitope mimic 25. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S66. HRMS analysis of epitope mimic 25. Calculated m/z for C 99 H 154 N 30 O 26 S 3: 759.3682 1/3[M + 3H+3; Found m/z 759.3712.
Figure S67. LRMS analysis of epitope mimic 25. Calculated m/z for C 99 H 154 N 30 O 26 S 3: 1138.55 ½[M + 2H]2+/ 759.37 1/3[M + 3H]+3; Found m/z 1139.08/ 759.67.
Figure S68. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 26, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S69. Analytical HPLC analysis of epitope mimic 26. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S70. HRMS analysis of epitope mimic 26. Calculated m/z for C 106 H 141 N 27 O 23 S 3: 1129.0006 ½[M + 2H]+2; Found m/z 1229.0049.
Figure S71. LRMS analysis of epitope mimic 26. Calculated m/z for C 106 H 141 N 27 O 23 S 3: 1129.00 ½[M + 2H]+2; Found m/z 1229.33.
Figure S72. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected cyclic epitope mimic 27, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S73. Analytical HPLC analysis of epitope mimic 27. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S74. HRMS analysis of epitope mimic 27. Calculated m/z for C 109 H 147 N 27 O 26 S 3: 1174.0165 ½[M + 2H]+2; Found m/z 1174.0163.
Figure S75. LRMS analysis of epitope mimic 27. Calculated m/z for C 109 H 147 N 27 O 26 S 3: 1174.02 ½[M + 2H]+2; Found m/z 1174.42.
Figure S76. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 28, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S77. Analytical HPLC analysis of epitope mimic 28. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S78. HRMS analysis of epitope mimic 28. Calculated m/z for C 109 H 144 N 24 O 23 S 3: 1127.5077 ½[M + 2H]+2; Found m/z 1127.5071.
Figure S79. LRMS analysis of epitope mimic 28. Calculated m/z for C 109 H 144 N 24 O 23 S 3: 1127.51 ½[M + 2H]+2; Found m/z 1127.92.
Figure S80. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 29, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S81. LRMS analysis of trityl‐protected epitope mimic 29. Calculated m/z for C 117 H 157 N 27 O 32 S 3: 1275.04 ½[M + 2H]+2; Found m/z 1275.08.
Figure S82. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected epitope mimic 30, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S83. Analytical HPLC analysis of epitope mimic 30. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S84. HRMS analysis of epitope mimic 30. Calculated m/z for C 101 H 149 N 27 O 35 S 3: 1220.9834 ½[M + 2Na]+2; Found m/z 1220.9838.
Figure S85. LRMS analysis of epitope mimic 30. Calculated m/z for C 101 H 149 N 27 O 35 S 3: 1199.00 ½[M + 2H]+2; Found m/z 1199.42.
Figure S86. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected scrambled negative control 31, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S87. Analytical HPLC analysis of scrambled negative control 31. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S88. HRMS analysis of scrambled negative control 31. Calculated m/z for C 107 H 143 N 27 O 23 S 3: 1136.0084 ½[M + 2H]+2; Found m/z 1136.0112.
Figure S89. LRMS analysis of scrambled negative control 31. Calculated m/z for C 107 H 143 N 27 O 23 S 3: 1136.01 ½[M + 2H]+2 / 757.67 1/3[M + 3H]3+; Found m/z 1136.50 / 758.00.
Figure S90. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected scrambled negative control 32, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S91. Analytical HPLC analysis of scrambled negative control 32. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S92. HRMS analysis of scrambled negative control 32. Calculated m/z for C 110 H 149 N 27 O 26 S 3: 1181.0243 ½[M + 2H]+2; Found m/z 1181.0253.
Figure S93. LRMS analysis of scrambled negative control 32. Calculated m/z for C 110 H 149 N 27 O 26 S 3: 1181.02 ½[M + 2H]+2 / 787.68 1/3[M + 3H]3+; Found m/z 1181.58/ 788.17.
Figure S94. Analytical HPLC analysis overlay of individual fraction(s) of trityl‐protected scrambled negative control 33, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S95. Analytical HPLC analysis of scrambled negative control 33. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S96. HRMS analysis of scrambled negative control 33. Calculated m/z for C 110 H 146 N 24 O 23 S 3: 1134.5156 ½[M + 2H]+2; Found m/z 1134.5160.
Figure S97. LRMS analysis of scrambled negative control 33. Calculated m/z for C 110 H 146 N 24 O 23 S 3: 1134.52 ½[M + 2H]+2/ 756.68 1/3[M + 3H]3+; Found m/z 1135.00/ 756.92.
Figure S98. Analytical HPLC analysis overlay of individual fraction(s) of cyclic epitope mimic 34, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S99. Analytical HPLC analysis of cyclic epitope mimic 34. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S100. HRMS analysis of cyclic epitope mimic 34. Calculated m/z for C 96 H 151 N 27 O 26 S 3: 1098.0321 ½[M + 2H]+2; Found m/z 1098.0355.
Figure S101. LRMS analysis of cyclic epitope mimic 34. Calculated m/z for C 96 H 151 N 27 O 26 S 3: 1098.03 ½[M + 2H]+2/732.36 1/3[M + 3H]3+; Found m/z 1098.50/732.50.
Figure S102. Analytical HPLC analysis overlay of individual fraction(s) of cyclic epitope mimic 35, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S103. Analytical HPLC analysis of cyclic epitope mimic 35. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S104. HRMS analysis of cyclic epitope mimic 35. Calculated m/z for C 106 H 141 N 21 O 23 S 3: 1086.9914 ½[M + 2H]+2; Found m/z 1086.9949.
Figure S105. LRMS analysis of cyclic epitope mimic 35. Calculated m/z for C 106 H 141 N 21 O 23 S 3: 1086.99 ½[M + 2H]+2; Found m/z 1087.25.
Figure S106. Analytical HPLC analysis overlay of individual fraction(s) of cyclic epitope mimic 36, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S107. Analytical HPLC analysis of cyclic epitope mimic 36. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S108. HRMS analysis of cyclic epitope mimic 36. Calculated m/z for C 98 H 143 N 21 O 32 S 3: 1109.9607 ½[M‐2H]−2; Found m/z 1109.9435.
Figure S109. LRMS analysis of cyclic epitope mimic 36. Calculated m/z for C 98 H 143 N 21 O 32 S 3: 1111.98 ½[M + 2H]+2; Found m/z 1112.33.
Figure S110. Analytical HPLC analysis overlay of individual fraction(s) of cyclic epitope mimic 37, purified by preparative HPLC. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S111. Analytical HPLC analysis of cyclic epitope mimic 37. Column: Dr. Maisch Reprosil gold 200 C18, 5 μm 250 x 4.6 mm; Gradient: 0 to 100% buffer B in 30 minutes.
Figure S112. HRMS analysis of cyclic epitope mimic 37. Calculated m/z for C 107 H 143 N 21 O 23 S 3: 1093.9992 ½[M + 2H]+2; Found m/z 1093.9997.
Figure S113. LRMS analysis of cyclic epitope mimic 37. Calculated m/z for C 107 H 143 N 21 O 23 S 3: 1094.00 ½[M + 2H]+2; Found m/z 1094.58.