

Abstract
Respiratory illnesses are prevalent around the world, and inhalation-based therapies provide an attractive, noninvasive means of directly delivering therapeutic agents to their site of action to improve treatment efficacy and limit adverse systemic side effects. Recent trends in medicine and nanoscience have prompted the development of inhalable nanomedicines to further enhance effectiveness, patient compliance, and quality of life for people suffering from lung cancer, chronic pulmonary diseases, and tuberculosis. Herein, we discuss recent advancements in the development of inhalable nanomaterial-based drug delivery systems and analyze several representative systems to illustrate their key design principles that can translate to improved therapeutic efficacy for prevalent respiratory diseases.
Keywords: asthma, chronic obstructive pulmonary disease, inhalation delivery, lung cancer, nanomaterials, nanomedicine, tuberculosis
1 |. INTRODUCTION
Local delivery via inhalation is the means of delivering therapeutics and/or other excipients directly into the lungs with their preferential accumulation in specific lung areas or regions, particularly those with afflicted or diseased cells (Kuzmov & Minko, 2015; Lam, Liang, & Chan, 2012; Resnier, Mottais, Sibiril, Le Gall, & Montier, 2016), which is predominantly useful for treating various lung diseases (Roy & Vij, 2010). For the active component(s) to be available in a respirable form, they are delivered in a liquid or solid that is suspended in a gaseous medium to form an aerosol, which is facilitated by different devices such as nebulizers or dry powder inhalers (DPIs) (Rogueda & Traini, 2007; Rubin & Williams, 2014; Ruge, Kirch, & Lehr, 2013; Zhou, Tang, Shui Yee Leung, Gar Yan Chan, & Chan, 2014). The lungs are an attractive delivery target for treating both respiratory and systemic diseases due to their very large surface area for the deposition of therapeutics (Patton & Byron, 2007; Sahay, Alakhova, & Kabanov, 2010), high vascularization to promote systemic delivery (Kleinstreuer, Zhang, & Donohue, 2008; Schneeberger-Keeley & Karnovsky, 1968), and ability to bypass degradation in the gastrointestinal (GI tract and first pass metabolism in the liver (Pison, Welte, Giersig, & Groneberg, 2006), while additionally providing a noninvasive alternative to intravenous (IV) or intramuscular (IM) injections (Z. Liang, Ni, Zhou, & Mao, 2015). Ideally, drug formulations designed for inhalation delivery must penetrate and deposit into the lungs at diseased cells, limit exposure to healthy cells, and either restrict or promote uptake into circulation for respiratory or systemic disease, respectively (X. Li, Vogt, Hayes, & Man-sour, 2014; O-H Sham, Zhang, Finlay, Roa, & Löbenberg, 2004; Thorley & Tetley, 2013). Key functional considerations for inhalation and systemic administration (like injections and oral formulations) for disease treatment are summarized in Table 1.
TABLE 1.
Functional considerations for inhalation and systemic delivery for the treatment of disease
| Inhalation delivery | Systemic delivery |
|---|---|
|
|
Our ability to manipulate and manufacture materials on the nanoscale has provided an impetus for the development of new formulation strategies for improving therapeutic efficacy and mitigating adverse side effects (Chakroun et al., 2018; Y. Li, Wang, & Cui, 2016; Monroe, Flexner, & Cui, 2018; Stern & Cui, 2019). Since many therapeutic agents, such as small molecule drugs, nucleic acids, and peptides, exhibit poor stability alone for aerosolization, nanomaterial-based inhalation delivery systems have been developed to enhance their transport in aerosols and increase lung deposition and retention (Abdelaziz et al., 2018; Azarmi, Roa, & Löbenberg, 2008; Hu et al., 2013; Thorley, Ruenraroengsak, Potter, & Tetley, 2014; Yacobi et al., 2010). Many small molecule drugs have poor water solubility and short residence times, where direct inhalation of free drugs (both small and macromolecular) can lead to bolus release and severe lung toxicity; however, nanomaterials directly overcome these limitations and can provide added benefits through engineering of their size, shape, and surface chemistry (Beck-Broichsitter, Merkel, & Kissel, 2012). Nanomaterials improve inhalation delivery and therapeutic efficacy by enhancing drug bioavailability, mitigating effects of clearance mechanisms and degradation, controlling drug release, providing targeted delivery, and reducing necessary dosage and frequency, which leads to greater patient compliance and outcomes (Abdelaziz et al., 2018; Al-Hallak, Azarmi, Anwar-Mohamed, Roa, & Löbenberg, 2010; Haque, Whittaker, McIntosh, Pouton, & Kaminskas, 2016; Loira-Pastoriza, Todoroff, & Vanbever, 2014), which is illustrated in Figure 1. When compared to microparticles, nanoparticles (NPs) are more effectively internalized by cells but are too small to deposit sufficiently in the lungs alone (Gratton et al., 2008). Therefore, many nanomaterial systems have been extensively studied and developed for the treatment of respiratory diseases for administration with common aerosolization devices.
FIGURE 1.
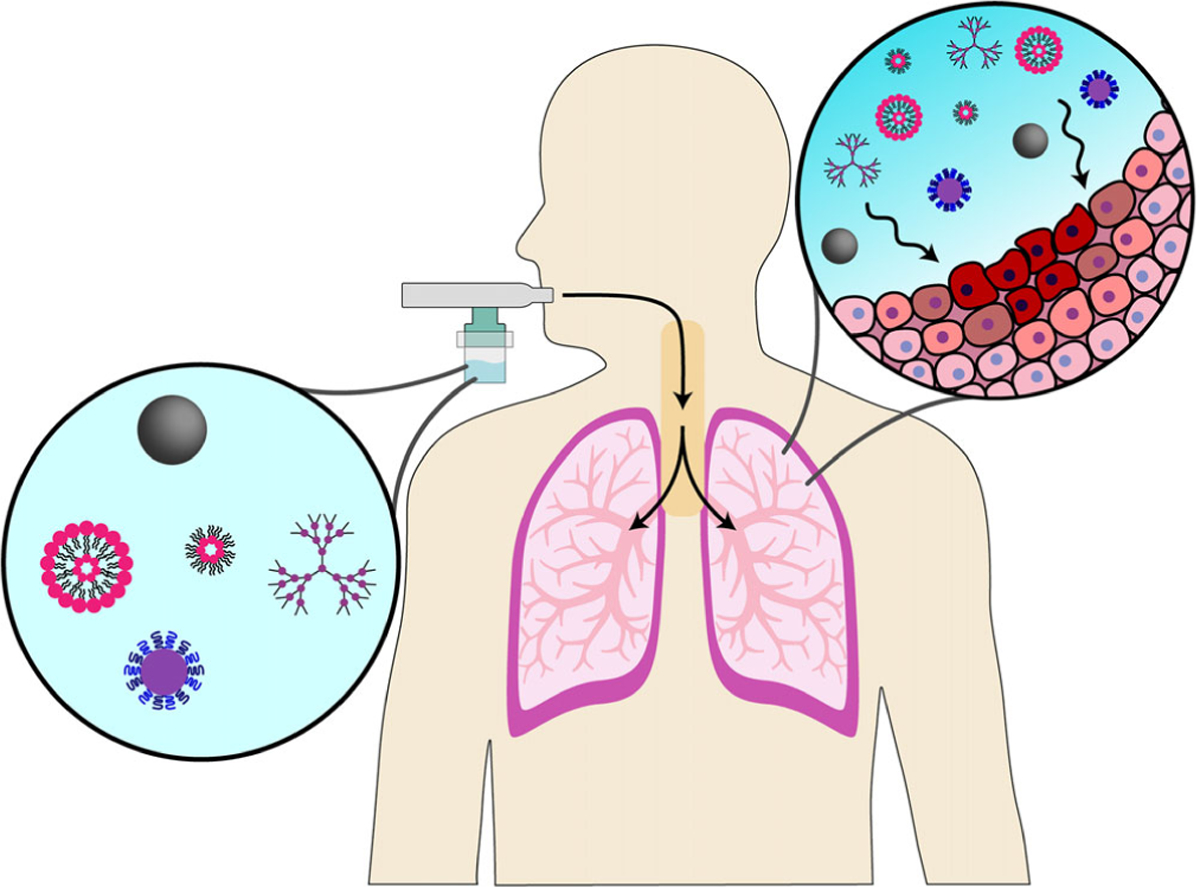
A wide variety of nanomaterial systems have been employed to develop inhalable nanomedicine systems (depicted here are liposomes, micelles, polymeric nanoparticles, dendrimers, and magnetic nanoparticles as examples of designs explored for this application). After administration with various aerosolization devices (jet nebulizer represented here), these systems are noninvasively delivered directly to lung tissues to passively target diseased cells. For a variety of respiratory diseases, this is advantageous as it enhances therapeutic efficacy and mitigates systemic toxicity, which can be further improved by decorating nanomaterials with targeting moieties for a particular lung disease
A wide range of materials have been employed in the design of nanoscale drug delivery systems for treating lung diseases, each with their own advantages and drawbacks (Madni et al., 2017). Generally, inorganic and nonbiodegradable NPs have been shown to induce inflammation, whereas biodegradable systems are relatively innocuous and induce fewer adverse side effects (Driscoll et al., 1996; Grabowski et al., 2013; Mura et al., 2011; Rytting, Nguyen, Wang, & Kissel, 2008). Polymeric systems typically exhibit high stability during nebulization and storage, induce minimal toxicity, and have easily tailored surface properties and degradation rates, but their small size allows for easy penetration into systemic circulation, reducing retention in the lungs (Dailey et al., 2006; Fiegel, Fu, & Hanes, 2004; Mura et al., 2011; Rodrigues et al., 2015; Ryan et al., 2013). Lipid-based systems are commonly amphiphilic in nature, which allows for incorporation of both hydrophilic and hydrophobic drugs in one particle, and are composed of biocompatible lipids (closely mimicking components of the lung surface lining), thereby limiting their toxicity and prolonging their retention in the lung (Cipolla, Shekunov, Blanchard, & Hickey, 2014; Cryan, Devocelle, Moran, Hickey, & Kelly, 2006; Èller, Èder, & Gohla, 2000; Haque et al., 2018; Lehofer et al., 2014; Nassimi et al., 2010; Weber, Zimmer, & Pardeike, 2014). However, lipid-based systems (for example, liposomes) have been found in some cases to suffer from burst release kinetics and instability during nebulization (Taylor, Taylor, Kellaway, & Stevens, 1990). Inorganic particles usually induce adverse effects to lung tissues (like oxidative stress), but their toxicity can be controlled by adjusting exposure time and dose (Ahmad et al., 2015; R. Liang, Wei, Evans, & Duan, 2014). Magnetic particles can be physically targeted to diseased sites in the lungs through an applied magnetic field for improved efficacy, while many metal NPs also display both imaging and therapeutic effects, yielding additional diagnostic and response-monitoring utility (Dames et al., 2007; Hasenpusch et al., 2012; Kumar Sukumar et al., 2013). While holistic comparative studies of different carriers have demonstrated that lipid-based systems have shown the best retention and accumulation in the lungs, the modification of nanocarrier surfaces with targeting moieties for diseased cells diminishes the differences of these systems for pulmonary delivery (Box 1) (Garbuzenko, Mainelis, Taratula, & Minko, 2014; Saad et al., 2008).
BOX 1. PARTICLE SIZING IN RELATION TO LUNG DEPOSITION.
The lungs are composed of a series of branching airways, divided into the conducting zone (upper airways, including the trachea, bronchi, and bronchioles) and the respiratory zone (lower airways, including the respiratory bronchioles, alveolar ducts, and alveolar sacs) (Crowder, Rosati, Schroeter, Hickey, & Martonen, 2002; Y. Lin, Wong, Qu, Chan, & Zhou, 2015; S. S. Park & Wexler, 2007). Particulate properties such as geometric size, shape, and density determine a particle’s inertia and thus influences where it will deposit along the respiratory tract (characterized as the aerodynamic diameter, AD) (Fu, Fiegel, Krauland, & Hanes, 2002; Joachim Heyder, 2004; Yeh, Phalen, & Raabe, 1976). There are three main mechanisms of airway deposition: inertial impaction, gravitation sedimentation, and diffusion (and interception for filamentous particles). Particles with an AD >5 μm deposit mostly in the upper airways via inertial impaction (as particles are unable to change their trajectory with tidal air); particles with an AD between 1 and 5 μm deposit mostly in the lower airways via sedimentation; and particles with an AD <1 μm remain mostly suspended in the airstream and are exhaled (with minor deposition usually through diffusion) (Balásházy, Martonen, & Hofmann2, W., 1990; Byron, 1986; Gagnadoux et al., 2008; Heyder, Gebhart, Rudolf, Schiller, & Stahlhofen, 1986; Jaques & Kim, 2000). Typically, nanomedicines systems are suspended in liquid or dry powder aerosol droplets, and it is the size and properties of these droplets that dictate deposition patterns.
Regardless of their material properties, all nanomedicine systems face the same challenge of navigating the respiratory tract and overcoming the clearance mechanisms of the lungs for effective deposition and retention to yield improved therapeutic outcomes (Todoroff & Vanbever, 2011; Yang, Peters, & Williams Iii, 2008a; J. Zhang, Wu, Chan, & Watanabe, 2011). Mucociliary clearance is the dominant clearance mechanism of the upper airways, where secreted mucus traps particles and the beating cilia of the epithelium propel particles out to then be coughed out or swallowed (Möller et al., 2008; Patton et al., 2010). Particle size and zeta potential are not critical particle parameters in influencing their clearance rate via the mucociliary escalator, whereas material properties and surface chemistry have a significant impact (Henning et al., 2010). Mucus itself is highly viscoelastic and adhesive and thus can serve as a barrier by immobilizing NPs, but particle surface properties of nanomaterials can be tuned to promote mucus penetration (Boylan et al., 2012; Lai et al., 2011; Lai, Wang, & Hanes, 2008; Sigurdsson, Kirch, & Lehr, 2013; Suk et al., 2009; Tang, Fu, Watkins, & Hanes, 2009). In the lower airways, phagocytosis by alveolar macrophages (AMs) is the dominant clearing mechanism, where after internalization, particles are digested in lysosomes or particle-loaded macrophages are removed into the lymph or by the mucociliary escalator (Brain, 1992; Lombry, Edwards, Préat, & Vanbever, 2004). Size, shape, and charge density properties of particles influence their uptake by macrophages, where small (<200 nm diameter) particles with high aspect ratios and low charge density are most likely to be best suited for avoiding macrophages (Champion & Mitragotri, 2009; Chono, Tanino, Seki, & Morimoto, 2006; Geiser et al., 2008; Makino et al., 2003). However, in the case of certain infections (like pneumonia and tuberculosis [TB]), the macrophages are the relevant drug target and nanomedicines should be designed to promote macrophage uptake (Lee, Loo, Traini, & Young, 2015b). Lastly, the pulmonary surfactant (PS) layer is a mixture of lipids and proteins that line the alveolar epithelium that are essential for breathing; nanomedicines can absorb PS-related components on their surface, which strongly influences their biological activity and the integrity of the PS layer (Geiser, Schürch, & Gehr, 2003; Guagliardo, Pérez-Gil, De Smedt, & Raemdonck, 2018; Raesch et al., 2015). Particle properties such as size, hydrophobicity, and surface charge influence surfactant stability and thereby toxicity, but nanomaterials can be decorated with different PS components to improve safety and delivery (De Backer, Cerrada, Pérez-Gil, De Smedt, & Raemdonck, 2015; Hidalgo, Cruz, & Pérez-Gil, 2015, 2017; Nag, Vidyashankar, Devraj, Garcia, & Panda, 2010). These physiological barriers must be considered when designing inhalable nanomedicines to maximize lung deposition and retention to increase therapeutic efficacy.
In this review, we highlight several examples of inhalable nanomedicine systems, focusing on their design and material properties that make them suited for pulmonary delivery and increase their therapeutic efficacy. We focus on the recent advances in the development of polymeric, lipid-based, and inorganic material systems in the context of inhalable treatments for some of the most common lung diseases, emphasizing lung cancer while also discussing chronic obstructive pulmonary disease (COPD), asthma, and TB. While inhalation is also an attractive strategy for the treatment of systemic diseases, and for some therapeutics, this route provides high absorption into the blood with a faster onset of action, such as insulin (Bi, Shao, Wang, & Zhang, 2008; Siekmeier & Scheuch, 2009), we note our discussion focuses solely on the treatment lung-resident diseases.
2 |. INHALABLE THERAPEUTICS FOR LUNG CANCER
Lung cancer represents the second most common type of cancer worldwide and leading cause of cancer death, with 250,000 new cases and 150,000 deaths projected in 2019 (Siegel, Miller, & Jemal, 2019). Lung cancer is classified into two types: non-small cell lung cancer (NSCLC) representing ~85% of cases and small cell lung cancer (SCLC) representing ~15% of cases (Mittal et al., 2016). Typical treatment options include surgical resection for nonmetastatic lung cancers (only roughly 10–20% of NSCLC patients) and otherwise chemotherapy (Lemjabbar-Alaoui, Hassan, Yang, & Buchanan, 2015). However, early diagnosis is very challenging and thus most lung cancers are diagnosed in advanced metastatic stages, leading to chemotherapy as the primary means of prolonging survival, controlling symptoms, and improving the quality of life of lung cancer patients (Mangal et al., 2017). Therefore, research efforts in nanomedicine have been focused on improving the therapeutic efficacy and safety of chemotherapy agents for lung cancer with an increased interest in pulmonary administration.
Pulmonary delivery of nanomedicines enhances lung cancer treatment by transporting encapsulated poorly water-soluble, potent anticancer drugs directly to their intended site of action, facilitating targeted and controlled release at tumor sites and reducing the necessary dosage and mitigating off-target side-effects (Hitzman, Wattenberg, & Wiedmann, 2006; Koshkina et al., 2001; Tomoda et al., 2009); however, inhalation distributes these drugs evenly around the lungs, potentially exposing both healthy and diseased cells to chemotherapy and inducing undesirable adverse effects on the lungs (Lee, Loo, Traini, & Young, 2015a). Drug deposition and distribution is influenced by the physical occlusion of the respiratory track by tumors, where tumors reduce the cross-sectional area of the airway and thereby divert airflow to nonoccluded areas and reduce drug deposition on tumor tissue (Carvalho, Carvalho, & Mcconville, 2011; Mangal et al., 2017). Modeling of the effects of tumors in terms of size and location on particle transport has revealed that most drugs deposit on the frontal end of the tumor (Z. Zhang, Kleinstreuer, Kim, & Hickey, 2002); this alongside the heterogeneity of tumor tissues further emphasizes the advantages and necessity of nanoformulations for treating lung cancer. By instilling enhanced tumor penetration, targeting capabilities, and means of overcoming multidrug resistance, nanomedicines improve the therapeutic efficacy of chemotherapy drugs and improve lung cancer patient outcomes (I. Kim et al., 2013; Markman, Rekechenetskiy, Holler, & Ljubimova, J. Y., 2013; N. R. Patel, Pattni, Abouzeid, & Torchilin, 2013).
Due to the potency of chemotherapy drugs, improving the targeting and accumulation in tumor tissues to enhance antitumor efficacy and reduce adverse side effects serves as an impetus for nanomedicine design. For cancer, the enhanced permeability and retention (EPR) effect is often accredited for nanomedicine accumulation in tumor tissues with systemic delivery, due to the tumor’s irregular vasculature and poor lymphatic drainage, as a passive means of targeting (Maeda, 2001). However, this phenomenon may not play a role with inhalable delivery, but the localization of therapeutics directly in the lungs does provide a means of passively targeting tumor tissues (Azarmi et al., 2006). More active targeting mechanisms, such a pH-triggered release and enzymatic responsivity, are able to further improve the therapeutic efficacy of these systems (Anderson & Cui, 2017; Danhier, 2016). In regards to lung cancer, active targeting by designing nanomaterials to interact with tumor-specific ligands, such as the epithelial growth factor receptor (EGFR) (Rusch et al., 1997), folate receptor (Nogueira et al., 2015; O’shannessy et al., 2012), and the luteinizing hormone-releasing hormone (LHRH) receptor (Dharap et al., 2005), has been thoroughly studied and proven to enhance antitumor efficacy (Saad et al., 2008). By harnessing these targeting techniques, localized nanomedicine through inhalation is a viable, safe, and effective treatment strategy for lung cancer as has been shown in preclinical and clinical studies (Mangal et al., 2017).
In this section, we discuss the recent progress and advancements in the design and development of different nanomaterial systems for the treatment of lung cancer that address challenges faced in the field. We focus on the design of different polymeric, lipid-based, and inorganic material systems for pulmonary delivery and its translation to therapeutic efficacy for lung tumors.
2.1 |. Polymeric nanomaterials
Direct linking of drugs to polymers is an effective strategy for enhancing drug delivery by improving solubility and hindering premature degradation. Vanbever and coworkers designed a drug-polymer conjugate from the anticancer drug paclitaxel (PTX) and polyethylene glycol (PEG) that increased PTX retention in the lungs and thus improved its efficacy and reduced its pulmonary toxicity (Luo et al., 2016). PTX was modified with pentynoic acid and conjugated to hydrophilic PEG-N3 (either 6 kDA or 20 kDA molecular weight [MW]) via click chemistry to yield a hydrolyzable ester bond for PTX release and to increase PTX solubility. Following intratracheal (IT) instillation in healthy C57BL/6NRj mice, PEG conjugation was able to increase the maximum tolerated dose of PTX up to 100-fold compared to Taxol® (free PTX formulation), enhancing the safety of PTX to healthy cells. When tested in a murine Lewis lung carcinoma model (LL/2-luc-M38), both PEG sizes were able to enhance antitumor efficacy of PTX after IT instillation after a single dose compared to free PTX for both IT and IV delivery, but the PTX-PEG 20 kDa conjugate showed an equivalent efficacy as the 6 kDa conjugate delivered at a 2.5-fold higher dose, suggesting the polymer MW plays a role in anticancer activity. They also investigated the induction of inflammation following inhalation therapy by detecting biochemical markers in the bronchoalveolar lavage (BAL) fluid of mice, specifically for the presence of lactate dehydrogenase (LDH, indicator of cell injury) and pulmonary cell number and distributions. The high dosage of PTX-PEG 6 kDa (50 mg/kg PTX equivalent) increased LDH and neutrophil levels 24-hr postinhalation delivery; however, this was lower than that induced by equivalent doses of PTX and was reversible. When compared to an equivalent dosage of free PEG-N3, there was only a slight increase in LDH and neutrophil levels, highlighting the compatibility of PEG. PTX-PEG was able to retain longer in the lungs and provide a means of sustained release with lower toxicity and superior antitumor efficacy compared to free PTX administered intratracheally, emphasizing PEGylation of PTX as an effective delivery system for inhalation treatment of lung cancer (Luo et al., 2016).
Dendrimers are typically smaller in size than many NP systems, and thus exhibit better interstitial diffusion, absorption, and tumor penetration properties (Kaminskas et al., 2014). Kaminskas and colleagues developed a 56 kDa PEGylated (PEG1100) polylysine dendrimer conjugated to the anticancer drug doxorubicin (DOX) through an acid-labile 4-hydrazinosuphonyl benzoic acid linker as a system that controlled and prolonged exposure of DOX to lung-resident cancers (called D-DOX). In a Sprague Dawley rat model, the pharmacokinetics and biodistribution of DOX and the dendrimer itself (through 3H-labeling) were monitored. After 7 days, 15% of the D-DOX dose remained in the lungs after IT instillation (around 20% cleared from the mucociliary escalator and less than 1% recovered from AMs), whereas free DOX was rapidly cleared after 1 day, emphasizing the improved retention of the dendrimer system. In a MAT 13762 IIIB breast metastasized lung cancer model in female F334 rats, twice weekly administration (either IV or IT administration) of D-DOX or free DOX showed a >95% tumor burden reduction after 2 weeks for IT D-DOX compared to the IV DOX which averaged around a 30–50% reduction; inhalation of free DOX solution lead to extensive lung-related toxicity and death. Their results suggest that this dendrimer system as an inhalable drug delivery system prolongs chemotherapy drug exposure to lung-resident cancers and improves overall anticancer therapeutic efficacy, and this group has further highlighted the advantages of this system with other animal models as well (Kaminskas et al., 2014; Leong et al., 2018; Ryan et al., 2016).
Combinations of different chemotherapy drugs have exhibited greater antitumor effects and can potentially lead to improved treatment outcomes. Chen and team showcased that combining multiple anticancer agents in a single copolymer NP system lead to enhanced lung cancer therapy after pulmonary delivery (Xu et al., 2019). They synthesized a methoxy poly(ethylene glycol)-poly(ethyenimine)-poly-(L-glutamate) (mPEG-OEI-PLG, 1:1:39 M composition ratio) copolymer carrier to codeliver DOX and cisplatin (CDDP), where the Co-NPs were formed via electrostatic interactions with DOX loading and chelate interactions with CDDP loading with the polymer, as detailed in Figure 2a. The Co-NPs ranged in size from 43 to 73 nm (depending on the loading of only one drug or both) and exhibited good dispersion stability. In phosphate buffer saline (PBS), the particles exhibited a pH-dependent release of DOX and CDDP with accelerated release at lower pH values, where DOX released faster than CDDP due to the protonation of carboxylic acid groups in mPEG-OEI-PLG and the increased hydrophilicity of DOX in acidic conditions, as seen in Figure 2b. An in vitro cytotoxicity assay on B16F10 melanoma cells revealed that Co-NPs exhibit higher cytotoxicity than either drug alone (as seen in Figure 2c), and the copolymer itself showed low cytotoxicity and good biocompatibility. In a B16F10 tumor-bearing C57BL/6 mice model, the biodistribution of the NP systems were compared after systemic or pulmonary administration, as detailed in Figure 2d, where free DOX was captured more by the reticuloendothelial system and Co-NPs showed higher and longer lung retention via pulmonary delivery that lasted 7 days (attributed to the mucoadhesive and penetration properties of PEG). Regarding anticancer efficacy, Co-NPs exhibited the greatest antitumor effect with lung weights most comparable to healthy mice lungs, as represented in Figure 2e, and no obvious side effects. Their NP system represents a very promising strategy for an inhalable lung cancer treatment, and they have emphasized the efficaciousness of pH-responsive codelivery systems by developing several therapeutic systems (Xu et al., 2015, 2019; Xu, Tian, Wang, Wang, & Chen, 2016).
FIGURE 2.
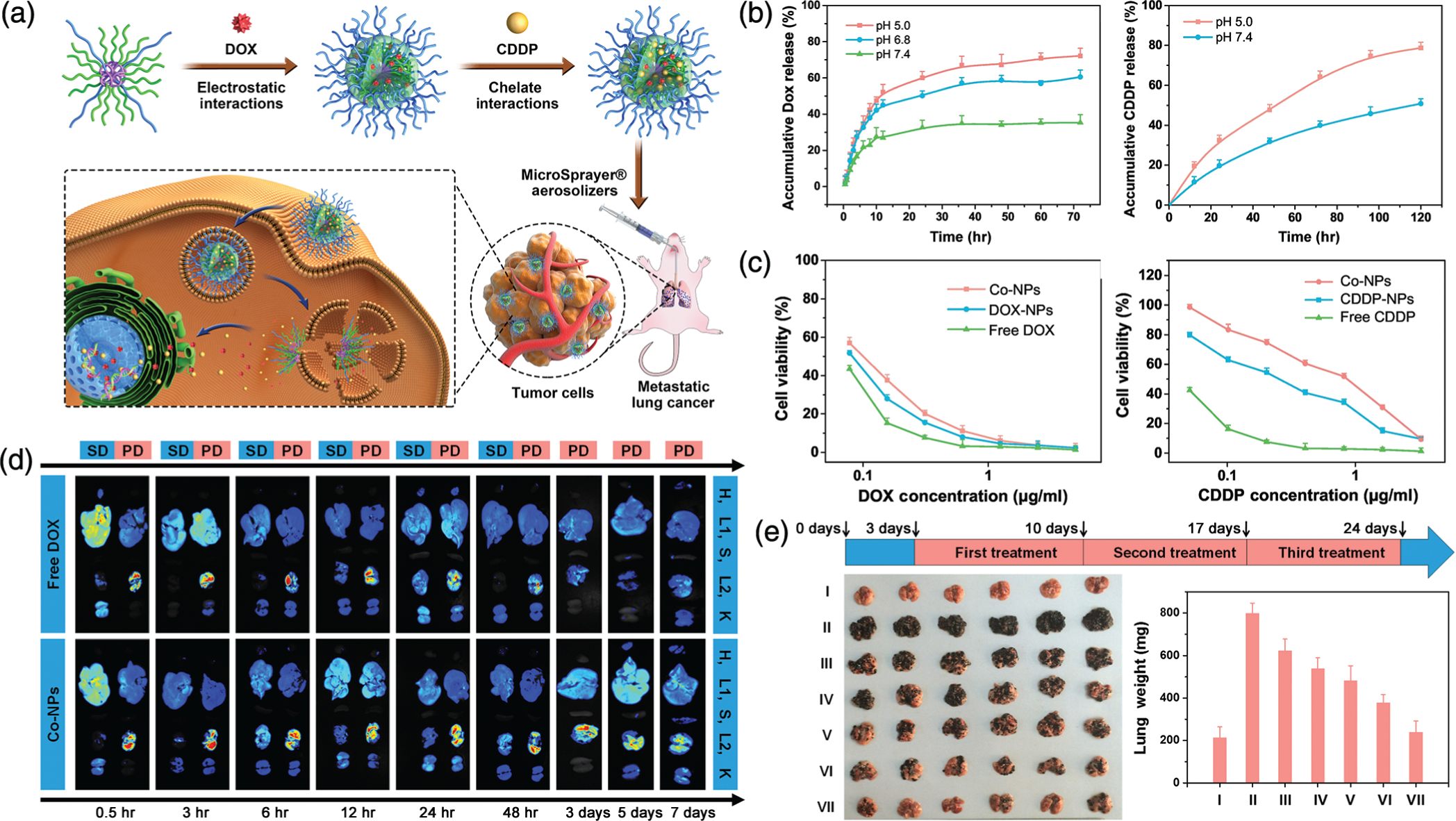
Copolymer nanoparticle (NP) system for the pulmonary codelivery of doxorubicin (DOX) and cisplatin (CDDP). (a) Schematic representation of the copolymer NP showing the incorporation of DOX through electrostatic interactions and CDDP through chelate interactions to create Co-NPs. The Co-NPs are aerosolized to be administered to mice bearing lung tumors, where within the Co-NPs penetrate and enter tumor cells and begin to rapidly release their contents to induce cytotoxicity. (b) The encapsulated drug release of DOX (left) and CDDP (right) from the Co-NPs in PBS, showing faster release at lower pH. (c) Cytotoxicity assay results against B16F10 melanoma cells in relation to DOX concentration (left) and CDDP concentration (right), indicating the enhanced cancer cell toxicity of the codelivery system compared to either drug alone. (d) The in vivo biodistribution of DOX in main organs of mice after either systemic delivery (SD) or pulmonary delivery (PD) of free DOX or Co-NPs (H, heart; L1, liver; S, spleen; L2, lung; K, kidney) at different time points over 72 hr, showing increased retention from Co-NPs in PD. (e) in vivo antitumor efficacy in a B16F10 metastatic lung cancer mice model with the treatment timeline (top). Surgically removed lungs (left) and their average weight (right) for each treatment group (I, healthy mice control; II, PBS; III, free CDDP; IV, free DOX; V, CDDP-NPs; VI, DOX-NPs; VII, Co-NPs) after pulmonary delivery. (Reprinted with permission from Xu et al. (2019). Copyright 2019 Elsevier)
In another combination therapy system, Youn and coworkers developed inhalable NPs self-assembled from human serum albumin (HSA) conjugated to DOX and octyl aldehyde (OA) and adsorbed with apoptotic tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) protein, called TRAIL/DOX HSA-NP, that enhanced apoptotic effects against drug-resistant lung tumors (I. Kim, Jun Byeon, et al., 2013). HSA is highly biocompatible, biodegradable, and stable and functions as a nontoxic and nonimmunogenic carrier; DOX and OA’s hydrophobicity facilitate the self-assembly of the particles. In insensitive or resistant cancer lines, TRAIL is not effective alone, but when combined with a chemotherapy agent, it helps improve antitumor efficacy. The TRAIL/DOX HSA-NP were observed to be almost spherical with a size around 340 nm and stable after aerosolization. Cytotoxicity of the NPs was evaluated in H226 human lung squamous cell carcinoma cells over the course of 3 days, which are resistant to TRAIL and DOX; the TRAIL/DOX HSA-NPs displayed synergistic cytotoxicity and apoptotic activity compared to DOX HSA-NPs and TRAIL HSA-NPs. Cytotoxic activity manifested 3 days after treatment, which was attributed to the delayed release of DOX from HSA. Using Cy5.5-labeling of TRAIL and DOX fluorescence, deposition in ICR mice was monitored after microspray aerosolization of TRAIL/DOX HSA-NPs, showing high deposition in the lungs for 3 days. In BALB/c nu/nu mice bearing H226 cell-induced metastatic tumors, the antitumor efficacy of TRAIL/DOX HSA-NPs was evaluated. In mice treated with TRAIL/DOX HSA-NPs, tumors were found to be much smaller and lighter compared to those treated with either solo drug NP, and excised lungs were found to have less malignant surface lesions and tumor tissues for the TRAIL/DOX HSA-NP treatment group. The increased antitumor efficacy was attributed to the synergistic apoptotic effects of DOX and TRAIL. While this system was not directly compared to other routes of administration (like IV injection), this work showcases an inhalable anti-lung cancer drug delivery system that can potentially reduce DOX doses and minimize side effects to provide more effective and safer lung cancer treatment, even for drug-resistant cancers (I. Kim, Jun Byeon, et al., 2013).
2.2 |. Lipid-based NPs
Many lipid-based material systems have been studied as drug carriers due to their high biocompatibility and ability to load multiple types of drugs in a single system. To improve cancer cell selectivity and tumor penetration, Rosière and team developed solid lipid nanoparticles (SLNs) modified with a folate-grafted copolymer to target lung cancer cell surfaces that proved to greatly increase tumor accumulation and retention (Rosière et al., 2018). The copolymer (F-PEG-HTCC) was composed of folate conjugated to polyethylene glycol (PEG) and N-[(2-hydroxy-3-trimethylammonium) propyl] chitosan chloride (HTCC), where chitosan is very biocompatible and prolongs lung retention due to its mucoadhesive properties. The F-PEG-HTCC was coated onto PTX-loaded SLNs (prepared via nanoprecipitation) to create spherical particles of around 250 nm. Using fluorescence microscopy to assess cell binding and uptake, folate receptor-expressing HeLa adenocarcinoma ovarian cells and M109-HiFR murine lung carcinoma cells were treated with SLNs, showing improved uptake or at least binding via folate receptor targeting and a reduction in the half-maximum inhibitory concentration of PTX loaded in SLNs compared to free PTX. In a M109 tumor model in CD1 and BALC/c mice, endotracheal administration via microsprayer of coated SLNs showed a 32-fold higher PTX concentration in the lungs and longer retention (after 6 hr postadministration) compared to pulmonary delivery and IV injection of free PTX. The coated SLNs exhibited a favorable pharmacokinetic profile after pulmonary delivery with enhanced tumor penetration and limited systemic exposure as measured as PTX plasma concentrations. This study demonstrates the enhanced efficacy of decorating nanomaterial surfaces with targeting moieties for improving lung cancer therapy and emphasizes inhalation as an advantageous route of administering PTX for treating tumors (Rosière et al., 2018).
To overcome pump and nonpump cellular drug resistance that can present itself in lung cancer, Minko and coworkers developed a lipid-based nanomaterial system that successfully delivered chemotherapy and resistance suppressors that improved lung cancer treatment (Taratula, Kuzmov, Shah, Garbuzenko, & Minko, 2013). They developed a nanostructured lipid carrier (NLC) using a modified melted ultrasonic dispersion method loaded with DOX (to evaluate uptake) or PTX (to assess anticancer efficacy) with small interfering RNA (siRNA) targeted to MRP1 and BCL2 mRNAs (suppressors of pump and nonpump resistance, respectively) to suppress drug resistance complexed at its surface via electrostatic interactions alongside a modified synthetic analog of LHRH conjugated to DSPE-PEG-COOH (DSPE-PEG-LHRH) to allow for targeting to receptors overexpressed on the surface of lung cancer cells, as represented in Figure 3a. Upon nebulization of the NLCs, they showed no change in size and distribution, making them stable and suitable carriers for pulmonary delivery. Against A549 human lung adenocarcinoma cells in vitro, the PEGylated NLC showed high cell viability after 48 hr incubation, suggesting the particles are safe carriers, as shown in Figure 3b. Compared to free TAX, the LHRH-NLC-TAX-siRNAs particles showed a 120-fold increased cytotoxicity and a 16-fold increase compared to particles without siRNAs, as depicted in Figure 3c, and the complexed siRNAs were able to effectively downregulate gene expression of MRP1 and BCL2. To assess the in vivo biodistribution and antitumor efficacy of the NLCs administered via pulmonary delivery, an orthotopic A549-Luc lung cancer model in athymic nu/nu mice was used with a nose-only chamber for delivery. As detailed in Figure 3d, inhalation lead to a high accumulation of the administered dose retained in the lungs, whereas IV injection lead to most of the NLCs to accumulate in the liver. The LHRH targeting moiety on the surface of the NLCs enhanced tumor localization with much less retention in healthy lung tissues, as depicted in Figure 3e and f. In regard to antitumor efficacy, the inhalation of LHRH-NLC-TAX-siRNAs lead to the greatest reduction in tumor volume as monitored over 25 days. This effect was greater compared to mice treated with IV-injected TAX and inhaled LHRH-NLC-TAX, as detailed in Figure 3g. This study highlights the advantages of inhalation delivery for lung cancer therapy with a means of primarily localizing in tumor tissues over healthy lung tissues and other organs, leading to sufficient tumor suppression and a reduced risk of adverse side effects (Taratula et al., 2013).
FIGURE 3.

Nanostructured lipid carrier (NLC) system for the pulmonary codelivery of paclitaxel (TAX) and siRNAs for resistance suppression to treat lung cancer. (a) Illustration of the NLC system, showing drugs loaded in the core (TAX), siRNAs complexed to cationic heads groups of the lipid particle (DOTAP), and lung cancer targeting moiety, luteinizing hormone-release hormone (LHRH) conjugated to DSPE-PEG on the surface. (b) Cell viability of A549 human lung adenocarcinoma cells after 48 hr incubation with blank NLC and PEGylated NLC, showing great biocompatibility of the carrier with increased safety after surface decoration with PEG. (c) in vitro cytotoxicity of the TAX-loaded NLCs and free TAX against A549 cells; the NLC systems showed enhanced cytotoxicity (lower IC50 value) compared to free TAX, where the addition of resistance suppressing siRNAs on the surface even further enhanced cytotoxic activity. (d) in vivo distribution of fluorescently-labeled (Cy5.5 dye) NLCs in mice after IV injection (left) and inhalation (right), showing increased accumulation and retention in lungs from pulmonary delivery. (e) Distribution of fluorescently-labeled NLCs in mice lungs bearing A549 tumors, highlighting improved targeting to tumor tissues by LHRH. (f) Bright-field and fluorescence microscope images of tumor tissues of mice treated with Cy5.5-loaded LHRH-NLCs, showing improved tumor targeting and retention with minimal accumulation in healthy lung tissues. (g) Tumor volume changes in mice after the beginning of treatment with repeated treatments every 3–5 days (x-axis), emphasizing the enhanced antitumor efficacy of LHRH-NLC-TAX-siRNAs system (1, untreated mice; 2, inhaled LHRH-NLC; 3, IV injected TAX; 4, inhaled LHRH-NLC-TAX; 5, inhaled LHRH-NLC-TAX-siRNAs). (Reprinted with permission from Taratula et al. (2013). Copyright 2013 Elsevier)
Surface-modification of nanomaterials with targeting moieties readily improves accumulation in lung tumor tissues, and Yang and colleagues decorated liposomes with antibodies that increased their selectivity to lung cancer cells after pulmonary delivery (C. Lin et al., 2017). Antibodies for carbonic anhydrase IX (CA IX), a hypoxia-inducible enzyme overexpressed on tumor cell surfaces in NSCLC, were conjugated (via thiol addition to maleimide of DSPE-PEG45-Mal) onto the surface of liposomes (composed of DSPE-PEG2000) loaded with the anticancer drug triptolide (TPL) to yield CA IX-TPL-Lips of around 160 nm in diameter. In PBS, the targeted and nontargeted liposomes displayed similar in vitro sustained release kinetics with about 25% of their contents released over 96 hr. The liposomes displayed high stability over 14 days with no changes in particle size or precipitation. To assess the in vitro cytotoxicity of the liposomes, CA IX-positive A549 human lung adenocarcinoma cells and A549 tumor spheroids were cultured in hypoxic environments and treated with liposomal and free TPL formulations. The targeted liposomes showed improved cellular uptake and a lower IC50 value than the nontargeted liposomes and free TPL, which was attributed to the targeted liposomes’ stronger penetration and binding affinity to NSCLC tumor cell surfaces. In an A549-Luc orthotopic BALB/c nude mouse model, liposomes and free TPL were administered endotracheally to observe biodistribution and assess antitumor efficacy. The CA IX-Lips showed prolonged residence in the lungs over 96 hr with no detection in other organs; however, this was not compared to nontargeted liposomes. Regarding anticancer efficacy, liposomes or free TPL were delivered via inhalation twice a week for 4 weeks; the targeted liposomes showed improved efficacy in reducing tumor burden and extending the survival time of mice bearing orthotopic lung tumors compared to free TPL and nontargeted liposomes, as confirmed with hematoxylin and eosin (H&E) staining and mouse body weight. This inhalable liposomal formulation improved therapeutic efficacy and reduced off-target toxicity in the treatment of lung cancer, highlighting the additional benefit imparted by target moieties in enhancing tumor accumulation.(C. Lin et al., 2017) The team has further improved the antitumor efficacy of their liposomal system by also decorating the surface also with a cell-penetrating peptide to improve tumor penetration (C. Lin et al., 2018).
2.3 |. Inorganic nanomaterials
Magnetic materials have been extensively explored as drug delivery systems, as they have both imaging and therapeutic properties. While magnetic hyperthermia can cause tumor ablation from heat generation from materials like superparamagnetic iron oxide nanoparticles (SPIO NPs), inadequate accumulation in tumor cells can induce resistance and damage healthy tissues. Panyam and coworkers developed a tumor-targeting SPIO NP system to overcome this challenge to provide an effective and noninvasive lung cancer therapy (Sadhukha, Wiedmann, & Panyam, 2013). They functionalized SPIO NPs with an EGFR-targeting peptide sequence (YHWYGYTPQNVI) to create a magnetic hyperthermia system that targets lung tumor cells after pulmonary administration. The resulting particles averaged around 369 nm in size after peptide functionalization and showed a concentration-dependent heating rate in the presence of an alternating magnetic field (AMF). When compared to SPIO NPs decorated with a scrambled peptide, the EGFR-targeted SPIO NPs showed 4.5-fold higher cellular uptake in A549 human adenocarcinoma lung cancer cells and more efficient cell killing via magnetic hyperthermia in the presence of an AMF. SPIO NP dispersions were administered in an aerosol using ultrasonic atomization to Fox Chase SCID mice bearing orthotopic A549-Luc-C8 tumors to assess biodistribution and antitumor efficacy. Targeting to EGFR alongside pulmonary delivery enhanced SPIO NPs retention in lung tissue over a week with very low accumulation in other organs, suggesting less likelihood of negative side effects. Regarding anticancer activity, the magnetic hyperthermia from the SPIO NPs reduced tumor burden, where the EGFR-targeted SPIO NP treatment lead to the greatest reduction in tumor size and weight compared to nontargeted SPIO-treated and untreated mice. This work showcases the increased efficacy and reduced off-target effects from pulmonary delivery of EGFR-targeting SPIO NPs for lung-resident tumor growth inhibition via magnetic hyperthermia, while additional studies concerning the long-term effects on lung tissues, such as inflammation, would further enhance the understanding of the safety of this system for long term treatment of lung tumors (Sadhukha et al., 2013).
Lung cancer detection and diagnosis remains a prevalent challenge in improving patient outcomes. In this context, Coll and team developed Gadolinium (Gd)-based ultra-small rigid platforms (USRPs) to serve as a theranostic agent with combined multimodal imaging and radiosensitizing properties that successfully provided inhalation-based simultaneous lung cancer therapy and response monitoring (Dufort et al., 2015). Through a top-down process, gadolinium oxide was encapsulated in a polysiloxane matrix with an near-infrared-fluorescent dye (Cy5.5) and a chelating species (DOTA) covalently grafted onto the surface of the particles (which complexed on average 10 Gd species), as detailed in Figure 4a. These small particles (less than 5 nm in diameter on average) can be detected in the lungs after administration through ultrashort echo-time magnetic resonance imaging (UTE-MRI) and fluorescence imaging while also acting as a radiosensitizing agent to kill cancer cells. To assess the theranostic properties of this system in vivo, USRPs were administered intratracheally using a microsprayer to nude mice bearing orthotopic H358-Luc human bronchioalveolar carcinoma tumors. Previous studies assessing the pharmacokinetics and biodistribution of the USRPs show the USRPs travel from the airways to lung tissues to the kidneys with negligible hepatic clearance and no observation of an increase in pulmonary and renal inflammation markers, suggesting the USRPs have suitable biocompatibility (Bianchi et al., 2014). Tumor tissues could be detected using the USRPs using UTE-MRI and fluorescence tomography, as detailed in Figure 4b, where nebulized USRPs showed stronger localization with tumor tissues compared to IV-injected USRPs and better lung retention over 24 hr. After nebulization, the USRP deposition around tumor nodules was sufficient for generating radiosensitization after subjecting mice to one dose of 10 Gy conventional radiation 1 day after inhalation of USRPs; this increased the mean survival time of mice treated with USRPs and irradiation compared to mice receiving irradiation alone (122 vs. 77 days). Additional studies assessing the effects on tumor burden would also shed more light on the therapeutic efficacy of the system. Considering the difficulties in diagnosing lung cancers, this USRP system provides a facile and noninvasive means of simultaneously detecting and treating lung tumors with negligible side effects or toxicity to healthy tissues. This system can be further optimized by the addition of targeting moieties which could help improve tumor accumulation and radiotherapy (Dufort et al., 2015; Morlieras et al., 2013).
FIGURE 4.
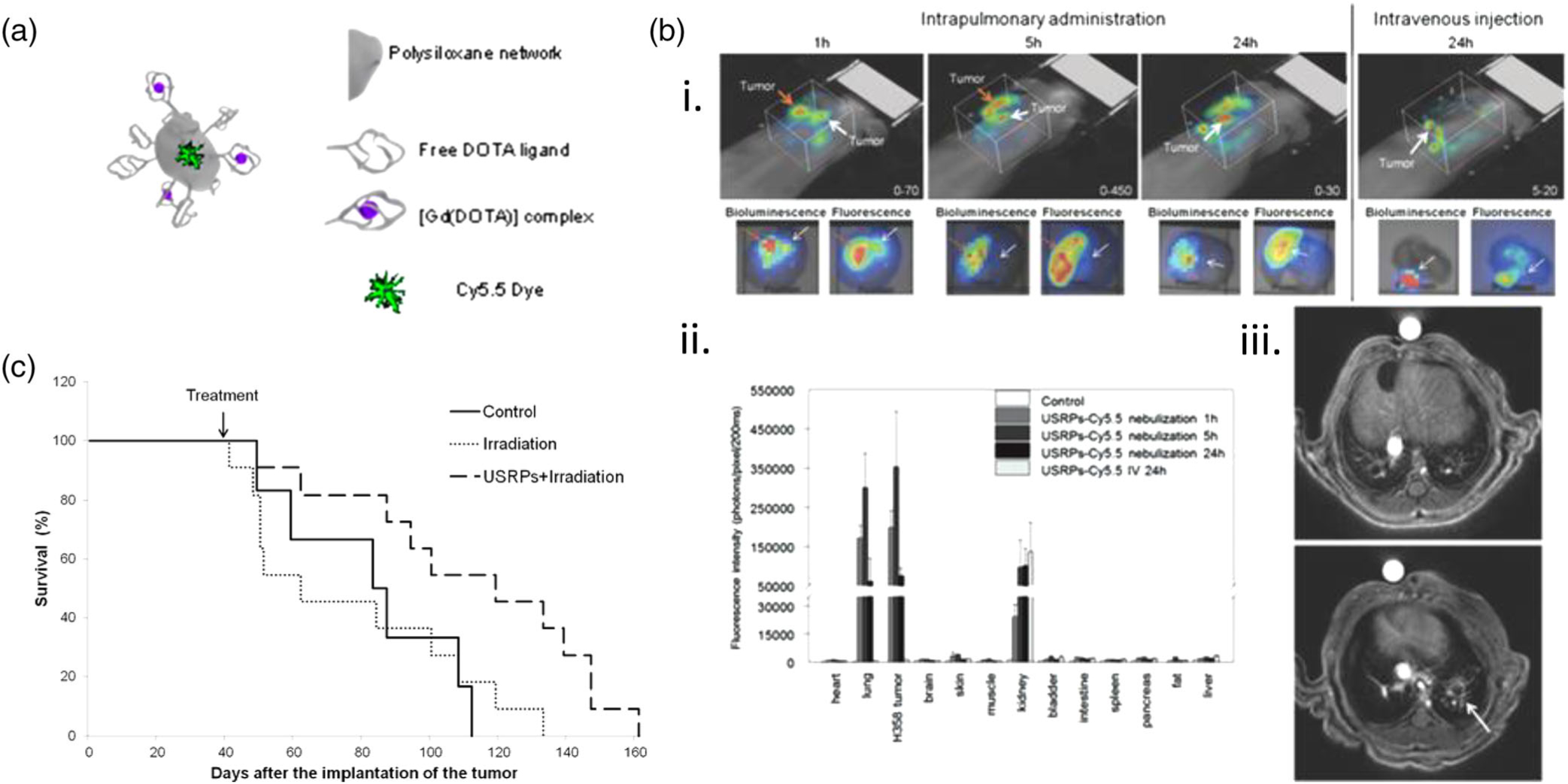
Gadolinium-based ultra-small rigid platform nanoparticles (USRPs) as a theranostic agent for the imaging and treatment of lung cancer administered via nebulization. (a) Schematic representation of the USRP system and its components, which allow for ultrashort echo time magnetic resonance and fluorescence imaging alongside radiotherapy. (b) Localization of USRPs after pulmonary delivery in orthotopic H358-Luc tumor mouse model. (i) Fluorescence tomography imaging of mice 1, 5, or 24 hr after pulmonary administration or IV injection of USRPs (top), with bioluminescence and fluorescence imaging performed on isolated lungs showcasing colocalization of tumor cells with the USRPs at the same time points (bottom). ii) Biodistribution of USRPs in mice 24 hr after pulmonary administration or IV injection, showing enhanced accumulation in lung and tissue tumors followed by kidneys with little retention comparatively in the liver. iii) Before (top) and after (bottom) pulmonary administration of USRPs allows for detection of lung cancer tumors via magnetic resonance imaging (MRI) imaging. (c) Survival curve of H358 tumor-bearing mice without treatment (n = 6), after one irradiation (n = 11), or after USRP administration followed by one irradiation 24 hr later (n = 11), showing longer mean survival time for mice treated with USRPs and irradiation. (Reprinted with permission from Morlieras et al. (2013) and Dufort et al. (2015). Copyright 2013 American Chemical Society and Copyright 2015 John Wiley and Sons)
To address challenges in selectivity and radiosensitivity for lung cancer therapies, Nyugen and colleagues developed a folate receptor-targeted multifunctional dual drug-loaded nanoparticle (MDNP) system for localized chemoradiotherapy that exhibited increased tumor accumulation and safety (Menon et al., 2017). The MDNPs are composed of a polylactic-co-glycolic acid (PLGA) core to allow for controlled release of its components with a poly(N-isopropylacrylamide)-carboxymethyl chitosan (PNIPAAm-CMC) copolymer shell, which forms a semi-interpenetrating network that allows for temperature- and pH-sensitivity (at slightly lower pH of tumor microenvironment) and degradability, and folic acid conjugated to the surface to target overexpressed folate receptor-α on lung cancer cell membranes. The particles (around 250–280 nm in diameter) are loaded with three agents: NU7441, a potent radiosensitizer; Gemcitabine (Gem), a chemotherapy agent; and SPIO NPs for magnetic resonance imaging (MRI) and hyperthermia therapy. When tested in vitro with healthy alveolar type-1 (AT1) cells and human dermal fibroblasts (HDFs), the nondrug-loaded MDNPs showed high cell viability (above 80%), indicating the particles alone are relatively nontoxic. Against folate receptor-positive lung cancer cells, A549 human lung adenocarcinoma cells and H460 human lung carcinoma cells, the MDNPs showed dose-dependent uptake which was further increased in the presence of a magnetic field. Loading with SPIOs allowed for the detection of H460 tumor-bearing athymic nude mice, showing a greater T2 signal intensity with folate-targeted particles compared to nontargeted particles. Regarding the system’s therapeutic efficacy, mice treated with drug-loaded MDNPs via nebulization and radiotherapy exhibited the slowest tumor growth and greatest reduction in tumor volume over 10 days compared to control mice receiving radiotherapy or NU7441 and Gem alone. Histopathology of tissues from animals treated with nondrug loaded MDNPs showed no signs of toxicity, highlighting again the safety of the carrier. Altogether, these results highlight the synergistic effect of combined chemotherapy and radiation for lung cancer treatment alongside improved localization through the addition of a targeting moiety on the MDNP surface (Menon et al., 2017).
3 |. INHALABLE THERAPEUTICS FOR CHRONIC PULMONARY DISEASES (CPDS)
Asthma and COPD are two of the most globally common and heterogeneously distributed CPDs (Soriano et al., 2017). Asthma is a currently incurable condition in which the upper airways become inflamed and produce excess mucus, causing shortness of breath, loss of aerobic function, and decreased quality of life (Beasley, Keil, von Mutius, & Pearce, 1998). COPD is also a chronic and currently incurable condition classified into two types: chronic bronchitis and emphysema. Chronic bronchitis results in inflammation, swelling, and mucus overproduction within the secondary bronchioles, while emphysema results in loss of shape and function of the alveoli in the lungs (Kessler et al., 2011). Both conditions result in stifling a person’s ability to breathe, eventually leading to long-term disability and significant impairments in quality of life. The prevalence of these two CPDs worldwide is significant; COPD is the third leading cause of death worldwide (Lozano et al., 2012; Quaderi & Hurst, 2018). Although asthma is not as mortal as COPD, the disease has greater morbidity and its own other deleterious effects; over 26 million Americans are impaired by asthmatic symptoms (Akinbami et al., 2012). Across the world, 339 million people live with asthma and 328 million live with COPD, either knowingly or possibly unknowingly (Ehteshami-Afshar, FitzGerald, Doyle-Waters, & Sadatsafavi, 2016).
Within modern pulmonary medicine, CPDs have commonly been treated with orally or systemically delivered adrenergic stimulants, oral and inhaled corticosteroids, and targeted treatments such as antileukotrienes and cromones (Chu & Drazen, 2005). However, current treatments only temporarily alleviate symptoms of CPDs and do not fully mitigate the impairments on aerobic function that these diseases impose. Furthermore, the overuse of corticosteroids is documented to result in systemic side effects—including impaired growth in children, decreased bone mineral density, skin thinning and bruising, and cataracts (Dahl, 2006). Therefore, recent work in nanotherapeutics designed to treat CPDs have focused on creating nanomaterial systems that can reach the target site locally and mitigate off-target side effects through low cytotoxicity and high pharmacological potency (Lopes Da Silva, Ferreira Cruz, Rieken, Rocco, & Morales, 2017; Sadikot, 2018). Inhaled pulmonary delivery of nanomedicines enhances CPD treatment by transporting encapsulated poorly water-soluble, potent treatments, directly to their intended site of action, facilitating targeted pulmonary release and reducing the necessary dosage while mitigating systemic side effects (Blank et al., 2017). Inhalation therapy poses a distinct advantage in distributing these drugs evenly and locally within the lungs, reaching all CPD-afflicted tissues in a controllable manner (Sahib, Abdulameer, Darwis, Peh, & Tan, 2012). Inhalation of nanotherapeutic systems is currently being extensively studied and has great potential for targeted drug delivery in the treatment of CPDs (Ratemi, Sultana Shaik, Al Faraj, & Halwani, 2016; Yhee, Im, & Nho, 2016).
In this section, we discuss the recent work in the development of different nanotherapeutics for the treatment of CPDs, while also highlighting challenges to still be solved. We focus on the design of different polymeric, lipid-based, and inorganic material systems for pulmonary delivery and its translation to pharmaceutical and therapeutic efficacy for diseases such as asthma and COPD.
3.1 |. Polymeric Nanomaterials
Gene therapies can provide long-term control of symptoms and significantly improve the quality of life of those suffering from lung diseases; many systems have been demonstrated via inhalation and injection to effectively localize in lung tissues with minimal side effects (Beck et al., 2002; Kaczmarek et al., 2016; A. K. Patel et al., 2019). In an attempt to improve patient compliance by lowering dosing frequency, Hanes and coworkers developed an inhalable long-lasting gene therapy composed of DNA-thymulin polymeric particles that were capable of inducing anti-inflammatory effects in asthma (da Silva et al., 2014). The biologically active thymulin-analog gene, methionine serum thymus factor (MSTF), prevents lung inflammation and remodeling in an allergic asthma model. The DNA NPs were composed of a single molecule of plasmid DNA compacted with block copolymers of poly-L-lysine and polyethylene glycol (CK30PEG), which have high biocompatibility, to yield CK30PEG-DNA NPs (encoding either MSTF or green fluorescent protein [GFP]) around 89 nm in diameter, as shown in Figure 5a. To confirm whether CK30PEG-DNA NPs mediate airway gene transfer in vivo, ovalbumin (OVA)-challenged BALB/c mice were intratracheally dosed with DNA NPs carrying either GFP plasmid DNA (CK30PEG-GFP) or MSTF plasmid DNA (CK30PEG-MSTF). Thymulin plasmids were detected in the lungs of OVA-challenged asthmatic mice up to 27 days after administration of DNA NPs. The alveolar collapse induced by OVA was mitigated in the lungs of mice treated with CK30PEG-MSTF compared with naked MSTF plasmids. A single dose of CK30PEG-MSTF NPs prevented lung inflammation (as depicted in Figure 5b–d), collagen deposition, and smooth muscle hypertrophy in the lungs of the allergic asthma murine model, leading to improved lung mechanics. The total number of cells, such as eosinophils, neutrophils, and monocytes, in the bronchoalveolar lavage fluid (BALF) were higher in the OVA-saline control group than in the OVA-challenged group treated with the CK30PEG-MSTF NPs, as shown in Figure 5e. Mice in the OVA-saline group exhibited considerable subepithelial fibrosis and smooth muscle hypertrophy, hallmarks of the lung remodeling presented in patients afflicted with asthma; in comparison, CK30PEG-MSTF NPs attenuated these ultrastructural changes. Moreover, the CK30PEG-MSTF NPs did not elevate lung static elastance nor airway resistance, as depicted in Figure 5f and g, respectively. These findings showcase a long-acting system for asthma treatment through pulmonary delivery of therapeutic genes for CPDs (da Silva et al., 2014).
FIGURE 5.
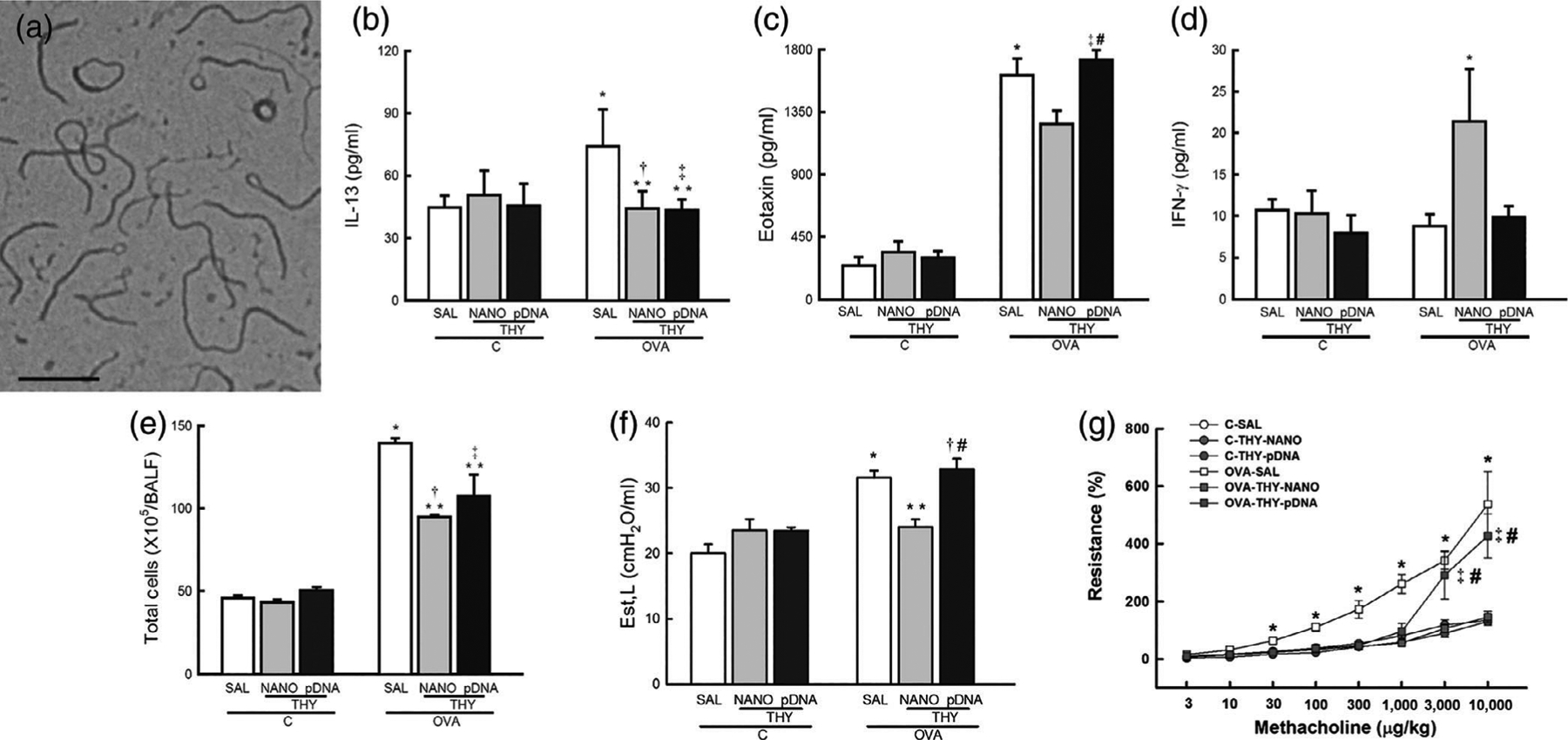
Thymulin gene therapy in allergic asthma provided via DNA-nanoparticles (NPs) that mitigate airway remodeling and inflammation after pulmonary delivery. (a) Transmission electron micrograph of the CK30PEG-MSTF NPs where scalebar represents 200 nm. Inflammatory cytokine concentrations, (b) interleukin 13 (IL-13), (c) Eotaxin, and (d) interferon-γ (IFN-γ) in lung tissues of healthy control or ovalbumin-challenged mice, highlighting anti-inflammatory effect of the DNA NPs. (e) Total number of cells counted in bronchoalveolar lavage fluid of healthy or ovalbumin-challenged mice, showing reduction of cell numbers representative of an inflammatory response after treatment with DNA NPs. Lung function of mice as represented by (f) lung static elastance and (g) airway hyper-responsiveness, highlighting reduction in elastance and resistance after treatment with DNA NPs. C, control mice challenged with saline; ovalbumin (OVA), mice challenged with ovalbumin; SAL, mice treated with saline; THY-NANO, mice treated with DNA NPs; THY-pDNA, mice treated with naked plasmid DNA. Statistically significant (p < .05) results from C-SAL (*), C-THY-NANO (†), C-THY-pDNA (‡), OVA-SAL (**), or OVA-THY-NANO (#). (Reprinted with permission from da Silva et al. (2014). Copyright 2014 Elsevier)
To overcome challenges in treating corticosteroid-resistant asthma, Hahn and colleagues designed Flt1 peptide-hyaluronic acid (HA) conjugate nanoparticles (Flt1-HA NPs) that treated neutrophilic pulmonary inflammation indicative of steroid resistance (H. Kim et al., 2013). These NPs were either loaded with dexamethasone (Dexa), a corticosteroid, or 5-carboxyfluorescein diacetate N-succinimidyl ester (CFSE), a green fluorescent dye, and labeled with HiLyte 647 amine, a red fluorescent dye. The Flt1 peptide (GGNQWFI) serves as a key mediator in regulating neutrophilic inflammation, while HA is biodegradable and interfaces with HA receptors on the lung epithelium (Jiang et al., 2005; Varghese, Sun, Ns Hilborn, & Ossipov, 2009). Flt1-HA conjugates self-assemble into spherical NPs of around 110 nm in diameter after loading with Dexa upon dissolution. These particles exhibited an initial burst release in vitro over 6 hr followed by a sustained linear release pattern. In vitro bioimaging showed efficient internalization of FITC-conjugated Flt1-HA NPs into A549 lung epithelial cells by HA-receptor mediated endocytosis, and cytotoxicity assays showed high cell viability after treatment with Flt1-HA NPs. Ex vivo imaging for the biodistribution in ICR mice revealed long-term retention (up to 72 hr) of CFSE-loaded Flt1-HA NPs in deep lung tissues, likely due to the mucoadhesive property of HA. Furthermore, the NPs also reduced inflammation pathway-related cytokine levels of lipopolysaccharide (LPS)-stimulated cells more efficiently than free Dexa. Moreover, according to the BAL cellularity and histological analysis, Dexa-loaded Flt1-HA NPs showed improved therapeutic effects in both eosinophilic and neutrophilic asthma mice models. Flt1-HA NPs overall showed successful and effective delivery and uptake into lung tissue in vivo with prolonged residence time as a means of treating steroid-resistant asthma (H. Kim, Park, et al., 2013).
To provide a means of targeting cells of the immune system responsible for pulmonary inflammation, Vij and coworkers developed an inhalable PEGylated immuno-conjugated PLGA nanoparticles (PINPs) system that were able to target neutrophils and modulate inflammatory responses associated with CPDs (Vij, Min, Bodas, Gorde, & Roy, 2016). The PINPs were prepared via emulsification of DSPE-PEG2000/DSPE-mPEG2000 with PLGA dissolved with either ibuprofen (IBF), a nonsteroidal anti-inflammatory drug, and/or Nile Red (NR) fluorescent dye and then incubated with NIMP-R14 antibodies for neutrophil targeting to yield PINP-NIMP-IBF NPs of around 344 nm in size. To assess the efficacy of neutrophil-targeted PINPs in transporting through the airway followed by specific binding and release of drug to neutrophils, C57BL/6 mice were intratracheally treated with Pseudomonas aeruginosa lipopolysaccharide (Pa-LPS) to induce pulmonary obstruction 12 hr before intranasal PINP-NIMP-IBF instillation. After administration, quantification of NIMP-R14+ BALF cells and NF-κB (pro-inflammatory response-regulating transcription factor) activities showed that treatment with the PINP-NIMP-IBF decreased Pa-LPS-induced NF-κB activity and the number of NIMP-R14-PE positive neutrophils, demonstrating its ability to control the inflammatory response. Further study of biodistribution of PINPs in lung tissue is necessary; however, this model serves as an inhaled therapeutic system that treats symptoms of obstructive pulmonary diseases (Vij et al., 2016).
3.2 |. Lipid-based nanomaterials
To prolong the effects of bronchodilating drugs in asthmatic patients, Yang and coworkers developed a liposomal formulation to deliver salbutamol sulfate (SBS), a highly water-soluble bronchodilator, that increased lung retention to mitigate bronchoconstriction (Chen et al., 2012). SBS was encapsulated into soybean phosphatidylcholine (PC) liposomes, measuring around 57 nm in diameter, which formed into a vesicular phospholipid gel (VPG). IT administration of liposomes loaded with the fluorescent dye 1,1-dioctadecyl tetramethyl indotricarbocyanine iodide (DiR) in Sprague Dawley rat model was assessed by a small animal imaging system and pharmacokinetic analysis, which showed that the liposomes were effectively distributed in the respiratory tract and lungs and sustained release of SBS for 48 hr. Guinea pigs were treated intratracheally with SBS liposome suspensions through a spray instillator, where SBS liposomes enhanced persistence of antiasthmatic effect of SBS for up to 18 hr, whereas free SBS solution lasted less than 8 hr. The results demonstrated that liposomes could increase the concentration and retention time of SBS in the lungs and therefore prolong its therapeutic effects to alleviate bronchoconstriction symptomatic of CPDs (Chen et al., 2012).
In a means of enhancing selectivity and minimizing premature drug release, Lanza and coworkers developed inhalable antiangiogenic Sn2 lipase-labile prodrug αVβ3-micelles that effectively suppress microvascular expansion and airway hyperresponsiveness, hallmarks of angiogenic inflammation of CPDs (Lanza et al., 2017). Because lung epithelial cells often overexpress αVβ3 integrins on their membranes in asthma due to increased angiogenesis, the micelles were designed to target these receptors for improved selectivity using an αVβ3-peptidomimetic homing ligand (αVβ3-PEG2000-PE). Figure 6a and b illustrate the synthesis of fumagilin and docetaxel prodrugs (Fum-PD and Dxtl-PD, respectively) through saponification and esterification with 1-palmitoyl-2-azelaoyl-sn-glycero-3-phosphocholine (PAzPC). The αVβ3-prodrug-micelles formed particles around 16 nm in size. The antiangiogenic effectiveness of αVβ3-mixed micelles incorporating Dxtl-PD and/or Fum-PD were shown to suppress neovascular expansion in the bronchi of house dust mite-challenged (HDM) rats after intranasal delivery of the micelles, compared to an empty-micelle control, as shown in Figure 6c. Less vascularity was observed in prodrug micelle treatment groups than in empty-micelle-treated rats, as depicted in Figure 6d. Respiratory resistance (Rrs) and respiratory system compliance (Crs) are two measures of resistance-to- or ease-of-airflow during inhalation, respectively, and were measured after application of αVβ3-prodrug-micelles, as depicted in Figure 6e. Methacholine (MCh)-induced Rrs was higher in HDM rats receiving empty micelles than those treated with αVβ3-Dxtl-PD and αVβ3-Fum-PD micelles, which markedly and equivalently attenuated airway hyper-responsiveness. Additionally, Crs was improved after treatment with both prodrug micelles. Furthermore, inflammatory cell numbers within bronchoalveolar sites were reduced by both Fum and Dxtl nanotherapies, as represented in Figure 6f. These results demonstrate the potential of an inhaled antiangiogenic nanotherapeutic to ameliorate symptoms of asthma; assessments of biodistribution and any systemic toxicity would further validate the use of this system (Lanza et al., 2017).
FIGURE 6.
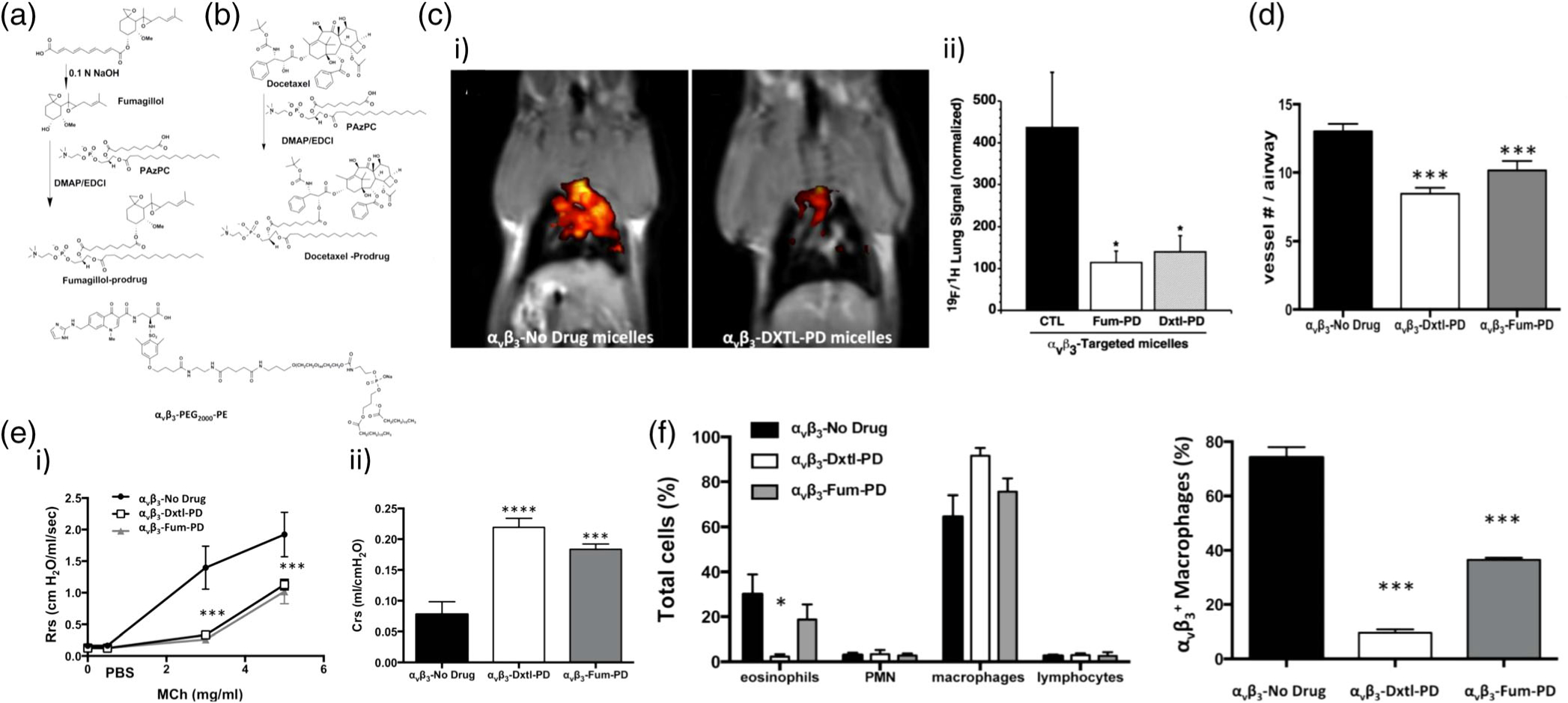
Synthesis and antiangiogenic effects of Sn2 lipase-liable prodrug micelles for the treatment of asthma. (a) Synthesis of fumagillin PAzPC prodrug and through saponification and esterification alongside structure of αVβ3-peptidomimetic-PEG2000-PE. (b) Synthesis of docetaxel prodrug. (c) i) 19F/1H magneitc resonance (MR) tomographic molecular imaging following antiangiogenesis treatment in HDM rats with αVβ3-Dxtl-PD micelles (right) revealed markedly reduced airway neovascular MR signal when compared to the control asthmatic animals receiving αVβ3-empty micelles (left). ii) Normalized lung signal quantification of 19F/1H MR tomographic molecular imaging (*p < 0.01). (d) Histogram of 19F/1H MR tomographic molecular imaging results showing equivalent reduction in neovascularity with anti-angiogenesis micelles. (e) Pulmonary functional changes in HDM rats receiving αVβ3-Dxtl-PD, αVβ3-Fum-PD, or αVβ3-no-drug micelles. i) Respiratory system resistance (Rrs) was measured after increasing methacholine (MCh) concentrations showing attenuated reactivity among rats treated with αVβ3-Dxtl-PD or αVβ3-Fum-PD micelles(***p < 0.001). ii) Respiratory system compliance (Crs) in HDM rats following the MCh challenge showing greater compliance after the treatment with drug-loaded micelles (***p < 0.001). (f) Bronchoalveolar (BAL) cell profiles showing a lower percentage of eosinophils (EOS) in HDM rats receiving αVβ3-Dxtl-PD and αVβ3-Fum-PD micelles (left) and flow cytometry of BAL fluid (right) revealing that αVβ3-Fum-PD and αVβ3-Dxtl-PD micelles decreased αVβ3+ macrophage/monocyte numbers versus empty micelles(*p < 0.05, ***p < 0.001). (Reprinted with permission from Lanza et al. (2017). Copyright 2017 Ivyspring International Publisher)
In an attempt to increase lung retention and mitigate cytotoxicity of a potent asthma drug, Pokharkar and coworkers developed Montelukast-loaded nanostructured lipid carriers (NLCs) to form Montelukast-NLCs (MNLCs) (Patil-Gadhe, Kyadarkunte, Patole, & Pokharkar, 2014). Montelukast (MTK) is a cysteinyl leukotriene receptor antagonist (c-LTRA) generally prescribed to asthma patients for use during exercise or to prevent aspirin-induced asthma (Schäper et al., 2011). Melt-emulsification-ultrasonication techniques of homogenization were used to prepare MNLCs with a Precirol (solid lipid) to Capryol (liquid lipid) ratio of 7:3, to yield particles of around 185 nm in diameter. MNLC-DPI particles were then formed for DPI by lyophilization using mannitol as a cryoprotectant and carrier. An in vitro cytotoxicity assessment was performed on A549 human lung adenocarcinoma cells, which revealed better cell viability of MNLCs compared to the free drug. The MNLCs were delivered intratracheally to Wistar rats to assess biodistribution, demonstrating improved bioavailability and longer residence of MTK with the MNLC system as compared to delivery of free MTK. Thus, these results demonstrate that lipidic nanoparticulate formulations improve lung MTK retention with reduced toxicity, which could lead to a potentially more efficacious asthma treatment utilizing MNLC-DPIs once therapeutic efficacy is assessed for this system against an in vivo asthma model (Patil-Gadhe et al., 2014).
3.3 |. Inorganic nanomaterials
Silver is generally a nontoxic and hypoallergenic metal, capable of cytoprotective and prohealing abilities, and has gained attention recently as a nanotherapeutic system (Franci et al., 2015; Modak & Fox, 1973). Jung and coworkers found silver nanoparticles (AgNPs) mitigate bronchial inflammation and hyper-responsiveness, which are common characteristics of CPDs (H. S. Park et al., 2010). Powders of free silver were dissolved in PBS and formed solubilized NPs of approximately 6 nm in size. OVA is a frequently used allergen that induces asthmatic pulmonary inflammation in laboratory mice (Nials & Uddin, 2008), and OVA-challenged C57BL/6 mice were administered silver NPs through jet nebulization every 24 hr for 5 days to assess the NPs anti-inflammatory effect. After collection of BAL, they found that the increased levels of inflammatory cells and markers that are typical of asthma and airway hyper-responsiveness were reduced by the pulmonary delivery of AgNPs. Th2 cytokine expression, a primary signal for asthma-induction immunology, was also decreased. Reactive oxygen species (ROS) result in bronchial hyper-reactivity and smooth muscle precontraction, and the increased intracellular ROS levels in BAL fluid collected after OVA challenge was decreased following the administration of AgNPs, suggesting AgNPs are capable of attenuating oxidative stress. Their results illustrate the potential of AgNPs as a promising therapeutic strategy, due to their antioxidant and anti-inflammatory properties, for inhalation therapy for CPDs like asthma (H. S. Park et al., 2010).
To address drug resistance and steroidal side effects, Faraj and coworkers developed an antibody-conjugated, polymer-coated SPIO NP system that hinders the inflammatory pathway triggered by interleukin-4 receptor α (IL4Rα) of the immune system to control asthmatic inflammation (Halwani et al., 2016). SPIO NPs have low intrinsic toxicity, easy surface functionalization, and are readily detected using MRI. The anti-IL4Rα-nanoparticles (anti-IL4Rα-NPs) are composed of a SPIO NP with a shell of dextran cross linked with polyethylene glycol (PEG2000) chains conjugated to anti-IL4Rα monoclonal antibodies and measure around 133 nm in diameter, as illustrated in Figure 7a. BALB/c OVA-sensitized mice were treated with anti-IL4Rα-NPs for in vivo characterization through intrapulmonary delivery using a microsprayer aerosolizer. Decreased levels of proinflammatory cytokine release in the lung tissue and BALF, as well as number of inflammatory cells, were observed, as shown in Figure 7b, d, and e. Moreover, anti-IL4Rα NPs dropped CD4+ and CD8+ T cell activity in lung tissue and inhibited their ability to release proinflammatory cytokines to greater extent than the free antibody, as shown in Figure 7c. The anti-IL4Rα-NPs localized within areas of lung tissue enriched in inflammatory cells, as confirmed by immunohistological staining. Moreover, they induced a sustained low level of lung inflammation for 1 week following the last instillation compared with the treatment with free anti-IL4Rα antibodies Overall, this NP system enhanced lung tissue penetrability and sustainability of the anti-IL4α antibodies after pulmonary administration to provide long-lasting anti-inflammatory effects without the use of corticosteroids (Halwani et al., 2016).
FIGURE 7.
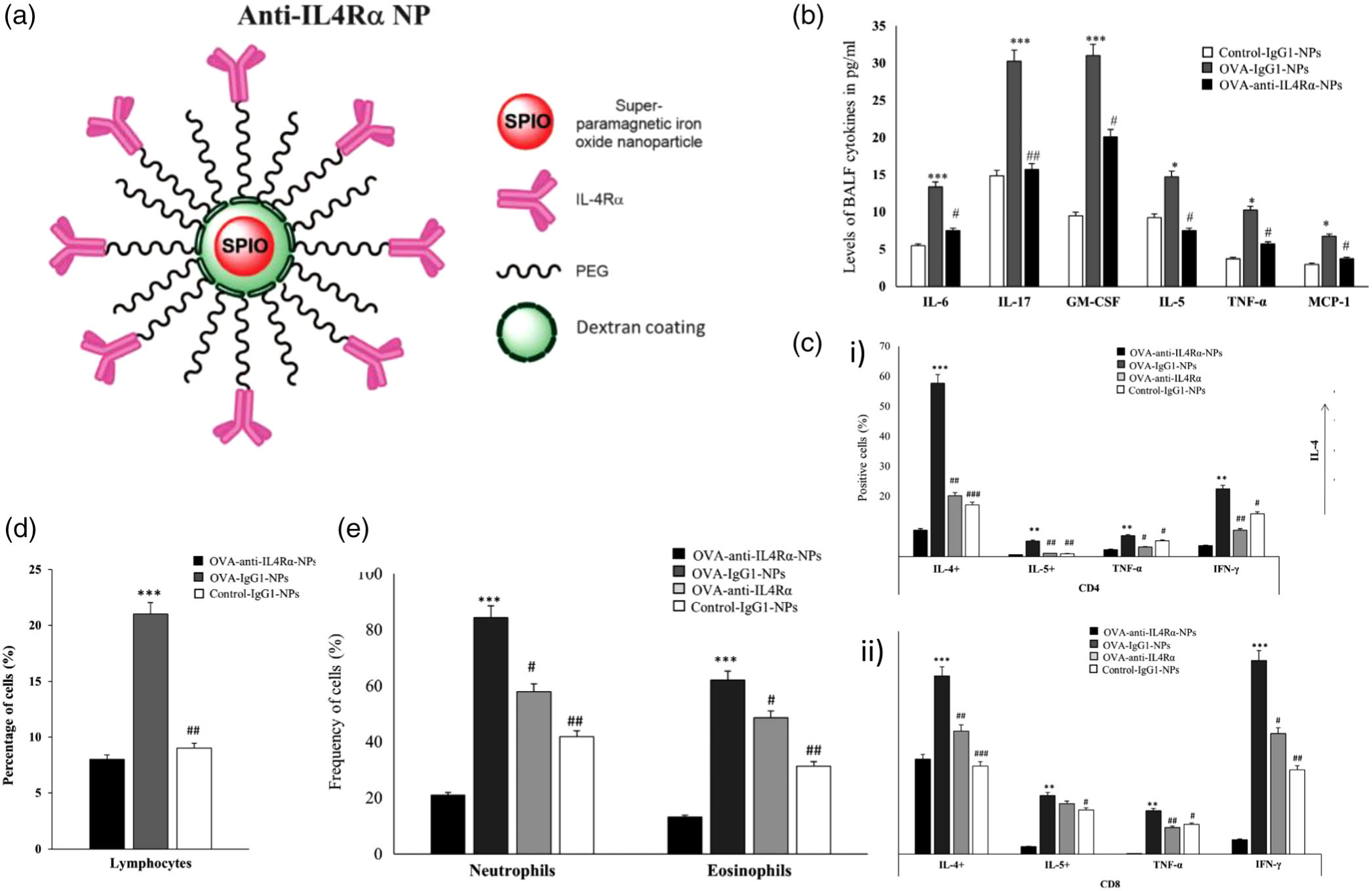
Design, inflammatory cell response, cytokine release, and immunohistological imaging of an anti-IL4Rα-NP system. (a) An illustration representing the design of the anti-IL4Rα NPs. (b) BALF levels of pro-inflammatory cytokines in mice treated with or without the anti-IL4Rα-NPs, where cytokine levels decreased in mice treated with anti-IL4Rα-NPs. (c) i) CD4 and ii) CD8 T cells isolated from the lungs of ovalbumin (OVA)-challenged mice treated with or without the anti-IL4Rα-NPs, highlighting attenuated inflammatory marker expression from anti-IL4Rα-NP treatment. (d) Effect of the anti-IL4Rα-NPs on the BALF inflammatory cells in the treated OVA-challenged mice showing observed decrease in lymphocyte frequency. (e) frequencies of neutrophils and eosinophils isolated from the lungs of OVA-challenged mice treated with or without anti-IL4Rα-NPs, showing decreased levels of both cell types from the anti-interleukin-4 receptor α (IL4Rα)-NPs (for control vs OVA: *p < 0.05, **p < 0.01, ***p < 0.001; for OVA vs anti-IL4α-NPs: #p < 0.05, ##p < 0.01). (Reprinted with permission from Halwani et al. (2016). Copyright 2016 Springer Nature)
Gold NPs have been extensively studied in drug delivery for their easily tunable optical and photodynamic properties for disease treatment and imaging. Geiser and coworkers developed an inhaled gold nanoparticle (AuNPs) system that validated the use of AuNPs as carriers for pulmonary delivery for COPD treatment (Geiser et al., 2013). The prepared AuNPs measured around 21 nm in diameter and were deliverable in an aerosol produced using a spark generator. Scnn1b-transgenic (Tg) murine models were employed, as they develop spontaneous chronic bronchitis, mucus hypersecretion, and emphysema, to recapitulate COPD in vivo. Scnn1b-Tg mice were administered AuNP aerosols for 2 hr, and AuNPs were mostly found as singlets or small agglomerates of less than 100 nm in diameter at the surface of and within the lung epithelium and as agglomerates greater than 100 nm in macrophages. Inhaled AuNPs accumulated in the alveolar epithelium of both Wt and Scnn1b-Tg mice; however, the Tg mice showed slower macrophagic uptake of AuNPs and higher epithelial internalization of the AuNPs. These results suggest inhalable AuNPs serve as a means of directly delivering to alveolar epithelial cells and macrophages in COPD via enhanced cellular uptake. However, more studies are needed to assess AuNP interactions at an immunological level and how they contribute to inflammation levels in Scnn1b-transgenic mice (Geiser et al., 2013).
4 |. INHALABLE THERAPEUTICS FOR TB
TB is a prevalent respiratory infection, infecting approximately one fourth of the world’s population, and led to 1.3 million deaths in 2017 (World Health Organization, 2018). Given the complexity of current drug treatment protocols for TB treatment—which often require multidrug cocktails (e.g., rifampicin [RIF], pyrazinamide [PYZ], isoniazid [INH], and ethambutol) taken for prolonged periods of time (e.g., 6 to 9 months)—patient compliance is a major obstacle in achieving successful eradication of the disease. Moreover, TB is especially difficult to eliminate, as Mycobacterium tuberculosis (MTB) persists within AMs (Natarajan, Kundu, Sharma, & Basu, 2011), and rather than becoming phagocytosed and eliminated in the AMs (as foreign particles are typically handled), M. tuberculosis is able to prevent phagosome-lysosome fusion within the AMs and uses them as a site for incubation (Zahrt, 2003). Current treatment with orally delivered drugs often fails to achieve sufficient drug concentrations within the AMs of pulmonary tissues to eradicate M. tuberculosis. Aerosolized nanomaterials have significant promise as drug delivery systems to treat TB owing to their high stability, high carrying capacity, and ability to deliver both water soluble and insoluble drugs (Gelperina, Kisich, Iseman, & Heifets, 2005; Yang, Peters, & Williams Iii, 2008a). Delivery directly to pulmonary tissues also has the advantage of requiring lower doses and minimizing side effects, as well as requiring less frequent administration, which can result in better patient compliance (Rojanarat, Nakpheng, Thawithong, Yanyium, & Srichana, 2012). Further surface modification with targeting moieties makes inhalable nanomedicine delivery systems attractive as they can target AMs, mitigate systemic side effects, and reduce frequent dosing requirements (Natarajan et al., 2011).
In this section, we discuss the recent examples of different inhalable nanomedicine systems for the treatment of TB, highlighting the material design and efficacy of these systems and the present challenges still faced in the field. We focus on the design of various polymeric, lipid-based, and inorganic material systems for pulmonary delivery and its translation to therapeutic efficacy against TB.
4.1 |. Polymeric nanomaterials
Polymersomes are nanosized vesicles made from amphiphilic block copolymers that have been extensively studied for their ability to encapsulate poorly water-soluble drugs and prevent their premature degradation. Moretton and colleagues developed polymersomes capable of delivering the antibiotic RIF (Moretton, Cagel, Bernabeu, Gonzalez, & Chiappetta, 2015). Polymersomes were formed with poly(ethylene glycol)-poly(ε-caprolactone) (PEG-PCL) that self-assembled and to encapsulate RIF. The polymersomes were round in morphology, measured around 60 nm in size, and demonstrate suitable colloidal stability. In vitro release studies showed that all polymersomes exhibited a sustained release profile, with about 50% of the drug released after 6 hr. Furthermore, in an assessment of polymersome uptake in murine macrophages, the RIF concentration within macrophages was twofold higher with the polymersomes compared to free RIF. The polymersome design allowed for improved macrophage uptake, and further investigation of the suitability of the system as a respirable therapy for the treatment of TB is necessary (Moretton et al., 2015).
As nanosized materials are too small to deposit into the lungs through inhalation as dry powders, Goyal and colleagues developed nanoembedded microparticle formulations that deliver anti-TB drugs, RIF and INH, through pulmonary delivery that enhance antitubercular efficacy (Goyal, Garg, Rath, Datta Gupta, & Gupta, 2015). Four different materials were used to yield chitosan-, guar gum-, mannan-, or guar gum-coated chitosan-based NPs, which were then spray dried with mannitol to form microparticles to carry in NPs in an aerosol. An example of the scanning electron microscopy images of the chitosan particles and guar gum-coated chitosan particles can be seen in Figure 8a. In vitro release studies in PBS revealed faster release for INH compared to RIF, which was attributed to differences in solubility, as seen in Figure 8b. A macrophage (J-774) uptake study revealed mannan and guar gum systems displayed highest uptake, which is attributed to targeting by these materials to mannan receptors on AMs, which is shown in Figure 8c. A cell viability study with J-774 macrophages showed that the drug-loaded formulations were less toxic than the free drugs, as shown in Figure 8d. After pulmonary administration of the particles in female BALB/c, the nanoembedded microparticle formulations resulted in increased drug retention in the lungs as compared to free drug, where the mucoadhesive properties of guar gum further improved retention, as depicted in Figure 8e. Moreover, in a TB mouse model, mice treated with the drug-loaded particles had less microbial loads as compared to free RIF-treated mice, where the greater antitubercular activity of the guar gum particles was attributed to their prolonged residence and macrophage targeting capabilities. This study highlights the advantages of utilizing inhalable nanoembedded microparticles for enhanced targeting to AMs to improve the efficacy of antitubercular drugs (Goyal et al., 2015).
FIGURE 8.
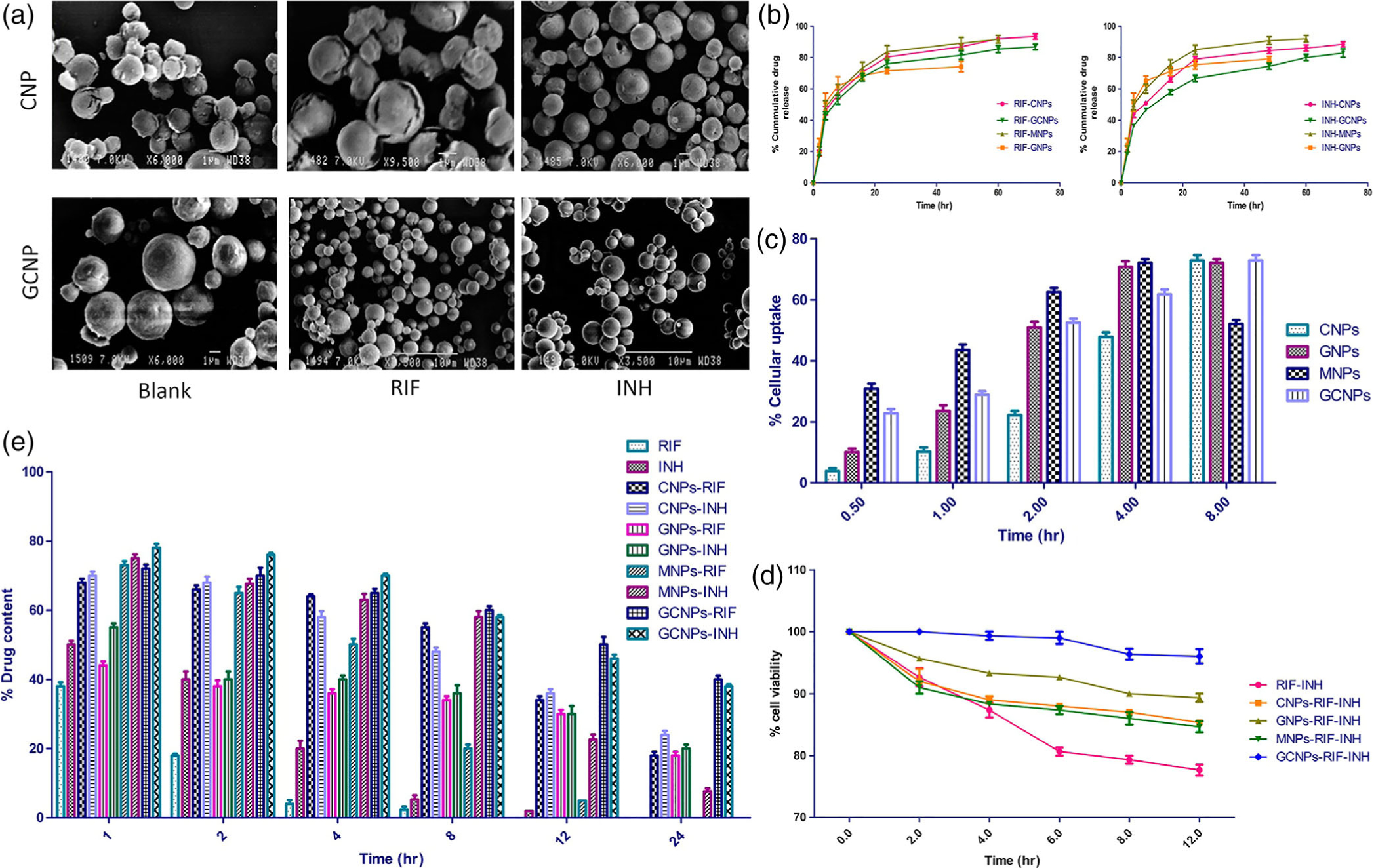
Nanoembedded microparticles for the delivery of rifampicin (RIF) and isoniazid (INH) via pulmonary route for the treatment of tuberculosis (TB) (a) Scanning electron microscopy images of chitosan and guar gum chitosan nanoembedded microparticles, where the nanoembedded microparticles are either blank, loaded with RIF, or loaded with INH. (b) in vitro release study in PBS of different nanoembedded microparticle formulations loaded with either RIF or INH, where RIF-loaded formulations released drug at a slower rate than INH-loaded formulations. (c) In vitro cellular uptake of different formulations into J-774 macrophage over time. Chitosan formulations displayed delayed cellular uptake, while mannan formulations and both guar gum formulations performed comparably, likely due to mannose moieties that allowing for targeting of mannan receptors on macrophages. (d) In vitro cell viability study in J-774 macrophages, showing reduced toxicity of nanoembedded microparticles as compared to free drug. (e) In vivo study to investigate drug distribution in the lungs of BALB/c mice over time of different formulations, where the various nanoembedded microparticles resulted in greater retention of drug as compared to free drug, with guar gum chitosan formulations exhibiting the longest retention time. (Reprinted with permission from Goyal et al. (2015). Copyright 2015 American Chemical Society)
4.2 |. Lipid-based nanomaterials
To address challenges in macrophage targeting and drug resistance in TB, Bhardwaj and coworkers designed liposomes with improved targeting efficacy that increase residence of antitubercular drugs to reduce necessary dosing frequency (Bhardwaj, Kumar, Narang, & Murthy, 2012). The investigated liposomes were composed of egg PC and cholesterol (CH) and appended with mannan, a targeting ligand for receptors on AMs, to actively target infected cells (Yu, Liu, Liu, Zhang, & Xu, 2010). The liposomes were loaded with the antibiotic drugs RIF, INH, or PYZ, ranged in size from around 300 to 630 nm, and were formed into a DPI formulation with sucrose. The lyophilized DPI formulas exhibited a controlled release profile of the antibiotics from the liposomes in vitro. Compared to free drug powders, the DPI liposomal formulations were still detectable in the lungs in Wistar albino rats 24 hr after IT instillation, highlighting the enhanced retention imparted by the liposomes. The AM-targeting mannan ligand increased drug concentrations and retention in the lungs compared to the neutral liposomes. This system highlights the advantages of pulmonary delivery to increase antibiotic drug retention in the lungs by targeting AMs; however, studies using infected animal models are needed to further investigate the efficacy of the DPI liposomal formulations to treat TB (Bhardwaj et al., 2012).
In an attempt to mitigate incidence of antibiotic resistance in TB treatment, Fraziano and coworkers designed asymmetric liposomes that resemble apoptotic bodies that kill M. tuberculosis bacteria without antibiotic drugs. (Greco et al., 2012). The two-faced liposomes were composed of a combination of either L-α-phosphatidylserine (PS), L-α-PC, or L-α-phosphatidic acid (PA). PS was used for its ability to mimic apoptotic bodies and thus promote uptake by macrophages, and PA was used to modulate vesicle trafficking and fusion inside macrophages to overcome blocked transport of phagosomes by MTB to lysosomes. The liposomes had a mean hydrodynamic radius of around 1,000 nm, as depicted in the fluorescence confocal microscopy images in Figure 9a. The apoptotic body-like (ABL) liposomes were found to be efficiently internalized by macrophages and induce an influx of Ca2+, which is associated with an increase in ROS, inhibition of bacterial growth, and inhibition of inflammatory responses. Preferential uptake into macrophages over alveolar epithelial cells was observed, as shown in Figure 9b, highlighting the enhanced internalization into macrophages via the PS outer layer. Furthermore, as shown in Figure 9c, ABL liposomes are also able to reduce the proinflammatory response caused by MTB in infected dTHP-1 macrophages. As depicted in Figure 9d, MTB colony forming units (CFUs) were reduced upon treatment with the ABL/PA liposomes. MTB-infected BALB/c mice were treated intranasally with liposomes, and INH, a common TB antibiotic, was incorporated in the drinking water of the mice for combined therapy. Within the lungs of treated mice, both the APL/PA formulation and APL/PA with INH resulted in a 100-fold decrease in CFU concentrations in comparison to untreated mice, while INH alone resulted in a two-fold decrease, as shown in Figure 9e. This system offers an approach to treating TB with an aerosolized liposomal formulation that does not require the incorporation of antitubercular drugs; the formulation was both effective at eliminating MTB CFUs and mitigating inflammatory responses (Greco et al., 2012).
FIGURE 9.
Asymmetric liposomes for the inhalable treatment of tuberculosis (TB). (a) Image of L-α-phosphatidic acid (PA) liposomes via fluorescence confocal microscopy characterized with fluorescent phosphatidic acid to show asymmetric distribution of phospholipids (scale bar = 16.3 μm). (b) In vitro uptake of apoptotic body-like (ABL) liposomes into different cell types, showing that ABL liposomes are more efficiently internalized into macrophages as compared to alveolar epithelial cells. (c) The effect of ABL liposomes on different cytokine secretion in dTHP-1 cells indicating that ABL liposomes reduce TB-infected cell inflammatory response. (d) The effect of different liposomal formulations on Mycobacterium tuberculosis (MTB)-infected dTHP-1 cells where the greatest colony forming units (CFU) reduction was observed with the ABL/PA formulation. (e) In vivo study with MTB-infected BALB/c mice treated with intranasally delivered ABL/PA, with or without isoniazid (INH) incorporated in drinking water, to see effects on CFUs in lung, spleen, and liver. In all organs, reduction of CFUs was observed with combined ABL/PA and INH therapy (in comparison to MTB-infect control mice: *p < 0.001, **p < 0.05, #p = not significant). (Reprinted with permission from Greco et al. (2012). Copyright 2012 National Academy of Sciences)
Expanding to other common lung-resident bacterial infections, Meers and colleagues developed inhalable liposomal amikacin, an antibiotic, that enhanced antibacterial efficacy against P. aeruginosa (Meers et al., 2008). Pseudomonas aeruginosa, like TB and other bacteria, frequently becomes a chronic infection in cystic fibrosis patients due to impaired mucociliary clearance; P. aeruginosa is particularly problematic for cystic fibrosis patients as it produces a hard-to-penetrate biofilm and that can result in increased tolerance to antibiotics and reduced phagocytic activity (Høiby, Ciofu, & Bjarnsholt, 2010). Liposomes composed of saturated lipid dipalmitoyl phosphatidylcholine (DPPC) and CH were prepared and loaded with amikacin to yield particles around 300 nm in size. Using sputum samples from cystic fibrosis patients, liposomes exhibited enhanced mucus and biofilm penetration. In vivo studies with Sprague–Dawley rats showed free drug (both amikacin and tobramycin, an antibiotic drug currently administered clinically to patients) is cleared within a few hours, whereas amikacin-loaded liposomes were found to have two release phases: a rapid release during the first 2 hr, and a larger (more than half of the administered drug), slower release over the course of 24 hr. Furthermore, a rat infection model was dosed three times a week over 14 days with amikacin-loaded liposomes, which reduced CFUs by two orders of magnitude and even resulted in undetectable bacteria concentrations in many of the treated mice. This study highlights the enhanced penetrative capabilities afforded by liposomal formulations to improve delivery of antibiotics across thick mucus and biofilm layers that are symptomatic of cystic fibrosis to increase antibacterial efficacy (Meers et al., 2008).
4.3 |. Inorganic nanomaterials
Inhalable inorganic particles have not been extensively researched to treat TB. Much of the research in this area is currently at a preliminary stage, focusing instead on the feasibility of inorganic particles as a means of treating bacterial infections. The area is promising, however, with studies evidencing the ability to load inorganic nanomaterials with antitubercular drugs (Mohseni, Gilani, & Mortazavi, 2015). Horwitz and colleagues studied two systems of silica NPs that enhanced drug uptake into AMs (Clemens et al., 2012). The first system equipped mesoporous silica nanoparticles (MSNPs) with a polyethyleneimine (PEI) coating. PEI, being positively-charged, interacts with the negatively-charged MSNP surface and serves to enhance cellular uptake of its loaded RIF. The second equips the MSNPs with cyclodextrin-based, pH-operated valves which release INH only under acidic conditions; this feature allows only MSNPs internalized into macrophages by phagocytosis to release INH once inside the acidified endosomes. The NPs measured approximately 100 nm in diameter for both systems, with hexagonal pores about 2 nm in size. In vitro testing with human macrophage-like THP-1 cells and with human peripheral blood monocyte-derived macrophages showed that both types of MSNPs were effectively internalized. The PEI coating promoted the release of RIF into the cytosol by causing osmotic rupture of phagosomes. Moreover, the PEI-coated, RIF-loaded MSNPs killed more M. tuberculosis within macrophages than either free drug or naked RIF-loaded MSNPs. The INH-loaded, pH-gated MSNPs eliminated more M. tuberculosis from human macrophages as compared to free INH. Future work to investigate stability upon aerosolization as well as lung tissue safety and side effects are needed to apply this as an inhalable therapy, but the MSNP system highlights key design features that promote uptake into macrophages to better TB treatment (Clemens et al., 2012).
5 |. CONCLUSION
In the treatment of common respiratory diseases, inhalable nanomaterials have become a promising and attractive strategy over conventional free drug formulations as it provides a noninvasive means of maximizing therapeutic efficacy, reducing systemic toxicity, and mitigating off-target effects. Furthermore, nanomaterials can increase retention of therapeutic agents in lung tissues, prevent their premature degradation, and allow for sustained release in lung tissue. By decorating these systems with various targeting moieties, drug uptake to diseased cells is promoted while mitigating side effects to healthy cells. This is particularly advantageous for pulmonary delivery for treating lung cancer, CPDs, and bacterial infections, whereby nanomedicines can reduce necessary drug dosages and lower the dosing frequency to improve patient compliance and quality of life. Based on the material choices for these systems, their design features will ultimately influence their performance as inhalable therapies. Polymeric systems typically exhibit high nebulization stability with minimal toxicity to lung tissues but have lower retention times, as they often penetrate more easily into the systemic circulation. Lipid-based systems closely resemble the surfactant lining of the lungs and thus have better retention and safety in the lungs; however, some systems have shown nonideal nebulization stability and drug release. Inorganic systems often induce adverse side effects such as inflammation and oxidative stress, but many metallic systems have dual imaging and therapeutic properties that allows for response monitoring. For successful design of a inhalable nanomaterial system, the system must: (1) be readily aerosolized and stable in a liquid or solid aerosol; (2) effectively deposit on tissues throughout the lung at appropriate depths; (3) overcome clearance barriers in the lungs such as mucus, the mucociliary escalator, and AM uptake; (4) target diseased cells over healthy cells for preferential accumulation; and only then can the system function to (5) promote cellular uptake and induce biological effects.
Despite the advantages inhalable nanomaterials present for treating a variety of respiratory illnesses, there has been poor translation of these systems to the clinic, where there are no active or successful clinical trials currently. Generally, researchers developing inhalable nanomaterial formulations need to design systems that yield longer retention in the lungs with extended release patterns to maintain effective drug concentrations without inducing severe side effects. Many factors influence the toxicological potential of nanomaterial systems, such as their size, shape, surface charge, and biodegradability, and thus is it increasingly important to thoroughly evaluate the safety of these systems, particularly with respect to inflammatory and immunological responses these systems may elicit in the lungs. Most in vivo studies have been almost exclusively conducted in rodents, which have different respiratory system dimensions compared to humans and thus make it difficult to accurately mimic the pulmonary delivery of nanomedicine systems in humans using rodent models. Furthermore, depending on the methodology used to administer nanomedicines to the lungs (liquid instillation, IT instillation, nose-only exposure chambers), deposition patterns and plasma concentrations of drugs can be heterogeneous, thus it is important to develop highly accurate in vitro models to recapitulate human lung environments that are representative of the disease model. Moreover, much of our understanding on lung clearance kinetics and mechanisms is founded on studies conducted that follow the entrapped drugs as opposed to its carrier or carrier components; these patterns may be different for the nanomaterial and are important to evaluate to assess tissue accumulation, permeation, and adverse side effects. Alongside improvements to nanomedicines, advances in aerosolization technology to enhance control over dosing and require easier-to-use devices will improve repeatability and patient compliance. Future translation of inhalable nanomaterial therapies to the clinic is achievable through addressing these challenges and rests on the design of targeting systems which will preferentially accumulate in and treat diseased cells over healthy cells for a variety of common respiratory diseases.
ACKNOWLEDGMENT
C.A. acknowledges the support of the NIH Predoctoral and Postdoctoral Training Program in Nanotechnology for Cancer Research (5T32CA153952).
Funding information
NIH Predoctoral and Postdoctoral Training Program in Nanotechnology for Cancer Research, Grant/Award Number: 5T32CA153952
Footnotes
CONFLICT OF INTEREST
The authors have declared no conflicts of interest for this article.
REFERENCES
- Abdelaziz HM, Gaber M, Abd-Elwakil MM, Mabrouk MT, Elgohary MM, Kamel NM, … Elzoghby AO (2018). Inhalable particulate drug delivery systems for lung cancer therapy: Nanoparticles, microparticles, nanocomposites and nanoaggregates. Journal of Controlled Release, 269, 374–392. 10.1016/j.jconrel.2017.11.036 [DOI] [PubMed] [Google Scholar]
- Ahmad J, Akhter S, Rizwanullah M, Amin S, Rahman M, Zaki Ahmad M, … Jalees Ahmad F (2015). Nanotechnology-based inhalation treatments for lung cancer: State of the art. Nanotechnology, Science and Applications, 8, 55–66. 10.2147/NSA.S49052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinbami LJ, Moorman JE, Bailey C, Zahran HS, King M, Johnson CA, & Liu X (2012). Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010. NCHS Data Brief, 94, 1–8. [PubMed] [Google Scholar]
- Al-Hallak KMHD, Azarmi S, Anwar-Mohamed A, Roa WH, & Löbenberg R (2010). Secondary cytotoxicity mediated by alveolar macrophages: A contribution to the total efficacy of nanoparticles in lung cancer therapy? European Journal of Pharmaceutics and Biopharmaceutics, 76, 112–119. 10.1016/j.ejpb.2010.05.002 [DOI] [PubMed] [Google Scholar]
- Anderson CF, & Cui H (2017). Protease-sensitive nanomaterials for cancer therapeutics and imaging. Industrial & Engineering ChemistryResearch, 56(20), 5761–5777. 10.1021/acs.iecr.7b00990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azarmi S, Roa WH, & Löbenberg R (2008). Targeted delivery of nanoparticles for the treatment of lung diseases. Advanced Drug Delivery Reviews, 60, 863–875. 10.1016/j.addr.2007.11.006 [DOI] [PubMed] [Google Scholar]
- Azarmi S, Tao X, Chen H, Wang Z, Finlay WH, Löbenberg R, & Roa WH (2006). Formulation and cytotoxicity of doxorubicin nanoparticles carried by dry powder aerosol particles. International Journal of Pharmaceutics, 319, 155–161. 10.1016/j.ijpharm.2006.03.052 [DOI] [PubMed] [Google Scholar]
- Balásházy I, Martonen TB, & Hofmann2 W (1990). Fiber deposition in airway bifurcations. Journal of Aerosol Medicine, 3(4), 243–260. 10.1089/jam.1990.3.243 [DOI] [Google Scholar]
- Beasley R, Keil U, von Mutius E, & Pearce N (1998). Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The Lancet, 351, 1225–1232. [PubMed] [Google Scholar]
- Beck SE, Laube BL, Barberena CI, Fischer AC, Adams RJ, Chesnut K, … Guggino WB (2002). Deposition and expression of aerosolized rAAV vectors in the lungs of rhesus macaques. Molecular Therapy, 6(4), 546–554. 10.1006/mthe.2002.0698 [DOI] [PubMed] [Google Scholar]
- Beck-Broichsitter M, Knuedeler M-C, Schmehl T, & Seeger W (2013). Following the concentration of polymeric nanoparticles during nebulization. Pharmaceutical Research, 30, 16–24. 10.1007/s11095-012-0819-0 [DOI] [PubMed] [Google Scholar]
- Beck-Broichsitter M, Merkel OM, & Kissel T (2012). Controlled pulmonary drug and gene delivery using polymeric nano-carriers. Journal ofControlled Release, 161, 214–224. 10.1016/j.jconrel.2011.12.004 [DOI] [PubMed] [Google Scholar]
- Bhardwaj A, Kumar L, Narang RK, & Murthy RSR (2012). Development and characterization of ligand-appended liposomes for multiple drug therapy for pulmonary tuberculosis. Artificial Cells, Nanomedicine, and Biotechnology, 41, 52–59. 10.3109/10731199.2012.702316 [DOI] [PubMed] [Google Scholar]
- Bi R, Shao W, Wang Q, & Zhang N (2008). Spray-freeze-dried dry powder inhalation of insulin-loaded liposomes for enhanced pulmonary delivery. Journal of Drug Targeting, 16(9), 639–648. 10.1080/10611860802201134 [DOI] [PubMed] [Google Scholar]
- Bianchi A, Dufort S, Lux F, Courtois A, Tillement O, Coll J-L, & Crémillieux Y (2014). Quantitative biodistribution and pharmacokinetics of multimodal gadolinium-based nanoparticles for lungs using ultrashort TE MRI. Magnetic Resonance Materials in Physics, 27, 303–316. 10.1007/s10334-013-0412-5 [DOI] [PubMed] [Google Scholar]
- Blank F, Fytianos K, Seydoux E, Rodriguez-Lorenzo L, Petri-Fink A, Von Garnier C, & Rothen-Rutishauser B (2017). Interaction of biomedical nanoparticles with the pulmonary immune system. Journal of Nanobiotechnology, 15(6), 1–9. 10.1186/s12951-016-0242-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank F, Stumbles PA, Seydoux E, Holt PG, Fink A, Rothen-Rutishauser B, … Von Garnier C (2013). Size-dependent uptake of particles by pulmonary antigen-presenting cell populations and trafficking to regional lymph nodes. American Journal of Respiratory Cell and Molecular Biology, 49(1), 67–77. 10.1165/rcmb.2012-0387OC [DOI] [PubMed] [Google Scholar]
- Boylan NJ, Suk JS, Lai SK, Jelinek R, Boyle MP, Cooper MJ, & Hanes J (2012). Highly compacted DNA nanoparticles with low MW PEG coatings: In vitro, ex vivo and in vivo evaluation. Journal of Controlled Release, 157, 72–79. 10.1016/j.jconrel.2011.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain JD (1992). Mechanisms, measurement, and significance of lung macrophage function. Environmental Health Perspectives, 97, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breeze R, & Turk M (1984). Cellular structure, function and organization in the lower respiratory tract. Environmental Health Perspectives, 55, 3–24. Retrieved from https://ehp.niehs.nih.gov/doi/pdf/10.1289/ehp.84553,3,24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bur M, Henning A, Hein S, Schneider M, & Lehr C-M (2009). Inhalative nanomedicine-opportunities and challenges. Inhalation Toxicology, 21(S1), 137–143. 10.1080/08958370902962283 [DOI] [PubMed] [Google Scholar]
- Byron PR (1986). Prediction of drug residence times in regions of the human respiratory tract following aerosol inhalation. Journal of Pharmaceutical Sciences, 75(5), 433–438. 10.1002/jps.2600750502 [DOI] [PubMed] [Google Scholar]
- Card JW, Zeldin DC, Bonner JC, & Nestmann ER (2008). Pulmonary applications and toxicity of engineered nanoparticles. American Journal of Physiology: Lung Cellular and Molecular Physiology, 295(3), L400–L411. 10.1152/ajplung.00041.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho TC, Carvalho SR, & Mcconville JT (2011). Formulations for pulmonary Administration of Anticancer Agents to treat lung malignancies. Journal of Aerosol Medicine and Pulmonary Drug Delivery, 24(2), 61–80. 10.1089/jamp.2009.0794 [DOI] [PubMed] [Google Scholar]
- Chakroun RW, Zhang P, Lin R, Schiapparelli P, Quinones-Hinojosa A, & Cui H (2018). Nanotherapeutic systems for local treatment of brain tumors. WIREs: Nanomedicine and Nanobiotechnology, 10(1), e1479 10.1002/wnan.1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion JA, & Mitragotri S (2009). Shape induced inhibition of phagocytosis of polymer particles. Pharmaceutical Research, 26(1), 244–249 Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2810499/pdf/nihms165018.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Blenda WH, Kwan C, Linlin W, Yuen Y, Wong F, … Yang Z (2012). Liposomes prolong the therapeutic effect of anti-asthmatic medication via pulmonary delivery. International Journal of Nanomedicine, 7, 1139–1148. 10.2147/IJN.S28011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Ashitate Y, Lee JH, Kim SH, Matsui A, Insin N, … Tsuda A (2010). Rapid translocation of nanoparticles from the lung airspaces to the body. Nature Biotechnology, 28(12), 1300–1304. 10.1038/nbt.1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chono S, Tanino T, Seki T, & Morimoto K (2006). Influence of particle size on drug delivery to rat alveolar macrophages following pulmonary administration of ciprofloxacin incorporated into liposomes. Journal of Drug Targeting, 14(8), 557–566. 10.1080/10611860600834375 [DOI] [PubMed] [Google Scholar]
- Chu EK, & Drazen JM (2005). Centennial review asthma one hundred years of treatment and onward. American Journal of Respiratory CriticalCare Medicine, 171, 1202–1208. 10.1164/rccm.200502-257OE [DOI] [PubMed] [Google Scholar]
- Cipolla D, Shekunov B, Blanchard J, & Hickey A (2014). Lipid-based carriers for pulmonary products: Preclinical development and case studies in humans. Advanced Drug Delivery Reviews, 75, 53–80. 10.1016/j.addr.2014.05.001 [DOI] [PubMed] [Google Scholar]
- Clemens DL, Lee B-Y, Xue M, Thomas CR, Meng H, Ferris D, … Horwitz MA (2012). Targeted intracellular delivery of anti-tuberculosis drugs to Mycobacterium tuberculosis-infected macrophages via functionalized mesoporous silica nanoparticles. Antimicrobial Agents and Chemotherapy, 56, 2535–2545. 10.1128/AAC.06049-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder TM, Rosati JA, Schroeter JD, Hickey AJ, & Martonen TB (2002). Fundamental effects of particle morphology on lung delivery: Predictions of stokes’ law and the particular relevance to dry powder inhaler formulation and development. Pharmaceutical Research, 19(3), 239–245 Retrieved from https://link.springer.com/content/pdf/10.1023%2FA%3A1014426530935.pdf [DOI] [PubMed] [Google Scholar]
- Cryan S-A, Devocelle M, Moran PJ, Hickey AJ, & Kelly JG (2006). Increased intracellular targeting to airway cells using octaarginine-coated liposomes: in vitro assessment of their suitability for inhalation. Molecular Pharmaceutics, 3(2), 104–112. 10.1021/mp050070i [DOI] [PubMed] [Google Scholar]
- da Silva AL, Martini SV, Abreu SC, Samary CDS, Diaz BL, Fernezlian S, … Morales MM (2014). DNA nanoparticle-mediated thymulin gene therapy prevents airway remodeling in experimental allergic asthma. Journal of Controlled Release, 180(1), 125–133. 10.1016/j.jconrel.2014.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl R (2006). Systemic side effects of inhaled corticosteroids in patients with asthma. Respiratory Medicine, 100, 1307–1317. 10.1016/j.rmed.2005.11.020 [DOI] [PubMed] [Google Scholar]
- Dailey LA, Jekel N, Fink L, Gessler T, Schmehl T, Wittmar M, … Seeger W (2006). Investigation of the proinflammatory potential of biodegradable nanoparticle drug delivery systems in the lung. Toxicology and Applied Pharmacology, 215, 100–108. 10.1016/j.taap.2006.01.016 [DOI] [PubMed] [Google Scholar]
- Dailey LA, Schmehl T, Gessler T, Wittmar M, Grimminger F, Seeger W, & Kissel T (2003). Nebulization of biodegradable nanoparticles: Impact of nebulizer technology and nanoparticle characteristics on aerosol features. Journal of Controlled Release, 86, 131–144 Retrieved from www.elsevier.com [DOI] [PubMed] [Google Scholar]
- Dames P, Gleich B, Flemmer A, Hajek K, Seidl N, Wiekhorst F, … Rudolph C (2007). Targeted delivery of magnetic aerosol droplets to the lung. Nature Nanotechnology, 2, 495–499. 10.1038/nnano.2007.217 [DOI] [PubMed] [Google Scholar]
- Danhier F (2016). To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? Journal of Controlled Release, 244, 108–121. 10.1016/j.jconrel.2016.11.015 [DOI] [PubMed] [Google Scholar]
- De Backer L, Cerrada A, Pérez-Gil J, De Smedt SC, & Raemdonck K (2015). Bio-inspired materials in drug delivery: Exploring the role of pulmonary surfactant in siRNA inhalation therapy. Journal of Controlled Release, 220, 642–650. 10.1016/j.jconrel.2015.09.004 [DOI] [PubMed] [Google Scholar]
- Dharap SS, Wang Y, Chandna P, Khandare JJ, Qiu B, Gunaseelan S, … Minko T (2005). Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. PNAS, 102(36), 12962–12967. Retrieved from. www.ncbi.nlm.nih [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll KE, Carter JM, Howard BW, Hassenbein DG, Pepelko W, Baggs RB, & Oberdo N (1996). Pulmonary inflammatory, chemokine, and mutagenic responses in rats after subchronic inhalation of carbon black. Toxicology and Applied Pharmacology, 136). Retrieved from https://ac.els-cdn.com/S0041008X96900459/1-s2.0-S0041008X96900459-main.pdf?_tid=38720394-03a9-450b-bb4f-3422aa5f5d90&acdnat=1548786064_a57f1022164f78a0fed3ae9a59794661, 372–380. [DOI] [PubMed] [Google Scholar]
- Dufort S, Bianchi A, Henry M, Lux F, Le Duc G, Josserand V, … Coll JL (2015). Nebulized gadolinium-based nanoparticles: A theranostic approach for lung tumor imaging and radiosensitization. Small, 11(2), 215–221. 10.1002/smll.201401284 [DOI] [PubMed] [Google Scholar]
- Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-jebria A, Eskew ML, … Langer R (1997). Large porous particles for pulmonary drug delivery. Science, 276(5320), 1868–1872. 10.1126/science.276.5320.1868 [DOI] [PubMed] [Google Scholar]
- Ehteshami-Afshar S, FitzGerald JM, Doyle-Waters MM, & Sadatsafavi M (2016). The global economic burden of asthma and chronic obstructive pulmonary disease. International Journal of Tuberculosis and Lung Disease, 20(1), 11–23. 10.5588/ijtld.15.0472 [DOI] [PubMed] [Google Scholar]
- Èller RHM, Èder KM, & Gohla S (2000). Solid lipid nanoparticles (SLN) for controlled drug delivery—A review of the state of the art.European Journal of Pharmaceutics and Biopharmaceutics, 50, 161–177 Retrieved from www.elsevier.com/locate/ejphabio [DOI] [PubMed] [Google Scholar]
- Fiegel J, Fu J, & Hanes J (2004). Poly(ether-anhydride) dry powder aerosols for sustained drug delivery in the lungs. Journal of Controlled Release, 96, 411–423. 10.1016/j.jconrel.2004.02.018 [DOI] [PubMed] [Google Scholar]
- Franci G, Falanga A, Galdiero S, Palomba L, Rai M, Morelli G, & Galdiero M (2015). Silver nanoparticles as potential antibacterial agents. Molecules, 20(5), 8856–8874. 10.3390/molecules20058856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Fiegel J, Krauland E, & Hanes J (2002). New polymeric carriers for controlled drug delivery following inhalation or injection. Biomaterials, 23, 4425–4433. [DOI] [PubMed] [Google Scholar]
- Gagnadoux F, Hureaux J, Vecellio L, Urban T, Le Pape A, Valo I, … Lemarie E (2008). Aerosolized chemotherapy. Journal of Aerosol Medicine and Pulmonary Drug Delivery, 21(1), 61–70. 10.1089/jamp.2007.0656 [DOI] [PubMed] [Google Scholar]
- Garbuzenko OB, Mainelis G, Taratula O, & Minko T (2014). Inhalation treatment of lung cancer: The influence of composition, size and shape of nanocarriers on their lung accumulation and retention. Cancer Biology & Medicine, 11, 44–55. 10.7497/j.issn.2095-3941.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser M, Casaulta M, Kupferschmid B, Schulz H, Semmler-Behnke M, & Kreyling W (2008). The role of macrophages in the clearance of inhaled ultrafine titanium dioxide particles. American Journal of Respiratory Cell and Molecular Biology, 38(3), 371–376. 10.1165/rcmb.2007-0138OC [DOI] [PubMed] [Google Scholar]
- Geiser M, Quaile O, Wenk A, Wigge C, Eigeldinger-Berthou S, Hirn S, … Kreyling WG (2013). Cellular uptake and localization of inhaled gold nanoparticles in lungs of mice with chronic obstructive pulmonary disease. Particle and Fibre Toxicology, 10(19), 1–10. 10.1186/1743-8977-10-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser M, Schürch S, & Gehr P (2003). Influence of surface chemistry and topography of particles on their immersion into the lung’s surface-lining layer. Journal of Applied Physiology, 94(5), 1793–1801. 10.1152/japplphysiol.00514.2002 [DOI] [PubMed] [Google Scholar]
- Gelperina S, Kisich K, Iseman MD, & Heifets L (2005). The potential advantages of nanoparticle drug delivery systems in chemotherapy of tuberculosis. Pulmonary Perspective, 172, 1487–1490. 10.1164/rccm.200504-613PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal AK, Garg T, Rath G, Datta Gupta U, & Gupta P (2015). Development and characterization of nanoembedded microparticles for pulmonary delivery of antitubercular drugs against experimental tuberculosis. Molecular Pharmaceutics, 12, 3839–3850. 10.1021/acs.molpharmaceut.5b00016 [DOI] [PubMed] [Google Scholar]
- Grabowski N, Hillaireau H, Vergnaud J, Santiago A, Kerdine-Romer S, Pallardy M, … Fattal E (2013). Toxicity of surface-modified PLGA nanoparticles toward lung alveolar epithelial cells. International Journal of Pharmaceutics, 454, 686–694. 10.1016/j.ijpharm.2013.05.025 [DOI] [PubMed] [Google Scholar]
- Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, & Desimone JM (2008). The effect of particle design on cellular internalization pathways. PNAS, 105(33), 11613–11618 Retrieved from www.pnas.org/cgi/content/full/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco E, Quintiliani G, Santucci MB, Serafino A, Ciccaglione AR, Marcantonio C, … Fraziano M (2012). Janus-faced liposomes enhance antimicrobial innate immune response in Mycobacterium tuberculosis infection. PNAS, 21, E1360–E1368. 10.1073/pnas.1200484109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene CM, & McElvaney NG (2009). Proteases and antiproteases in chronic neutrophilic lung disease - relevance to drug discovery. British Journal of Pharmacology, 158(4), 1048–1058. 10.1111/j.1476-5381.2009.00448.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guagliardo R, Pérez-Gil J, De Smedt S, & Raemdonck K (2018). Pulmonary surfactant and drug delivery: Focusing on the role of surfactant proteins. Journal of Controlled Release, 291(July), 116–126. 10.1016/j.jconrel.2018.10.012 [DOI] [PubMed] [Google Scholar]
- Halwani R, Sultana Shaik A, Ratemi E, Afzal S, Kenana R, Al-Muhsen S, & Al Faraj A (2016). A novel anti-IL4Rα nanoparticle efficiently controls lung inflammation during asthma. Experimental & Molecular Medicine, 262, e262 10.1038/emm.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque S, Whittaker M, McIntosh MP, Pouton CW, Phipps S, & Kaminskas LM (2018). A comparison of the lung clearance kinetics of solid lipid nanoparticles and liposomes by following the 3 H-labelled structural lipids after pulmonary delivery in rats. European Journal of Pharmaceutics and Biopharmaceutics, 125, 1–12. 10.1016/j.ejpb.2018.01.001 [DOI] [PubMed] [Google Scholar]
- Haque S, Whittaker MR, McIntosh MP, Pouton CW, & Kaminskas LM (2016). Disposition and safety of inhaled biodegradable nanomedicines: Opportunities and challenges. Nanomedicine: Nanotechnology, Biology, and Medicine, 12, 1703–1724. 10.1016/j.nano.2016.03.002 [DOI] [PubMed] [Google Scholar]
- Hasenpusch G, Geiger J, Wagner K, Mykhaylyk O, Wiekhorst F, Trahms L, … Rudolph C (2012). Magnetized aerosols comprising superparamagnetic iron oxide nanoparticles improve targeted drug and gene delivery to the lung. Pharmaceutical Research, 29, 1308–1318. 10.1007/s11095-012-0682-z [DOI] [PubMed] [Google Scholar]
- Henning A, Schneider M, Nafee N, Muijs L, Rytting E, Wang X, … Lehr C-M (2010). Influence of particle size and material properties on mucociliary clearance from the airways. Journal of Aerosol Medicine and Pulmonary Drug Delivery, 23(4), 233–241. Retrieved from. www.liebertpub.com [DOI] [PubMed] [Google Scholar]
- Heyder J, Gebhart J, Rudolf G, Schiller CF, & Stahlhofen W (1986). Deposition of particles in the human respiratory tract in the size range 0.005–15 um. Journal of Aerosol Science, 17, 811–825. Retrieved from https://ac.els-cdn.com/0021850286900352/1-s2.0-0021850286900352-main.pdf?_tid=8079a208-3b64-46ae-872d-a66d724a1d73&acdnat=1548715781_174705a93f45e7ff2780f287b2547314 [Google Scholar]
- Heyder J (2004). Deposition of inhaled particles in the human respiratory tract and consequences for regional targeting in respiratory drug delivery. Proceedings of the American Thoracic Society, 1, 315–320. 10.1513/pats.200409-046TA [DOI] [PubMed] [Google Scholar]
- Hidalgo A, Cruz A, & Pérez-Gil J (2015). Barrier or carrier? Pulmonary surfactant and drug delivery. European Journal of Pharmaceutics and Biopharmaceutics, 95, 117–127. 10.1016/j.ejpb.2015.02.014 [DOI] [PubMed] [Google Scholar]
- Hidalgo A, Cruz A, & Pérez-Gil J (2017). Pulmonary surfactant and nanocarriers: Toxicity versus combined nanomedical applications. Biochimica et Biophysica Acta - Biomembranes, 1859(9), 1740–1748. 10.1016/j.bbamem.2017.04.019 [DOI] [PubMed] [Google Scholar]
- Hitzman CJ, Wattenberg LW, & Wiedmann TS (2006). Pharmacokinetics of 5-fluorouracil in the hamster following inhalation delivery of lipid-coated nanoparticles. Journal of Pharmaceutical Sciences, 95, 1196–1211. 10.1002/jps [DOI] [PubMed] [Google Scholar]
- Høiby N, Ciofu O, & Bjarnsholt T (2010, November). Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiology, 5, 1663–1674. 10.2217/fmb.10.125 [DOI] [PubMed] [Google Scholar]
- Hu G, Jiao B, Shi X, Valle RP, Fan Q, & Zuo YY (2013). Physicochemical properties of nanoparticles regulate translocation across pulmonary surfactant monolayer and formation of lipoprotein Corona. ACS Nano, 7, 10525–10533. 10.1021/nn4054683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaques PA, & Kim CS (2000). Measurement of total lung deposition of inhaled ultrafine particles in healthy men and women. Inhalation Toxicology, 12(8), 715–731. 10.1080/08958370050085156 [DOI] [PubMed] [Google Scholar]
- Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, … Noble PW (2005). Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nature Medicine, 11(11), 1173–1179. 10.1038/nm1315 [DOI] [PubMed] [Google Scholar]
- Kaczmarek JC, Patel AK, Kauffman KJ, Fenton OS, Webber MJ, Heartlein MW, … Anderson DG (2016). Polymer–lipid nanoparticles for systemic delivery of mRNA to the lungs. Angewandte Chemie—International Edition, 55(44), 13808–13812. 10.1002/anie.201608450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminskas LM, Mcleod VM, Ryan GM, Kelly BD, Haynes JM, Williamson M, … Porter CJH (2014). Pulmonary administration of a doxorubicin-conjugated dendrimer enhances drug exposure to lung metastases and improves cancer therapy. Journal of Controlled Release, 183, 18–26. 10.1016/j.jconrel.2014.03.012 [DOI] [PubMed] [Google Scholar]
- Kessler R, Partridge MR, Miravitlles M, Cazzola M, Vogelmeier C, Leynaud D, & Ostinelli J (2011). Symptom variability in patients with severe COPD: A pan-European cross-sectional study. European Respiratory Journal, 37(2), 264–272. 10.1183/09031936.00051110 [DOI] [PubMed] [Google Scholar]
- Kim H, Park HT, Kong H, Sung DK, Woo Hwang B, Kim KS, … Kwang Hahn S (2013). Bioimaging and pulmonary applications of self-assembled Flt1 peptideehyaluronic acid conjugate nanoparticles. Biomaterials, 34, 8478–8490. 10.1016/j.biomaterials.2013.07.062 [DOI] [PubMed] [Google Scholar]
- Kim I, Jun Byeon H, Hyung Kim T, Seong Lee E, Taek Oh K, Soo Shin B, … Seok Youn Y (2013). Doxorubicin-loaded porous PLGA microparticles with surface attached TRAIL for the inhalation treatment of metastatic lung cancer. Biomaterials, 34, 6444–6453. 10.1016/j.biomaterials.2013.05.018 [DOI] [PubMed] [Google Scholar]
- Kleinstreuer C, Zhang Z, & Donohue JF (2008). Targeted drug-aerosol delivery in the human respiratory system. Annual Review of Biomedical Engineering, 10, 195–220. 10.1146/annurev.bioeng.10.061807.160544 [DOI] [PubMed] [Google Scholar]
- Koshkina NV, Gilbert BE, Waldrep JC, Seryshev A, & Knight V (1999). Distribution of camptothecin after delivery as a liposome aerosol or following intramuscular injection in mice. Cancer Chemotherapy and Pharmacology, 44, 187–192 Retrieved from https://link.springer.com/content/pdf/10.1007%2Fs002800050966.pdf [DOI] [PubMed] [Google Scholar]
- Koshkina NV, Waldrep JC, Roberts LE, Golunski E, Melton S, & Knight V (2001). Paclitaxel liposome aerosol treatment induces inhibition of pulmonary metastases in murine renal carcinoma model. Clinical Cancer Research, 7, 3258–3262 Retrieved from http://clincancerres.aacrjournals.org/content/clincanres/7/10/3258.full.pdf [PubMed] [Google Scholar]
- Kumar Sukumar U, Bhushan B, Dubey P, Matai I, Sachdev A, & Packirisamy G (2013). Emerging applications of nanoparticles for lung cancer diagnosis and therapy. International Nano Letters, 3(45), 1–17. 10.1186/2228-5326-3-45 [DOI] [Google Scholar]
- Kuzmov A, & Minko T (2015). Nanotechnology approaches for inhalation treatment of lung diseases. Journal of Controlled Release, 219,500–518. 10.1016/j.jconrel.2015.07.024 [DOI] [PubMed] [Google Scholar]
- Lai SK, Suk JS, Pace A, Wang Y-Y, Yang M, Mert O, … Hanes J (2011). Drug carrier nanoparticles that penetrate human chronic rhinosinusitis mucus. Biomaterials, 32, 6285–6290. 10.1016/j.biomaterials.2011.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai SK, Wang Y-Y, & Hanes J (2008). Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Advanced Drug Delivery Reviews, 61, 158–171. 10.1016/j.addr.2008.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam JK-W, Liang W, & Chan H-K (2012). Pulmonary delivery of therapeutic siRNA. Advanced Drug Delivery Reviews, 64, 1–15. 10.1016/j.addr.2011.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza GM, Jenkins J, Schmieder AH, Moldobaeva A, Cui G, Zhang H, … Langenberg OM (2017). Anti-angiogenic nanotherapy inhibits airway remodeling and hyper-responsiveness of dust mite triggered asthma in the Brown Norway rat. Theranostics, 7(2), 377–389. 10.7150/thno.16627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W-H, Loo C-Y, Traini D, & Young PM (2015a). Inhalation of nanoparticle-based drug for lung cancer treatment: Advantages and challenges. Asian Journal of Pharmaceutical Sciences, 10, 481–489. 10.1016/j.ajps.2015.08.009 [DOI] [Google Scholar]
- Lee W-H, Loo C-Y, Traini D, & Young PM (2015b). Nano-and micro-based inhaled drug delivery systems for targeting alveolar macrophages. Expert Opinion on Drug Delivery, 12(6), 1009–1026. 10.1517/17425247.2015.1039509 [DOI] [PubMed] [Google Scholar]
- Lehofer B, Bloder F, Jain PP, Marsh LM, Leitinger G, Olschewski H, … Prassl R (2014). Impact of atomization technique on the stability and transport efficiency of nebulized liposomes harboring different surface characteristics. European Journal of Pharmaceutics and Biopharmaceutics, 88, 1076–1085. 10.1016/j.ejpb.2014.10.009 [DOI] [PubMed] [Google Scholar]
- Lemjabbar-Alaoui H, Hassan OU, Yang Y-W, & Buchanan P (2015). Lung cancer: Biology and treatment options. Biochimica et Biophysica Acta, 1856, 189–210. 10.1016/j.bbcan.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong NJ, Mehta D, Mcleod VM, Kelly BD, Pathak R, Owen DJ, … Kaminskas LM (2018). Doxorubicin conjugation and drug linker chemistry alter the intravenous and pulmonary pharmacokinetics of a PEGylated generation 4 polylysine dendrimer in rats. Journal of Pharmaceutical Sciences, 107, 2509–2513. 10.1016/j.xphs.2018.05.013 [DOI] [PubMed] [Google Scholar]
- Li X, Vogt FG, Hayes D, & Mansour HM (2014). Design, characterization, and aerosol dispersion performance modeling of advanced spray-dried microparticulate/nanoparticulate mannitol powders for targeted pulmonary delivery as dry powder inhalers. Journal of Aerosol Medicine and Pulmonary Drug Delivery, 27(2), 81–93. 10.1089/jamp.2013.1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang F, & Cui H (2016). Peptide-based supramolecular hydrogels for delivery of biologics. Bioengineering & Translational Medicine, 1(3), 306–322. 10.1002/btm2.10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Wei M, Evans DG, & Duan X (2014). Inorganic nanomaterials for bioimaging, targeted drug delivery and therapeutics. Chemical Communications, 50, 14071–14081. 10.1039/c4cc03118k [DOI] [PubMed] [Google Scholar]
- Liang Z, Ni R, Zhou J, & Mao S (2015). Recent advances in controlled pulmonary drug delivery. Drug Discovery Today, 20(3), 380–389. 10.1016/j.drudis.2014.09.020 [DOI] [PubMed] [Google Scholar]
- Lin C, Chi B, Wong K, Chen H, Bian Z, Zhang G, … Yang Z (2017). Pulmonary delivery of triptolide-loaded liposomes decorated with anti-carbonic anhydrase IX antibody for lung cancer therapy. Scientific Reports, 7(1097), 1097 10.1038/s41598-017-00957-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Zhang X, Chen H, Bian Z, Zhang G, Kashif Riaz M, … Yang Z (2018). Dual-ligand modified liposomes provide effective local targeted delivery of lung-cancer drug by antibody and tumor lineage-homing cell-penetrating peptide. Drug Delivery, 25(1), 256–266. 10.1080/10717544.2018.1425777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Wong J, Qu L, Chan H, & Zhou QT (2015). Powder production and particle engineering for dry powder inhaler formulations. Current Pharmaceutical Design, 21, 3902–3916. 10.2174/1381612821666150820111134 [DOI] [PubMed] [Google Scholar]
- Loira-Pastoriza C, Todoroff J, & Vanbever R (2014). Delivery strategies for sustained drug release in the lungs. Advanced Drug Delivery Reviews, 75, 81–91. 10.1016/j.addr.2014.05.017 [DOI] [PubMed] [Google Scholar]
- Lombry C, Edwards DA, Préat V, & Vanbever R (2004). Alveolar macrophages are a primary barrier to pulmonary absorption of macromolecules. American Journal of Physiology, 286(5), L1002–L1008. 10.1152/ajplung.00260.2003 [DOI] [PubMed] [Google Scholar]
- Lopes Da Silva A, Ferreira Cruz F, Rieken P, Rocco M, & Morales MM (2017). New perspectives in nanotherapeutics for chronic respiratory diseases. Biophysical Reviews, 9, 793–803. 10.1007/s12551-017-0319-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano R, Naghavi M, Lim SS, Ahn MPH SY, Alvarado MB, Andrews MPH KG, … L Murray CJ (2012). Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. The Lancet (Vol. 380). 10.1016/S0140-6736(12)61728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo T, Loira-Pastoriza C, Patil HP, Ucakar B, Muccioli GG, Bosquillon C, & Vanbever R (2016). PEGylation of paclitaxel largely improves its safety and anti-tumor efficacy following pulmonary delivery in a mouse model of lung carcinoma. Journal of Controlled Release, 239, 62–71. 10.1016/j.jconrel.2016.08.008 [DOI] [PubMed] [Google Scholar]
- Madni A, Batool A, Noreen S, Maqbool I, Rehman F, Kashif M, … Raza A (2017). Novel nanoparticulate systems for lung cancer therapy: An updated review. Journal of Drug Targeting, 25(6), 499–512. 10.1080/1061186X.2017.1289540 [DOI] [PubMed] [Google Scholar]
- Maeda H (2001). The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Advances in Enzyme Regulation, 41, 189–207. [DOI] [PubMed] [Google Scholar]
- Makino K, Yamamoto N, Higuchi K, Harada N, Ohshima H, & Terada H (2003). Phagocytic uptake of polystyrene microspheres by alveolar macrophages: Effects of the size and surface properties of the microspheres. Colloids and Surfaces B: Biointerfaces, 27, 33–39 Retrieved from www.elsevier.com/locate/colsurfb [Google Scholar]
- Mangal S, Gao W, & Zhou Q (2017). Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: Challenges and opportunities. Acta Pharmacologica Sinica, 38, 782–797. 10.1038/aps.2017.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markman JL, Rekechenetskiy A, Holler E, & Ljubimova JY (2013). Nanomedicine therapeutic approaches to overcome cancer drug resistance. Advanced Drug Delivery Reviews, 65, 1866–1879. 10.1016/j.addr.2013.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meers P, Neville M, Malinin V, Scotto AW, Sardaryan G, Kurumunda R, … Perkins WR (2008). Biofilm penetration, triggered release and in vivo activity of inhaled liposomal amikacin in chronic Pseudomonas aeruginosa lung infections. Journal of Antimicrobial Chemotherapy, 61, 859–868. 10.1093/jac/dkn059 [DOI] [PubMed] [Google Scholar]
- Menon JU, Kuriakose A, Iyer R, Hernandez E, Gandee L, Zhang S, … Nguyen T (2017). Dual-drug containing core-shell nanoparticles for lung cancer therapy. Scientific Reports, 7(13249), 1–13. 10.1038/s41598-017-13320-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal V, El Rayes T, Narula N, Mcgraw TE, Altorki NK, Barcellos-Hoff MH, … Rayes TE (2016). The microenvironment of lung cancer and therapeutic implications In Lung cancer and personalized medicine (Vol. 890, pp. 75–110). Switzerland: Springer International Publishing; 10.1007/978-3-319-24932-2_5 [DOI] [PubMed] [Google Scholar]
- Modak SM, & Fox CL (1973). Binding of silver sulfadiazine to the cellular components of Pseudomonas aeruginosa Biochemical Pharmacology (Vol. 22, pp. 2391–2404). Oxford, England: Pergamon Press; Retrieved from https://pdf.sciencedirectassets.com/271311/1-s2.0-S0006295200X05990/1-s2.0-0006295273903419/main.pdf?x-amz-security-token=AgoJb3JpZ2luX2VjEM7%2F%2F%2F%2F%2F%2F%2F%2F%2F%2FwEaCXVzLWVhc3QtMSJGMEQCIEAS%2F26i6FGY7aRH93MmBCR3WgmRMOIFVkDoPTl0NgLOAiBZm9NtwTOOCRm [DOI] [PubMed] [Google Scholar]
- Mohseni M, Gilani K, & Mortazavi SA (2015). Preparation and characterization of rifampin loaded mesoporous silica nanoparticles as a potential system for pulmonary drug delivery. Iranian Journal of Pharmaceutical Research, 14(1), 27–34. [PMC free article] [PubMed] [Google Scholar]
- Möller W, Felten K, Sommerer K, Scheuch G, Meyer G, Meyer P, … Kreyling WG (2008). Deposition, retention, and translocation of ultrafine particles from the central airways and lung periphery. American Journal of Respiratory and Critical Care Medicine, 177(4), 426–432. 10.1164/rccm.200602-301OC [DOI] [PubMed] [Google Scholar]
- Monroe M, Flexner C, & Cui H (2018). Harnessing nanostructured systems for improved treatment and prevention of HIV disease. Bioengineering & Translational Medicine, 3(2), 102–123. 10.1002/btm2.10096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretton MA, Cagel M, Bernabeu E, Gonzalez L, & Chiappetta DA (2015). Nanopolymersomes as potential carriers for rifampicin pulmonary delivery. Colloids and Surfaces B: Biointerfaces, 136, 1017–1025. 10.1016/j.colsurfb.2015.10.049 [DOI] [PubMed] [Google Scholar]
- Morlieras J, Dufort S, Sancey L, Truillet C, Mignot A, Rossetti F, … Cedex V (2013). Functionalization of small rigid platforms with cyclic RGD peptides for targeting Tumors overexpressing α v β 3-Integrins. Bioconjugate Chemistry, 24, 1584–1597. 10.1021/bc4002097 [DOI] [PubMed] [Google Scholar]
- Mura S, Hillaireau H, Nicolas J, Le Droumaguet B, Gueutin C, Zanna S, … Fattal E (2011). Influence of surface charge on the potential toxicity of PLGA nanoparticles towards Calu-3 cells. International Journal of Nanomedicine, 6, 2591–2605. 10.2147/IJN.S24552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mura S, Nicolas J, Kerdine-Ro S, Le Droumaguet B, Delome C, Nicolas R, … Fattal E (2011). Biodegradable nanoparticles meet the bronchial airway barrier: How surface properties affect their interaction with mucus and epithelial cells. Biomacromolecules, 12, 4136–4143. 10.1021/bm201226x [DOI] [PubMed] [Google Scholar]
- Nag K, Vidyashankar S, Devraj R, Garcia F, & Panda AK (2010). Physicochemical studies on the interaction of serum albumin with pulmonary surfactant extract in films and bulk bilayer phase. Journal of Colloid and Interface Science, 352, 456–464. 10.1016/j.jcis.2010.08.058 [DOI] [PubMed] [Google Scholar]
- Nassimi M, Schleh C, Lauenstein HD, Hussein R, Hoymann HG, Koch W, … Müller-Goymann C (2010). A toxicological evaluation of inhaled solid lipid nanoparticles used as a potential drug delivery system for the lung. European Journal of Pharmaceutics and Biopharmaceutics, 75, 107–116. 10.1016/j.ejpb.2010.02.014 [DOI] [PubMed] [Google Scholar]
- Natarajan K, Kundu M, Sharma P, & Basu J (2011). Innate immune responses to M. tuberculosis infection. Tuberculosis, 91, 427–431. 10.1016/j.tube.2011.04.003 [DOI] [PubMed] [Google Scholar]
- Nials AT, & Uddin S (2008). Mouse models of allergic asthma: Acute and chronic allergen challenge. Disease Models and Mechanisms, 1(4–5), 213–220. 10.1242/dmm.000323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira EE, Mangialavori IC, Loureiro A, Azoia NG, Sárriasárria MP, Nogueira P, … Cavaco-Paulo A (2015). Peptide anchor for folate-targeted liposomal delivery. Biomacromolecules, 16, 2904–2910. 10.1021/acs.biomac.5b00823 [DOI] [PubMed] [Google Scholar]
- O-H Sham J, Zhang Y, Finlay WH, Roa WH, & Löbenberg R (2004). Formulation and characterization of spray-dried powders containing nanoparticles for aerosol delivery to the lung. International Journal of Pharmaceutics, 269, 457–467. 10.1016/j.ijpharm.2003.09.041 [DOI] [PubMed] [Google Scholar]
- Olsson B, Bondesson E, Borgström L, Edsbäcker S, Eirefelt S, Ekelund K, … Hegelund-Myrbäck T (2011). Pulmonary drug methabolism, clearance and absorption In Controlled pulmonary drug delivery: Advances in delivery science and technology (pp. 21–50). New York, NY: Springer; 10.1007/978-1-4419-9745-6 [DOI] [Google Scholar]
- O’shannessy DJ, Yu G, Smale R, Fu Y-S, Singhal S, Thiel RP, … Vachani A (2012). Folate receptor alpha expression in lung cancer: Diagnostic and prognostic significance. Oncotarget, 3(4), 414–425 Retrieved from www.impactjournals.com/oncotarget [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HS, Kim KH, Jang S, Park JW, Cha HR, Lee JE, … Jung SS (2010). International journal of nanomedicine dovepress attenuation of allergic airway inflammation and hyperresponsiveness in a murine model of asthma by silver nanoparticles. International Journal of Nanomedicine, 5, 510–515. Retrieved from www.dovepress.com [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SS, & Wexler AS (2007). Particle deposition in the pulmonary region of the human lung: A semi-empirical model of single breath transport and deposition. Aerosol Science, 38, 228–245. 10.1016/j.jaerosci.2006.11.009 [DOI] [Google Scholar]
- Patel AK, Kaczmarek JC, Bose S, Kauffman KJ, Mir F, Heartlein MW, … Anderson DG (2019). Inhaled nanoformulated mRNA polyplexes for protein production in lung epithelium. Advanced Materials, 31(8), 1–7. 10.1002/adma.201805116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NR, Pattni BS, Abouzeid AH, & Torchilin VP (2013). Nanopreparations to overcome multidrug resistance in cancer. Advanced DrugDelivery Reviews, 65, 1748–1762. 10.1016/j.addr.2013.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil-Gadhe A, Kyadarkunte A, Patole M, & Pokharkar V (2014). Montelukast-loaded nanostructured lipid carriers: Part II pulmonary drug delivery and in vitro-in vivo aerosol performance. European Journal of Pharmaceutics and Biopharmaceutics, 88(1), 169–177. 10.1016/j.ejpb.2014.07.007 [DOI] [PubMed] [Google Scholar]
- Patton JS (1996). Mechanisms of macromolecule absorption by the lungs. Advanced Drug Delivery Reviews, 19, 3–36. Retrieved from https://ac.els-cdn.com/0169409X9500113L/1-s2.0-0169409X9500113L-main.pdf?_tid=8c634288-8f5a-469a-a53f-d709bb3c5101&acdnat=1548718443_a3e53efa6f80c090504376b97283a27e [Google Scholar]
- Patton JS, Brain JD, Davies LA, Fiegel J, Gumbleton M, Kim K-J, … Ehrhardt C (2010). The particle has landed-characterizing the fate of inhaled pharmaceuticals. Journal of Aerosol Medicine and Pulmonary Drug Delivery, 23(2), 71–81. 10.1089/jamp.2010.0836 [DOI] [PubMed] [Google Scholar]
- Patton JS, & Byron PR (2007). Inhaling medicines: Delivering drugs to the body through the lungs. Nature Reviews Drug Discovery, 6(1),67–74. 10.1038/nrd2153 [DOI] [PubMed] [Google Scholar]
- Pison U, Welte T, Giersig M, & Groneberg DA (2006). Nanomedicine for respiratory diseases. European Journal of Pharmacology, 533,341–350. 10.1016/j.ejphar.2005.12.068 [DOI] [PubMed] [Google Scholar]
- Quaderi SA, & Hurst JR (2018). The unmet global burden of COPD. Global Health, Epidemiology and Genomics, 3, 1–3. 10.1017/gheg.2018.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raesch SS, Tenzer S, Storck W, Rurainski A, Selzer D, Ruge CA, … Lehr C-M (2015). Proteomic and lipidomic analysis of nanoparticle corona upon contact with lung surfactant reveals differences in protein, but not lipid composition. ACS Nano, 9(12), 11872–11885. 10.1021/acsnano.5b04215 [DOI] [PubMed] [Google Scholar]
- Ratemi E, Sultana Shaik A, Al Faraj A, & Halwani R (2016). Alternative approaches for the treatment of airway diseases: Focus on nanoparticle medicine. Clinical and Experimental Allergy, 46(8), 1033–1042. 10.1111/cea.12771 [DOI] [PubMed] [Google Scholar]
- Reed MD, Tellez CS, Grimes MJ, Picchi MA, Tessema M, Cheng YS, … Belinsky SA (2013). Aerosolised 5-azacytidine suppresses tumour growth and reprogrammes the epigenome in an orthotopic lung cancer model. British Journal of Cancer, 109, 1775–1781. 10.1038/bjc.2013.575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnier P, Mottais A, Sibiril Y, Le Gall T, & Montier T (2016). Challenges and successes using nanomedicines for aerosol delivery to the airways. Current Gene Therapy, 16(1), 34–46. 10.2174/1566523216666160104142013 [DOI] [PubMed] [Google Scholar]
- Rodrigues S, Cordeiro C, Seijo N, Remuñán C, Remuñán-López R, & Grenha A (2015). Hybrid nanosystems based on natural polymers as protein carriers for respiratory delivery: Stability and toxicological evaluation. Carbohydrate Polymers, 123, 369–380. 10.1016/j.carbpol.2015.01.048 [DOI] [PubMed] [Google Scholar]
- Rogueda PG, & Traini D (2007). The nanoscale in pulmonary delivery. Part 2: Formulation platforms. Expert Opinion on Drug Delivery, 4(6),607–620. 10.1517/17425247.4.6.607 [DOI] [PubMed] [Google Scholar]
- Rojanarat W, Nakpheng T, Thawithong E, Yanyium N, & Srichana T (2012). Inhaled pyrazinamide proliposome for targeting alveolar macrophages. Drug Delivery, 19(7), 334–345. 10.3109/10717544.2012.721144 [DOI] [PubMed] [Google Scholar]
- Rosière R, Van Woensel M, Gelbcke M, Mathieu VV, Hecq J, Mathivet T, … Wauthoz N (2018). New folate-grafted chitosan derivative to improve delivery of paclitaxel-loaded solid lipid nanoparticles for lung tumor therapy by inhalation. Molecular Pharmaceutics, 15, 899–910. 10.1021/acs.molpharmaceut.7b00846 [DOI] [PubMed] [Google Scholar]
- Roy I, & Vij N (2010). Nanodelivery in airway diseases: Challenges and therapeutic applications. Nanomedicine: Nanotechnology, Biology, and Medicine, 6, 237–244. 10.1016/j.nano.2009.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BK, & Williams RW (2014). Emerging aerosol drug delivery strategies: From bench to clinic *. Advanced Drug Delivery Reviews, 75,141–148. 10.1016/j.addr.2014.06.008 [DOI] [PubMed] [Google Scholar]
- Ruge CA, Kirch J, & Lehr C-M (2013). Pulmonary drug delivery: From generating aerosols to overcoming biological barriers-therapeutic possibilities and technological challenges. Lancet Respiratory Medicine, 1, 402–413. 10.1016/S2213-2600(13)70072-9 [DOI] [PubMed] [Google Scholar]
- Rusch V, Klimstra D, Venkatraman E, Pisters PWT, Langenfeld J, & Dmitrovsky E (1997). Overexpression of epidermal growth factor receptor and its ligand transforming growth factor alpha is frequent in resectable non-small cell lung cancer but does not predict tumor progression. Clinical Cancer Research, 3, 515–522. [PubMed] [Google Scholar]
- Ryan GM, Bischof RJ, Enkhbaatar P, Mcleod VM, Chan LJ, Jones SA, … Kaminskas LM (2016). A comparison of the pharmacokinetics and pulmonary lymphatic exposure of a generation 4 PEGylated dendrimer following intravenous and aerosol administration to rats and sheep. Pharmaceutical Research, 33, 510–525. 10.1007/s11095-015-1806-z [DOI] [PubMed] [Google Scholar]
- Ryan GM, Kaminskas LM, Kelly BD, Owen DJ, Mcintosh MP, & Porter CJH (2013). Pulmonary administration of PEGylated polylysine dendrimers: Absorption from the lung versus retention within the lung is highly size-dependent. Molecular Pharmaceutics, 10, 2986–2995. 10.1021/mp400091n [DOI] [PubMed] [Google Scholar]
- Rytting E, Nguyen J, Wang X, & Kissel T (2008). Biodegradable polymeric nanocarriers for pulmonary drug delivery. Expert Opinion on Drug Delivery, 5(6), 629–639. 10.1517/17425247.5.6.629 [DOI] [PubMed] [Google Scholar]
- Saad M, Garbuzenko OB, Ber E, Chandna P, Khandare JJ, Pozharov VP, & Minko T (2008). Receptor targeted polymers, dendrimers, liposomes: Which nanocarrier is the most efficient for tumor-specific treatment and imaging? Journal of Controlled Release, 130, 107–114. 10.1016/j.jconrel.2008.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhukha T, Wiedmann TS, & Panyam J (2013). Inhalable magnetic nanoparticles for targeted hyperthermia in lung cancer therapy. Biomaterials, 34, 5163–5171. 10.1016/j.biomaterials.2013.03.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadikot RT (2018). The potential role of nanomedicine in lung diseases. Medical Research Archives, 6(5), 1–9. 10.18103/mra.v6i5.1723 [DOI] [Google Scholar]
- Sahay G, Alakhova DY, & Kabanov AV (2010). Endocytosis of nanomedicines. Journal of Controlled Release, 145, 182–195. 10.1016/j.jconrel.2010.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahib MN, Abdulameer SA, Darwis Y, Peh KK, Tan YT. (2012). Solubilization of beclomethasone dipropionate in sterically stabilized phospholipid nanomicelles (SSMs): Physicochemical and in vitro evaluations. Drug Design, Development and Therapy, 6, 29–42. 10.2147/DDDT.S28265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäper C, Noga O, Koch B, Ewert R, Felix SB, Gläser S, … Gustavus B (2011). Anti-infl ammatory properties of Montelukast, a leukotriene receptor antagonist in patients with asthma and nasal polyposis. Journal of Investigational Allergology & Clinical Immunology (Vol. 21, pp. 51–58). Retrieved from https://pdfs.semanticscholar.org/6bc5/34311b6bf44160009ee7c0d31fde4f4a062b.pdf [PubMed] [Google Scholar]
- Schneeberger-Keeley EE, & Karnovsky MJ (1968). The ultrastructural basis of alveolar-capillary membrane permeability to peroxidase used as a tracer. The Journal of Cell Biology, 37(3), 781 Retrieved from http://jcb.rupress.org/content/37/3/781.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seydoux E, Rodriguez-Lorenzo L, Blom RAM, Stumbles PA, Petri-Fink A, Rothen-Rutishauser BM, … Von Garnier C (2016). Pulmonary delivery of cationic gold nanoparticles boost antigen-specific CD4 + T cell proliferation. Nanomedicine: Nanotechnology, Biology and Medicine, 12, 1815–1826. 10.1016/j.nano.2016.02.020 [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, & Jemal A (2019). Cancer statistics, 2019. CA: A Cancer Journal for Clinicians, 69(1), 7–34. 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- Siekmeier R, & Scheuch G (2009). Treatment of systemic diseases by inhalation of biomolecule aerosols. Journal of Physiology and Pharmacology, 60(5), 15–26. [PubMed] [Google Scholar]
- Sigurdsson HH, Kirch J, & Lehr C-M (2013). Mucus as a barrier to lipophilic drugs. International Journal of Pharmaceutics, 453, 56–64. 10.1016/j.ijpharm.2013.05.040 [DOI] [PubMed] [Google Scholar]
- Smith PL (1997). Peptide delivery via the pulmonary route: A valid approach for local and systemic delivery. Journal of Controlled Release, 46, 99–106. Retrieved from https://ac.els-cdn.com/S0168365996015799/1-s2.0-S0168365996015799-main.pdf?_tid=325a8c72-2c5f-4857-ae3d-20f632f447c8&acdnat=1552343426_504c981f1fe879f3ff7c9be533eb8e69 [Google Scholar]
- Smola M, Vandamme T, & Sokolowski A (2008). Nanocarriers as pulmonary drug delivery systems to treat and to diagnose respiratory and non respiratory diseases. International Journal of Nanomedicine, 3, 1–19. [PMC free article] [PubMed] [Google Scholar]
- Soriano JB, Abajobir AA, Abate KH, Abera SF, Agrawal A, Ahmed MB, … Vos T (2017). Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: A systematic analysis for the global burden of disease study 2015. The Lancet Respiratory Medicine, 5(9), 691–706. 10.1016/S2213-2600(17)30293-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern D, & Cui H (2019). Crafting polymeric and peptidic hydrogels for improved wound healing. Advanced Healthcare Materials, 1900104, 1–17. 10.1002/adhm.201900104 [DOI] [PubMed] [Google Scholar]
- Suk JS, Lai SK, Wang Y-Y, Ensign LM, Zeitlin PL, Boyle MP, & Hanes J (2009). The penetration of fresh undiluted sputum expectorated by cystic fibrosis patients by non-adhesive polymer nanoparticles. Biomaterials, 30, 2591–2597. 10.1016/j.biomaterials.2008.12.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JC, Pulliam BL, & Edwards DA (2007). Nanoparticles for drug delivery to the lungs. Trends in Biotechnology, 25(12), 563–570. 10.1016/j.tibtech.2007.09.005 [DOI] [PubMed] [Google Scholar]
- Tang BC, Fu J, Watkins DN, & Hanes J (2009). Enhanced efficacy of local etoposide delivery by poly(ether-anhydride) particles against small cell lung cancer in vivo. Biomaterials, 31, 339–344. 10.1016/j.biomaterials.2009.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taratula O, Kuzmov A, Shah M, Garbuzenko OB, & Minko T (2013). Nanostructured lipid carriers as multifunctional nanomedicine platform for pulmonary co-delivery of anticancer drugs and siRNA. Journal of Controlled Release, 171, 349–357. 10.1016/j.jconrel.2013.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumura T, Koyama S, Tsujimoto M, Kitagawa M, & Kagamimori S (1993). Further study of nebulisation chemotherapy, a new chemotherapeutic method in the treatment of lung carcinomas: Fundamental and clinical. British Journal of Cancer, 68, 1146–1149. New York, NY: Macmillan Press Ltd. Retrieved from https://www.nature.com/articles/bjc1993495.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KMG, Taylor G, Kellaway IW, & Stevens J (1990). The stability of liposomes to nebulisation. International Journal of Pharmaceutics, 58, 57–61. Retrieved from https://ac.els-cdn.com/037851739090287E/1-s2.0-037851739090287E-main.pdf?_tid=2b3978e9-77b3-49cdb128-2090f882bafd&acdnat=1548792244_97e94afa9773aafa9182a8a3bf315536 [Google Scholar]
- Thorley AJ, Ruenraroengsak P, Potter TE, & Tetley TD (2014). Critical determinants of uptake and translocation of nanoparticles by the human pulmonary alveolar epithelium. ACS Nano, 8(11), 11778–11789. 10.1021/nn505399e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorley AJ, & Tetley TD (2013). New perspectives in nanomedicine. Pharmacology and Therapeutics, 140, 176–185. 10.1016/j.pharmthera.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Todoroff J, & Vanbever R (2011). Fate of nanomedicines in the lungs. Current Opinion in Colloid and Interface Science, 16(3), 246–254. 10.1016/j.cocis.2011.03.001 [DOI] [Google Scholar]
- Tomoda K, Ohkoshi T, Hirota K, Sonavane GS, Nakajima T, Terada H, … Makino K (2009). Preparation and properties of inhalable nanocomposite particles for treatment of lung cancer. Colloids and Surfaces B: Biointerfaces, 71, 177–182. 10.1016/j.colsurfb.2009.02.001 [DOI] [PubMed] [Google Scholar]
- Varghese OP, Sun W, Ns Hilborn J, & Ossipov DA (2009). In situ cross-linkable high molecular weight hyaluronan-bisphosphonate conjugate for localized delivery and cell-specific targeting: A hydrogel linked prodrug approach. JACS Communications, 131, 8781–8783. 10.1021/ja902857b [DOI] [PubMed] [Google Scholar]
- Vij N, Min T, Bodas M, Gorde A, & Roy I (2016). Neutrophil targeted nano-drug delivery system for chronic obstructive lung diseases. Nanomedicine: Nanotechnology, Biology, and Medicine, 12(8), 2415–2427. 10.1016/j.nano.2016.06.008 [DOI] [PubMed] [Google Scholar]
- Weber S, Zimmer A, & Pardeike J (2014). Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) for pulmonary application: A review of the state of the art. European Journal of Pharmaceutics and Biopharmaceutics, 86, 7–22. 10.1016/j.ejpb.2013.08.013 [DOI] [PubMed] [Google Scholar]
- World Health Organization. (2018). Global Tuberculosis Report, 2018, 1. [Google Scholar]
- Xu C, Tian H, Wang P, Wang Y, & Chen X (2016). The suppression of metastatic lung cancer by pulmonary administration of polymer nanoparticles for co-delivery of doxorubicin and Survivin siRNA. Biomaterials Science, 4, 1646 10.1039/c6bm00601a [DOI] [PubMed] [Google Scholar]
- Xu C, Wang P, Zhang J, Tian H, Park K, & Chen X (2015). Pulmonary codelivery of doxorubicin and siRNA by pH-sensitive nanoparticles for therapy of metastatic lung cancer. Small, 11(34), 4321–4333. 10.1002/smll.201501034 [DOI] [PubMed] [Google Scholar]
- Xu C, Wang Y, Guo Z, Chen J, Lin L, Wu J, … Chen X (2019). Pulmonary delivery by exploiting doxorubicin and cisplatin co-loaded nanoparticles for metastatic lung cancer therapy. Journal of Controlled Release, 295, 153–163. 10.1016/j.jconrel.2018.12.013 [DOI] [PubMed] [Google Scholar]
- Yacobi NR, Malmstadt N, Fazlollahi F, DeMaio L, Marchelletta R, Hamm-Alvarez SF, … Crandall ED (2010). Mechanisms of alveolar epithelial translocation of a defined population of nanoparticles. American Journal of Respiratory Cell and Molecular Biology, 42(5), 604–614. 10.1165/rcmb.2009-0138OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Peters JI, & Williams Iii RO (2008a). Inhaled nanoparticles—A current review. International Journal of Pharmaceutics, 356,239–247. 10.1016/j.ijpharm.2008.02.011 [DOI] [PubMed] [Google Scholar]
- Yeh HC, Phalen RF, & Raabe OG (1976). Factors influencing the deposition of inhaled particles. Environmental Health Perspectives, 15, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yhee J, Im J, & Nho R (2016). Advanced therapeutic strategies for chronic lung disease using nanoparticle-based drug delivery. Journal of Clinical Medicine, 5(9), 82 10.3390/jcm5090082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Liu C, Liu Y, Zhang N, & Xu W (2010). Mannan-modified solid lipid nanoparticles for targeted gene delivery to alveolar macrophages. Pharmaceutical Research, 27, 1584–1596. 10.1007/s11095-010-0149-z [DOI] [PubMed] [Google Scholar]
- Zahrt TC (2003). Molecular mechanisms regulating persistent Mycobacterium tuberculosis infection. Microbes and Infection, 5, 159–167.Retrieved from www.elsevier.com/locate/micinf [DOI] [PubMed] [Google Scholar]
- Zarogoulidis P, Chatzaki E, Porpodis K, Domvri K, Hohenforst-Schmidt W, Goldberg EP, … Zarogoulidis K (2012). Inhaled chemotherapy in lung cancer: Future concept of nanomedicine. International Journal of Nanomedicine, 7, 1551–1572. 10.2147/IJN.S29997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wu L, Chan H-K, & Watanabe W (2011). Formation, characterization, and fate of inhaled drug nanoparticles. Advanced Drug Delivery Reviews, 63, 441–455. 10.1016/j.addr.2010.11.002 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Kleinstreuer C, Kim CS, & Hickey AJ (2002). Aerosol transport and deposition in a triple bifurcation bronchial airway model with local tumors. Inhalation Toxicology, 14(11), 1111–1133. 10.1080/08958370290084809 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Tang P, Shui Yee Leung S, Gar Yan Chan J, & Chan H-K (2014). Emerging inhalation aerosol devices and strategies: Where are we headed? Advanced Drug Delivery Reviews, 75, 3–17. 10.1016/j.addr.2014.03.006 [DOI] [PubMed] [Google Scholar]