

Abstract
The prostate is the only organ in a man that continues to grow with age. John McNeal proposed, 40 years ago, that this BPH is characterized by an age-related reinitiation of benign neoplastic growth selectively in developmentally abortive distal ducts within the prostate transition–periurethral zone (TPZ), owing to a reawakening of inductive stroma selectively within these zones. An innovative variant of this hypothesis is that, owing to its location, the TPZ is continuously exposed to urinary components and/or autoantigens, which produces an inflammatory TPZ microenvironment that promotes recruitment of bone marrow-derived mesenchymal stem cells (MSCs) and generates a paracrine-inductive stroma that reinitiates benign neoplastic nodular growth. In support of this hypothesis, MSCs infiltrate human BPH tissue and have the ability to stimulate epithelial stem cell growth. These results provide a framework for defining both the aetiology of BPH in ageing men and insights into new therapeutic approaches.
Prostatic embryonic development, subsequent pubertal and adult growth, and homeostatic maintenance (for example, cell turnover) are tightly regulated via androgen-sensitive reciprocal paracrine interactions between stromal and epithelial cell compartments1. However, the prostate is unique in that it is the only glandular organ in the human body that continues to undergo net benign neoplastic growth as a man ages (FIG. 1a). This BPH is a progressive condition of ageing men that John McNeal characterized as a selective benign neoplastic overgrowth within the transition–periurethral zone (TPZ) proximal to the verumontanum (FIG. 1b), which can clinically result in bladder outlet obstruction (BOO) and lower urinary tract symptoms (LUTS)2–6. This overgrowth translates into the reality that ~25% of men will develop clinical symptoms of BPH during their lifetime7. Estimated direct costs for the medical management of LUTS exceed US$1 billion annually in the USA alone, and costs are anticipated to continue rising8. Current treatment for LUTS secondary to BPH consists of α-blockers or 5α-reductase inhibitors (5-ARIs) for mild to moderate symptoms and surgical intervention, such as transurethral resection of the prostate (TURP), for more symptomatic disease9. These treatments are not without shortcomings: oral agents often offer limited symptomatic relief and surgical treatments have a risk of causing sexual dysfunction or impotency9. On the basis of these shortcomings, many men choose to forgo invasive treatment when oral agents become ineffective10. Thus, millions of men suffer in silence with this disease10.
Fig. 1 |. Anatomical changes to the prostate with age.
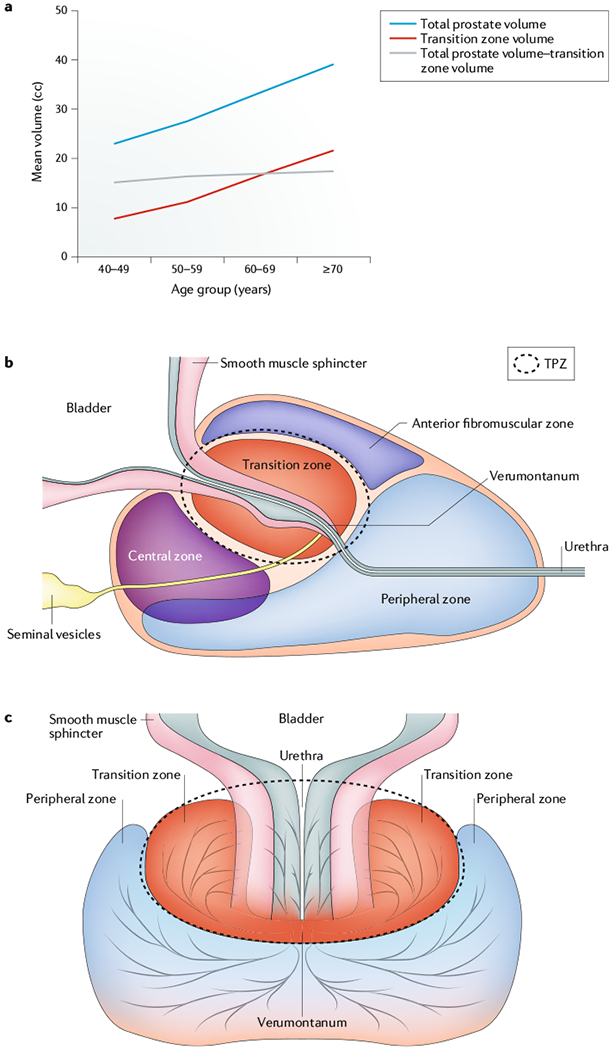
a | The volume of the prostate transition-periurethral zone (TPZ) increases with age and is the primary site of BPH. b | Zonal organization of the human prostate in a prone position. BPH characteristically originates in the TPZ (dashed circle) that envelops the urethra, which is a source of inflammatory stimuli. The TPZ is also adjacent to the periurethral smooth muscle sphincter, which suppresses the full proliferative potential but not the overall stem cell number in the distal ducts within the TPZ during ontogeny. Age-related increase in the volume of the transition zone drives the increase in the total prostate volume during ageing11,12. c | Zonal organization of the human prostate in the upright position.
LUTS have many causes, one of which is BOO caused by mechanical compression that is a result of prostatic enlargement9. In men with clinically symptomatic BPH, the prostate frequently increases from a normal size of ~20 g in a young adult (<30 years old) to sizes of ≥40 g over the next four decades (FIG. 1a). This benign neoplastic volume increase is not uniform throughout the gland and is selectively restricted to the TPZ, which increases at a modest rate of ~3.5% per year from an initial size of <2 g in a young adult (<30 years old) to ≥20 g by the sixth decade of life11,12 (FIG. 1a).
In 1978, John McNeal proposed that this selectivity in benign neoplastic expansion is related to abortive ductal development (the suppression of full epithelial stem cell hierarchical expansion) during initial adult ontogeny of the TPZ caused by its unique intimate relationship with the periurethral smooth muscle sphincter4. Subsequently, during ageing, a reawakening of the inductive prostate stroma occurs selectively within this zone that induces these abortive ducts to undergo renewed focal stem cell hierarchical expansion, producing BPH nodules1,4 (FIG. 1c). Since McNeal’s seminal observations and hypothesis nearly 40 years ago4, major increases have occurred in our understanding of how the prostate is functionally organized by androgen-sensitive reciprocal paracrine interactions between prostatic stromal and epithelial stem cell units, and how this functional organization is affected in an inflammatory tissue microenvironment. These new insights have led to an innovative variant of McNeal’s hypothesis, described in this Opinion article. Specifically, that, owing to its unique anatomical location, the TPZ is continuously exposed to urinary components and/or autoantigens, which produce an inflammatory microenvironment that induces recruitment of bone marrow-derived uncommitted stromal progenitors (mesenchymal stem cells (MSCs)) to this periurethral area. This environment results in a paracrine-inductive stroma that stimulates hierarchical benign neoplastic expansion of epithelial stem cells in the initially abortive ducts in the TPZ to produce BPH nodules1.
This Opinion focuses on the data supporting this new hypothesis and the implications for both prevention and treatment of LUTS, providing an overview of the different normal and abnormal growth phases of the ageing human prostate and how androgen-sensitive regulation of the reciprocal paracrine interactions between stromal and epithelial stem cell units affects these growth phases.
Androgen-dependent prostate growth
In humans, unlike most other animal species, development of the prostate occurs during two temporally distinct growth phases (FIG. 2). The first is initiated during embryogenesis, when the primitive prostate begins to develop from the urogenital sinus, an embryonic structure that gives rise to the urinary bladder, prostate, bulbourethral glands, and urethra in men (FIG. 2a). The urogenital sinus comprises a mesoderm-derived urogenital mesenchyme, which is responsible for dictating tissue-specific differentiation of an endoderm-derived urogenital epithelium13–15 (FIG. 2b). The urogenital sinus in human male and female fetuses is indistinguishable until ~10 weeks gestation, when the fetal testes in the male begin producing testosterone16. This testosterone is converted to dihydrotestosterone (DHT), a tenfold more potent androgen, by stromal cells that express 5α-reductase in the urogenital mesenchyme1 (FIG. 3). Over the next few weeks, binding of DHT to this androgen receptor (AR)-positive, 5α-reductase-expressing subset of stromal cells stimulates secretion of paracrine growth and survival factors, which are collectively known as andromedins (first proposed by Wallace McKeehan17), including insulin-like growth factor 1 (IGF1), fibroblast growth factors (FGFs), and vascular endothelial growth factor (VEGF), among others1,16–20. These stromal-derived andromedins diffuse throughout the immediate microenvironment, including the urogenital mesenchyme and epithelial compartments, where binding to their respective cognate receptors on specific cell types within the epithelium and stroma induces compartment-specific differentiation programmes1 (FIG. 3).
Fig. 2 |. Embryonic development of the human prostate.
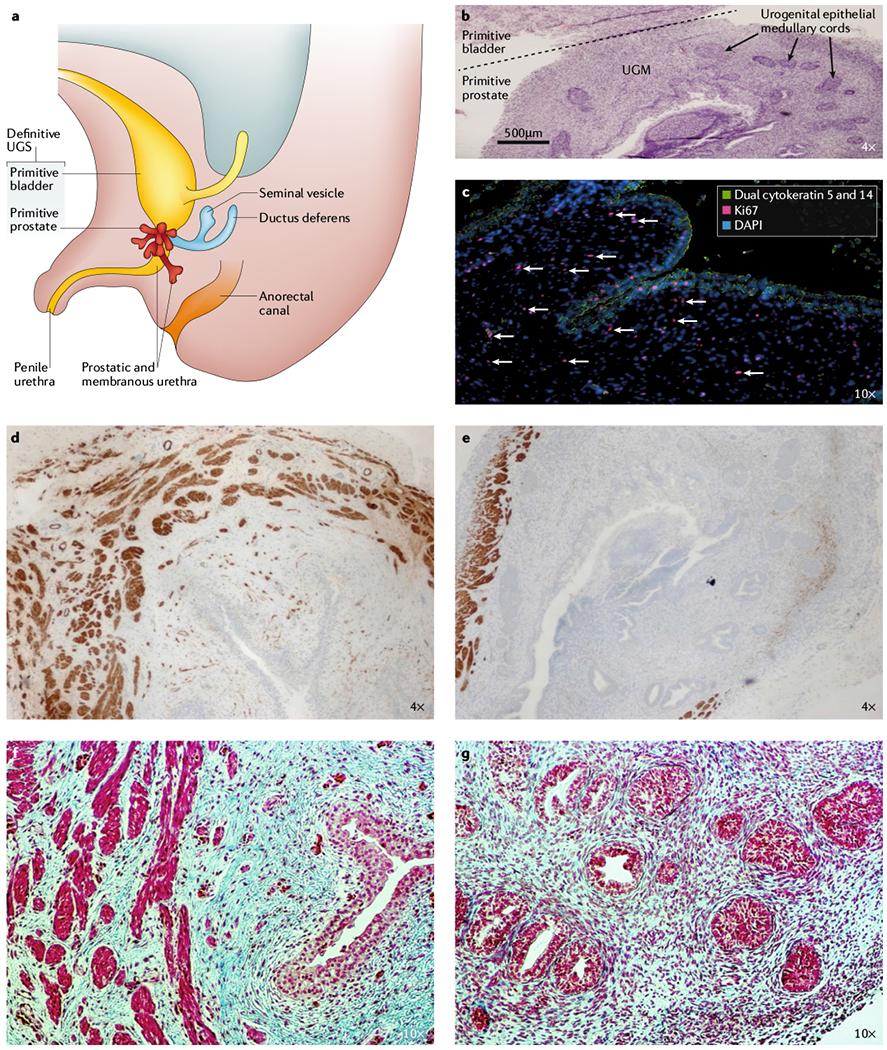
a | The prostate is derived from the embryonic urogenital sinus (UGS), depicted here at ~16 weeks of development. b | Haematoxylin and eosin staining of human UGS (16 weeks) depicting urogenital epithelial (UGE) medullary cords embedded in a densely cellular urogenital mesenchyme (UGM), 4 x magnification. c | UGE identified by positive immunofluorescent staining for basal cytokeratins (green) invading a Loosely connected, highly proliferative UGM (antigen Ki67-positive (red); white arrows). Nuclei are stained with 4’,6-diamidino-2-phenylindole (DAPI; blue), 10 x magnification. d | Myosin heavy chain, smooth muscle isoform (SMMHC; also known as MYH11) staining in the primitive bladder at 16 weeks, 4 × magnification. e | SMMHC staining in the primitive prostate at 16 weeks, 4 × magnification. f | Masson’s trichrome staining (MTC; smooth muscle (bright red), collagen (blue), and epithelium (red)) in primitive bladder at 16 weeks, 10 × magnification. g | MTC staining in primitive prostate at 16 weeks, 10 × magnification. Staining shows that the primitive bladder in the human UGS has a well-developed smooth muscle Layer compared with the primitive prostate at 16 weeks, which only has a small amount of smooth muscle localized to the periphery of the tissue.
Fig. 3 |. Reciprocal crosstalk between epithelium and stroma in the human prostate.
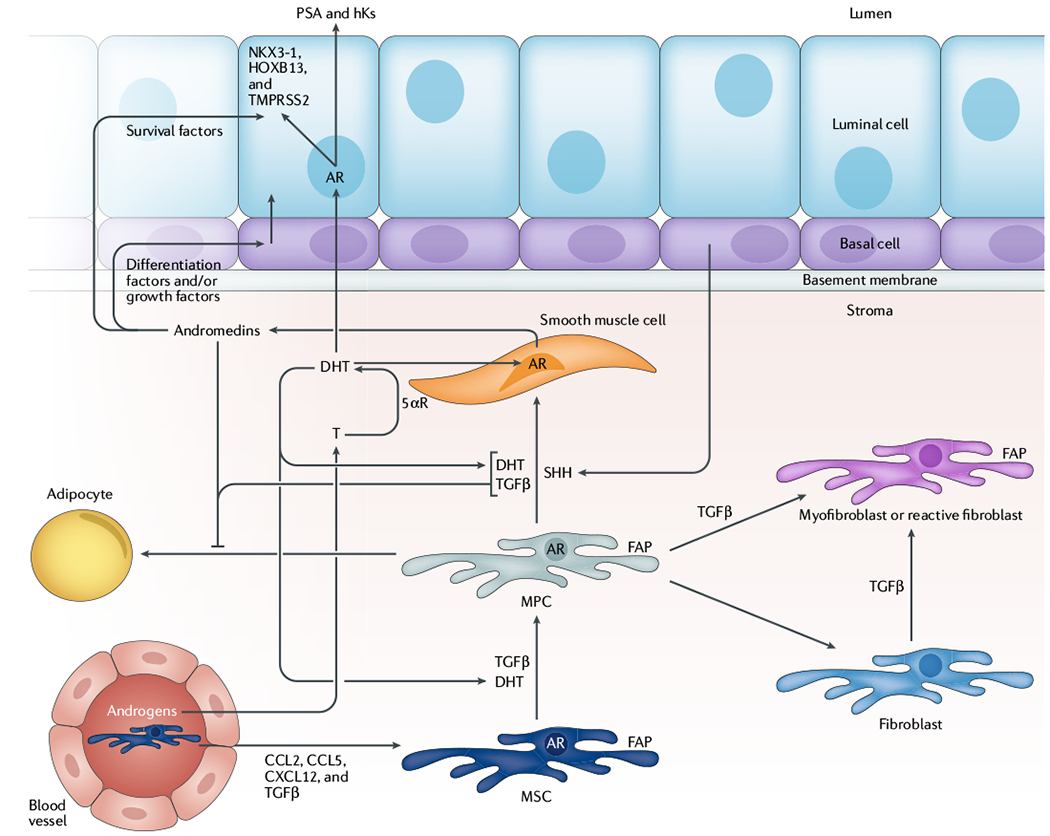
Androgens diffusing from the circulation through prostate tissue are converted from testosterone (T) to dihydrotestosterone (DHT), a tenfold more potent androgen, by 5α-reductase (5αR)-expressing, androgen receptor (AR)-positive smooth muscle cells in the stroma. DHT-dependent AR signalling drives expression of andromedins (such as insulin-like growth factor 1, fibroblast growth factor, and vascular endothelial growth factor) that promote differentiation of prostate epithelial basal cells into PSA-expressing secretory luminal cells, the survival of which is dependent on stromal-derived andromedins. Additionally, DHT, transforming growth factor-β (TGFβ), and paracrine factors from the epithelium, such as sonic hedgehog (SHH), promote differentiation of fibroblast activation protein (FAP)-expressing mesenchymal stem cells (MSCs) and mesenchymal progenitor cells (MPCs) into smooth muscle cells in a reciprocal fashion during normal ontogeny. A consequence of this myogenic differentiation is the suppression of the adipogenic differentiation potential via reciprocal feedback pathways regulating key transcription factors. In response to chemokine signals (such as CC-chemokine ligand 2 (CCL2), CCL5, CXC-chemokine ligand 12 (CXCL12), and TGFβ), additional FAP-positive MSCs can be mobilized from the bone marrow and recruited to sites of inflammation and tissue damage, which are commonly present in the transition-periurethral zone (TPZ) of the prostate as a result of uric acid, aberrant protease localization, and other inflammatory stimuli. Importantly, recruitment of these uncommitted MSCs to the TPZ can promote embryonic reawakening via disrupting and/or displacing periurethral smooth muscle, which induces benign neoplastic expansion of dormant epithelial stem cells within these developmentally abortive distal ducts to produce BPH nodules. hKs, hexokinases; HOXB13, homeobox protein Hox-B13; NKX3-1, homeobox protein Nkx-3.1; TMPRSS2, transmembrane protease serine 2.
In the stroma, these andromedins in combination with other paracrine factors produced by the epithelium (such as sonic hedgehog (SHH)) stimulate myogenesis and suppress adipogenesis in undifferentiated MSCs and mesenchymal progenitor cells (MPCs) present within the urogenital mesenchyme1,21–24 (FIG. 3). The bladder has a fairly well-developed smooth muscle layer by gestational week 16 (FIG. 2d,f), but smooth muscle is only present in the periphery of the primitive prostate at this developmental stage (FIG. 2e,g). Cunha and colleagues15 demonstrated that this discontinuous layer of smooth muscle enables the epithelium to respond to inductive andromedin signals from the undifferentiated mesenchyme (such as MSCs) to promote continued ductal growth and branching morphogenesis of the epithelium. Experimental evidence suggests that the differentiation and thickness of this smooth muscle layer in the stromal compartment is regulated by androgen and transforming growth factor-β (TGFβ) signalling14,15,25. Notably, the TGFβ promoter region contains an androgen response element (ARE), providing evidence for coordinated crosstalk between the two pathways to mediate smooth muscle differentiation of stromal progenitors in the prostate26–30.
In the epithelial compartment, stromal-derived andromedins induce the budding and invasion of solid medullary cords with no canalization from the initially AR-negative urogenital epithelium into the surrounding undifferentiated urogenital mesenchyme (FIG. 2b,c). These stromal-derived andromedins bind their cognate receptors on epithelial adult stem cells, which drives the maturation of a subset of these cells into a simple stratified glandular epithelium1. This stratified epithelium consists of a basal layer of cuboidal cells (expressing a low to undetectable level of AR), upon which sits a second layer of columnar secretory luminal cells that express a reproducibly detectable level of AR adjacent to a now patent lumen1,16,31,32. As organogenesis continues, an increasing subset of mesenchymal cells undergoes smooth muscle differentiation in response to epithelial-derived paracrine factors (such as SHH) and progressively envelops each prostatic acini in a sheath of smooth muscle, which acts as a barrier that limits andromedin diffusion into the epithelial compartment, inhibiting continuous epithelial growth and branching15,22–24. During this initial developmental phase, circulating testosterone levels are sufficient to enable these reciprocal paracrine interactions between the stromal and epithelial compartments that are required for embryonic and fetal prostate development1. However, by the end of the first year of life, levels of circulating testosterone concentrations in boys decrease to a level insufficient to promote the continued production of necessary amounts of andromedins for continued prostate growth, thereby stopping net prostate growth until puberty1. At puberty, circulating testosterone concentration again rises to a sufficient level to restimulate adequate secretion of stromal andromedins and reinduce prostate growth1. This second growth phase results in the prostate ultimately reaching its normal adult size of ~20 g by 20 years of age. By contrast, if a boy is castrated before the age of 10 years, which prevents the rise in circulating testosterone at puberty, this secondary growth phase is completely blocked1,33. Furthermore, if no exogenous androgen replacement is ever given to prepubertal castrated males, the prostate remains <5 g in size, and BPH does not develop in men with chronic androgen ablation as they age33.
In noncastrated men, once androgen-induced full adult maturation of the epithelium and stromal compartment of the prostate occurs by the second decade of life, net growth of prostate tissue, excluding the TPZ, essentially stops despite the continuous high levels of circulating testosterone and the resulting high andromedin tissue levels1 (FIG. 1a). During ageing, levels of circulating androgen need to continue to be adequate for prostatic levels of paracrine andromedin survival and growth factors to remain sufficient to suppress regression of the gland. These factors act by inhibiting activation of the apoptotic cell death pathway in prostatic epithelial cells while enabling sufficient proliferation to replace cell loss in both the stromal and epithelial compartments of all prostate zones other than the TPZ1. Thus, cellular turnover occurs in prostatic zones other than the TPZ, but as the rate of proliferation is balanced by the rate of cell loss, neither overgrowth nor regression transpires in these zones with ageing (FIG. 1a). By contrast, Schulze et al.34 demonstrated that the proliferation rate in BPH tissue is elevated in both the stromal and epithelial compartments relative to the other zones of the prostate (~40-fold and ~9-fold in the stroma and epithelium, respectively). The overall proliferative index (PI) in the two compartments is nearly equal (PI = 0.121% in the stroma and 0.142% in the epithelium), but this proliferation is balanced by the rate of apoptosis in the epithelium but not in the stroma35. The net result produces an increase in the area ratio of stroma to epithelium, from 3:1 in a nonhyperplastic TPZ in young men to 5:1 in BPH tissue36. These data show that age-related perturbation occurs in androgen-dependent homeostatic cell turnover within the benign neoplastic TPZ.
Regulation of stem cell organization
To determine how androgen-dependent homeostatic regulation is disrupted in the benign neoplastic TPZ, an understanding of stem cell organization within the adult prostate is required. The normal prostate can undergo successive cycles of androgen deprivation and replacement without diminishing its ability for continued regeneration, as was demonstrated <30 years ago7. Since then, a number of independent groups have clarified how the prostate of a man is organized into stromal and epithelial adult stem cell units (FIG. 4) and how androgen-sensitive reciprocal paracrine interactions enable this profound cyclic regenerative growth capacity7,37–43. In the stromal compartment, adult MSCs respond to paracrine factors (such as SHH) that are secreted by prostate basal epithelial cells. These paracrine factors promote MSC self-renewal and generation of stromal progeny cells, which have a limited proliferative ability before differentiating into a variety of proliferatively quiescent stromal cell types (for example, smooth muscle cells, adipocytes, fibroblasts, and pericytes)22,24,38. Studies using human fetal prostate stromal cells recombined with human adult prostate epithelial cells in mice have shown that, when adequate levels of androgen are present to efficiently bind to and activate AR transcriptional signalling, these AR-expressing stromal cells secrete andromedins44. These secreted andromedins then diffuse throughout the immediate microenvironment until binding to their cognate receptors on specific cell types within the epithelial and stromal compartments, inducing compartment-specific differentiation programmes1,37 (FIG. 3).
Fig. 4 |. Reciprocal hierarchical expansion of human epithelial and stromal adult stem cells in the prostate.
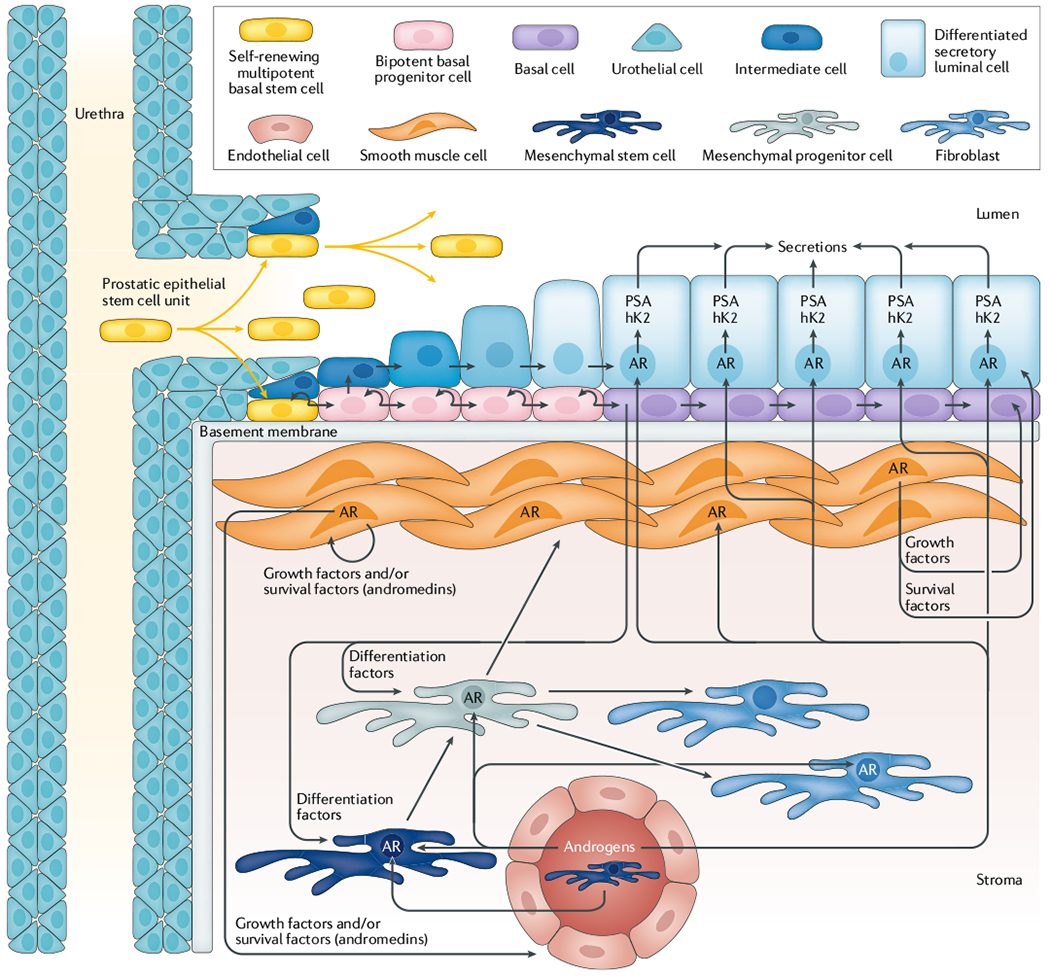
Prostate epithelial stem cells located in niches in the basal layer of the proximal ducts are induced to undergo hierarchical expansion in response to androgen receptor (AR)-dependent stromal-derived andromedins. This hierarchical expansion produces a stratified epithelium consisting of a continuous layer of ΔNp63-positive, basal cytokeratin-positive, and AR-negative basal cells under a layer of ΔNp63-negative, luminal cytokeratin-positive, AR-positive, PSA-expressing secretory luminal cells. hK2, hexokinase 2. Figure adapted with permission from REF.1, Wiley-VCH and REF43, Cell Press, CC BY 4.0.
The human prostate epithelial compartment is a tubuloalveolar structure containing a simple stratified epithelium composed of a continuous basal layer underneath a luminal layer. Functionally, this epithelial compartment is organized into epithelial adult stem cell units1. These prostate epithelial adult stem cells are located in niches in the basal layer in the proximal epithelial ducts1,41,43 (FIG. 4) and are androgen independent, as has been shown in tissue recombination studies. Cunha and Lung37 reported that epithelial morphogenesis and growth occurs even when AR is not expressed by any prostate epithelial cells (including adult stem cells) as long as ligand-dependent AR paracrine signalling occurs in the supporting stromal cells37. The mechanism for this epithelial growth in the absence of epithelial AR expression is related to the hierarchical expansion and maturation of AR-negative epithelial adult stem cells and their progeny (FIG. 4). Wilson et al.41,43 documented that these androgen-insensitive epithelial stem cells are located in niches positioned in the basal epithelial layer at the opening of the proximal ducts as they enter the urethra, which control their survival and self-renewal. AR-negative epithelial adult stem cells within these proximal duct basal niches undergo self-renewal division in which one daughter remains in the niche as a stem cell and the other daughter cell migrates out of the niche and undergoes differentiation1,43. This migrating daughter cell occasionally differentiates into a non-proliferating, AR-negative neuroendocrine cell, but more frequently it differentiates into a ΔNp63-positive, basal cytokeratin (cytokeratin 5 (CK5); also known as KRT5)-positive, and AR-negative proliferating progenitor cell.
Using in situ lineage tracing in human prostate tissues, Heer et al.43 document that these basal progenitor cells undergo a limited number of amplifying proliferations while they migrate in streams in the basal layer along the proximal-distal ductal axis (FIG. 4). This basal progenitor proliferation requires the androgen-regulated production and secretion of diffusible stromal-derived paracrine andromedins7.
These secreted paracrine andromedins diffuse from the stroma into the epithelial compartment, where they bind to cognate receptors and stimulate progenitor cell proliferation and subsequent maturation of a subset of these cells into AR-negative, ΔNp63-negative, CK5-positive, and luminal cytokeratin (CK18)-positive intermediate basal cells initially documented by Schalken and colleagues40. These intermediate basal cells begin to express increasing levels of AR as they migrate from the basal layer to the luminal layer to become luminal intermediate cells during their movement to the more distal ducts. Ligand-dependent AR binding to the AREs in the enhancer and promoter regions of AR target genes (such as PSA (also known as KLK3)) induces differentiation of these luminal intermediate cells into ΔNp63-negative, CK18-positive, AR-positive, PSA-expressing secretory luminal cells39,40,42. Coupled with this differentiation, androgen-dependent, andromedin-driven growth stimulation is counterbalanced by androgen-induced AR binding to nuclear β-catenin-transcription factor 4 (TCF4) complexes that inhibit MYC transcription in these secretory luminal cells45,46. This MYC downregulation results in terminal G0 growth arrest of the fully differentiated secretory luminal cells, thereby preventing continuous prostatic epithelial hyperplasia despite chronic high andromedin levels45,46.
Unfortunately, this normal androgen-sensitive homeostatic reciprocal paracrine balance between stromal and epithelial stem cells is selectively disrupted in the TPZ in the vast majority of men, starting sometime during the third or fourth decade of life1,4. Once initiated, this imbalance results in continuing benign neoplastic growth of the TPZ, eventually resulting in BOO and clinical LUTS7. Androgen deprivation therapy (ADT) is commonly misconceived to target only the prostate epithelial compartment47. The epithelial response induced by androgen deprivation is initiated by the decrease in production of andromedins by prostate stromal cells1. In addition, androgen deprivation results in loss of VEGF production and secretion by both prostate stromal smooth muscle cells and secretory luminal epithelial cells, which results in a decrease in prostate blood vessel density and blood flow18,48. This decrease in blood flow means that ADT for BPH actually targets the stromal compartment to disrupt reciprocal stromal–epithelial paracrine interactions, thereby restricting the hierarchical expansion of both stromal and epithelial stem cells1,47. Although the development of clinical LUTS occurs over several decades, Peters and Walsh47 demonstrated that it only takes 6 months of exposure to a castration level of circulating testosterone (induced by a luteinizing hormone-releasing hormone (LHRH; also known as GNRH) analogue) to occur. This low testosterone level leads to a decrease in TPZ volume by ~40%, which involves a reduction in both the stromal and epithelial compartments of the TPZ and relief of BOO symptoms and LUTS in patients47. Notably, this study also demonstrated that it takes only 2 months after cessation of LHRH-therapy-induced androgen deprivation for full restoration of the abnormal overgrowth in both the stroma and epithelium in the TPZ and the return of BOO and LUTS47. These clinical results show that androgen ablation does not reduce the number of stem cell units within nodular BPH tissue; it only restricts their hierarchical proliferative expansion. This observation strongly implies that BPH growth is driven by an age-related disturbance involving either an increase in total adult stem cell number and/or an increase in the proliferative expansion of adult stem cell-derived progenitor cells in the TPZ1.
Embryonic stromal reawakening theory and nodular BPH.
Experimental studies by Cunha et al.13,15,38 showed that, where present, sheets of smooth muscle cells within the prostate stromal compartment shield the prostatic epithelium from andromedins secreted by the stromal cells, restricting epithelial proliferation. This observation provides an explanation for the finding by McNeal2–6 that the TPZ undergoes abortive distal ductal development during its ontogeny owing to its unique intimate relationship with the periurethral smooth muscle sphincter that extends downward from the bladder neck (FiG. 1b,c). The location of prostatic epithelial adult stem cell niches outside but adjacent to this periurethral smooth muscle sphincter, in the basal layer at the junction between proximal ducts and the urethra, provides strong support for the embryonic stromal reawakening theory as a major driver of nodular BPH development with age. The periurethral smooth muscle sphincter restricts full proliferative expansion of the epithelial progenitors in the TPZ, causing abortive distal ductal development during normal ontogeny, but it does not decrease the overall number of adult stem cells at the junction located between the proximal ducts and urethra within the TPZ15. These correlative clinical observations are consistent with the experimental results showing that ductal morphogenesis and expansive proliferative growth of the stroma and epithelium occur when human prostate epithelial adult stem cells are combined with an appropriately inductive mesenchyme44. These results support the theory that ectopic acquisition of adult mesenchymal cells within the TPZ to produce new stroma and disrupt and/or displace the periurethral smooth muscle sphincter results in the production of characteristic BPH nodules. Once a nodule is produced, its expansion further disrupts and/or displaces the periurethral smooth muscle sphincter, stimulating additional nodular development, and providing an explanation for the fact that BPH tissue is characteristically composed of multiple nodules of varying size. In addition, this theory suggests that the ectopic acquisition of an embryonic stromal reawakening phenotype by mesenchymal cells in the TPZ is not necessarily continuous with ageing, but might occur episodically with repeated cycles of MSC recruitment and stromal displacement.
Inflammation in BPH.
The crucial question of what induces this episodic reawakening of an inductive stroma within the TPZ of ageing men is currently unanswered. A strong candidate for this age-related inducer is the inflammatory microenvironment in the TPZ. Inflammatory effector molecules (including IFNγ, tumour necrosis factor (TNF), IL-1β, and IL-6) can activate important developmental signalling networks such as the SHH, IGF, Wnt, and Notch pathways via nuclear factor-κB and other downstream mediators of these effector molecules49–57. The reported association between inflammation and BPH was first proposed as a causative factor in 1937 by Moore33. Infiltration of immune cells is commonly observed in tissue from open prostatectomy samples from men undergoing surgery for symptomatic BPH (FIG. 5a,b). Histological inflammation is present in the majority of BPH specimens, with one study showing inflammation in >98% of samples58–61. Marberger and colleagues59 showed a nearly 28-fold increase in the inflammatory infiltrate present in BPH compared with normal prostate tissue. This infiltrate mostly consists of T cells, but B cells, macrophages, mast cells, and other cells are also present59, providing robust evidence for the widespread recruitment of immune cells to BPH tissue from systemic sources. Elevated levels of IL-4 and IL-13 are associated with this inflammatory infiltrate62. Furthermore, the infiltrating T cell population is dominated by memory and suppressor phenotypes59, consistent with repeated antigenic exposure and exhaustion during chronic inflammation over extended periods of time.
Fig. 5 |. Mesenchymal stem cells infiltrate BPH nodules along with other immune cells.
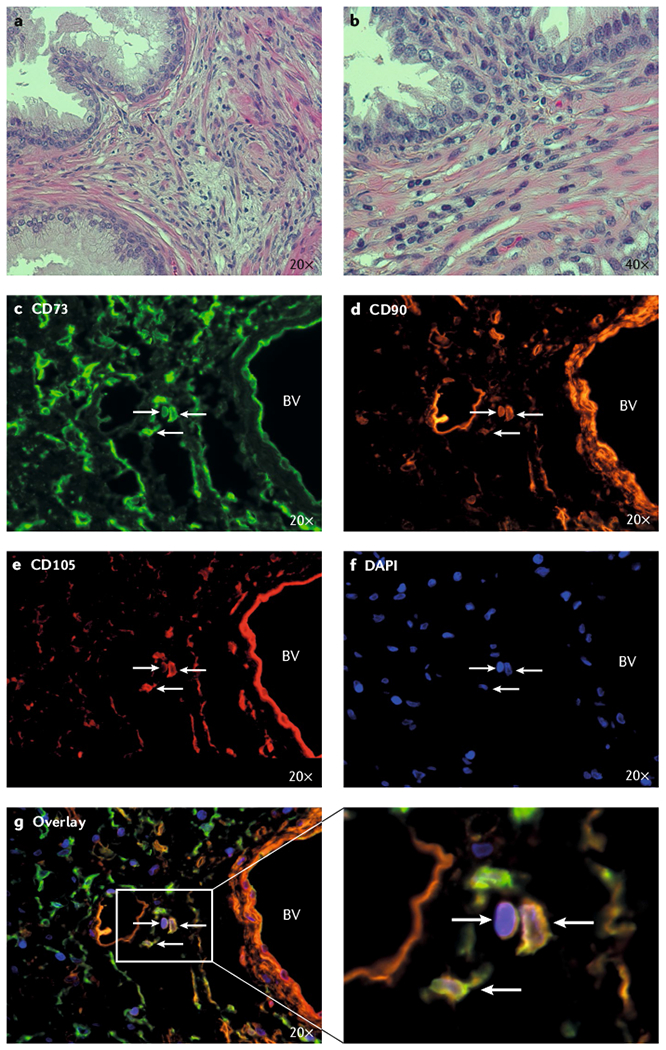
Infiltrating cells identified by their non-elongated nuclei (characteristic of smooth muscle cells and fibroblasts) are frequently present in the transition zone stroma from men undergoing open prostatectomy for symptomatic BPH. a | Haematoxylin and eosin staining of human BPH tissue from open prostatectomy tissue, 20 × magnification. b | Haematoxylin and eosin staining of human BPH tissue from open prostatectomy tissue, 40 × magnification. c | Immunofluorescent staining for CD73. d | Immunofluorescent staining for CD90. e | Immunofluorescent staining for CD105. f | Nuclei stained with 4’,6-diamidino-2-phenylindole (DAPI). g | Identification of mesenchymal stem cells (MSCs) in human BPH tissue from an open prostatectomy using a triple-label immunofluorescence assay based on positive staining for CD73, CD90, and CD105. BV, blood vessel.
A relationship between inflammation and increased prostate volume has been reported in multiple clinical studies63. The presence of inflammation is associated with increased prostate volumes and risk of acute urinary retention59,64. This relationship was even more pronounced when stratified by severity assessed by histological markers of inflammation61. In men with low-grade inflammation, mean prostate volume was 62 ml and men with low-grade inflammation had an average International Prostate Symptom Score (IPSS) of 12 compared with men with high-grade inflammation, who had a mean prostate volume of 77 ml and an average IPSS score of 21 (REF.61). These observations provide strong evidence that immune cells are actively recruited to the prostate as a function of BPH pathogenesis, and their presence is associated with increased prostate volume and clinical symptoms.
Candidate age-related inducers
Induction of a stromal inflammatory microenvironment in the TPZ is probably multifactorial, and several candidates could be involved in this induction. For example, exogenous agents such as microorganisms are capable of producing prostatitis65. BPH is associated with metabolic aberrations including metabolic syndrome, obesity, dyslipidaemia, and diabetes66. These aberrations lead to local inflammation and elevated systemic levels of adipokines and pro-inflammatory cytokines, such as adiponectin, leptin, TNF, IL-6, and CC-chemokine ligand 2 (CCL2)67. Another endogenous physiological candidate is intraprostatic urinary reflux into prostatic ducts, which occurs during micturation and has been suggested to initiate an inflammatory response in the prostatic stroma owing to enhanced tissue exposure to urinary urate68,69. This possibility is consistent with the fact that the proximal ducts in the TPZ are the first to be exposed to urine in the urethra68. As humans are upright bipeds, the TPZ in men experiences the greatest urinary pressure during the downstream flow from the bladder, which maximizes intraprostatic urinary reflux70.
In addition, PSA and kallikrein 2 (KLK2) are endogenous candidate inducers of inflammation because they are characteristically leaked into the stroma of BPH nodules owing to alterations in cellular junctions and/or increased epithelial barrier permeability, rather than being secreted restrictively into the acinar lumen of the epithelial compartment71,72. Both of these proteins have proteolytic enzymatic activity (chymotrypsin-like activity for PSA and trypsin-like activity for KLK2), and once in the TPZ stroma, they can proteolyse soluble and matrix proteins to liberate and/or activate peptide growth factors (such as FGFs and TGFβs), in addition to producing inappropriate peptide fragments or neoantigens, which could drive the generation of an immune response. Generation of neoantigens has been shown by Mikolajczyk and colleagues73, who identified a specific molecular form of PSA that is clipped between Lys182 and Ser183 and termed BPSA. This form of PSA was increased specifically within the prostatic TPZ of patients exhibiting nodular BPH73.
Infiltrating MSCs as drivers of embryonic reawakening.
Since the inception of the embryonic reawakening theory, the working model has assumed that the endogenous TPZ mesenchyme is inappropriately reawakened in a man through an unknown process that drives activation of developmental pathways, potentially via dedifferentiation4. On the basis of an improved understanding of how inflammatory tissue damage is repaired, we propose an alternative hypothesis in which the recruitment of stromal progenitors (such as MSCs) from the bone marrow in response to inflammation is the initiating factor for symptomatic BPH. MSCs are highly plastic, multipotent cells residing in the bone marrow that have the ability to differentiate into cells of mesodermal lineage, including osteoblasts, adipocytes, chondrocytes, and smooth muscle cells, among others74. In addition to this functional multipotency, MSCs are defined analytically by the co-expression of CD73, CD90, and CD105, but they do not express markers of the haematopoietic lineage such as CD14, CD20, CD34, CD45, and HLA-DR74. MSCs have been identified in tissues throughout the body75–77, including the prostate21,78–82; however, distinct differences exist in differentiation potential and expression profiles between those isolated from different sources21,83–92. These differences suggest that microenvironment-induced epigenetic reprogramming occurs and promotes tissue-specific functions and further suggest a differentiation hierarchy consisting of true multipotent stem cells and lineage-committed progenitors.
Canonical MSCs reside in the bone marrow under homeostatic conditions, but are mobilized to sites of inflammation in response to chemokine gradients owing to the expression of multiple cytokine and chemokine receptors on their cell surface, including CC-chemokine receptor 2 (CCR2), CCR5, CXC-chemokine receptor 4 (CXCR4), and TGFβ receptor (TGFβR)93–98. For example, granulocyte colony-stimulating factor induces proliferation of MSCs within the bone marrow and promotes their mobilization into the peripheral blood99. Notably, the ligands for these cytokine receptors are highly overexpressed in BPH tissue49,96,100,101. This high expression would explain why bone marrow-derived MSCs have been shown to incorporate into the bladder and prostate stroma in chimeric mouse models of tissue injury, including collagen-induced BOO 97,98— a phenomenon that has been independently supported in a rat model of transurethral lipopolysaccharide-induced prostate injury102. MSCs have also been shown to infiltrate into rat prostates containing sites of turpentine-induced acute inflammation103. Infiltrating MSCs have also been shown to contribute to the periglandular smooth muscle layer of the developing prostate21 using a tissue recombination model in which human urogenital sinus was implanted under the renal capsule of nude rats104. In addition, aberrant TGFβ signalling has been shown to recruit nestin-positive MSCs from the bone marrow to the prostate stroma, where they gave rise to fibroblasts and smooth muscle cells, which resulted in stromal hyperplasia that was blocked via depletion of TGFβR2 in the nestin-positive cells96.
Once at sites of active inflammation, recruited MSCs contribute to tissue repair by replenishing local reservoirs of stromal progenitors and stimulating angiogenesis82,94,105–107. Additionally, MSCs limit immune-driven tissue fibrosis via a broad array of immunomodulatory properties that are thought to serve as a rheostat for regulating the overall immune response94,105–107. This observation suggests that bone marrow-derived MSCs are recruited as an adaptive response to episodic cycles of intraprostatic inflammation produced by urinary reflux and other inflammatory inducers over a man’s lifetime21.
Identification of MSCs in human BPH.
A key prediction of the proposed model is that MSCs are actually present in human BPH tissue. A subset of prostate stromal cells derived from BPH cultures was previously found by Lin et al.78 to have strong proliferative potential, express MSC markers, and differentiate along the myogenic, adipogenic, and osteogenic lineages. Additionally, MSCs have been observed in primary tissue samples from men undergoing open prostatectomy for symptomatic BPH, using both multiparameter flow cytometry21 and a triple-label immunofluorescence assay based on positive co-staining for CD73, CD90, and CD105 (FIG. 5c–g). The number of MSCs identified is fairly low in most cases (mean, 0.5% of total cells; median, 0.1%)21, but this finding is not surprising for two reasons. First, BPH is a slow-growing, progressive disease that begins many years, if not decades, before it becomes clinically symptomatic7,108. In fact, pathological BPH has been identified in men as young as 25 years of age in autopsy studies7,109,110. Second, MSCs undergo fibroblast and smooth muscle differentiation in response to paracrine signals in the prostate microenvironment21,79–81,111–119 (FIG. 3). Thus, accumulation of a large number of uncommitted MSCs late in the clinical course of BPH at the time of open prostatectomy is not anticipated in most instances, whereas an expansion of their differentiated progeny is predicted21.
Consistent with the alternative hypothesis of BPH initiation and progression, we have documented the selective expansion of MSCs in primary cultures of prostate stromal cells grown under standard tissue culture conditions81. Furthermore, consistent with a stem or progenitor cell population, these cultures have considerable clonogenic and proliferative potential, undergoing ≥30 population doublings before senescence over a 2–3 month period, with population doubling times as fast as 30–35 h in the early phases of growth81,90,120. Importantly, the selective expansion of MSCs is entirely consistent with in vivo phenotypic observations upon which various dedifferentiation and embryonic reawakening models of BPH have been based. Specifically, the downregulation of smooth muscle myosin, calponin, and other differentiation-specific markers in BPH tissue121,122 can be explained by the infiltration of uncommitted stromal progenitors or MSCs rather than the loss of these markers on endogenous prostate stromal cells. Rowley and colleagues122–124 reported the accumulation of myofibroblasts in the reactive stroma of BPH and prostate cancer. Notably, a growing body of literature suggests reactive fibroblasts are derived from MSCs79–81,96,113,114,125,126, suggesting that the two models are complementary rather than mutually exclusive.
Implications for therapy
At present, α1A-adrenergic receptor antagonists (α-blockers) with or without 5-ARIs are standard-of-care treatments for the medical management of clinical LUTS owing to BPH. This management strategy is rationally based on the ability of the α-blockers to relax prostatic smooth muscle and the ability of 5-ARIs to prevent formation of the more potent androgen DHT from testosterone, therefore, decreasing andromedin production9. This reduced andromedin production decreases prostate volume, BOO, and the associated LUTS9. Unfortunately, these therapies must be chronically maintained and often lose their clinical efficacy with time10, raising the question of whether more effective and permanent relief of symptoms not requiring chronic therapy is possible on the basis of this new hypothesis for the role of infiltrating MSCs in driving the progressive development of nodular BPH. The rationale for a new approach is provided by two seminally important studies47,127. The first is the clinical study by Peters and Walsh47 that showed that 1–2 months of reversible LHRH-induced full testicular androgen deprivation disrupts the reciprocal interactions between the prostate stroma and epithelium, decreasing the weight of both compartments and relieving BOO and/or LUTS. Furthermore, this study showed that once BPH develops, androgen deprivation does not decrease the number of stem cell units in either the stromal or epithelial compartments, as once LHRH treatment is stopped, the subsequent androgen restoration fully restores the enlarged gland to its pretreatment size within 2–4 months47. The second is a preclinical study by Coffey et al.127 that demonstrated that androgen deprivation disrupts the stromal–epithelial interactions within prostate stem cell niches, which renders the initially radiation-resistant prostate stem cells acutely sensitive to radiation-induced killing127. More specifically, subsequent focal radiation of the prostate produces a permanent inability of exogenous androgen restoration to induce full regrowth of the gland regardless of time or androgen dose127. Clinical precedents also support this observation in which the disruption of stromal–epithelial interactions induced by hormonal deficiency in humans enables subsequent focal radiotherapy to permanently prevent hormone-induced glandular growth127–131. For example, similar stromal-epithelial interactions are involved in the mechanism of action of oestrogens in the breast128. The mammary glands in the breast tissue of adult men are atrophic unless estrogen is administered, as demonstrated in men with metastatic prostate cancer who develop gynaecomastia after estrogen treatment; however, if these men are given a single dose of focal breast irradiation (as low as 9 Gy) before initiation of estrogen therapy, gynaecomastia is prevented for ≥4 years after radiotherapy129. Notably, if the radiotherapy is delayed for even 1–4 months after the start of estrogen treatment, this preventive effect on gynaecomastia is significantly abrogated (P < 0.05)130, suggesting a short therapeutic window to capitalize on this hormone-induced radiation sensitivity.
With regard to focal prostate radiotherapy, patients with BPH-induced BOO have been treated with transurethral brachytherapy to deliver 16 Gy with no acute toxic effects while producing BOO relief with no recurrence of symptoms within 12–18 months131. Similarly, 3D conformal stereotactic radiotherapy at a dose of 14 Gy to spontaneous BPH in canines significantly decreased prostate volume, the epithelial cell PI, and smooth muscle content (P < 0.05) in the long term (years, not months) with minimal adverse effects132. Combining these clinical and preclinical results raises the possibility that more effective definitive therapy for symptomatic BPH might be possible by combining acute short-term LHRH analogue treatment with reversible androgen deprivation for a limited (1–2 months) period to disrupt stromal-epithelial interactions in prostate stem cell niches. This regimen could enhance the stem cell killing induced by subsequent local treatment with either a single dose of conformal external beam radiotherapy to the prostate or local injection of a selective cytotoxin into the TPZ before allowing the man to recover his normal serum testosterone.
Topsalysin (also known as PRX302) is a candidate for this latter cytotoxic approach. Topsalysin is a genetically engineered recombinant form of the bacterial protein proaerolysin, which has been modified to replace its native furin activation site with a sequence that is highly specific for enzymatically active PSA133. Importantly, PSA is enzymatically active only in the extracellular fluid of prostate tissue, particularly in the TPZ, and once it enters the bloodstream, its enzymatic activity is totally inhibited by ubiquitous serum protease inhibitors (such as α2-macroglobulin and α1-antichymotrypsin)133. Thus, topsalysin remains inactive and, therefore, noncytotoxic outside of prostate tissue133. Additionally, topsalysin is not immediately activated by PSA when injected into BPH tissue, unlike other focal tissue-ablative agents (such as ethanol and cryosurgery), which are immediately destructive and, therefore, limited in their ablative effect to only a small radius around the injection site134. Thus, topsalysin can diffuse throughout the extracellular fluid beyond the immediate injection site to reach a larger portion of the injected nodular tissue than other focal tissue-ablative agents, before it is eventually proteolysed by PSA. The proteolysed topsalysin monomers can, therefore, spontaneously oligomerize and form a stable transmembrane heptameric pore, which leads to cell death via disruption of membrane integrity133. On this basis, topsalysin has entered clinical development as a highly targeted, localized therapy for symptomatic BPH134–136. This therapy is administered in the outpatient setting as a single, brief intraprostatic injection into BPH nodules where PSA-dependent activation of topsalysin eventually ablates tissue over several days and alleviates LUTS134–136. The favourable safety profile and efficacy of topsalysin in previous phase I and II, open-label clinical trials led to a randomized placebo-controlled evaluation of intraprostatic injection of topsalysin as a short, outpatient-based procedure in men with moderate-to-severe LUTS secondary to BPH135,136. The results of the latter phase III study showed that the procedure was well tolerated in patients with LUTS owing to BOO and that topsalysin also produces clinically meaningful and statistically significant improvement in patient subjective (IPSS) and quantitative objective (peak urine flow) measures that are sustained for ≥12 months (P < 0.05)134. The adverse effect profile of topsalysin is favourable and mostly attributed to the injection itself rather than drug-related toxic effects135,136. These encouraging clinical results suggest that combining acute local therapy using a cytotoxic agent (such as focal topsalysin injections) with reversible androgen deprivation for a limited period of time (1–2 months) to disrupt stromal–epithelial interactions in prostate stem cell niches is very reasonable. This strategy is designed to sensitize these normally quiescent stem cells to subsequent local therapy and enhance killing in order to produce permanent suppression of nodular regrowth and, therefore, permanent relief of LUTS.
A possible limitation to this PSA-based approach is that PSA production is androgen sensitive133 and administering a focal topsalysin injection subsequent to short-term (1 month), reversible, LHRH-induced androgen ablation could limit activation of topsalysin. Alternatively, a decrease in the level of PSA in the extracellular fluid in BPH nodular tissue might actually improve tissue distribution of topsalysin before being activated by the subsequent restoration of enzymatically active PSA in the extracellular fluid, which occurs rapidly once LHRH treatment is stopped47.
If, in fact, short-term, reversible, LHRH-induced androgen ablation does limit activation of topsalysin, an additional strategy would be to directly target the stromal cells that are responsible for andromedin production or their progenitors (MSCs and MPCs) and/or the molecular pathways operating in these cells that are required for benign neoplastic epithelial stem cell expansion. One example of this approach is the use of the unique enzymatic activity of fibroblast activation protein (FAP) to activate a prodrug or protoxin. FAP is selectively expressed by MSCs and reactive fibroblasts in areas of tissue repair and/or remodelling and is not androgen sensitive137–139. This androgen insensitivity is important as androgen deprivation is known to increase the infiltration of bone marrow-derived MSCs into the prostate98. FAP is a serine protease in the dipeptidyl peptidase family with unique post-prolyl endopeptidase activity that has been exploited to develop FAP-activated thapsigargin-based prodrugs and melittin-based protoxins that are only active in the presence of enzymatically active FAP140–142, similar to topsalysin. Thus, local therapy with a FAP-activated prodrug or protoxin could be used to further disrupt stromal-epithelial interactions regulating the stem cell niche and combined with short-term androgen deprivation for potentially permanent LUTS relief.
Conclusions
In summary, we propose a new hypothesis for the nearly universal development of BPH in ageing men in which episodic periods of prostatic inflammation lead to the recruitment of MSCs to the TPZ via chemokine-induced expansion and mobilization from the bone marrow to promote tissue repair and replenish pools of local progenitors. Unfortunately, these recruited MSCs disrupt and/or displace the periurethral smooth muscle sphincter in the TPZ, which enables hierarchical benign neoplastic expansion of epithelial stem cells within the developmentally abortive distal ducts to produce BPH nodules. Episodic influx of MSCs over many years of repeated tissue injury occurs, resulting from inflammatory insults combined with aberrant localization of proteases and excessive liberation and/or activation of growth and pro-inflammatory factors within the TPZ stroma. This process generates a feedforward mechanism that stimulates recruitment of additional MSCs and an inductive stroma that promotes benign neoplastic expansion of epithelial adult stem cells via paracrine andromedin production and the development of additional BPH nodules (FIG. 6). This hypothesis is an innovative alternative mechanistic explanation for the embryonic stromal reawakening theory of BPH proposed by John McNeal 40 years ago. Importantly, this hypothesis is consistent with existing observational clinical data, including the loss of smooth-muscle-specific differentiation markers and the accumulation of reactive fibroblasts, which have well-documented roles in tissue repair and accumulating evidence suggests are themselves derived from MSCs. Importantly, this model suggests novel interventional strategies for the treatment of LUTS secondary to BPH-induced BOO.
Fig. 6 |. Heuristic model of mesenchymal stem cell-dependent episodic inductive reawakening in BPH pathogenesis.
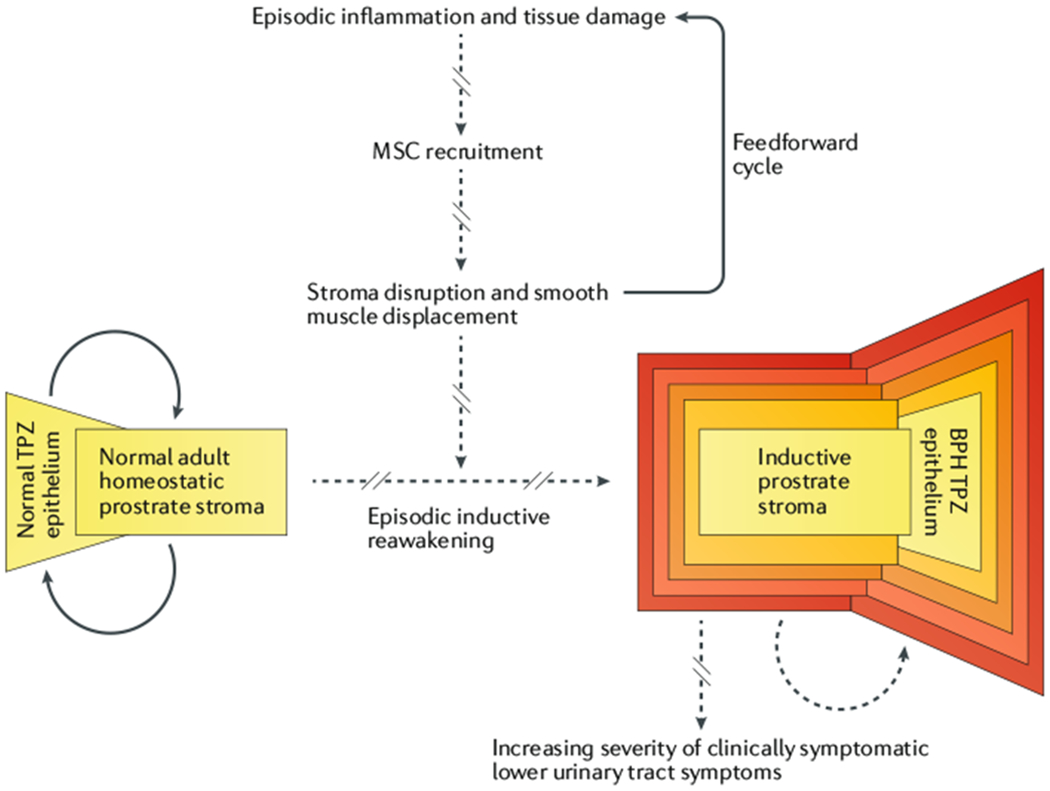
Repeated cycles of tissue damage and inflammation produced by urine reflux, aberrant protease localization, and other candidate inducers promote the episodic recruitment of mesenchymal stem cells (MSCs) to the transition–periurethral zone (TPZ). These recruited MSCs displace endogenous periurethral smooth muscle cells with uncommitted progenitors, which induces benign neoplastic expansion of dormant epithelial stem cells within the developmentally abortive distal ducts to produce BPH nodules. This disruption of normal epithelial-stromal interactions that regulate tissue homeostasis leads to further tissue damage and inflammation, which drives additional MSC recruitment resulting in more stromal disruption. Thus, a feedforward cycle of episodic inductive reawakening induces progressive expansion of BPH nodules and increasing severity of clinical symptoms.
Acknowledgements
The authors thank B. Zhang and I. P. Garraway (University of California-Los Angeles) for generously providing urogenital sinus tissue, A. Meeker for his assistance in developing the immunofluorescence assays, and the Sidney Kimmel Comprehensive Cancer Center (SKCCC) Immunohistochemistry Core supported by the SKCCC Cancer Center Support Grant (CCSG; P30 CA006973). The authors also acknowledge the following sources of financial support: the Prostate Cancer Foundation Young Investigator Award (W.N.B.), SKCCC CCSG developmental funds (P30 CA006973 (W.N.B.)), the U.S. Department of Defense (W81XWH-17-1-0528 (W.N.B.)), and the US NIH Prostate SPORE Grant (P50 CA058236 (J.T.I.)).
Footnotes
Competing interests
J.T.I. declares financial interest with Sophiris Bio related to topsalysin (also known as PRX302). W.N.B. declares no competing interests.
References
- 1.Isaacs JT Prostate stem cells and benign prostatic hyperplasia. Prostate 68, 1025–1034 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McNeal JE Regional morphology and pathology of the prostate. Am. J. Clin. Pathol 49, 347–357 (1968). [DOI] [PubMed] [Google Scholar]
- 3.McNeal JE The prostate and prostatic urethra: a morphologic synthesis. J. Urol 107, 1008–1016 (1972). [DOI] [PubMed] [Google Scholar]
- 4.McNeal JE Origin and evolution of benign prostatic enlargement. Invest. Urol 15, 340–345 (1978). [PubMed] [Google Scholar]
- 5.McNeal JE The zonal anatomy of the prostate. Prostate 2, 35–49 (1981). [DOI] [PubMed] [Google Scholar]
- 6.McNeal JE Anatomy of the prostate and morphogenesis of BPH. Prog. Clin. Biol. Res 145, 27–53 (1984). [PubMed] [Google Scholar]
- 7.Isaacs JT & Coffey DS Etiology and disease process of benign prostatic hyperplasia. Prostate Suppl 2, 33–50 (1989). [DOI] [PubMed] [Google Scholar]
- 8.Hollingsworth JM & Wilt TJ Lower urinary tract symptoms in men. BMJ. 349, g4474 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sausville J & Naslund M Benign prostatic hyperplasia and prostate cancer: an overview for primary care physicians. Int. J. Clin. Pract 64, 1740–1745 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Cindolo L et al. Drug adherence and clinical outcomes for patients under pharmacological therapy for lower urinary tract symptoms related to benign prostatic hyperplasia: population-based cohort study. Eur. Urol 68, 418–425 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Bosch JL, Tilling K, Bohnen AM, Bangma CH & Donovan JL Establishing normal reference ranges for prostate volume change with age in the population-based Krimpen-study: prediction of future prostate volume in individual men. Prostate 67, 1816–1824 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Park J et al. Establishment of reference ranges for prostate volume and annual prostate volume change rate in Korean adult men: analyses of a nationwide screening population. J. Kor. Med. Sci 30, 1136–1142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayward SW, Cunha GR & Dahiya R Normal development and carcinogenesis of the prostate. A unifying hypothesis. Ann. NY Acad. Sci 784, 50–62 (1996). [DOI] [PubMed] [Google Scholar]
- 14.Hayward SW et al. Interactions between adult human prostatic epithelium and rat urogenital sinus mesenchyme in a tissue recombination model. Differentiation 63, 131–140 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Thomson AA, Timms BG, Barton L, Cunha GR & Grace OC The role of smooth muscle in regulating prostatic induction. Development 129, 1905–1912 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Marker PC, Donjacour AA, Dahiya R & Cunha GR Hormonal, cellular, and molecular control of prostatic development. Dev. Biol 253, 165–174 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Yan G, Fukabori Y, Nikolaropoulos S, Wang F & McKeehan WL Heparin-binding keratinocyte growth factor is a candidate stromal-to-epithelial-cell andromedin. Mol. Endocrinol 6, 2123–2128 (1992). [DOI] [PubMed] [Google Scholar]
- 18.Richard C et al. Androgens modulate the balance between VEGF and angiopoietin expression in prostate epithelial and smooth muscle cells. Prostate 50, 83–91 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Litvinov IV, De Marzo AM & Isaacs JT Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J. Clin. Endocrinol. Metab 88, 2972–2982 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Ohlson N, Bergh A, Stattin P & Wikstrom P Castration-induced epithelial cell death in human prostate tissue is related to locally reduced IGF-1 levels. Prostate 67, 32–40 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Brennen WN et al. Mesenchymal stem cell infiltration during neoplastic transformation of the human prostate. Oncotarget 8, 46710–46727 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamm ML et al. Sonic hedgehog activates mesenchymal Gli1 expression during prostate ductal bud formation. Dev. Biol 249, 349–366 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Karhadkar SS et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 431, 707–712 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Peng YC, Levine CM, Zahid S, Wilson EL & Joyner AL Sonic hedgehog signals to multiple prostate stromal stem cells that replenish distinct stromal subtypes during regeneration. Proc. Natl Acad. Sci. USA 110, 20611–20616 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peehl DM & Sellers RG Basic FGF, EGF, and PDGF modify TGFbeta-induction of smooth muscle cell phenotype in human prostatic stromal cells. Prostate 35, 125–134 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Zhu ML & Kyprianou N Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr. Relat. Cancer 15, 841–849 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoon G, Kim JY, Choi YK, Won YS & Lim IK Direct activation of TGF-beta1 transcription by androgen and androgen receptor complex in Huh7 human hepatoma cells and its tumor in nude mice. J. Cell. Biochem 97, 393–411 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Kanda T, Jiang X & Yokosuka O Androgen receptor signaling in hepatocellular carcinoma and pancreatic cancers. World J. Gastroenterol 20, 9229–9236 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF & Bhasin S Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology 144, 5081–5088 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Singh R et al. Regulation of myogenic differentiation by androgens: cross talk between androgen receptor/ beta-catenin and follistatin/transforming growth factor-beta signaling pathways. Endocrinology 150, 1259–1268 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Timms BG Prostate development: a historical perspective. Differentiation 76, 565–577 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Lowsley O Embryology, anatomy and surgery of the prostate gland with report of operative results. Am. J. Surg 8, 526–541 (1930). [Google Scholar]
- 33.Moore RA Benign hypertrophy and carcinoma of the prostate, occurence and experimental production in animals. Surgery 16, 152–167 (1944). [Google Scholar]
- 34.Claus S, Wrenger M, Senge T & Schulze H Immunohistochemical determination of age related proliferation rates in normal and benign hyperplastic human prostates. Urol. Res 21,305–308 (1993). [DOI] [PubMed] [Google Scholar]
- 35.Claus S, Berges R, Senge T & Schulze H Cell kinetic in epithelium and stroma of benign prostatic hyperplasia. J. Urol 158, 217–221 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Shapiro E, Becich MJ, Hartanto V & Lepor H The relative proportion of stromal and epithelial hyperplasia is related to the development of symptomatic benign prostate hyperplasia. J. Urol 147, 1293–1297 (1992). [DOI] [PubMed] [Google Scholar]
- 37.Cunha GR & Lung B The possible influence of temporal factors in androgenic responsiveness of urogenital tissue recombinants from wild-type and androgen-insensitive (Tfm) mice. J. Exp. Zool 205, 181–193 (1978). [DOI] [PubMed] [Google Scholar]
- 38.Cunha GR, Hayward SW, Dahiya R & Foster BA Smooth muscle-epithelial interactions in normal and neoplastic prostatic development. Acta Anat. (Basel) 155, (63–72 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Hayward S, Cao M, Thayer K & Cunha G Cell differentiation lineage in the prostate. Differentiation 68, 270–279 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Schalken JA & van Leenders G Cellular and molecular biology of the prostate: stem cell biology. Urology 62, 11–20 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Goto K et al. Proximal prostatic stem cells are programmed to regenerate a proximal-distal ductal axis. Stem Cells 24, 1859–1868 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Vander Griend DJ et al. The role of CD133 in normal human prostate stem cells and malignant cancer-initiating cells. Cancer Res 68, 9703–9711 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moad M et al. Multipotent basal stem cells, maintained in localized proximal niches, support directed long-ranging epithelial flows in human prostates. Cell Rep. 20, 1609–1622 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo C et al. Epcam, CD44, and CD49f distinguish sphere-forming human prostate basal cells from a subpopulation with predominant tubule initiation capability. PLOS ONE 7, e34219 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antony L, van der Schoor F, Dalrymple SL & Isaacs JT Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/beta-catenin/TCF-4 complex inhibition of c-MYC transcription. Prostate 74, 1118–1131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vander Griend DJ, Litvinov IV & Isaacs JT Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int. J. Biol. Sci 10, 627–642 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peters CA & Walsh PC The effect of nafarelin acetate, a luteinizing-hormone-releasing hormone agonist, on benign prostatic hyperplasia. N. Engl. J. Med 317, 599–604 (1987). [DOI] [PubMed] [Google Scholar]
- 48.Joseph IB, Nelson JB, Denmeade SR & Isaacs JT Androgens regulate vascular endothelial growth factor content in normal and malignant prostatic tissue. Clin. Cancer Res 3, 2507–2511 (1997). [PubMed] [Google Scholar]
- 49.Macoska JA Chemokines and BPH/LUTS. Differentiation 82, 253–260 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McLaren ID, Jerde TJ & Bushman W Role of interleukins, IGF and stem cells in BPH. Differentiation 82, 237–243 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quillard T & Charreau B Impact of notch signaling on inflammatory responses in cardiovascular disorders. Int. J. Mol. Sci 14, 6863–6888 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hahn AM, Myers JD, McFarland EK, Lee S & Jerde TJ Interleukin-driven insulin-like growth factor promotes prostatic inflammatory hyperplasia. J. Pharmacol. Exp. Ther 351,605–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edeling M, Ragi G, Huang S, Pavenstadt H & Susztak K Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol 12, 426–439 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fazio C & Ricciardiello L Inflammation and Notch signaling: a crosstalk with opposite effects on tumorigenesis. Cell Death Dis 7, e2515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shang Y, Smith S & Hu X Role of Notch signaling in regulating innate immunity and inflammation in health and disease. Protein Cell 7, 159–174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Balistreri CR, Madonna R, Melino G & Caruso C The emerging role of Notch pathway in ageing: focus on the related mechanisms in age-related diseases. Ageing Res. Rev 29, 50–65 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Ma B & Hottiger MO Crosstalk between Wnt/beta-catenin and NF-kappaB signaling pathway during Inflammation. Front. Immunol 7, 378 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kohnen PW & Drach GW Patterns of inflammation in prostatic hyperplasia: a histologic and bacteriologic study. J. Urol 121, 755–760 (1979). [DOI] [PubMed] [Google Scholar]
- 59.Kramer G, Mitteregger D & Marberger M Is benign prostatic hyperplasia (BPH) an immune inflammatory disease? Eur. Urol 51, 1202–1216 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Nickel JC Inflammation and benign prostatic hyperplasia. Urol. Clin. North Amer 35, 109–115 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robert G et al. Inflammation in benign prostatic hyperplasia: a 282 patients’ immunohistochemical analysis. Prostate 69, 1774–1780 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steiner GE et al. Cytokine expression pattern in benign prostatic hyperplasia infiltrating T cells and impact of lymphocytic infiltration on cytokine mRNA profile in prostatic tissue. Lab Invest. 83, 1131–1146 (2003). [DOI] [PubMed] [Google Scholar]
- 63.De Nunzio C, Presicce F & Tubaro A Inflammatory mediators in the development and progression of benign prostatic hyperplasia. Nat. Rev. Urol 13, 613–626 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Nickel JC et al. The relationship between prostate inflammation and lower urinary tract symptoms: examination of baseline data from the REDUCE trial. Eur. Urol 54, 1379–1384 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu H et al. Urinary microbiota in patients with prostate cancer and benign prostatic hyperplasia. Arch. Med. Sci 11,385–394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vignozzi L, Gacci M & Maggi M Lower urinary tract symptoms, benign prostatic hyperplasia and metabolic syndrome. Nat. Rev. Urol 13, 108–119 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Tilg H & Moschen AR Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol 6, 772–783 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Persson BE & Ronquist G Evidence for a mechanistic association between nonbacterial prostatitis and levels of urate and creatinine in expressed prostatic secretion. J. Urol 155, 958–960 (1996). [PubMed] [Google Scholar]
- 69.Motrich RD et al. Uric acid crystals in the semen of a patient with symptoms of chronic prostatitis. Fertil Steril. 85, 751 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Kirby RS, Lowe D, Bultitude MI & Shuttleworth KE Intra-prostatic urinary reflux: an aetiological factor in abacterial prostatitis. Br. J. Urol 54, 729–731 (1982). [DOI] [PubMed] [Google Scholar]
- 71.Luo J et al. Gene expression signature of benign prostatic hyperplasia revealed by cDNA microarray analysis. Prostate 51, 189–200 (2002). [DOI] [PubMed] [Google Scholar]
- 72.O’Malley KJ et al. Proteomic analysis of patient tissue reveals PSA protein in the stroma of benign prostatic hyperplasia. Prostate 74, 892–900 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mikolajczyk SD et al. “BPSA,” a specific molecular form of free prostate-specific antigen, is found predominantly in the transition zone of patients with nodular benign prostatic hyperplasia. Urology 55, 41–45 (2000). [DOI] [PubMed] [Google Scholar]
- 74.Horwitz EM et al. Clarification of the nomenclature for MSC: the international society for cellular therapy position statement. Cytotherapy 7, 393–395 (2005). [DOI] [PubMed] [Google Scholar]
- 75.Zuk PA et al. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 13, 4279–4295 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.da Silva Meirelles L, Chagastelles PC & Nardi NB Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci 119, 2204–2213 (2006). [DOI] [PubMed] [Google Scholar]
- 77.Crisan M et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3, 301–313 (2008). [DOI] [PubMed] [Google Scholar]
- 78.Lin VK et al. Prostatic stromal cells derived from benign prostatic hyperplasia specimens possess stem cell like property. Prostate 67, 1265–1276 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Brennen WN, Chen S, Denmeade SR & Isaacs JT Quantification of mesenchymal stem cells (MSCs) at sites of human prostate cancer. Oncotarget 4, 106–117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim W et al. RUNX1 is essential for mesenchymal stem cell proliferation and myofibroblast differentiation. Proc Natl Acad. Sci. USA 111, 16389–16394 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brennen WN, Kisteman LN & Isaacs JT Rapid selection of mesenchymal stem and progenitor cells in primary prostate stromal cultures. Prostate 76, 552–564 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brennen WN et al. Assessing angiogenic responses induced by primary human prostate stromal cells in a three-dimensional fibrin matrix assay. Oncotarget 7, 71298–71308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Colter DC, Sekiya I & Prockop DJ Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc Natl Acad. Sci. USA 98, 7841–7845 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sakaguchi Y, Sekiya I, Yagishita K & Muneta T Comparison of human stem cells derived from various mesenchymal tissues: superiority of synovium as a cell source. Arthritis Rheum 52, 2521–2529 (2005). [DOI] [PubMed] [Google Scholar]
- 85.Musina RA, Bekchanova ES, Belyavskii AV & Sukhikh GT Differentiation potential of mesenchymal stem cells of different origin. Bull. Exp. Biol. Med 141, 147–151 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Park HW, Shin JS & Kim CW Proteome of mesenchymal stem cells. Proteomics 7, 2881–2894 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Noel D et al. Cell specific differences between human adipose-derived and mesenchymal-stromal cells despite similar differentiation potentials. Exp. Cell Res 314, 1575–1584 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Petrini M et al. Identification and purification of mesodermal progenitor cells from human adult bone marrow. Stem Cells Dev. 18, 857–866 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jansen BJ et al. Functional differences between mesenchymal stem cell populations are reflected by their transcriptome. Stem Cells Dev. 19, 481–490 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Russell KC et al. Clonal analysis of the proliferation potential of human bone marrow mesenchymal stem cells as a function of potency. Biotechnol. Bioeng 108, 2716–2726 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Strioga M, Viswanathan S, Darinskas A, Slaby O & Michalek J Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev. 21, 2724–2752 (2012). [DOI] [PubMed] [Google Scholar]
- 92.Pacini S et al. Mesangiogenic progenitor cells derived from one novel CD64(bright)CD31(bright)CD14(neg) population in human adult bone marrow. Stem Cells Dev 25, 661–673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Spaeth E, Klopp A, Dembinski J, Andreeff M & Marini F Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 15, 730–738 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Brennen WN, Denmeade SR & Isaacs JT Mesenchymal stem cells as a vector for the inflammatory prostate microenvironment. Endocr. Relat. Cancer 20, R269–R290 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lourenco S et al. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J. Immunol 194, 3463–3474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang L et al. Aberrant transforming growth factor-beta activation recruits mesenchymal stem cells during prostatic hyperplasia. Stem Cells Transl Med. 6, 394–404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tanaka ST et al. Recruitment of bone marrow derived cells to the bladder after bladder outlet obstruction. J. Urol 182, 1769–1774 (2009). [DOI] [PubMed] [Google Scholar]
- 98.Placencio VR, Li X, Sherrill TP, Fritz G & Bhowmick NA Bone marrow derived mesenchymal stem cells incorporate into the prostate during regrowth. PLOS One 5, e12920 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garcia NP et al. Kinetics of mesenchymal and hematopoietic stem cells mobilization by G-CSF and its impact on the cytokine microenvironment in primary cultures. Cell. Immunol 293, 1–9 (2015). [DOI] [PubMed] [Google Scholar]
- 100.Begley L, Monteleon C, Shah RB, Macdonald JW & Macoska JA CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell 4, 291–298 (2005). [DOI] [PubMed] [Google Scholar]
- 101.Fujita K et al. Monocyte chemotactic protein-1 (MCP-1/CCL2) is associated with prostatic growth dysregulation and benign prostatic hyperplasia. Prostate 70, 473–481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nakata W et al. Bone marrow-derived cells contribute to regeneration of injured prostate epithelium and stroma. Prostate 75, 806–814 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Sokolova IB, Zin’kova NN, Shvedova EV, Kruglyakov PV & Polyntsev DG Distribution of mesenchymal stem cells in the area of tissue inflammation after transplantation of the cell material via different routes. Bull. Exp. Biol. Med 143, 143–146 (2007). [DOI] [PubMed] [Google Scholar]
- 104.Saffarini CM et al. Maturation of the developing human fetal prostate in a rodent xenograft model. Prostate 73, 1761–1775 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Caplan AI Why are MSCs therapeutic? New data: new insight. J. Pathol 217, 318–324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Newman RE, Yoo D, LeRoux MA & Danilkovitch-Miagkova A Treatment of inflammatory diseases with mesenchymal stem cells. Inflamm Allergy Drug Targets 8, 110–123 (2009). [DOI] [PubMed] [Google Scholar]
- 107.English K & Mahon BP Allogeneic mesenchymal stem cells: agents of immune modulation. J. Cell. Biochem 112, 1963–1968 (2011). [DOI] [PubMed] [Google Scholar]
- 108.Guess HA, Arrighi HM, Metter EJ & Fozard JL Cumulative prevalence of prostatism matches the autopsy prevalence of benign prostatic hyperplasia. Prostate 17, 241–246 (1990). [DOI] [PubMed] [Google Scholar]
- 109.Berry SJ, Coffey DS, Walsh PC & Ewing LL The development of human benign prostatic hyperplasia with age. J. Urol 132, 474–479 (1984). [DOI] [PubMed] [Google Scholar]
- 110.Franks LM Benign nodular hyperplasia of the prostate: a review. Ann. R. Coll. Surg. Engl 14, 92–106 (1954). [PMC free article] [PubMed] [Google Scholar]
- 111.Ross JJ et al. Cytokine-induced differentiation of multipotent adult progenitor cells into functional smooth muscle cells. J. Clin. Invest 116, 3139–3149 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Narita Y, Yamawaki A, Kagami H, Ueda M & Ueda Y Effects of transforming growth factor-beta 1 and ascorbic acid on differentiation of human bone-marrow-derived mesenchymal stem cells into smooth muscle cell lineage. Cell Tissue Res 333, 449–459 (2008). [DOI] [PubMed] [Google Scholar]
- 113.Mishra PJ et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res 68, 4331–4339 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Paunescu V et al. Tumour-associated fibroblasts and mesenchymal stem cells: more similarities than differences. J. Cell. Mol. Med 15, 635–646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao L & Hantash BM TGF-beta1 regulates differentiation of bone marrow mesenchymal stem cells. Vitam. Horm 87, 127–141 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Liu J, Wang Y, Wu Y, Ni B & Liang Z Sodium butyrate promotes the differentiation of rat bone marrow mesenchymal stem cells to smooth muscle cells through histone acetylation. PLOS ONE 9, e116183 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shi N, Guo X & Chen SY Olfactomedin 2, a novel regulator for transforming growth factor-beta-induced smooth muscle differentiation of human embryonic stem cell-derived mesenchymal cells. Mol. Biol. Cell 25, 4106–4114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Visweswaran M et al. Multi-lineage differentiation of mesenchymal stem cells - to Wnt, or not Wnt. Int. J. Biochem. Cell Biol 68, 139–147 (2015). [DOI] [PubMed] [Google Scholar]
- 119.Almalki SG & Agrawal DK Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation 92, 41–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Russell KC et al. In vitro high-capacity assay to quantify the clonal heterogeneity in trilineage potential of mesenchymal stem cells reveals a complex hierarchy of lineage commitment. Stem Cells 28, 788–798 (2010). [DOI] [PubMed] [Google Scholar]
- 121.Lin VK et al. Myosin heavy chain gene expression in normal and hyperplastic human prostate tissue. Prostate 44, 193–203 (2000). [DOI] [PubMed] [Google Scholar]
- 122.Schauer IG, Ressler SJ, Tuxhorn JA, Dang TD & Rowley DR Elevated epithelial expression of interleukin-8 correlates with myofibroblast reactive stroma in benign prostatic hyperplasia. Urology 72, 205–213 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schauer IG & Rowley DR The functional role of reactive stroma in benign prostatic hyperplasia. Differentiation. 82, 200–210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tuxhorn JA et al. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res 8, 2912–2923 (2002). [PubMed] [Google Scholar]
- 125.Kidd S et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLOS ONE 7, e30563 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shangguan L et al. Inhibition of TGF-beta/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells 30, 2810–2819 (2012). [DOI] [PubMed] [Google Scholar]
- 127.Sufrin G, Heston WD, Hazra T & Coffey DS The effect of radiation on prostatic growth. Invest. Urol 13, 418–423 (1976). [PubMed] [Google Scholar]
- 128.Mallepell S, Krust A, Chambon P & Brisken C Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc. Natl Acad. Sci. USA 103, 2196–2201 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fass D, Steinfeld A, Brown J & Tessler A Radiotherapeutic prophylaxis of estrogen-induced gynecomastia: a study of late sequela. Int. J. Radiat. Oncol. Biol. Phys 12, 407–408 (1986). [DOI] [PubMed] [Google Scholar]
- 130.Alfthan O & Kettunen K The effect of roentgen ray treatment of gynecomastia in patients with prostatic carcinoma treated with estrogenic hormones: a preliminary communication. J. Urol 94, 604–606 (1965). [DOI] [PubMed] [Google Scholar]
- 131.Koukourakis M et al. Transurethral radiotherapy for benign prostatic hypertrophy-related urethral obstruction. Dosimetry, ethics, and preliminary results. Med. Dosim 19, 67–72 (1994). [DOI] [PubMed] [Google Scholar]
- 132.Zhao P, Lu S, Yang Y, Shao Q & Liang J Three-dimensional conformal radiation therapy of spontaneous benign prostatic hyperplasia in canines. Oncol. Res 19, 225–235 (2011). [DOI] [PubMed] [Google Scholar]
- 133.Williams SA et al. A prostate-specific antigen-activated channel-forming toxin as therapy for prostatic disease. J. Natl Cancer Inst 99, 376–385 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Magistro G et al. Emerging minimally invasive treatment options for male lower urinary tract symptoms. Eur. Urol 72, 986–997 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Denmeade SR et al. Phase 1 and 2 studies demonstrate the safety and efficacy of intraprostatic injection of PRX302 for the targeted treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia. Eur. Urol 59, 747–754 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Elhilali MM et al. Prospective, randomized, double-blind, vehicle controlled, multicenter phase IIb clinical trial of the pore forming protein PRX302 for targeted treatment of symptomatic benign prostatic hyperplasia. J. Urol 189, 1421–1426 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Scanlan MJ et al. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc. Natl Acad. Sci. USA 91, 5657–5661 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brennen WN, Isaacs JT & Denmeade SR Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Mol. Cancer Ther 11, 257–266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bae S et al. Fibroblast activation protein alpha identifies mesenchymal stromal cells from human bone marrow. Br. J. Haematol 142, 827–830 (2008). [DOI] [PubMed] [Google Scholar]
- 140.Aggarwal S et al. Fibroblast activation protein peptide substrates identified from human collagen I derived gelatin cleavage sites. Biochemistry 47, 1076–1086 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brennen WN, Rosen DM, Wang H, Isaacs JT & Denmeade SR Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J. Natl Cancer Inst 104, 1320–1334 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.LeBeau AM, Brennen WN, Aggarwal S & Denmeade SR Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Mol. Cancer Ther 8, 1378–1386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]