

Abstract
Background
Chorionic villus sampling (CVS) is the method of choice for obtaining fetal tissue for prenatal diagnosis before 15 weeks of pregnancy. CVS can be performed using either a transabdominal or transcervical approach. The type of instrument and technique used could have a significant impact on the outcome of the procedure. An ability to manoeuvre the instrument within the uterine cavity without puncturing the gestational sac, to see the tip of the instrument on ultrasound scanning and to minimise the number of instrument passes into the uterus are particularly important.
Objectives
To compare the efficacy and safety of different instruments and techniques used to obtain chorionic tissue in early pregnancy by the transabdominal or transcervical route. Primary outcomes included failure to obtain an adequate sample (greater than 5 mg of chorionic villi), need for reinsertion of the instrument, pain, and miscarriage following the procedure. Secondary outcomes included mean weight of tissue obtained, successful culture, difficult instrument insertion, poor visualisation of instrument, vaginal bleeding following the procedure and cost per procedure.
Search methods
We searched the Cochrane Pregnancy and Childbirth Group's Trials Register (31 August 2012).
Selection criteria
Randomised trials comparing different instruments (forceps, cannula, needle) or techniques for CVS using either transabdominal or transcervical approach.
Data collection and analysis
Two review authors assessed eligibility and trial quality.
Main results
For transcervical CVS, forceps and cannulae were evaluated in five trials involving 472 women. When a cannula was used, operators failed to obtain an adequate sample (greater than 5 mg of chorionic villi) more often (average risk ratio (RR) 3.81; 95% confidence interval (CI) 1.52 to 9.56). There was no difference in the need for reinsertion of instruments (average RR 2.44; 95% CI 0.83 to 7.20). However, inserting a cannula was more painful (RR 1.93; 95% CI 1.11 to 3.37). There was no difference in spontaneous miscarriage when the use of a cannula was compared with biopsy forceps (RR 1.00; 95% CI 0.14 to 6.96). One study reported the cost of the procedures and found CVS with a cannula to be more expensive (mean difference (MD) $183.7; 95% CI 152.62 to 214.78).
When different types of cannulae for transcervical CVS were compared, a Portex cannula was more likely to result in an inadequate sample (RR 2.23; 95% CI 1.25 to 3.98) compared with the silver cannula and to result in a difficult (RR 3.26; 95% CI 1.38 to 7.67) or painful (RR 5.81; 95% CI 1.41 to 23.88) procedure when compared with the aluminium cannula.
For transabdominal CVS, two trials comparing different needle techniques were included involving 285 women. One study using an ex vivo system of term placentae was excluded. The included trials compared different continuous negative pressure aspiration techniques with a discontinuous negative pressure system created by a syringe attached to a 20 gauge needle. The studies produced discrepant results. One study found there was no significant difference between groups in the mean weight of chorionic villi obtained (MD 0.40; 95% CI ‐2.25 to 3.05) or in failure to obtain an adequate sample (more than 5 mg of chorionic villi) on the first attempt (RR 1.02; 95% CI 0.54 to 1.93), whereas the other study found both of these outcomes to be significantly less favourable with the standard discontinuous technique using a syringe (mean weight of chorionic villi obtained: MD ‐14.80; 95% CI ‐21.71 to ‐7.89; failure to obtain an adequate sample on the first attempt: RR 2.73; 95% CI 1.08 to 6.92). There was no difference in rate of miscarriage following the procedure in either study (RR 7.15; 95% CI 0.37 to 136.50; RR 2.93; 95% CI 0.12 to 70.00). Perceived pain by the patient was similar between groups (MD 0.00; 95% CI ‐0.04 to 0.04) as was success of culture (no failed cases).
Authors' conclusions
For transcervical CVS, although there is some evidence to support the use of small forceps instead of cannulae, the evidence is not strong enough to support change in practice for clinicians who have become familiar with a particular technique. For transabdominal CVS, based on current evidence, there is no difference in clinically important outcomes with the use of a continuous compared with a discontinuous negative pressure needle aspiration system.
Keywords: Female; Humans; Pregnancy; Abortion, Spontaneous; Abortion, Spontaneous/etiology; Biopsy, Fine‐Needle; Biopsy, Fine‐Needle/instrumentation; Biopsy, Fine‐Needle/methods; Catheterization; Catheterization/adverse effects; Catheterization/instrumentation; Chorionic Villi Sampling; Chorionic Villi Sampling/adverse effects; Chorionic Villi Sampling/instrumentation; Chorionic Villi Sampling/methods; Randomized Controlled Trials as Topic; Surgical Instruments
Plain language summary
Instruments to use for chorionic villus sampling for prenatal diagnosis
More research is needed to identify the best surgical instruments and techniques to use for chorionic villus sampling testing during early pregnancy (before 15 weeks).
Chorionic villus sampling (CVS) is a test where a small piece of placental tissue (chorion) is removed from the pregnant woman's womb (uterus) and used for testing for genetic disorders. The tissue can be removed through the cervix or through the abdomen using plastic or metal tubes (cannulae), biopsy forceps or fine needles. The woman's risk of miscarriage increases if several attempts are needed to obtain sufficient placental tissue or the gestational sac is damaged during the procedure using ultrasound guidance.
The review found there is some evidence that small forceps may be more effective and less painful for the woman than using an aspiration cannula for sampling through the cervix (transcervical CVS). However, there was no difference in risk of miscarriage. When sampling was obtained through the abdomen (transabdominal CVS) using a needle with either a continuous or discontinuous negative pressure aspiration system, there was no difference in the effectiveness of the techniques, the amount of pain or in the risk of miscarriage. Forceps and cannulae were evaluated in five transcervical CVS trials involving 472 women. Two trials involving 285 women compared different needle techniques for transabdominal CVS. These randomised trials are too small to provide reliable evidence in favour of one particular instrument or technique for transcervical or transabdominal chorionic villus sampling.
Background
Despite its drawbacks, chorionic villus sampling (CVS) remains the method of choice when prenatal diagnosis of possible genetic disorders is required before 15 weeks of pregnancy. Early amniocentesis has been suggested as an alternative, but complication rates (miscarriage rate, talipes equinovarus (club foot)) are too high (Alfirevic 1999; CEMAT 1998). CVS can be performed transabdominally or transcervically. Ultrasound guidance is essential for both techniques. Transabdominal CVS is very similar to amniocentesis; a needle or small forceps is inserted into the uterus through the abdominal wall using an aseptic technique. Fibroids, obesity and low posterior placenta may cause technical difficulties. The transcervical route involves the passage of an instrument (cannula, forceps) through the cervical canal. Instruments are usually slightly bigger than the ones used for the transabdominal approach and the risk of infection using the vaginal route is likely to be higher.
When needles and cannulae are used, the placental tissue (chorionic villi) required for cytogenetic analysis is obtained by aspiration using negative pressure created by a syringe or a vacuum aspirator. Alternatively, a tissue forceps is passed through a guide needle and a small amount of placental tissue is grasped and removed. The current evidence suggests that the transabdominal approach to sampling is safer (Smidt‐Jensen 1992), but the transcervical route continues to be used, particularly to allow easy access to the low‐lying posterior placentae.
The type of instrument and technique used could have a significant impact on the success rate of the procedure. An ability to manoeuvre the instrument within the uterine cavity without puncturing the gestational sac, and easy visualisation of the tip of the instrument on ultrasound scanning are particularly important. Also, the risk of miscarriage is increased with multiple uterine insertions. It is, therefore, important for an instrument to allow adequate tissue sample to be obtained with as few attempts as possible (preferably one). Several types of instruments (needles, plastic or metal aspiration catheters, biopsy forceps) have been used since CVS was first described in the early 1970s but the choice is still made on the basis of personal preference rather than scientific evidence.
Objectives
To compare the efficacy and safety of different instruments and techniques used for transabdominal or transcervical chorionic villus sampling. More specifically, to compare the quality and quantity of obtained chorionic tissue, technical difficulties during the procedure, maternal side‐effects, pregnancy outcomes and cost between different instruments and techniques.
Methods
Criteria for considering studies for this review
Types of studies
All known randomised trials comparing different methods of chorionic villus sampling (CVS), using either the transabdominal approach, transcervical approach or both. We anticipated that early trials may not have used strictly random allocation and, therefore, we planned to include trials that used a quasi‐randomised method of treatment allocation such as alternation by hospital number or the woman's date of birth.
Types of participants
Pregnant women.
Types of interventions
Transabdominal or transcervical CVS using different instruments and techniques.
Types of outcome measures
In general, quality and quantity of obtained chorionic tissue, technical difficulties during the procedure, maternal side‐effects, pregnancy outcome and cost were assessed.
Primary outcomes
Failure to obtain an adequate sample (failure to obtain greater than 5 mg of chorionic villi)
Need for reinsertion of the instrument (failure to obtain an adequate sample on the first attempt)
Pain
Miscarriage following the procedure
Secondary outcomes
Mean weight of tissue obtained
Culture successful
Difficult insertion of the instrument
Poor visualisation of the instrument
Vaginal bleeding following procedure (transcervical CVS)
Cost per procedure
Other outcomes, not specified here, have been included if they are reported in the studies and are considered clinically relevant.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Pregnancy and Childbirth Group’s Trials Register by contacting the Trials Search Co‐ordinator (31 August 2012).
The Cochrane Pregnancy and Childbirth Group’s Trials Register is maintained by the Trials Search Co‐ordinator and contains trials identified from:
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
weekly searches of MEDLINE;
weekly searches of EMBASE;
handsearches of 30 journals and the proceedings of major conferences;
weekly current awareness alerts for a further 44 journals plus monthly BioMed Central email alerts.
Details of the search strategies for CENTRAL, MEDLINE and EMBASE, the list of handsearched journals and conference proceedings, and the list of journals reviewed via the current awareness service can be found in the ‘Specialized Register’ section within the editorial information about the Cochrane Pregnancy and Childbirth Group.
Trials identified through the searching activities described above are each assigned to a review topic (or topics). The Trials Search Co‐ordinator searches the register for each review using the topic list rather than keywords.
We did not apply any language restrictions.
Data collection and analysis
For methods used to assess trials included in previous versions of this review (Barkai 1989; Chalkiadakis 1993; MacKenzie 1986; Pons 1989; von Dadelszen 2005), seeAppendix 1.
The following methods were used to assess Battagliarin 2009; Buyukkurt 2010; Cochrane 2003; von Dadelszen 2005.
Selection of studies
Two review authors (CY and PVD) independently assessed for inclusion of all the potential studies we identified as a result of the search strategy. We resolved any disagreement through discussion or by consulting a third person.
Data extraction and management
Two review authors (CY and PVD) extracted the data and discrepancies were resolved through discussion or by consulting a third person. Data were entered into Review Manager software (RevMan 2011) and checked for accuracy.
When information regarding any of the above was unclear, we attempted to contact authors of the original reports to provide further details.
Assessment of risk of bias in included studies
Two review authors (CY and PVD) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved any disagreement by discussion or by involving a third assessor.
(1) Random sequence generation (checking for possible selection bias)
We described for each included study the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups.
We assessed the method as:
low risk of bias (any truly random process, e.g. random number table; computer random number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital or clinic record number);
unclear risk of bias.
(2) Allocation concealment (checking for possible selection bias)
We described for each included study the method used to conceal allocation to interventions prior to assignment and assessed whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment.
We assessed the methods as:
low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes);
high risk of bias (open random allocation; unsealed or non‐opaque envelopes, alternation; date of birth);
unclear risk of bias.
(3.1) Blinding of participants and personnel (checking for possible performance bias)
We described for each included study the methods used, if any, to blind study participants and personnel from knowledge of which intervention a participant received. We considered that studies were at low risk of bias if they were blinded, or if we judged that the lack of blinding would be unlikely to affect results. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed the methods as:
low, high or unclear risk of bias for participants;
low, high or unclear risk of bias for personnel.
(3.2) Blinding of outcome assessment (checking for possible detection bias)
We described for each included study the methods used, if any, to blind outcome assessors from knowledge of which intervention a participant received. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed methods used to blind outcome assessment as:
low, high or unclear risk of bias.
(4) Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
We described for each included study, and for each outcome or class of outcomes, the completeness of data including attrition and exclusions from the analysis. We have stated whether attrition and exclusions were reported and the numbers included in the analysis at each stage (compared with the total randomised participants), reasons for attrition or exclusion were reported, and whether missing data were balanced across groups or were related to outcomes. Where sufficient information was reported, or could be supplied by the trial authors, we re‐included missing data in the analyses which we undertook.
We assessed methods as:
low risk of bias (e.g. no missing outcome data; missing outcome data balanced across groups);
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups; ‘as treated’ analysis done with substantial departure of intervention received from that assigned at randomisation);
unclear risk of bias.
(5) Selective reporting (checking for reporting bias)
We described for each included study how we investigated the possibility of selective outcome reporting bias and what we found.
We assessed the methods as:
low risk of bias (where it is clear that all of the study’s pre‐specified outcomes and all expected outcomes of interest to the review have been reported);
high risk of bias (where not all the study’s pre‐specified outcomes have been reported; one or more reported primary outcomes were not pre‐specified; outcomes of interest are reported incompletely and so cannot be used; study fails to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias.
(6) Other bias (checking for bias due to problems not covered by (1) to (5) above)
We described for each included study any important concerns we have about other possible sources of bias.
We assessed whether each study was free of other problems that could put it at risk of bias:
low risk of other bias;
high risk of other bias;
unclear whether there is risk of other bias.
(7) Overall risk of bias
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Handbook (Higgins 2011). With reference to (1) to (6) above, we assessed the likely magnitude and direction of the bias and whether we considered it likely to impact on the findings. We explored the impact of the level of bias through undertaking sensitivity analyses, if indicated ‐ seeSensitivity analysis.
Measures of treatment effect
Dichotomous data
For dichotomous data, we have presented results as summary risk ratio with 95% confidence intervals.
Continuous data
For continuous data, we used the mean difference if outcomes were measured in the same way between trials. We planned to use the standardised mean difference to combine trials that measured the same outcome, but used different methods.
Unit of analysis issues
Cluster‐randomised trials
We did not identify any eligible cluster‐randomised trials.
Cross‐over trials
There were problems with incorporating data from Pons 1989, which was a cross‐over trial, and MacKenzie 1986, in which more than one CVS technique was performed on each randomised woman. Each study presented the data as if the results for each technique were independent. However, this may not be the case, i.e. there is a possibility that performing the first technique may affect the second. The numbers included in the analysis are the numbers of procedures, not numbers of women, and the sample sizes are therefore overestimated.
Dealing with missing data
For included studies, we noted levels of attrition. For all outcomes, we carried out analyses, as far as possible, on an intention‐to‐treat basis, i.e. we attempted to include all participants randomised to each group in the analyses, and all participants were analysed in the group to which they were allocated, regardless of whether or not they received the allocated intervention. The denominator for each outcome in each trial was the number randomised minus any participants whose outcomes were known to be missing.
Assessment of heterogeneity
We assessed statistical heterogeneity in each meta‐analysis using the T², I² and Chi² statistics. We regarded heterogeneity as substantial if the I² was greater than 30% and either the T² was greater than zero, or there was a low P value (less than 0.10) in the Chi² test for heterogeneity.
Assessment of reporting biases
Due to the small number of studies included in the meta‐analysis, we did not investigate reporting bias.
Data synthesis
We carried out statistical analysis using the Review Manager software (RevMan 2011). We used fixed‐effect meta‐analysis for combining data where it was reasonable to assume that studies were estimating the same underlying treatment effect: i.e. where trials were examining the same intervention, and the trials’ populations and methods were judged sufficiently similar. If there was clinical heterogeneity sufficient to expect that the underlying treatment effects differed between trials, or if substantial statistical heterogeneity was detected, we used random‐effects meta‐analysis to produce an overall summary if an average treatment effect across trials was considered clinically meaningful. The random‐effects summary was treated as the average range of possible treatment effects and we discuss the clinical implications of treatment effects differing between trials. If the average treatment effect was not clinically meaningful due to heterogeneity, we did not combine trials. When, despite strict inclusion criteria the heterogeneity was likely to be caused by clinical differences between techniques for participants, we did not pool the results.
Where we used random‐effects analyses, the results were presented as the average treatment effect with 95% confidence intervals, and the estimates of T² and I².
Subgroup analysis and investigation of heterogeneity
In the presence of substantial heterogeneity, we planned to investigate it, where appropriate, using sensitivity analyses. We planned to consider whether an overall summary was meaningful, and if it was, used random‐effects analysis to produce it. No subgroup analysis was planned a priori.
Sensitivity analysis
We planned to conduct sensitivity analyses to explore the effect of risk of bias in the included studies. Given the small number of studies in this update, we were not able to perform any sensitivity analyses. In future updates, we plan to use both adequate labelled sequence generation and adequate allocation concealment as essential criteria for sensitivity analyses. Only studies assessed as low risk of bias for both of these domains will be included in the analyses, in order to see whether this makes any difference to the overall result.
Results
Description of studies
Results of the search
Eight studies were identified of which seven met the inclusion criteria and involved 757 women ‐ see Characteristics of included studies. Five studies compared instruments for transcervical CVS, of which none were excluded. Three studies compared needle techniques for transabdominal CVS, one of which was excluded.
Included studies
Five studies compared instruments for transcervical CVS involving 472 women. von Dadelszen 2005 report data from the same study in two separate publications, an abstract and a paper respectively, of which the data have been reported in detail in the review under von Dadelszen 2005. Three studies randomised women with ongoing pregnancies (Barkai 1989; Chalkiadakis 1993; von Dadelszen 2005) and two studies included women before termination of pregnancy (MacKenzie 1986; Pons 1989). Obviously, CVS before termination is not clinically indicated, unless it is important to have cytogenetic diagnosis of an aborted fetus (post‐termination analyses often fail to provide adequate tissue for analysis). However, CVS is sometimes performed, with the consent from women, for training and research purposes. The same women in the study by Pons 1989 had four attempts to obtain chorionic villi (two with a catheter and two with a forceps). MacKenzie 1986 also used two different instruments in the same women. This does not reflect clinical practice in ongoing pregnancies and posed considerable problems in the data analysis (see Methodological quality).
Two studies comparing needle techniques for transabdominal CVS were included (Battagliarin 2009; Buyukkurt 2010), involving 285 women undergoing CVS for standard indications in ongoing, singleton pregnancies.
Excluded studies
The Cochrane 2003 study was excluded as it compared the effect of needle and syringe size on the amount of villi obtained in ex‐vivo term placentae (37 weeks' gestation) of women who had delivered that day. Term placentae following delivery may not accurately mimic a first trimester placenta in utero. However, the study did find that when sampling term placentae, a large syringe (20 mL versus 5 mL) and needle size(18 gauge versus 20 gauge) yields a larger sample of chorionic villi, with the needle size being of greatest significance. The authors suggest using an 18 gauge needle with a 20 mL syringe as a control group in future studies comparing transabdominal CVS techniques.
Risk of bias in included studies
See Figure 1; Figure 2 for summaries of 'Risk of bias' assessments.
1.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
2.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Based on risk of bias, there are two groups of included studies, those of high quality and those of low quality. The study by von Dadelszen 2005, a prospective randomised single‐blinded trial involving 200 women in ongoing pregnancies, is the only high quality study. This study compared biopsy forceps with the Portex cannula. There is a low risk of bias for all categories, with the exception of performance bias, as neither the patient nor the clinician were blinded. However, it would be difficult if not impossible to blind the clinician.
The remaining studies, including the four studies by Barkai 1989, Chalkiadakis 1993, MacKenzie 1986 and Pons 1989 comparing transcervical CVS instruments as well as the two studies by Battagliarin 2009 and Buyukkurt 2010 comparing transabdominal CVS techniques, were of low quality due to considerable or unclear risk of bias.
Allocation
The methods of randomisation used in the transabdominal study by Battagliarin 2009 and of the four earlier transcervical CVS studies (Barkai 1989; MacKenzie 1986; Pons 1989; Chalkiadakis 1993) are not clear and there may have been a selection bias. The study by Buyukkurt 2010 used quasi‐randomisation (participants were randomised by the last number of their national identification number, whether odd or even), increasing the risk of selection bias. von Dadelszen 2005 used an adequate randomisation process (computer‐generated random number table).
The methods of allocation concealment are not clear in three studies (Battagliarin 2009; Chalkiadakis 1993; Pons 1989). Barkai 1989 used alternation and Buyukkurt 2010 used the last number of the national identification number, either odd or even to allocate women, and so both studies are at high risk of bias for allocation concealment. MacKenzie 1986 was also assessed as being at high risk of bias for allocation concealment. von Dadelszen 2005 used an adequate method of allocation concealment (consecutively numbered opaque envelopes).
Blinding
In three of the studies (Buyukkurt 2010; Battagliarin 2009; von Dadelszen 2005), the operators and patients were not blinded to the instrument used, but all other clinicians involved (i.e. cytogeneticists) were not aware of the instrument used until the data analysis was complete. Information regarding blinding was not indicated for the other studies (Barkai 1989; Chalkiadakis 1993; MacKenzie 1986; Pons 1989).
Incomplete outcome data
In the study by von Dadelszen 2005 there was one post‐randomisation withdrawal. Final pregnancy outcome data were missing for two patients in this study and eight patients (four from each study group) in the study by Battagliarin 2009 and were not included in the pregnancy outcomes analysis. Outcome data were complete for the studies by Barkai 1989; Buyukkurt 2010; MacKenzie 1986; Pons 1989. For the study by Chalkiadakis 1993, it could not be determined if outcome data were complete.
Selective reporting
Except for the study by von Dadelszen 2005, the risk of reporting bias was high for the remaining studies (Barkai 1989; Battagliarin 2009; Buyukkurt 2010; Chalkiadakis 1993; MacKenzie 1986; Pons 1989), primarily because primary and secondary outcomes were not clearly specified. Furthermore, the study by Barkai 1989 did not report the data on complications in such a way that they could be included in the meta‐analysis.
Other potential sources of bias
There were problems with incorporating data from Pons 1989 and MacKenzie 1986. Both studies performed more than one technique on each randomised woman and presented the data as if these were independent. However, this may not be the case, i.e. there is a possibility that performing the first technique may affect the second. The numbers included in the analysis are the numbers of procedures, not numbers of women, and the sample sizes are therefore overestimated. There is a low risk of other potential sources of bias for the study by von Dadelszen 2005. The risk of other potential sources of bias is unclear for the remaining studies, Barkai 1989; Battagliarin 2009; Buyukkurt 2010; and Chalkiadakis 1993.
Effects of interventions
(i) Transcervical approach
Aspiration cannula versus small forceps
Pons 1989 randomised 30 women before the termination of the pregnancy and von Dadelszen 2005 randomised 200 women who requested prenatal diagnosis in an ongoing pregnancy. Pons 1989 reported the data on instrument visualisation, failure to obtain villi and failure to obtain a sample of more than 5 mg 'per instrument insertion' (n = 120) rather than per randomised woman (n = 30). This also applies for the outcome 'failure to obtain sample in the first attempt' for which 60 'first attempt' were reported in 30 randomised women (see 'Methodological quality of included studies'). The sample size of the pooled analysis for these four outcomes is somewhat 'inflated' and the results should, therefore, be interpreted with caution. With this proviso, operators failed to obtain an adequate sample (greater than 5 mg of chorionic villi) more often when a cannula was used (average risk ratio (RR) 3.81; 95% confidence interval (CI) 1.52 to 9.56; random‐effects model T² = 0.17 , I² = 34%), Analysis 1.1. Although there was a trend for re‐insertion of a cannula more often compared with forceps, the result is not significant (average RR 2.44; 95% CI 0.83 to 7.20; random‐effects model T² = 0.37 , I² = 59%), Analysis 1.2. Insertion of a cannula was more painful (RR 1.93; 95% CI 1.11 to 3.37), Analysis 1.3, than insertion of forceps. Risk of spontaneous miscarriage following the procedure was similar in both groups (RR 1.00; 95% CI 0.14 to 6.96), Analysis 1.4.
1.1. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 1 Failure to obtain sample > 5 mg.
1.2. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 2 Need for reinsertion of instrument (Failure to obtain sample in the first attempt).
1.3. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 3 Moderate‐to‐severe pain during procedure.
1.4. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 4 Spontaneous miscarriage.
With respect to secondary outcomes, the culture was successful for all procedures regardless of the instrument used, Analysis 1.5. There was no difference in difficulty of instrument insertion or poor visualisation of the instrument (RR 0.58; 95% CI 0.34 to 1.00; RR 1.00; 95% CI 0.15 to 6.87), Analysis 1.6; Analysis 1.7 respectively, whether a cannula or biopsy forceps were used. Neither bleeding during the procedure nor heavy vaginal bleeding less than two days post‐CVS differed between instruments (RR 0.33; 95% CI 0.04 to 2.85; RR 0.20; 95% CI 0.01 to 4.11), Analysis 1.8; Analysis 1.9 respectively. The use of biopsy forceps was associated with cost savings (mean difference (MD) $183.70, 95% CI 152.62 to 214.78), Analysis 1.10. Mean weight of tissue obtained was not assessed.
1.5. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 5 Culture unsuccessful.
1.6. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 6 Difficult insertion.
1.7. Analysis.
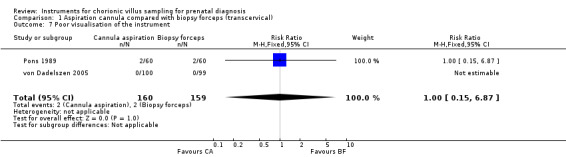
Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 7 Poor visualisation of the instrument.
1.8. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 8 Bleeding.
1.9. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 9 Heavy vaginal bleeding (less than 2 days post‐CVS).
1.10. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 10 Cost per procedure.
Both instruments caused similar degrees of placental trauma (rise in maternal serum alpha‐feto protein) (MD ‐3.40; 95% CI ‐24.48 to 17.68), Analysis 1.19, and were similarly effective in producing successful direct karyotypes (RR 0.95; 95% CI 0.90 to 1.00), Analysis 1.18.
1.19. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 19 Rise in maternal serum aFP [ug/L] (1 hour post‐CVS).
1.18. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 18 Direct karyotype successful.
Portex cannula versus silver cannula
Two instruments were compared in a study by Barkai 1989 (168 women). The operator obtained an inadequate sample of less than 5 mg more often with the Portex cannula (RR 2.23; 95% CI 1.25 to 3.98), Analysis 2.1. The other primary and secondary outcomes were not assessed.
2.1. Analysis.

Comparison 2 Portex cannula compared with silver cannula (transcervical), Outcome 1 Failure to obtain sample greater than 5 mg.
Portex cannula versus malleable cannula versus aluminium cannula
These three instruments were compared in one study by MacKenzie 1986. Each of the 50 randomised women had two procedures before termination of pregnancy. Two types of cannulae were used in 31 women and the same type of cannula twice in the remaining 19 women. The data were not presented per randomised woman (intention‐to‐treat) and the results should, therefore, be interpreted with caution (see 'Methodological quality on included studies').
Of the primary and secondary outcomes assessed, the only differences identified were that the Portex cannula was more difficult (RR 3.26; 95% CI 1.38 to 7.67), Analysis 4.1, and more painful to insert (RR 5.81; 95% CI 1.41 to 23.88), Analysis 4.2 than the aluminium cannula.
4.1. Analysis.

Comparison 4 Portex cannula compared with aluminium cannula (transcervical), Outcome 1 Difficult insertion.
4.2. Analysis.

Comparison 4 Portex cannula compared with aluminium cannula (transcervical), Outcome 2 Pain during insertion.
Flexible catheter versus catheter with stiff guide probe
The Chalkiadakis 1993 trial failed to show clear differences between the two methods.
(ii) Transabdominal approach
Two trials involving 285 women were included comparing different needle techniques for transabdominal CVS.
Standard discontinuous suction using a 20 mL syringe (held with a hand‐grip device) versus continuous negative pressure 4 mL vacutainer aspiration system
These two needle techniques were compared in a study by Battagliarin 2009. There was no difference in failure to obtain an adequate sample (greater than 5 mg of villi) on the first attempt (RR 1.02; 95% CI 0.54 to 1.93), Analysis 7.1, when the standard syringe technique with a hand‐grip device was compared with the vacutainer. Despite this, a second needle insertion was significantly less likely with the standard syringe approach (RR 0.11; 95% CI 0.01 to 0.88), Analysis 7.2. There was no difference in perceived pain experienced between women in whom the standard syringe or the vacutainer technique was used (MD 0.00; 95% CI ‐0.04 to 0.04), Analysis 7.3, based on a visual analogue scale assessment performed immediately after the CVS procedure. The rate of fetal loss with a normal karyotype was 1.7% with all fetal losses occurring with the standard syringe (RR 7.15; 95% CI 0.37 to 136.50), Analysis 7.4.
7.1. Analysis.

Comparison 7 Standard syringe with hand‐grip device compared with vacutainer needle techniques (transabdominal), Outcome 1 Failure to obtain sample > 5 mg on first attempt.
7.2. Analysis.
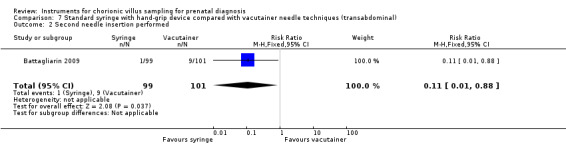
Comparison 7 Standard syringe with hand‐grip device compared with vacutainer needle techniques (transabdominal), Outcome 2 Second needle insertion performed.
7.3. Analysis.

Comparison 7 Standard syringe with hand‐grip device compared with vacutainer needle techniques (transabdominal), Outcome 3 Perceived pain.
7.4. Analysis.

Comparison 7 Standard syringe with hand‐grip device compared with vacutainer needle techniques (transabdominal), Outcome 4 Fetal loss with normal karyotype.
There was no significant difference in the mean weight of chorionic villi obtained between groups (MD 0.40; 95% CI ‐2.25 to 3.05), Analysis 7.5. The other secondary outcomes were not assessed.
7.5. Analysis.

Comparison 7 Standard syringe with hand‐grip device compared with vacutainer needle techniques (transabdominal), Outcome 5 Mean weight of tissue obtained.
Standard discontinuous suction using a syringe versus continuous negative pressure system using syringe with fixed piston (held by a metallic clip and controlled with a three‐way stopcock)
Buyukkurt 2010 randomised 85 women to one of these two needle techniques in ongoing, singleton gestations. The experimental technique is described in detail in Buyukkurt 2009. Failure to obtain an adequate sample (more than 5 mg of villi) on the first sample was significantly greater with the traditional syringe (RR 2.73; 95% CI 1.08 to 6.92), Analysis 8.1. Pregnancy outcomes were assessed up to four weeks following the CVS procedure and there was only one spontaneous miscarriage identified which occurred in the standard syringe group (RR 2.93; 95% CI 0.12 to 70.00), Analysis 8.2. Other primary outcomes were not assessed.
8.1. Analysis.

Comparison 8 Standard syringe compared with fixed piston syringe needle techniques (transabdominal), Outcome 1 Failure to obtain sample > 5 mg on first attempt.
8.2. Analysis.

Comparison 8 Standard syringe compared with fixed piston syringe needle techniques (transabdominal), Outcome 2 Spontaneous miscarriage (up to 4 weeks post‐procedure).
The mean weight of chorionic tissue obtained was significantly lower in the standard syringe group (MD ‐14.80 mg; 95% CI ‐21.71 to ‐7.89), Analysis 8.3. However, the culture was successful in all cases for both groups, Analysis 8.4. Other secondary outcomes were not assessed.
8.3. Analysis.

Comparison 8 Standard syringe compared with fixed piston syringe needle techniques (transabdominal), Outcome 3 Mean weight of tissue obtained.
8.4. Analysis.

Comparison 8 Standard syringe compared with fixed piston syringe needle techniques (transabdominal), Outcome 4 Culture unsuccessful.
Discussion
The identified randomised trials are too small to provide reliable evidence in favour of one particular instrument or technique for transcervical or transabdominal chorionic villus sampling (CVS). It is possible that the type of instrument or technique used can have profound effects on the sampling success and subsequent risk of pregnancy loss.
Only three trials (Barkai 1989; Chalkiadakis 1993; von Dadelszen 2005) studying transcervical CVS techniques enrolled women with ongoing pregnancies (n = 392). Therefore, although the current data are reassuring and there appears to be no difference in the risk of miscarriage with the use of a cannula compared with biopsy forceps, the impact of various instruments for transcervical CVS on important clinical outcomes remains unknown.
There were only two trials (Battagliarin 2009; Buyukkurt 2010) that enrolled women who had transabdominal CVS in ongoing pregnancies (n = 285), each of which studied different continuous negative pressure needle aspiration systems compared with the more common discontinuous negative pressure needle technique created by a syringe attached to a 20 gauge needle. There was no difference in the rate of miscarriage following the procedure in either study. However, the studies produced discrepant findings with the study by Battagliarin 2009 finding no significant difference between groups in the mean weight of chorionic villi obtained or in failure to obtain an adequate sample (more than 5 mg of villi) on the first attempt, whereas the study by Buyukkurt 2010 found both of these outcomes to be significantly more favourable with the continuous negative pressure technique. Possible reasons for this heterogeneity include:
i) the difference in operator experience between the two studies and the potential learning curve associated with using the new technique. In the study by Battagliarin 2009, there were four operators with different levels of experience, two with intermediate and two with advanced experience, but all were new to the continuous negative pressure technique. In the study by Buyukkurt 2010, only one operator performed the procedure, who had experience with the standard technique. His experience with the novel technique included 10 procedures on pregnant sheep as well as practice using a model.); ii) use of a hand‐grip device with the standard syringe control in the study by Battagliarin 2009 but not in the study by Buyukkurt 2010; and iii) the difference in technique used to estimate the weight of the sample (a visual analogue scale set to a photographic standard in the Battagliarin 2009 study compared with a laboratory microbalance to 0.001 mg used in the Buyukkurt 2010 study).
Authors' conclusions
Implications for practice.
On balance, the data from identified randomised trials favour small forceps for transcervical chorionic villus sampling (CVS). Overall, the data on transabdominal CVS suggest there is no clinically important difference in outcomes between the continuous and discontinuous negative pressure needle techniques. However, the methodological weakness of the included studies and the small number of participants make any firm conclusions unreliable for either CVS approach.
Implications for research.
Larger trials are required if differences in the success rate and complications are to be estimated reliably. For example, an increase of 2% in the total perinatal loss with one particular instrument would encourage most clinicians to use a safer alternative. Assuming that the total perinatal loss in women who request prenatal diagnosis in the first trimester is around 10% (Alfirevic 1999), around 6500 women would need to be recruited to ensure that a 2% difference (if true) is detected. This is relevant to transabdominal CVS for which two different instruments (single needle and double needle) are used regularly, with potentially important differential effects on perinatal loss.
Of greatest need is a large, randomised controlled trial directly comparing transcervical and transabdominal CVS procedures as it is not known if there is a difference in safety and efficacy between these approaches and often the clinical scenario allows for either.
What's new
| Date | Event | Description |
|---|---|---|
| 31 August 2012 | New citation required but conclusions have not changed | Conclusions not changed, but now trials comparing instruments for transabdominal chorionic villus sampling incorporated. |
| 31 August 2012 | New search has been performed | Review updated. Three new studies included (Battagliarin 2009; Buyukkurt 2010; von Dadelszen 2005) and one excluded (Cochrane 2003). Methods updated. |
History
Protocol first published: Issue 2, 1996 Review first published: Issue 2, 1996
| Date | Event | Description |
|---|---|---|
| 3 November 2008 | Amended | Converted to new review format. |
| 1 May 2002 | New search has been performed | A new trial, the largest so far, has been added. The trial compared biopsy forceps with cannula in women who had transvaginal chorionic villus sampling. |
Acknowledgements
The first systematic review on this topic was compiled by Professor Adrian Grant. We are also grateful to Professor Jim Neilson for his input into earlier versions of this review.
As part of the pre‐publication editorial process, this review has been commented on by four peers (an editor and three referees who are external to the editorial team) and the Group's Statistical Adviser.
Appendices
Appendix 1. Methods used to assess trials included in previous versions of this review
The following methods were used to assess Barkai 1989; Chalkiadakis 1993; MacKenzie 1986; Pons 1989; von Dadelszen 2005.
All trials were assessed for methodological quality using the criteria in the Cochrane Handbook (Clarke 2000), with a grade allocated to each trial on the basis of allocation concealment. Allocation concealment was scored as A (adequate) for telephone randomisation and use of consecutively numbered sealed envelopes; B (unclear) for trials where randomisation is not clearly described or prone to bias (e.g. open cards, toss of a coin) or C for quasi‐randomised designs, such as alternate allocation and use of record numbers. No other formal or informal qualitative analysis was planned as there were no planned exclusions based on quality.
The data were extracted on the 'hard‐copy' data sheets, entered onto the Review Manager computer software (RevMan 2000), checked for accuracy, and analysed using the RevMan software. The data were extracted by allocated intervention, irrespective of compliance with the allocated intervention, in order to allow an 'intention‐to‐treat' analysis. Women who were randomised and then either excluded or lost to follow‐up were assumed to have no event in the main analysis.
In the presence of significant heterogeneity, a subgroup analysis was planned based on the quality of allocation concealment.
Data and analyses
Comparison 1. Aspiration cannula compared with biopsy forceps (transcervical).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to obtain sample > 5 mg | 2 | 319 | Risk Ratio (M‐H, Random, 95% CI) | 3.81 [1.52, 9.56] |
| 2 Need for reinsertion of instrument (Failure to obtain sample in the first attempt) | 2 | 259 | Risk Ratio (M‐H, Random, 95% CI) | 2.44 [0.83, 7.20] |
| 3 Moderate‐to‐severe pain during procedure | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [1.11, 3.37] |
| 4 Spontaneous miscarriage | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.14, 6.96] |
| 5 Culture unsuccessful | 1 | 194 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Difficult insertion | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.34, 1.00] |
| 7 Poor visualisation of the instrument | 2 | 319 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.15, 6.87] |
| 8 Bleeding | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.04, 2.85] |
| 9 Heavy vaginal bleeding (less than 2 days post‐CVS) | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 4.11] |
| 10 Cost per procedure | 1 | 189 | Mean Difference (IV, Fixed, 95% CI) | 183.7 [152.62, 214.78] |
| 11 Failure to obtain villi | 2 | 319 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.47, 2.61] |
| 12 Intrauterine haematoma | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 96.13] |
| 13 Total time to aspirate sample > 90s | 1 | 199 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.68 [1.36, 2.08] |
| 14 Uterine sound required | 1 | 199 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 8.01] |
| 15 Tenaculum required | 1 | 199 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.63, 3.85] |
| 16 Procedure difficult | 1 | 199 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.75 [1.03, 2.96] |
| 17 Laboratory sample preparation time | 1 | 185 | Mean Difference (IV, Fixed, 95% CI) | 2.80 [0.55, 5.05] |
| 18 Direct karyotype successful | 1 | 165 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.90, 1.00] |
| 19 Rise in maternal serum aFP [ug/L] (1 hour post‐CVS) | 1 | 199 | Mean Difference (IV, Fixed, 95% CI) | ‐3.40 [‐24.48, 17.68] |
| 20 Rise in maternal serum hCG [kIU/L] (1 hour post‐CVS) | 1 | 199 | Mean Difference (IV, Fixed, 95% CI) | 1.10 [‐3.13, 5.33] |
| 21 Rise in Betke‐Kleihauer [mL] (1 hour post‐CVS) | 1 | 199 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.01, 0.01] |
| 22 Rise in HbF‐positive cells in maternal circulation [1%] (1 hour post‐CVS) | 1 | 199 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐0.09, 0.03] |
| 23 Procedure information given unsatisfactory for women | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.11, 3.90] |
| 24 Subjectively poor control during procedure for patient | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.88, 2.15] |
| 25 Sampling time seemed too long | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [1.01, 6.18] |
| 26 Sampling embarrassing | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.0 [1.38, 26.11] |
| 27 Moderate‐to‐severe pain (less than 2 days post‐CVS) | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 28 Vaginal fluid loss (less than 2 days post‐CVS) | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.11, 3.90] |
| 29 Pyrexia [greater than 37.5C] (less than 2 days post‐CVS) | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 30 Total pregnancy loss | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.58, 3.27] |
| 31 Termination of pregnancy for aneuploidy | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.25 [0.72, 7.07] |
| 32 Perinatal mortality | 1 | 183 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.01, 4.34] |
| 33 Prelabour rupture of the membranes (greater than 2 days post‐CVS) | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 34 IUGR | 1 | 183 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.17 [0.34, 29.90] |
| 35 Pre‐eclampsia | 1 | 183 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.07, 1.70] |
| 36 Delivery gestation | 1 | 183 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐0.34, 0.94] |
| 37 Birthweight | 1 | 183 | Mean Difference (IV, Fixed, 95% CI) | ‐143.0 [‐340.80, 54.80] |
| 38 Birthweight centile (gestational age‐ and sex‐corrected) | 1 | 183 | Mean Difference (IV, Fixed, 95% CI) | ‐1.70 [‐8.68, 5.28] |
| 39 Level 3 neonatal intensive care unit/special care baby unit admission | 1 | 183 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.5 [0.52, 173.95] |
| 40 Congenital malformation (maternal report by 3 weeks postnatally) | 1 | 183 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.22, 5.10] |
1.11. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 11 Failure to obtain villi.
1.12. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 12 Intrauterine haematoma.
1.13. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 13 Total time to aspirate sample > 90s.
1.14. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 14 Uterine sound required.
1.15. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 15 Tenaculum required.
1.16. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 16 Procedure difficult.
1.17. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 17 Laboratory sample preparation time.
1.20. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 20 Rise in maternal serum hCG [kIU/L] (1 hour post‐CVS).
1.21. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 21 Rise in Betke‐Kleihauer [mL] (1 hour post‐CVS).
1.22. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 22 Rise in HbF‐positive cells in maternal circulation [1%] (1 hour post‐CVS).
1.23. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 23 Procedure information given unsatisfactory for women.
1.24. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 24 Subjectively poor control during procedure for patient.
1.25. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 25 Sampling time seemed too long.
1.26. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 26 Sampling embarrassing.
1.27. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 27 Moderate‐to‐severe pain (less than 2 days post‐CVS).
1.28. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 28 Vaginal fluid loss (less than 2 days post‐CVS).
1.29. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 29 Pyrexia [greater than 37.5C] (less than 2 days post‐CVS).
1.30. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 30 Total pregnancy loss.
1.31. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 31 Termination of pregnancy for aneuploidy.
1.32. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 32 Perinatal mortality.
1.33. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 33 Prelabour rupture of the membranes (greater than 2 days post‐CVS).
1.34. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 34 IUGR.
1.35. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 35 Pre‐eclampsia.
1.36. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 36 Delivery gestation.
1.37. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 37 Birthweight.
1.38. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 38 Birthweight centile (gestational age‐ and sex‐corrected).
1.39. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 39 Level 3 neonatal intensive care unit/special care baby unit admission.
1.40. Analysis.

Comparison 1 Aspiration cannula compared with biopsy forceps (transcervical), Outcome 40 Congenital malformation (maternal report by 3 weeks postnatally).
Comparison 2. Portex cannula compared with silver cannula (transcervical).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to obtain sample greater than 5 mg | 1 | 168 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.23 [1.25, 3.98] |
| 2 Failure to obtain sample after 3 attempts | 1 | 168 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.38 [1.10, 5.12] |
2.2. Analysis.

Comparison 2 Portex cannula compared with silver cannula (transcervical), Outcome 2 Failure to obtain sample after 3 attempts.
Comparison 3. Portex cannula compared with malleable cannula (transcervical).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Difficult insertion | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.87 [1.00, 3.51] |
| 2 Pain during insertion | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.88 [0.84, 4.17] |
| 3 Bleeding during insertion | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.13, 1.66] |
| 4 Poor visualisation of cannula tip | 1 | 59 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.74 [0.66, 4.58] |
| 5 Failure to obtain sample greater than 5 mg | 1 | 69 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.80, 1.53] |
3.1. Analysis.

Comparison 3 Portex cannula compared with malleable cannula (transcervical), Outcome 1 Difficult insertion.
3.2. Analysis.

Comparison 3 Portex cannula compared with malleable cannula (transcervical), Outcome 2 Pain during insertion.
3.3. Analysis.

Comparison 3 Portex cannula compared with malleable cannula (transcervical), Outcome 3 Bleeding during insertion.
3.4. Analysis.

Comparison 3 Portex cannula compared with malleable cannula (transcervical), Outcome 4 Poor visualisation of cannula tip.
3.5. Analysis.

Comparison 3 Portex cannula compared with malleable cannula (transcervical), Outcome 5 Failure to obtain sample greater than 5 mg.
Comparison 4. Portex cannula compared with aluminium cannula (transcervical).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Difficult insertion | 1 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.26 [1.38, 7.67] |
| 2 Pain during insertion | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.81 [1.41, 23.88] |
| 3 Bleeding during insertion | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.18, 2.99] |
| 4 Poor visualisation of cannula tip | 1 | 55 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.47, 2.46] |
| 5 Inadequate sample (less than 5 mg) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.49 [0.97, 2.27] |
4.3. Analysis.

Comparison 4 Portex cannula compared with aluminium cannula (transcervical), Outcome 3 Bleeding during insertion.
4.4. Analysis.

Comparison 4 Portex cannula compared with aluminium cannula (transcervical), Outcome 4 Poor visualisation of cannula tip.
4.5. Analysis.

Comparison 4 Portex cannula compared with aluminium cannula (transcervical), Outcome 5 Inadequate sample (less than 5 mg).
Comparison 5. Malleable cannula compared with aluminium cannula (transcervical).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Difficult insertion | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.74 [0.66, 4.60] |
| 2 Pain during insertion | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.1 [0.69, 13.83] |
| 3 Bleeding during insertion | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.50, 4.80] |
| 4 Poor visualisation of cannula tip | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.22, 1.70] |
| 5 Inadequate sample (less than 5 mg) | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [0.87, 2.07] |
5.1. Analysis.

Comparison 5 Malleable cannula compared with aluminium cannula (transcervical), Outcome 1 Difficult insertion.
5.2. Analysis.

Comparison 5 Malleable cannula compared with aluminium cannula (transcervical), Outcome 2 Pain during insertion.
5.3. Analysis.

Comparison 5 Malleable cannula compared with aluminium cannula (transcervical), Outcome 3 Bleeding during insertion.
5.4. Analysis.

Comparison 5 Malleable cannula compared with aluminium cannula (transcervical), Outcome 4 Poor visualisation of cannula tip.
5.5. Analysis.

Comparison 5 Malleable cannula compared with aluminium cannula (transcervical), Outcome 5 Inadequate sample (less than 5 mg).
Comparison 6. Flexible catheter compared with catheter with a stiff guided probe (transcervical).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 First insertion failed | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.86] |
| 2 Miscarriage | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
6.1. Analysis.

Comparison 6 Flexible catheter compared with catheter with a stiff guided probe (transcervical), Outcome 1 First insertion failed.
6.2. Analysis.

Comparison 6 Flexible catheter compared with catheter with a stiff guided probe (transcervical), Outcome 2 Miscarriage.
Comparison 7. Standard syringe with hand‐grip device compared with vacutainer needle techniques (transabdominal).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to obtain sample > 5 mg on first attempt | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.54, 1.93] |
| 2 Second needle insertion performed | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 0.88] |
| 3 Perceived pain | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.04, 0.04] |
| 4 Fetal loss with normal karyotype | 1 | 192 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.15 [0.37, 136.50] |
| 5 Mean weight of tissue obtained | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [‐2.25, 3.05] |
Comparison 8. Standard syringe compared with fixed piston syringe needle techniques (transabdominal).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to obtain sample > 5 mg on first attempt | 1 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.73 [1.08, 6.92] |
| 2 Spontaneous miscarriage (up to 4 weeks post‐procedure) | 1 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.93 [0.12, 70.00] |
| 3 Mean weight of tissue obtained | 1 | 85 | Mean Difference (IV, Fixed, 95% CI) | ‐14.80 [‐21.71, ‐7.89] |
| 4 Culture unsuccessful | 1 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Total pregnancy loss | 1 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.93 [0.32, 27.06] |
| 6 Termination of pregnancy for abnormal results | 1 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.95 [0.18, 20.74] |
8.5. Analysis.

Comparison 8 Standard syringe compared with fixed piston syringe needle techniques (transabdominal), Outcome 5 Total pregnancy loss.
8.6. Analysis.

Comparison 8 Standard syringe compared with fixed piston syringe needle techniques (transabdominal), Outcome 6 Termination of pregnancy for abnormal results.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Barkai 1989.
| Methods | Method of randomisation not stated. | |
| Participants | 168 women between 9 and 12 weeks of gestation in ongoing pregnancies. | |
| Interventions | Sampling was done using either Silver cannula (Down's surgical, UK) or the Portex catheter (Portex, Hythe, Kent, UK). In cases of failure after 3 attempts, the procedure was repeated using the other device. | |
| Outcomes | Successful outcome, amount of trophoblast obtained, complications (nonfatal and miscarriages). | |
| Notes | Data on complications are not presented according to randomised group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated. |
| Allocation concealment (selection bias) | High risk | Alternation used. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data complete. |
| Selective reporting (reporting bias) | High risk | Did not clearly specify primary and secondary outcomes. The complications (miscarriage and nonfatal outcomes) were presented in such a way that they can not be included in the meta‐analysis. |
| Other bias | Unclear risk | Not clear. |
Battagliarin 2009.
| Methods | Prospective randomised single‐blinded trial. Sequentially numbered sealed envelopes. | |
| Participants | 200 women between 10 weeks 2 days and 16 weeks 2 days gestation in ongoing, singleton pregnancies. | |
| Interventions | Both sampling approaches used a transabdominal technique with a 20‐guage needle. Sampling was done using either a standard technique with discontinuous negative pressure applied with a 20 mL syringe and attached hand‐grip device OR a continuous negative pressure technique created by a 4 mL vacutainer aspiration system connected to the needle. | |
| Outcomes | Reported outcomes include maximum depth of needle insertion, angle between the needle and the chorionic plate, number of passes of the needle within the chorion, weight of the first sample and of the total sample obtained which was estimated using a VAS set to a photographic standard, number of needle insertions, pain assessment immediately following the CVS procedure using a VAS, pregnancy outcomes (fetal loss), in vitro assessment of the maximum negative pressures obtained with different syringe sizes and a 4 mL vacutainer. | |
| Notes | Pregnancy outcome data missing for 8 patients (4%), 4 from each treatment group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided. |
| Allocation concealment (selection bias) | Unclear risk | "Sequentially numbered sealed envelopes". Does not state if opaque envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Neither the patient nor the clinician were blinded. This could impact the patients' perceived pain or the number of uterine needle insertions by the clinician. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The technician assessing the chorionic villi sample size was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The 8 patients (4%) who were lost to follow‐up (4 from each study group) were not included in the pregnancy outcomes analysis. |
| Selective reporting (reporting bias) | High risk | Did not clearly specify primary and secondary outcomes. |
| Other bias | Unclear risk | Not clear. |
Buyukkurt 2010.
| Methods | Prospective randomised trial. The last number of the participants' national identification number, either odd or even, determined the group to which they were placed. | |
| Participants | 85 women between 11 weeks 0 days and 13 weeks 6 days gestation in ongoing, singleton pregnancies. | |
| Interventions | Both sampling techniques used a transabdominal approach with a 20‐guage needle. Sampling was done using either a standard technique with discontinuous negative pressure created by a 20 mL syringe OR continuous negative pressure maintained by fixing the 20 mL syringe piston with a metallic clip and controlled with a 3 stopcock. The standard technique required an assistant to hold the ultrasound probe whereas the experimental technique allowed the clinician to self‐guide by holding the ultrasound probe. | |
| Outcomes | Reported outcomes included the relationship between the volume in the syringe and the corresponding negative pressure created, need for > 1 needle insertion, the weight of chorionic tissue obtained (assessed by a technician using a microbalance in the laboratory), success of cytogenetic evaluation, and pregnancy outcomes (fetal loss) up to 4 weeks following the procedure. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Patients were placed into two groups based on the last number of their national identification number, either odd or even (quasi‐randomisation). |
| Allocation concealment (selection bias) | High risk | Patients were placed into 2 groups based on the last number of their national identification number, either odd or even. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Neither the patient nor the clinician were blinded. This could impact the patients' perceived pain or the number of uterine needle insertions by the clinician. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The technician assessing the chorionic villi sample size was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data complete. |
| Selective reporting (reporting bias) | High risk | Did not clearly specify primary and secondary outcomes. |
| Other bias | Unclear risk | Not clear. |
Chalkiadakis 1993.
| Methods | Method of randomisation not described. | |
| Participants | 24 women randomised. Gestational age not given. | |
| Interventions | A flexible transcervical catheter was compared with transcervical flexible Holzgrave catheter which was inserted through a hollow special probe. | |
| Outcomes | Number of successful first insertions and number of miscarriages was given 'per randomised patient'. Number of insertions and amount of villi were given as 'total' per randomised group. The amount of villi per insertion was given as mean but without standard deviation. | |
| Notes | Intention‐to‐treat analysis was possible only for 2 outcomes: successful first insertion and miscarriage rate. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Inadequate information presented. |
| Selective reporting (reporting bias) | High risk | Inadequate information presented. Did not clearly specify primary and secondary outcomes. |
| Other bias | Unclear risk | Not clear. |
MacKenzie 1986.
| Methods | Method of randomisation not stated. | |
| Participants | 50 women between 8 and 12 weeks of gestation before planned termination of pregnancy. | |
| Interventions | 3 different devices were tested (Portex cannula, malleable stainless steel cannula and aluminium cannula). Each woman had 2 procedures (cannula was passed twice per procedure). In the majority of women, 2 different cannulae were used but in 38% of women 2 procedures were performed with the same type of cannula (3/50 with Portex, 8/50 with malleable cannula and 8/50 with aluminium cannula). | |
| Outcomes | Success rate, karyotype, quantity of villi, ease of insertion, visualisation, pain, bleeding, suction pressures of cannulae. | |
| Notes | Not every woman is included in every analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. |
| Allocation concealment (selection bias) | High risk | Prior to the study, 1 of the 3 cannula types was "randomly selected" and assigned to the procedure. The next two consecutive cannulas were used on each patient as each patient had two procedures performed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data complete. |
| Selective reporting (reporting bias) | High risk | Did not clearly specify primary and secondary outcomes. |
| Other bias | High risk | |
Pons 1989.
| Methods | Method of randomisation not stated. | |
| Participants | 30 women between 7 and 12 weeks of gestation before planned termination of pregnancy. | |
| Interventions | In all 30 participants, 2 biopsies were taken with the CVS catheter (1.5 mm in diameter) and 2 with a Storz tissue biopsy forceps (1.5 mm in diameter). After randomisation 1 group of 15 women had 2 aspirations followed by 2 forceps biopsies. The other 15 women had 2 forceps biopsies followed by 2 aspirations. | |
| Outcomes | Reported outcomes include successful biopsies and karyotyping, amount of tissue removed, ease of instrument insertion, echographic visualisation of biopsy instrument and intraoperative complications. | |
| Notes | Denominators vary. Some outcomes are reported per woman (n = 15), some per first 2 instrument insertions (n = 30) and some per total number of instrument insertions (n = 60). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data complete. |
| Selective reporting (reporting bias) | High risk | Did not clearly specify primary and secondary outcomes. |
| Other bias | High risk | |
von Dadelszen 2005.
| Methods | Prospective randomised single‐blinded trial. Consecutively numbered opaque envelopes, computer‐generated random number tables, blocks of 6. | |
| Participants | 200 women between 10 and 13 weeks of gestation in ongoing pregnancies. | |
| Interventions | Sampling was done using either Rodeck biopsy forceps or the Portex catheter (Portex, Hythe, Kent, UK). In cases of failure after 3 attempts, the procedure was abandoned. Operators permitted to change instruments. | |
| Outcomes | Primary: rise in aFP; secondary include rise in HCG, fetal red cells, early pregnancy complications, cytogenetics laboratory outcomes, final pregnancy outcomes, patient satisfaction, operator satisfaction, and cost analysis. | |
| Notes | 1 post‐randomisation withdrawal in the biopsy forceps arm; data missing from primary analysis, but pregnancy outcome data analysed by intention‐to‐treat. Final pregnancy outcome data missing for 2 cases. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random number tables in blocks of 6. |
| Allocation concealment (selection bias) | Low risk | Consecutively numbered opaque envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Neither the patient nor the clinician were blinded. This could impact the patients' perceived pain or the number of uterine needle insertions by the clinician. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Cytogenetics, biochemistry and flow cytometry staff were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Final pregnancy outcome data missing for 2 cases. |
| Selective reporting (reporting bias) | Low risk | Data complete. |
| Other bias | Low risk | |
aFP: alpha‐fetoprotein CVS: chorionic villus sampling HCG: human chorionic gonadotropin VAS: visual analogue scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Cochrane 2003 | The study used an ex vivo system of term placentae (37 weeks' gestation) following delivery which may not accurately mimic chorionic villus sampling performed in utero on first trimester placentae. |
Differences between protocol and review
For the 2012 update of the review, primary and secondary outcomes were specified.
Contributions of authors
Zarko Alfirevic and Peter von Dadelszen prepared the 2003 review and Carmen Young, with the other two authors, updated the review in 2011‐12. Zarko Alfirevic maintains the review.
Sources of support
Internal sources
University of Liverpool, UK.
Division of Maternal‐Fetal Medicine, Department of Obstetrics and Gynaecology, University of Toronto, Canada.
BC Research Institute for Children's and Women's Health, Vancouver, BC, Canada.
External sources
Physicians' Services Incorporated, Ontario, Canada.
British Columbia Ministry of Health, Canada.
Declarations of interest
Peter von Dadelszen is the first author of one of the trials (von Dadelszen 2005) included in this review. Zarko Alfirevic is the principal investigator of the study which was excluded (Cochrane 2003).
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Barkai 1989 {published data only}
- Barkai G, Rabinovici, Chaki R, Shalev J, Katznelson MBM, Mashiach S, et al. Transcervical chorionic villi sampling: a comparison between the silver cannula and the portex catheter. Gynecologic and Obstetric Investigation 1989;27:70‐3. [DOI] [PubMed] [Google Scholar]
- Rabinovici J, Barkai G, Maschiach S, Chaki R, Goldman B. Transcervical CVS. A comparison between the Portex canula and the silver needle. Proceedings of 11th European Congress of Perinatal Medicine; 1988 April 10‐13; Rome, Italy. 1988:143.
Battagliarin 2009 {published data only}
- Battagliarin G, Lanna M, Coviello D, Tassis B, Quarenghi A, Nicolini U. A randomized study to assess two different techniques of aspiration while performing transabdominal chorionic villus sampling. Ultrasound in Obstetrics and Gynecology 2009;33(2):169‐72. [DOI] [PubMed] [Google Scholar]
Buyukkurt 2010 {published data only}
- Buyukkurt S, Seydaoglu G, Demir C, Ozgunen FT, Evruke C, Guzel AB, et al. Evaluation of the feasibility of a new method for performing chorion villus sampling. Clinical and Experimental Obstetrics & Gynecology 2010;37(3):190‐2. [PubMed] [Google Scholar]
Chalkiadakis 1993 {published data only}
- Chalkiadakis G, Menton M, Wiest E, Schrage R. Catheter guided chorionic villus sampling technique. A prospective randomised study [Sondergesteurte Choriozottenbiopsietechnik ‐ eine prospektive randomisierte Studie]. Archives of Gynecology and Obstetrics 1993;254(1‐4):1243‐4. [Google Scholar]
MacKenzie 1986 {published data only}
- MacKenzie WE, Holmes DS, Webb T, Whitehouse C, Newton JR. A randomized study of three cannulas for transcervical chorionic villus sampling. American Journal of Obstetrics and Gynecology 1986;154:34‐9. [DOI] [PubMed] [Google Scholar]
Pons 1989 {published data only}
- Pons JC, Fernandez H, Eydoux P, Diallo A, Doumerc S, Frydman R, et al. Chorionic villus sampling (CVS). Randomized study of efficacy of two transcervical biopsy methods: aspiration cannulas and small forceps. European Journal of Obstetrics and Gynecology and Reproductive Biology 1989;32:187‐94. [DOI] [PubMed] [Google Scholar]
von Dadelszen 2005 {published and unpublished data}
- Dadelszen P, Sermer M, Hillier J, Allen L, Fernandes B, Johnson J, et al. Randomised‐controlled trial (RCT) of instruments for transcervical chorionic villus sampling (CVS). American Journal of Obstetrics and Gynecology 2000;182(1 Pt 2):S187. [Google Scholar]
- Dadelszen P, Sermer M, Hillier J, Allen L, Fernandes B, Johnson J, et al. A randomised controlled trial of biopsy forceps and cannula aspiration for transcervical chorionic villus sampling. BJOG: an international journal of obstetrics and gynaecology 2005;112:559‐66. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Cochrane 2003 {published data only}
- Cochrane L, Ainscough M, Alfirevic Z. The influence of needle and syringe size on chorionic villus sampling of term placentae: a randomised trial. Prenatal Diagnosis 2003;23:1049‐51. [DOI] [PubMed] [Google Scholar]
Additional references
Alfirevic 1999
- Alfirevic Z. Early amniocentesis versus transabdominal chorionic villus sampling for prenatal diagnosis. Cochrane Database of Systematic Reviews 1999, Issue 1. [DOI: 10.1002/14651858.CD000077] [DOI] [PMC free article] [PubMed] [Google Scholar]
Buyukkurt 2009
- Buyukkurt S, Evruke C, Demir C, Ozgunen FT, Kadayifci O. A new device to facilitate the chorion villus sampling. Journal of Perinatal Medicine 2009;37:425. [DOI] [PubMed] [Google Scholar]
CEMAT 1998
- CEMAT Group. Randomised trial to assess safety and fetal outcome of early and midtrimester amniocentesis. The Canadian Early and Mid‐trimester Amniocentesis Trial (CEMAT) Group. Lancet 1998;351(9098):242‐7. [PubMed] [Google Scholar]
Clarke 2000
- Clarke M, Oxman AD, editors. Cochrane Reviewers' Handbook 4.1 [updated June 2000]. In: Review Manager (RevMan) [Computer program]. Version 4.1 for Windows. Oxford, England: The Cochrane Collaboration, 2000.
Higgins 2011
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
RevMan 2000 [Computer program]
- The Cochrane Collaboration. Review Manager (RevMan). Version 4.1 for Windows. Oxford, England: The Cochrane Collaboration, 2000.
RevMan 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Smidt‐Jensen 1992
- Smidt‐Jensen S, Permin M, Philip J, Lundsteen C, Zachary JM, Fowler SE, et al. Randomised comparison of amniocentesis and transabdominal and transcervical chorionic villus sampling. Lancet 1992;340:1237‐44. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Alfirevic 2003
- Alfirevic Z, Dadelszen P. Instruments for chorionic villus sampling for prenatal diagnosis. Cochrane Database of Systematic Reviews 2003, Issue 1. [DOI: 10.1002/14651858.CD000114] [DOI] [PubMed] [Google Scholar]