

ABSTRACT
“In the field of observation, chance favours only the prepared mind” (Louis Pasteur). This motto seems to have guided our unexpected results published recently in Nature Communications, where we describe an epigenetic rheostat that regulates expression of the constituents of the lysosomal and autophagic systems.
KEY WORDS: Lysosome and autophagy gene transcription, intermittent drainage, Epigenetic regulation, Acetylation, MYC, MiT/TFE
Serendipity often leads to the most rewarding scientific findings. An accidental set of experiments brought Christian de Duve to the discovery of lysosomes, heterogeneous and ubiquitous organelles tasked with digesting and recycling macromolecules in an acidic environment.1 De Duve also introduced the word “autophagy” to describe the degradation of defective or aging cellular constituents by lysosomes. The fundamental role of the lysosomal system is epitomized in the devastating effects of impaired lysosomal catabolism occurring in the large group of inherited pediatric lysosomal storage diseases (LSDs).2 Curiously, the first LSD linked to deficiency of α-glucosidase, glycogenosis type II or Pompe disease, was again discovered serendipitously by a coworker of de Duve, Henri-Gery Hers.3 He identified α-glucosidase as a lysosomal enzyme and introduced the concept of LSDs, arguing that substrates of deficient lysosomal enzymes would store in lysosomes.
In the following decades, investigators identified mechanisms of lysosomal enzymes’ biosynthesis, post-translational modifications, inter-organellar routing and processing in lysosomes. These studies were performed to understand how this sequence of events was altered in LSDs. In 1967 Elizabeth Neufeld discovered again by accident the phenomenon of cross-correction of lysosomal enzymes, which set the basis for enzyme replacement therapy for LSDs4 These milestone discoveries were followed by the cloning of lysosomal hydrolase cDNAs, which enabled studies of disease-causing mutations in LSDs, and how they affect mutant enzymes’ activities. Later, the generation of genetically engineered animal models of different LSDs provided a platform to understand mechanisms of pathogenesis associated with lysosomal dysfunction, and to investigate therapeutic modalities.
Despite the steady progress made in this field, it remains a conundrum as to how the lysosomes undergo expansion in LSDs, and how they respond to specific metabolic cues. In 2009 Andrea Ballabio’s group began to answer these questions when they discovered that TFEB (transcription factor EB)5 a well-characterized member of the MiT/TFE (microphthalmia-associated-basic helix-loop-helix (bHLH) leucine zipper) family of transcription factors, regulates the coordinated transcription of lysosomal and autophagic genes.5 This function branded TFEB as the master regulator of lysosomal biogenesis and autophagy. It was later shown that two other family members, TFE3 and MITF, also recognize the same E-box motif, renamed CLEAR for “coordinated lysosomal expression and regulation”, within the proximal promoters of lysosomal and autophagy genes and, in turn, activate their transcription.5,6 What came as a surprise was that these transcription factors respond to nutrient availability downstream of the mTORC1 complex tethered at the lysosomal membrane. Thus, the lysosome has also been regarded as a nutrient sensing organelle.
Given that MiT/TFE are differentially expressed in tissues/cell types and change localization under stress conditions, one could ask which factors, besides nutrients, influence their levels and activity. In our publication, we addressed in part this question by demonstrating that MiT/TFE are regulated by an epigenetic and transcriptional network, another serendipitous finding.7 We wanted to understand how the gene encoding for the lysosomal sialidase, neuraminidase 1 (NEU1), was regulated transcriptionally. NEU1 functions in complex with two other lysosomal hydrolases, β-galactosidase and cathepsin A (PPCA), and deficiency of NEU1 leads to sialidosis, a pediatric, neurosomatic LSD. We have shown that sialidosis mice develop neuropathological features of sporadic Alzheimer’s disease, and a generalized fibrosis that in muscle leads to severe myopathy.8,9 Furthermore, Neu1 haploinsufficiency favors the occurrence of invasive, aggressive tumors, specifically sarcomas, in a cancer prone mouse.10 Together these results establish NEU1 loss-of-function as a predisposing, contributing factor to aging-associated diseases. Seeking for an epigenetic component that regulates NEU1 gene expression, we discovered that inhibition of histone deacetylases (HDACi) potently increases NEU1 transcription, enzyme levels and activity. HDACs, including the 11 Zn2+-dependent, classical members, regulate cellular pathways by repressing metabolic gene transcription. Given the interdependency of PPCA, NEU1 and β-galactosidase in complex, we also tested, their transcriptional levels upon HDACi, and used additional lysosomal genes as controls for the assay. Surprisingly, the transcription of many lysosomal genes increased upon HDACi inhibition, affecting lysosomal number and function. We further demonstrated that HDAC2, one of the most widely expressed classical HDACs, occupies the promoters of lysosomal genes in combination with E-box bHLH transcription factors. Realizing that c-MYC (MYC), the master regulator of metabolism whose transcription is regulated by HDACs, recognizes the same E-box/CLEAR sequence bound by MiT/TFE, we tested whether MiT/TFE would compete with MYC for occupancy of the E-box/CLEAR motif at the promoters of lysosomal genes. We discovered that not only MYC binds to lysosomal gene promoters, but also, together with HDAC2, engages in an epigenetic rheostat with MiT/TFE members. Upon HDACi, downregulation of MYC grants promoter occupancy to TFEB and TFE3 leading to increased lysosomal biogenesis and function (Figure 1). In addition, we showed that the MYC/HDAC2 complex occupies the promoters of TFEB and TFE3. We took the story a step further by showing that MYC and HDAC2 engage in a similar epigenetic rheostat with a new autophagy transcription factor, FOXH1, which regulates expression of autophagy genes and FOXH1, itself. (Figure 1).
Figure 1.
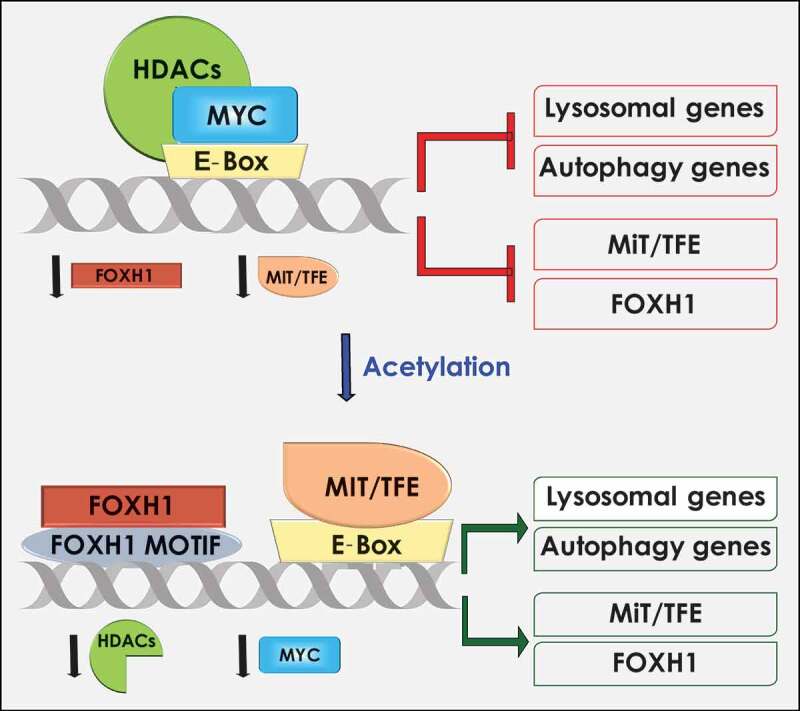
Proposed model for HDAC/MYC regulation of the lysosomal and autophagic systems. MYC and HDAC occupy the E-box–binding sites in the proximal promoters of lysosomal, autophagy, MiT/TFE, and FOXH1 genes, thereby inhibiting their expression. Inhibition of HDAC reduces MYC levels and allows for the binding of the E-boxes/CLEAR and/or FOXH1 motifs by MiT/TFE and FOXH1 transcription factors, which results in the transcriptional activation of lysosomal and autophagy genes.
Given the robust response of NEU1 mRNA expression to HDACi, we tested whether this treatment would increase the levels of mutant NEU1 mRNA in fibroblasts from patients with sialidosis. Remarkably, HDACi enhanced NEU1 mRNA expression and the residual activity of the mutant enzymes, creating a window of opportunity for therapy of this disease.
Our model (Figure 1) predicts that under physiological or pathological conditions the concentration of MYC determines the accessibility to lysosomal and autophagy gene promoters for occupancy by MiT/TFE or FOXH1. To further test this paradigm in biological systems where cell proliferation is dependent on high levels of MYC, we used a set of mouse group 3 medulloblastoma cell lines overexpressing Myc. In these samples, the expression of Mit/Tfe members was significantly downregulated, as was that of all tested lysosomal and autophagic genes. The latter was accompanied by reduced lysosomal volume and function, as well as decreased autophagy. These results were then confirmed in human cancer specimens and patient-derived tumor xenografts. We showed that in cancer cells the localization of MYC and HDAC2 in nuclei confines MiT/TFE factors to the cytoplasm, where they remain inactive. Another intriguing application of this epigenetic switch was observed in human induced pluripotent stem cells (hiPSCs). When compared to their parental fibroblasts, hiPSCs show increased levels of both MYC and HDAC2 proteins, consequently repressing TFEB expression and reducing lysosomal and autophagic protein levels. These results suggest that cells use these competing transcription factors to control their stemness and differentiation status.
In conclusion, this newly discovered epigenetic rheostat provides another tier of complexity to the fine-tuned regulation of lysosomal and autophagy genes in response to the myriad of physiologic and pathologic stimuli cells are exposed to. It is intuitive that this is not the end of the story but the beginning of a new chapter.
Funding Statement
A.d’A. holds the Jewelers for Children Endowed Chair in Genetics and Gene Therapy. This work was supported, in part, by NIH grants GM104981, and CA021764, the Assisi Foundation of Memphis and by the American Lebanese Syrian Associated Charities (ALSAC).
Acknowledgments
We thank Gerard Grosveld and Leigh Ellen Fremuth for critical reading of the manuscript and help in editing it, and Diantha van de Vlekkert for help in preparing the schematic figure.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.De Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F.. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 1955;60:1–3. doi: 10.1042/bj0600604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Platt FM, d’Azzo A, Davidson BL, Neufeld EF, Tifft CJ.. Lysosomal storage diseases. Nat Rev Dis Primers. 2018;4:27. doi: 10.1038/s41572-018-0025-4. [DOI] [PubMed] [Google Scholar]
- 3.Hers HG. alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease). Biochem J. 1963;86:11–16. doi: 10.1042/bj0860011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. 1968;162:570–572. doi: 10.1126/science.162.3853.570. [DOI] [PubMed] [Google Scholar]
- 5.Sardiello M, Palmieri M, Di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 6.Slade L, Pulinilkunnil T. The MiTF/TFE Family of Transcription Factors: master Regulators of Organelle Signaling, Metabolism, and Stress Adaptation. Mol Cancer Res. 2017;15:1637–1643. doi: 10.1158/1541-7786.MCR-17-0320. [DOI] [PubMed] [Google Scholar]
- 7.Annunziata I, van de Vlekkert D, Wolf E, Finkelstein D, Neale G, Machado E, Mosca R, Campos Y, Tillman H, Roussel MF, et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat Commun. 2019;10:3623. doi: 10.1038/s41467-019-11568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Annunziata I, Patterson A, Helton D, Hu H, Moshiach S, Gomero E, Nixon R, d'Azzo A. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-beta secretion via deregulated lysosomal exocytosis. Nat Commun. 2013;4:2734. doi: 10.1038/ncomms3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van de Vlekkert D, Demmers J, Nguyen XX, Campos Y, Machado E, Annunziata I, et al. Excessive exosome release is the pathogenic pathway linking a lysosomal deficiency to generalized fibrosis. Sci Adv. 2019;5:eaav3270. doi: 10.1126/sciadv.aav3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Machado E, White-Gilbertson S, van de Vlekkert D, Janke L, Moshiach S, Campos Y, Finkelstein D, Gomero E, Mosca R, Qiu X, et al. Regulated lysosomal exocytosis mediates cancer progression. Sci Adv. 2015;1:e1500603. doi: 10.1126/sciadv.1500603. [DOI] [PMC free article] [PubMed] [Google Scholar]