

ABSTRACT
Acquired resistance to anti-HER2 therapy is a significant clinical challenge in breast cancer. We recently discovered that during acquisition of resistance to HER2 inhibition, upregulation of the fatty acid transporter CD36 takes place, playing a key role in metabolic rewiring and resistance to anti-HER2 therapy.
KEYWORDS: FASN, FATP, lapatinib, lipid metabolism, trastuzumab
Cancer drug resistance is a significant problem. It appears that cancer cells almost always find a way to somehow escape any therapy as resistance could arise from multiple molecular mechanisms. In particular, when cancer cells become resistant to therapy targeting an oncogenic kinase, two mechanisms are largely accepted to play a role. First, a mutation in the targeted kinase could nullify the action of kinase inhibitors. Imatinib-resistant mutations of BCR-ABL in chronic myeloid leukemia are well known in this regard.1 Second, activation of a compensatory signaling pathway could help cancer cells to override inhibition of a kinase. For instance, inhibition of the receptor tyrosine kinase (RTK) epidermal growth factor receptor (EGFR) in non-small cell lung cancer could be overcome by amplification of another RTK, hepatocyte growth factor receptor (MET).2 In a recent study, our lab reported the unexpected discovery that CD36, a fatty acid (FA) transporter, plays a key role in acquired resistance to anti-HER2 therapy in breast cancer.3
HER2 (ERBB2, best known as HER2) is an RTK, which is often overexpressed in breast cancer. HER2 inhibitors, such as the humanized-antibody trastuzumab and the small molecule kinase inhibitor lapatinib, are widely used to treat patients with HER2+ breast cancer. Despite their initial effectiveness, however, acquired resistance to HER2-targeted inhibitors unavoidably develops.4 Previously, we generated multiple lapatinib-resistant breast cancer cell lines through chronic lapatinib exposure in formerly responsive lines and showed that lapatinib still completely inhibits autophosphorylation of HER2 in the lapatinib-resistant cells,5 suggesting that unlike imatinib-resistant leukemic cells, lapatinib resistance is not secondary to the insufficient inhibition of HER2. Indeed, activating mutations in HER2 are rare in breast cancer cells.6 Thus, it is more likely that an alternative pro-survival pathway might be activated in the resistant cells.
In the new study, we sought to identify such a compensatory mechanism and compared gene expression patterns between a naïve HER2+ breast cancer cell line and its lapatinib-resistant counterpart.3 A list of 304 differentially expressed genes was identified. Interestingly, none of those gene products appeared to directly compensate the inhibition of HER2 kinase activity as they were not upstream or downstream of the AKT and ERK pathways. Rather, gene enrichment analyses revealed that signaling pathways related to lipid metabolism were among the most significantly enriched pathways in resistant cells. Of 18 lipid metabolism-related gene transcripts, the FA transporter CD36 was most enriched in the resistant cells. Indeed, expression of CD36 was significantly enhanced in lapatinib-resistant cells at both the mRNA and protein levels. The transmembrane protein CD36 is an FA translocase that facilitates cellular uptake of exogenous FAs.7 Accordingly, we observed an increase in the presence of lipid droplets in lapatinib-resistant cells as compared to their sensitive counterparts. Moreover, lapatinib-resistant cells displayed a significantly enhanced rate of uptake of a fluorescently tagged fatty acid as compared to the parental line. Importantly, siRNA-mediated CD36 knockdown alone induced apoptotic cell death in lapatinib-resistant cells, but not in sensitive cells.
FAs are lipids essential for signal transduction, energy production, and membrane biogenesis. In general, cells acquire FAs from exogenous sources via CD36-mediated uptake and/or endogenously through de novo lipogenesis involving FA synthase (FASN) (Figure 1). FASN is critical in lipid metabolism by converting precursors derived from aerobic glycolysis and glutamine anaplerosis into FAs. Although normally expressed at low levels in tissues other than liver and adipose, FASN is highly expressed in many cancers, including breast cancer, and is associated with poor prognosis.8 Interestingly, FASN inhibitors (e.g., (-)-C75) trigger apoptosis in breast cancer cells.8 In contrast, however, our new study shows that lapatinib-resistant cells are markedly resistant to (-)-C75.3 These results suggest that there is a metabolic shift in lapatinib-resistant cells toward reliance on CD36-mediated FA uptake over de novo FA synthesis for maintaining the cellular FA pool (Figure 1).
Figure 1.
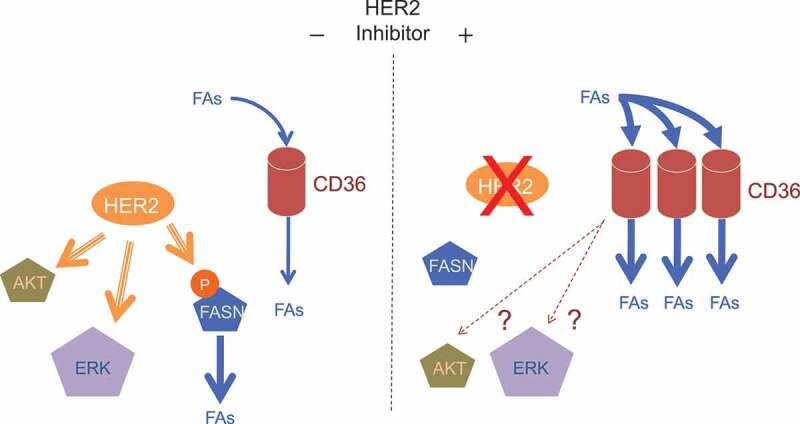
CD36 is induced by HER2-targeted therapy and is required for HER2+ breast cancer cells to acquire resistance to HER2 inhibitors. In general, HER2+ breast cancer cells generate fatty acids (FAs) endogenously through FA synthase (FASN) and exogenously via CD36. HER2 activates FASN by phosphorylation and also by transcriptional induction (left). Inhibition of HER2 by lapatinib or trastuzumab results in suppression of FASN activity. To compensate for the suppression of de novo FA synthesis, the CD36-mediated pathway is activated as the major source of FAs acquisition (right). Whether CD36 is also able to compensate for the loss of kinase signaling (e.g., activation of AKT and ERK kinases) remains to be elucidated (right).
The critical role of CD36 in lapatinib resistance was also supported by our mouse tumor xenograft experiment, in which a function-blocking anti-CD36 antibody markedly sensitized resistant tumors to lapatinib.3 Moreover, mammary gland-specific Cd36 knockout suppressed tumor growth and extended survival in a HER2/neu mammary tumor mouse model.3 We also showed that in breast cancer patients, CD36 expression is induced after anti-HER2 therapy, including not only lapatinib treatment but also trastuzumab treatment.3 Furthermore, high CD36 expression significantly correlates with a poor prognosis in HER2+ breast cancer patients, but not in HER2− breast cancer patients.3 Taken together, our studies indicate that CD36 plays a critical role in therapeutic resistance to anti-HER2 therapy.
How can inhibition of HER2 kinase be compensated by a FA transporter? Importantly, HER2 activates FASN through phosphorylation9 and also induces FASN gene expression indirectly10 (Figure 1). Thus, it is tempting to speculate that CD36 upregulation may be developed to compensate chronic FASN inhibition. However, other FA transporters, such as fatty acid transport proteins (FATP), were not found to be upregulated and linked to poor prognosis in our study.3 Why would breast cancer cells choose CD36 over other FA transporters? What is so special about CD36? CD36 is a multifunctional protein that regulates angiogenesis and cellular adhesion in addition to FA uptake.7 It may be that one of such functions of CD36, unrelated to FA uptake, is also critical for the survival of breast cancer cells. Further research remains to be done to elucidate the signaling downstream of CD36.
Funding Statement
The work was supported by an NIH Career Development Award R00 CA140948, an NIH R03 CA208384, and a Mary Kay Foundation research grant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.An X, Tiwari AK, Sun Y, Ding PR, Ashby CR Jr, ZS C.. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res. 2010;34:1–2. doi: 10.1016/j.leukres.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 3.Feng WW, Wilkins OM, Bang S, Ung M, Li J, An J, Del Genio CL, Canfield K, DiRenzo J, Wells W, et al. CD36-mediated metabolic rewiring of breast cancer cells promotes resistance to HER2-targeted therapies. Cell Rep. 2019;29:3405–3420.e5. doi: 10.1016/j.celrep.2019.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ.. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269–280. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 5.Kurokawa M, Kim J, Geradts J, Matsuura K, Liu L, Ran X, Xia W, Ribar TJ, Henao R, Dewhirst MW, et al. A network of substrates of the E3 ubiquitin ligases MDM2 and HUWE1 control apoptosis independently of p53. Sci Signal. 2013;6:ra32. doi: 10.1126/scisignal.2003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rexer BN, Engelman JA, Arteaga CL. Overcoming resistance to tyrosine kinase inhibitors: lessons learned from cancer cells treated with EGFR antagonists. Cell Cycle. 2009;8:18–22. doi: 10.4161/cc.8.1.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 9.Jin Q, Yuan LX, Boulbes D, Baek JM, Wang YN, Gomez-Cabello D, Hawke DH, Yeung SC, Lee MH, Hortobagyi GN, et al. Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010;12:R96. doi: 10.1186/bcr2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar-Sinha C, Ignatoski KW, Lippman ME, Ethier SP, Chinnaiyan AM. Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer Res. 2003;63:132–139. [PubMed] [Google Scholar]