

ABSTRACT
The oncoprotein transcription factor MYC is overexpressed in most cancers and is responsible for hundreds of thousands of cancer deaths worldwide every year. MYC is also a highly validated – but currently undruggable – anti-cancer target. We recently showed that breaking the interaction of MYC with its chromatin co-factor WD repeat-containing protein 5 (WDR5) promotes tumor regression in mouse xenografts, laying the foundation for a new strategy to inhibit MYC in the clinic.
KEYWORDS: Cancer, MYC, WDR5, Cancer therapy
The rise of molecularly targeted therapies has introduced an era of precision oncology that promises more effective cancer treatments with less harmful side effects. The number of targets for such therapies has increased considerably in recent years, fueled by an increase in understanding of the key processes and vulnerabilities in specific cancer settings and evolving views of what is considered druggable via pharmacologic means. Ultimately, it may be possible for clinicians to give patients a cocktail of targeted therapies that are precisely tuned to the molecular drivers of their disease. But given the disparate nature of the more than 100 human cancer types, and diminishing returns associated with pursuing pinpointed therapies against more and more specific cancer drivers, it is also important that we do not lose sight of those targets that would have shared therapeutic value across the majority of malignancies. And when thinking about common targets that fit the bill, one of the most important and potentially impactful is the emperor of all oncogenes: MYC.
The term “MYC” refers to a family of three DNA-binding transcription factors (c-, N-, and L-MYC) that dimerize with their obligate partner MAX to modulate the expression of genes linked to growth and proliferation.1 In normal cells, MYC proteins are tightly controlled by mechanisms that limit their expression or activity. But in cancer cells, mutations – either within the MYC genes or in other oncogene or tumor suppressor pathways – override these restrictive mechanisms, unleashing the potential of MYC to drive the cell cycle, promote protein synthesis, reprogram metabolism, induce genome instability, and defeat critical tumor surveillance pathways.1 Because loss of any one of a number of regulatory mechanisms can induce or dysregulate MYC, and due to the profound pro-tumorigenic consequences of MYC induction, loss of control of MYC is considered a hallmark of cancer.2 Indeed, most cancers overexpress at least one MYC family member, and conservative estimates suggest that one-third of all cancer deaths can be directly attributed to MYC activation.1 Based on these metrics, and what is known about the proteins themselves, a strong argument can be made for the case that it is impossible to establish, progress, or maintain the malignant state without the involvement of MYC.
Accordingly, there is great interest in the notion that targeting MYC could form the basis of a broadly-effective anti-cancer therapy. Time and time again, experimental inactivation of MYC in preclinical mouse models promotes tumor regression,3 even in cases where MYC is not the primary oncogenic lesion.4 On one hand, therefore, MYC is considered a highly-validated anti-cancer target. On the other hand, however, the absence of well-structured surfaces on MYC that are amenable to small molecule inhibition also renders it currently undruggable. Some advances have been made in protein-based approaches toward MYC inhibition,5 but in terms of routes that could produce drug-like MYC inhibitors the outlook is poor, and the goal of pharmacologically inhibiting MYC remains frustratingly out of reach … Or does it?
In 2015, we reported the identification of WD repeat-containing protein 5 (WDR5) – a component of multiple histone modifying complexes6 – as a critical factor that facilitates target gene recognition and tumorigenesis by MYC (Figure 1).7 We solved the X-ray crystal structure of part of MYC in complex with WDR5, and proposed that small molecules that bind the “MYC site” of WDR5 could be developed that would block the MYC–WDR5 interaction, displacing MYC from chromatin and disabling its tumorigenic function. If targeting the MYC–WDR5 nexus is to become a viable anti-cancer approach, however, several obstacles need to be overcome. We need to expose the gene networks controlled by MYC and WDR5 and ask if they are connected to the core tumorigenic functions of MYC. We need to know if disrupting the MYC–WDR5 connection in the context of an existing cancer will promote tumor regression. And we need some way to pharmacologically target WDR5. Our recent studies have begun to overcome these obstacles.8,9,10
Figure 1.
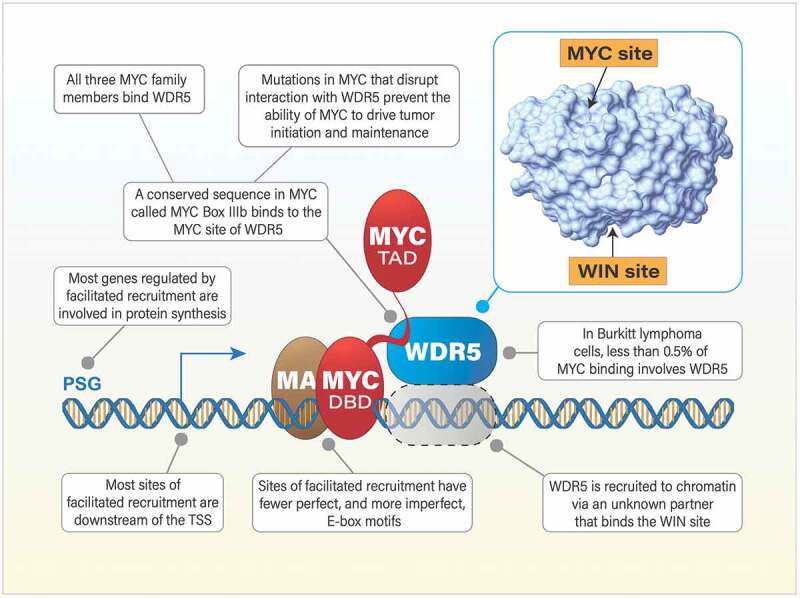
Facilitated recruitment of MYC to chromatin by WD repeat-containing protein 5 (WDR5) provides a new opportunity to therapeutically target MYC. In the facilitated recruitment paradigm, target gene recognition by MYC is an avidity-driven process that depends on (a) the inherent affinity of the DNA-binding domain (DBD) of MYC and MAX for a DNA sequence and (b) interaction of MYC with a pre-bound and proximal molecule of WDR5. In Burkitt lymphoma cells, there are fewer than 100 sites of facilitated recruitment, but most of these occur at “protein synthesis genes” (PSG), which are known to be important for the oncogenicity of MYC. The dependency of MYC on WDR5 to bind to and regulate these genes creates the opportunity to target MYC through WDR5. Small molecule inhibitors against the MYC site of WDR5 should leave WDR5 on chromatin but displace MYC. This prediction has not yet been tested. Small molecule inhibitors against the WIN (WDR5-interaction) site are known to displace WDR5 from chromatin, and our recent paper7 reports that MYC is also displaced from chromatin at PSGs. ‘TAD’ refers to the transcriptional activation domain of MYC. ‘TSS’ is transcriptional start site. E-box motifs are variations on the sequence ‘CACGTG”.
In our most recent paper,10 we studied the MYC–WDR5 connection in the context of Burkitt lymphoma, which is driven by a chromosomal translocation that places MYC expression under control of a potent immunoglobulin heavy chain enhancer. We learned that MYC and WDR5 co-bind to a fairly small group of genes that are manifestly connected to protein synthesis, including those encoding more than half of the proteins in the ribosome. This is an important point of intersection for MYC and WDR5, because the ability of MYC to drive biomass accumulation has long been recognized as a fundamental part of its oncogenic repertoire.1 By establishing a system to exchange wild-type for mutant MYC proteins in the Burkitt lymphoma setting, we confirmed that interaction with WDR5 is required to recruit MYC to chromatin at these genes. These MYC/WDR5 co-bound sites display a paucity of perfect E-boxes – MYC’s preferred binding sequence – consistent with the idea that WDR5 facilitates MYC recruitment to chromatin at sites with sub-prime DNA sequence information. Importantly, we also showed that exchanging wild-type for WDR5-interaction defective MYC in the context of an established tumor promotes rapid and profound tumor regression. Thus, from a therapeutic perspective, these data demonstrate that disrupting the MYC–WDR5 interaction has real anti-cancer potential.
We have discovered small molecules that bind the MYC site on WDR5 and disrupt the MYC–WDR5 interaction.9 These molecules require further refinement before extensive cell- or animal-based testing can begin. But for those who read our Burkitt lymphoma study closely,10 there is a strong sign that MYC-site inhibitors may not be absolutely needed to target MYC through WDR5.
In separate studies,8 we discovered potent small molecule inhibitors against a second site on WDR5 known as the “WIN (WDR5-interaction) site”6 (Figure 1). WIN site inhibitors were originally discovered with the intention of treating leukemias bearing MLL1 gene rearrangements, and every indication is that they will have utility in that context. In the course of characterizing these inhibitors, we learned that they act by promoting the wholesale eviction of WDR5 from chromatin. We do not know how the WIN site tethers WDR5 to chromatin. But if WDR5 is evicted from chromatin by our WIN site inhibitors, we reasoned, then MYC should be evicted with it at co-bound genes. Indeed, we showed that WIN site inhibition is as effective at displacing MYC from chromatin at protein synthesis genes as genetic disruption of the MYC–WDR5 interaction10 – a manipulation that, as mentioned above, causes rapid and complete tumor loss. The effect of WIN site inhibitors on recruitment of MYC to chromatin is a notable advance with real practical consequences: The WIN site of WDR5 is much more druggable than the MYC site, and there are several groups that have distinct WIN site inhibitors that – in light of our recent work – could be repurposed for targeting MYC.
We certainly have a long row to hoe in terms of bringing WDR5 inhibitors to the clinic as a way to thwart MYC function in cancer cells. But the foundation laid by our recent work – particularly the unexpected potential of WIN site inhibitors as anti-MYC agents – brings us one step closer to breaking the decades long stalemate and chaperones MYC into the druggable universe.
Funding Statement
This project has been funded in part with Federal funds from the National Cancer Institute, under Chemical Biology Consortium Contract No. HHSN261200800001E and grants CA200709, CA211305, CA148950, CA056036, and CA68485. This work was also supported by grants from the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation, The TJ Martell Foundation, St. Baldrick’s Foundation, Alex’s Lemonade Stand Foundation, and the Edward P. Evans Foundation.
Disclosure of potential conflicts of interest
S.W.F., S.R. Stauffer, W.P.T., E.T. Olejniczak, J. Phan, F. Wang, K. Jeon, and R.D. Gogliotti. were granted US Patent 10,160,763, “WDR5 Inhibitors and Modulators,” on December 25, 2018.
S.W.F., S.R. Stauffer, J.M. Salovich, W.P.T, F. Wang, J. Phan, and E.T. Olejniczak were granted US Patent 10,501,466 “WDR5 Inhibitors and Modulators,” on December 10, 2019.
References
- 1.Tansey WP. Mammalian MYC proteins and cancer. New J Sci. 2014;Article ID 757534:1. doi: 10.1155/2014/757534. [DOI] [Google Scholar]
- 2.Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4(6):a014241–3. doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. doi: 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, Swigart LB, Evan GI. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27(5):504–513. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beaulieu ME, Jauset T, Massó-Vallés D, Martínez-Martín S, Rahl P, Maltais L, Zacarias-Fluck MF, Casacuberta-Serra S, Serrano Del Pozo E, Fiore C, et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci Transl Med. 2019;11(484):eaar5012. doi: 10.1126/scitranslmed.aar5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guarnaccia AD, Tansey WP. Moonlighting with WDR5: a cellular multitasker. J Clin Med. 2018;7(2):21. doi: 10.3390/jcm7020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas LR, Wang Q, Grieb B, Phan J, Foshage A, Sun Q, Olejniczak E, Clark T, Dey S, Lorey S, et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell. 2015;58(3):440–452. doi: 10.1016/j.molcel.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aho ER, Wang J, Gogliotti RD, Howard GC, Phan J, Acharya P, Macdonald JD, Cheng K, Lorey SL, Lu B, et al. Displacement of WDR5 from chromatin by a WIN site inhibitor with picomolar affinity. Cell Rep. 2019;26(11):2916–2928 e13. doi: 10.1016/j.celrep.2019.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macdonald JD, Chacón Simon S, Han C, Wang F, Shaw JG, Howes JE, Sai J, Yuh JP, Camper D, Alicie BM, et al. Discovery and optimization of salicylic acid-derived sulfonamide inhibitors of the WD repeat-containing protein 5-MYC protein-protein interaction. J Med Chem. 2019. December 5;62:11232–11259. [Epub ahead of print]. doi: 10.1021/acs.jmedchem.9b01411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas LR, Adams CM, Wang J, Weissmiller AM, Creighton J, Lorey SL, Liu Q, Fesik SW, Eischen CM, Tansey WP. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc Natl Acad Sci U S A. 2019. November 25. pii: 201910391. [Epub ahead of print]. doi: 10.1073/pnas.1910391116. [DOI] [PMC free article] [PubMed] [Google Scholar]