

Abstract
BACKGROUND AND PURPOSE: MR imaging findings in adult-onset autosomal dominant leukodystrophy (ADLD) with autonomic symptoms have been described in the brain, but no descriptions of MR imaging findings in the spinal cord have been published. Here, we describe MR imaging findings in the spinal cord in adult-onset ADLD with autonomic symptoms and histopathologic investigations of the spinal cord.
MATERIALS AND METHODS: Twelve subjects from 2 families with adult-onset ADLD with autonomic symptoms identified by clinical investigation underwent MR imaging examination of the spinal cord. Sagittal and transverse sections were obtained. MR imaging examination of the brain was performed in 11 patients. One of the patients underwent postmortem examination, and the spinal cord was subjected to histopathologic analysis.
RESULTS: In all family members with adult-onset ADLD with autonomic symptoms, even in the asymptomatic person, the spinal cord was thin. All examined family members also had a slight general white matter signal intensity (SI) increase in the whole spinal cord, mainly visible in T2-weighted transverse images. The pathologic examination revealed a discrete demyelination in the spinal cord. Brain MR imaging also showed increased T2 SI in the white matter.
CONCLUSIONS: The spinal cord is affected in adult-onset ADLD with autonomic symptoms. Findings consist of atrophy and a diffuse T2 SI increase in the white matter. Transverse images are needed to assess these findings. The typical SI changes of the spinal cord are also present in subjects without clinical symptoms of the disease and with very limited changes in the brain.
Leukodystrophies are disorders affecting white matter by failed development or degeneration of myelin. These effects are the results of genetically determined errors of metabolism, and in most cases, they have an onset in early childhood. Inheritance of the more common forms is autosomal recessive or X-linked recessive.1
Adult-onset autosomal dominant leukodystrophy (ADLD; Online Mendelian Inheritance in Man accession number 169500) is thus a rarity and was initially reported in 1984 in an Irish-American kindred.2 The same disease was also described in 2 Swedish families and named adult-onset ADLD with autonomic symptoms.3 Similar disorders have been described in Japan and Italy, though these may be separate clinical entities.4-6
A detailed description of the MR imaging and histologic changes of the brain in adult-onset ADLD with autonomic symptoms has been published.3 To our knowledge, there are no reports on the radiologic changes outside the brain. Histopathologic examination of the spinal cord has been reported in 1 patient.7 In this case, there were vacuolar changes and loss of myelin in the white matter, whereas gross visual inspection had shown changes in the anterior, lateral, and dorsal columns. In another patient, no macroscopic changes were found in the spinal cord at postmortem examination.2
In this study, we describe MR imaging findings of the spinal cord of 12 members of 2 families with adult-onset ADLD with autonomic symptoms. We also describe spinal cord neuropathology in 1 patient. The findings are compared with those described in literature concerning other leukodystrophies, primarily adult-onset types.
Materials and Methods
Subjects
Twelve persons from 2 unrelated families with adult-onset ADLD with autonomic symptoms were enrolled in this study. The disease was identified by clinical characteristics, a pattern of autosomal dominant inheritance and white matter changes visualized on brain MR imaging, as previously described in a report on these families.3 Fig 1 shows the pedigree of the larger family. The members affected in family 2 were a mother and her son, the latter examined in this study. Autonomic nervous system evaluation was based on a history of micturition, bowel motion, and erectile problems. Ability to sweat and symptoms related to postural hypotension were evaluated. The blood pressure was measured at rest and after standing for 3 minutes. Neurophysiologic assessment of the autonomic nervous system included sympathetic skin response and R-R interval measurements on normal breathing, deep breathing, and a tilt test. One patient belonging to family 1 did not undergo brain MR imaging. He had symptoms and signs of the disease, including bladder and bowel dysfunction, preceding pyramidal signs and ataxia.
Fig 1.

Pedigree of family 1 with adult-onset ADLD with autonomic symptoms. Filled circle indicates affected female; filled square, affected male; arrows, family members examined in this study.
Genetic studies in family 1 confirmed linkage to chromosome 5q23,8 the genetic region harboring the LMNB1 gene.9
The study was approved by the local medical research ethics committee. All subjects gave their informed consent to enter the study.
One individual, 40 years of age, was asymptomatic, and clinical examination showed no signs of disease. Two individuals presented only with autonomic symptoms of disease, pathologic R-R intervals in the electrocardiogram and erectile dysfunction (in a 43-year-old man) and micturition urgency (in a 53-year-old woman). Three others (ages, 40–71 years) had signs of slight spasticity in the lower limbs in combination with autonomic symptoms. A further 6 subjects (ages, 45–61 years) had autonomic dysfunction, pyramidal signs, and ataxia. Ten subjects were able to walk independently, whereas 2 were wheelchair-bound.
MR Imaging Examinations
All subjects were examined with sagittal spin-echo (SE) T1- and fast SE T2-weighted sequences through the entire spinal cord. Transverse T2-weighted images were obtained by using a 3D SE sequence (TR/TE, 2500/120 ms) at the levels of C2, C5, T3, T6, T9, and the conus medullaris in 8 subjects. In 4 subjects, the transverse images were obtained with different T2-weighted SE or gradient-echo sequences in varying levels (the retrospective part of the study).
Signal intensity (SI) was assessed visually by an experienced neuroradiologist. When the SI was higher in white than in gray matter, the white matter SI was considered pathologic. Spinal cord anteroposterior and coronal diameters and cross-sectional areas were measured at the C2, T6, and T11 levels on the screen of a workstation, as described by Krabbe et al,10 and the measures were compared with those in their healthy controls (n = 15; ages; 24–61 years). The measures out of the range of 2 SDs were considered pathologic.
An MR imaging examination of the brain was performed with a routine technique on the same day as the spinal examination. In 1 patient in the retrospective material, the brain was not studied with MR imaging.
Histopathology
The spinal cord of a female patient who died at the age of 56 due to a postoperative lung embolus was investigated. She had a 3-year history of gait difficulties, as well as micturition urgency, ataxia, and pyramidal signs. The upper part of the cervical spinal cord was available for neuropathologic examination. Paraffin sections were stained for basic histopathology with hematoxylin-eosin and for myelin with Luxol fast blue-cresyl violet. Astrocytes were demonstrated immunohistochemically with antibodies to glial fibrillary acidic protein (GFAP); Dako, Glostrup, Denmark). Neurons and their processes were identified with neurofilament antibody (Euro/DPC, Gwynedd, UK), and phagocytic cells (microglial cells and macrophages) were identified with CD 68 antibody (Dako). The bound primary antibodies were visualized by using peroxidase-labeled secondary antibodies (Vector Laboratories, Burlingame, Calif), with diaminobenzidine as the chromogen and hematoxylin as the counterstain.
Results
Measurements of the Spinal Cord
In all patients and the clinically unaffected family member, a severe thinning of the spinal cord was manifest (Figs 2–4). All the anteroposterior and coronal diameters and the cross-sectional areas were significantly smaller than those in the control material (ie, out of the 2 SD range). The measurements and the control values from the literature10 are presented in the Table.
Fig 2.
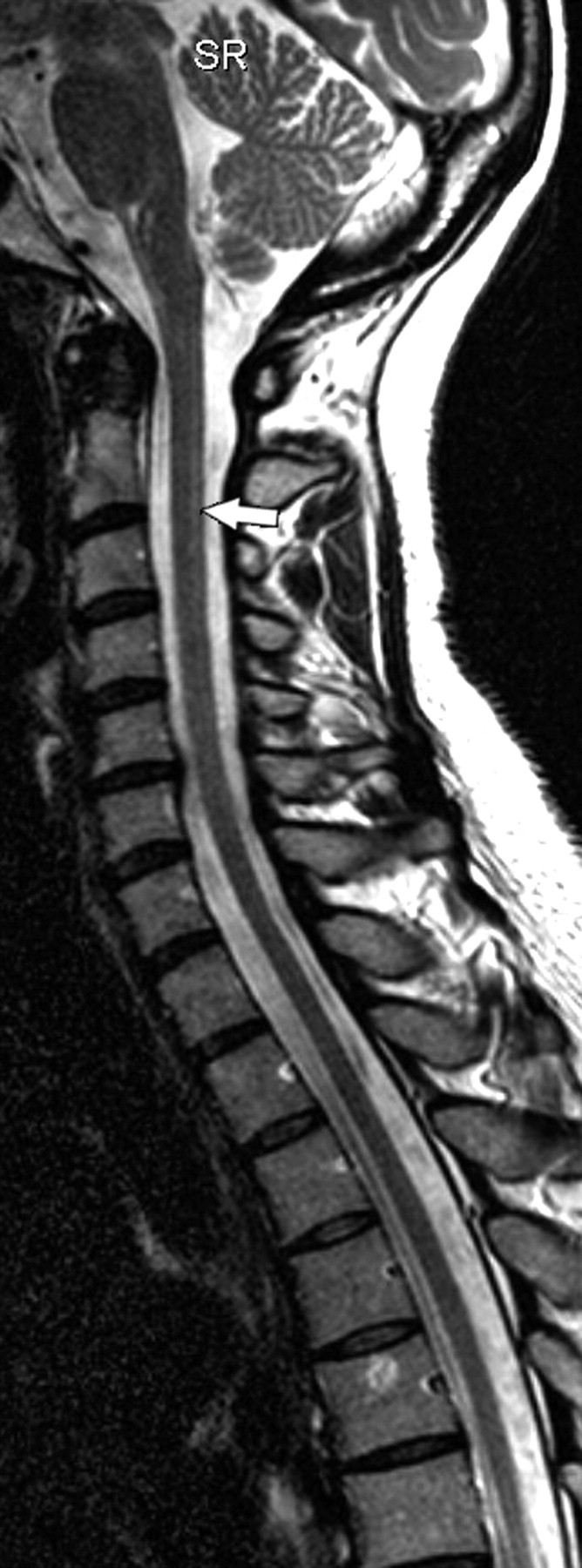
Sagittal T2-weighted image of the cervical spine in a 49-year-old woman with adult-onset ADLD with autonomic symptoms. Her symptoms consisted of micturition urgency and slight pyramidal affection. The spinal cord is thin, and slight SI changes in the posterior part of the spinal cord, easily mistaken for truncation artifacts, are seen (arrow).
Fig 4.
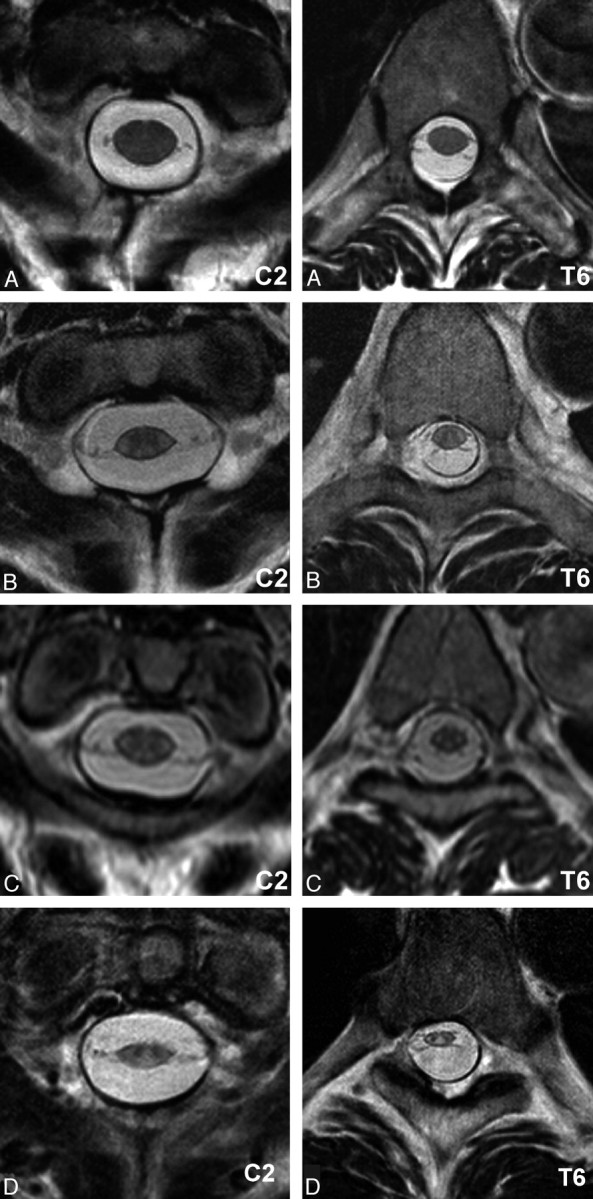
Transverse T2-weighted images of the spinal cord at the C2 and T6 levels of a healthy control subject (A), along with images of the same levels in an asymptomatic ADLD family member (B) and in the same patients as seen in Figs 2 and 3 (C and D, respectively). In all ADLD family members, a general white matter SI increase is seen and a reduction in the cross-sectional area. The thinning of the spinal cord seems to worsen with progression of symptoms. D, Parts of the dorsal columns have an increased SI indistinguishable from that in white matter SI.
Measurements of the spinal cord*
| Patients |
Controls† |
P Value | ||
|---|---|---|---|---|
| Mean ± SD | 95% CL | Mean ± SD | ||
| Anteroposterior diameter (mm) | ||||
| C2 | 5.6 ± 1.0 | 5.0–6.2 | 9.5 ± 0.6 | <.001 |
| T6 | 4.4 ± 0.7 | 4.0–4.9 | 7.8 ± 0.7 | <.001 |
| T11 | 4.8 ± 0.9 | 4.3–5.3 | 8.3 ± 1.1 | <.001 |
| Coronal diameter (mm) | ||||
| C2 | 10.2 ± 0.8 | 9.7–10.7 | 13.3 ± 1.1 | <.001 |
| T6 | 6.8 ± 0.6 | 6.5–7.2 | 10.2 ± 0.7 | <.001 |
| T11 | 8.1 ± 0.9 | 7.5–8.7 | 10.6 ± 1.5 | <.001 |
| Level cross-sectional area (mm2) | ||||
| C2 | 46.5 ± 8.4 | 40.7–52.3 | 93.9 ± 11.4 | <.001 |
| T6 | 24.4 ± 4.9 | 21.2–27.7 | 59.9 ± 7.4 | <.001 |
| T11 | 31.7 ± 7.1 | 26.5–37.0 | 78.4 ± 21.1 | <.001 |
Note:—CL indicates confidence limit.
Student 2-tailed t test was used for comparisons.
Measurements of the controls from Krabbe et al.10
SI of the Spinal Cord
A subtle increase in SI was observed along the posterior part of the spinal cord in sagittal T2-weighted images of the cervical and/or thoracic spine in 4 of the 12 patients (Fig 2). In the clinically most severely affected patient, 1 of the 2 patients who were wheelchair-bound, (see the case history below), distinct but diffuse SI changes were seen (Fig 3).
Fig 3.
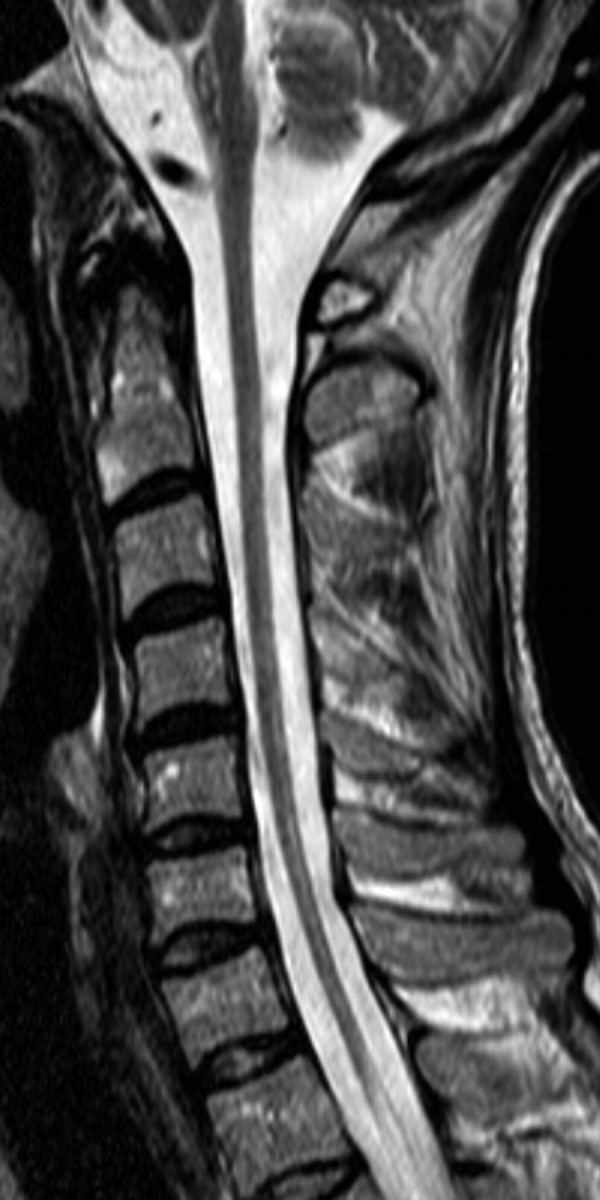
T2-weighted MR image of the cervical spine in the most severely affected patient, a man 51 years of age with paraparesis, severe dysautonomia, and ataxia. Severe atrophy of the spinal cord is seen, and diffuse SI changes are not confined to the posterior part of the spinal cord.
T2-weighted transverse images suggested a homogeneous increase of the SI of white matter along the whole spinal cord in all patients and in the asymptomatic family member (Fig 4B−D). Images of the conus medullaris were difficult to interpret; and also in stray sections elsewhere, motion (pulsation) artifacts disturbed evaluation of SI homogeneity, even when the overall quality of all 12 examinations was good.
MR imaging of the brain showed the same characteristic features as described previously in detail in the same families.3 The subjects included in this study presented varying severity grades of the radiologic brain involvement. Images of a patient with characteristic but not very advanced changes are shown in Fig 5. In all patients and in the asymptomatic family member, increased SI was seen in the cerebral white matter beneath the motor cortex and along the corticospinal tract down to the medulla oblongata. Images of the clinically affected patients also showed large frontoparietal white matter changes. In this leukodystrophy, myelin is better preserved close to the lateral ventricles than in the surrounding white matter (Fig 5). Seven patients had white matter changes also in the upper and middle cerebellar peduncles. Some atrophy was found in 10 subjects, most often in the corpus callosum (7 subjects) and in the medulla oblongata (8 subjects).
Fig 5.
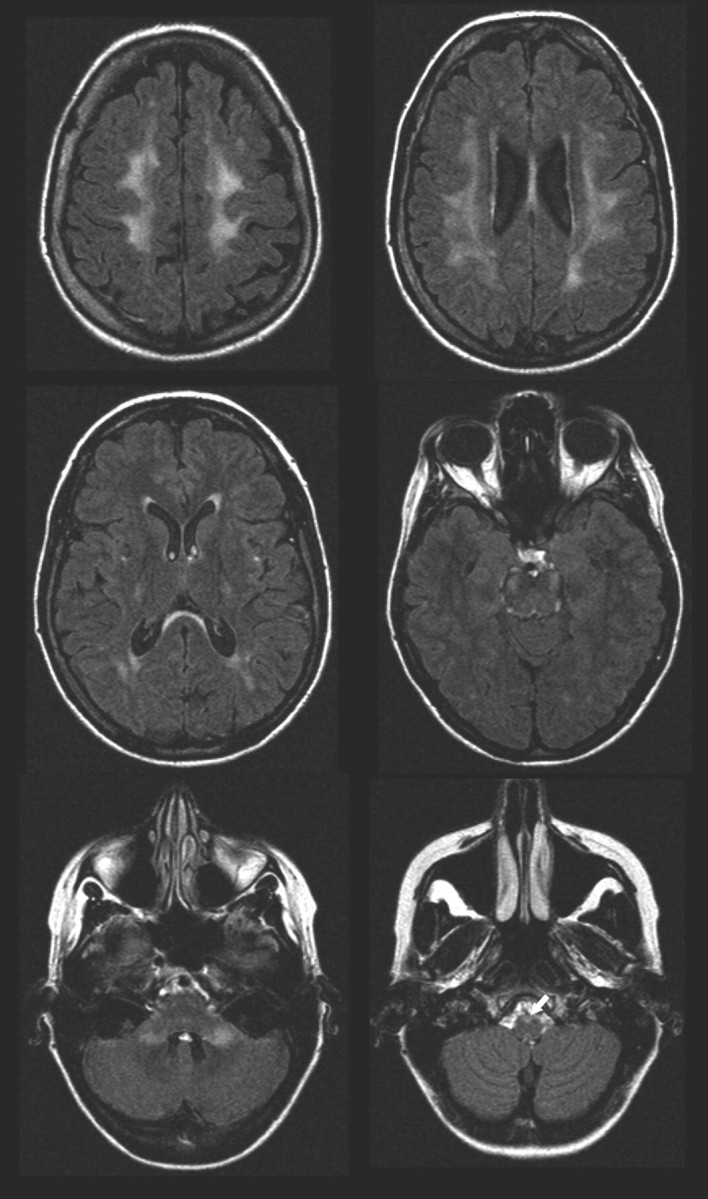
Fluid-attenuated inversion recovery images of the brain in the same patient as in Fig 2. Increased white matter SI can be seen beneath the motor cortex and in the upper part of the parietal lobes. The SI changes can be followed along the corticospinal tract including the pyramids (arrow) and in the cerebellar peduncles. A periventricular rim with less increased signal intensity is characteristic of adult-onset ADLD with autonomic symptoms.
Case History and MR Imaging Findings of the Most Severely Affected Patient
The most severely affected patient, a man 51 years of age, had paraparesis of the legs, postural and intentional tremor, bladder dysfunction, dry skin, slow bowel movements, and postural hypotension. The clinical examination also showed slight spasticity of all extremities, atactic eye movements, brisk tendon reflexes, and extensor plantar signs. MR imaging of the brain showed increased SI on T2-weighted images in almost all cerebral white matter. The optic radiations were spared. The corticospinal tract in the brain stem, the cerebellar peduncles, and the cerebellum adjacent to the fourth ventricle were also affected. The patient had marked general cerebral and cerebellar atrophy. The coronal diameter of the medulla oblongata was reduced. MR imaging examination of the spine revealed hyperintense white matter on T2-weighted images along the whole spinal cord (Figs 3 and 4D). In addition, parts of the posterior horns were partially indistinguishable from white matter, due to increased SI (Fig 4D). A marked thinning of the spinal cord was manifest in sagittal images (Fig 3) and when cross-sectional areas were measured. The cross-sectional area was 36 mm2 (mean ± 2 SD; in healthy individuals:8 93.9 ± 11.4 mm2) at the C2 level, 18 mm2 (normal: 59.9 ± 7.4 mm2) at the T6 level, and 23 mm2 (normal: 78.4 ± 21.1 mm2) at the T11 level. This was the thinnest spinal cord in all the patients examined, regardless of age or sex.
Case History and MR Imaging Findings of the Asymptomatic Family Member
The asymptomatic family member was 40 years old with an unremarkable medical history except for an episode of vertigo 2 months before the investigation. He had no autonomic symptoms or signs on clinical examination. MR imaging of the brain showed increased signal intensities on T2-weighted images in cerebral white matter below the motor and premotor cortices and in the posterior limb of the internal capsule. Small separate changes were seen parietally. Slight SI changes could also be seen in the cerebral and cerebellar peduncles and the pyramids. The coronal diameter of the medulla oblongata was slightly reduced. Of all subjects, his brain was the least affected on MR imaging. MR imaging of the spinal cord showed increased T2 SI in the whole white matter on transverse images (Fig 4B), though this was uncertain in the conus medullaris. No SI abnormalities were obvious in sagittal images. Thinning of the spinal cord was seen. The cross-sectional area was 59 mm2 at the C2 level, 33 mm2 at the T6 level, and 38 mm2 at the T11 level.
Histopathologic Findings
In the cervical spinal cord of the patient, no obvious loss of myelinated fibers was visible at a low magnification at postmortem examination (Fig 6A, -B). However, at a higher magnification, such a loss was observed subpially at the periphery of the spinal cord with both myelin and neurofilament stainings (Fig 7A, -B). The loss of nerve fibers had induced reactive astrogliosis (Fig 7C), whereas the number of microglial cells was not increased. The MR images of this patient showed atrophy and white matter changes consistent with MR imagingfindings in the other patients. We were not able to see any SI difference between the most peripheral and the other parts of white matter.
Fig 6.

In the spinal cord, no obvious loss of myelinated fibers, neither in the myelin-stained (A) nor in the silver-stained for axons (B), is detectable at a low magnification. A, Luxol fast blue-cresyl violet. B, Bielschowsky silver. Ventral part down.
Fig 7.

At a higher magnification, a loss of myelinated fibers can be seen subpially at the periphery of the spinal cord on the right (A and B), associated with moderate reactive astrocytosis (C). A, Luxol fast blue-cresyl violet. B and C, Immunostainings for neurofilament (B) and GFAP (C) with hematoxylin counterstaining. Original magnification: A and B, 40×; C, 20×. The bar in A and B is 100 μm.
Parietal sections of the brain from the same patient were also examined. Myelin was reduced, rarefied, and vacuolated with better preservation around blood vessels. Despite a prominent reduction of myelin, there was only minimal reactive gliosis.
Discussion
Six kindreds with adult-onset ADLD with the first symptoms from the autonomic nervous system have been described.2,3,7-9,11-14 Autonomic symptoms, manifesting during the fifth decade, usually precede cerebellar and pyramidal symptoms and consist of urinary incontinence, dry skin, orthostatic hypotension, and bowel dysfunction. The genetic cause underlying adult-onset ADLD with autonomic symptoms has been located at chromosome 5q8,14 and consists of a duplication in the LMNB1 gene, leading to an overexpression of lamin B1 in brain tissue of affected individuals.9
In the initial kindred, CT findings comprise signs of leukodystrophy most prominent in the frontoparietal and cerebellar white matter.12 MR imaging has revealed findings also in the occipital and temporal lobes in more advanced disease and white matter changes in the whole length of the corticospinal tract in the brain and the corpus callosum.3,12 Other typical findings in this disease entity were sparing or less severe alteration of the immediate periventricular white matter and slight atrophy of the corpus callosum. Relatively slight atrophy in the cerebrum and brain stem was also found. Similar findings were also revealed in the brains of asymptomatic members of a family with adult-onset ADLD with autonomic symptoms. Some asymptomatic individuals had changes only in the uppermost portion of the corticospinal tract below the cortex.3 Histopathologic examinations of brain tissue have revealed vacuolation and loss of myelin in affected white matter, with preservation of the oligodendroglia.3,7,14
We postulate that the white matter in the spinal cord is affected in adult-onset ADLD with autonomic symptoms and, further, that spinal cord white matter is affected even before the onset of clinical symptoms. A pattern of early involvement of the corticospinal tract appeared in the brain, but in the spinal cord, the entire white matter was affected in all patients. The whole spinal cord seems to be affected at a time when changes in the brain are still very limited. The SI changes in the spinal cord can be very subtle and easily overlooked in sagittal images. This finding agrees with the slight demyelinization seen histopathologically.
In the earlier study of the brain in adult-onset ADLD with autonomic symptoms, some atrophy was seen in 16 of 18 patients with SI changes, most markedly in measurements of the coronal diameter of the medulla oblongata and in thinning of the corpus callosum. Some subtle cerebral atrophy was seen in 11 of 14 symptomatic subjects.3 Compared with the atrophy of the brain, that in the spinal cord must be considered significantly more marked. This is somewhat surprising because there was a slighter degree of demyelination in the spinal cord than in the brain in the histopathologically examined patient, and a possible reason for the atrophy seen in the central nervous system of patients with adult-onset ADLD with autonomic symptoms could be loss of myelin. Histopathologic findings in patients with the disease are consistent with myelin involvement. It is interesting to note that the pathologic findings in the spinal cord were slighter than those in the brain when myelin loss was assessed, whereas a greater degree of reactive gliosis was seen. This finding suggests that part of the reason for the SI increase in the spinal cord is the reactive gliosis, something that perhaps could explain the slighter degree of SI increase in the spinal cord compared with the brain. Also, some vacuolization was seen in the cerebral white matter, whereas this was not seen in the spinal cord of our patient.
Pathologic findings in the spinal cord have been described in the Irish-American kindred, and changes in the anterior, lateral, and dorsal columns on gross visual assessment were reported along with loss of myelin and vacuolar changes seen on the histopathologic examination. These vacuoles became confluent and created a “cracked” appearance.7 It is possible that the slighter histopathologic changes in the spinal cord of our patient represent an earlier stage of the disease. The patient examined by Schwankhaus et al7 had a disease duration of >10 years and had been admitted to a care facility, whereas the patient examined in our study had experienced symptoms for only 3 years and was still completely independent. The patient described by Schwankhaus et al7 had marked pathologic changes in the dorsal columns of the spinal cord corresponding reasonably well with the SI changes seen in the dorsal columns of the most severely affected patient in our study.
Differential Diagnosis
MR imaging findings in the spinal cord have been described in leukoencephalopathy with brain stem and spinal involvement and high lactate (LBSL). No atrophy was described, but high T2 SI was seen in both the lateral corticospinal tracts and the dorsal columns along the whole spinal cord.15 This disorder is inherited in an autosomal recessive pattern and is caused by a mutation in DARS2. Clinically, the patients show slowly progressive pyramidal, cerebellar, and dorsal column dysfunction.16,17 Three adult-onset cases have been described, though these did not have elevated lactate.18,19 MR imaging investigation of the spinal cord revealed a pattern distinct from that in adult-onset ADLD with autonomic symptoms, with prominent SI increase that was very high and easily detected also in sagittal images.15,16,18,20
Leukoencephalopathy with vanishing white matter was initially described in children,21 but adult-onset cases have also been reported.22-25 The disease is inherited in an autosomal recessive pattern and is linked to mutations in the genes encoding the 5 subunits of the eukaryotic translation initiation factor EIF2B.24 Both the brain stem and the cervical spinal cord were atrophied in an adult-onset case, but no SI abnormalities were mentioned.25
Krabbe disease (globoid cell leukodystrophy) is caused by a genetic defect of the lysosomal enzyme galactocerebrosidase and inherited in an autosomal recessive pattern. Thinning of the spinal cord has been reported in 1 adult-onset case,26 but SI changes have not been described. In a childhood-onset case, a diffuse gadolinium enhancement of the lumbosacral nerve roots and cauda equina was reported.27
Metachromatic leukodystrophy is inherited in an autosomal recessive pattern and is caused by reduced activity of arylsulfatase A. Cases usually present in childhood, but disease onset shows great variability.28 Spinal MR imaging findings have been described in a late childhood−onset case, with abnormal enhancement of the cauda equina and lumbosacral nerve roots as earlier described in Krabbe disease.29
The X-linked leukodystrophies are Pelizaeus-Merzbacher disease (PMD) and adrenoleukodystrophy. In 1 adult-onset case of PMD, histopathology of the spinal cord showed patchy demyelination with no atrophy mentioned.30 Adrenoleukodystrophy, caused by mutations in the ABCD1 gene and subsequent accumulation of very long-chain fatty acids, has an adult-onset variant in adrenomyeloneuropathy.31 In adrenomyeloneuropathy, severe spinal cord atrophy has been described without SI changes.32,33
As of now, 2 distinct leukodystrophies with adult-onset forms and autosomal dominant inheritance have been described, Alexander disease and adult-onset ADLD with autonomic symptoms. Alexander disease, a leukodystrophy caused by mutations in the GFAP gene, is most frequently found in infants and children and is often sporadic.34 Adult-onset (≥13 years) forms with clinical and radiologic findings different from those of the infantile type have been reported.35-45 Ataxia,35,36,38-40,42-44 pyramidal signs,35,38-41,43,44 and autonomic dysfunction39,41,45 overlap the clinical features of ADLD with autonomic symptoms. However, palatal tremor and palatal myoclonus, common in adult-onset Alexander disease,35,38-40,43,45 have not been reported in adult-onset ADLD with autonomic symptoms. Due to genetic characterization and diagnosis of the disease, new radiologic patterns have been described, even in asymptomatic family members.43,44 In families with adult-onset Alexander disease, atrophy of the medulla oblongata and the upper spinal cord can be the most important MR imaging findings.35,37-44 In adult-onset ADLD with autonomic symptoms, the entire spinal cord is atrophic. Atrophy of the medulla oblongata occurs but is not very severe, as shown in this and the previous study.3 In adult-onset Alexander disease, SI changes in the spinal cord and medulla oblongata are most often small foci and are found in different locations,36,41,43 whereas the whole spinal cord white matter and the pyramids in the medulla oblongata are affected in adult-onset ADLD with autonomic symptoms. Large central SI increase in a swollen spinal cord has been described in 1 patient with a juvenile type of Alexander disease.34 Microscopic cavitations were found in central areas of the spinal cord in adult-onset Alexander disease,35 but in our histologically studied patient, white matter only was affected.
Imaging of the brain is beneficial in the differential diagnosis. An increased SI has been seen in the hilus of the dentate nucleus in adult-onset Alexander disease36,37,40,43 but has not been reported in ADLD with autonomic symptoms. The cerebellar peduncles can be affected in both diseases.3,6,41,43,44 The supratentorial neuroimaging features are very different in these 2 conditions. In adult-onset ADLD with autonomic symptoms, the corticospinal tract is affected, and in most cases, there are large supratentorial white matter changes that are less intense immediately around the lateral ventricles.3 In adult-onset Alexander disease, the cerebrum may be intact or there are thin periventricular hyperintensities around the lateral ventricles.36,38,40,41,43-45
In leukoencephalopathies with autosomal dominant inheritance and adult onset, such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), autosomal dominant retinal vasculopathy with cerebral leukodystrophy,46 and autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids,47 spinal cord abnormalities have been described only in CADASIL. Patients with CADASIL show a normal conventional MR imaging finding of the spinal cord with a reduced peak height of the magnetization transfer ratio histogram.48
The neuroradiologic features of untreated vitamin B12 deficiency may include extensive areas of SI changes in the periventricular matter and SI changes along the posterior columns of the spinal cord.49 The distribution of SI changes differs from that observed in our patients, and the clinical presentation is different. The most severely affected patient in our material was found to have normal vitamin B12 levels.
The partly overlapping clinical features and radiologic findings suggest that the most important differential consideration perhaps should be adult-onset Alexander disease, which also presents marked spinal cord atrophy and may show SI changes. The type of SI changes and the distinct features of brain MR imaging should enable a diagnosis of adult-onset ADLD with autonomic symptoms. Alexander disease has been excluded in our material. Genetic linkage of family 1 was reported at chromosome 5q23,8 which coincides with the genetic locus reported for the other families with adult-onset ADLD with autonomic symptoms.9,14
The clinical and neuroradiologic features of the patient from family 2 are very similar to those in the affected members in family 1. The genetic locus of GFAP, the gene responsible for most cases of Alexander disease, is located on chromosome 17q21.50 Furthermore, the morphologic hallmark of Alexander disease, Rosenthal fibers, was not detected in the brains of the patients with adult-onset ADLD with autonomic symptoms.3
SI changes in the spinal cord may be easily missed on MR imaging examinations because they may be difficult to see and differentiate from truncation artifacts in sagittal images. Transverse sections of good quality are needed for detection. A higher SI in white matter than in gray matter is a reliable sign of pathology in transverse images.
Conclusions
Atrophy and SI changes in the spinal cord are manifest in adult-onset ADLD with autonomic symptoms. The changes in the spinal cord are observable even before the onset of clinical symptoms and at a time when the MR imaging changes in the brain are still very limited. The fact that the SI increase is less marked in the spinal cord than in the brain may reflect a lesser degree of demyelination. To assess the SI changes in the spinal cord, one needs to obtain transverse images. The extent of changes and atrophy may hint at the grade of clinical severity.
The main differential diagnosis is adult-onset Alexander disease, which also may be inherited as an autosomal dominant trait. Both conditions show atrophy of the medulla oblongata and the cervical spinal cord and partly overlapping SI changes in the cerebellar peduncles. However, these 2 conditions are most convincingly distinguished radiologically by very different patterns of SI changes in the spinal cord and supratentorial white matter.
Footnotes
This work was supported by grants from the Selander Foundation, Hedberg Foundation for Medical Research, the Swedish Medical Research Council, and the Swedish Association of Persons with Neurological Disabilities.
Paper previously presented in part at: 32nd Annual Meeting of the European Society of Neuroradiology, September 20–23, 2007; Genoa, Italy.
References
- 1.Garbern J. Leukodystrophies. In: Lynch DR, ed. Neurogenetics: Scientific and Clinical Advances. New York, NY: Taylor & Francis;2006. :469–554
- 2.Eldridge R, Anayiotos CP, Schlesinger S, et al. Hereditary adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. N Engl J Med 1984;311:948–53 [DOI] [PubMed] [Google Scholar]
- 3.Melberg A, Hallberg L, Kalimo H, et al. MR characteristics and neuropathology in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. AJNR Am J Neuroradiol 2006;27:904–11 [PMC free article] [PubMed] [Google Scholar]
- 4.Abe K, Ikeda M, Watase K, et al. A kindred of hereditary adult-onset leukodystrophy with sparing of the optic radiations. Neuroradiology 1993;35:281–83 [DOI] [PubMed] [Google Scholar]
- 5.Quattrocolo G, Leombruni S, Vaula G, et al. Autosomal dominant late-onset leukoencephalopathy: clinical report of a new Italian family. Eur Neurol 1997;37:53–61 [DOI] [PubMed] [Google Scholar]
- 6.Bergui M, Bradac GB, Leombruni S, et al. MRI and CT in an autosomal-dominant adult-onset leukodystrophy. Neuroradiology 1997;39:423–26 [DOI] [PubMed] [Google Scholar]
- 7.Schwankhaus JD, Katz DA, Eldridge R, et al. Clinical and pathological features of an autosomal dominant, adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. Arch Neurol 1994;51:757–66 [DOI] [PubMed] [Google Scholar]
- 8.Marklund L, Melin M, Melberg A, et al. Adult-onset autosomal dominant leukodystrophy with autonomic symptoms restricted to 1.5 Mbp on chromosome 5q23. Am J Med Genet B Neuropsychiatr Genet 2006;141:608–14 [DOI] [PubMed] [Google Scholar]
- 9.Padiath Q, Saigoh K, Schiffmann R, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet 2006;38:1114–23 [DOI] [PubMed] [Google Scholar]
- 10.Krabbe K, Nielsen JE, Fallentin E, et al. MRI of autosomal dominant pure spastic paraplegia. Neuroradiology 1997;39:724–27 [DOI] [PubMed] [Google Scholar]
- 11.Laxova R, Hogan K, Haun J. A new autosomal dominant adult onset leukodystrophy. Am J Hum Genet 1985;37:A65 [Google Scholar]
- 12.Schwankhaus JD, Patronas N, Dorwart R, et al. Computed tomography and magnetic resonance imaging in adult-onset leukodystrophy. Arch Neurol 1988;45:1004–08 [DOI] [PubMed] [Google Scholar]
- 13.Asahara H, Yoshimura T, Sada S, et al. A Japanese family with probably autosomal dominant adult onset leukodystrophy [in Japanese]. Rinsho Shinkeigaku 1996;36:968–72 [PubMed] [Google Scholar]
- 14.Coffeen C, McKenna C, Koeppen A, et al. Genetic localization of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. Hum Mol Genet 2000;9:787–93 [DOI] [PubMed] [Google Scholar]
- 15.Távora DG, Nakayama M, Gama RL, et al. Leukoencephalopathy with brainstem and spinal cord involvement and high brain lactate: report of three Brazilian patients. Arq Neuropsiquiatr 2007;65:506–11 [DOI] [PubMed] [Google Scholar]
- 16.van der Knaap M, van der Voorn P, Barkhof F, et al. A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann Neurol 2003;53:252–58 [DOI] [PubMed] [Google Scholar]
- 17.Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 2007;39:534–39 [DOI] [PubMed] [Google Scholar]
- 18.Labauge P, Roullet E, Boespflug-Tanguy O, et al. Familial, adult onset form of leukoencephalopathy with brain stem and spinal cord involvement: inconstant high brain lactate and very slow disease progression. Eur Neurol 2007;58:59–61 [DOI] [PubMed] [Google Scholar]
- 19.Petzold GC, Bohner G, Klingebiel R, et al. Adult onset leukoencephalopathy with brain stem and spinal cord involvement and normal lactate. J Neurol Neurosurg Psychiatry 2006;77:889–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linnankivi T, Lundbom N, Autti T, et al. Five new cases of a recently described leukoencephalopathy with high brain lactate. Neurology 2004;63:688–92 [DOI] [PubMed] [Google Scholar]
- 21.van der Knaap M, Barth P, Gabreels F, et al. A new leukoencephalopathy with vanishing white matter. Neurology 1997;48:845–55 [DOI] [PubMed] [Google Scholar]
- 22.van der Knaap MS, Kamphorst W, Barth PG, et al. Phenotypic variation in leukoencephalopathy with vanishing white matter. Neurology 1998;51:540–47 [DOI] [PubMed] [Google Scholar]
- 23.Biancheri R, Rossi A, Di Rocco M, et al. Leukoencephalopathy with vanishing white matter: an adult onset case. Neurology 2003;61:1818–19 [DOI] [PubMed] [Google Scholar]
- 24.Leegwater PA, Pronk JC, van der Knaap M. Leukoencephalopathy with vanishing white matter: from magnetic resonance imaging pattern to five genes. J Child Neurol 2003;18:639–45 [DOI] [PubMed] [Google Scholar]
- 25.Gallo A, Rocca MA, Falini A, et al. Multiparametric MRI in a patient with adult-onset leukoencephalopathy with vanishing white matter. Neurology 2004;62:323–26 [DOI] [PubMed] [Google Scholar]
- 26.Wang C, Melberg A, Weis J, et al. The earliest MR imaging and proton MR spectroscopy abnormalities in adult-onset Krabbe disease. Acta Neurol Scand 2007;116:268–72 [DOI] [PubMed] [Google Scholar]
- 27.Vasconcellos E, Smith M. MRI nerve root enhancement in Krabbe disease. Pediatr Neurol 1998;19:151–52 [DOI] [PubMed] [Google Scholar]
- 28.Gieselmann V. What can cell biology tell us about the heterogeneity in lysosomal storage disorders? Acta Paediatr Suppl 2005;94:80–86 [DOI] [PubMed] [Google Scholar]
- 29.Toldo I, Carollo C, Battistella PA, et al. Spinal cord and cauda equina MRI findings in metachromatic leukodystrophy: case report. Neuroradiology 2005;47:572–75 [DOI] [PubMed] [Google Scholar]
- 30.Saito Y, Ando T, Doyo M, et al. An adult case of classical Pelizaeus-Merzbacher disease: magnetic resonance images and neuropathological findings [in Japanese]. Rinsho Shinkeigaku 1993;33:187–93 [PubMed] [Google Scholar]
- 31.Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol 2007;3:140–51 [DOI] [PubMed] [Google Scholar]
- 32.Kumar AJ, Köhler W, Kruse B, et al. MR findings in adult-onset adrenoleukodystrophy. AJNR Am J Neuroradiol 1995;16:1227–37 [PMC free article] [PubMed] [Google Scholar]
- 33.Israel H, Ostendorf F, Stiepani H, et al. Spinal cord atrophy in adrenomyeloneuropathy. Arch Neurol 2005;62:1157. [DOI] [PubMed] [Google Scholar]
- 34.van der Knaap M, Ramesh V, Schiffmann R, et al. Alexander disease: ventricular garlands and abnormalities of the medulla and spinal cord. Neurology 2006;66:494–98 [DOI] [PubMed] [Google Scholar]
- 35.Schwankhaus JD, Parisi JE, Gulledge WR, et al. Hereditary adult-onset Alexander's disease with palatal myoclonus, spastic paraparesis, and cerebellar ataxia. Neurology 1995;45:2266–71 [DOI] [PubMed] [Google Scholar]
- 36.Martidis A, Yee R, Azzarelli B, et al. Neuro-ophthalmic, radiographic, and pathologic manifestations of adult-onset Alexander disease. Arch Ophthalmol 1999;117:265–67 [DOI] [PubMed] [Google Scholar]
- 37.Namekawa M, Takiyama Y, Aoki Y, et al. Identification of GFAP gene mutation in hereditary adult-onset Alexander's disease. Ann Neurol 2002;52:779–85 [DOI] [PubMed] [Google Scholar]
- 38.Okamoto Y, Mitsuyama H, Jonosono M, et al. Autosomal dominant palatal myoclonus and spinal cord atrophy. J Neurol Sci 2002;195:71–76 [DOI] [PubMed] [Google Scholar]
- 39.Stumpf E, Masson H, Duquette, et al. Adult Alexander disease with autosomal dominant transmission. Arch Neurol 2003;60:1307–12 [DOI] [PubMed] [Google Scholar]
- 40.Thyagarajan D, Chataway T, Li R, et al. Dominantly-inherited adult-onset leukodystrophy with palatal tremor caused by a mutation in the glial fibrillary acidic protein gene. Mov Disord 2004;19:1244–48 [DOI] [PubMed] [Google Scholar]
- 41.Romano S, Salvetti M, Ceccherini I, et al. Brainstem signs with progressing atrophy of medulla oblongata and upper cervical spinal cord. Lancet Neurol 2007;6:562–70 [DOI] [PubMed] [Google Scholar]
- 42.Ohnari K, Yamano M, Uozumi T, et al. An adult form of Alexander disease: a novel mutation in glial fibrillary acidic protein. J Neurol 2007;254:1390–94 [DOI] [PubMed] [Google Scholar]
- 43.Farina L, Pareyson D, Minati L, et al. Can MR imaging diagnose adult-onset Alexander disease? AJNR Am J Neuroradiol 2008;29:1190–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balbi P, Seri M, Ceccherini I, et al. Adult-onset Alexander disease: report on a family. J Neurol 2008;255:24–30 [DOI] [PubMed] [Google Scholar]
- 45.Howard KL, Hall DA, Moon M, et al. Adult-onset Alexander disease with progressive ataxia and palatal tremor. Mov Disord 2008;23:118–22 [DOI] [PubMed] [Google Scholar]
- 46.Richards A, van den Maagdenberg AM, Jen JC, et al. C-terminal truncations in human'3-'5 DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet 2007;39:1068–70. Epub 2007 Jul 29 [DOI] [PubMed] [Google Scholar]
- 47.van der Knaap MS, Naidu S, Kleinschmidt-Demasters BK, et al. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology 2000;54:463–68 [DOI] [PubMed] [Google Scholar]
- 48.Rocca MA, Filippi M, Herzog J, et al. A magnetic resonance imaging study of the cervical cord of patients with CADASIL. Neurology 2001;56:1392–94 [DOI] [PubMed] [Google Scholar]
- 49.Scherer K. Images in clinical medicine: neurologic manifestations of vitamin B12 deficiency. N Engl J Med 2003;348:2208. [DOI] [PubMed] [Google Scholar]
- 50.Brenner M, Johnson AB, Boespflug-Tanguy O, et al. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 2001;27:117–20 [DOI] [PubMed] [Google Scholar]