

Abstract
The OPRM1 A118G single nucleotide polymorphism (SNP rs1799971) gene variant encoding the N40D ¼-opioid receptor (MOR) has been associated with dependence on opiates and other drugs of abuse but its mechanism is unknown. The frequency of G-allele carriers is ~40% in Asians, ~16% in Europeans, and ~3% in African-Americans. With opioid abuse-related deaths rising at unprecedented rates, understanding these mechanisms may provide a path to therapy. Here we generated homozygous N40D subject-specific induced inhibitory neuronal cells (iNs) from seven human induced pluripotent stem (iPS) cell lines from subjects of European descent (both male and female) and probed the impact of N40D MOR regulation on synaptic transmission. We found that D40 iNs exhibit consistently stronger suppression (versus N40) of spontaneous inhibitory postsynaptic currents (sIPSCs) across multiple subjects. To mitigate the confounding effects of background genetic variation on neuronal function, the regulatory effects of MORs on synaptic transmission were recapitulated in two sets of independently engineered isogenic N40D iNs. In addition, we employed biochemical analysis and observed differential N-linked glycosylation of human MOR N40D. This study identifies neurophysiological and molecular differences between human MOR variants that may predict altered opioid responsivity and/or dependence in this subset of individuals.
INTRODUCTION
Well over 72,000 Americans died of opioid overdose in 2017, with a sharp increase in 2014 – 2017 due to synthetic opioids 1, prompting a public health crisis whose biological underpinnings are poorly understood. The ¼-opioid receptor (MOR) mediates the most powerful addictive properties of abused opiates and much research has identified chemically diverse ligands of varying efficacies for pain relief or treatment of addiction. Because of its substantive role in mediating reward and positive reinforcement, the MOR is also an indirect target of alcohol, nicotine, and other drugs of abuse 2, 3. MOR-mediated synaptic alterations in reward-associated brain regions may represent a key underlying mechanism of reinforcement in drug abuse 4, but our understanding of this process in human neurons is limited.
Human genetic studies suggest that OPRM1 (encodes MOR) gene variants play key roles in susceptibility to opioid addiction in humans. Most prominently, an A118G single nucleotide polymorphism (SNP) in OPRM1, rs1799971, is a non-synonymous gene variant which replaces asparagine at position 40 (N40) with aspartate (D40), occurs at a frequency of ~3% in African, 39-42% in Asian and ~16% in European ancestry 5, and is associated with drug dependence phenotypes 6, 7. There have been numerous investigations into the functional consequences of the MOR D40 variant on receptor activation in overexpression models or knock-in models with mouse, primates, and humans 6, 8–16. Importantly, human clinical studies found that MOR A118G alters hypothalamic-pituitary-adrenal (HPA) axis activation, and human subjects (of both sexes) harboring A118G-allelilc variants were shown to have a relatively greater tonic inhibition at HPA sites through the MOR 17, 18. In addition, monkeys carrying OPRM1 C77G SNP (thought to be analogous to A118G, but not on A118G per se) demonstrated a higher MOR affinity to β-endorphin and significantly lower basal adrenocorticotropic hormone (ACTH) stimulated plasma cortisol levels associated with aggression16. Bioinformatic analyses and animal studies reveal that the N40D substitution likely destroys an N’-terminal glycosylation site and reduces the surface expression of MORs 19–21. Nevertheless, another study using a humanized mouse model of N40D (i.e., the first exon of mouse OPRM1 was replaced with the first of human OPRM1 harboring N40D) is not in total agreement with this finding 22. Thus, understanding how the D40 variant affects MOR signaling and synaptic function when expressed at normal levels in human neurons may provide insight into mechanisms underlying drug abuse, at least in people carrying this variant.
In order to fill the gap in studies performed in the mouse and heterologous systems, we generated human induced neuronal (iN) cells from induced pluripotent stem (iPS) cells derived from subjects carrying homozygous alleles for either MOR N40 or D40 in order to better dissect the role of MOR N40D in a physiologically relevant and human-specific model system. The aim of this project is to unravel the cellular/synaptic mechanism(s) of MOR N40D gene variants in a human neuronal cell context but we do not intend to elucidate the etiology of N40D MOR variants here because the main readout is patch clamp synaptic physiology (which is rather labor intensive and low in throughput). We found that MOR modulation of synaptic function is affected by N40D substitutions in human neurons in donor iPS cells (3 N40, 4 D40 homozygous subjects) as well as in two pairs of isogenic N40D neurons generated using CRISPR gene targeting. However, we believe an analysis of 3 vs 4 unrelated donors, varying at only a single SNP, will be difficult to justify any detectable effects at the population level. Nevertheless, elucidating the molecular/cellular/synaptic mechanism(s) of N40D will reveal potential contributions to neuropsychiatric disorders, such as alcohol use disorders (AUDs) and drug use disorders (DUDs). This study, however, exemplifies the use of patient-specific iPS cells as well as gene targeted isogenic stem cell lines to advance our understanding of the fundamental cellular and synaptic alterations associated with addiction risk gene variants in a human neuronal context.
METHODS AND MATERIALS
Generation of human iPS cells from lymphocytes of subjects carrying MOR N40D
The original selection criteria we requested from the Collaborative Genetic Study of Nicotine Dependence (COGEND) group was to include subjects with similar backgrounds, including both sexes, and availability of frozen cells in the repository. We did not request additional SNP data, nor did we receive ages of the subjects. We specifically chose to draw a line between the genetics group performing genome wide associated studies (GWAS) and our study, since we could never achieve the power necessary to assess additional genetic markers in such a small number of subjects, and we wished to maintain subject anonymity since we used deidentified repository specimens. All cells in this collection are consented for cell line construction and are exempt from IRB review under 45 CFR part 46 exemption 4.
Human iPS cell lines were generated by RUCDR Infinite Biologics ® from human primary lymphocytes carrying either MOR N40 or D40 genotypes using Sendai viral vectors (CytoTune™, ThermoFisher Scientific), as previously described 23. Human iPS cells were cultured and maintained as described previously 24. Human iPS cells were cultured in 37°C, 5% CO2 on Matrigel® Matrix (Corning Life Sciences)- coated plates in mTeSR medium (Stem Cell Technologies). For passaging and differentiation, done weekly, iPS cells were dissociated using Accutase (Stem Cell Technologies), spun down at 1000 rpm for 5 minutes, and re-plated at a density of 20,000 cells/cm2 for maintenance cultures and 50,000 cells/cm2 for differentiation.
Generations of isogenic human stem cell lines carrying N40D MOR gene variants
Two pairs of isogenic N40D MOR human stem cells lines were generated using CRISPR/Cas9 genome editing. Briefly, to convert H1 embryonic stem (ES) cells carrying homozygous AA118 major allele to GG118 homozygous minor alleles, a sgRNA designed from Optimized CRISPR Design Tool (http://crispr.mit.edu/) and Cas9 were expressed using the PX459 vector (Addgene plasmid #62988) and was transfected using the Lipofectamine 3000 reagent (ThermoFisher Scientific, L300015) along with a single stranded oligodeoxynucleotide (ssODN) of 140 base pairs with homology arms flanking the mutation site carrying mutations for G118, a BamHI restriction enzyme site for screening, along with a mutation to mutate the PAM sequence. Individual clones were hand-picked for expansion and screening by PCR and sequencing. Heterozygous clone 9-2 was expanded and transfected for targeting the second allele of OPRM1 Exon 1. The two homozygous G118 knock-in clones were further subcloned before expansion and freezing.
To convert rs1799971 in the 03SF subject iPS cell line from homozygous minor allele (GG) to major allele (AA), a slightly different strategy was used. First, a CRISPR targeting site was found using ZiFit software 25. The target site (GGCAACCTGTCCGACCCATG) included the major allele sequence so the guide RNA (gRNA) was designed to incorporate the minor allele (GGCgACCTGTCCGACCCATG). A 200 nt homologous recombination donor oligo was designed to convert minor to major allele, inactivate the CRISPR site, and introduce a HpaI site for screening. The gRNA was synthesized by PCR and in vitro transcription (GeneArt Precision gRNA Synthesis Kit, Life Technologies) 26, mixed with synthetic Cas9 protein (Life Technologies), donor oligo, and the mixture was electroporated into iPS cells (Amaxa nucleofector, Lonza) along with a GFP expression plasmid (pGFP-Max, Lonza). One day later, cells were dissociated with Accutase and GFP-expressing cells were collected by FACS and plated at about 5,000 cells per well in a 6-well plate on irradiated mouse embryonic fibroblasts (MEFs). By 7-10 days, colonies were visible and hand-picked for screening. Three iPS cell clones were selected: C12, which had no evidence of editing to be used as a negative control; D11 and A10, which both had homozygous edits to produce rs1799971 major allele (AA). In all gene-targeted cell lines, sequencing confirmed these edits and that all predicted off-target sites were unchanged.
Generation of GABAergic iN cells from human ES and iPS cells
The protocol of generating GABAergic human iN cells was described recently 27. Briefly, iPS cells and ES cells were plated as dissociated cells on Matrigel ® Matrix (Corning Life Sciences)-coated dishes in mTeSR (Stem Cell Technologies) medium with 2 ¼M Y-27632 (Stemgent). The following day, the cells were infected with Ascl1, Dlx2 and rtTA lentiviruses for 10-12 hours upon which culture medium was replaced with Neurobasal medium (GIBCO by Life Technologies) with B27 and L-Glutamine supplemented with 2 ¼g/mL of doxycycline (MP Biomedicals) and 2 ¼M Y-compound to induce TetO expression. The protocol for generating lentiviruses expressing different transcription factors was previously described 27. Puromycin and hygromycin selection was conducted for the following 2 days, and on day 5, the iN cells were dissociated with Accutase and plated on glass coverslips with a monolayer of passage three primary astrocytes isolated from p1-3 pups, as described previously 24, 27, 28. Following plating, 50% of the culture medium was replaced every 2-3 days with fresh Neurobasal media containing B27, L-Glutamine, 100 ng/ml of BDNF, NT3 and GDNF.
Real-time RT-PCR (qPCR)
Total neuronal RNA from three independently generated batches of iN cells for each cell line was prepared using TRIzol ® Reagent (Thermo Fisher Scientific), human-specific Taqman probes were purchased for OPRM1, MAP2, Tuj1, VGAT, GAD1, TH and PCR reaction conditions followed the manufacturer’s recommendations. Undifferentiated iPS cells, ES cells, and mouse astrocytes were used as negative controls. A sample of total RNA of a healthy human brain as well as human Thalamus from Biochain ® was used as a positive control. Relative RQ values were obtained by normalizing expression levels to the C12 iN condition. Student’s t-test was used to compare grouped N40 and D40 means. Evaluation of qPCR was performed blind to the genotype. See Supplemental Table 1 for a comprehensive list of primers and probes used in this study.
Immunocytochemistry and confocal imaging
Inhibitory human neurons were fixed for 15 minutes in 4% paraformaldehyde in phosphate buffered saline (PBS) and permeabilized using 0.1% Triton X-100 in PBS for 10 minutes at room temperature. Cells were then incubated in blocking buffer (4% bovine serum albumin (BSA) with 1% normal goat serum in PBS) for 1 hour at room temperature and then incubated with primary antibodies diluted in blocking buffer for 1 hour at room temperature, washed with PBS three times, and subsequently incubated in secondary antibodies for 1 hour at room temperature. Confocal imaging analysis was performed using a Zeiss LSM700. Primary Antibodies used include: mouse anti Oct4 (Millipore Sigma MAB4401, 1:2000), mouse anti Tra-1-60 (Millipore Sigma MAB4360, 1:1000), mouse anti MAP2 (Sigma-Aldrich M1406, 1:500), rabbit anti MAP2 (Sigma-Aldrich M3696, 1:500), chicken anti MAP2 (Millipore AB5543, 1:1000), rabbit anti Synapsin (e028, 1:3000), rabbit anti VGAT (Millipore Sigma AB5062P, 1:2000), mouse anti Gad-67 (Abcam ab26116, 1:500), mouse anti β3 Tubulin (BioLegend 801201, 1:2000), mouse anti Calbindin (Abcam ab82812, 1:500), guinea pig anti Parvalbumin (Swant GP72, 1:2500), rat anti Somatostatin (Millipore MAB354, 1:50).
Electrophysiology
Functional analyses of iN cells were conducted using whole cell patch-clamp as described elsewhere 24, 29. Briefly, a K-Gluconate internal solution was used, which consisted of (in mM): 126 K-Gluconate, 4 KCl, 10 HEPES, 4 ATP-Mg, 0.3 GTP-Na2, 10 Phosphocreatine. The pH was adjusted to 7.2 and osmolarity was adjusted to 270-290 mOsm. The bath solution consisted of (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 10 Glucose. The pH was adjusted to 7.4. Spontaneous inhibitory postsynaptic currents (sIPSCs) were recorded either at a holding potential of 0 mV under voltage-clamp mode or at a −70 mV holding potential with 20 ¼M CNQX added to the perfusion solution. Miniature IPSCs (mIPSCs) were recorded in the presence of tetrodotoxin (1 ¼M). Intrinsic action potential firing properties of the iN cells were recorded in a bath solution containing 50 ¼M Picrotoxin and 20 ¼M CNQX. Evoked synaptic currents were elicited using an extracellular concentric bipolar stimulating electrode positioned approximately 100 ¼m away from the cell soma. All recordings in cultured human neurons were performed at room temperature because recordings made at higher temperature in cultured neurons resulted in poor recording quality and short time period of recordings. Cells were excluded from analysis if access resistance changed by more than 20% along the duration of the recording. In addition, any iNs whose access resistance was greater than 35 MΩ was also excluded from analysis. The following MOR agonists/antagonist were used for the recordings: DAMGO (Hellobio): 0.1-10 ¼M; Naltrexone hydrochloride (Tocris): 5 ¼M; Morphine sulphate: 10 ¼M; Data presentation: All data are presented as mean ± S.E.M. Student’s t-test or 2-way ANOVAs were used to assess statistical significance.
Expression of recombinant N’-terminal fragment of human MOR and glycosylation assay
The N-terminal fragments carrying N40D variants were expressed by using an expression vector for extracellular secretion in mammalian cells. Briefly, the soluble N-terminal region of MOR N40 and D40 fused with a C-terminal IgG Fc domain to permit affinity chromatography was inserted in expression vector p-cDNA 3.1. This construct has been further modified to introduce a 3Cpro cleavage site upstream from the Fc domain as described previously 30. Both N-terminal MOR constructs (N40 and D40) contained the original expression vector, an N-terminal FLAG tag, signal peptide sequence and Fc sequence. They were expressed in HEK293 and HEK293 GnT1- (lacking the N-acetylglucosaminyltransferase I gene needed for high-order N-linked glycosylation processing) cells.
Transfected HEK cells were maintained at 37℃ and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) containing up to 10% fetal bovine serum (FBS). Media was collected 48 hours following transfection. Cell lysates from HEK-293 cells expressing N40D MOR N-terminal peptides were collected and centrifugation was performed to remove all non-soluble components from the cell culture media. 5% SDS and 1 M DTT were added to the lysate and heated to 95℃ for 5 minutes to denature total proteins contained within the soluble fraction. Denatured proteins were incubated with recombinant PNGase F (2.5 U/μL) overnight at 37℃. To stop the reactions, 4x SDS-PAGE loading buffer was added to each sample and heated at 95℃ for 5 minutes. Samples were analyzed with Western blotting using anti-FLAG tag antibody (1:2500) (Sigma Aldrich; F3165) and horseradish peroxidase-conjugated anti-mouse secondary antibody (1:20,000) (Life Technologies; 31430).
Statistics
For electrophysiological and qPCR analyses we employed at least 3 independent infection batches of iNs (biological replicates). The effects of MOR agonists (DAMGO or morphine) were normalized to basal activity in the absence of the drugs, i.e. drug effects are defined as the relative changes to the baseline values (in the absence of drug). The effect of drugs before and after the application in the same group of cells were compared using paired t-tests. On normalized post-treatment data, 1-way ANOVA tests were used to evaluate the effects between genotypes.
RESULTS
Generation of human inhibitory neurons carrying N40D MOR variants
To investigate the functional role of the MOR N40D variant in a human neuronal context, we obtained iPS cells from multiple individuals of European descent carrying homozygous alleles for either MOR N40 (n=4) or MOR D40 (n=3) (Supplemental Fig. 1A). The rs1799971 genotype and the pluripotency of all seven iPS cell lines were confirmed by sequencing and colocalized immunocytochemistry (ICC) for OCT4 and Tra-1-60 (Fig. 1A–B).
Figure 1 |. MOR N40D expressing inhibitory human neurons exhibit more robust suppression of inhibitory synaptic transmission by DAMGO.
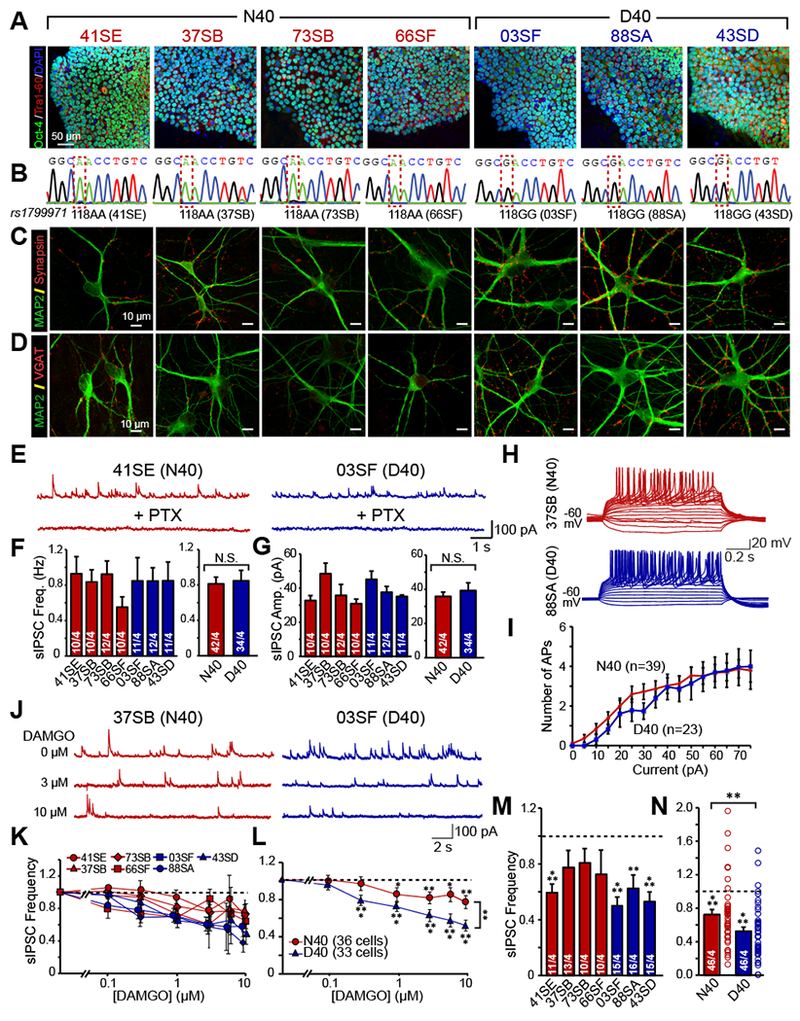
(A) Oct4 (green), Tra 1-60 (red), and DAPI (blue) ICC for N40 and D40 subject iPS cells depicting pluripotency (B) Sequencing confirming homozygous A118 or G118 genotype of human iPS cell lines (C) MAP2 (green) and Synapsin (red) ICC of iN cells generated from N40 and D40 subject iPS cells (D) MAP2 (green) and VGAT (red) ICC of induced inhibitory neuronal (iN) cells generated from N40 and D40 subject iPS cells (E-G) Both N40 and D40 iN cells exhibit PTX sensitive spontaneous IPSCs whose frequency (N40 vs D40: N.S.) and amplitude (N40 vs D40: N.S.) are unaffected by MOR N40D substitution (H) Representative traces of action potentials induced by step current injections (from −20 to +75 pA, 5 pA increments) during current clamp recordings from one N40 and D40 cell line (I) Quantification of induced action potentials in inhibitory iNs cells illustrating that neuronal excitability is unchanged as a consequence of MOR N40D (N40 vs D40: N.S. at all current injections) (J) Representative traces of sIPSCs recorded to increasing concentrations of DAMGO in N40 and D40 iN cells (K) Quantification of inhibition of sIPSC frequency in individual subject derived N40 and D40 iN cells (L) Merged data of the four N40 and three D40 subject lines illustrates that D40 iN cells exhibit stronger suppression of IPSC frequency compared to N40 iN cells (M-N) sIPSC frequency response to a single concentration of 10 ¼M DAMGO; data is normalized to control (N40 vs control: p<0.001, D40 vs control: p<0.001). Summary graphs are shown as individual cell lines or merged data of either four N40 patients (red bars) and three D40 patients (blue bars) (N40 vs D40: p <0.01). Data are depicted as means ± SEM. Numbers of cells/Number of independently generated cultures analyzed are depicted in bars. Paired t-test was used to evaluate within genotype statistical differences and one-way ANOVA was used to evaluate between genotype statistical differences (*p <0.05, **p <0.01, ***p <0.001).
It has been shown that activation of MOR signaling causes suppression of inhibitory neurons which finally leads to excitation of midbrain dopaminergic (DA) neurons, a disinhibition mechanism 31, 32. We therefore derived inhibitory induced neuronal (iN) cells from all 7 iPS cell lines by lentiviral mediated ectopic expression of the transcription factors Ascl1 and Dlx2 27. These induced human neuronal cells express pan-neuronal markers including MAP2, βIII-tubulin, and Synapsin (Fig. 1C, Supplemental Fig. 1B) as well as inhibitory neuronal markers GAD67 and VGAT (Fig. 1D, Supplemental Figs. 1C, 1N–O). They also exhibit OPRM1 of similar expression levels (Supplemental Fig. 1M). Thus, the N40D SNP has no impact on MOR expression or inhibitory neuronal identity. To examine whether the N40 and D40 iN cells are functionally comparable under baseline conditions, we performed whole cell patch-clamp recordings of iN cells after 5-6 weeks of re-plating onto a monolayer of mouse glia. The iN cells of both genotypes exhibit similar intrinsic membrane properties (Supplemental Fig. 1D–F), can fire repetitive spontaneous action potentials (APs) at baseline levels (Supplemental Fig. 1G–I), and exhibit similar intrinsic excitability at baseline conditions (Fig. 1H–I). Similarly, no significant differences in spontaneous or miniature inhibitory postsynaptic currents (sIPSCs and mIPSCs, respectively) were observed by genotype (Figs. 1E–G, Supplemental Fig. 1J–L), indicating that the N40D variant does not affect passive or active membrane properties and that the neurons generated from subject iPS cell lines are of similar functional maturation and differentiation status.
MOR D40 iN cells exhibit altered sensitivity to the MOR agonist DAMGO
There have been numerous studies 6, 8–15 examining the functional consequences of MOR N40D on receptor activation in overexpression models and in knock-in mice harboring MOR N40D, but no functional or electrophysiological analyses on cultured neurons have been conducted, specifically not in a human neuronal context. To gauge whether N40 and D40 iN cells respond differently to MOR activation, we used a MOR-specific agonist DAMGO ([D-Ala2, N-MePhe4, Gly-ol]-enkephalin) to study its role on modulating synaptic release. In both N40 and D40 iN cells, DAMGO suppressed sIPSCs in a dose-dependent manner (Fig. 1J). However, the suppression of sIPSC frequency was more robust in D40 iN cells compared to N40 iN cells in multiple repeated experiments and multiple iPS cell lines (Fig. 1K–L), with no difference in sIPSC amplitude by genotype. To confirm that the observation is not due to a residual effect of prolonged agonist exposure, we applied a single concentration of 10 ¼M DAMGO (Fig. 1M–N) and similarly found that D40 iN cells respond more robustly to MOR activation compared to N40 iN cells, illustrating genotype-dependent regulation of MOR signaling.
Generation of isogenic human pluripotent stem cell lines carrying MOR N40D SNP
To directly compare the two MOR genotypes in identical genetic backgrounds, eliminating the impact of secondary genetic variation, we generated two sets of isogenic human stem cell lines using CRISPR/Cas9 gene targeting. We first targeted the MOR locus in a well-characterized human H1 ES cell line, which is homozygous for the major allele (A118), using a sgRNA targeting the antisense DNA strand along with a 140bp single stranded oligodeoxynucleotide carrying G118 (Fig. 2A–B). Simultaneously, we converted one patient iPS cell line which is homozygous for the G118 allele to a homozygous A118 genotype with an alternative strategy utilizing direct transduction of guide RNA and Cas9 protein into the subject cell line (Fig. 2C–D). We isolated two clones (Supplemental Fig. 2A–C) with no detectable off-target effects from each targeting scheme. Inhibitory iN cells generated from isogenic lines are positive for MAP2, Synapsin and VGAT (Fig. 2E–F) and exhibit similar intrinsic membrane properties (Supplemental Fig. 2D–E) as well as sIPSC and AP properties in both genotypes (Supplemental Figs. 2F–G, Supplemental Figs. 3A–C).The densities and sizes of synapses were also not significantly different between genotypes (Supplemental Fig. 2H–I). To examine the overall homogeneity as well as to demonstrate similar maturation stages of isogenic iNs we performed qPCR against several diagnostic mRNA targets (Summary is at Supplemental Table 1, Supplemental Fig. 4). We measured the expression levels of all four classes of opioid receptors, various subunits of GABAA and GABAB receptors, and classic markers to identify specific subclasses of GABAergic neurons. We found no significant differences between mRNA levels (with the exception of the mRNA encoding the Kappa Opioid Receptor, OPRK1) between N40- and D40-containing iNs. To further validate our qPCR data, we also performed IHC against 3 of the major GABAergic neuronal subtypes: parvalbumin (PV), calbinidin-1 (Calb1), and somatostatin (SST). Consistent with the qPCR we observed a strong SST signal in both genotypes (Supplemental Fig. 4A). These data illustrate that the N40D SNP does not alter synaptogenesis and functional maturation as well as the patterning of isogenic human neurons. These data further suggest no difference in baseline neuronal function in N40D human iNs.
Figure 2 |. Human neurons from two sets of independently targeted isogenic human stem cell lines for OPRM1 A118G validate differential DAMGO response observed in patient cell lines.

(A) OPRM1 Targeting Strategy 1: Structure of OPRM1 gene on chromosome 6 and schematic overview of CRISPR/Cas9 gene targeting strategy to knock-in homozygous G118 alleles into human H1 embryonic stem cell (H1ES) in which sgRNA targets donor strand. In the 140bp ssODN, we inserted a T to C mutation to incorporate OPRM1 GG118, synonymous G to A for PAM mutation, and synonymous G to C and G to A mutations to create a BamHI restriction enzyme site. (B) Sequencing of original H1ES control cell line carrying homozygous A118 (N40) alleles, and two isolated clones carrying homozygous OPRM1 G118 (D40) alleles (Clone 9-2-17, Clone 9-2-18). (C) OPRM1 Targeting Strategy 2: Structure of OPRM1 gene on chromosome 6 and an independent CRISPR/Cas9 gene targeting strategy to correct 03SF patient line (originally homozygous G118 expressing MOR D40) to homozygous A118 (N40). We designed a 200 nt template strand to knock-in homozygous A118 alleles, containing mutations to generate a HpaI restriction enzyme site for screening (D) Sequencing of passage-matched, uncorrected 03SF patient cell line carrying homozygous D40 alleles (C12) and two gene-corrected clones (Clone A10, D11) carrying homozygous OPRM1 A118 (N40) alleles after subcloning. (E) ICC of MAP2 (green) and Synapsin (red) of iN cells produced from gene-targeted ES cells and iPS cells. (F) Immunofluorescence of MAP2 (green) and VGAT (red) of iN cells produced from gene-targeted ES cells and iPS cells. (G-I) Relative mRNA levels of OPRM1 as well as markers for inhibitory subtype specificity (GAD1, VGAT) measured by quantitative RT-PCR; mRNA levels are normalized to Synapsin I. Data are represented as means of three independently differentiated batches of iNs from each patient iPS cell line. (J) Representative traces of sIPSCs recorded to increasing concentrations of DAMGO in N40 and D40 iN isogenic iN cells derived from ES cells. (K-L) Quantification of sIPSC frequency (H1 vs control: p <0.05, Clone 17 vs control: p <0.001, Clone 18 vs control: p <0.001, N40 vs control: p <0.05, D40 vs control: p <0.001, N40 vs D40: p <0.05) and amplitude in response to 6 ¼M DAMGO (H1 vs control: N.S., Clone 17 vs control: N.S., Clone 18 vs control: N.S., N40 vs control: N.S., D40 vs. control: N.S., N40 vs D40: N.S.) (M) Representative traces of sIPSCs recorded to increasing concentrations of DAMGO in N40 and D40 iN isogenic iN cells derived from iPS cells. (N-O) Quantification of sIPSC frequency (A10: DAMGO vs control: p <0.01, D11: DAMGO vs control: N.S., C12: DAMGO vs control: p <0.001, N40: DAMGO vs control: p <0.01, D40: DAMGO vs control: p <0.001, N40 vs D40: p <0.05) and amplitude in response to 6 ¼M DAMGO (A10: DAMGO vs control: N.S., D11: DAMGO vs control: N.S., C12: DAMGO vs control: N.S., N40: DAMGO vs control: N.S., D40: DAMGO vs. control: N.S., N40 vs D40: N.S.). Data are depicted as means ± SEM. Numbers of cells/Number of independently generated cultures analyzed are depicted in bars. Paired t-test was used to evaluate within genotype statistical differences and one-way ANOVA was used to evaluate between genotype statistical differences (*p <0.05, **p <0.01, ***p <0.001).
Isogenic human neurons recapitulate differential DAMGO response phenotype and exhibit altered synaptic function
In this highly controlled system of isogenic iN cells, we observed less culture-to-culture variability than the subject cell lines for OPRM1 mRNA and inhibitory neuronal markers (Fig. 2G–I). Moreover, because the patient derived A118 and G118 iN cells exhibited the most robust difference in sIPSC frequency at a DAMGO concentration of 6 ¼M, we examined whether the isogenic iN cells exhibit a similar difference in DAMGO mediated synaptic responses at this DAMGO concentration. We observed a similar decrease in sIPSC frequency compared to subject iN cells following acute DAMGO application, with a stronger inhibition in D40 versus N40 iN cells, and no effect on amplitude (Fig. 2J–O). Furthermore, to determine whether the effect of DAMGO was mediated by the MOR, we applied Naltrexone, a broad spectrum MOR antagonist, and found that the DAMGO-induced synaptic suppression could be reversed (Supplemental Fig. 2J–K). Importantly, administration of Naltrexone alone does not affect synaptic transmission in either genotype (Supplemental Fig. 5). Thus, the reproducibility of the DAMGO response phenotype illustrates that the D40 variant alone explains the differential signaling and the effect is unlikely due to the impact of intrinsic genetic variability between individuals.
We focused the remaining analyses on one pair of isogenic cell lines, C12 (D40) and A10 (N40), on the basis of their consistent differentiation and maturation. We found that DAMGO application more robustly decreased mIPSC frequency in D40 versus N40 iN cells, which no change in mIPSC amplitude (Fig. 3A–C), which suggests a DAMGO mediated decrease in synaptic release. Consistent with the decrease in mIPSC frequency, we observed that DAMGO decreased evoked IPSC amplitude more robustly in D40 iN cells compared to N40 iNs (Fig. 3D–E). This is consistent with the hypothesis that MOR activation by DAMGO in human iN cells more robustly decreases neurotransmitter release probability in D40 iN cells compared to N40 iN cells, suggesting that the A118G SNP directly regulates synaptic function.
Figure 3 |. D40 iN cells exhibit greater inhibition of synaptic release and intrinsic excitability.
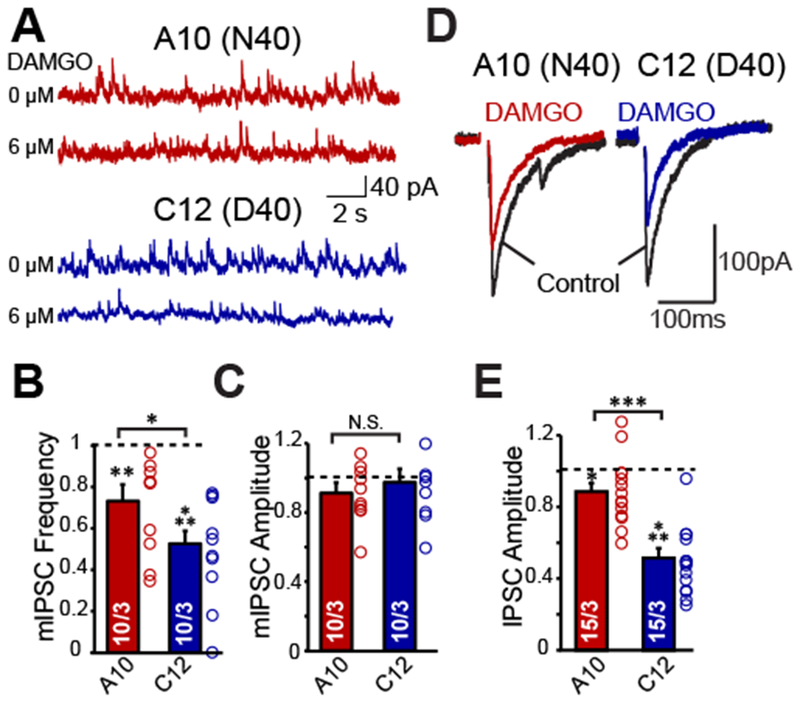
(A) Representative traces of mIPSCs in one N40 (A10) and one D40 (C12) cell line derived iN cells recorded at a 0 mV holding potential and their response to DAMGO. (B-C) Quantification of mIPSC frequency (A10: DAMGO vs control p <0.01, C12: DAMGO vs control p <0.001) and amplitude (A10 and C12: DAMGO vs control p >0.05) in A10 and C12 iN cells normalized to before DAMGO application. (DAMGO effect on frequency: A10 vs C12: p < 0.05, DAMGO effect on amplitude: A10 vs C12: N.S.) (D) Representative traces of evoked IPSCs from one N40 (A10) and one D40 (C12) cell line derived iN cells (E) Quantification of evoked IPSC amplitude in A10 and C12 iN cells normalized to before DAMGO application (A10: DAMGO vs control p <0.05, C12: DAMGO vs control p <0.001, A10 vs C12: p <0.001). Data are depicted as means ± SEM. Numbers of cells/Number of independently generated cultures analyzed are depicted in bars. Paired t-test was used to evaluate within genotype statistical differences and one-way ANOVA was used to evaluate between genotype statistical differences (*p <0.05, **p <0.01, ***p <0.001).
To determine whether the increased sensitivity of D40 iN cells is a DAMGO-specific effect or whether it can be observed across other MOR-specific agonists, we studied the effect of morphine in modulating synaptic release in both N40 and D40 iN cells. In response to 100 ¼M morphine, we observed a robust suppression of both sIPSC frequency and amplitude in both N40 and D40 iN cells with a significantly stronger sIPSC suppression in D40 iN cells compared to N40 iN cells (Supplemental Fig. 6). We also performed morphine wash out experiments to demonstrate that sIPSC suppression was reversible (Supplemental Fig. 6). These data substantiate the D40-specific increased sensitivity observed in human iN cells.
D40 MOR-expressing neurons exhibit a more robust decrease in excitability following DAMGO compared to N40 iN cells
To understand whether the decreased synaptic release in response to DAMGO is compounded by decreased intrinsic excitability, we examined the effect of DAMGO on induced action potential (AP) firing in N40 and D40 iN cells. Although no significant differences of AP firing rate, amplitudes and thresholds were observed between N40 (A10 clone) and D40 (C12 clone) (Supplemental Fig. 3), we observed that 10 ¼M DAMGO induced D40 versus N40 iN cells to fire significantly fewer APs (Fig. 4A–B) with no effect on AP amplitude and firing threshold (Fig. 4C–D). This is supported by an immediate and more robust decrease in spontaneous AP firing frequency following DAMGO application in D40 versus N40 iN cells (Fig. 4E), an effect which is sustained over the course of several minutes (Fig. 4F). This sustained decrease in AP frequency is paralleled by a rapid hyperpolarization of N40 and D40 iN cells (Fig. 4G). This effect was found to be significantly more robust in D40 versus N40 iN cells in the first minute following DAMGO application. The immediate drop in both AP firing frequency and membrane potential in iN cells suggests that this may be occurring through a G-protein mediated signaling mechanism, which is activated immediately following agonist binding 33. No differences in AP rise time, decay time or half width were detected by DAMGO application (data not shown). However, we observed a slight increase in the after-hyperpolarization potential (AHP) in the D40 versus N40 iN cells (Fig. 4H–I), with no significant difference in firing threshold or AP half width (Fig. 4J–K). These data indicate the functional differences between the two genotypes are at least partly mediated by a preferential decrease in excitability in D40 versus N40 iN cells, likely mediated by alterations in the G-protein coupled signaling cascade. Overall, these data suggest that the DAMGO-induced decrease in excitability is superimposed by a synapse-specific effect, i.e., a stronger reduction in synaptic release probability mediated by presynaptic MOR at the nerve terminal in D40 MOR inhibitory neurons.
Figure 4 |. D40 iN cells exhibit a sustained decrease in intrinsic excitability.
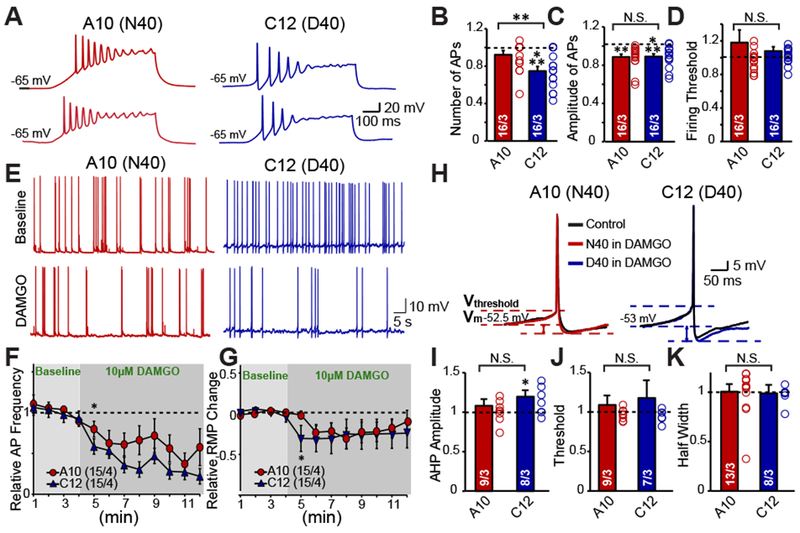
(A) Representative traces of repetitive action potentials generated from depolarizing current injections in one N40 (A10) cell line derived iN and one D40 (C12) cell line derived iN, and their response to DAMGO (B-D) Summary graphs of DAMGO effect on AP Number (A10: DAMGO vs control: N.S., C12: DAMGO vs control p <0.001), amplitude (A10: DAMGO vs control p <0.01, C12: DAMGO vs control p <0.001) and firing threshold (A10: DAMGO vs control: N.S., C12: DAMGO vs control: N.S.). Data normalized to before DAMGO application reveals DAMGO preferentially decreases intrinsic excitability of D40 iNs but not N40 iNs (DAMGO effect on frequency: A10 vs C12 p <0.01) with no effect on amplitude (A10 vs C12: N.S.) or firing threshold (A10 vs C12: N.S.) (E) Representative traces depicting the effect of DAMGO on spontaneous action potential firing in one D40 (C12) and one N40 (A10) cell line derived iN (F) Quantification of number of spontaneous action potentials fired before and after DAMGO application in N40 and D40 iNs represented as a time course (G) Quantification of resting membrane potential before and after DAMGO application in N40 and D40 iNs represented as a time course (H) Representative traces of individual Action Potentials before and after DAMGO in N40 and D40 iN cells (J-K) Summary graphs depicting that DAMGO causes a trending increase in the after hyperpolarization potential (AHP) amplitude in D40 iN cells compared to N40 iN cells (A10: DAMGO vs control: N.S., C12: DAMGO vs control: p <0.05, A10 vs C12: N.S.) with no effect on firing threshold (A10: DAMGO vs control: N.S., C12: DAMGO vs control: N.S., A10 vs C12: N.S.) or half width (A10: DAMGO vs control: N.S., C12: DAMGO vs control: N.S., A10 vs C12: N.S.). Data are depicted as means ± SEM. Numbers of cells/Number of independently generated cultures analyzed are depicted in bars. Paired t-test was used to evaluate within genotype statistical differences and one-way ANOVA was used to evaluate between genotype statistical differences (*p <0.05, **p <0.01, ***p <0.001).
Differential N-glycosylation of human MOR carrying N40D variants
It has been shown that the A112G substitution in mouse MOR causes a deficit in N-glycosylation 19–21, which might underlie the differential sensitivities of MOR modulation on synaptic transmission. However, this has not been shown in human N40D MORs. To address this, we used HEK293 cells to express FLAG-tagged N-terminal (amino acids 1-67) domain of human MORs. Media was collected ~ 5 days post transfection from FLAG-MOR (1-67) N40 or FLAG-MOR (1-67) D40 containing HEK cells and subjected to SDS-PAGE and Western Analysis (Fig. 5). Anti-FLAG immunostaining revealed the molecular mass of recombinant MORN40 was higher than that in MORD40 cell lysates (Fig. 5A, left side lane 1 compared to lane 2). We next tested whether the differences observed in the masses of human MOR in N40 and D40 are due to variations in N-linked glycosylation from the predicted loss of one putative glycosylation site (Asn40) in D40 MOR peptide. To this end, we treated cellular lysates with PNGase F, an enzyme which removes all N-linked glycans. Western blotting of FLAG-MOR using an anti-FLAG antibody showed that MOR N40 and D40 peptides migrated at approximately the same molecular weight (Fig. 5A, lanes 3-6). This indicates the differences in molecular masses observed for N40 and D40 human peptides is due to differential N-linked glycosylation.
Figure 5 |. Differential N-glycosylation of human MOR carrying N40D variants.
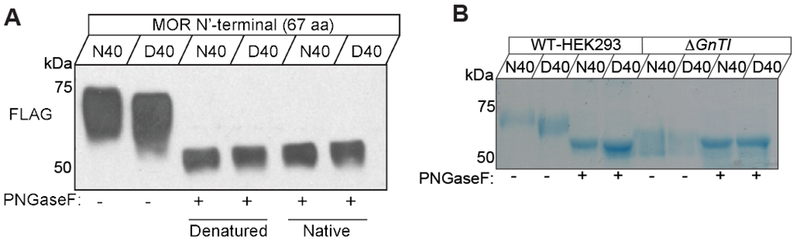
(A) Immunoblot (IB) of recombinant human FLAG-MOR N-terminal region-human Fc (amino acids 1-67) expressed as a soluble entity in HEK 293 cells. Both peptides (N40 and D40) were separated by size on an SDS-PAGE gel and detection was accomplished using an anti-FLAG antibody. Note that the untreated D40 sample (lane 2) is already of lower molecular weight compared to the N40 (lane 1) and after PNGase F digestion (lanes 3-6) both samples show an identical migration pattern, indicating complete removal of the N-linked glycans. PNGase reactions were carried out in two ways. Specifically, either the peptide was denatured prior to enzyme application (lane 3 and 4) or the native peptide was treated with PNGase F (lanes 5 and 6). Glycan removal was equally effective both ways tested. (B) Recombinant human FLAG-MOR N-terminal region-human Fc peptide was purified from the culture media of HEK293S (wild-type) (lanes 1-4) or HEK293S GnT1- (lanes 5-8). SDS-PAGE followed by Coomassie staining revealed that under conditions where glycosylation is defective or abolished N40 and D40 MOR peptide migrate at the same molecular weight (lanes 5 and 6) regardless of PNGase F treatment.
To further validate our findings, both FLAG-tagged MOR N-terminal constructs were expressed in the culture medium of HEK293S GnT1- cells. These cells lack N-acetylglucosaminyl-transferase I (GnT1) activity, and consequently lack complex N-glycans. Expressed N-linked MOR peptides were purified from the culture media and separated on SDS-PAGE followed by Coomassie brilliant blue staining to visualize purified MOR peptide (Fig. 5B). The profile of N40 and D40 MOR was similar to that of the anti-FLAG immunoblotting shown in Figure 5A, when isolated from wild-type HEK293 cells (Fig. 5B, lanes 1-4). The molecular mass of MORD40 was slightly smaller compared to MORN40 (Fig. 5B, lanes 1 and 2), which is due to a difference in N-linked glycosylation (Fig. 5B, lanes 3 and 4). Moreover, when constructs were collected from the culture medium of HEK293S GnT1- cells both N40 and D40 MOR N-terminal peptide run at roughly the same molecular weight before (Fig. 5B, lanes 5 and 6) and after PNGase F treatment (Fig. 5B, lanes 7 and 8). Thus, further supporting that the differences in mobility shift observed for MOR N40 and D40 is due to differential N-linked glycosylation.
DISCUSSION
Our study provides the first experimental evidence detailing the functional consequences of the N40D SNP on MOR activation in a human neuronal construct. First, we generated iN cells from human subject-derived stem cells carrying homozygous alleles for N40 MOR or D40 MOR and found that D40 MOR expressing iN cells exhibit stronger inhibitory effects of MOR activation on synaptic release. Second, to validate the functional consequences of the SNP in a system highly controlled for background genetic variation, we used CRISPR/Cas9 mediated gene targeting to: 1) knock-in homozygous D40 alleles into H1ES cells; 2) correct the homozygous D40 alleles in the 03SF iPS cell subject line into N40 alleles, and thus generated two sets of isogenic stem cell lines for highly controlled mechanistic analyses. Third, we demonstrated that the isogenic iN cells not only recapitulated the DAMGO response phenotype of the patient iN cells, but also revealed that the N40D SNP mediates a more robust decrease in excitability and synaptic release, which was accompanied with disruption of a putative N-linked glycosylation motif observed in human D40 MOR 34. The MOR is a GPCR and its activation has been shown to impact calcium as well as potassium influx in neurons 35, both processes affect the calcium dependency of synaptic vesicle release. The primary effect of MOR activation is a dampening in neuronal excitability. The A118G SNP does not affect baseline synaptic function per se, which can be seen based on our comparison of baseline sIPSC responses in N40 and D40 neurons (sIPSCs, Figs.1&2). However, it does affect the response of iN cells to MOR agonists (DAMGO and morphine). Based on the results of our study, the N40D SNP causes a more robust suppression of neuronal excitability upon MOR activation by DAMGO and also causes a stronger suppression of synaptic release. These data will help to elucidate the synaptic consequences of the N40D SNP in human neurons.
The novelty of our approach using human iN cells to investigate the synaptic impact of a major functional OPRM1 SNP is that these cells carry the genetic signatures of the subjects from whom they were derived. Identifying identical DAMGO-mediated responses across multiple subject-derived and CRISPR-edited cell lines generated using independently executed targeting strategies clearly demonstrates that the observed effect is a direct consequence of only the MOR N40D variant. In fact, the use of human stem cell derived neurons provides novel information about human specific mechanisms of a highly prevalent OPRM1 SNP, A118G, in response to opioids 36. Thus, we show the utility of disease modeling using stem cell derived disease-relevant cell types as a framework to the field of addiction for conducting future mechanistic analyses.
Despite previous studies in knock-in mouse models and heterologous expression systems, the precise molecular and cellular consequences of MOR N40D have remained unclear, primarily due to species-specific and context-specific mechanisms in the modulation of MOR signaling. For instance, overexpression systems have suggested that the D40 allele confers a “gain-of-function” effect by causing an increased potency for DAMGO and other MOR agonists 9, 13, 14, 37. Similarly, human studies (heterologous expression systems) and rodent models suggest that the D40 allele carriers exhibit altered reward to nicotine 38, alcohol 39–48, and heroin 49. However, subsequent studies have reported that the D40 allele is associated with reduced mRNA and protein expression in multiple brain regions of knock-in mice 12, 15 along with reduced antinociceptive responses to morphine 10, providing support for a “loss-of-function” phenotype. These contradictory results in the literature strongly suggest not only species-specific but also context-specific mechanisms in the modulation of MOR signaling. For example, overexpression studies were primarily done in cell models in order to elucidate differences in signaling pathways relevant to the human condition. However, it is also possible that this “gain of function” effect of MOR N40D observed in overexpression systems is an artifact of an increased receptor density at the cell surface and not due to true differences in signaling efficacy from MOR N40D, suggesting the need for a model system that recapitulates proper receptor expression levels. Moreover, it is possible that the “loss-of-function” phenotype observed in the A112G and A118G humanized mouse models are likely a consequence of rodent-specific regulation in receptor expression levels. Both these results necessitate the need for a human neuronal model to elucidate the appropriate mechanisms, which would represent endogenous levels of MOR in a disease-relevant cell type.
This study not only represents a significant advance in understanding the neurobiological mechanisms underlying the N40D MOR variant in a human neuronal context, but also highlight the benefits of using human cell reprograming technology for in depth analysis of gene variants that have been associated with certain human disease. With our model we were able to generate homogeneous iN lines with MOR expression comparable to human thalamus. Currently we have not examined the effect of DAMGO on excitatory postsynaptic current (EPSC) frequency in other human neuronal subtypes. However, we hypothesize that NGN2 excitatory iN cells will likely exhibit a DAMGO mediated suppression of EPSC frequency assuming they express OPRM1. Whether or not the genotype-specific effect is observed in excitatory iN cells or whether it is characteristic to the inhibitory neuronal subtype can only be determined experimentally. However, considering our interest in understanding the role of N40D in inhibitory neurons that mimic those in the reward circuitry, characterizing the role of N40D in other neuronal subtypes will need further investigation.
The observed effect of MOR N40D in human iN cells is consistent with a number of genome wide association studies (GWAS) in humans linking OPRM1 A118G with increased alcohol response, etc. However, in the humanized mouse model, a morphine mediated decrease in mIPSC frequency in OPRM118AA mice but not in 118GG mice was observed 50. These results are likely mediated by species-specific differences in receptor expression levels or signaling mechanisms, making the elucidation of human-specific phenotypes key to understanding the role of MOR N40D in reward and addiction pathogenesis.
The use of human iN cells enables the study of synaptic perturbations that may be involved in addiction and reward pathogenesis. We observed increased sensitivity in D40 MOR iNs to the MOR-specific agonist, DAMGO, in a number of patient-derived and gene-edited iN cells. We also tested another MOR agonist, opiate morphine, and observed similar responses which we observed with the synthetic opioid peptide DAMGO. Our data indicate that MOR activation affects the frequency of spontaneous IPSCs (sIPSCs) and miniature IPSCs (mIPSCs) but not their amplitudes. These data suggest that the MOR is expressed at the presynaptic inhibitory nerve terminals to exert presynaptic modulation. It is traditionally believed that in synaptic physiology, a change in the frequency of events (and not the amplitude) is due to a presynaptic alteration of neurotransmitter release. Modulation of the number of events is likely a consequence of a change in presynaptic release of neurotransmitter. This result could be explained by differences seen in N-linked glycosylation of human MOR which could impact agonist binding affinity and/or receptor trafficking 51 .
This study identifies neurophysiological differences between common variation in human OPRM1, which may explain altered opioid responsivity and/or dependence in humans. This model not only enables studies that help clarify the mechanistic discrepancies observed in the literature, but also enables a platform to understand human-specific regulation of MOR signaling. Overall the use of patient-derived stem cells to unravel the impact of OPRM1 gene variants may ultimately provide the necessary insight to develop patient-specific, precision medical interventions for drug and alcohol dependence.
Supplementary Material
ACKNOWLEDGMENTS
We thank RUCDR Infinite Biologics for generating the iPS cells from human subjects and assisting with CRISPR/Cas9 gene targeting on 03SF iPS cell line. Research is supported by grants from NIH-NIAAA R01 AA023797 as well as Collaborative Studies on the Genetics of Alcoholism/COGA 5U10AA008401-26. AH is supported by NIH-NIAAA NRSA F31AA024033. We are grateful to the members of the Collaborative Genetic Study of Nicotine Dependence (COGEND) for the selection of human subjects, and we are grateful to the de-identified individuals who contributed tissue to the study. Pang laboratory at CHINJ is partly supported by a grant from the RWJ Foundation.
Footnotes
DISCLOSURES
The authors declare they have no competing financial interests.
REFERENCES
- 1.Centers for Disease Control and Prevention. Opioid overdose: understanding the epidemic. 2017.
- 2.Contet C, Kieffer BL, Befort K. Mu opioid receptor: a gateway to drug addiction. Curr Opin Neurobiol 2004; 14(3): 370–378. [DOI] [PubMed] [Google Scholar]
- 3.Gerrits MA, Lesscher HB, van Ree JM. Drug dependence and the endogenous opioid system. Eur Neuropsychopharmacol 2003; 13(6): 424–434. [DOI] [PubMed] [Google Scholar]
- 4.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci 2007; 8(11): 844–858. [DOI] [PubMed] [Google Scholar]
- 5.Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S et al. The Ensembl gene annotation system. Database (Oxford) 2016; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mague SD, Blendy JA. OPRM1 SNP (A118G): involvement in disease development, treatment response, and animal models. Drug Alcohol Depend 2010; 108(3): 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LaForge KS, Yuferov V, Kreek MJ. Opioid receptor and peptide gene polymorphisms: potential implications for addictions. Eur J Pharmacol 2000; 410(2-3): 249–268. [DOI] [PubMed] [Google Scholar]
- 8.Kroslak T, Laforge KS, Gianotti RJ, Ho A, Nielsen DA, Kreek MJ. The single nucleotide polymorphism A118G alters functional properties of the human mu opioid receptor. J Neurochem 2007; 103(1): 77–87. [DOI] [PubMed] [Google Scholar]
- 9.Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L et al. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci U S A 1998; 95(16): 9608–9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mague SD, Isiegas C, Huang P, Liu-Chen LY, Lerman C, Blendy JA. Mouse model of OPRM1 (A118G) polymorphism has sex-specific effects on drug-mediated behavior. Proc Natl Acad Sci U S A 2009; 106(26): 10847–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Befort K, Filliol D, Decaillot FM, Gaveriaux-Ruff C, Hoehe MR, Kieffer BL. A single nucleotide polymorphic mutation in the human mu-opioid receptor severely impairs receptor signaling. J Biol Chem 2001; 276(5): 3130–3137. [DOI] [PubMed] [Google Scholar]
- 12.Beyer A, Koch T, Schroder H, Schulz S, Hollt V. Effect of the A118G polymorphism on binding affinity, potency and agonist-mediated endocytosis, desensitization, and resensitization of the human mu-opioid receptor. J Neurochem 2004; 89(3): 553–560. [DOI] [PubMed] [Google Scholar]
- 13.Mahmoud S, Thorsell A, Sommer WH, Heilig M, Holgate JK, Bartlett SE et al. Pharmacological consequence of the A118G mu opioid receptor polymorphism on morphine- and fentanyl-mediated modulation of Ca(2)(+) channels in humanized mouse sensory neurons. Anesthesiology 2011; 115(5): 1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Margas W, Zubkoff I, Schuler HG, Janicki PK, Ruiz-Velasco V. Modulation of Ca2+ channels by heterologously expressed wild-type and mutant human micro-opioid receptors (hMORs) containing the A118G single-nucleotide polymorphism. J Neurophysiol 2007; 97(2): 1058–1067. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Wang D, Johnson AD, Papp AC, Sadee W. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J Biol Chem 2005; 280(38): 32618–32624. [DOI] [PubMed] [Google Scholar]
- 16.Miller GM, Bendor J, Tiefenbacher S, Yang H, Novak MA, Madras BK. A mu-opioid receptor single nucleotide polymorphism in rhesus monkey: association with stress response and aggression. Mol Psychiatry 2004; 9(1): 99–108. [DOI] [PubMed] [Google Scholar]
- 17.Ducat E, Ray B, Bart G, Umemura Y, Varon J, Ho A et al. Mu-opioid receptor A118G polymorphism in healthy volunteers affects hypothalamic-pituitary-adrenal axis adrenocorticotropic hormone stress response to metyrapone. Addict Biol 2013; 18(2): 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wand GS, McCaul M, Yang X, Reynolds J, Gotjen D, Lee S et al. The mu-opioid receptor gene polymorphism (A118G) alters HPA axis activation induced by opioid receptor blockade. Neuropsychopharmacology 2002; 26(1): 106–114. [DOI] [PubMed] [Google Scholar]
- 19.Wang YJ, Huang P, Ung A, Blendy JA, Liu-Chen LY. Reduced expression of the mu opioid receptor in some, but not all, brain regions in mice with OPRM1 A112G. Neuroscience 2012; 205: 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang YJ, Huang P, Blendy JA, Liu-Chen LY. Brain region- and sex-specific alterations in DAMGO-stimulated [ S]GTPgammaS binding in mice with Oprm1 A112G. Addiction biology 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray R, Ruparel K, Newberg A, Wileyto EP, Loughead JW, Divgi C et al. Human Mu Opioid Receptor (OPRM1 A118G) polymorphism is associated with brain mu-opioid receptor binding potential in smokers. Proc Natl Acad Sci U S A 2011; 108(22): 9268–9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bilbao A, Robinson JE, Heilig M, Malanga CJ, Spanagel R, Sommer WH et al. A Pharmacogenetic Determinant of Mu-Opioid Receptor Antagonist Effects on Alcohol Reward and Consumption: Evidence from Humanized Mice. Biological psychiatry 2014. [DOI] [PubMed] [Google Scholar]
- 23.Moore JC, Sheldon MH, Hart RP. Biobanking in the Era of the Stem Cell: A Technical and Operational Guide. Morgan & Claypool Life Sciences 2012, 78pp. [Google Scholar]
- 24.Oni EN, Halikere A, Li G, Toro-Ramos AJ, Swerdel MR, Verpeut JL et al. Increased nicotine response in iPSC-derived human neurons carrying the CHRNA5 N398 allele. Sci Rep 2016; 6: 34341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK, Dobbs D. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res 2010; 38(Web Server issue): W462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of biotechnology 2015; 208: 44–53. [DOI] [PubMed] [Google Scholar]
- 27.Yang N, Chanda S, Marro S, Ng YH, Janas JA, Haag D et al. Generation of pure GABAergic neurons by transcription factor programming. Nat Methods 2017; 14(6): 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maximov A, Pang ZP, Tervo DG, Sudhof TC. Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J Neurosci Methods 2007; 161(1): 75–87. [DOI] [PubMed] [Google Scholar]
- 29.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010; 463(7284): 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comoletti D, Miller MT, Jeffries CM, Wilson J, Demeler B, Taylor P et al. The macromolecular architecture of extracellular domain of alphaNRXN1: domain organization, flexibility, and insights into trans-synaptic disposition. Structure 2010; 18(8): 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci 1992; 12(2): 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Margolis EB, Hjelmstad GO, Fujita W, Fields HL. Direct bidirectional mu-opioid control of midbrain dopamine neurons. J Neurosci 2014; 34(44): 14707–14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S et al. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 2013; 65(1): 223–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang P, Chen C, Mague SD, Blendy JA, Liu-Chen LY. A common single nucleotide polymorphism A118G of the mu opioid receptor alters its N-glycosylation and protein stability. Biochem J 2012; 441(1): 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011; 115(6): 1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scarnati MS, Halikere A, Pang ZP. Using human stem cells as a model system to understand the neural mechanisms of alcohol use disorders: Current status and outlook. Alcohol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez Soto EJ, Raingo J. A118G Mu Opioid Receptor polymorphism increases inhibitory effects on CaV2.2 channels. Neurosci Lett 2012; 523(2): 190–194. [DOI] [PubMed] [Google Scholar]
- 38.Perkins KA, Lerman C, Grottenthaler A, Ciccocioppo MM, Milanak M, Conklin CA et al. Dopamine and opioid gene variants are associated with increased smoking reward and reinforcement owing to negative mood. Behav Pharmacol 2008; 19(5-6): 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bart G, Kreek MJ, Ott J, LaForge KS, Proudnikov D, Pollak L et al. Increased attributable risk related to a functional mu-opioid receptor gene polymorphism in association with alcohol dependence in central Sweden. Neuropsychopharmacology 2005; 30(2): 417–422. [DOI] [PubMed] [Google Scholar]
- 40.Ehlers CL, Lind PA, Wilhelmsen KC. Association between single nucleotide polymorphisms in the mu opioid receptor gene (OPRM1) and self-reported responses to alcohol in American Indians. BMC Med Genet 2008; 9: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Enoch MA. Genetic influences on the development of alcoholism. Curr Psychiatry Rep 2013; 15(11): 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Enoch MA. Genetic influences on response to alcohol and response to pharmacotherapies for alcoholism. Pharmacol Biochem Behav 2014; 123: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SG, Kim CM, Kang DH, Kim YJ, Byun WT, Kim SY et al. Association of functional opioid receptor genotypes with alcohol dependence in Koreans. Alcohol Clin Exp Res 2004; 28(7): 986–990. [DOI] [PubMed] [Google Scholar]
- 44.Koller G, Zill P, Rujescu D, Ridinger M, Pogarell O, Fehr C et al. Possible association between OPRM1 genetic variance at the 118 locus and alcohol dependence in a large treatment sample: relationship to alcohol dependence symptoms. Alcohol Clin Exp Res 2012; 36(7): 1230–1236. [DOI] [PubMed] [Google Scholar]
- 45.Miranda R, Ray L, Justus A, Meyerson LA, Knopik VS, McGeary J et al. Initial evidence of an association between OPRM1 and adolescent alcohol misuse. Alcohol Clin Exp Res 2010; 34(1): 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishizawa D, Han W, Hasegawa J, Ishida T, Numata Y, Sato T et al. Association of mu-opioid receptor gene polymorphism A118G with alcohol dependence in a Japanese population. Neuropsychobiology 2006; 53(3): 137–141. [DOI] [PubMed] [Google Scholar]
- 47.Ray LA, Hutchison KE. A polymorphism of the mu-opioid receptor gene (OPRM1) and sensitivity to the effects of alcohol in humans. Alcohol Clin Exp Res 2004; 28(12): 1789–1795. [DOI] [PubMed] [Google Scholar]
- 48.Rommelspacher H, Smolka M, Schmidt LG, Samochowiec J, Hoehe MR. Genetic analysis of the mu-opioid receptor in alcohol-dependent individuals. Alcohol 2001; 24(2): 129–135. [DOI] [PubMed] [Google Scholar]
- 49.Szeto CY, Tang NL, Lee DT, Stadlin A. Association between mu opioid receptor gene polymorphisms and Chinese heroin addicts. Neuroreport 2001; 12(6): 1103–1106. [DOI] [PubMed] [Google Scholar]
- 50.Robinson JE, Vardy E, DiBerto JF, Chefer VI, White KL, Fish EW et al. Receptor Reserve Moderates Mesolimbic Responses to Opioids in a Humanized Mouse Model of the OPRM1 A118G Polymorphism. Neuropsychopharmacology 2015; 40(11): 2614–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gupta A, Rozenfeld R, Gomes I, Raehal KM, Decaillot FM, Bohn LM et al. Post-activation-mediated changes in opioid receptors detected by N-terminal antibodies. J Biol Chem 2008; 283(16): 10735–10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.