

Graphical abstract

Keywords: Diabetes; PPAR-γ; Thiazolidine-2,4-diones; Pioglitazone; Rosiglitazone
Abbreviations: ADDP, 1,1′-(Azodicarbonyl)dipiperidine; AF, activation factor; ALT, alanine transaminase; ALP, alkaline phosphatase; AST, aspartate transaminase; Boc, Butyloxycarbonyl; DNA, deoxyribonucleic acid; DBD, DNA-binding domain; DM, diabetes mellitus; DCM, dichloromethane; DMF, dimethylformamide; DMSO, dimethyl sulfoxide; E, Entgegen; ECG, electrocardiogram; FDA, food and drug administration; FFA, free fatty acid; GAL4, Galactose transporter type; GLUT4, glucose transporter type 4; GPT, glutamic pyruvic transaminase; HCl, Hydrochloric Acid; HDL, high-density lipoprotein; HEp-2, Human epithelial type 2; HFD, high-fat diet; HEK, human embryonic kidney; i.m, Intramuscular; INS-1, insulin-secreting cells; IL-β, interlukin-beta; IDF, international diabetes federation; K2CO3, Potassium carbonate; LBD, ligand-binding domain; LDL, low-density lipoprotein; MDA, malondialdehyde; mCPBA, meta-chloroperoxybenzoic acid; NBS, N-bromosuccinimide; NaH, Sodium Hydride; NA, nicotinamide; NO, nitric oxide; NFκB, nuclear factor kappa-B; OGTT, oral glucose tolerance test; PPAR, peroxisome-proliferator activated receptor; PPRE, peroxisome proliferator response element; Pd, Palladium; PDB, protein data bank; PTP1B, protein-tyrosine phosphatase 1B; KOH, potassium hydroxide; QSAR, quantitative structure-activity relationship; RXR, retinoid X receptor; STZ, streptozotocin; SAR, structure-activity relationship; T2DM, type 2 diabetes mellitus; THF, tetrahydrofuran; TZD, thiazolidine-2,4-dione; TFA, trifluoroacetic acid; TFAA, trifluoroacetic anhydride; TG, triglycerides; TNF-α, tumor necrosis factor-alpha; WAT, white adipose tissue; Z, Zusammen
Highlights
-
•
TZDs, an important pharmacophore in the treatment of diabetes.
-
•
Various analog-based synthetic strategies and biological significance are discussed.
-
•
Clinical studies using TZDs along with other antidiabetic agents are also highlighted.
-
•
SAR has been discussed to suggest the interactions between derivatives and receptor sites.
-
•
Pyrazole, chromone, and acid-based TZDs can be considered as potential lead molecules.
Abstract
Diabetes or diabetes mellitus is a complex or polygenic disorder, which is characterized by increased levels of glucose (hyperglycemia) and deficiency in insulin secretion or resistance to insulin over an elongated period in the liver and peripheral tissues. Thiazolidine-2,4-dione (TZD) is a privileged scaffold and an outstanding heterocyclic moiety in the field of drug discovery, which provides various opportunities in exploring this moiety as an antidiabetic agent. In the past few years, various novel synthetic approaches had been undertaken to synthesize different derivatives to explore them as more potent antidiabetic agents with devoid of side effects (i.e., edema, weight gain, and bladder cancer) of clinically used TZD (pioglitazone and rosiglitazone). In this review, an effort has been made to summarize the up to date research work of various synthetic strategies for TZD derivatives as well as their biological significance and clinical studies of TZDs in combination with other category as antidiabetic agents. This review also highlights the structure-activity relationships and the molecular docking studies to convey the interaction of various synthesized novel derivatives with its receptor site.
Introduction
In this modernized industrial world, the ever-growing population rate along with physical inactivity of people has put the life of mankind on an edge of being targeted by various diseases among which diabetes is the most common one. According to the International Diabetes Federation (IDF), the morbidity rate of this insidious disease has been estimated to show an increase from 425 million in 2017 to 629 million by 2045 [1]. Diabetes or diabetes mellitus (DM) is a complex or polygenic disorder which is characterized by increased levels of glucose (hyperglycemia) resulting from defects in insulin secretion, action or both (resistance) to insulin over an elongated period in the liver and peripheral tissues. DM is classified as type 1 i.e. insulin-dependent, type 2 i.e. non-insulin dependent and gestational diabetes (in pregnant women) [2], [3]. The symptoms include polyuria, tiredness, dehydration, polyphagia, and polydipsia [4]. Therefore, it is necessary to maintain the proper blood glucose level, mainly during the early stages of diabetes. Several types of anti-hyperglycaemic agents are used as monotherapy or combination therapy to treat DM. These include meglitinides, biguanides, sulphonylurea, and α-glucosidase inhibitors. In addition to these, sesquiterpenoids have also been reported as potential anti-diabetic agents by virtue of protecting β-pancreatic cells and improving insulin secretion [5]. The treatment of type 2 diabetes mellitus (T2DM) has been reformed with the origin of thiazolidine-2,4-diones (TZDs) class of molecules that bring down the increased levels of blood glucose to normal [6].
TZDs also called as glitazones are the heterocyclic ring system consisting of five-membered thiazolidine moiety having carbonyl groups at 2 and 4 positions. Various substitutions can only be done at third and fifth positions. A comprehensive research has been done on TZDs resulting in various derivatives [7]. Though, substantial evidence reported with TZDs but none of them have reported up to date review and clinical studies of TZD [7], [8], [9]. In this review, we aimed to present the information from synthetic, in vitro, and in vivo studies that had been carried out on various TZD derivatives by collecting research journals published from the date of discovery of TZD in the early 1980s. In addition, we have discussed their molecular target (peroxisome proliferator-activated receptors, PPAR-γ), toxicity profiling (hepatotoxicity and cardiotoxicity) and their structure–activity relationship (SAR). Further, we have compiled clinical studies of TZDs that had been done in combination with other categories as antidiabetic agents. We believe that this review will provide sound knowledge, and guidance to carry out further research on this scaffold to mitigate the problems of clinically used TZDs.
The general procedure for synthesizing TZDs has been shown in S1. TZDs (3) has been synthesized by refluxing thiourea (1) with chloroacetic acid (2) for 8–12 h at 100–110 °C, using water and conc. HCl as a solvent [10].
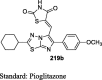
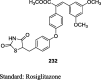
Antiquity of TZDs
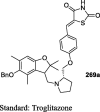
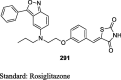
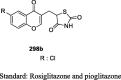
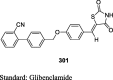
The antihyperglycemic activity of TZDs came into notice by the entry of first drug, ciglitazone in the early 1980s but later on, it was withdrawn due to its liver toxicity. Then, troglitazone was discovered and developed by Sankyo Company in the year 1988. However, it caused hepatotoxicity, as a result, it was banned in 2000. In 1999, Takeda and Pfizer developed two drugs, pioglitazone, and englitazone. However, englitazone was discontinued due to its adverse effects on the liver. Conversely, pioglitazone was described to be safe on the hepatic system. Meanwhile, rosiglitazone and darglitazone developed by Smithkline and Pfizer. However, darglitazone was terminated in the year 1999. Reports in 2001 revealed that rosiglitazone had shown to cause heart failure due to fluid retention and was first restricted by Food and Drug Administration (FDA) in 2010, later on in 2013 in a trial, it fails to show any effect on heart attack, and therefore restriction was removed by FDA (Fig. 1). The structure of various clinically reported TZDs is shown in Fig. 2 [11], [12], [13] and the studies, which were carried out in diabetic patients are presented in Table 1 [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61].
Fig. 1.

The history of TZDs (modified and).
adapted from [13].
Fig. 2.

Chemical structures of clinically used thiazolidine-2, 4-dione compounds (structures are original and made by using chem draw ultra 12.0).
Table 1.
Efficacy of TZDs in diabetes in clinical trials.
| Clinical Trial No. | Population Size | Status | Interventions | Phase | End Point | Reference |
|---|---|---|---|---|---|---|
| NCT00396227 | 2665 | Completed | 1. Vildagliptin add- on to metformin 2. TZD (pioglitazone, rosiglitazone) add on to metformin |
Phase 3 | • Mean change in HbA(1c) was −0.68 ± 0.02% in the vildagliptin group and −0.57 ± 0.03% in the TZD group. • Body weight increased in the TZD group (0.33 ± 0.11 kg) and decreased in the vildagliptin group (−0.58 ± 0.09 kg). • Adverse events were similar in both groups (vildagliptin: 39.5% and TZD: 36.3%). |
[14] |
| NCT02653209 | 600 | Undergoing | 1. Sitagliptin, 2. Canagliflozin 3. Pioglitazone |
Phase 4 | • HbA(1c) in obese patients (BMI > 30 kg/m2) was compared to non-obese patients. • Test the hypothesis that the patients with BMI > 30 kg/m2 respond well to pioglitazone, and less well to sitagliptin in comparison to non-obese patients or not. • On treatment HbA(1c) levels in patients with an eGFR < 90 mL/min/1.73 m2 compared to patients with an eGFR > 90 mL/min/1.73 m2. • Test the hypothesis that the patients with modestly reduced eGFR (60–90 mL/min/1.73 m2) respond poorly to canagliflozin, and well to sitagliptin in comparison to eGFR > 90 mL/min/1.73 m2 eGFR or not. • Prevalence of side effects: weight gain, hypoglycemia, edema, genital tract infection and discontinuation of therapy. • HbA(1c) therapy vs. predefined test of gender heterogeneity (i.e., Females are likely to show an improved response relative to males for pioglitazone). |
[15] |
| NCT00743002 | 87 | Completed | 1. TT223 with Metformin and/or TZD 2. Placebo with Metformin and/or TZD |
Phase 2 | • The safety and tolerability of TT223 was evaluated at 1 mg, 2 mg and 3 mg. • The efficacy of TT223 was evaluated in terms of changes in HbA(1c) value, fasting glucose levels vs. placebo group. • Determining the pharmacokinetic parameter of TT223 in patients. |
[26] |
| NCT01026194 | 204 | Completed | 1. Placebo/Teneligliptin + pioglitazone 2. Teneligliptin/Teneligliptin + pioglitazone |
Phase 3 | • The changes in HbA(1c) were greater (−0.9 ± 0.0%) in the teneligliptin group than that in the placebo group (−0.2 ± 0.0%). • The change in FPG was greater in the teneligliptin group than that in the placebo group. |
[37] |
| NCT00879970 | 1332 | Terminated | 1. Pioglitazone 2. Rosiglitazone 3. Placebo 4. Vitamin D placebo 5. Vitamin D |
Phase 4 | • Cardiovascular outcome (MI, stroke or cardiovascular death) is more in the placebo than in the treatment groups [TZD arm (0.4%) than Vitamin D arm (0.3%)]. • Hospitalization due to cancer is more in the placebo vs. Vitamin D arm. |
[48] |
| NCT00676338 | 820 | Completed | 1. Exenatide (once weekly) 2. Metformin 3. Sitagliptin 4. Pioglitazone 5. Placebo |
Phase 3 | • Exenatide was non-inferior to metformin but superior to sitagliptin, and pioglitazone with regard to HbA(1c) reduction. • Exenatide and metformin provided similar improvements in glycemic control along with the benefit of weight reduction and no increased risk of hypoglycemia. • Weight gain was observed in the pioglitazone group. |
[57] |
| NCT00683878 | 972 | Completed | 1. Dapagliflozin (5 mg) + TZD 2. Dapagliflozin (10 mg) + TZD 3. Placebo matching dapagliflozin + pioglitazone |
Phase 3 | • The mean reduction in HbA(1c) was higher for arm 1 and 2 groups (−0.82 and −0.97%) vs. placebo (−0.42%). • Pioglitazone alone had greater weight gain (3 kg) than those receiving plus pioglitazone in combination with dapagliflozin (0.7–1.4 kg). • Events of genital infection were reported with dapagliflozin (8.6–9.2%). |
[58] |
| NCT01135394 | 134 | Completed | 1. Pioglitazone | Phase 4 | • Characterize the changes at the physiological, cellular and molecular levels after TZD treatment. • Define genes that are regulated by TZD response. • Identify the SNPs and haplotypes genes that are influenced by TZD. • Glycemic, lipoprotein profile, and weight were monitored. |
[59] |
| NCT00481429 | 12 | Completed | 1. Rosiglitazone 2. Diet control + metformin |
NA | • The performance of baseline biochemical biomarkers (plasma and urine) in patients who respond to TZD therapy from those do not, through the changes in HbA(1c) at 12 weeks. • Changes in baseline levels of key biochemical markers. • Effect of treatment on various novel predictive biomarkers and markers of insulin sensitivity. |
[60] |
| NCT00295633 | 565 | Completed | 1. Saxagliptin 2.5 mg + Pioglitazone 30 mg + Rosiglitazone 4 mg + Metformin 500–2500 mg 2. Saxagliptin 5 mg + Pioglitazone 30 mg + Rosiglitazone 4 mg + Metformin 500–2500 mg 3. Placebo + Pioglitazone + Rosiglitazone + Metformin |
Phase 3 | • Mean changes from baseline HbA(1c) was more in saxagliptin (−0.66% and −0.94% for 2.5 and 5 mg, respectively) than that in placebo group (−0.30%). • Plasma glucose level was also significantly reduced in the saxagliptin group than that in the placebo group. • Hypoglycemic events were similar between groups. |
[61] |
| NCT00308373 | 73 | Completed | 1. Metformin 2. Pioglitazone |
NA | • Impact of TZD on the levels of cortisol. • Effect of TZD on breathing or sleepiness in patients with type 2 diabetes. |
[16] |
| NCT01055223 | 98,483 | Completed | 1. TZD only (rosiglitazone or pioglitazone or troglitazone) 2. TZD + spironolactone 3. TZD + amiloride |
NA | • Impact on the fracture number/number of fracture of hand/foot/upper arm/wrist fracture and hip in both males and females after 6 and 12-months treatment. | [17] |
| NCT00637273 | 514 | Completed | 1. Exenatide (once weekly) 2. Sitagliptin 3. Pioglitazone 4. Placebo tablet 5. Placebo once weekly |
Phase 3 | • Greater reduction of HbA(1c) in exenatide (−1.5%) than sitagliptin (−0.9%) or pioglitazone (−1.2%). • Weight loss was greater with exenatide (−2.3 kg) than sitagliptin (−1.5 kg) or pioglitazone (−5.1 kg). • Major adverse events were nausea and diarrhea with exenatide and sitagliptin. |
[18] |
| NCT00953498 | 40 | Completed | 1. Pioglitazone 2.Rosiglitazone |
Phase 4 | • HDL from control subjects had significantly shown to reduce the inhibitory effect of oxidised LDL on vasodilatation (Emax = 77.6 ± 12.9 vs. 59.5 ± 7.7%), whereas HDL from type 2 diabetic patients had no effect (Emax = 52.4 ± 20.4 vs 57.2 ± 18.7%). | [19] |
| NCT02315287 | 190 | Recruiting | 1.Metformin + Sitagliptin + Pioglitazone 2. Metformin + Sitagliptin + Lobeglitazone |
Phase 4 | • Change in the level of HbA(1c). • Changes in β-cell function and insulin resistance after 1-year treatment. • Changes in FBS after 5 and 12 months. |
[20] |
| NCT01147627 | 416 | Completed | 1. Exenatide injection 2. Mixed protamine zinc recombinant human Insulin Lispro 25R 3. Pioglitazone |
NA | • Changes in baseline value of HbA(1c) after 48-weeks • Percentage of patients achieving HbA(1c) (<6.5–7) and effect on fasting and postprandial plasma glucose concentration, blood pressure, lipid profiles. • Safety and tolerability in various groups. |
[21] |
| NCT00700856 | 3371 | Active, not recruiting | 1. Metformin + pioglitazone 2. Metformin + sulphonylureas (glibenclamide or gliclazide glimepiride) |
Phase 4 | • Hypoglycemia occurred less in the pioglitazone group (10%) than in the sulfonylurea group (34%). • Moderate weight gain (<2 kg) occurred in both groups. • Rate of adverse events such as heart failure, bladder cancer, and fractures was similar in both groups. |
[22] |
| NCT00329225 | 630 | Completed | 1. Rosiglitazone | Phase 4 | • The decrease in HbA(1c), C-reactive protein, fibrinogen and matrix metalloproteinase 9 levels upon addition of rosiglitazone to insulin. • Adverse events were mild to moderate. |
[23] |
| NCT03646292 | 60 | Not yet recruiting | 1. Pioglitazone 2. Empagliflozin 3. Pioglitazone + empagliflozin |
Phase 4 | • Changes in liver fat through MRI-PDFF and liver fibrosis through MRE. • Changes in lipid profile, liver enzyme, glucose metabolism and inflammation status (CRP) were monitored. |
[24] |
| NCT02426294 | 154 | Recruiting | 1. Pioglitazone 2. Glimepiride |
Phase 4 | • Evidence of efficacy of glycemic control by HbA(1c). • Changes in insulin resistance by HOMA and lipid profile from baseline value after 26-weeks treatment. |
[25] |
| NCT00333723 | 245 | Completed | 1. Rosiglitazone | Phase 4 | • Efficacy of rosiglitazone combined with glyburide to glyburide monotherapy upon FPG, c-peptide, HOMA and in reducing HbA(1c) after 24-weeks of the treatment period. | [27] |
| NCT02954692 | 111 | Completed | 1. Insulin glargine 2. Metformin 3. Sulfonylurea 4. Meglitinides 5. TZDs 6. α-glucosidase inhibitors 7. GLP1 receptor agonist 8. DPP-4 inhibitors 9. SGLT-2 inhibitors |
Phase 4 | • Changes from baseline in the levels of HbA(1c), SMBG, FPG and DTSQ scores at 12 and 24-weeks. • Percentage of patients reaching targeted fasting SMBG (80–130 mg/dL) at 12 and 24-weeks. |
[28] |
| NCT02475499 | 886,172 | Completed | 1. DPP-4 inhibitors 2. GLP-1 analogs 3. Sulfonylureas 4. Biguanides 5. TZDs 6. α-glucosidase inhibitors 7. Meglitinides |
NA | • Number of increased risk of pancreatic cancer was measured while using incretin-based drugs in comparison with sulfonylureas. |
[29] |
| NCT01030679 | 214 | Completed | 1. CKD-501 (Lobeglitazone) (0.5, 1 and 2 mg) 2. Placebo |
Phase 2 | • Changes from baseline in the levels of FPG, glycemic and lipid parameters at 8-weeks. • Profiling of adverse events at 8-weeks. |
[30] |
| NCT01593371 | 98 | Completed | 1. Metformin 2. Pioglitazone |
NA | • No changes in BMI while using pioglitazone and metformin. • Improvements in glycemia and insulin resistance. • Increase in chemerin levels. • Indices of glycemic control and insulin resistance were significantly improved by both groups after 3-months. • Both treatments are equally effective in reducing chemerin concentrations, a novel member of the adipokine family. • Did not alter waist circumference, weight or BMI by both drugs. |
[31] |
| NCT01223196 | 29 | Completed | 1. Pioglitazone 2. Placebo |
Phase 4 | • Improvements in glycaemic control, β-cell function and inflammatory indices (MCP-1, IL-6, FRK, hsCRP, and PAI) at low-dose of pioglitazone (15 mg/day) in obese patients with type 2 diabetes. • Adiponectin levels and TACE enzymatic activity is significantly decreased by pioglitazone than in the placebo group. |
[32] |
| NCT00367055 | 84 | Completed | 1. Rosiglitazone + metformin 2. Metformin 3. Metformin + gliclazide |
Phase 4 | • Changes from baseline in the insulin secretory capacity, insulin resistance index (HOMA-IR) and β-cell function index (HOMA-beta) • Changes from baseline in HbA(1c), FBG, CPP total and incremental AUC and • Changes from baseline in CPP concentration peak and incremental concentration peak at the month of 36. |
[33] |
| NCT02476760 | 1,417,914 | Completed | 1. DPP-4 inhibitors 2. GLP-1analogs 3. Insulin 4. Biguanides 5. Sulfonylureas 6. TZDs 7. α-glucosidase inhibitors 8. Meglitinides |
NA | • No signs of acute pancreatitis while using incretin-based as compared to other oral antidiabetic drugs. | [34] |
| NCT01468181 | 394 | Completed | 1. LY2189265 (Dulaglutide) 2. Sulfonylureas 3. Biguanides 4. α-glucosidase inhibitor 5. TZD 6. Glinides |
Phase 3 | • Percentage of participants with TEAE and hypoglycemic episodes from baseline to 52-weeks. • Changes from baseline in HbA(1c), FBG, SMBG, body weight, and HOMA2. |
[35] |
| NCT02027103 | 102 | Completed | 1.Metformin 2. Pioglitazone |
NA | • Both medications were equally effective in reducing FBG, HbA(1c), fetuin-A and osteoprotegerin levels in both diabetic women and men. | [36] |
| NCT02887625 | 410 | NA | 1. Pioglitazone + exenatide 2. Insulin glargine 3. Insulin Aspart |
NA | • A great decrease in HbA(1c) (6.1 ± 0.1% or 43 ± 0.7 mmol/mol) by combination therapy as compared to insulin therapy (7.1 ± 0.1% or 54 ± 0.8 mmol/mol). • More weight gain and a higher rate of hypoglycemia in insulin therapy than in the combination therapy. |
[38] |
| NCT00373178 | 100 | Completed | 1. Rosiglitazone 2. Metformin 3. Antidiabetic medications |
Phase 4 | • Similar improvement in glycemic profile and apelin levels, whereas lipid parameters, fat mass, and visfatin remained almost unaffected by both rosiglitazone and metformin. • Significant improvement in plasma ghrelin level and reduction in HOMA-IR, hs-CRP and systolic blood pressure from baseline values in the rosiglitazone group than in the metformin group. • Improvement in cardiovascular risk profile. |
[39] |
| NCT01777282 | 374 | Completed | 1. Albiglutide + Sulfonylurea 2. Albiglutide + Biguanide 3. Albiglutide + Glinide 4. Albiglutide + TZD 5. Albiglutide + α-glucosidase inhibitor |
Phase 3 | • Common adverse effects were nasopharyngitis (32.6%), constipation (7.2%), and diabetic retinopathy (5.3%). • Hypoglycemia occurred in 6.4% of patients in the first and third groups. • More reduction from baseline in HbA(1c) was observed when albiglutide added to TZD than in the other groups, whereas, reductions in FBG levels were observed in all groups. • The slight increase from baseline in body weight was observed with the addition of albiglutide to TZD. |
[40] |
| NCT00225225 | 45 | Terminated | 1. Rosiglitazone 2. Rosiglitazone + dietary recommendation for weight maintenance |
NA | • Change in weight from 270 +/− 54 lbs to 244 +/− 61 lbs was observed with a low-calorie diet and behavioral modification in patients treated with TZDs and is associated with glycemic and blood pressure control. | [41] |
| NCT00482183 | 38 | Completed | 1. Pioglitazone 2. Sirolimus-eluting stent |
Phase 3 | • No significant differences in glycemic control levels, lipid levels, and restenosis. • The HOMA-IR was significantly lowered and the incidence of major adverse cardiac events tended to be lower in the pioglitazone than in the sirolimus group after 1-yr therapy. |
[42] |
| NCT02285205 | 38 | Completed | Lobeglitazone | Phase 4 | • Significant decrease in controlled attenuation parameter values (313.4 dB/m at baseline vs. 297.8 dB/m) at 24-weeks. • Improvements in HbA(1c) values (6.56%), as well as the lipid and liver profiles and reduction in intrahepatic fat content, was observed in the treated patients. |
[43] |
| NCT00123643 | 36 | Completed | 1. Rosiglitazone 2. Glyburide |
Phase 4 | • Changes from baseline on flow-mediated dilation as a measure of endothelial function after 6-months of treatment. | [44] |
| NCT02365233 | 5 | Terminated | 1. DPP4inhibitor 2. Pioglitazone 3. Lantus insulin |
Phase 4 | • Change in hepatic lipid content from baseline to 6-month follow up. | [45] |
| NCT00575471 | 250 | Completed | 1. Rivoglitazone HCl (0.5, 1 and 1.5 mg) 2. Placebo |
Phase 2 | • Change in HbA(1c) and FPG from baseline for rivoglitazone as compared to placebo at 12-weeks. | [46] |
| NCT02456428 | 1,499,650 | Completed | 1. DPP4 inhibitor 2. GLP-1 analogs 3. Insulins 4. Biguanides 5. Sulfonylureas 6. TZDs 7. α-glucosidase inhibitors 8. Meglitinides |
NA | • The rate of hospitalization for heart failure did not increase with the use of incretin-based drugs as compared with oral antidiabetic-drug combinations among patients with heart failure. | [47] |
| NCT00819325 | 50 | Completed | 1. Pioglitazone + Oral hypoglycemic agents (sulfonylurea or metformin) 2. Oral hypoglycemic agents |
Phase 4 | • Change in 3D-neointimal plaque volume at 6-months compared to baseline. • Change in the 2D-neointimal area within the stent at 6-months compared to baseline. |
[49] |
| NCT00994682 | 176 | Completed | 1. Pioglitazone study drug 2. Placebo 3. Pioglitazone open label |
Phase 4 | • Pioglitazone treatment caused a significant improvement in individual fibrosis score (−0.5); reduced hepatic triglyceride content (7%) and improved adipose tissue, hepatic, and muscle insulin sensitivity. • The resolution of NASH was observed a greater number of patients treated with active drug treatment. • The rate of adverse events was similar between the groups, although weight gain was more in the pioglitazone group. |
[50] |
| NCT02730377 | 1994 | Active, not recruiting | 1. Liraglutide add on to metformin 2. Oral antidiabetics (α-glucosidase inhibitors+ DPP4 inhibitor + Meglitinides + SGLT2 inhibitor + Sulphonylurea + TZDs) + metformin |
Phase 4 | • A number of subjects who achieve HbA(1c) below or equal to 6.5% (48 mmol/mol). • A number of subjects who achieve HbA(1c) below or equal to 7.0% (53 mmol/mol) without weight gain. • Changes from baseline in FPG and body weight gain. |
[51] |
| NCT00006305 | 2368 | Completed | 1, 2. Revascularization with intensive medical therapy (1. Insulin, sulfonylurea; 2. Biguanides, TZDs) along with ACEIs, ARBs, beta-blockers and CCBs) 3, 4. Intensive medical therapy with delayed revascularization (3. Insulin, sulfonylurea, and 4. Biguanides, TZDs) along with ACEIs, ARBs, beta-blockers and CCBs. |
Phase 3 | • The baseline health status was improved significantly at 1-year in the treatment group. • Compared with medical therapy, revascularization was associated with significant improvement in the Duke Activity Status Index and was maintained over a 4-year follow-up. • Duke Activity Status Index was significantly larger in the patients intended for coronary artery bypass surgery than in the patients intended for percutaneous coronary intervention. |
[57] |
| NCT00575874 | 150 | Completed | 1, 2 and 3. Rivoglitazone HCl (0.5, 1.0, and 1.5 mg, respectively) 4. Pioglitazone HCl 5. Placebo |
Phase 2 | • Change from baseline in HbA(1c) for rivoglitazone HCl vs. placebo. • Change from baseline in FPG for rivoglitazone HCl vs. placebo. • Change from baseline in HbA(1c) for pioglitazone HCl. |
[53] |
| NCT00549874 | 27 | Completed | 1. Rosiglitazone 2. Glyburide |
NA | • Rosiglitazone significantly reduced plasma nitrotyrosine, hs-CRP, and von Willebrand antigen and significantly increased plasma adiponectin but no significant changes in these parameters were observed with glyburide. • Significant deterioration in both resting and stress myocardial blood flow in the glyburide group but not in the rosiglitazone group. |
[54] |
| NCT02231021 | 216 | Completed | 1. Alogliptin 2. Pioglitazone 3. Alogliptin + pioglitazone |
Phase 4 | • Change from baseline in HbA(1c), glycated albumin, GA/HbA(1c) ratio, FPG, HOMA-IR, PAI, hs-CRP, BNP, TC, and TGs. • Incidence of hyperglycemia rescue. • Proportion of subjects achieving HbA(1c) < 7.0 and 6.5%. • A number of hypoglycemic event rates. • A number of subjects with adverse events of special interest. |
[55] |
| NCT01001611 | 173 | Completed | 1. CKD-501 (Lobeglitazone) (0.5 mg) 2. Placebo |
Phase 3 | • HbA(1c) < 7% was achieved significantly more in the lobeglitazone group. • Lobeglitazone treatment significantly improved markers of insulin resistance, TGs, HDL cholesterol, small dense LDL cholesterol, FFA, and apolipoprotein B/CIII levels. • More weight gain was in the lobeglitazone group than in the placebo. |
[56] |
ACEI: angiotensin-converting-enzyme inhibitor; ARB: angiotensin receptor blocker; AUC: area under curve; BMI: body mass index; BNP: brain natriuretic peptide; CCBs: calcium channel blocker; CPP: cerebral perfusion pressure; DTSQ: diabetes treatment satisfaction questionnaire; DPP: dipeptidyl peptidase; eGFR: estimated glomerular filtration rate; FBG: fasting blood glucose; FBS: fasting blood sugar; FPG: fasting plasma glucose; FRK: fractalkine; FFA: free fatty acid; GLP-1: glucagon-like peptide 1; GA: glycated albumin; HbA(1c): glycated hemoglobin; HDL: high-density lipoproteins; hs-CRP: high sensitivity C-reactive protein; HOMA: homeostatic model assessment; IR: insulin resistance; IL: interleukin; LDL: low-density lipoproteins; MRE: magnetic resonance elastography; MRF: magnetic resonance fingerprinting; MRI-PDFF: magnetic resonance imaging proton density fat fraction; MCP-1: monocyte chemoattractant protein-1; MI: myocardial infarction; NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; NA: not applicable; PAI: plasminogen activator inhibitor; SMBG: self-monitoring of blood glucose; SNPs: single nucleotide polymorphisms; SGLT-2: sodium-glucose cotransporter-2; TC: total cholesterol; TACE: trans arterial chemoembolization; TEAE: treatment-emergent adverse events; TGs: triglycerides.
Structure and biological functions of PPAR-γ in diabetes
Peroxisome proliferator-activated receptors (PPARs) are the transducer proteins belonging to the superfamily of steroid/thyroid/retinoid receptors, which is involved in many processes when activated by a specific ligand. These receptors were recognized in the 1990s in rodents. PPARs help in regulating the expression of various genes that are essential for lipid and glucose metabolism [62], [63].
The structure of PPAR consists of four domains, namely A/B, C, D and E/F (Fig. 3A). The NH2-terminal A/B domain consists of ligand-independent activation function 1 (AF-1) liable for the phosphorylation of PPAR. The C domain is the DNA binding domain (DBD) having 2-zinc atoms responsible for the binding of PPAR to the peroxisome proliferator response element (PPRE) in the promoter region of target genes. The D site is responsible for the modular union of the DNA receptor and its corepressors. The E/F domain is the ligand-binding domain (LBD) consists of the AF-2 region used to heterodimerize with retinoid X receptor (RXR), thereby regulating the gene expression [64], [65]. There are three major isoforms of PPAR: PPAR-α, PPAR-δ/β, and PPAR-γ. Their distribution in tissues, biological functions, and their agonists are shown in Table 2 [62], [63], [64], [65].
Fig. 3A.

General structure of PPAR (modified and).
adapted from [64].
Table 2.
Isoforms of PPAR.
| Isoforms | Location | Biological Functions | Agonists |
|---|---|---|---|
| PPAR-α | Hepatocytes, cardiomyocytes, kidney cortex, skeletal muscles, and enterocytes | Fatty acid oxidation, mainly in the liver and heart and to a lesser extent in muscles. Reduces inflammation both in the vascular wall and the liver. Regulates energy homeostasis. |
Unsaturated fatty acids, 8-(S) hydroxyl eicosatetraenoic acid, fibrates (clofibrate, fenofibrate, and bezafibrate), B4 leucotriene, prostaglandin E, or farnesol |
| PPAR-δ/β | In almost all the tissues, mainly higher levels in the brain, adipose tissue, and skin | Regulator of fat oxidation, lipoprotein metabolism, glucose homeostasis. Regulates the genes involved in adipogenesis, cholesterol metabolism, inflammation, and atherosclerosis. |
Fatty acids |
| PPAR-γ | White and brown adipose tissue (major) Immune cells (monocytes, macrophages, and Peyer’s patches in the digestive tract), mucosa of the colon and cecum and in the placenta (lesser extent). | Insulin sensitization, adipogenesis, and adipocyte differentiation, inflammation, and cell growth | TZDs, unsaturated fatty acids such as oleate, linoleate, eicosapentaenoic, and arachidonic acids, and prostanoid. |
Effects of TZDs on PPAR-γ molecular pathways involved in diabetes
The efficacy of PPAR-γ agonists in the management of insulin resistance and T2DM has been confirmed by a number of important experimental assays with TZDs [62]. TZDs act as the selective agonists of PPAR-γ. PPARs regulate the gene transcription by two mechanisms: transactivation (DNA dependent) and transrepression (DNA independent) [65]. In transactivation, when TZDs bind to PPAR-γ, it gets activated and binds to 9-cis RXR, thereby forming a heterodimer [66]. This causes the binding of PPAR-γ-RXR complex to PPRE in target genes, which further regulates the genetic transcription and translation of various proteins that are indulged in cellular differentiation and glucose and lipid metabolism [67]. In transrepression, PPARs negatively interact with other signal-transduction pathways, such as nuclear factor kappa beta (NFκB) pathway that controls many genes involved in inflammation, thereby regulating various inflammatory mediators such as cytokines, leukocyte, etc. (Fig. 3B) [66], [68].
Fig. 3B.

Mechanistic action of TZDs (modified and).
adapted from [68].
In adipose tissues, when PPAR-γ gets activated by TZDs, it causes lipid uptake and triglycerides (TGs) storage. Free fatty acids (FFAs) are further taken up by white adipose tissues (WAT) and sequestered away from tissues (liver, skeletal muscle) where their growth leads to obstruction of insulin signaling called as lipid steal hypothesis. PPAR-γ also controls the adipocyte production from various signaling molecules like adipokines. PPAR-γ also gets directly activated by TZDs in macrophages which cause an anti-inflammatory M2 phenotype and thereby, decrease macrophage infiltration in WAT. TZDs also act on PPAR-γ in the parenchymal cells of steatosis liver or in Kupffer and stellate cells which cause a reduction in fibrosis and inflammation. TZDs also play a role in atherosclerosis by interfering with PPAR-γ action in macrophages [Fig. 4] [69].
Fig. 4.

Various targets of TZDs on PAAR-γ (modified and).
adapted from [69].
Chemistry and pharmacological profile of TZD derivatives
Alkoxy benzyl TZDs derivatives
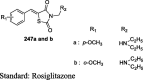
5-(4-Pyridylalkoxybenzylidene)-2,4-TZDs (8) analogs of pioglitazone were synthesized by Momose et al. through Knoevenagel condensation of aldehydes (7) with the corresponding thiazolidine-2,4-diones as shown in S2. The aldehydes (7) were synthesized from the coupling of pyridylethanols (4) with 4-fluorobenzonitrile to give 4-(2-(2-Pyridyl)ethoxy)benzonitriles (5) followed by either treatment with Raney Ni in HCO2H or with tosylchloride and 4-hydroxybenzaldehyde (6) in presence of phase transfer catalyst to give aldehydes (7). All the analogs were then evaluated for hypoglycemic and hypolipidemic activity in KKAy mice by administering as dietary admixture at a concentration of 0.005% or 0.01% for 4 days. The compound 8a-d reduced blood glucose level (38–48%) and plasma TG level (24–58%) and the effect was found to be equipotent to pioglitazone (Table 4) [70].
Table 4.
Summary of in vivo studies of TZDs on diabetes mellitus.
| Compound | Cell line | Dose | Effect | References |
|---|---|---|---|---|
| Alkoxy benzyl based TZDs | ||||
 |
KKAy mice | 0.005% or 0.01% as dietary admixture (4 days) |
Reduction in BG (38–48%) and TG (24–58%) | [70] |
 |
KKAy mice | 0.005% or 0.01% as dietary admixture (4 days) |
Reduction of PG and TG 100 times more than pioglitazone | [71] |
 |
KKAymice | 100 mg/kg (4 days) |
Reduction in BG (T/C = 0.39) | [72] |
 |
db/db mice ob/ob mice |
200 mg/kg 10 and 100 mg/kg (9 days) 1,3 and 10 mg/kg (14 days) |
Reduction in BG (74%) and TG (77%) Equipotent activity in reducing PG Reduction in PG (51–59%) but no reduction in TG |
[73] |
 |
db/db mice ob/ob mice |
100 mg/kg (6 days) 1, 3, 10 and 30 mg/kg (14–15 days) 3, 10, 30 and 100 mg/kg (15 days) |
Reduction in PG (57%) and TG (77.7%) Impressive improvement in glucose tolerance even at 10 mg/kg Dose-dependent reduction in PG |
[74] |
 |
KK mice | 1 mg/kg (1 day) 50 mg/kg (2 weeks) |
Reduction in BG (55.8%) and cardiac hypertrophy |
[75] |
 |
db/db mice Wistar rats |
10 mg/kg (6 days) 100 mg/kg (14 days) |
Reduction of PG (72%) and TG (68%) No significant change in body weight and food consumption |
[76] |
 |
db/db mouse | 30 or 100 mg/kg (6 days) 0.3, 3 and 10 mg/kg (15 days) 100 mg/kg (28 days) |
Reduction in PG (73%) and TG (85%) Better than standard in terms of reduction in PG levels Neither mortality nor any evidence of toxicity |
[77] |
 |
STZ-diabetic rats | 0.1 mmol/kg (3 days) |
Reduction in BG (47%) | [78] |
 |
db/db mouse | 5 and 10 mg/kg (11 days) |
Dose-dependent reduction in glucose (86%) and TG (78%) | [80] |
 |
KKAy mice | 1, 6 and 30 mg/kg (5 days) |
Reduction in BG and TG (ED25 = 0.020 and 2.5 mg/kg/day) | [81] |
 |
Wistar male rats | 3, 10, 30 and 100 mg/kg (14–15 days) |
Dose-dependent reduction in PG and TG | [82] |
 |
KKAy mice | 1,6 and 30 mg/kg (5 days) |
Drastically improved the hypoglycemic activity | [83] |
 |
Alloxan-induced diabetic male Wistar rats | 3,10,30 and 100 mg/kg (15 days) |
Reduction of PG and TG | [85] |
 |
STZ-induced diabetic Wistar rats | 35 μmol/ kg (15 days) |
Reduction in PG (44.7%) | [87] |
 |
Alloxan-induced diabetic rat model | 3 mg/kg (16 days) |
Reduction in BG (295.50 mg/dL), enhancement in HDL level (3.16 mg/dL) and HDL/LDL ratio (4.02) | [10] |
 |
Sucrose loaded rat model | 100 mg/kg (2 days) |
9.4% improvement in oral glucose tolerance | [88] |
| Pyrazole-based TZDs | ||||
 |
STZ-induced diabetic rat model Hepatotoxicity study |
36 mg/kg (15 days) 108 mg/kg |
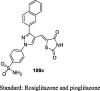
Reduction in BG (134.1 mg/dL) No bodyweight change Lower the levels of AST, ALT, and ALP and cause no damage to the liver |
[89] |
 |
STZ-induced diabetic rat model Hepatotoxicity study |
36 mg/kg (15 days) 108 mg/kg |
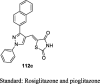
Reduction in BG (140.1 mg/dL) No bodyweight change Lower the levels of AST, ALT, and ALP and cause no damage to the liver |
[91] |
 |
C57BL/6J mice | 30 mg/kg (15 days) |
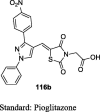
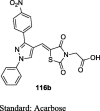
Compound 116b (134.46 mg/dL) exhibited significant blood glucose-lowering activity and were found to be similar to standard pioglitazone (136.56 mg/dL) | [92] |
| N-substituted TZDs | ||||
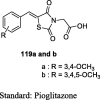 |
Sucrose loaded model | 100 mg/kg (1 day) |
Reduction in BG within 30 min and the effect was maintained till the duration of 120 min | [93] |
| Sulfonyl based TZDs | ||||
 |
db/db mice ob/ob mice Zucker rats |
100 and 20 mg/kg (4 days) 100 mg/kg (4 days) 20 mg/kg (4 days) |
Reduction in PG (52 and 21%) Reduction in glucose (40%) and insulin (65%) Significantly improved the glucose tolerance |
[96] |
 |
Alloxan-induced diabetic albino rats | 36 mg/kg (1 day) |
Moderate reduction in BG (95–180 mg/dL) | [4] |
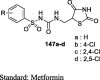 |
Sucrose loaded rat model | 100 mg/kg (1 day) |
Significantly inhibited the postprandial rise in BG (14.3–17.2%) |
[97] |
| Naphthyl based TZDs | ||||
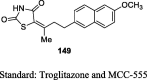 |
db/db mice | 30 mg/kg (6 days) |
Reduction in PG (16%) and TG (50%) | [98] |
| Phenothiazine based TZDs | ||||
 |
STZ-induced diabetic Wistar rats | 5 and 10 mg/kg (21 days) |
Stimulates insulin secretion by inhibiting K+ATP channels | [99] |
| Amide-based TZDs | ||||
 |
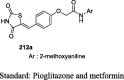
fa/fa Zucker rats | 100 mg/kg (4 weeks) |
No effect on BG and body weight but a marked reduction in serum insulin (78%) and TG (83%) | [100] |
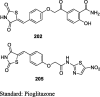 |
STZ-induced diabetic rat model | 20 mg/kg (14 days) |
Reduction in BG (64% and 56%) and TG (74% and 78%) | [103] |
 |
Albino Rats STZ- NA induced diabetic rat model |
100 and 500 mg/kg (14 days) 10 and 100 mg/kg (14 days) |
Well tolerated up to higher dose and cause no mortality Reduction in PG (271 and 304 mg/dL) and TG (97 and 94 mg/dL) |
[104] |
 |
DMS-induced Wistar albino mice | 30 mg/kg (7 days) |
Reduction in PG (113.7 mg/dL) | [105] |
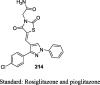 |
STZ-induced diabetic rat model Hepatotoxicity study |
36 mg/kg (15 days) 108 mg/kg |
Reduction in BG (142.4 mg/dL) No body weight change Lower the levels of AST, ALT, and ALP and cause no damage to the liver |
[106] |
| Imidazo-thiadiazole based TZDs | ||||
 |
Alloxan-induced diabetic Wistar rats | 3, 10, 30 and 100 mg/kg (15 days) |
Dose dependent reduction in PG (40, 44, 59 and 73%) and TG (36, 35, 38 and 44%) | [107] |
| Dispiropyrrolidines based TZDs | ||||
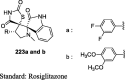 |
Alloxan-induced diabetic Wistar rats | 36 mg/kg (3 days) |
Reduction in BG (115.3 and 115.8 mg/dl) | [108] |
| Acid-based TZDs | ||||
 |
ob/ob mice | 10 mg/kg (6 days) 3.1, 6.3 and 12.5 mg/kg (19 days) 5 mg/kg (11 days) |
Reduction in BG (29%) All dose levels were effective in reducing BG No difference in body weight |
[109] |
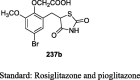 |
STZ-induced diabetic rat model Hepatotoxicity study |
36 mg/kg (15 days) 108 mg/kg |
Reduction in BG (158.8 mg/dL) No bodyweight change Lower the levels of AST, ALT, and ALP and cause no damage to the liver |
[110] |
 |
STZ-NA induced diabetic rats | 30 mg/kg (14 days) |
Reduction in PG (142 and144.4 mg/dL) | [111] |
| Benzylidene based TZDs | ||||
 |
DMS-induced diabetic rat | 50 mg/kg (5 days) |
Reduction in BG level (58 and 65%) | [112] |
 |
STZ-NA induced diabetic rat model | 10, 30 and 50 mg/kg (1 day) |
Dose-dependent reduction in BG was observed (39.83%, 44.62%, and 52.81%) | [113] |
 |
Wistar rats Alloxan-induced diabetic rats |
30 and 100 mg/kg (2 days) 100 mg/kg (14 days) |
No change in weight without toxic effects Reduction in BG (65%) |
[114] |
 |
Albino Wistar rats (oral glucose tolerance test) |
30 mg/kg (1 day) |
Exhibited potent antidiabetic activity (100–120 mg/dL) similar to pioglitazone (100 mg/dL) | [115] |
 |
Albino Wistar rats | 175, 350, 700, 1400 and 2000 mg/kg (14 days) |
Normal behavior and no physical changes were seen Fat deposits at a dose ≥ 350 mg/kg |
[116] |
 |
DMS-induced diabetic mice | 0.72 mg/kg (10 days) |
An unexpected decrease in BG level within 30 min and then decreased steadily | [117] |
| Benzo-fused TZDs | ||||
 |
db/db mice | 100 mg/kg (6 days) |
Reduction in PG (66%) and TG (52%) | [118] |
 |
Alloxan-induced diabetic albino rats | 36 mg/kg (1 day) |
Reduction in BG (116–123 mg/dL) | [120] |
 |
Alloxan-induced diabetic mice | 30 mg/kg (1 day) |
Reduction in serum glucose level (−30.62%) |
[122] |
| Chromones based TZDs | ||||
 |
STZ-induced diabetic rats | 15 days Hepatotoxicity study |
Reduction in BG (135.5%), no change in body weight Lower levels of AST, ALT, and ALP without causing any hepatotoxicity |
[124] |
| Miscellaneous | ||||
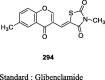
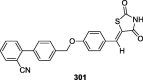
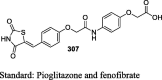
 |
STZ-NA induced diabetic rats | 50 mg/kg body weight | Decreased 32.36% glycemia level whereas glibenclamide reduced 43.6% levels | [125] |
 |
STZ-NA induced diabetic rats | 50 mg/kg body weight | Decreased 31% glycemia level whereas glibenclamide reduced 43.6% levels | [126] |
 |
STZ-NA induced diabetic rats | 50 mg/kg body weight | Decreased 34% glycemia level whereas glibenclamide reduced 43.6% levels | [127] |
K+ATP: adenosine triphosphate-sensitive potassium channel; ALT: alanine transaminase; ALP: alkaline phosphatase; AST: aspartate transaminase; BG: blood glucose; DMS: dexamethasone; HDL: high-density lipoprotein; LDL: low-density lipoprotein; NA: nicotinamide; PG: plasma glucose; STZ: streptozotocin; TG: triglycerides; T/C: treated group over control group.
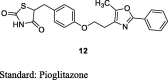
Sohda et al. prepared a series of 5-(4-(2- or 4-azolylalkoxy)benzyl-or- benzylidene)-2,4-TZDs by using S3 in which Meerwein arylation of aniline derivatives (9) give the 3-aryl-2-bromo-propionates (10), which were further reacted with thiourea (1) to give iminothiazolidinones (11) followed by acid hydrolysis of 11 give the resulted product (12). The synthesized compounds were evaluated for hypoglycemic and hypolipidemic activities in genetically obese and diabetic KKAy mice. The compounds were administered along with food as a dietary admixture at 0.005 or 0.001%. Among the compounds synthesized, 5-(4-(2-(5-methyl-2-phenyl-4-oxazolyl)ethoxy)-benzyl)-2,4-TZD (12) exhibited the most potent activity (>100 times) than that of pioglitazone (Table 4) [71].
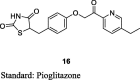
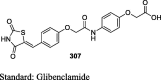
Tanis et al. have reported the synthesis of pioglitazone metabolites (15 and 16) by oxidizing pioglitazone (13) using m-chloroperoxybenzoic acid (mCPBA) to give N-oxide (14), which was then converted to alcoholic derivative (15) of pioglitazone using trifluoroacetic anhydride (TFAA) in methylene chloride which in turn upon oxidation gives putative metabolite (16) as shown in S4. The antihyperglycemic activity of these metabolites was determined in the KKAy mice in comparison to pioglitazone. The compounds were administered as a food admixture at a dose of 100 mg/kg for 4 days. The antihyperglycemic activity was determined from the ratio of glucose level for the treated over the control group (T/C). As a result, compound 16 has proven to be the most potent of these metabolites with a T/C value of 0.39 in comparison to pioglitazone (T/C = 0.49). Further, the compounds were evaluated for their ability to augment insulin-stimulated lipogenesis in vitro in 3T3-L1 cells. Again, compound 16 was proven to be effective in augmenting insulin-stimulated lipogenesis through its ability to provide high levels of [14C] acetate incorporation into lipids at different concentrations (1, 3 and 10 μM), while others were roughly equivalent to pioglitazone. These results implicate that compound 16 is considered as a congener of pioglitazone with greater potency elicited through the simpler metabolic pathway (Table 3, Table 4) [72].
Table 3.
Summary of in vitro studies of TZDs on diabetes mellitus.
| Compound | Cell line | Dose | IC50/EC50 | Effect | References |
|---|---|---|---|---|---|
 |
3T3-L1 cells | 0–10 µM | Stimulated insulin-mediated lipogenesis (via 14C-acetate incorporation). Comparable activity at 10 µM to that of pioglitazone. |
[72] | |
 |
3T3-L1 cells | 0.1% v/v | Increased adipocyte differentiation which is expressed as concentrations equivalent to the [1-14C] uptake counts (0.080 µM). | [75] | |
 |
HEK 293T cells | 0.25, 0.5, 1.0, 5.0 µM | Increased PPAR-γ transactivation in a dose-dependent manner (11 folds) in comparison to troglitazone (5.5 folds) and pioglitazone (6 folds). | [76] | |
 |
HEK 293T cells | 0.010, 0.050, 0.2, 1.0 and 5.0 µM | Increased PPAR-γ transactivation in a dose dependent manner (20 folds) in comparison to rosiglitazone (19 folds) and pioglitazone (6 folds) | [77] | |
 |
COS-1 cells | – | EC50 = 0.12 µM IC50 = 0.25 µM |
Increased PPAR-γ activation (10-fold) than standard | [80] |
 |
3T3-L1 cells | 3 × 10−5 – 3 × 10−11 M | EC50 = 0.00054 μM | Better TG accumulation activity was observed in comparison to rosiglitazone (0.047 μM) and pioglitazone (0.015 μM) | [81] |
 |
3T3-L1 cells | 3 × 10−5 – 3 × 10−11 M |
EC50 = 0.0012 μM (63a) and 0.00041 μM (63b) | Better TGs accumulation activity was observed in comparison to rosiglitazone (0.047 μM) and pioglitazone (0.015 μM) | [83] |
 |
1) CV1-cells 2) Murine macrophage cell line |
2 µM 100 μL |
Marginal PPAR-γ transactivation (21.2%) with no PPAR-α activity Inhibits NO production (51.5%) |
[84] | |
 |
1) Rat hemi-diaphragm 2) HEp-2 and A549cells |
2 mg 100 µL |
CTC50 is 80 µg/mL against HEp2 cells and no activity against A549 cells | Enhanced glucose uptake activity especially in the presence of insulin (38.0 mg/dL/45 min) Showed significant cytotoxic activity |
[86] |
 |
1) HEK 293 cells 2) 3T3-L1 cells |
10 μM 10 μM |
Increased PPAR-γ transactivation (61.2%) as compared to standard Increased expression of PPAR-γ significantly due to AMPK activation (1.9 folds) |
[89] | |
 |
1) HEK 293 cells 2) 3T3-L1 cells |
10 μM 10 μM |
Increased PPAR-γ transactivation (52.06%) as compared to standard Increased expression of PPAR-γ significantly due to AMPK activation (2.35-fold) |
[91] | |
 |
Alpha-amylase | 10 mg | 4.08 μg/mL | Better alpha-amylase inhibitory activity than the standard acarbose (8 μg/mL) | [92] |
 |
INS-1 cells | 1 and 10 μg/mL | Increased insulin release at higher concentration | [94] | |
 |
1) INS-1 cells 2) Aldose reducatse enzyme |
1 and 10 μg/mL 0.1 mL |
0.415 μg/mL against Aldose reductase | More insulintropic effect (128.6%) at higher concentration (10 μg/mL) Showed the highest aldose reductase inhibitory activity (86.57%) |
[95] |
 |
3T3-L1 cells | 0.1, 1.0 and 10 μM | Caused differentiation of 3T3-L1 preadipocyte fibroblasts into myoblast during terminal differentiation and increased lipid accumulation | [100] | |
 |
Rat hemi-diaphragm | 2 mg | Enhance the glucose uptake (36.25 mg/g/45 min) | [101] | |
 |
Rat hemi-diaphragm | 1 and 2 mg | Significant glucose uptake activity especially in the presence of insulin (42.16 mg/dL) | [102] | |
 |
1) HEK 293 cells 2) 3T3-L1 cells |
10 μM 10 μM |
Increased PPAR-γ transactivation (53.67%) as compared to standard Increased expression of PPAR-γ significantly due to AMPK activation (2.1 folds) |
[106] | |
 |
NIH3T3 cells | Different concentrations | EC50 = 280 nM | Significant PPAR-γ agonistic activity with 64% activation | [107] |
 |
HEK 293 cells | Between 0.1 and 30 | EC50 = 0.284 μM | Moderate PPAR-γ agonist activity | [109] |
 |
HEK 293 cells 3T3-L1 cells |
10 μM 10 μM |
EC50 = 0.77 μM | Increased PPAR-γ transactivation (48.35, 54.21%) but found to be PPAR-α and PPAR-δ inactive Increased expression of PPAR-γ significantly due to AMPK activation (2.0 folds) |
[110] |
 |
Yeast cells | 10, 20, 40, 80, 100 and 200 μL/mL | Increased glucose uptake by the cells (39.23 and 38.19%) | [111] | |
 |
CV-1 cells | – | Significant PPAR-γ activity (113.2%) without any PPARα activity. | [119] | |
 |
1) CV-1 cells 2) RAW 264.7 cells |
2 μM 5, 10 and 20 μM |
Significant PPAR-γ activity (120%) without any PPARα activity Inhibitory activity against NO production |
[121] | |
 |
INS-1 cells | 0.001 and 0.01 mg/mL | Increase the insulin release at lower concentration (120%) but more potent at higher concentration (152%) | [123] | |
 |
1) hERG 2) 3T3-L1 cells |
– 10 μM |
No cardiotoxic effect (135 μM) Increased PPAR-γ gene expression due to the activation of AMPK (45%) |
[124] | |
 |
3T3-L1 cells | – | 0.58 µM (hERG) | Significantly increased the levels of PPAR-γ, PPAR-α and GLUT4 | [125] |
 |
3T3-L1 cells | 10 µM | 0.01 µM (hERG) | Increased the relative expression of PPAR-γ and GLUT-4 (2-folds) but no change was observed in the expression of PPAR-α | [126] |
 |
PTP1B | 20 µM | 3.7 µM | The decrease in enzyme activity up to 85% | [127] |
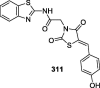
AMPK: adenosine monophosphate-activated protein kinase; EC: effective concentration; GLUT4: glucose transporter type 4; HEK cells: human embryonic kidney cells; HEp-2: human epithelial type 2 cells; INS-1 cells: insulin-secreting cell; NO: nitric oxide; PPAR: peroxisome proliferator-activated receptors; PTP1B: protein-tyrosine phosphatase 1B; TGs: triglycerides.
Lohray et al. have reported the synthesis of a series of [[(heterocyclyl)ethoxy]-benzyl]-2,4-TZDs (19) by the Knoevenagel condensation of aldehyde (17) and 2,4-TZD (3) in the presence of piperidinium benzoate to give benzylidenes (18) followed by catalytic reduction over Pd-C as shown in S5. Synthesized compounds were evaluated for antihyperglycemic and hypolipidemic activity and the effects were compared with troglitazone and rosiglitazone (BRL-49653) in db/db and ob/ob mice. The compound DRF-2189 (18) at 200 mg/kg have been shown to exhibit superior activity in terms of blood glucose (74%) and TG (77%) reduction than those in troglitazone (200 mg/kg) treated (24 and 50%, respectively) mice. Then, the efficacy of compound DRF-2189 (18) was compared with rosiglitazone in db/db mice. Compound DRF-2189 (18) at 10 and 100 mg/kg have shown to reduce plasma glucose whereas, rosiglitazone failed to show the activity at 10 mg/kg dose. Further, dose–response effects of DRF-2189 (18) (1, 3, 10 mg/kg) were carried out along with rosiglitazone (1, 3, 10 mg/kg) and troglitazone (100, 200 and 800 mg/kg). Both DRF-2189 (18) and rosiglitazone were shown to exhibit equipotent activity in reducing plasma glucose but troglitazone failed to show the activity even at a higher dose. In addition, compound DRF-2189 (18) and rosiglitazone failed to show the activity on the reduction of TG; however, compound DRF-2189 (18) at 3 and 10 mg/kg has been shown to reduce total cholesterol. In addition, both DRF-2189 and rosiglitazone have been shown to exhibit equipotency in oral glucose tolerance test (OGTT) after 9-days of treatment in db/db mice. Consequently, both the drugs were evaluated in ob/ob mice at 10 mg/kg for 14 days. The reduction in blood glucose level (51–59%) and TG levels (53–55%) were observed and the results were in accordance with db/db study. The indole analog DRF-2189 (18) was found to be a very potent insulin sensitizer, comparable to rosiglitazone in genetically induced diabetic models (i.e., ob/ob and db/db mice) (Table 4) [73].
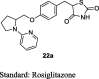
Lohray et al. synthesized a series of substituted pyridyl and quinolinyl containing 2,4-TZDs incorporated with an interesting cyclic amine as shown in S6. The aldehyde (20) underwent Knoevenagel condensation with TZD (3) to afford benzylidene derivatives (21) followed by reduction yielded final derivatives (22a and b). The synthesized compounds were evaluated for euglycemic and hypolipidemic effects in db/db mice by administering the synthesized derivatives at a dose of 100 mg/kg for 6 days. The compounds synthesized were then compared with unsaturated rosiglitazone. As a result, compound 22a showed very good euglycemic and hypolipidemic activities measured in terms of percentage reduction in plasma glucose (57%) and TG (77.75%) level in comparison to unsaturated rosiglitazone (55% and 35%, respectively). On the other hand, quinoline based compound (22b) also had significantly shown to reduce plasma glucose than rosiglitazone, but failed to produce a significant result on plasma TG. Further, their saturated derivatives were prepared and evaluated in the same diabetic model at a dose of 30 mg/kg for 6 days in comparison to saturated rosiglitazone (BRL-49653). The results showed that the euglycemic and hypolipidemic activity were maintained for a saturated analog of compound 22a (52% plasma glucose reduction) similar to unsaturated analog. Surprisingly, quinoline based saturated analogs of TZD (22b) had shown to exhibit good hypolipemic activity in addition to euglycemic activity. Then, they prepared various salt (maleate, hydrochloride or sodium salt) forms of TZD and evaluated at 30 mg/kg for 6 days in the same animal model. It was found that HCl and maleate salt form of compound 22a exhibited euglycemic (70% and 63.6%, respectively) and hypolipidemic (31% and 66.4%, respectively) activities. Further, dose-dependent studies were carried out in db/db and ob/ob mice at different doses of 3, 10, 30 mg/kg and 1, 3, 10 mg/kg, respectively for 14 days. The results in db/db mice revealed that maleate salts of compound 22a (10 and 30 mg/kg) were equipotent to rosiglitazone in terms of euglycemic activity but superior to rosiglitazone in terms of hypolipidemic activity at higher doses. The maleate salt of compound 22a also exhibited excellent plasma glucose and TG lowering activities in ob/ob mice. The OGTT was also performed in both models (db/db and ob/ob). Maleate salt of unsaturated compound 22a had shown an impressive improvement in glucose tolerance (10 mg/kg). In order to understand the mechanism, PPAR-γ (0.1, 1.0 and 10 μM) and PPAR-α (50 μM) transactivation assay was performed at different concentrations. The maleate form of unsaturated compound 22a did not show any significant PPAR-γ or PPAR-α transactivation (Table 4) [74].
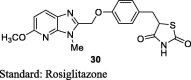
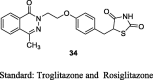
Oguchi et al. reported a series of imidazopyridine TZDs and synthesized them from their corresponding pyridines. 2,6-dichloro-3-nitropyridine (23) was substituted with methylamine to give 6-chloro-2-methylamino-3-nitropyrdine (24) and was then reacted with sodium alkoxide to give 25, which then reduced to give amino derivative (26). Imidazopyridine (27) was obtained through cyclization of 26 with glycolic acid followed by reaction with 28 gave compound 29 and the final product (30) was then obtained by removing trityl group (S7). The synthesized compounds were evaluated for its hypoglycemic activities, both in vitro and in vivo. The in vitro adipocyte differentiation activity of synthesized derivatives was carried out in the preadipocyte cell line (3T3-L1) at the concentration of 0.1% (v/v). The in vivo activity was carried out in KK mice for one day and one week by administering test compounds at a dose of 1 mg/kg and administering along with food as an admixture, respectively. Further, toxicity studies were also carried out for 2-weeks at a dose of 50 mg/kg. On the basis of evaluation, firstly they identified compounds as a potent hypoglycemic agent through percent reduction in blood glucose and adipocyte differentiation; however, these compounds caused cardiac hypertrophy after multiple oral administrations and also caused high concentration in blood (i.e., tendency to accumulate over the course of administration). Then, they tried to reduce the drug accumulation by introducing the functional groups that can be metabolized in vivo easily, as a resulting compound 30 (1 mg/kg) with methoxy substitution at 5-position of imidazopyridine ring (5-[4-(5-methoxy-3-methyl-3H-imidazo[4,5-b]pyridine-2-ylmethoxy)benzyl]-TZD) showed relatively high adipocyte differentiation but did not reduce blood glucose level due to poor oral bioavailability. However, compound 30 had shown to reduce blood glucose (55.8%) when it was administered orally as an admixture with food for 1 week. On the other hand, compound 30 has shown to exhibit poor dissolution rate, hence they improved the solubility of compound 30 by converting them into salt form (HCl and fumaric acid). As a result, HCl salt of compound 30 improved hypoglycemic effect with ED25 value of 0.02 mg/kg/day in comparison to that of rosiglitazone maleate (0.39 mg/kg/day). On the basis of above results, TZD-HCl salt of compound 30 was selected as the candidate for further studies (Table 3, Table 4) [75].
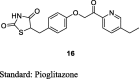
Madhavan et al. prepared a series of phthalazinones based TZD derivatives by treating phthalazinones (31) with 4-(2-bromoethoxy)benzaldehyde in the presence of K2CO3 in dimethylformamide (DMF) at 70 °C for 2–6 h to yield phthalazinones substituted aldehyde (32) which was further treated with TZD (3) in the presence of piperidine benzoate to furnish benzylidene TZD analogs (33) and was then reduced using 10% Pd/C catalyst to give 5-(4-[2-(4-methyl-1-oxo-1,2-dihydrophthalazin-2-yl)ethoxy)phenylmethyl)TZD (or 5-benzyl-TZDs) (34) as shown in S8. The synthesized compounds were screened for both in vivo (db/db mice) and in vitro (PPAR-γ transactivation in the human embryonic kidney (HEK) 293T cells) activity. From the synthesized series, compound 34 was the most potent PPAR-γ activator and also demonstrated to lower both glucose (72%) and TGs (68%) levels (Table 3, Table 4) [76].
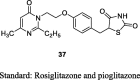
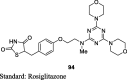
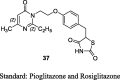
Madhavan et al. synthesized a series of pyrimidinone derivatives of TZDs by condensation of pyrimidinone substituted aldehydes (35) with TZDs to give unsaturated compounds (36), which was further reduced to give the final product (37) as shown in S9. Of note, compound 35 was synthesized through a coupling reaction between substituted pyrimidinone and 4-[2-bromoethoxy]benzaldehyde. The synthesized compounds were examined for plasma glucose and TGs lowering activity in db/db mouse and the effects were compared to both rosiglitazone and pioglitazone as a standard. In a preliminary study, compounds were evaluated for 6-days at a dose of 30 or 100 mg/kg in db/db mice. Among various compounds, 5-(4-(2-(2-ethyl-4-methyl-6-oxo-1,6-dihydro-1-pyrimidinyl)ethoxy)phenyl methyl)TZD (PMT13) (37) exhibited superior activity (73% reduction in plasma glucose and 85% reduction in TG level) than rosiglitazone (65% reduction in plasma glucose and 41% reduction in TG level). Other synthesized derivatives showed less effect even at a higher dose (100 mg/kg). Subsequently, dose-dependent study (0.3, 3 and 10 mg/kg) was carried out for 15 days in the same genetic models. As a result, PMT13 (37) showed a better reduction in hypoglycemic and hypolipidemic activity than rosiglitazone and pioglitazone. During subacute toxicity studies for 28 days, PMT13 (37) at 100 mg/kg produced neither mortality nor any evidence of toxicity measured through changes in the body weight or food consumption. The synthesized compounds were also evaluated for PPAR-γ transactivation study using HEK 293T cells. PMT13 at lower concentration (0.010 μM) showed better PPAR-γ activation than rosiglitazone but showed similar activation at higher concentration (5.0 μM). In contrast, PMT13 (37) was shown to exhibit better activation towards PPAR-γ than pioglitazone (Table 3, Table 4) [77].
Storr et al. synthesized a series of vanadium compounds (42) through chelation of ligand having TZD precursor (41) and vanadium sulphate as shown in S10. TZD precursor is prepared from compound (38), which was coupled with 4-hydroxybenzaldehyde (6) to afford the aldehyde (39). Next, Knoevenagel condensation was carried out using aldehyde (39) and TZD to get compound (40), on which deprotection was carried out in acidic condition to afford precursor (41). All the compounds were evaluated for insulin enhancing capability in streptozotocin (STZ)-induced diabetic rats through percent reduction in the blood glucose level and the effects were compared with rosiglitazone and BMOV. The compounds were administered at a dose of 0.1 mmol/kg. Complex 42 showed the most efficient hypoglycemic effects than the ligands without chelation. The hypoglycemic effects were superior and comparable to that of rosiglitazone and BMOV, respectively (Table 4) [78].
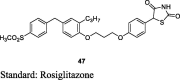
Koyama et al. synthesized a series of 5-aryl TZDs containing 4-phenoxyphenyl side chains (47) as shown in S11. Mandelates (43) were treated with 1,3-dibromopropane or 1,4-dibromobutane in DMF to give ether linkage derivative (44), which further underwent coupling reaction with 4-(4-(methylsulfonyl)phenoxy)-2-propylphenol (45) to give 46, then standard TZD protocol was applied to 46 to yield the final derivatives (47) and evaluated them for both in vivo (db/db mice) and in vitro (PPAR-γ) activities. Firstly, they evaluated functional activity (PPAR-γ) and pharmacokinetic profile of synthesized compounds. The PPAR-γ transactivation assay was performed as reported previously using COS-1 cells [79]. Then, compounds with good functional activity and pharmacokinetic profiles were evaluated in vivo. The synthesized compounds were administered as sodium salt at a dose of 5 and 10 mg/kg for 11 days. Among the synthesized compounds, compound 47 exhibited good oral bioavailability similar to rosiglitazone. Compound 47 showed a dose-dependent reduction in plasma glucose and TG in db/db mice despite its weak PPAR-γ activity than rosiglitazone. The reduced functional activity may be due to longer half-life (t1/2 = 2.8 h) than the reference compound (t1/2 = 1.2 h) (Table 3, Table 4) [80].
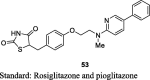
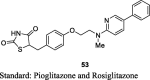
Kim et al. reported a series of substituted pyridines and purines having 2,4-TZDs moiety and synthesized them from their respective pyridines and purines in order to increase the hydrophobic properties as shown in S12. Firstly, 5-amino-2-chloropyridine (48) was converted into 5-substituted pyridines (49) using either simple aromatic hydrocarbon or substituted heteroaromatic compound in the presence of isoamyl nitrite and copper (I) oxide followed by amination with 2-methylaminoethanol yielded amino alcohols (50). Then, the compound 50 treated with 4-fluorobenzaldehyde in the presence of NaH to give aldehyde compounds (51), which underwent Knoevenagel condensation to give unsaturated TZD analogs (52) in turn reduced into desired compound (53) through using Pd(OH)2. The synthesized compounds were evaluated for their hypoglycemic and hypolipidemic activity (in vitro and in vivo). The in vitro effect was observed for the TGs accumulation in 3T3-L1 cells by keeping the concentration of test compounds in the range of 3 × 10−5 – 3 × 10−11 M. Among the various compounds, compound 53 (5-(4-{2-[N-methyl-(5-phenyl-pyridin-2yl)amino]ethoxy}benzyl)thi-azolidine-2,4-dione) increased insulin-induced TG accumulation in 3T3-L1 cells with EC50 value of 0.00054 μM in comparison to rosiglitazone and pioglitazone. Further, they evaluated the potent compounds in vivo based on the in vitro results. The in vivo activity was performed in KKAy mice for 5 days. The synthesized compounds were administered at a dose of 1, 6 and 30 mg/kg. The hypoglycemic and hypolipidemic activity were estimated through ED25 value. The compound 53 found to reduce 25% blood glucose and TG (ED25 = 0.020 and 2.5 mg/kg/day, respectively) in comparison to that of rosiglitazone and pioglitazone. These results suggested that TZD compound 53 showed to be more effective than the reference standards and was selected for further pharmacological studies (Table 3, Table 4) [81].
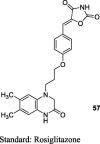
Gupta et al. reported a series of substituted 5-(4-(2-(6,7-dimethyl-3-oxo-3,4-dihydroquinoxalin-1(2H)-yl)ethoxy)benzylidene)TZDs (57) which were synthesized by the coupling of 6,7-dimethyl-1,2,3,4-tetrahydroquinoxalin-3-one (54) and 4-(2-bromoethoxy)benzaldehyde (55) in the presence of NaH in dry DMF to afford 4-(2-(6,7-dimethyl-3-oxo-3,4-dihydroquinoxalin-1(2H)-yl)ethoxy)benzaldehyde (56) which in turn underwent Knoevenagel condensation with TZD yielded 57 as shown in S13. These synthesized derivatives were screened for euglycemic and hypolipidemic activities in Wistar male rats and were administered at doses of 3, 10, 30 and 100 mg/kg for 14–15 days. OGTT was also carried out on the final day by loading glucose at a dose of 3 g/kg. Among these series, compound 57 showed a dose-dependent reduction in plasma glucose and TG in comparison to those in rosiglitazone (Table 4) [82].
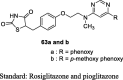
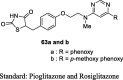
Lee et al. designed novel substituted pyrimidines containing TZD analogs by treating 4,6-dichloropyrimidine (58) and substituted aromatic alcohols in the presence of NaH to give 4-substituted pyrimidines (59), which underwent amination with 2-methylaminoethanol to afford substituted N-methylaminoethanol derivative (60). The compound 60 was further reacted with 4-fluorobenzaldehyde to yield benzaldehyde derivatives (61), which then underwent Knoevenagel condensation to give unsaturated TZD analogs (62) and were reduced further to give the product (63a and b) as depicted in S14. The compounds were tested for their glucose and lipid- lowering activity in KKAy mice. Test compounds were administered orally at a dose of 1, 6 and 30 mg/kg for 5 days. Among the synthesized analogs, compounds 63a and b have significantly attenuated the hyperglycemic activity when compared to pioglitazone and rosiglitazone. Compounds 63a and b showed comparable potency to pioglitazone in terms of reducing TG level, whereas rosiglitazone failed to show the hypolipidemic activity. Further, the compounds (3 × 10−5 – 3 × 10−11 M) were evaluated for the accumulation of TG in 3T3-L1 after insulin differentiation. The study results demonstrated that almost all the 4- or 5-substituted pyrimidine derivatives showed better results on TG accumulation in 3T3-L1 cells than that of rosiglitazone and pioglitazone except for the compound substituted with phenylamino at the 4th position of pyrimidine and 2-thiophenyl at the 5th position of pyrimidine. Whereas, 2-substituted pyrimidines showed inferior activity compared to 4 or 5-substituted pyrimidine derivatives (Table 3, Table 4) [83].
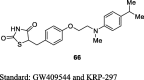
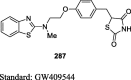
Gim et al. reported the synthesis of novel TZD derivative, 5-(4-(2-(methyl-p-substituted phenylamino)ethoxy)benzyl)TZD (66) by Mitsunobu reaction as shown in S15. The compound 66 was prepared by the reaction between 2-(methyl-p-substituted phenylamino) ethanols (64) and N-tritylated 5-(4-hydroxybenzyl)TZD (28) using tributylphosphine (Bu3P) and 1,1′-(azo-dicarbonyl)dipiperidine (ADDP) to afford the compound 65, then treatment of compound 65 with TFA yielded the final derivative 66. The compounds were evaluated for PPAR-γ agonistic activity in CV-1 cells and inhibition of nitric oxide (NO) production in the murine macrophage cell line (RAW 264.7). As a result, except compound 66, all the synthesized compounds found to exhibit very low PPAR-γ activity and NO inhibition. The compound 66 was reported to exhibit potent activity with 21.2% PPAR-γ activation in comparison with PPAR-γ activator (i.e., GW409544) and 51.5% NO inhibition compared to standard (Table 3) [84].
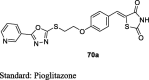
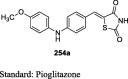
Iqbal et al. designed novel TZD derivatives that are structural analogs of pioglitazone, by introducing pharmacologically important heterocycles specifically, thiazole, triazole and oxadiazole nucleus linked to the middle phenyl ring through heteroatom linkage with one/two carbon atom. The compound of interest was obtained through base-catalyzed nucleophilic substitution reaction of 4-(2-bromoethoxy)benzaldehyde (55) with 2-mercaptotriazoles/2-mercaptooxadiazole (67) at room temperature to yield the intermediate (68). This intermediate was then refluxed with TZD in toluene, with a catalytic amount of piperidine to get the target compounds (69a and 70a and b) (S16). The synthesized compounds were evaluated for their in vivo hypoglycemic and hypolipidemic activities against alloxan-induced diabetic male Wistar rats. The compounds were treated orally to the rats for 15 days at different dose levels (3, 10, 30 and 100 mg/kg body weight) and the effects (i.e. percentage reduction of plasma glucose and TG) were compared with pioglitazone. The compounds (69a, 70a and b) showed comparable hypoglycemic and hypolipidemic activity with that of the standard pioglitazone. The compound containing 3-pyridyl substitution at the 5th position of oxadiazole (70a) was shown to be more potent in terms of percent reduction of glucose in comparison to that of pioglitazone in diabetic rats (Table 4) [85].
Kumar et al. reported a series of novel glitazones incorporated with phenols by using S17. 4-(2-bromoethoxy)benzaldehyde (55) was refluxed with various substituted phenols in dry acetone to give various phenol substituted 4-(2-bromoethoxy)benzaldehydes (71) which in turn underwent Knoevenagel condensation with TZD (3) or rhodamine to yield the final derivatives (72a, b and 73a, b). The synthesized derivatives were screened for in vitro insulin-induced glucose uptake using rat hemidiaphragm and also the cytotoxicity by sulforhodamine assay using human epithelial type 2 cells (HEp-2) and A549 cells. Compound 73a with TZD ring showed better glucose uptake activity (38.0 mg/dL/45 min) than others and the effect was comparable to that of standard (37.0 mg/dL/45 min). In the cytotoxicity assay, compound 72a with rhodamine ring showed cytotoxicity in both cells (Table 3) [86].
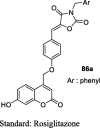
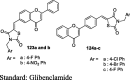
To circumvent TZD side effects like weight gain, hepatotoxicity, and fluid retention, Shukla et al. designed a new series of coumarin-based TZD analogs and its bioisosteres (oxazolidinedione and imidazolidinedione) (85, 86 and 87) as shown in S18. The resorcinol (74) was condensed with ethylacetoacetate (75) using Pechman reaction to afford 7-hydroxy-4-methyl coumarin (76), which was brominated by NBS to give 7-hydroxy-4-bromomethyl coumarin (77) followed by reaction with 4-hydroxy benzaldehyde to afford 4-(7-hydroxy-2-oxo-2H-chromen-4-yl)methoxy)benzaldehyde (78). This compound upon Knoevenagel condensation with TZD (3), 2,4-oxazolidinedione (79) and 2,4-imidazolidinedione (80) yielded coumarin-based bioisosteric analogs (81, 82 and 83) followed by refluxing with substituted benzyl chloride (84) afforded the desired products (85a-c, 86a-c, and 87a-c). The synthesized compounds were then evaluated for antidiabetic activity in STZ-induced diabetic rats. Firstly, the dose selection study was carried out using these three bioisosteric analogs (85, 86 and 87) at different dose level (15, 25, 35 and 45 μmol/kg) for the duration of 3, 7, 10 and 15 days. As a result, 35 μmol/kg dose was found to be most effective in reducing plasma glucose level and this dose was selected for the subsequent study for the different time periods (i.e., 3, 7, 10 and 15). It was found that compounds having N-substituted oxazolidinedione moiety (82 and 86a) showed a maximum reduction in plasma glucose level (40.25% and 44.67%, respectively) in comparison to other bioisosteres. Whereas, the standard drug rosiglitazone was more active in lowering plasma glucose levels (56.7%). Molecular docking studies were also carried out against PPAR-γ (PDB ID: 2PRG protein) and the results showed that compounds 82 and 86a showed hydrophilic interaction between the oxygen atom of oxazolidinedione and SER342 while hydroxyl group of coumarin moiety showed hydrophobic interaction with HID449 (Table 4) [87].
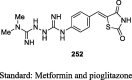
Ahmadi et al. reported the synthesis of two novel derivatives of rosiglitazone (94 and 98) as shown in S19a and 19b. 2,4-Bis(morpholino)-6-chloro-1,3,5-triazine (90) was synthesized from the reaction of cyanuric chloride (88) and morpholine (89) in DCM. The synthesized compound 90 was then refluxed with 2-methylaminoethanol (91) to afford 2-((4,6-dimorpholino-1,3,5-triazin-2-yl)(methyl)amino)ethanol (92). Then, the final compound 5-(4-(2-((4,6-dimorpholino-1,3,5-triazin-2-yl)(methyl)amino)ethoxy)benzyl))TZD (94) was synthesized by mixing of compounds 92 and 93 in DMSO. On the other hand, compound 5-(2-chloro-4-(2-(methyl(pyridin-2-yl)amino)ethoxy)benzyl)TZDs (98) was synthesized by mixing of compound 96 and 97 in DMSO. Compound 96, 2-(N-Methyl-N-2(pyridine-2-yl)amino)ethanol was synthesized by heating the mixture of 2-chloropyridine (95) and N-methyl ethanolamine (91) at 120 °C. Both the synthesized derivatives (94 and 98) were then screened for antihyperglycemic and antihyperlipidemic activity in the alloxan-induced diabetic rat model for 16 days. The synthesized derivatives and the standard drug rosiglitazone were administered at a dose of 3 mg/kg i.p. and the results were observed for the reduction in blood glucose levels and the lipid profile [TGs, total cholesterol, and low-density lipoprotein (LDL)]. As a result, compound 94 showed a significant reduction in blood glucose (295.5 mg/dL) as compared to control (411.0 mg/dL) and rosiglitazone (304.0 mg/dL). Also, compounds 94 and 98 were able to significantly increase the high-density lipoprotein (HDL) level and HDL/LDL ratio which is considered as a good indicator for the improvement of lipid profile in comparison to rosiglitazone (Table 4) [10].
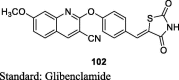
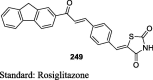
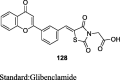
Deshmukh et al. carried out the Knoevenagal condensation of 2-(4-formyl-phenoxy) substituted quinoline-3-carbonitriles (101) and TZD to design a new series of (Z)-2-(4-((2,4-dioxothiazolidin-5-ylidene)methyl)phenoxy) substituted quinoline-3-carbonitriles (102) as shown in S20. The compound 102 was obtained by reacting substituted 2-chloroquinoline-3-carbaldehyde (99) with ammonia and molecular iodine in tetrahydrofuran (THF) thereby, yielding substituted 2-chloroquinoline-3-carbonitriles (100) which upon condensation with 4-hydroxy benzaldehyde (6) in DMF gave the compound 101. The synthesized compounds were screened for in vivo antidiabetic activity in sucrose (10 g/kg) loaded rat model. Compounds were administered orally at a dose of 100 mg/kg. As a result, compound 102 showed 9.4% improvement on oral glucose tolerance during 0–120 min but the effect was inferior to that of the standard drug i.e., metformin (19.1%) (Table 4) [88].
Structural activity relationship (SAR) studies of alkoxy benzyl analogs
The inclusion of thioethyloxy linkage connected to triazole and oxadiazole leads to more potent results. Incorporation of oxazole ring with the substitution of phenyl at the 2nd position is best for hydrophobic interaction with the active site and had a superior activity level. Substitution of X1 with CH(OH) on the ethoxy chain, showed higher or comparable activity than the substitution with CH2 but substitution with an oxo group (C O) decreases the antidiabetic activity. Incorporation of heterocyclic with an indole ring with no substitution exhibited more potent euglycemic and hypolipidemic activities. Incorporation of heterocyclic with tryptophan and carbazole ring completely abolished the activity. Substitution at the 2nd position of pyrimidinone with alkyl group reduced plasma glucose level whereas; increase in the alkyl chain reduced the antidiabetic activity. Replacement of oxygen at the 4-position of a 5-benzyl moiety with sulfur led to completely inactive for hypoglycemic effects. Substitution with electron-withdrawing groups at X position does not have much impact on the activity. The distance between the benzene ring and heterocyclic moiety should be of two carbon atoms. The unsaturated compounds showed lesser results than saturated compounds [71], [72], [74], [76], [78], [82], [86] (Fig. 5A).
Fig. 5A.

SAR of ethoxy benzyl based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Pyrazole based TZDs
Naim et al. carried out the synthesis and molecular docking studies (against PPAR-γ) (PDB ID: 1ZEO) of TZD based benzene sulphonamide derivatives containing pyrazole moiety as shown in S21. Firstly, compound, 4-hydrazinyl-benzenesulphonamide (104) has been synthesized from sulphanilamide (103) through diazotization followed by reduction with SnCl2. The imines were prepared by refluxing hydrazine (104) with substituted aryl ketones to yield corresponding substituted hydrazones (105). The compound 105 upon Vilsmeier-Haack reaction with DMF and POCl3 gave pyrazolecarbaldehydes having N,N-dimethyl formimidamide group (106) which on treatment with sodium hydroxide gave pyrazolecarbaldehydes having benzenesulphonamide group (107) followed by Knoevenagel condensation with TZD to yield the final product (108a-c). The compounds having glide scores > 10 were further evaluated in vitro for PPAR-γ transactivation assay in HEK-293 cells. Among the eight compounds, compounds 108a-c significantly increased PPAR-γ transactivation (57.3%, 60.5%, and 61.2%, respectively) and the effect was comparable with that of pioglitazone (68.3%) and less than that of rosiglitazone (83.6%). After that, compounds showed good PPAR-γ transactivation were further screened for in vivo antidiabetic activity in STZ-induced diabetic rats for 15 days by administering the compounds at a dose of 36 mg/kg. All the three compounds (108a-c) showed superior activity in terms of reduction in blood glucose level (138.7, 137.4 and 134.1 mg/dL respectively) than that of standard drug rosiglitazone (143.1 mg/dL) but exhibited comparable potency with that of pioglitazone (132.2 mg/dL). Further, compound (108c), which showed the more potent antihyperglycemic effect was evaluated for body weight gain for 1 and 15 days and as a resulting compound 108c did not show any significant change in body weight. Then, the compounds 108a-c were analyzed for liver toxicity through assessing the levels of biochemical parameters [aspartate transaminase (AST), alanine transaminase (ALT) and alkaline phosphatase (ALP)] and histological staining. As a result, compound 108c showed significant and maximal lowering of these biochemical parameters as compared to pioglitazone while compounds 108a and 108b were equipotent to pioglitazone. In the histopathological study, compound 108c showed no signs of toxicity whereas compound 108a, 108b and pioglitazone showed the least pathological changes. From the above results, the compound 108c was considered as the most potent compound and was further evaluated for PPAR-γ gene expression in 3T3-L1 cells. The results showed that compound 108c significantly increased PPAR-γ gene expression by 1.9 folds whereas expression was increased 1.2 and 1.5 folds for the standard drugs rosiglitazone and pioglitazone, respectively (Table 3, Table 4) [89].
Naim et al. synthesized a series of TZDs coupled with pyrazole as shown in S22. The compounds 110 were synthesized by refluxing aryl ketones with phenylhydrazine (109), which further underwent Vilsmeier-Haack reaction to afford pyrazole carbaldehydes (111) which in turn further reacted with TZD to furnish the final product (112a-c). Firstly, molecular docking studies were carried out against PPAR-γ (PDB ID: 2PRG) and the compounds with glide scores > −8 were selected further for PPAR-γ transactivation assay (HEK-293 cells) and in vivo antidiabetic activity in STZ-induced diabetic rats. As a result, compounds 112a-c showed significant PPAR-γ transactivation (48.65, 51.30 and 52.06%, respectively) and the results were comparable to those of standard drugs (pioglitazone (65.22%) and rosiglitazone (85.30%)). The in vivo study carried out on the synthesized compounds for 15 days at a dose of 36 mg/kg also produced similar results with that of in vitro. Compounds (112a-c) displayed significant reduction in blood glucose levels (150.7, 141.4 and 140.1 mg/dL), which were comparable to those of standard drugs pioglitazone (135.2 mg/dL) and rosiglitazone (141.1 mg/dL). Further, compound 112c was evaluated on body weight gain for 15 days, as a result, compound 112c did not show any significant change in body weight, which suggests that the weight neutral effect exhibited by the compound 112c. Then, the hepatotoxicity study was carried out with the compounds (112a-c) at 3 times higher dose (i.e., 108 mg/kg) that was used in the antidiabetic activity (36 mg/kg). As a result, compound 112c came out to be most potent in terms of lowering the levels of AST, ALT and ALP and did not cause any toxic effect to the liver but the compound 112a showed a small dilation of sinusoidal space along with pioglitazone. After that compound 112c was evaluated for PPAR-γ gene expression in 3T3-L1 cells and the results showed that compound 112c significantly increased the PPAR-γ gene expression (2.35 fold) in comparison to that of pioglitazone (1.6 fold) and rosiglitazone (1.27 fold) and also increased the levels of GLUT1 and GLUT4 [90]. Altogether, study results demonstrated that compound with naphthalene moiety (112c) showed a more potent antihyperglycemic activity with devoid of any side effect that was observed with the standard drugs (Table 3, Table 4) [91].
We have recently synthesized TZDs clubbed with pyrazoles analogues. The title compounds were synthesized using a synthetic procedure involving 4-steps and the synthetic strategy is outlined in Scheme S23. The first step involves the same procedure for the synthesis of TZD (3) as highlighted in Scheme S1. After that, substituted pyrazole carbaldehydes (111) were made through Vilsmeier-Haack reaction on substituted hydrazones (110), which in turn were synthesized by treating substituted acetophenones with phenylhydrazine (109) as shown in Scheme S22. Next, N-alkylation and acidification of TZD (3) was carried out with benzyl bromide (113) and bromoacetic acid (114), respectively to yield N-alkylated (115a) and acidified TZD (115b) followed by Knoevenagel condensation with synthesized carbaldehydes to yield TZD clubbed pyrazole adducts (116a and b). The compounds were docked against PPAR-γ (PDB ID: 2PRG) and alpha-amylase (PDB ID: 4GQR) and further evaluated for in vivo and in vitro antidiabetic activity, in addition to in vitro anti-inflammatory and antioxidant activities. Compound 116b exhibited significant blood glucose-lowering activity (134.46 mg/dL) and was found to be similar to standard pioglitazone (136.56 mg/dL). In addition, the compound (116b, IC50 4.08 μg/mL) was also found to be a potent inhibitor of alpha-amylase. These results were consistent with in vitro docking results with PPAR-γ and amylase. Compound 116a was found to exhibit anti-inflammatory and antioxidant activity to a greater extent through reducing inflammatory markers (TNF-α, and IL-β) and oxidative stress marker (MDA). These results suggest that compounds (116a and b) can be considered as a promising candidates for the discovery of new antidiabetics [92].
SAR of pyrazole based TZD analogs
Substitution with bulkier aryl groups at the N-position of pyrazole ring showed strong hydrophobic interactions with receptor site. Substitution with smaller halogens (X and X1) at meta and para positions of the phenyl ring of the 3rd position of pyrazole significantly reduced blood glucose levels but electron releasing groups lowers the antidiabetic activity. Replacement of aromatic ring (green colored) with thiophene reduces the activity but with large hydrophobic naphthalene moiety increases the antidiabetic activity. The acidic head derivatives resulted in more potent activity in comparison to benzylated TZD [89], [90], [91], [92] (Fig. 5B).
Fig. 5B.

SAR of pyrazole based TZD analogs (structures are original and made by using chem draw ultra 12.0).
N-substituted TZDs
Datar et al. synthesized 5-substituted benzylidene TZD-3-acetic acid derivatives based on 2D-QSAR studies as shown in S24. Firstly, benzylidene derivatives (117) upon N-alkylation with ethyl bromoacetate gave 5-substituted benzylidene-2,4-dioxothiazolidin-3-yl-acetic acid ethyl ester (118) followed by acidic hydrolysis with conc. HCl gave the final compound as acid derivatives (119a and b). The synthesized derivatives were then evaluated for their hypoglycemic effect using a sucrose loaded model. The compounds were administered orally at a dose of 100 mg/kg and the effects were observed as a percentage reduction in blood glucose level. The results showed that the analogs with di- and trimethoxy group (119a and b) on phenyl ring showed reduction in blood glucose level within 30 min and the effect was maintained till the duration of 120 min, whereas pioglitazone showed a decrease in blood glucose level in 30 min and later slightly increased levels of blood glucose in 60, 90 and 120 min (Table 4) [93].
SAR of N-substituted TZD analogs
Substitution with dimethoxy or trimethoxy at the benzene ring showed similar hypoglycemic activity as compared to pioglitazone but when replaced with the hydroxyl group lowers the activity. By maintaining the acidic nature of the TZD head, results in more potent activity [93] (Fig. 6A).
Fig. 6A.

SAR of N-substituted TZD analogs (structures are original and made by using chem draw ultra 12.0).
Flavonyl based TZDs
Tunçbilek et al. synthesized a new series of 3-substituted benzyl-5-[3′-(4H-4-oxo-1-benzopyran-2-yl)-benzylidene]-TZD derivatives as shown in S25 by carrying out Knoevenagel condensation of N-substituted derivative (120) with flavone-6-carboxaldehyde (121) and flavone-4′-carboxaldehyde (4-(4-oxo-4H-chromen-2-yl)benzaldehyde) (122) to yield 3-(p-substituted benzyl)-5-(6- and 4′-flavonyl) TZD derivatives (123a and b and 124a-c, respectively). The compounds were then evaluated for insulinotropic activity in vitro using insulin-secreting cells (INS-1) cells. The effects were observed at different concentrations (1 and 10 μg/mL) in comparison to the reference standard glibenclamide (1 μg/mL). It was found that compounds 123a and b and 124a-c were able to increase the insulin release (105–140%) more than the other derivatives at higher concentration (10 μg/mL) whereas the glibenclamide was able to increase the insulin release (210.4%) at 1 μg/mL (Table 3) [94].
Bozdag-Dundar et al. designed a new series of flavonyl-2,4-TZDs by carrying out Knoevenagel condensation of flavone-aldehyde (125) with ethyl 2,4-dioxothiazolidine-3-ylacetate (126) thereby, furnishing flavonyl-2,4-TZD acetic acid ethyl ester derivatives (127) followed by acid hydrolysis to yield carboxylic acid derivatives (128) (S26). The synthesized compounds were screened for insulinotropic activities in INS-1 cells by comparing them with glibenclamide at two different concentrations (0.01 and 0.001 mg/mL). As a result, compound 128 showed the most potent insulin-releasing ability (128.6%) at higher concentration (0.01 mg/mL) in comparison to other synthesized derivatives whereas glibenclamide even at lower concentration (0.001 mg/mL) showed more potent insulin-releasing ability (179.6%) (Table 3) [95].
SAR of flavonyl based TZD analogs
Flavonyl substituted TZDs at 4′ position will result in better activity than substituted at 3′ or 6 positions. The substitution of N with the acetic acid group showed better hypoglycemic activity than the aryl halides [94], [95] (Fig. 6B).
Fig. 6B.

SAR of flavonyl based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Sulfonyl based TZDs
Wrobel et al. prepared the novel 5-(3-aryl-2-propynyl)-5-(arylsulfanyl)TZDs (133) and 5-(3-aryl-2-propynyl)-5-(arylsulfonyl)TZDs (136) as shown in S27. Precursor (arylsulfanyl)TZDs (131) were firstly synthesized from aryl thiols (129) and 5-bromoTZDs (130) and the resulted arylsulfanyl TZD upon reaction with 3-arylprop-2-ynyl-bromides (132) and sodium hydride furnished the target compound (133). The compound 129 also converted to arylsulfinic acid upon oxidation with hydrogen peroxide followed by treatment with the base to yield arylsulfinate which in turn underwent treatment with 5-bromoTZDs (130) yielded another precursor (arylsulfonyl)TZDs (134). The resulted arylsulfonyl TZDs upon reaction with 3-arylprop-2-ynyl-bromides (132) and sodium hydride furnished the target compound (136). Compound 136 can also be synthesized from 134 by firstly protecting the nitrogen with trityl chloride to give trityl derivative (135) followed by treatment with 3-arylprop-2-ynyl-bromides (132) and deprotection to give the final compound 136. Firstly, the synthesized derivatives (133 and 136) were evaluated for oral antihyperglycemic effect in db/db mice model for 4 days by administering the compounds at a dose of 20 and 100 mg/kg. The results showed that sulfonyl TZDs (136) were found to be more potent than the sufanyl derivatives (133) and most of the sulfanyl derivatives (133) did not show any significant reduction in plasma glucose levels even at a dose of 100 mg/kg. In arylsulfonyl derivatives (136), the compounds substituted with 4-halogens exhibited more potent results at 100 mg/kg and were further evaluated in ob/ob mice model for 4 days at a dose of 50 or 100 mg/kg. It was found that compound 136 (65%) surpassed the reference standards, ciglitazone (39%) and BM13.907 (54%) in terms of percentage reduction in insulin. On the other hand, ciglitazone and compound 136 showed equipotent results in terms of percentage reduction in glucose. Then the compound 136 was further screened for glucose tolerance test by subcutaneous administration in fa/fa rats for 4 days at a dose of 20 and 100 mg/kg and the reference standard (ciglitazone) at 100 mg/kg. It was found that compound 136 considerably improved glucose tolerance at 20 mg/kg and did not exhibit any effect at 100 mg/kg. The overall results showed that arylsulfonyl TZD derivatives (136) were more potent as compared to arylsulfanyl TZD derivatives (129) (Table 4) [96].
Pattan et al. designed a series of TZD derivatives through microwave-assisted reaction of 5-(4-chlorosulfonyl benzylidene)-TZD (138) with the various aromatic/heteroaromatic amines (139) to give the final product (140a–f) as shown in S28. Of note, compound 138 was synthesized by refluxing benzylidene-TZD (137) with chlorosulphonic acid. All the synthesized compounds were then evaluated for antidiabetic activity in alloxan-induced diabetic albino rats by administering the test compounds at a dose of 36 mg/kg. Among all the synthesized compounds, compounds 140a-f produced a significant lowering of blood glucose levels in diabetic rats when compared with standard drug glibenclamide (Table 4) [4].
Jawale et al. designed various TZD derivatives with aryl sulfonylurea nucleus as shown in S29. The condensation reaction of maleic anhydride (141) with thiourea (1) yielded 2-imino-4-oxo-5-thiazolidine acetic acid (142) which upon hydrolysis gave 2,4-TZD acetic acid (143) followed by reaction with thionyl chloride and sodium azide afforded acylazide (144) which underwent Curtius rearrangement to yield 5-(isocyanatomethyl)TZD (145) which upon condensation with aryl sulfonamides (146) furnished the final compound 1-((2,4-dioxothiazolidin-5-yl)methyl)-3-benzenesulfonylureas (147a–d). The newly synthesized derivatives were then screened for antihyperglycemic activity in the sucrose loaded rat model. The synthesized compounds and the reference drug (metformin) were given at a dose of 100 mg/kg. As a result, only four compounds (147a–d) repressed the postprandial increase in blood glucose level showing 15.8, 17.2, 14.3 and 16.5% activity, respectively in comparison to that of standard i.e., metformin (27.0%) (Table 4) [97].
SAR of sulfonyl based TZDs
Introduction with sulfonyl group showed good antihyperglycemic activity than the sulfanyl group. Substitution of R with heterocyclic moieties or aryl halides showed better antidiabetic activity than the simple alkyl groups [4], [96], [97] (Fig. 6C).
Fig. 6C.

SAR of sulfonyl based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Naphthyl based TZD
Prabhakar et al. synthesized novel TZD derivatives by combining two active pharmacophores, namely a TZD nucleus and a methoxy naphthyl moiety of nabumetone (148), an anti-inflammatory drug. Knoevenagel condensation of nabumetone (148) with TZD (3) yields an unsaturated mixture of E & Z (149) which upon hydrogenation with Pd/C gives a mixture of diastereoisomers (150). Further, they synthesized modified analogs of nabumetone based TZDs by reacting the enol-ketone (151/152) with TZD gives Z (153) and E (154) compounds. Similar reactions of TZD (3) with acetyl naroline (155), 4-(6-hydroxy-2-naphthyl)-butan-2-one (156), 2-acetyl thiophene (157) and 2-acetyl furan (158) gave their corresponding unsaturated compounds (159–162) (S30a-c). The synthesized compounds were then evaluated for antidiabetic activity in db/db mice for 6 days and compared against a phase-II antidiabetic candidate, 6-(2-flourobenzyloxy)-2-naphthyl derivative of TZD (MCC-555) and troglitazone. The derivatives were administered at a dose of 30 mg/kg. The results showed that unsaturated compound (149) exhibited a better reduction in plasma glucose (16%) and TG (50%) level in comparison to saturated compound (150) (8% and 44% respectively). The Z-isomer (153) also showed a better reduction in plasma glucose (16%) as compared to E-isomer (154) which did not show any effect. Also, the compounds 159, 160 and 162 exhibited good antidiabetic activity as they showed 26, 20 and 16% plasma glucose reduction, respectively but did not show any reduction in TG levels (Table 4) [98].
SAR of naphthyl based TZD analogs
The introduction of naphthalene moiety showed better antidiabetic activity along with some marginal anti-inflammatory activity. Unsaturated compounds showed better antidiabetic activity than the saturated one. For better antidiabetic activity, Z-isomer is preferred as compared to E-isomer. Replacement of naphthalene with thiophene moiety completely abolished the activity [98] (Fig. 7A).
Fig. 7A.

SAR of naphthyl based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Phenothiazine based TZDs
Saini et al. synthesized phenothiazine based TZD as antidiabetic analogs as shown in S31. Firstly, 10H-phenothiazine (164) has been synthesized from diphenylamine (163) using sulphur and a catalytic amount of iodine followed by stirring with acetyl chloride gives N-acetyl phenothiazine (165) which upon Vilsmeier-Haack reaction yielded N-acetyl phenothiazinal (166). The compound 166 on reaction with substituted aniline (167) gives corresponding imines (168) followed by reaction with TZD and thioglycolic acid to give the final product 4-(10-acetyl-10H-phenothiazin-3-yl)-3-phenylTZD (169a and b). The synthesized compounds were then evaluated for antidiabetic activity in STZ-induced diabetic rat model. The compound 169a was administered for a period of 21 days at a dose of 5 mg/kg and compound 169b at a dose of 10 mg/kg and was compared to that of rosiglitazone (8 mg/kg). It was found that both the compounds (169a and b) exhibited equipotent antidiabetic activity to that of standard therapy (Table 4) [99].
SAR of phenothiazine based TZD analogs
By combining phenothiazine moiety with TZD results in good antidiabetic activity. Substitution with chloro group at the para position of phenyl rings showed better antidiabetic activity than with bromo or nitro groups [99] (Fig. 7B).
Fig. 7B.

SAR of phenothiazine based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Amide based TZDs
Chittiboyina et al. discovered potent antidiabetic compounds including a series of [1], [2]-dithiolanepentamide TZDs as shown in S32. Firstly, 4-hydroxybenzaldehyde (6) was coupled with Boc-protected alcohol (170) under Mitsunobu conditions to give O-substituted aldehyde (171) followed by condensation with TZD yielded benzylidene derivative (172) which was further reduced with Mg/MeOH to give compound 173. After that, deprotection of Boc was carried out using HCl in dioxane to give the amine hydrochloride compound (174) which was then condensed with dithiolane-substituted acid (175) to give the amide, N-(2-(4-((2,4-dioxothiazolidin-5-yl)methyl)phenoxy)ethyl)-2-(1,2-dithiolan-3-yl)-N-methylpentanamide (176) followed by reduction of dithiolane ring to give dithiane compound (177). Compound 177 on further reaction with Boc-glycine (178) gives bis-Boc-glycinates (179) which upon exposure to anhydrous HCl gives another amide derivative, 2,2′-((8-((2-(4-((2,4-dioxo-1,3-thiazolidin-5-yl)methyl]phenoxy}ethyl)(methyl)amino)-8-oxooctane-1,3-diyl}bis(thio))bis(2-oxoethanaminiu- m)dichloride (180). The synthesized derivatives (176 and 180) were then evaluated for adipocyte differentiation and adipogenesis using 3T3-L1 cells. It was found that compounds 176, 180 and rosiglitazone equally promoted the differentiation of 3T3-L1 cells, which indicate potent PPAR-γ agonistic activity. Further, the compound 176 (100 mg/kg) was evaluated for its antidiabetic and hypolipidemic activity in fa/fa rat for 1 month. The results showed that there was no change in blood glucose level but there was 78% and 83% reduction in serum insulin and TGs level, respectively while rosiglitazone caused only 40% reduction in TG levels. These results suggest that compound 176 has the potential to treat not only T2DM but also hyperlipidemic state associated with atherosclerosis (Table 3, Table 4) [100].
Kumar and Nanjan designed various novel glitazones as PPAR-γ agonists and synthesized them as shown in S33. Firstly, acylated amines (181) were reacted with vanillin to give compound 182 which upon condensation with TZD gives the final compound (183). Compound 182 on further reduction with Pd over charcoal gave the saturated product (184). The synthesized compounds were evaluated for their in vitro antihyperglycemic activity by measuring glucose uptake using rat hemidiaphragm model, in the presence and absence of insulin. Although some of the synthesized derivatives showed good glucose uptake activity but the compound, 2-(4-((2,4-dioxothiazolidin-5-ylidene)methyl)-2-methoxy-phenoxy)-N-(4-methoxyphenyl)acetamide (183) exhibited the highest glucose uptake activity (36.25 mg/g/45 min) as compared to rosiglitazone (35.25 mg/g/45 min) (Table 3) [101].
Kumar et al. synthesized a series of novel glitazones by adding glycine, aromatic and alicyclic amine through two carbon linkers using both conventional and microwave methods as shown in schemes S34a–c. In S34a, firstly, the C-terminal of glycine (185) was converted to ethyl ester (186) followed by N-acylation with chloroacetyl chloride to give 187. Side by, compound 188 was further converted to potassium salt (189) with the help of KOH. Then, compound 189 was coupled with 187 in the presence of DMF to get the final compound, (Z)-ethyl-2-(2-(5(4-methoxybenzylidene)-2,4-dioxothiazolidin-3-yl)acetamido)acetate (190). In S34b, firstly, aromatic/alicyclic amines (191) was treated with the chloroacetyl chloride to get acylated amines (192), which were then reacted with p-hydroxybenzaldehyde (6) to obtain compound 193 which upon condensation with TZD yield the final product, (Z)-2-(4-((2,4-dioxothiazolidin-5-ylidine)methyl)phenoxy)-N-phenylacetamide (194). In S34c, firstly, aniline (196) was acylated to form 2-chloro-N-phenylacetamide (197) which was then coupled with potassium salt of TZD (195) to form 2-(2,4-dioxothiazolidin-3-yl)-N-phenylacetamide (198) followed by Knoevenagel condensation with aryl aldehydes to get the final product (199a–e). The synthesized compounds were tested in vitro at two different dose levels (1 and 2 mg) for glucose uptake activity using rat hemidiaphragm model. As a result, compounds 190, 194 and 199a–e depicted significant glucose uptake activity but the compound 199a was the most potent among all the glitazones having 42.16 mg/dL/45 min of glucose uptake level and the effect was comparable to that of standard drug rosiglitazone (48.34 mg/dL/45 min). The results also showed that increasing the concentration of drugs from 1 mg to 2 mg, did not produce a significant change in glucose uptake level (Table 3) [102].
Munj and Ghosh reported the synthesis of two novel benzylidene TZD derivatives namely (Z)-5-(2-(4-((2,4-dioxothiazolidin-5-ylidene)methyl)phenoxy)-acetyl)-2-hydroxybenzamide (202) and (Z)-5-(2-(4-((2,4-dioxothiazolidin-5ylidene)methyl)phenoxy)-N-(5-nitro-thiazol-2-yl)acetamide (205) as shown in S35a and S35b. In S35a, the final derivative (202) was synthesized by the condensation of compound 200 and 5-(2-bromoacetyl)-2-hydroxybenzamide (201) in the presence of basic condition (K2CO3 in DMF). In S35b, intermediate 204 was synthesized by the base-catalyzed bromoacetylation of 5-nitro-2-aminothiazole (203) with bromoacetylbromide which was then condensed with compound 200 to get the final product (205). The newly synthesized derivatives were screened for hypoglycemic and hypolipidemic activity against high-fat diet (HFD) and STZ-induced T2DM in Sprague-Dawley rats. The synthesized compounds were administered at a dose of 20 mg/kg and pioglitazone hydrochloride at a dose of 10 mg/kg for 14 days. The results showed that compounds 202 and 205 exhibited significant antihyperglycemic (64% and 56% reduction in the blood glucose) and hypolipidemic activity (74% and 78% reduction in TG and 89% and 92% reduction in cholesterol levels, respectively) and the effect was comparable to that of standard drug (Table 4) [103].
Shrivastava et al. reported the synthesis of a few methylphenylbenzamide derivatives having an amide linkage between the central aryl ring and the hydrophobic tail as shown in S36. Firstly, compound 5-(4-nitrobenzylidene)TZD (206) was reduced to obtain the amino intermediate 207 which was further reacted with various substituted benzoyl chlorides (208) to furnish the final amide compounds (2,4-dioxo-1,3-thiazolidine-5-yl)methylphenylbenzamide (209a and b). After that, synthesized derivatives were screened for acute oral toxicity test at a dose of 100 mg/kg in healthy albino rats for 14 days. As a result, compounds did not show any signs of mortality even at a dose of 500 mg/kg after 14 days of administration. Further, compounds were evaluated in STZ-nicotinamide (NA)-induced diabetic rats by administering at a dose of 1, 10 and 100 mg/kg. The results showed that compounds (209a and b) significantly decreased plasma glucose and TG levels in a dose-dependent manner after the 1-week duration of treatment in comparison to diabetic control and showed comparable potency to that of pioglitazone. However, chloro-substituted derivative (209a) at 10 and 100 mg/kg exhibited more potent activity than that of standard therapy at the same dose. Molecular docking studies were also carried out against PPAR-γ and it was found that both the compounds (209a and b) showed noteworthy binding energies (−6.1 and −6.3, respectively), close to that of pioglitazone (−7.75) (Table 4) [104].
Dadasaheb et al. carried out the reaction between 5-(4-hydroxybenzylidene)-TZD (200)/5-(4-hydroxy-3-methoxybenzylidene)-TZD (210) and aromatic 2-chloro-N-substituted acetamide (211) at room temperature to synthesize their corresponding TZD derivates (212a and b and 213a and b, respectively) as shown in S37. Of note, compounds 200 and 210 were synthesized by the Knoevenagel condensation of TZD with their respective benzaldehydes. The synthesized compounds were then screened for antidiabetic activity at a dose of 30 mg/kg in dexamethasone (1 mg/kg)-induced diabetic-mice for 7 days and were compared against pioglitazone (30 mg/kg) and metformin (30 mg/kg). The plasma glucose levels were assessed at a different time interval after 7 days of treatment. It was found that compound 212a and b and 213a and b significantly decreased the blood glucose level (113.7 mg/dL and 129.9 mg/dL) in comparison to diabetic control (330.1 mg/dL). The compound 212a and b were more potent in comparison to the standard drugs (pioglitazone, 120.2 mg/dL, and metformin, 122.7 mg/dL) in terms of reducing plasma glucose level whereas compound 213a and b showed comparable potency to that of standard. Molecular docking studies were also carried out against PPAR-γ (PDB ID: 2PRG) and the most active compound was found to be 212a because of its increased contact with receptor active site due to the introduction of an extra hydrophobic group (Table 4) [105].
Naim et al. synthesized a series of TZD based amide derivatives (214) by reacting the compound 112 with 2-chloroacetamide as shown in S38. The compound 112 was synthesized as shown in S22. Firstly, molecular docking studies were carried out against PPAR-γ (PDB ID: 3TY0) and as a result, compounds having glide score in the range of −9.0 to 10.5 were further screened for in vivo antidiabetic activity in STZ-induced diabetic rats. The compounds were administered at a dose of 36 mg/kg for 15 days and the effects were observed for their reduction in blood glucose level in comparison to standard drugs (pioglitazone and rosiglitazone). It was found that compound 214 showed a significant reduction in blood glucose level (142.4 mg/dL) in comparison to that of diabetic control (325 mg/dL) and the effect was comparable to that of pioglitazone (134.8 mg/dL) and rosiglitazone (144.6 mg/dL). The selected compounds were then further evaluated for PPAR-γ transactivation assay using HEK-293 cells. Compound 214 significantly increased PPAR-γ transactivation (53.65%) in comparison to that of control but the effect was not as much of pioglitazone (62.21%) and rosiglitazone (86.4%). Further, compound 214 was screened for PPAR-γ gene expression studies using 3T3-L1 cells. As a result, compound 214 exhibited a 2.1 folds increase in PPAR-γ gene expression in comparison to that of pioglitazone (1.15 folds) and rosiglitazone (1.64-fold). In addition, the toxicity study was carried out with compound 214 through assessing body weight gain and biochemical parameters and morphological changes in the liver. As a result, compound 214 did not show any signs of toxicity (no change in weight gain and decreased the levels of AST, ALT, and ALP), whereas pioglitazone exhibited mild dilation and inflammation in the sinusoidal space of central vein and in the portal triad, respectively. From all the studies, it was revealed that compound 214 exhibited a potent antidiabetic activity both in terms of in vivo and in vitro studies (Table 3, Table 4) [106].
SAR of amide based TZD analogs
The introduction of an extra hydrophobic group increases the binding efficacy with the receptor site. The chain length of two carbon atoms between the terminal nitrogen of amide and the oxygen next to the benzene ring is significant for antidiabetic activity. Substitution with electron releasing groups on the benzene ring does not have much effect on activity. The polar TZDs as their head group showed good activity than its bioisosteres (2-thioxo-thiazolidine-4-ones or rhodanine). The activity got reduced on decreasing the ring size from benzene to furan [100], [101], [102], [106] (Fig. 8A, Fig. 8B).
Fig. 8A.

SAR of amide-based analogs substituted at N-position of TZD (structures are original and made by using chem draw ultra 12.0).
Fig. 8B.

SAR of amide-based analogs substituted at 5th position of TZD (structures are original and made by using chem draw ultra 12.0).
Imidazo-thiadiazole based TZDs
Khazi et al. synthesized a series of eighteen 5-(2-alkyl/aryl-6-arylimidazo[2,1-b][1,3,4]thiazdiazol-5-yl)methylene-1,3-TZDs as shown in S39. The condensation of substituted 2-aminothiadiazole (215) with α-haloaryl ketones (216) under reflux furnished a fused imidazo[2,1-b][1,3,4]thiadiazole nucleus (217) which upon Vilsmeier-Haack reaction gives carbaldehyde (218) followed by Knoevenagel condensation with TZD to give the final product (219a–c). All the synthesized derivatives were then evaluated for in vivo hypoglycaemic and hypolipidemic activity in alloxan-induced male Wistar rats by administering at a dose of 3, 10, 30 and 100 mg/kg, and the results were compared against pioglitazone. After the study period of 15 days, it was found that only three compounds (219a–c) showed the most significant and dose-dependent reduction in plasma glucose and TGs level comparable with those in pioglitazone. These three compounds were further evaluated for PPAR-γ agonistic activity using NIH3T3-cells. As a result, compound 219b showed the maximum PPAR-γ agonistic activity (64%) in comparison to that of pioglitazone (100%) (Table 3, Table 4) [107].
SAR of Imidazo-thiadiazole based TZD analogs
Replacement of central benzyl ring with imidazo-thiadiazole ring did not show promising results. Substitution on this ring with cyclohexyl or thiophene analogs may result in better anti-hyperglycemic activity. Further, substitution with the p-methoxyphenyl group on 6th position resulted in good activity [107] (Fig. 9A).
Fig. 9A.

SAR of imidazo-thiadiazole based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Dispiropyrrolidines based TZDs
Murugan et al. reported a series of new dispiropyrrolidines by carrying out 1,3-dipolar cycloaddition reaction as shown in S40. Firstly, azomethine ylides (222) were obtained by the reaction of isatin (220) with sarcosine (221), which were further reacted with the double bond of 5-arylidene-TZD derivatives to give the corresponding cycloadducts (223a and b). The synthesized derivatives were evaluated for antidiabetic activity in alloxan-induced diabetic male Wistar rats by administering the compounds at a dose of 36 mg/kg. As a result, compounds (223a and b) had shown a better reduction in blood glucose (115.3 and 115.8 mg/dL) levels as compared to rosiglitazone (131.0 mg/dL). Further, compound 223b also showed the best score (71.18) among all the molecules when docked against PPAR-γ (PDB ID: 1FM9). Also, the compounds were tested for some change in biochemical parameters [serum glutamic pyruvic transaminase (GPT), creatinine, blood urea, liver GPT, and ALP] in the same diabetic model for 15 days. The results showed that levels of these biochemical parameters reduced by the administration of these compounds (Table 4) [108].
SAR of dispiropyrrolidines based TZD analogs
The cyclic adduct derivatives resulted in more potent derivatives than rosiglitazone. Electron releasing groups at the tail position resulted in interactions with the active site [108] (Fig. 9B).
Fig. 9B.

SAR of dispiropyrrolidines based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Acid-based TZDs
Neogi et al. synthesized a series of 2,4-TZD derivatives of phenyl substituted cinnamic acid as shown in S41. The first step was to synthesize α-phenyl substituted cinnamic acid (226) via Perkin condensation of 3,5-dimethoxybenzaldehyde (224) with 4-hydroxyphenylacetic acid (225) followed by esterification to give an intermediate (227) which upon further condensation with 4-fluorobenzaldhyde furnished compound 228. Knoevenagel condensation of 228 with TZD gave an intermediate 229 followed by hydrogenation with Pt over carbon afforded the final product, (Z)-methyl 3-(3,5-dimethoxyphenyl)-2-(4-(4-((2,4-dioxothiazolidin-5-yl)methyl)phen- oxy)phenyl)acrylate (232). The compound 232 has also been synthesized by reducing aldehyde (228) to alcohol (230) followed by treatment with PBr3 yielded bromo compound (231) which upon Knoevenagel condensation with TZD (3) gave the final compound (232). In the final step, the free acid analog of the final compound (233) has been synthesized by alkaline hydrolysis of 232. The synthesized compounds were then evaluated for PPAR-γ agonist activity using HEK-293 cells at different concentrations (0.1 – 30 μM). It was found that compound 232 came out to be the most potent (EC50 = 0.28 μM) amongst all as compared to rosiglitazone (EC50 = 0.009 μM) whereas compound 233 showed less activity than 232 (EC50 = 0.69 μM). Further, compound 232 was evaluated for its antihyperglycemic activity in ob/ob mice for 9 days at a dose of 10 mg/kg in comparison to pioglitazone (20 mg/kg), rosiglitazone (10 mg/kg) and troglitazone (100 mg/kg). As a result, compound 232, pioglitazone, rosiglitazone, and troglitazone exhibited 29, 25, 42 and 26% reduction in blood glucose levels, respectively. Furthermore, a dose-dependent study was carried out on compound 232 in the same diabetic model at doses of 3.1, 6.3 and 12.5 mg/kg in comparison to troglitazone (12.5 mg/kg) for 19 days. The results showed that the most effective blood glucose-lowering (55%) activity occurred at a dose of 6.3 mg/kg whereas troglitazone caused only a 23% reduction in blood glucose level. In addition, compounds 232 and 233 were evaluated at a dose of 5 mg/kg for 11 days in the same diabetic model. As a result, a similar reduction in blood glucose levels was observed for both the compounds in comparison to vehicle-treated diabetic control mice. These results suggest that the presence of geometry in the cinnamic acid derivative is important for PPAR-γ agonistic activity and thus cinnamic acid- based TZDs can be considered as a lead molecule to discover new antidiabetic agents (Table 3, Table 4) [109].
Nazreen et al. synthesized a series of phenoxy acetic acid-based TZDs as shown in S42. Initially, different hydroxybenzaldehydes (234) were treated with chloroacetic acid (2) to furnish different substituted formyl phenoxy acetic acids (235) which upon Knoevenagel condensation with TZD (3) gives an intermediate (236) followed by reduction with sodium borohydride gives the target compounds i.e. 2-(4-((2,4-dioxothiazolidin-5-yl)methyl)-2-methoxyphenoxy)acetic acid (237a), 2-(4-bromo-2-((2,4-dioxothiazolidin-5-yl)methyl)phenoxy)acetic acid (237b) and 2-(4-bromo-2-((2,4-dioxothiazolidin-5-yl)methyl)-6-methoxyphenoxy)acetic acid (237c). Firstly, molecular docking studies were carried out against PPAR-γ (PDB ID: 3CS8) and the compounds with glide scores > −6.50 were selected further for PPAR-γ transactivation (HEK-293 cells) by in vitro and in vivo antidiabetic activity (STZ-induced diabetic rats; 60 mg/kg). The transactivation of PPAR-γ was confirmed through luciferase activity. As a result, compounds 237a–c showed significant PPAR-γ transactivation (48.35, 54.21 and 55.41%, respectively) than that in control (7%) but the effect of transactivation was not as much of standard drugs, pioglitazone (65.94%) and rosiglitazone (82.21%). The in vivo study was carried out using synthesized compounds at a dose of 36 mg/kg for 15 days and revealed the same results as that of in vitro. Compounds (237a-c) displayed significant reduction in blood glucose levels (157.5, 158.8 and 159.2 mg/dL), and the effects were comparable to that of standard drugs pioglitazone (134.2 mg/dL) and rosiglitazone (142.2 mg/dL). Further, compound 237c was evaluated for body weight gain for 15 days and as a resulting compound 237c showed a significant change in body weight. Then, compounds (237a–c) were evaluated for hepatotoxic effects by administering the compounds at 3-times higher (108 mg/kg) than that used in the antidiabetic activity (36 mg/kg). As a result, compounds 237b and c found to be most potent in terms of lowering the levels of AST, ALT, and ALP and did not cause any toxicity/damage to the liver as compared to pioglitazone. Since the compound 237c showed more potent activity on PPAR-γ transactivation, it was further evaluated for PPAR-γ gene expression in 3T3-L1 cells and the results showed that compound 237c significantly increased the PPAR-γ gene expression by 2.0-fold in comparison to that of pioglitazone (1.5 fold) and rosiglitazone (1.0 fold) and also increased the levels of GLUT1 and GLUT4. These results demonstrate that 237b and c can be considered as a potential lead molecule for the development of new antidiabetic agents (Table 3, Table 4) [110].
Maji and Samanta synthesized a series of TZD-5-acetic acid peptide hybrids as shown in S43. The first step was to synthesize 2-(2,4-dioxothiazolidin-5-yl)acetic acid (238) from the reaction of maleic anhydride (141) with thiourea (1) which was then dissolved in dioxane:water (1:1) and the acid was converted to the acid chloride by stirring with SOCl2. Then the resulting solution was treated with different esters [single amino acid esters (239), dipeptide methyl ester (240) and tripeptide methyl ester (241)], which was synthesized in the lab to get their corresponding peptide hybrids (242–244). The synthesized hybrids were then evaluated for antidiabetic and cardioprotective activities. Firstly, compounds were evaluated for antidiabetic activity in vitro by measuring glucose uptake using yeast cells at different concentrations (10, 20, 40, 80, 100 and 200 μL/mL). As a result, compounds 242–244 had shown to increase glucose uptake (39.23, 38.19 and 38.80%, respectively), which was similar to that of pioglitazone (42.87%). In addition, these compounds (30 mg/kg) were evaluated in vivo for antidiabetic activity using STZ-NA-induced diabetic rats for 14 days. It was found that compounds 242–244 showed a significant reduction in blood glucose level (142, 144.4 and 156 mg/dL, respectively) similar to that of standard pioglitazone (137.8 mg/dL). Further, these hybrids underwent in vivo cardioprotective and ECG studies, which demonstrated that 242 and 243 were more effective in maintaining the cardiac function thereby preventing diabetic cardiomyopathy (Table 3, Table 4) [111].
SAR of acid-based TZD analogs
The lowering of blood glucose levels was more when substitution with o-phenoxy acetic acid in comparison to p-substitution. The activity was lowered as the number of methoxy or alkyl groups increases. The presence of cinnamic acid double bond as well as the geometry of the molecule plays an important role in PPAR agonism [110], [111] (Fig. 10A).
Fig. 10A.

SAR of acid-based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Benzylidene based TZDs
Jiwane et al. carried out the reaction on compound 245 with different dialkyl/diarylamines (246) and formaldehyde in the presence of DMF to yield the final product 3-dialkyl/diaryl amino methyl-5-(o/p-substituted benzylidine)-TZDs (247a and b) (S44). All the synthesized compounds were screened for their glucose-lowering ability in dexamethasone (1 mg/kg, for 5 days)-induced diabetic rats. Compounds were administered at a dose of 50 mg/kg and the results were compared against rosiglitazone. It was found that compounds (247a and b) showed significant reduction in blood glucose level (58 and 65%), whereas rosiglitazone reduced blood glucose level up to 88%, which suggested that substitution with α-amino methyl group at 3rd position of TZD showed variations in activity in comparison to rosiglitazone (88%) (Table 4) [112].
Avupati et al. synthesized some novel 2,4-TZDs as shown in S45. Reaction of (Z)-4-((2,4-dioxo-1,3-thiazolidin-5-ylidene)methyl)benzaldehyde (248) was carried out by base-catalyzed condensation with substituted aromatic/heteroatomic ketones to yield the final compound, (Z)-5-(4-((Z)-3-(9H-fluoren-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-TZD (249). The title compounds were then screened for antihyperglycemic activity in STZ-NA-induced diabetic rat model. The compounds were administered at different doses (10, 30 and 50 mg/kg) for 1 day and the results were compared with rosiglitazone (10, 30 and 50 mg/kg). The results showed that compound 249 exhibited dose-dependent reduction in plasma glucose levels (39.83%, 44.62% and 52.81% for 10, 30 and 50 mg/kg, respectively) in comparison to rosiglitazone (38.57%, 14.83% and 12.74%, respectively). Further, molecular docking studies were carried out against PPAR-γ (PDB ID: 3CS8) and the compound 249 showed −170 dock score as compared to rosiglitazone (−131), which suggested that compound 249 exhibited better binding affinity towards PPAR-γ (Table 4) [113].
Patil et al. synthesized various TZD derivatives as shown in S46. The first step was to synthesize (Z)-5-(4-chlorobenzylidene)-TZD (250) by the Knoevenagel condensation followed by refluxing with metformin (251) in the presence of K2CO3 to give the final derivative, (Z)-N1-(4-((2,4-dioxothiazolidin-5-ylidene)methyl)phenyl)-N2,N2-dimethylhydrazine-1,2-bis(carboximidamide) (252). The final derivatives were first evaluated for acute oral toxicity study at a dose of 30 and 100 mg/kg for 2 days and as a result; compounds did not show any signs of toxicity even at 100 mg/kg. So, compounds were further screened for antidiabetic activity in alloxan-induced diabetic rats at 100 mg/kg for 1 day (acute study) and 14 days (subacute study). The effects were observed for the reduction in blood glucose levels in comparison to metformin (100 mg/kg) and pioglitazone (100 mg/kg). The results from the acute study revealed that compound 252 exhibited a significant reduction in blood glucose level (231.4 mg/dL) similar to that of pioglitazone (232.2 mg/dL). The results from the subacute study displayed that compound 252 showed a 65% reduction in blood glucose level to the value close to that of pioglitazone (69%). The most potent compound from the synthesized derivatives was found to be 252, which showed nearly the same activity as that of the standard compound (pioglitazone) (Table 4) [114].
Patel et al. reported a series of novel 5-[4-(substituted) benzylidine]thiazolidine-2,4-dione along with the evaluation of an antidiabetic activity. Initially, TZD (3) was synthesized by 1, 3 dipolar cycloaddition of thiourea (1) and chloroacetic acid (2) in presence of water (S1). Next, 5-(4-chlorobenzylidene)-2,4-thiazolidinedione (250) was synthesized through Knoevenagel’s condensation. The title compounds 5-[4-(substituted) benzylidene]-2.4-thiazolidinediones (254a-c) were prepared by microwave-assisted reaction of 5-(4-chlorobenzylidene)-2,4-thiazolidinedione (250) with substituted primary aromatic amines (253) in presence of K2CO3 and acetonitrile (S47). The synthesized compounds (30 mg/kg) were evaluated for antidiabetic activity in male Wistar rats through OGTT using pioglitazone (30 mg/kg) as an internal standard. Compounds 254a–c exhibited potent antidiabetic activity (100–120 mg/dL) similar to pioglitazone (100 mg/dL) (Table 4) [115].
Duhart et al. carried out the in-silico study of 130 TZD derivatives, out of which only two were selected and synthesized in a solvent-free environment as shown in S48. Knoevenagel condensation was carried out between TZD (3) and variously substituted aldehydes (255) to achieve the desired 5-arylidene-2,4-thiazolidinediones (256a and b). The synthesized compounds were then tested for acute oral toxicity (14 days) in female Wistar rats by administering the compounds (256a and b) at a dose of 175, 350, 700, 1400 and 2000 mg/kg in ethanol. As a result, compounds (256a and b) showed normal behavior and no physical changes were seen but showed fat deposition in the abdominal cavity at a dose ≥350 mg/kg. Further, the same compounds were evaluated in the same model at the same doses but in a different vehicle (dimethyl sulfoxide). The results showed that compounds at a dose of 700 mg/kg exhibited severe tiredness along with sedation. One animal at a dose of 1400 mg/kg displayed lethargy for 5 h and hypnosis up to 10 h leading to coma and also showed stomach fundus hardening. Further, the animals receiving 2000 mg/kg were dead within 5 h post-administration on day 1 and showed stomach fundus hardening. So, the doses considered to be safe were 175–1400 mg/kg (Table 4) [116].
Rekha and Chandrashekhara synthesized a series of 5-[4-(substituted) benzylidene TZDs based on the 2D QSAR studies. Knoevenagel product (257) upon reaction with 1,4-dibromobutane (258) in presence of NaH/DMSO gave another intermediate (259) followed by reaction with various cyclic amines in K2CO3 afforded the final compounds, 5-(4-(4-(piperdin-1-yl)butoxy)benzylidene)-TZD (260a) and 5-(4-(4-(cyclohexylamino)butoxy)benzylidene)-TZD (260b) (S49). The final derivatives were then screened for anti-hyperglycemic effect in dexamethasone (0.7 mg/kg, i.m.) induced-diabetic rat model by administering the compounds at a dose of 0.72 mg/kg for 10 days. As a result, compounds (260a and b) showed a sudden lowering of blood glucose level within 30 min and then showed a constant decrease while the standard drug rosiglitazone showed a decrease in blood glucose level in 30 min (Table 4) [117].
SAR of benzylidene based TZD analogs
The substitution of R with electron-donating groups resulted in a better antidiabetic activity. Substitution of phenyl ring with fluorene moiety resulted in a significant increase in activity in comparison to other aromatic rings (pyridin-2-yl, naphthalene-2-yl). Substitution with bis-guanidine moiety at R position enhanced the activity due to H-bonding with the active site. Substitution on the aromatic ring at the 2nd and/or 4th position with an electron releasing group of the lipophilic site shows good antidiabetic activity than any other position [114], [115], [116] (Fig. 10B).
Fig. 10B.

SAR of benzylidene based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Benzofused TZDs
Reddy et al. synthesized several TZD derivatives having 5-hydroxy-2,3-dihydro-2,2,4,6,7-pentamethylbenzofuran moieties and their saturated analogs as shown in S50. The mesylate (261) was heated with (S)-prolinol to give pyrrolidine derivative (262) which upon reaction with thionyl chloride gave 3-chloropiperidine derivative (263). Compound 263 on reaction with 4-hydroxybenzaldehyde (6) yielded a mixture of 5- and 6-membered ring products (266 and 267). Finally, the mixture of aldehydes (266 and 267) was condensed with TZD to furnish a mixture of unsaturated compounds (269a and 270a) followed by reflux with AcOH-HCl to furnish the debenzylated products (269b and 270b). The saturated TZD analog (268a) was synthesized by treating prolinol derivative (262) with 4-fluoronitrobenzene to give nitro compound (264) followed by reduction gave the amino derivative (265), which upon reaction with TZD gave one of the final compounds (268a and b). These moieties were then evaluated for their euglycemic and hypolipidemic activity in db/db mice. The synthesized derivatives were administered at a dose of 100 mg/kg for 6 days and the reduction in plasma glucose and TG level was compared with troglitazone (200 mg/kg). The results showed that compound 269a with benzyl protecting group exhibited the most potent plasma glucose (66%) and TG (52%) lowering activities whereas, removing the benzyl protecting group (269b) showed poorer plasma glucose (19%) and TG (not active) lowering activities. The related trend was also seen for saturated TZD analogs (268a and b) but less efficient than compounds 269a and b. As a result, compounds (268a, 269a and 269b) were further evaluated in the same model at 30 mg/kg for 6 days. It was found that compounds 268a and 269a exhibited a good reduction in plasma glucose and TG level but troglitazone was not active at 30 mg/kg. After that, salt forms were prepared for the compounds 268a (maleate) and 269a (maleate and HCl) and the results showed that compound 269a maleate form was the most potent in lowering blood glucose (45%) and TG (42%) level. Further, the dose-dependent study was carried out using 269a maleate (30 and 100 mg/kg) and troglitazone (30, 100, 200 and 800 mg/kg) for 11 days. The results showed that compound 269a maleate reduced 70% plasma glucose level at 100 mg/kg whereas troglitazone even at 800 mg/kg showed only 52% plasma glucose reduction. Then, compounds (268a and b, 269a and b) were evaluated for GAL4-PPAR transactivation by luciferase assay and it was found that none of the compounds was efficient to activate PPAR-α and PPAR-γ as compared to troglitazone. The overall results conveyed that compound, 5-[4-[N-[3(R/S)-5-benzyloxy-2,3-dihydro-2,2,4,6,7-pentamethylbenzofuran-3-ylmethyl]-(2S)-pyrrolidin-2-ylmethoxy]phenylene]-TZD (269a) was the most potent and efficacious compound as compared to other synthesized derivatives (Table 4) [118].
Jeon and Park prepared TZDs containing benzoxazole moiety with different alkyl substituents. Initially, acetylation of saturated Knoevenagel product (271) was carried out with acetic anhydride to yield an intermediate (272). The compound 272 on N-tritylation gave compound 273 followed by deprotection of acetyl group, gave a tritylated derivative (274). Furthermore, 2-chlorobenzoxazole (275) was reacted with substituted alkyl-amino alcohol (91) to give an intermediate (276), which upon Mitsunobu reaction with derivative (274) furnished compound 277 followed by deprotection of trityl group in the presence of TFA gave the final derivative, 5-(3-(2-(benzo[d]oxazol-2-yl(methyl)amino)ethoxy)benzyl)TZDs (278) (S51). The synthesized derivatives were then screened for PPARα and PPARγ transactivation assay using CV-1 cells. As a result, compound 278 showed 113.2% PPARγ activation while reference standard (GW409544) showed 100% activation (Table 3) [119].
Pattan et al. synthesized a new series of 2-amino[5′(4-sulphonylbenzylidine)-2,4-TZD]-7-chloro-6-fluorobenzothiazole (283) as shown in S52. Initially, 2-amino-6-fluro-7-chlorobenzothiazole (280) was synthesized from 3-chloro-4-fluroaniline (279) in the presence of potassium thiocyanate. Furthermore, condensation of compound 280 and 281 was done in the presence of pyridine and acetic anhydride to synthesize derivative 282. Moreover, compound 282 was reacted with substituted aniline to achieve the final derivatives (283a-c). Of note, compound 281 was synthesized by carrying out the chlorosulfonation of benzylidene-TZD. The final derivatives were then evaluated for their antidiabetic activity in alloxan-induced diabetic rats at a dose of 36 mg/kg for one day. It was found that out of all the synthesized derivatives, only three compounds 283a–c showed the maximum antidiabetic activity in terms of blood glucose-lowering activity (116–123 mg/dL) (Table 4) [120].
Jeon et al. carried out a modified Mitsunobu reaction to synthesize benzothiazole derivatives of TZDs as shown in S53. 2-chlorobenzothiazole (284) was reacted with substituted alkylaminoethanols (91) to yield amino alcohols (285). Then, Mitsunobu reaction of 285 was carried out with 5-(4-hydroxybenzyl)TZD (274) in the presence of ADDP and tributylphosphine to yield compound 286 followed by removal of trityl group with the help of TFA furnished the final compound, 5-(4-(2-(benzo[d]thiazol-2-yl(methyl)amino)ethoxy)benzyl)TZD (287). The synthesized compounds were evaluated for PPAR transactivation assay and anti-inflammatory activity via NO production using CV-1 cells and RAW 264.7 cells, respectively. As a result, compound 287 which was substituted with methyl on exocyclic nitrogen showed 120% PPARγ activation as compared to standard i.e., GW409544 (100%) but compound 287 showed lowest anti-inflammatory activity (Table 3) [121].
Purohit and Veerapur carried out the designing, characterization and molecular docking of twelve benzisoxazole containing TZDs. Based on the molecular docking studies carried out against PPARγ (PDB ID: 2PRG) and Lipinski’s rule of five, nine compounds were selected and synthesized as shown in S54. 2-((3-phenylbenzo[c]isoxazol-5-yl)(propyl)amino)ethan-1-ol (290) was synthesized by stirring the mixture of 5-chloro-3-phenyl-2,1-benzisoxazole (288), 2-substituted aminoethanol (289) and triethylamine in THF followed by reaction with 5-arylidene TZD in tributylphosphine to get the final product (291). The final compounds were then screened for antidiabetic activity in alloxan-induced diabetic-mice at a dose of 30 mg/kg for one day. As a result, compound 291 exhibited the most potent activity in terms of reducing the serum glucose level (−30.62%) than the other synthesized derivatives and the standard drug rosiglitazone (−17.24%) (Table 4) [122].
SAR of benzofused TZD analogs
The substitution of exocyclic nitrogen with methyl enhances PPAR-γ activation. However, on increasing the N-alkyl chain, the activity of TZD analogs decreases; whereas, replacement of benzofuran with benzoxazole or benzisoxazole or benzothiazole selectively activates PPAR-γ [118], [119], [120], [121], [122] (Fig. 11A).
Fig. 11A.

SAR of benzofused TZD analogs (structures are original and made by using chem draw ultra 12.0).
Chromones based TZDs
Unlusoy et al. synthesized a series of (Z)-3-methyl-5-((6-methyl-4-oxo-4H-chromen-3-yl)methylene)TZDs (294) in order to improve the pharmacological index of insulinotropic activities. The Knoevenagel condensation of 3-formyl chromone (292) with 2,4-TZD (3) yielded 6-methylchromonyl-TZD (293) followed by alkylation with alkyl iodide to furnish the final compound (294) (S55). The synthesized compounds were screened for in vitro insulin-releasing activity using INS-1 cells at different concentrations (0.001 and 0.01 mg/mL). The results showed that among all the synthesized derivatives, the compound 294 was found to be the most potent at both the concentrations (0.001 mg/mL = 120.6% and 0.01 mg/mL = 152%) in terms of releasing insulin and the results were comparable to glibenclamide (145.7% at 0.001 mg/mL) (Table 3) [123].
Nazreen et al. synthesized a number of chromones based TZD derivatives as shown in S56. The compound 295 was used to synthesize 3-formyl chromones (296), which underwent Knoevenagel condensation with TZD (3) yielded chromonyl-2,4-TZDs (297) followed by catalytic hydrogenation to give the final products, 5((6-methoxy-4-oxo-4H-chromen-3-yl)methyl)TZD (298a) and 5((6-chloro-4-oxo-4H-chromen-3-yl)methyl)TZD (298b) along with by-products (299a and b). The synthesized compounds were first docked against PPAR-γ (PDB ID: 3CS8) and the compounds 298a and b showed −7.57 and −7.76 dock score in comparison to that of rosiglitazone (−5.77). Further, the compounds were evaluated for STZ-induced diabetic rats for 15 days. Compounds 298a and b showed 140% and 135.5% reduction in blood glucose level, respectively as compared to pioglitazone (120%) and rosiglitazone (122%). Compound 298b was further evaluated for body weight gain for 15 days, as a result, compound 298b did not show any significant change in body weight, which suggested it showed weight neutral effects. Then, the hepatotoxicity study was carried out with compounds (298a and b) and as a result, compound 298a and b came out to be most potent in terms of lowering the levels of AST, ALT, and ALP and did not cause any toxic effect to the liver. However, pioglitazone caused mild dilation of sinusoidal spaces. Since, the majority of drugs used for arrhythmia have been withdrawn due to their ability to cause prolongation of QT interval via blockade of human ether-a-go-go-related gene (hERG), which may lead to syncope and sudden death. Therefore, the compound 298b was evaluated to ensure whether it has any effect on QT prolongation or not and it was found that it did not cause cardiotoxicity because IC50 was found to be 135 μM. Further, compound 298a and b were evaluated for PPAR-γ gene expression using 3T3-L1 cells and the results showed that compound 298b significantly increased the PPAR-γ gene expression (45%) in comparison to that of pioglitazone (60%) and rosiglitazone (82%) and also increased the levels of GLUT1 and GLUT4 (Table 3, Table 4) [124].
SAR of chromones based TZD analogs
The substitution of the N-3 position of TZD with lipophilic groups resulted in an increased insulin-releasing activity. Reducing the olefinic bonds of chromone ring resulted in a reduced antidiabetic activity. Substitution on chromone ring at sixth position with halogens resulted in more potent compounds as compared to substitution on other positions [123], [124] (Fig. 11B).
Fig. 11B.

SAR of chromones based TZD analogs (structures are original and made by using chem draw ultra 12.0).
Miscellaneous targets
Hidalgo-Figueroa et al. carried out the synthesis of TZD derivatives as dual PPAR-α/γ modulator as shown in S57. The compound 300 and 302 underwent Knoevenagel condensation with TZD to produce corresponding derivatives (301 and 303). Of note, compounds 300 and 302 were synthesized from 4-bromomethylbiphenyl-2-carbonitrile and ethylbromoacetate with 4-hydroxybenzaldehyde, respectively. Subsequently, synthesized compounds were evaluated for the relative expression of PPAR-α and PPAR-γ using 3T3-L1 cells. As a result, compound 301 significantly increased the levels of PPAR-γ, PPAR-α and GLUT4. However, compound 303 lacks the activity. Then, compound 301 was docked against PPAR-α and PPAR-γ (PDB ID: 1I7G and 1I7I, respectively) and as a result, it gets bind into the active site of both isoforms (α andγ). After that, compound 301 was evaluated for in vivo antidiabetic effects in STZ-NA induced diabetic rats at a dose of 50 mg/kg body weight and the results were compared against glibenclamide (5 mg/kg). It was found that compound 301 decreased 32.36% glycemia and the results were comparable with glibenclamide (43.6%) (Table 3, Table 4) [125].
Navarrete-Vázquez et al. carried out the synthesis of (Z)-2-(4-(2-(4-((2,4-dioxothiazolidin-5-ylidene)methyl)phenoxy)acetamido)phenoxy)acetic acid (307) and evaluated in vivo for the relative expression of PPAR-γ, GLUT-4 and PPAR-α. Initially, Knoevenagel condensation of α-chloroacetamide (304) with 4-hydroxybenzladehyde (6) gave ether-aldehyde (305), which on reaction with TZD (3) yielded compound 306 followed by ethyl ester hydrolysis to give the final derivative (307) (S58). Subsequently, compounds (306 and 307) were evaluated in vitro for PPARs and GLUT4 expression using 3T3-L1 cells. As a result, compounds (306 and 307) both increased (2-folds) the relative expression of PPAR-γ and GLUT-4. However, no change was observed in the expression of PPAR-α. Successively, compound 306 (ester prodrug) was evaluated in vivo in STZ-NA induced diabetic rat model at a dose of 50 mg/kg by keeping glibenclamide (5 mg/kg) as a reference drug. It was found that compound 306 decreased 31% glycemia and the results were comparable with glibenclamide (43.6%). Next, the compound 307 was docked against the PPAR-γ (PDB ID: 1I7I) and as a result, the compound showed important interactions with residues Ser289, His323 and His449 in the active site. The compound 307 has been developed as a potential lead molecule for the treatment of diabetes (Table 3, Table 4) [126].
Hidalgo-Figueroa et al. designed two TZD-based derivatives as shown in S59 and evaluated them as antihyperglycemic agents. The final derivatives (311 and 312) were synthesized by reacting Knoevenagel product (308) with intermediates (309 and 310, respectively) in basic medium. Subsequently, the compounds were evaluated in vitro as PTP1B inhibitors at 20 µM and as a result, compound 311 decreased the enzyme activity up to 85% whereas compound 312 reduced the activity up to 50%. Therefore, the most active inhibitor was found to be compound 311 on which further concentration–response test has been performed. As a result, compound 311 had shown IC50 value of 9.6 ± 0.5 μM. In addition, the in vivo (STZ-NA induced diabetic rats) activity was performed for compound 311 at a dose of 50 mg/kg body weight and the reference drug was glibenclamide (5 mg/kg). It was found that compound 311 decreased 34% glycemia and the results were comparable with glibenclamide (43.6%). Furthermore, molecular docking studies were carried out against PTP-1B (PDB ID: 1C83) for both compounds 311 and 312. The compound 311 shown to have the highest affinity with PTP1B in comparison to compound 312, having a free binding energy of −8.94 Kcal/mol and −8.04 Kcal/mol, respectively (Table 3 and4) [127].
Conclusion and future perspectives
T2DM is considered as one of the major risk factors for cardiovascular morbidity and mortality. TZDs are reported to increase the transactivation of PPARs thereby, reduce insulin resistance (i.e., reduce gluconeogenesis and increase utilization of glucose and lipid metabolism in the peripheral tissues), which in turn leads to improve the effect of endogenous insulin to maintain the level of blood glucose. Unfortunately, clinically used TZD class of medications suffered from various serious side effects like hepatotoxicity, edema (fluid retention) and weight gain as a result of troglitazone and rosiglitazone were banned and the pioglitazone has shown to increase the risk of bladder cancer. This review emphasizes TZDs not only as a fortunate and potential scaffold in the field of medicinal chemistry but also outlined the chemistry and biological activities of the TZDs scaffold as antidiabetic agents. The synthetic methodologies signify simplicity and versatility, which offer the medicinal chemist to discover a complete range of novel derivatives. The study also highlighted the SAR studies as well as molecular docking studies in order to carry out future studies on this moiety. Based on this review report, pyrazole, chromone, and acid-based TZD impair the side effects and significantly reduce the blood glucose level than that of clinically used TZDs. Moreover, studies on various approaches such as virtual screening, in-silico drug design, docking etc. can be utilized to develop this class medication for targeting other molecular targets of diabetes to avoid unwanted side effects. Hence, this review will be valuable for the scientific world to develop lead compounds or clinical candidates in various biological areas. Future investigations of pyrazole, chromone, and acid-based TZD scaffold are warranted on other molecular targets of TZD, which can give us more encouraging results. Based on the available study results, TZDs can be considered as one of the promising classes of compounds that can overcome problems of the clinically used TZDs in the management of diabetes.
Compliance with ethics requirement
This article does not contain any studies with human or animal subjects.
Declaration of Competing Interest
The author has declared no conflict of interest.
Acknowledgement
This work was supported by ISF College of Pharmacy, Moga, Punjab, India.
Biographies

Garima Bansal was born in 1995 in the Moga district of Punjab, India. She has completed her bachelor’s degree from one of the top 5 best pharmacy colleges among India, ISF College of Pharmacy, Moga. She has completed her Master’s degree at ISF College in Pharmaceutical Chemistry, where she is working on the synthesis of thiazolidine-2,4-diones as anti-diabetic agents. Her area of interest is the development of new reactions and methodologies for various derivatives.

Dr. Punniyakoti Veeraveedu Thanikachalam worked as Professor-cum-HOD in the department of pharmaceutical chemistry, ISF College of Pharmacy, Moga, Punjab, India. He holds a Ph.D. in Pharmaceutical Sciences from the Niigata University of Pharmacy and Applied Life Sciences, Niigata Japan. His Professor career has included 7.9 years in academia and 6.1 years in the research field as a Postdoctoral fellow in Japan. He is a recipient of the prestigious JSPS Postdoctoral fellowship, Japan. He has published several research papers in peer-reviewed journals and presented his work in various scientific conferences of international and national repute. He has received various research grants from Malaysia while working at International Medical University, Kuala Lumpur. He is also a reviewer of various scientific journals. He has successfully guided several undergraduate, graduate and Ph.D. students in their research. His area of interest is drug discovery in diabetes, cardiovascular biology, and cancer.

Dr. Rahul K. Maurya earned his Ph.D. in Medicinal Chemistry in 2019 from CDRI Lucknow, India. He is an Assistant Professor in Amity Institute of Pharmacy, Amity University Uttar Pradesh, Lucknow Campus, India. He has received CSIR-CDRI incentive award in 2016. His career has included 6.0 years in academia and 6.0 years in research. He has published research papers in peer-reviewed journals and presented research papers in conferences of national and international repute. His area of research interest focuses on the design and synthesis of functionalized Indoles, indazoles, TZD, isoxazole and novel nitrogen-containing heterocyclic compounds for the treatment of diabetes, and infectious diseases.

Dr. Pooja Chawla is working as Professor, Department of Pharmaceutical Chemistry, ISF College of Pharmacy Moga, India. She has 18 years of experience in the research, discovery and development of biologically active pharmaceuticals. Dr. Chawla holds a Ph D degree in Medicinal Chemistry and has published 3 books and more than 45 papers in national/international journals and is serving as a reviewer for international journals of repute. She also presented a poster at FIP 2011 and CTDDR 2013 and delivered an oral talk at World Chemistry Congress, Dubai 2019. She has guided 47 post graduate projects and her research team consists of 3 Master and 2 doctoral research scholars. Her area of research interest focuses on the design and synthesis of various 4-Thiazolidinone and TZD derivatives for the treatment of diabetes, infectious diseases and inflammation.

Dr. Srinivasan Ramamurthy is an Assistant Professor in the College of Pharmacy & Health Sciences, Ajman University, Fujairah, UAE. He holds a Ph.D. in Pharmaceutical Chemistry and his professional career has included 12 years in academia and 4 years in the Pharmaceutical Industry. He has published several research papers in peer-reviewed journals and presented research papers in conferences of national and international repute. He is also a reviewer of various scientific journals. He has successfully supervised several undergraduate and graduate students in their research.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2020.01.008.
Contributor Information
Punniyakoti Veeraveedu Thanikachalam, Email: nspkoti2001@gmail.com.
Pooja Chawla, Email: pvchawla@gmail.com.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.IDF diabetes atlas - Home 2017. http://www.diabetesatlas.org/ (accessed March 8, 2019).
- 2.Rotella D.P. Novel “second-generation” approaches for the control of Type 2 diabetes. J Med Chem. 2004;47:4111–4112. doi: 10.1021/jm030626a. [DOI] [PubMed] [Google Scholar]
- 3.Sucheta Tahlan S., Verma P.K. Biological potential of thiazolidinedione derivatives of synthetic origin. Chem Cent J. 2017;11:130. doi: 10.1186/s13065-017-0357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pattan S.R., Kekare P., Patil A., Nikalje A., Kittur B.S. Studies on the synthesis of novel 2, 4-thiazolidinedione derivatives with antidiabetic activity. Iran J Pharm Sci. 2009;5:225–230. [Google Scholar]
- 5.Diabetes control and complications trial research group. Nathan D.M., Genuth S., Lachin J., Cleary P., Crofford O., et al. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 6.Saltiel A.R., Olefsky J.M. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes. 1996;45:1661–1669. doi: 10.2337/diab.45.12.1661. [DOI] [PubMed] [Google Scholar]
- 7.Naim M.J., Alam M.J., Ahmad S., Nawaz F., Shrivastava N., Sahu M., et al. Therapeutic journey of 2,4-thiazolidinediones as a versatile scaffold: An insight into structure activity relationship. Eur J Med Chem. 2017;129:218–250. doi: 10.1016/j.ejmech.2017.02.031. [DOI] [PubMed] [Google Scholar]
- 8.Kaur Manjal S., Kaur R., Bhatia R., Kumar K., Singh V., Shankar R., et al. Synthetic and medicinal perspective of thiazolidinones: A review. Bioorg Chem. 2017;75:406–423. doi: 10.1016/j.bioorg.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 9.Tomasic T., Masic L. Rhodanine as a privileged scaffold in drug discovery. Curr Med Chem. 2009;16:1596–1629. doi: 10.2174/092986709788186200. [DOI] [PubMed] [Google Scholar]
- 10.Ahmadi A., Khalili M., Samavat S., Shahbazi E., Nahri-Niknafs B. Synthesis and evaluation of the hypoglycemic and hypolipidemic activity of novel arylidene thiazolidinedione analogson a type 2 diabetes model. Pharm Chem J. 2016;50:165–171. [Google Scholar]
- 11.Day C. Thiazolidinediones: a new class of antidiabetic drugs. Diabet Med. 1999;16:179–192. doi: 10.1046/j.1464-5491.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- 12.Nanjan M.J., Mohammed M., Prashantha Kumar B.R., Chandrasekar M.J.N. Thiazolidinediones as antidiabetic agents: A critical review. Bioorg Chem. 2018;77:548–567. doi: 10.1016/j.bioorg.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Soccio R.E., Chen E.R., Lazar M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20:573–591. doi: 10.1016/j.cmet.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blonde L., Dagogo-Jack S., Banerji M.A., Pratley R.E., Marcellari A., Braceras R., et al. Comparison of vildagliptin and thiazolidinedione as add-on therapy in patients inadequately controlled with metformin: Results of the GALIANT trial - a primary care, type 2 diabetes study. Diabetes, Obes Metab. 2009;11:978–986. doi: 10.1111/j.1463-1326.2009.01080.x. [DOI] [PubMed] [Google Scholar]
- 15.TriMaster: study of a DPP4 inhibitor, SGLT2 inhibitor and thiazolidinedione as third line therapy in patients with type 2 diabetes n.d. https://clinicaltrials.gov/ct2/show/NCT02653209 (accessed March 8, 2019).
- 16.Effects of fats on blood glucose in people with and without type 2 diabetes mellitus n.d. https://clinicaltrials.gov/ct2/show/NCT00308373 (accessed March 8, 2019).
- 17.Fracture risk with thiazolidinediones n.d. https://clinicaltrials.gov/ct2/show/NCT01055223 (accessed March 8, 2019).
- 18.Bergenstal R.M., Wysham C., MacConell L., Malloy J., Walsh B., Yan P., et al. Efficacy and safety of exenatide once weekly versus sitagliptin or pioglitazone as an adjunct to metformin for treatment of type 2 diabetes (DURATION-2): A randomised trial. Lancet. 2010;376:431–439. doi: 10.1016/S0140-6736(10)60590-9. [DOI] [PubMed] [Google Scholar]
- 19.Perségol L., Vergès B., Foissac M., Gambert P., Duvillard L. Inability of HDL from type 2 diabetic patients to counteract the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia. 2006;49:1380–1386. doi: 10.1007/s00125-006-0244-1. [DOI] [PubMed] [Google Scholar]
- 20.Comparison of lobeglitazone with pioglitazone as initial triple therapy for diabetes management n.d. https://clinicaltrials.gov/ct2/show/NCT02315287 (accessed March 8, 2019).
- 21.Effect of different interventions on glycemic control and β-cell function in newly diagnosed type 2 diabetic patients n.d. https://clinicaltrials.gov/ct2/show/NCT01147627 (accessed March 8, 2019).
- 22.Vaccaro O., Masulli M., Nicolucci A., Bonora E., Del Prato S., Maggioni A.P., et al. Effects on the incidence of cardiovascular events of the addition of pioglitazone versus sulfonylureas in patients with type 2 diabetes inadequately controlled with metformin (TOSCA.IT): a randomised, multicentre trial. Lancet Diabetes Endocrinol. 2017;5:887–897. doi: 10.1016/S2213-8587(17)30317-0. [DOI] [PubMed] [Google Scholar]
- 23.Hollander P., Yu D., Chou H.S. Low-dose rosiglitazone in patients with insulin-requiring type 2 diabetes. Arch Intern Med. 2007;167:1284–1290. doi: 10.1001/archinte.167.12.1284. [DOI] [PubMed] [Google Scholar]
- 24.Comparison of the effects of thiazolidinediones(TZD), sodium- glucose cotransporter 2 inhibitors(SGLT2i) alone and TZD / SGLT2i combination therapy on non-alcoholic fatty liver disease in type 2 diabetic patients with fatty liver n.d. https://clinicaltrials.gov/ct2/show/NCT03646292 (accessed March 8, 2019).
- 25.Comparison of pioglitazone versus glimepiride in type 2 diabetes inadequately controlled with metformin plus alogliptin n.d. https://clinicaltrials.gov/ct2/show/NCT02426294 (accessed March 8, 2019).
- 26.A trial of TT223 in patients with type 2 diabetes who are taking metformin and/or thiazolidinedione n.d. https://clinicaltrials.gov/ct2/show/NCT00743002 (accessed March 8, 2019).
- 27.AVANDIA with glyburide in African American and hispanic patients with type 2 diabetes not controlled by glyburide alone n.d. https://clinicaltrials.gov/ct2/show/NCT00333723 (accessed March 9, 2019).
- 28.A study to evaluate the effectiveness and safety initiation and titration of insulin glargine (U300) in insulin-naïve patients with type 2 diabetes mellitus (T2DM) controlled on oral antidiabetic drug treatment in Turkey n.d. https://clinicaltrials.gov/ct2/show/NCT02954692 (accessed March 8, 2019).
- 29.Azoulay L., Filion K.B., Platt R.W., Dahl M., Dormuth C.R., Clemens K.K., et al. Incretin based drugs and the risk of pancreatic cancer: International multicentre cohort study. BMJ. 2016;352:i581. doi: 10.1136/bmj.i581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evaluate the glycemic control of CKD-501 in type 2 diabetes mellitus n.d. https://clinicaltrials.gov/ct2/show/NCT01030679 (accessed March 8, 2019).
- 31.Esteghamati A., Ghasemiesfe M., Mousavizadeh M., Noshad S., Nakhjavani M. Pioglitazone and metformin are equally effective in reduction of chemerin in patients with type 2 diabetes. J Diabetes Investig. 2014;5:327–332. doi: 10.1111/jdi.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tripathy D., Daniele G., Fiorentino T.V., Perez-Cadena Z., Chavez-Velasquez A., Kamath S., et al. Pioglitazone improves glucose metabolism and modulates skeletal muscle TIMP-3–TACE dyad in type 2 diabetes mellitus: A randomised, double-blind, placebo-controlled, mechanistic study. Diabetologia. 2013;56:2153–2163. doi: 10.1007/s00125-013-2976-z. [DOI] [PubMed] [Google Scholar]
- 33.Rosiglitazone-metformin combination versus metformin-sulfonylurea combination on beta-cell function in type 2 diabetes n.d. https://clinicaltrials.gov/ct2/show/NCT00367055 (accessed March 8, 2019).
- 34.Azoulay L., Filion K.B., Platt R.W., Dahl M., Dormuth C.R., Clemens K.K., et al. Association between incretin-based drugs and the risk of acute pancreatitis. JAMA Intern Med. 2016;176:1464–1473. doi: 10.1001/jamainternmed.2016.1522. [DOI] [PubMed] [Google Scholar]
- 35.A study of LY2189265 in Japanese participants with type 2 diabetes mellitus n.d. https://clinicaltrials.gov/ct2/show/NCT01468181 (accessed March 8, 2019).
- 36.Esteghamati A., Afarideh M., Feyzi S., Noshad S., Nakhjavani M. Comparative effects of metformin and pioglitazone on fetuin-A and osteoprotegerin concentrations in patients with newly diagnosed diabetes: A randomized clinical trial. Diabetes Metab Syndr. 2015;9:258–265. doi: 10.1016/j.dsx.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 37.Kadowaki T., Kondo K. Efficacy and safety of teneligliptin in combination with pioglitazone in Japanese patients with type 2 diabetes mellitus. J Diabetes Investig. 2013;4:576–584. doi: 10.1111/jdi.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdul-Ghani M., Migahid O., Megahed A., Adams J., Triplitt C., DeFronzo R.A., et al. Combination therapy with exenatide plus pioglitazone versus basal/bolus insulin in patients with poorly controlled type 2 diabetes on sulfonylurea plus metformin: The qatar study. Diabetes Care. 2017;40:325–331. doi: 10.2337/dc16-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kadoglou N.P.E., Tsanikidis H., Kapelouzou A., Vrabas I., Vitta I., Karayannacos P.E., et al. Effects of rosiglitazone and metformin treatment on apelin, visfatin, and ghrelin levels in patients with type 2 diabetes mellitus. Metabolism. 2010;59:373–379. doi: 10.1016/j.metabol.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 40.Okuda I., Wilson T.H., Yue L., Nakajima H., Carr M.C., Tsuboi M., et al. Albiglutide, a weekly GLP-1 receptor agonist, improves glycemic parameters in Japanese patients with type 2 diabetes over 1 year when added to single oral antidiabetic drugs. Curr Med Res Opin. 2017;33:431–438. doi: 10.1080/03007995.2016.1261817. [DOI] [PubMed] [Google Scholar]
- 41.Asnani S., Richard B.C., Desouza C., Fonseca V. Is weight loss possible in patients treated with thiazolidinediones? Experience with a low-calorie diet. Curr Med Res Opin. 2003;19:609–613. doi: 10.1185/030079903125002306. [DOI] [PubMed] [Google Scholar]
- 42.Nishio K., Shigemitsu M., Kodama Y., Konno N., Katagiri T., Kobayashi Y. Comparison of bare metal stent with pioglitazone versus sirolimus-eluting stent for percutaneous coronary intervention in patients with type 2 diabetes mellitus. Cardiovasc Revascularization Med. 2009;10:5–11. doi: 10.1016/j.carrev.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 43.Lee Y., Kim J.H., Kim S.R., Jin H.Y., Rhee E.-J., Cho Y.M., et al. Lobeglitazone, a novel thiazolidinedione, improves non-alcoholic fatty liver disease in type 2 diabetes: Its efficacy and predictive factors related to responsiveness. J Korean Med Sci. 2017;32:60–69. doi: 10.3346/jkms.2017.32.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vascular effects of rosiglitazone versus glyburide in type 2 diabetic patients n.d. https://clinicaltrials.gov/ct2/show/NCT00123643 (accessed March 8, 2019).
- 45.Fatty liver study in patients with type II diabetes n.d. https://clinicaltrials.gov/ct2/show/NCT02365233 (accessed March 8, 2019).
- 46.Placebo controlled dose-response study of rivoglitazone in type 2 diabetes n.d. https://clinicaltrials.gov/ct2/show/NCT00575471 (accessed March 8, 2019).
- 47.Filion K.B., Azoulay L., Platt R.W., Dahl M., Dormuth C.R., Clemens K.K., et al. A multicenter observational study of incretin-based drugs and heart failure. N Engl J Med. 2016;374:1145–1154. doi: 10.1056/NEJMoa1506115. [DOI] [PubMed] [Google Scholar]
- 48.Punthakee Z., Bosch J., Dagenais G., Diaz R., Holman R., Probstfield J.L., et al. Design, history and results of the thiazolidinedione intervention with vitamin D evaluation (TIDE) randomised controlled trial. Diabetologia. 2012;55:36–45. doi: 10.1007/s00125-011-2357-4. [DOI] [PubMed] [Google Scholar]
- 49.Prevention of instent renarrowing with aggressive glucose lowering with pioglitazone in diabetic patients n.d. https://clinicaltrials.gov/ct2/show/NCT00819325 (accessed March 8, 2019).
- 50.Cusi K., Orsak B., Bril F., Lomonaco R., Hecht J., Ortiz-Lopez C., et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus. Ann Intern Med. 2016;165:305–315. doi: 10.7326/M15-1774. [DOI] [PubMed] [Google Scholar]
- 51.Efficacy in controlling glycaemia with Victoza® (liraglutide) as add-on to metformin vs. OADs as add-on to metformin after up to 104 weeks of treatment in subjects with type 2 diabetes n.d. https://clinicaltrials.gov/ct2/show/NCT02730377 (accessed March 8, 2019).
- 52.Bypass angioplasty revascularization investigation 2 diabetes study group Baseline characteristics of patients with diabetes and coronary artery disease enrolled in the bypass angioplasty revascularization investigation 2 diabetes (BARI 2D) trial. Am Heart J. 2008;156:528–536. doi: 10.1016/j.ahj.2008.05.015. e1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Placebo and active comparator controlled dose response study of rivoglitazone in type 2 diabetes n.d. https://clinicaltrials.gov/ct2/show/NCT00575874 (accessed March 8, 2019).
- 54.Pop-Busui R., Oral E., Raffel D., Byun J., Bajirovic V., Vivekanandan-Giri A., et al. Impact of rosiglitazone and glyburide on nitrosative stress and myocardial blood flow regulation in type 2 diabetes mellitus. Metabolism. 2009;58:989–994. doi: 10.1016/j.metabol.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 55.The practical evidence of antidiabetic combination therapy in Korea n.d. https://clinicaltrials.gov/ct2/show/NCT02231021 (accessed March 8, 2019).
- 56.Kim S.G., Kim D.M., Woo J.-T., Jang H.C., Chung C.H., Ko K.S., et al. Efficacy and safety of lobeglitazone monotherapy in patients with type 2 diabetes mellitus over 24-weeks: A multicenter, randomized, double-blind, parallel-group, placebo controlled trial. PLoS ONE. 2014;9:e92843. doi: 10.1371/journal.pone.0092843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Russell-Jones D., Cuddihy R.M., Hanefeld M., Kumar A., Gonzalez J.G., Chan M., et al. Efficacy and safety of exenatide once weekly versus metformin, pioglitazone, and sitagliptin used as monotherapy in drug-naive patients with type 2 diabetes (DURATION-4): A 26-week double-blind study. Diabetes Care. 2012;35:252–258. doi: 10.2337/dc11-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosenstock J., Vico M., Wei L., Salsali A., List J.F. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA1c, body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35:1473–1478. doi: 10.2337/dc11-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pharmacogenomics of thiazolidinediones n.d. https://clinicaltrials.gov/ct2/show/NCT01135394 (accessed March 8, 2019).
- 60.Predictive markers of glitazone efficacy in diabetic patients - pilot study n.d. https://clinicaltrials.gov/ct2/show/NCT00481429 (accessed March 8, 2019).
- 61.Hollander P., Li J., Allen E., Chen R. CV181-013 Investigators. Saxagliptin added to a thiazolidinedione improves glycemic control in patients with type 2 diabetes and inadequate control on thiazolidinedione alone. J Clin Endocrinol Metab. 2009;94:4810–4819. doi: 10.1210/jc.2009-0550. [DOI] [PubMed] [Google Scholar]
- 62.Kota B., Huang T., Roufogalius B. An overview on biological mechanisms of PPARs. Pharmacol Res. 2005;51:85–94. doi: 10.1016/j.phrs.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 63.Bermúdez V., Finol F., Parra N., Parra M., Pérez A., Peñaranda L., et al. PPAR-γ agonists and their role in type 2 diabetes mellitus management. Am J Ther. 2010;17:274–283. doi: 10.1097/MJT.0b013e3181c08081. [DOI] [PubMed] [Google Scholar]
- 64.Ferré P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53:S43–S50. doi: 10.2337/diabetes.53.2007.s43. [DOI] [PubMed] [Google Scholar]
- 65.Muralidaran S., Roy A. The role of PPAR agonists in diabetes mellitus. J Pharm Sci Res. 2016;8:864–866. [Google Scholar]
- 66.Greenfield J.R., Chisholm D.J. Thiazolidinediones - mechanisms of action. Aust Prescr. 2004;27:67–70. [Google Scholar]
- 67.Hauner H. The mode of action of thiazolidinediones. Diabetes Metab Res Rev. 2002;18:S10–S15. doi: 10.1002/dmrr.249. [DOI] [PubMed] [Google Scholar]
- 68.Yki-Järvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 69.Cariou B., Charbonnel B., Staels B. Thiazolidinediones and PPARγ agonists: Time for a reassessment. Trends Endocrinol Metab. 2012;23:205–215. doi: 10.1016/j.tem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 70.Momose Y., Meguro K., Ikeda H., Hatanaka C., Oi S., Sohda T. Studies on antidiabetic agents. X. Synthesis and biological activities of pioglitazone and related compounds. Chem Pharm Bull (Tokyo) 1991;39:1440–1445. doi: 10.1248/cpb.39.1440. [DOI] [PubMed] [Google Scholar]
- 71.Sohda T., Mizuno K., Momose Y., Ikeda H., Fujita T. Meguro K. Studies on antidiabetic agents. 11. Novel thiazolidinedione derivatives as potent hypoglycemic and hypolipidemic agents. J Med Chem. 1992;35:2617–2626. doi: 10.1021/jm00092a012. [DOI] [PubMed] [Google Scholar]
- 72.Tanis S.P., Parker T.T., Colca J.R., Fisher R.M., Kletzein R.F. Synthesis and biological activity of metabolites of the antidiabetic, antihyperglycemic agent pioglitazone. J Med Chem. 1996;39:5053–5063. doi: 10.1021/jm9605694. [DOI] [PubMed] [Google Scholar]
- 73.Lohray B.B., Bhushan V., Rao B.P., Madhavan G.R., Murali N., Rao K.N., et al. Novel euglycemic and hypolipidemic agents. J Med Chem. 1998;41:1619–1630. doi: 10.1021/jm970444e. [DOI] [PubMed] [Google Scholar]
- 74.Lohray B.B., Bhushan V., Reddy A.S., Rao P.B., Reddy N.J., Harikishore P., et al. Novel euglycemic and hypolipidemic agents. 4. Pyridyl- and quinolinyl- containing thiazolidinediones. J Med Chem. 1999;42:2569–2581. doi: 10.1021/jm980622j. [DOI] [PubMed] [Google Scholar]
- 75.Oguchi M., Wada K., Honma H., Tanaka A., Kaneko T., Sakakibara S., et al. Molecular design, synthesis, and hypoglycemic activity of a series of thiazolidine-2, 4-diones. J Med Chem. 2000;43:3052–3066. doi: 10.1021/jm990522t. [DOI] [PubMed] [Google Scholar]
- 76.Madhavan G.R., Chakrabarti R., Kumar S.K.B., Misra P. Novel phthalazinone and benzoxazinone containing thiazolidinediones as antidiabetic and hypolipidemic agents. Eur J Med Chem. 2001;36:627–637. doi: 10.1016/s0223-5234(01)01257-0. [DOI] [PubMed] [Google Scholar]
- 77.Madhavan G.R., Chakrabarti R., Vikramadithyan R.K., Mamidi R.N.V.S., Balraju V., Rajesh B.M., et al. Synthesis and biological activity of novel pyrimidinone containing thiazolidinedione derivatives. Bioorganic Med Chem. 2002;10:2671–2680. doi: 10.1016/s0968-0896(02)00107-4. [DOI] [PubMed] [Google Scholar]
- 78.Storr T., Mitchell D., Buglyó P., Thompson K.H., Yuen V.G., McNeill J.H., et al. Vanadyl-thiazolidinedione combination agents for diabetes therapy. Bioconjug Chem. 2003;14:212–221. doi: 10.1021/bc025606m. [DOI] [PubMed] [Google Scholar]
- 79.Berger J., Leibowitz M.D., Doebber T.W., Elbrecht A., Zhang B., Zhou G., et al. Novel peroxisome proliferator-activated receptor (PPAR) gamma and PPAR delta ligands produce distinct biological effects. J Biol Chem. 1999;274:6718–6725. doi: 10.1074/jbc.274.10.6718. [DOI] [PubMed] [Google Scholar]
- 80.Koyama H., Boueres J.K., Han W., Metzger E.J., Bergman J.P., Gratale D.F., et al. 5-aryl thiazolidine-2, 4-diones as selective PPAR agonists. Bioorg Med Chem Lett. 2003;13:1801–1804. doi: 10.1016/s0960-894x(03)00257-9. [DOI] [PubMed] [Google Scholar]
- 81.Kim B.Y., Ahn J.B., Lee H.W., Kang S.K., Lee J.H., Shin J.S., et al. Synthesis and biological activity of novel substituted pyridines and purines containing 2,4-thiazolidinedione. Eur J Med Chem. 2004;39:433–447. doi: 10.1016/j.ejmech.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 82.Gupta D., Ghosh N.N., Chandra R. Synthesis and pharmacological evaluation of substituted 5-[4-[2-(6, 7-dimethyl-1, 2, 3, 4-tetrahydro-2-oxo-4-quinoxalinyl) ethoxy] phenyl] methylene] thiazolidine-2, 4-dione derivatives as potent euglycemic and hypolipidemic agents. Bioorg Med Chem Lett. 2005;15:1019–1022. doi: 10.1016/j.bmcl.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 83.Lee H.W., Bok Y.K., Joong B.A., Sung K.K., Lee J.H., Jae S.S., et al. Molecular design, synthesis, and hypoglycemic and hypolipidemic activities of novel pyrimidine derivatives having thiazolidinedione. Eur J Med Chem. 2005;40:862–874. doi: 10.1016/j.ejmech.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 84.Gim H.J., Kang B., Jeon R. Synthesis and biological activity of 5- 4- [2- (Methyl- p -substi- tuted phenylamino) ethoxy ] benzyl thiazolidine-2, 4-diones. Arch Pharm Res. 2007;30:1055–1061. doi: 10.1007/BF02980237. [DOI] [PubMed] [Google Scholar]
- 85.Mohammed Iqbal A.K., Khan A.Y., Kalashetti M.B., Belavagi N.S., Gong Y.D., Khazi I.A.M. Synthesis, hypoglycemic and hypolipidemic activities of novel thiazolidinedione derivatives containing thiazole/triazole/oxadiazole ring. Eur J Med Chem. 2012;53:308–315. doi: 10.1016/j.ejmech.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 86.Kumar B.R.P., Kumar S.S., Viral P., Wadhwani A., Vadivelan R., Kumar M., et al. Novel glitazones : glucose uptake and cytotoxic activities, and structure – activity relationships. Med Chem Res. 2012;21:2689–2701. [Google Scholar]
- 87.Shukla S., Kumar P., Das N., HariNarayanaMoorthy N.S., KumarShrivastava S., Trivedi P., et al. Synthesis, Characterization, Biological Evaluation and Docking of Coumarin Coupled Thiazolidinedione Derivatives and its Bioisosteres as PPARγ Agonists. med chem. 2012;8(5):834–845. doi: 10.2174/157340612802084388. [DOI] [PubMed] [Google Scholar]
- 88.Deshmukha Amarsinh R., Dhumala Sambhaji T., Bhaleraoa Mahendra B., Mishrab Akansha, Srivastavab Arvind K., et al. Design synthesis and antidiabetic evaluation of new cyanoquinoloxy benzylidenyl 2,4-thiazolidinediones. Chem Biol Interface. 2016;6:189–197. [Google Scholar]
- 89.Naim M.J., Alam O., Alam M.J., Hassan M.Q., Siddiqui N., Naidu V.G.M., et al. Design, synthesis and molecular docking of thiazolidinedione based benzene sulphonamide derivatives containing pyrazole core as potential anti-diabetic agents. Bioorg Chem. 2018;76:98–112. doi: 10.1016/j.bioorg.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 90.LeBrasseur N.K., Kelly M., Tsao T.-S., Farmer S.R., Saha A.K., Ruderman N.B., et al. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Metab. 2006;291:E175–E181. doi: 10.1152/ajpendo.00453.2005. [DOI] [PubMed] [Google Scholar]
- 91.Naim M.J., Alam O., Alam M.J., Shaquiquzzaman M., Alam M.M., Naidu V.G.M. Synthesis, docking, in vitro and in vivo antidiabetic activity of pyrazole-based 2,4-thiazolidinedione derivatives as PPAR-γ modulators. Arch Pharm (Weinheim) 2018;351:1700223. doi: 10.1002/ardp.201700223. [DOI] [PubMed] [Google Scholar]
- 92.Bansal G., Singh S., Monga V., Thanikachalam P.V., Chawla P. Synthesis and biological evaluation of thiazolidine-2,4-dione-pyrazole conjugates as antidiabetic, anti-inflammatory and antioxidant agents. Bioorg Chem. 2019;92 doi: 10.1016/j.bioorg.2019.103271. [DOI] [PubMed] [Google Scholar]
- 93.Datar P.A., Aher S.B. Design and synthesis of novel thiazolidine-2,4-diones as hypoglycemic agents. J Saudi Chem Soc. 2016;20:S196–S201. [Google Scholar]
- 94.Tunçbilek M., Bozdag-Dündar O., Ayhan-Kilcigil G., Ceylan M., Waheed A., Verspohl E.J., et al. Synthesis and hypoglycemic activity of some substituted flavonyl thiazolidinedione derivatives - Fifth communication: Flavonyl benzyl substituted 2,4-thiazolidinediones. Farmaco. 2003;58:79–83. doi: 10.1016/S0014-827X(02)01241-7. [DOI] [PubMed] [Google Scholar]
- 95.Bozdaǧ-Dündar O., Verspohl E.J., Daş-Evcimen N., Kaup R.M., Bauer K., Sarikaya M., et al. Synthesis and biological activity of some new flavonyl-2,4-thiazolidinediones. Bioorganic Med Chem. 2008;16:6747–6751. doi: 10.1016/j.bmc.2008.05.059. [DOI] [PubMed] [Google Scholar]
- 96.Wrobel J., Li Z., Dietrich A., McCaleb M., Mihan B., Sredy J., et al. Novel 5-(3-aryl-2-propynyl)-5-(arylsulfonyl)thiazolidine-2,4-diones as antihyperglycemic agents. J Med Chem. 1998;41:1084–1091. doi: 10.1021/jm9706168. [DOI] [PubMed] [Google Scholar]
- 97.Jawale D.V., Pratap U.R., Rahuja N., Srivastava A.K., Mane R.A. Synthesis and antihyperglycemic evaluation of new 2,4-thiazolidinediones having biodynamic aryl sulfonylurea moieties. Bioorganic Med Chem Lett. 2012;22:436–439. doi: 10.1016/j.bmcl.2011.10.110. [DOI] [PubMed] [Google Scholar]
- 98.Prabhakar C., Madhusudhan G., Sahadev K., Reddy C.M., Sarma M.R., Reddy G.O., et al. Synthesis and biological activity of novel thiazolidinediones. Bioorg Med Chem Lett. 1998;8:2725–2730. doi: 10.1016/s0960-894x(98)00485-5. [DOI] [PubMed] [Google Scholar]
- 99.Saini P., Farooqui N.A., Easwari T.S. Synthesis and biological evaluation of some 5-substituted phenothiazine based thiazoli- dine-2, 4-dione derivatives. MIT Int J Pharm Sci. 2018;4:13–17. [Google Scholar]
- 100.Chittiboyina A.G., Venkatraman M.S., Mizuno C.S., Desai P.V., Patny A., Benson S.C., et al. Design and synthesis of the first generation of dithiolane thiazolidinedione- and phenylacetic acid-based PPARy agonists. J Med Chem. 2006;49:4072–4084. doi: 10.1021/jm0510880. [DOI] [PubMed] [Google Scholar]
- 101.Kumar B.R.P., Nanjan M.J. Novel glitazones : Design, synthesis, glucose uptake and structure – activity relationships. Bioorg Med Chem Lett. 2010;20:1953–1956. doi: 10.1016/j.bmcl.2010.01.125. [DOI] [PubMed] [Google Scholar]
- 102.Kumar B.R.P., Soni M., Kumar S.S., Singh K., Patil M., Baig R.B.N., et al. Synthesis, glucose uptake activity and structure e activity relationships of some novel glitazones incorporated with glycine, aromatic and alicyclic amine moieties via two carbon acyl linker. Eur J Med Chem. 2011;46:835–844. doi: 10.1016/j.ejmech.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 103.Mehendale-Munj S., Ghosh R., Ramaa C.S. Synthesis and evaluation of the hypoglycemic and hypolipidemic activity of novel 5-benzylidene-2,4-thiazolidinedione analogs in a type-2 diabetes model. Med Chem Res. 2011;20:642–647. [Google Scholar]
- 104.Shrivastava S.K., Batham A., Sinha S.K., Parida T.K., Garabadu D., Choubey P.K. Design, synthesis and evaluation of novel thiazolidinedione derivatives as anti-hyperglycemic and anti-hyperlipidemic agents. Med Chem Res. 2016;25:2258–2266. [Google Scholar]
- 105.Dadasaheb K., Jain N., Vijay J., Kashid G. Design synthesis and evaluation of novel thiazolidinedione derivatives as antidiabetic agents. Pharma Innov J. 2017;6:390–398. [Google Scholar]
- 106.Naim M.J., Alam M.J., Nawaz F., Naidu V.G.M., Aaghaz S., Sahu M., et al. Synthesis, molecular docking and anti-diabetic evaluation of 2,4-thiazolidinedione based amide derivatives. Bioorg Chem. 2017;73:24–36. doi: 10.1016/j.bioorg.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 107.Khazi M.I.A., Belavagi N.S., Kim K.R., Gong Y.D., Khazi I.A.M. Synthesis, hypoglycaemic, hypolipidemic and ppar-y agonist activities of 5-(2-alkyl/aryl-6-arylimidazo[2,1-b][1,3,4]thiadiazol-5-yl)methylene-1,3-thiazolidinediones. Chem Biol Drug Des. 2013;82:147–155. doi: 10.1111/cbdd.12140. [DOI] [PubMed] [Google Scholar]
- 108.Murugan R., Anbazhagan S., Sriman Narayanan S. Synthesis and in vivo antidiabetic activity of novel dispiropyrrolidines through [3 + 2] cycloaddition reactions with thiazolidinedione and rhodanine derivatives. Eur J Med Chem. 2009;44:3272–3279. doi: 10.1016/j.ejmech.2009.03.035. [DOI] [PubMed] [Google Scholar]
- 109.Neogi P., Lakner F.J., Medicherla S., Cheng J., Dey D., Gowri M., et al. Synthesis and structure-activity relationship studies of cinnamic acid-based novel thiazolidinedione antihyperglycemic agents. Bioorganic Med Chem. 2003;11:4059–4067. doi: 10.1016/s0968-0896(03)00393-6. [DOI] [PubMed] [Google Scholar]
- 110.Nazreen S., Alam M.S., Hamid H., Yar M.S., Dhulap A., Alam P., et al. Design, synthesis, and biological evaluation of thiazolidine-2,4-dione conjugates as PPAR-γ agonists. Arch Pharm (Weinheim) 2015;348:421–432. doi: 10.1002/ardp.201400280. [DOI] [PubMed] [Google Scholar]
- 111.Maji D., Samanta S. Novel thiazolidinedione-5-acetic-acid-peptide hybrid derivatives as potent antidiabetic and cardioprotective agents. Biomed Pharmacother. 2017;88:1163–1172. [Google Scholar]
- 112.Jiwane S.K., Singh V.K., Namdeo K.P., Prajapati S.K. Synthesis of some novel 2, 4-thiazolidinedione derivatives and their biological screening as antidiabetic agents. Asian J Chem. 2009;21:5068–5072. [Google Scholar]
- 113.Avupati V.R., Yejella R.P., Akula A., Guntuku G.S., Doddi B.R., Vutla V.R., et al. Synthesis, characterization and biological evaluation of some novel 2,4-thiazolidinediones as potential cytotoxic, antimicrobial and antihyperglycemic agents. Bioorganic Med Chem Lett. 2012;22:6442–6450. doi: 10.1016/j.bmcl.2012.08.052. [DOI] [PubMed] [Google Scholar]
- 114.Patil S.D., Nawale S.L., Balasubramaniyan V. Evaluation of thiazolidinedione derivatives for acute toxicity and potential antidiabetic activity. Der Pharma Chem. 2015;7:216–223. [Google Scholar]
- 115.Patel K.D., Patel C.N., Patel G.M. Microwave Assisted Synthesis and Antidiabetic Activity of Novel 5-[4-(Substituted) Benzylidine]Thiazolidine-2,4-Dione. Med Chem (Los Angeles) 2016;6:647–651. doi: 10.4172/2161-0444.1000409. [DOI] [Google Scholar]
- 116.Alemán-González-Duhart D., Tamay-Cach F., Correa-Basurto J., Padilla-Martínez I.I., Álvarez-Almazán S., Mendieta-Wejebe J.E. In silico design, chemical synthesis and toxicological evaluation of 1,3-thiazolidine-2,4-dione derivatives as PPARγ agonists. Regul Toxicol Pharmacol. 2017;86:25–32. doi: 10.1016/j.yrtph.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 117.S. R, S. C. Novel substituted thiazolidinedione derivatives as anti-diabetic agents. Eur J Pharm Med Res 2017; 4: 643–52.
- 118.Reddy K.A., Lohray B.B., Bhushan V., Bajji A.C., Reddy K.V., Reddy P.R., et al. Novel antidiabetic and hypolipidemic agents. 3. Benzofuran-containing thiazolidinediones. J Med Chem. 1999;42:1927–1940. doi: 10.1021/jm980549x. [DOI] [PubMed] [Google Scholar]
- 119.Jeon R., Park S.Y. Synthesis and biological activity of benzoxazole containing thiazolidinedione derivatives. Arch Pharm Res. 2004;27:1099–1105. doi: 10.1007/BF02975111. [DOI] [PubMed] [Google Scholar]
- 120.Pattan S., Suresh C., Pujar V., Reddy V., Rasal V., Koti B. Synthesis and antidiabetic activity of 2-amino[5(4-sulphonylbenzylidine)-2,4-thiazolidinedione]-7-chloro-6- fluorobenzothiazole. Indian J Chem. 2005;44:2404–2408. [Google Scholar]
- 121.Jeon R., Kim Y., Cheon Y., Ryu J. Synthesis and biological activity of [[(heterocycloamino) alkoxy ] benzyl ] -2, 4-thiazolidinediones as PPARy agonists. Arch Pharm Res. 2006;29:394–399. doi: 10.1007/BF02968589. [DOI] [PubMed] [Google Scholar]
- 122.Purohit S.S., Veerapur V.P. Benzisoxazole containing thiazolidinediones as peroxisome proliferator activated receptor- γ agonists : Design, molecular docking, synthesis & anti - diabetic studies. Sch Acad J Pharm. 2014;3:26–37. [Google Scholar]
- 123.Unlusoy M.C., Kazak C., Bayro O., Verspohl E.J., Ertan R., Dundar O.B. Synthesis and antidiabetic activity of 2,4-thiazolidindione, imidazolidinedione and 2-thioxo-imidazolidine-4-one derivatives bearing 6-methyl chromonyl pharmacophore. J Enzyme Inhib Med Chem. 2013;28:1205–1210. doi: 10.3109/14756366.2012.723207. [DOI] [PubMed] [Google Scholar]
- 124.Nazreen S., Alam M.S., Hamid H., Yar M.S., Dhulap A., Alam P., et al. Thiazolidine-2,4-diones derivatives as PPAR-γ agonists: Synthesis, molecular docking, in vitro and in vivo antidiabetic activity with hepatotoxicity risk evaluation and effect on PPAR-γ gene expression. Bioorganic Med Chem Lett. 2014;24:3034–3042. doi: 10.1016/j.bmcl.2014.05.034. [DOI] [PubMed] [Google Scholar]
- 125.Hidalgo-Figueroa S., Ramírez-Espinosa J.J., Estrada-Soto S., Almanza-Pérez J.C., Román-Ramos R., Alarcón-Aguilar F.J., et al. Discovery of thiazolidine-2,4-dione/biphenylcarbonitrile hybrid as dual PPAR α/γ modulator with antidiabetic effect: In vitro, in silico and in vivo approaches. Chem Biol Drug Des. 2013;81:474–483. doi: 10.1111/cbdd.12102. [DOI] [PubMed] [Google Scholar]
- 126.Navarrete-Vázquez G., Torres-Gómez H., Hidalgo-Figueroa S., Ramírez-Espinosa J.J., Estrada-Soto S., Medina-Franco J.L., et al. Synthesis, in vitro and in silico studies of a PPARγ and GLUT-4 modulator with hypoglycemic effect. Bioorg Med Chem Lett. 2014;24:4575–4579. doi: 10.1016/j.bmcl.2014.07.068. [DOI] [PubMed] [Google Scholar]
- 127.Hidalgo-Figueroa S., Estrada-Soto S., Ramírez-Espinosa J.J., Paoli P., Lori G., León-Rivera I., et al. Synthesis and evaluation of thiazolidine-2,4-dione/benzazole derivatives as inhibitors of protein tyrosine phosphatase 1B (PTP-1B): Antihyperglycemic activity with molecular docking study. Biomed Pharmacother. 2018;107:1302–1310. doi: 10.1016/j.biopha.2018.08.124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.