

Abstract
Designing functional proteins that can withstand extreme heat is beneficial for industrial and protein therapeutic applications. Thus, elucidating the atomic-level determinants of thermostability is a major interest for rational protein design. To that end, we compared the structure and dynamics of a set of previously designed, thermostable proteins based on the activation domain of human procarboxypeptidase A2 (AYEwt). The mutations in these designed proteins were intended to increase hydrophobic core packing and inter-secondary-structure interactions. To evaluate whether these design strategies were successfully deployed, we performed all-atom, explicit-solvent molecular dynamics (MD) simulations of AYEwt and three designed variants at both 25 and 100°C. Our MD simulations agreed with the relative experimental stabilities of the designs based on their secondary structure content, Cα root-mean-square deviation/fluctuation, and buried-residue solvent accessible surface area. Using a contact analysis, we found that the designs stabilize inter-secondary structure interactions and buried hydrophobic surface area, as intended. Based on our analysis, we designed three additional variants to test the role of helix stabilization, core packing, and a Phe → Met mutation on thermostability. We performed the additional MD simulations and analysis on these variants, and these data supported our predictions.
Keywords: molecular dynamics, protein design, protein thermostability
Introduction
Computational de novo protein design, the engineering of proteins using computers to determine a new amino acid sequence, has increased in prevalence over the last two decades. Initial steps in this field involved the optimization of algorithms to create de novo sequences that would adopt template backbone folds. Successful designs of this type are often more thermostable than the wild type template upon which they are based (Dahiyat and Mayo, 1997; Bryson et al., 1998; Kuhlman et al., 2003; Shah et al., 2007; Koga et al., 2012; McCully and Daggett, 2012; Childers and Daggett, 2017; Rocklin et al., 2017; Baker, 2019). Thermostability is not an explicit goal of these algorithms’ scoring functions, and protein engineers often do not know why designed proteins exhibit such thermostability. Knowledge of the design strategies that promote thermostability will allow protein designers to employ or avoid them as needed depending on the desired application. Here, we used molecular dynamics (MD) simulations to identify the atomic-level stabilizing factors in a set of engineered proteins.
We investigated a set of de novo designed proteins engineered by Dantas et al. (2003, 2007) based on the 70-residue activation domain of human procarboxypeptidase A2 (AYEwt). This protein was originally chosen to be the template in the protein design study due to its small, globular fold and simple secondary structure, two α-helices and a four-stranded β-sheet (Dantas et al., 2003) (Fig. 1a). The amino acid sequence of AYEdes was designed using RosettaDesign to fold to the same backbone topology as AYEwt. Relative to AYEwt, AYEdes has a ΔΔG of −10.3 kcal/mol, ΔTm > 30°C, 32% sequence identity, and a Cα root-mean-square deviation (RMSD) of 1.5 Å (Dantas et al., 2007). In the 2003 study, the authors noted that designed proteins with greater thermostability than their template counterparts contained more hydrophobic residues. Dantas et al. (2007) further investigated the role of specific residues in stabilizing AYEdes by designing and expressing several minimal mutants that they believed would enhance packing between elements of secondary structure. They predicted that Glu7Val/His44Val/Arg46Leu would improve inter-strand packing, Phe32Trp would improve helix-strand packing, and Ala54Trp/Val55Phe would improve inter-helical packing. They designed and expressed two variants combining these sets of mutations, AYEwt-4mut (Glu7Val/His44Val/Arg46Leu, Phe32Trp) and AYEwt-5mut (Glu7Val/His44Val/Arg46Leu, Ala54Trp/Val55Phe) (Fig. 1b), and found that both proteins folded stably with ΔΔGs of −5.2 and −4.1 kcal/mol, respectively, relative to AYEwt.
Fig. 1.
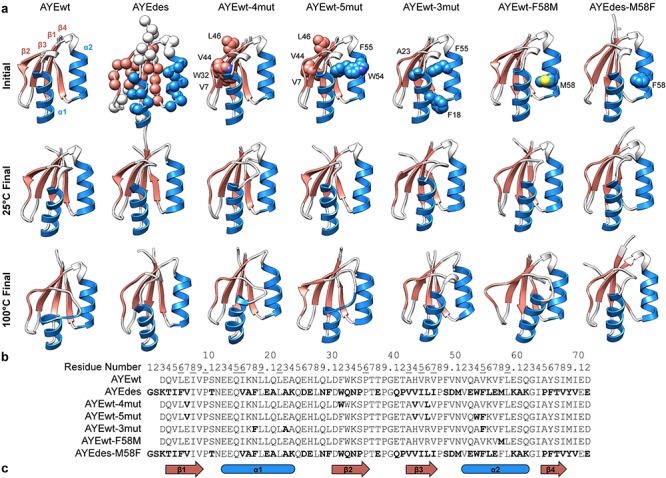
Protein structures and sequences. (a) Initial and final structures from simulation shown for all seven proteins. Representative final structures were chosen as the frame with the median Core Cα RMSD among the five replicate simulations. On the initial structures, mutated residues relative to the template structure were shown in spheres. For simplicity, hydrogen atoms were not displayed throughout, and only Cα atoms of mutated residues were shown for AYEdes. (b) Sequences for all seven proteins with mutations relative to AYEwt shown in bold and residue numbers of buried residues underlined. All sequences have been aligned and numbered relative to the AYEdes structure. (c) Secondary structure elements shown for β1 (residues 4–9), α1 (13–24), β2 (31–36), β3 (43–47), α2 (52–62), and β4 (65–68).
To identify the atomic-level interactions responsible for the thermostability of AYEdes, AYEwt-4mut, and AYEwt-5mut, we performed 4 μs of all-atom, explicit solvent MD simulations of these three designed proteins along with AYEwt at both 25 and 100°C. We analyzed secondary structure content, Cα RMSD and root-mean-square fluctuation (RMSF), and buried residue solvent accessible surface area (SASA) as measures of protein stability. Additionally, we determined whether the stabilization strategies predicted by Dantas et al. (2003, 2007) were realized in their designs by quantifying contacts between secondary structure elements and among buried residues, including identifying critical contact residue pairs. Our MD simulations agreed with the relative stabilities of AYEwt, AYEdes, AYEwt-4mut, and AYEwt-5mut as measured experimentally, and NOEs calculated from simulation for AYEdes also agree well with those measured experimentally (Dantas et al., 2007). Subsequent contact analyses validated the increases in inter-secondary structure packing and reduced buried-residue solvent exposure in the three designs, though the increased packing was not quite as predicted.
With these data, we designed three additional, minimally modified variants, AYEwt-3mut (Asn18Phe, Glu23Ala, and Val55Phe), AYEwt-F58M, and AYEdes-M58F (Fig. 1), to test our own predictions of residues that contributed to or detracted from the thermostability of AYEdes. We predicted that Phe18 and Ala23 from AYEdes would stabilize AYEwt-3mut’s α1, which was particularly unstable in AYEwt, and that Phe55 would enhance core packing. We designed AYEwt-F58M and AYEdes-M58F to test our prediction that phenylalanine would be more stabilizing than methionine at position 58. Finally, we performed an additional 3 μs of MD simulation and subsequent coordinate analysis for these three proteins. Our simulations confirmed stabilization of α1 in AYEwt-3mut and showed a small stabilizing effect for phenylalanine at position 58 relative to methionine.
Methods
Protein structure preparation and homology modeling
The activation domain of pancreatic human procarboxypeptidase A2 (chain A, residues 10–79), AYEwt, was isolated from the 1.8 Å-resolution crystal structure of the peptidase (PDB ID 1AYE) (Garcίa‐Sáez et al., 1997). Similarly, AYEdes was isolated from the NMR structure (PDB ID 2GJF, chain A, model 1) (Dantas et al., 2007). Four additional designed variants, AYEwt-4mut, AYEwt-5mut, AYEwt-3mut, and AYEwt-F58M, were built by homology modeling with ModeLLer (Eswar et al., 2006) using AYEwt as the template, and AYEdes-M58F was similarly built using AYEdes as a template. The automodel function was used to thread the target protein sequence onto the template structure. Of the 25 structures generated, the model with the lowest DOPE score was selected (Shen and Sali, 2006). All proteins were renumbered to align with AYEdes (Fig. 1b).
Molecular dynamics simulations
All-atom, explicit-solvent MD simulations were performed using NAMD 2.11 (Phillips et al., 2005) with the CHARMM36m force field (MacKerell et al., 1998, 2004; Best et al., 2012). Visual Molecular Dynamics (VMD) (Humphrey et al., 1996) was used to generate PSF files, and His44 was specified as HSE (protonated Nε) when present. Structures were minimized for 1000 steps of conjugate gradient minimization then solvated in a 50 × 50 × 50 Å periodic box of TIP3P water (Jorgensen et al., 1998), and KCl ions were added to a concentration of 150 mM, neutralizing the system (Beglov and Roux, 1998). The system was minimized for an additional 100 steps, heated incrementally to either 25 or 100°C with an integration time step of 2 fs and then equilibrated for 5 ps. A Langevin thermostat and barostat were used to maintain an NPT (constant number of atoms, pressure, and temperature) system. Nonbonded interactions were treated with a smooth switching function at 8 Å for van der Waals interactions and Particle Mesh Ewald (PME) for electrostatics. Five independent, 100-ns simulations were performed at both temperatures starting from the equilibrated system for all seven proteins, with structures saved every 1 ps, totaling 7 μs of simulation time and 7 000 000 frames.
Simulation analysis
The MD simulation and analysis software package, in lucem molecular mechanics (ilmm), was used to analyze the resulting simulations (Beck et al., 2000). The dictionary of protein secondary structure (DSSP) algorithm (Kabsch and Sander, 1983) implemented in ilmm was used to identify the secondary structure of each residue at each simulation time point. Residues in AYEwt that spent > 50% of simulation time in α-helix or β-sheet at 25°C were used to define standard secondary structure residue ranges for our set of proteins, and β2 was extended from two residues to six in order to align better with other strands in the β-sheet (β1: 4–9, α1: 13–24, β2: 31–36, β3: 43–47, α2: 52–62, and β4: 65–68, Fig. 1c). The average fraction of simulation time that these residues adopted either α-helix or β-sheet was calculated and averaged across five simulations at both temperatures for each protein.
The RMSF of the Cα atoms from the mean structure was calculated across all residues for all five simulations at a given temperature. Cα RMSD from the minimized starting structure was calculated for the globular core (residues 4–68) to avoid overcontribution of floppy terminal residues and limit variation caused by the difference in the number of residues. SASA was calculated using the Lee and Richards algorithm (Lee and Richards, 1971) implemented in ilmm to analyze the degree of solvent exposure of the side chains of 13 previously identified buried residues (6, 8, 10, 15, 16, 19, 22, 36, 43, 45, 47, 55 and 59, Fig. 1b) (Dantas et al., 2007). Both of these values were calculated as an average per simulation, and the average and standard deviation of these values for all five simulations at both temperatures were reported.
NOE data for AYEdes (MR block ID 425459) (Dantas et al., 2007) were downloaded from the Biological Magnetic Resonance Data Bank NMR Restraints Grid (Doreleijers et al., 2005). A total of 1801 intramolecular NOEs were reported for chain A. NOEs were calculated from our trajectories using an r−6-weighted average distance among all structures at each simulation temperature. NOEs were considered satisfied if this distance was greater than 5.5 Å, the longest rfar reported experimentally. Long-range NOEs were defined as those between residues with a contact order less than 5.
We quantified interactions using a distance- and angle-based cutoff in ilmm, with contact groupings defined by secondary structure element (α1, α2, and β-sheet) or buried classification. Hydrophobic contacts were defined between CHx (x > 1) groups where carbon atoms were <5.4 Å apart. Hydrogen bonds were defined by donor–hydrogen–acceptor angles between 135° and 225° and donor–acceptor distances of <2.6 Å. ‘Other’ contacts were defined as pairs of nonhydrogen atoms that did not satisfy either the hydrophobic or the hydrogen bond criteria with distances <4.6 Å. Fraction time in contact was calculated for pairs of residues that contained at least one hydrogen bond, hydrophobic interaction, or other interaction among residue groups, α1–α2, α1–β-sheet, α2–β-sheet, buried. Inter-residue contacts that were present for on average > 95% of the final 50 ns of simulation time were considered ‘in-contact.’ The number of in-contact residue pairs for each residue group during simulations at 25°C was defined as ‘starting contacts.’ Similarly, the number of in-contact residue pairs at 100°C was defined as ‘ending contacts.’ ‘Missing contacts’ were those that were starting contacts for AYEwt and not ending contacts for the designed variant. Contacts that were not ending contacts for AYEwt were classified as ‘rescued contacts’ if they were in-contact only at 25°C in the designed variants and as ‘fully rescued contacts’ if in-contact at both temperatures.
Results and Discussion
Relative stabilities in silico of previously designed proteins agreed with experiment
We used MD simulations to identify key structural characteristics that contribute to thermostability in a set of engineered proteins. Our set included the activation domain from human procarboxypeptidase A2 (AYEwt), which was used as the backbone template for de novo design using RosettaDesign resulting in AYEdes (Dantas et al., 2003). Dantas et al. (2007) additionally designed two minimal mutants, AYEwt-4mut and AYEwt-5mut, with four and five mutations relative to the wild type that RosettaDesign predicted were most critical to the stability of AYEdes due to increased packing between secondary structure elements and burial of hydrophobic surface area upon folding. When these four proteins were previously expressed, the authors found that AYEdes was the most stable (ΔΔG = −10.3 kcal/mol relative to AYEwt), followed by AYEwt-4mut (ΔΔG = −5.2 kcal/mol) and AYEwt-5mut (ΔΔG = −4.1 kcal/mol) (Dantas et al., 2007).
We employed high-temperature MD simulations to assess the stabilities of these four proteins. Stability at high temperature does not reproduce ΔG, but protein melting temperatures and free energies of folding tend to be correlated (Becktel and Schellman, 1987; Schellman, 1987; Razvi and Scholtz, 2006). Here, we used average structural data to infer protein stability. Our MD simulations reproduced the relative experimental stabilities of AYEwt, AYEdes, AYEwt-4mut, and AYEwt-5mut and supported the design strategy used by Dantas et al. (2007) in designing AYEwt-4mut and AYEwt-5mut.
NOE satisfaction
Experimental NOE data were available only for AYEdes. We calculated NOEs from our simulations of AYEdes at both 25 and 100°C. At 25°C, 92% of NOEs were satisfied, including 90% of long-order NOEs. Similarly, at 100°C, 93% of all and 91% of long-range NOEs were satisfied. AYEdes adopted conformations in our simulations that were consistent with the native state observed experimentally, and it was as stable at 100°C as at 25°C.
Backbone motion
One of the benefits of using MD simulations to study protein dynamics is the ability to observe visually protein dynamics at picosecond- and angstrom-level resolutions. We qualitatively analyzed simulation trajectories to assess each protein’s stability at 25 and 100°C (Fig. 1a). At 25°C, no protein displayed motions consistent with denaturation, and AYEdes and AYEwt-4mut were particularly rigid with fewer thermal vibrations than the other variants. Although AYEdes was more dynamic at 100°C than at 25°C, it was still more rigid and packed than the other variants at high temperature. However, at 100°C, AYEwt showed signs of denaturation, with prominent deformations in the C-terminus of α1 and heightened motion in the termini (Fig. 1a). AYEwt-4mut and AYEwt-5mut were more stable than AYEwt at 100°C, but for both, α1 and the α1-β2 loop were most mobile.
In agreement with our qualitative assessment, Cα RMSF showed AYEdes to be the most rigid protein at 25°C and AYEwt to be the most flexible (Fig. 2). At 100°C, all proteins were more dynamic than they were at 25°C. AYEwt and AYEwt-5mut were the most dynamic at 100°C, and AYEdes was the least. Additionally, in agreement with our qualitative analysis, the N-terminal halves of the proteins were the most dynamic, with the C-terminus of α1 and β2 being the most flexible structural elements. To quantify the dependence of protein dynamics on simulation temperature, we averaged the Cα RMSD for the core amino acids across the five replicates (Fig. 3a). At 25°C, motion was fairly consistent across the proteins. However, at 100°C, AYEwt’s core Cα RMSD was the highest at 2.4 ± 0.4 Å (average ± SEM), followed by AYEwt-5mut and then AYEwt-4mut, and AYEdes was the most stable at 1.1 ± 0.03 Å. These relative RMSDs at 100°C agree with the relative stabilities of the four proteins measured experimentally.
Fig. 2.
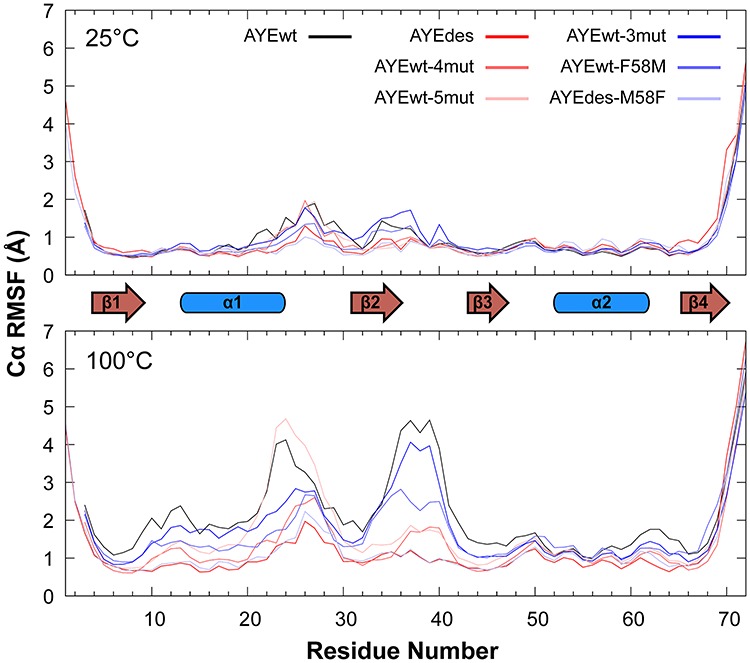
Backbone dynamics at low and high temperature. Cα RMSF is reported for all proteins at both temperatures. Secondary structure elements are shown between the two plots.
Fig. 3.
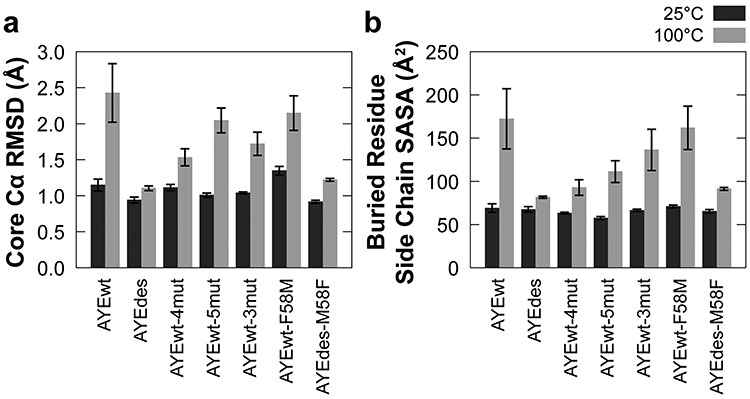
Core Cα RMSD and side chain SASA of buried residues. Average (a) core (residues 4–68) Cα RMSD and (b) SASA of buried residue side chains (residues 6, 8, 10, 15, 16, 19, 22, 36, 43, 45, 47, 55 and 59) for all seven proteins at 25 (dark) and 100°C (light). Error bars represent SEM, n = 5.
Secondary structure consistency
For each of the defined secondary structure elements (Fig. 1c), we averaged the percentage of residues across the simulation that adopted their expected structural element, α-helix or β sheet (Fig. 4). Note that at 25°C, the average percentage of β2 residues in β-sheet was between 30 and 34% for all variants due to our purposeful over-estimation of the length of β2 (see Methods).
Fig. 4.
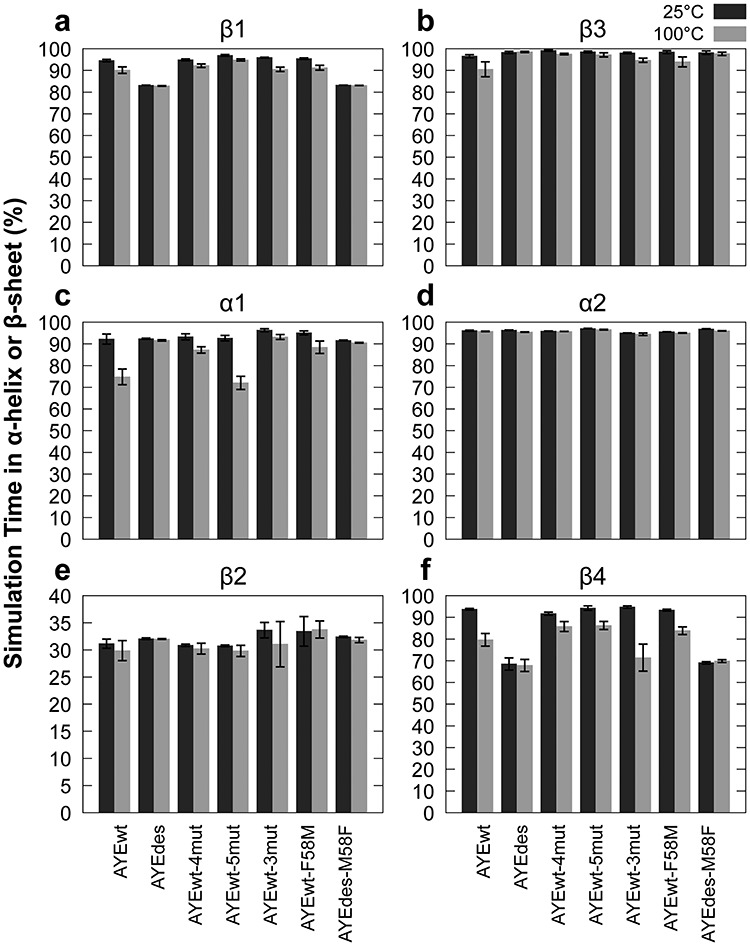
Persistence of secondary structure elements. Average percent of simulation time that all residues in each secondary structure element spent in either α-helix or β-sheet as defined by DSSP for all seven proteins at 25 (dark) and 100°C (light). See Fig. 1c for residue secondary structure assignments. Error bars represent SEM, n = 5.
The elements that suffered the greatest loss of secondary structure at 100°C relative to 25°C were α1, β1, and β4. At 25°C, all four proteins had the same α-helical content in α1 (Fig. 4c). However, at 100°C, AYEwt and AYEwt-5mut lost the most α-helical content (75 ± 4 and 72 ± 3%, respectively), AYEwt-4mut lost a moderate amount (87 ± 1%), and AYEdes lost the least (92 ± 0.3%), in agreement with their relative stabilities. AYEdes had the lowest β-sheet content in β1 and β4 at 25 °C, but it did not lose any β-sheet content when the temperature was raised to 100°C (Fig. 4a and f). At 25°C, β-sheet content in both β1 and β4 was highest in AYEwt, AYEwt-4mut and AYEwt-5mut, but all three proteins lost β-sheet content at 100°C. α2, β2, and β3 showed little variation among the proteins and temperatures (Fig. 4b, d and e).
Burial of hydrophobic surface area
Decreasing the solvent exposure of hydrophobic residues stabilizes proteins (Pace, 1992), so we quantified the SASA of residue side chains that were defined by Dantas et al. (2007) as buried (Fig. 3b). All variants had similar SASA at 25°C, ranging from 57 to 69 Å2. SASA increased the most at 100°C for AYEwt (to 172 ± 35 Å2), an intermediate amount for AYEwt-5mut (to 111 ± 13 Å2) and AYEwt-4mut (to 93 ± 9 Å2), and least for AYEdes (to 82 ± 2 Å2). The relative ΔSASAs at 100°C agree with the relative stabilities of the four proteins.
Secondary structure interactions and buried residue packing
Dantas et al. (2007) selected specific residue mutations in AYEwt-4mut and AYEwt-5mut that were extracted from AYEdes’s de novo design. Both variants’ combinations of sparse mutations were expected to confer thermostability through a combination of (1) enhanced residue packing, specifically between secondary structure elements and (2) increased buried hydrophobic surface area in the native state. We quantified contacts between secondary structure elements (α1–α2, α1–β-sheet, and α2—β-sheet) to determine whether the sets of mutations in AYEwt-4mut and AYEwt-5mut that were chosen to increase inter-helix (AYEwt-5mut) and helix-sheet (AYEwt-4mut) packing were successful (Fig. 5a–c). To compare the packing of the buried residues, we quantified their hydrophobic contacts and again averaged the summations across the five replicate simulations at low and high temperature (Fig. 5d).
Fig. 5.
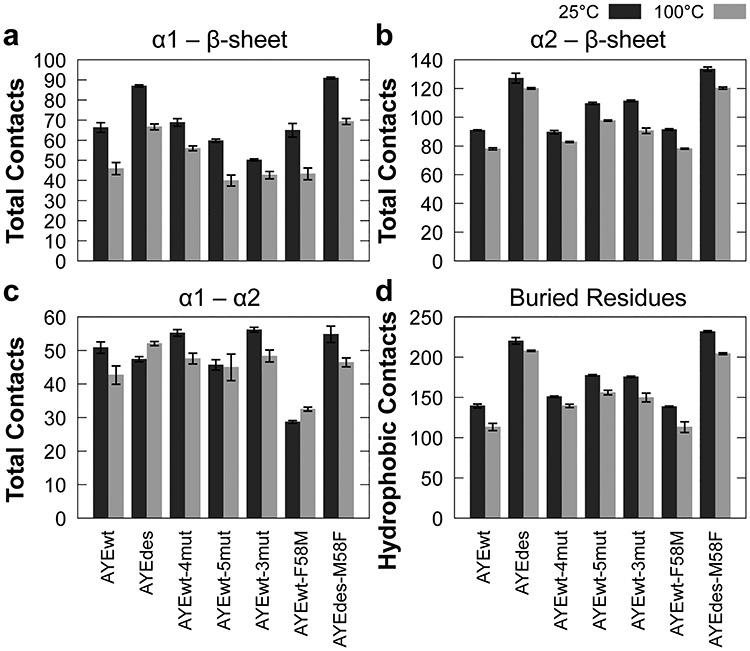
Contacts between secondary structure elements and buried residues. Average number of hydrogen bonds, hydrophobic interactions and other interactions between (a) α1 and the β-sheet, (b) α2 and the β-sheet, (c) α1 and α2 and (d) average number of hydrophobic interactions among buried residues (6, 8, 10, 15, 16, 19, 22, 36, 43, 45, 47, 55 and 59) for all seven proteins at 25 (dark) and 100°C (light). Error bars represent SEM, n = 5.
The packing between secondary structure elements was successfully increased in AYEdes relative to AYEwt, based on the number of contacts at 25°C, and AYEdes lost slightly fewer contacts between elements than AYEwt at 100°C as well (Fig. 5a–c). The intermediately stable minimal mutants, AYEwt-4mut and AYEwt-5mut, were not as successful as AYEdes at increasing contacts between secondary structure elements. Relative to AYEwt, AYEwt-5mut increased the number of α2–β-sheet contacts, and AYEwt-4mut increased the number of α1–α2 contacts at 25°C, but otherwise the number of contacts between elements was the same or lower. The number of contacts between secondary structure elements nearly always decreased at 100°C relative to 25°C, but the biggest decreases were seen between α1 and both the β-sheet and α2, especially for AYEwt.
AYEwt had the lowest number of hydrophobic contacts among buried residues, while AYEdes maintained the highest number of contacts at both 25 and 100°C (Fig. 5d). AYEwt-4mut and AYEwt-5mut both had a similar and intermediate number of buried hydrophobic contacts. This pattern, once again, agrees with the relative stabilities of the four proteins as measured experimentally. Every protein lost hydrophobic contacts among buried residues at 100°C relative to 25°C, though AYEwt lost the most.
Inter-secondary-structure contact pairs
In order to determine which residues were responsible for the changes in the number of contacts between secondary structure elements, we identified the pairs of residues between elements that were in contact > 95% of the time during the second half of each simulation. We identified residue pairs that gained or lost contact at 100°C relative to 25°C, and we identified contact pairs in the variants that were ‘missing’ (not present at 100°C or both temperatures), ‘rescued’ (present at 25°C), or ‘fully rescued’ (present at both temperatures) relative to AYEwt.
Of the 12 rescued contact pairs in AYEdes, all were rescued at both 25 and 100°C (fully rescued), and 11 incorporated at least one mutant amino acid (Table I). All of the mutated residues involved in these contacts were hydrophobic, with the exception of Glu20, and six of the rescued pairs involved one of three phenylalanines, Phe6, Phe18, and Phe55. Two polar/charged-to-hydrophobic mutations, Lys56Leu and Asn18Phe, resulted in surface residues in AYEwt packing into the core of AYEdes. The core of AYEwt is largely packed with branched-chain aliphatic residues, but AYEdes’s core also includes aromatic phenylalanine residues (Fig. 6a and b). These bulkier residues increased the number of contacts between the helices (Fig. 5c), but they also caused the two α-helices to splay apart (Fig. 1a), resulting in the loss of contacts between smaller residues.
Table I.
Fraction time in contact for residue pairs that were rescued or missing in AYEdes
| Contact group | AYEwt | AYEdes | Status | ||||
|---|---|---|---|---|---|---|---|
| Contact pair | 25°C | 100°C | Contact pair | 25°C | 100°C | ||
| α1–α2 | Asn18–Leu59 | 0.00 | 0.01 | Phe18a–Leu59 | 0.99 | 0.98 | FRb |
| Leu19– Val55 | 0.01 | 0.87 | Leu19–Phe55a | 0.98 | 0.96 | FR | |
| Leu19– Phe58 | 0.99 | 1.00 | Leu19– Met58a | 0.18 | 0.05 | Mb | |
| Gln21–Phe58 | 0.96 | 0.70 | Ala21a–Met58a | 0.00 | 0.00 | M | |
| Leu22–Ala54 | 0.00 | 0.43 | Leu22–Trp54a | 0.99 | 0.99 | FR | |
| Leu22–Val55 | 1.00 | 0.93 | Leu22–Phe55a | 1.00 | 1.00 | FR | |
| Leu22–Leu59 | 1.00 | 0.74 | Leu22–Leu59 | 0.38 | 0.68 | M | |
| α1–β-sheet | Gln15–Val9 | 1.00 | 0.91 | Gln15–Val9 | 1.00 | 1.00 | FR |
| Gln15–Ala65 | 0.14 | 0.20 | Gln15–Pro65a | 0.99 | 0.99 | FR | |
| Leu19–Ala43 | 1.00 | 0.91 | Leu19–Val43a | 1.00 | 1.00 | FR | |
| Leu20–Ser35 | 0.99 | 0.66 | Leu20–Pro35a | 0.00 | 0.05 | M | |
| Leu20–Pro36 | 1.00 | 0.80 | Glu20a–Pro36 | 1.00 | 1.00 | FR | |
| Glu23–Pro36 | 0.65 | 0.03 | Ala23a–Pro36 | 1.00 | 0.99 | FR | |
| α2–β-sheet | Val52–Ile68 | 0.99 | 1.00 | Val52–Val68a | 0.42 | 0.32 | M |
| Val55–Ile8 | 0.67 | 0.28 | Phe55a–Ile8 | 1.00 | 1.00 | FR | |
| Val55–Val45 | 0.88 | 0.75 | Phe55a–Ile45a | 1.00 | 1.00 | FR | |
| Lys56–Leu6 | 1.00 | 0.94 | Leu56a–Phe6a | 1.00 | 1.00 | FR | |
| Lys56–Ile68 | 1.00 | 0.97 | Leu56a–Val68a | 0.94 | 0.72 | M | |
aResidues mutated in AYEdes relative to AYEwt.
bFR = Fully rescued, M = Missing.
Fig. 6.
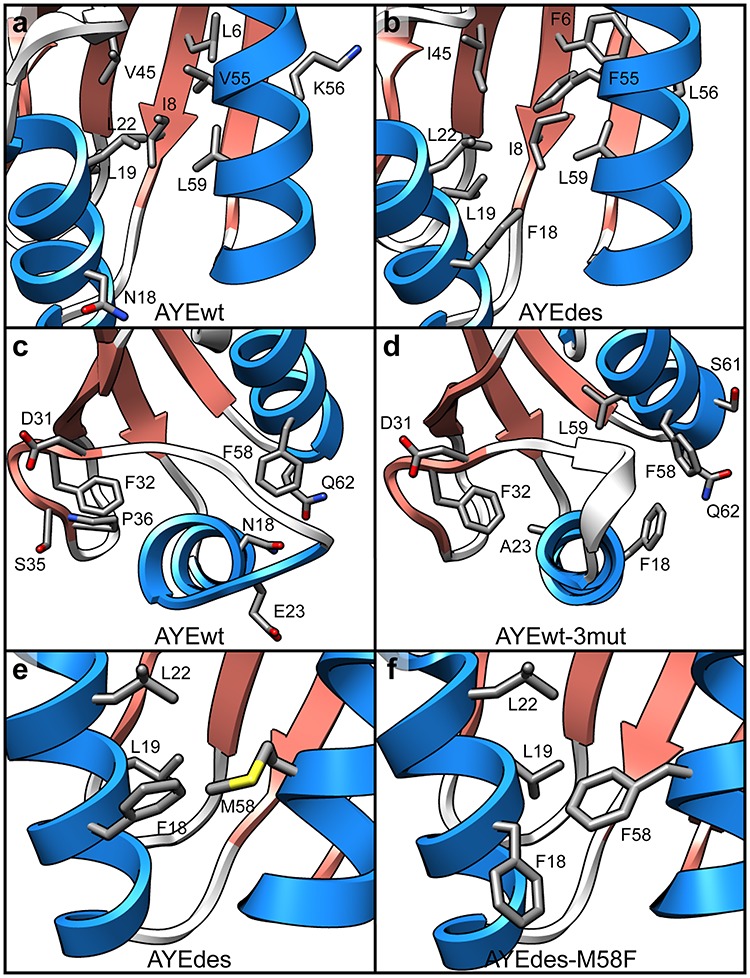
Inter-secondary-structure contacts. Representative final structures from simulations at 25°C showing Phe residues and their rescued contacts in sticks for (a) AYEwt and (b) AYEdes. Representative final structures from simulations at 100°C showing residues 18 and 23 and all residues they contacted for any amount of simulation time in the last 50 ns at 25°C in sticks for (c) AYEwt and (d) AYEwt-3mut. Representative final structures from simulations at 25°C showing residue 58 and all residues it contacted for any amount of simulation time in the last 50 ns in sticks for (e) AYEdes and (f) AYEdes-M58F.
AYEwt-4mut and AYEwt-5mut both had seven fully rescued contacts (Tables II and III). None of AYEwt-4mut’s rescued pairs directly incorporated one of its mutated residues (Val7, Trp32, Val44, and Leu46), but AYEwt-5mut had five fully rescued contact pairs that involved mutated residues. For AYEwt-5mut, four of the fully rescued mutations included Val55Phe and one included Ala54Trp.
Table II.
Fraction time in contact for residue pairs fully rescued in AYEwt-4mut
| Contact group | AYEwt | AYEwt-4mut | ||||
|---|---|---|---|---|---|---|
| Contact pair | 25°C | 100°C | Contact pair | 25°C | 100°C | |
| α1–α2 | Gln21–Phe58 | 0.96 | 0.70 | Gln21–Phe58 | 1.00 | 0.99 |
| Leu22–Val55 | 1.00 | 0.93 | Leu22–Val55 | 1.00 | 1.00 | |
| Leu22–Leu59 | 1.00 | 0.74 | Leu22–Leu59 | 1.00 | 1.00 | |
| α1–β-Sheet | Gln15–Val9 | 1.00 | 0.91 | Gln15–Val9 | 1.00 | 1.00 |
| Leu19–Ala43 | 1.00 | 0.91 | Leu19–Ala43 | 1.00 | 1.00 | |
| Leu20–Pro36 | 1.00 | 0.80 | Leu20–Pro36 | 1.00 | 0.98 | |
| Leu22–Ile8 | 0.92 | 0.54 | Leu22–Ile8 | 0.99 | 0.96 | |
Table III.
Fraction time in contact for residue pairs fully rescued in AYEwt-5mut
| Contact group | AYEwt | AYEwt-5mut | ||||
|---|---|---|---|---|---|---|
| Contact pair | 25°C | 100°C | Contact pair | 25°C | 100°C | |
| α1–α2 | Leu19–Val55 | 0.01 | 0.87 | Leu19–Phe55a | 1.00 | 1.00 |
| Leu22–Ala54 | 0.00 | 0.43 | Leu22–Trp54a | 1.00 | 0.99 | |
| Leu22–Val55 | 1.00 | 0.93 | Leu22–Phe55a | 1.00 | 1.00 | |
| α1–β-Sheet | Leu20–Pro36 | 1.00 | 0.80 | Leu20–Pro36 | 1.00 | 0.99 |
| α2–β-Sheet | Val55–Ile8 | 0.67 | 0.28 | Phe55a–Ile8 | 1.00 | 1.00 |
| Val55–Val45 | 0.88 | 0.75 | Phe55a–Val45 | 1.00 | 1.00 | |
| Lys56–Leu6 | 1.00 | 0.94 | Lys56–Leu6 | 1.00 | 0.98 | |
aResidues mutated in AYEwt-5mut relative to AYEwt.
Summary
In simulations at 100°C, AYEwt had a consistent increase in average core Cα-RMSD from 0 to 100 ns and the highest average core Cα RMSD and buried residue SASA across the entire simulation (Fig. 3). Qualitatively, AYEwt began denaturing at elevated temperature, evidenced by heightened dynamics and melting of secondary structure elements. In particular, the C-terminus of α1 melted at high temperature (Figs 1a and 4c). For AYEdes, on the other hand, the core Cα RMSD, α1 helical content and buried residue SASA were the same at 25 and 100°C (Figs 1a, 3 and 4c). Experimentally, AYEwt was the least stable of the four proteins, and our MD simulations reproduced this behavior.
AYEdes adopted conformations at both 25 and 100°C that were consistent with experimentally observed NOEs collected at 25°C. The relative stabilization of AYEdes compared to AYEwt at 100°C in MD simulations agrees with AYEdes’s experimental ΔΔG of −10.3 kcal/mol (Dantas et al., 2007) as well as previous findings that designed proteins tend to be more thermostable than their backbone template counterparts (Dahiyat and Mayo, 1997; Bryson et al., 1998; Kuhlman et al., 2003; Shah et al., 2007; Koga et al., 2012; McCully and Daggett, 2012; Childers and Daggett, 2017; Rocklin et al., 2017; Baker, 2019). As secondary structure propensity is a discerning factor in thermostable protein design algorithms, stabilizing α1 in AYEdes would be expected to increase thermostability (Malakauskas and Mayo, 1998).
AYEwt-4mut and AYEwt-5mut were originally designed to test the contributions of specific mutations in AYEwt to the stability of AYEdes. AYEwt-4mut (Glu7Val, Phe32Trp, His44Val, and Arg46Leu) and AYEwt-5mut (Glu7Val, His44Val, Arg46Leu, and Ala54Trp Val55Phe) were both found by Dantas et al. to be more stable than AYEwt, but not as stable as AYEdes, with ΔΔG = −5.2 kcal/mol for AYEwt-4mut and ΔΔG = −4.1 kcal/mol for AYEwt-5mut relative to AYEwt (Dantas et al., 2007). Based on core Cα RMSD at 100°C, buried residue SASA and α1 content, AYEwt-4mut was indeed more stable than AYEwt-5mut, and both variants had stabilities between AYEwt and AYEdes (Figs 3 and 4c). However, given the small difference in ΔΔG between AYEwt-4mut and AYEwt-5mut, relative variations in metrics like core Cα RMSD and SASA may be fortuitous. AYEwt-4mut increased α1–α2 contacts (Fig. 5c), though none of its mutated residues made contacts that were not already present in AYEwt (Table II). AYEwt-5mut increased α2–β-sheet contacts but not α1–β-sheet contacts, and it had the same number of α1–α2 contacts as AYEwt but did not lose any at 100°C (Fig. 5b and c). Its Ala54Trp and Val55Phe mutations were involved in increasing these interactions (Table III). Curiously, Trp32 in AYEwt-4mut was supposed to increase helix-sheet packing, and Trp54 and Phe55 in AYEwt-5mut were predicted to increase α1–α2 packing. However, we found that AYEwt-4mut had more α1–α2 contacts, and AYE-5mut had more α2–β-sheet contacts.
Stabilization of α1 increased thermostability in AYEwt-3mut
MD simulations at high temperature can identify the most unstable regions of proteins. The denaturation of these regions may be the first step of a protein’s unfolding pathway, and stabilization of these regions by mutation has been shown experimentally to increase protein thermostability (Pikkemaat et al., 2002; Childers and Daggett, 2017; Zimmerman et al., 2017; Korendovych, 2018). In simulations of AYEwt, the C-terminus of α1 was the most dynamic region of the protein (Fig. 2), and we observed melting of it at high temperature (Figs 1a and 4c). We designed AYEwt-3mut to stabilize this region, selecting Asn18Phe and Glu23Ala, both of which were involved in rescued contacts involving α1 in AYEdes (Table I). We also included Val55Phe, which was consistently involved in rescued contacts in AYEdes, AYEwt-4mut and AYEwt-5mut (Tables I–III).
Relative stability of AYEwt-3mut
Based on backbone motion, AYEwt-3mut was as stable as the four Dantas-designed proteins at 25°C and was similarly stable to AYEwt-4mut and AYEwt-5mut at 100°C (Fig. 3a). Based on buried hydrophobic surface area, AYEwt-3mut was stably folded at 25°C, but had more solvent exposure than the intermediately stable AYEwt-4mut and AYEwt-5mut though not as much as the least-stable AYEwt (Fig. 3b). If we were to express AYEwt-3mut, we would predict that it would be similarly stable to AYEwt-4mut and AYEwt-5mut, but not as stable as AYEdes.
Stabilization of α1
Based on RMSF, α1 in AYEwt-3mut was less dynamic than in AYEwt at both 25 and 100°C but not as stable as in AYEdes (Fig. 2). What AYEwt-3mut gained in stability in α1, it seemed to lose in the next-most dynamic region, β2. Indeed, α1 α-helical content was highest at 25°C in AYEwt-3mut among all of the proteins investigated, and it maintained its helicity nearly as well as AYEdes did at 100°C (Fig. 4c). While β2 was highly dynamic in AYEwt-3mut, it had the most β-sheet content at 25°C among all of the proteins investigated, though it was more variable at 100°C (Fig. 4e). AYEwt-3mut had as much β-sheet content in β4 as AYEwt, but it also had the greatest loss at 100°C (Fig. 4f).
In designing AYEwt-3mut by incorporating select residues from AYEdes, we attempted to stabilize the C-terminal end of α1 with Asn18Phe/Glu23Ala and stabilize core packing with Val55Phe. AYEwt-3mut had seven fully rescued contacts (Table IV). As with AYEwt-5mut, four of AYEwt-3mut’s seven fully rescued pairs included Val55Phe. Residues that interacted with residues 18 and 23 are shown in Fig. 6c and d in structures from the 100°C simulations. The hydrophobic packing of Ala23 and Phe18 maintained α1 in AYEwt-3mut, whereas the polar Asn18 and charged Glu23 residues did not maintain contacts as α1 unwound in AYEwt at high temperature.
Table IV.
Fraction time in contact for residue pairs fully rescued in AYEwt-3mut
| Contact group | AYEwt | AYEwt-3mut | ||||
|---|---|---|---|---|---|---|
| Contact pair | 25°C | 100°C | Contact pair | 25°C | 100°C | |
| α1–α2 | Leu19–Val55 | 0.01 | 0.87 | Leu19–Phe55a | 1.00 | 1.00 |
| Leu22–Val55 | 1.00 | 0.93 | Leu22–Phe55a | 1.00 | 1.00 | |
| α1–β-Sheet | Gln15–Val9 | 1.00 | 0.91 | Gln15–Val9 | 1.00 | 0.98 |
| Leu19–Ala43 | 1.00 | 0.91 | Leu19–Ala43 | 1.00 | 0.97 | |
| Leu20–Phe32 | 0.34 | 1.00 | Leu20–Phe32 | 1.00 | 1.00 | |
| α2–β-Sheet | Val55–Ile8 | 0.67 | 0.28 | Phe55a–Ile8 | 1.00 | 1.00 |
| Val55–Val45 | 0.88 | 0.75 | Phe55a–Val45 | 1.00 | 1.00 | |
aResidues mutated in AYEwt-3mut relative to AYEwt.
Summary
Based on our contact analysis, we designed an additional variant, AYEwt-3mut, which contained three mutations from AYEdes that we predicted would provide stability to the C-terminus of α1. AYEwt-3mut was more stable than AYEwt, less stable than AYEdes and similarly stable to AYEwt-4mut and AYEwt-5mut. In agreement with our design strategy, α1 did not melt in AYEwt-3mut as it did in AYEwt at 100°C, and it maintained the same amount of α-helical content as in AYEdes. AYEwt-3mut showed the importance of the C-terminus of α1 and hydrophobic packing in the core to the stability of the protein.
Wild-type phenylalanine imparts little more stability than designed methionine at position 58
Investigating minimally modified proteins with MD simulations allows for relatively rapid analysis of their structural dynamics and assessment of the stabilization imparted by their mutations. However, such variants might also be engineered to study the opposite: the destabilizing effects of mutations. Although the investigation of stabilizing effects has been a major focus of de novo computational protein design, there is still a need for quantitative methods to differentiate between ‘stabilizing’ and ‘destabilizing’ residues for the continual improvement of design algorithms (Johnson et al., 2015). A structural understanding of mutations that are destabilizing will aid in engineering similar motifs in designed proteins, for which a degree of instability may be necessary for proper function (Dill, 1990; Fersht, 1998).
Of the missing contact pairs in AYEdes, two of them involved Phe58Met, an unusual mutation away from a phenylalanine in AYEwt (Table I). Leu19–Phe58 was present 99% of the time at 25°C in AYEwt, but Leu19–Met58 was present only 18% of the time in AYEdes. Gln21–Phe58 was present 96% of the time in AYEwt, but Ala21–Met58 was present only 18% of the time in AYEdes. Residues that were mutated to phenylalanine in AYEdes and the minimal mutants were commonly involved in rescued contacts, so we tested whether Met58 would be destabilizing in AYEwt, or Phe58 could further stabilize AYEdes by creating the mutants AYEwt-F58M and AYEdes-M58F.
Met58 was not destabilizing in AYEwt-F58M
We predicted that Met58 would destabilize AYEwt-F58M relative to the wild type AYE-wt, containing Phe58. AYEwt-F58M deviated more from its starting structure at 25°C than AYEwt based on core Cα RMSD, and both deviated equally at 100°C (Fig. 3a). However, AYEwt-F58M had slightly lower backbone fluctuations than AYEwt at 25°C and much lower fluctuations at 100°C (Fig. 2). Both α1 and β4 were more stable in AYEwt-F58M compared with the wild type (Fig. 4c and f), but there was no difference in burial of hydrophobic surface area (Fig. 3b). AYEwt-F58M had far fewer contacts between α1 and α2 than AYEwt and even fewer than AYEdes (Fig. 5c), though, curiously, the number of contacts increased slightly when the temperature was raised. However, the number of buried hydrophobic contacts did not decrease in AYEwt-F58M compared to the wild type (Fig. 5d). Indeed, AYEwt-F58M lost six contacts relative to AYEwt, three of which directly involved Met58 (Table V). However, there were also seven rescued contacts in AYEwt-F58M, none of which involved Met58.
Table V.
Fraction time in contact for residue pairs rescued or missing in AYEwt-F58M
| Contact group | AYEwt | AYEwt-F58M | Status | ||||
|---|---|---|---|---|---|---|---|
| Contact pair | 25°C | 100°C | Contact pair | 25°C | 100°C | ||
| α1–α2 | Asn18–Phe58 | 1.00 | 1.00 | Asn18–Met58a | 0.61 | 0.77 | Mb |
| Leu19–Phe58 | 0.99 | 1.00 | Leu19–Met58a | 0.25 | 0.94 | M | |
| Gln21–Phe58 | 0.96 | 0.70 | Gln21–Met58a | 0.22 | 0.47 | M | |
| Leu22–Val55 | 1.00 | 0.93 | Leu22–Val55 | 0.90 | 0.95 | M | |
| Leu22–Leu59 | 1.00 | 0.74 | Leu22– Leu59 | 1.00 | 0.97 | FRb | |
| α1–β-Sheet | Gln15–Val9 | 1.00 | 0.91 | Gln15–Val9 | 1.00 | 0.96 | FR |
| Leu19–Ala43 | 1.00 | 0.91 | Leu19–Ala43 | 1.00 | 0.97 | FR | |
| Leu19–Val45 | 0.37 | 0.91 | Leu19–Val45 | 0.98 | 0.81 | R | |
| Leu22–Phe32 | 1.00 | 0.97 | Leu22–Phe32 | 0.77 | 0.92 | M | |
| Glu23–Asp31 | 0.36 | 0.12 | Glu23–Asp31 | 0.95 | 0.80 | R | |
| α2–β-Sheet | Val55–Val45 | 0.88 | 0.75 | Val55–Val45 | 0.99 | 0.91 | R |
| Lys56–Leu6 | 1.00 | 0.94 | Lys56–Leu6 | 1.00 | 0.96 | FR | |
| Lys56–Ile68 | 1.00 | 0.97 | Lys56–Ile68 | 0.99 | 0.93 | M | |
aResidue mutated in AYEwt-F58M relative to AYEwt.
bR = Rescued, FR = Fully rescued, M = Missing.
We observed destabilization of AYEwt-F58M relative to AYEwt in the decreased number of contacts between α1 and α2 and contacts pairs with Met58, and the slightly higher core Cα RMSD at 25°C. However, both Cα RMSF and secondary structure content in α1 and β4 actually pointed to increased stability. All other measures showed the proteins to be equally stable. We concluded that the Phe58Met mutation did result in fewer contacts and decreased α1–α2 packing, as predicted, but it was not destabilizing in AYEwt overall.
Phe58 was slightly stabilizing in AYEdes-M58F
We predated that Met58Phe would stabilize AYEdes-M58F relative to AYEdes, containing Met58. AYEdes-M58F had very slightly lower backbone motion, both as measured by core Cα RMSD and Cα RMSF (Figs 2 and 3a). Secondary structure content and buried residue SASA were similar (Figs 2 and 3b). Where we did see stronger evidence of stabilization was in the contacts. There were increased contacts between α1 and α2 at 25°C and among buried residues (Fig. 5c and d). AYEdes–M58F restored one contact that did not involve Phe58 (Table VI). The Leu19–Met58 contact that was missing in AYEdes was present more frequently as Leu19–Phe58 in AYEdes-M58F (0.18 and 0.94 fraction time in contact at 25°C, respectively). However, it was just below the 0.95 cutoff to be classified as ‘in contact’ or ‘rescued’ in our analysis, and thus was not included in Table VI. On the other hand, the missing Ala21–Phe58 contact was no more likely to be present in AYEdes-M58F than Ala21–Met58 in AYEdes (0.04 and 0.00, respectively). Met58 is less bulky than Phe58, but the space in AYEdes that Met58 did not occupy was packed by another phenylalanine, Phe18 (Fig. 6e). In AYEdes-M58F, Phe18 rotated out of the way to accommodate Phe58 (Fig. 6f). While Phe58 provided more hydrophobic surface area to bury than Met58, the stabilization observed in AYEdes-M58F may have been minimal due to Phe18’s ability to pack into the hole left by Met58 in AYEdes.
Table VI.
Fraction time in contact for residue pairs rescued or missing in AYEdes-M58F
| Contact group | AYEdes | AYEdes-M58F | Status | ||||
|---|---|---|---|---|---|---|---|
| Contact pair | 25°C | 100°C | Contact pair | 25°C | 100°C | ||
| α1–α2 | Phe18–Leu59 | 0.99 | 0.98 | Phe18–Leu59 | 0.94 | 0.92 | Ma |
| Leu19–Phe55 | 0.98 | 0.96 | Leu19–Phe55 | 0.97 | 0.78 | M | |
| Leu19–Leu59 | 0.99 | 0.99 | Leu19–Leu59 | 0.97 | 0.93 | M | |
| Leu22–Trp54 | 0.99 | 0.99 | Leu22–Trp54 | 1.00 | 0.81 | M | |
| α2–β-Sheet | Leu56–Val68 | 0.94 | 0.72 | Leu56–Val68 | 0.99 | 0.90 | Ra |
aR = Rescued, M = Missing.
We observed stabilization due to Met58Phe in AYEdes-M58F in the number of contacts, especially between α1 and α2 and among buried residues. Phe58 restored the contact between residues 19 and 58 that was present in AYEwt but not AYEdes. We did not observe many differences between the overall motions of the proteins, but the lower backbone motion observed in AYEdes-M58F suggests a small stabilizing effect of Phe58. We concluded that Met58Phe increased the number of contacts, as predicted, but only had a minor stabilizing effect on the protein overall.
Summary
To broaden our investigation to both stabilizing and destabilizing mutants in AYEdes’s engineered sequence, we designed AYEwt-F58M with a mutation that we predicted to be destabilizing based on its inter-secondary-structure contacts in AYEdes (Table I). We also designed AYEdes-M58F to investigate whether the wild type Phe58 was stabilizing in AYEdes. We observed little effect when changing between Met and Phe at position 58 in either AYEwt or AYEdes. Phe58 was slightly stabilizing in the context of AYEdes, but Met58 was not destabilizing in AYEwt. The biggest effects were that Met58 resulted in fewer contacts between α1 and α2 in AYEwt-F58M (Fig. 5c), and Phe58 restored the contact between residues 19 and 58 in AYEdes-M58F that was present in AYEwt but not AYEdes (Table I).
Hydrophobic core packing was a reliable measure of thermostability
The contribution of core hydrophobicity and buried nonpolar surface area to protein stability has been consistently reported (Pace, 1992; Malakauskas and Mayo, 1998; Magliery and Regan, 2004; Rocklin et al., 2017; Nguyen et al., 2019). In designing AYEwt-4mut and AYEwt-5mut, Dantas et al. chose mutant residues that they predicted would increase burial of nonpolar surface area in the variants upon folding. When considering the dynamics of these proteins, rather than simply the static structures predicted by RosettaDesign, we found that the core hydrophobic packing was maintained as predicted.
We quantified the packing of each variant’s hydrophobic core with two measures, SASA of buried residues and contacts among buried residues. The burial of these often-hydrophobic residues helps drive the process of protein folding, and less solvent exposure for buried residues is thus an indication of proper, native folding and protein stability. AYEwt had the greatest buried solvent exposure at 100°C, AYEdes had the least, and AYEwt-5mut, and AYEwt-4mut were in between (Fig. 3b). For the second measure of core hydrophobicity, we quantified the number of hydrophobic contacts among buried residues. In agreement with the SASA data, AYEdes had the greatest number of buried hydrophobic contacts, AYEwt had the least, and AYEwt-4mut and AYEwt-5mut had an intermediate number (Fig. 5d). Thus, Dantas et al. (2007) met their structural goal of stabilizing proteins by increasing buried hydrophobic surface area in their designed mutants of AYEwt.
Inter-secondary-structure interactions increased protein thermostability
Quantifying inter-residue contacts is a common technique for assessing which residue interactions are critical for maintaining a protein’s folded structure. The importance of optimal inter-residue interaction networks for the rapid, reliable design of stable proteins has also been explored (specifically for hydrogen bonds), and designed proteins have been shown to engage in stabilizing interactions that are not seen in naturally occurring proteins (Boyken et al., 2016; Baker, 2019). In choosing mutations to implement in their minimally modified variants, Dantas et al. (2007) also selected residues that would promote interactions between specific secondary structure elements even at high temperature, based on structures and scoring data from RosettaDesign.
Nearly all of the fully rescued contact pairs in AYEdes (12 total) involved at least one of its mutated residues (Table I), and the vast majority of these were hydrophobic amino acids, with Phe present in half of AYEdes’s rescued contact pairs (Fig. 5a and b). A similar pattern was noted for AYEwt-3mut and AYEwt-5mut, with seven and six fully rescued pairs each (Tables III and IV). Although they have fewer total rescued contacts than AYEdes, more than half of their rescued pairs also involved one or multiple of their mutated amino acids. For AYEwt-3mut, the sole mutation involved was Val55Phe, while for AYEwt-5mut, both Ala54Trp and Val55Phe were involved in rescued pairs. Thus, in designed variants, bulky hydrophobic residues rescued the most contact pairs that were lost in AYEwt at 100°C.
However, not all of the rescued contacts in AYEdes, AYEwt-5mut, and AYEwt-3mut directly involved mutated residues, and AYEwt-4mut, despite containing four bulky hydrophobic mutations, had none of its mutated residues counted among its seven rescued pairs. Considering the thermostability of AYEwt-4mut in simulation, its rescued contact pairs were likely pivotal for achieving that stability, even without direct involvement in rescuing inter-secondary-structure interactions.
Limitations
The structural properties we calculated here provide insight into the relative stabilities of the AYE family of proteins and suggest mechanisms for further stabilizing or destabilizing additional designs. However, these biophysical properties are by no means a measure of the proteins’ relative stabilities, or ΔΔGs. In many cases, relative stabilities inferred from structural properties correlate well with the experimentally determined ΔΔGs, but this is not always to be expected. Free energy calculations may be performed using MD simulation data to calculate ΔΔG directly using techniques such as free energy perturbation (Straatsma and Berendsen, 1988), enveloping distribution sampling (Christ and van Gunsteren, 2007) or λ-dynamics (Hayes et al., 2018). The major challenge when applying these techniques to calculate free energies of folding in proteins is sufficiently sampling both the folded and denatured states with the available computational resources. Many ΔΔG calculations operate on the assumption that the unfolded, transition and any intermediate states are unperturbed while only the native state is affected by the mutations. However, this assumption is not always valid, particularly in the case of thermophilic proteins (Sawle et al., 2017), and nonnative states may need to be considered (Pan and Daggett, 2001). Coarse-grained models or umbrella-sampling methods may be applied in cases where the nonnative states must be considered, including structure-based models (also known as Gō-models) in combination with the weighted histogram analysis method (Ferrenberg and Swendsen, 1988; Wang et al., 2012). We have previously applied these methods alongside all-atom MD simulations for thermodynamic and structural insights in a family of three-helix bundle proteins (Nguyen et al., 2019). Applying such methods in this study could offer further evidence that our simulations reproduced the proteins’ relative stabilities as observed by experiment and better assess the success of our designed variants in de/stabilizing the proteins as predicted.
Conclusions
We performed MD simulations for a wild type protein (AYEwt) and its engineered variant (AYEdes) at 25 and 100°C, along with five minimally modified proteins (AYEwt-4mut, AYEwt-5mut, AYEwt-3mut, AYEwt-F58M, and AYEdes-M58F). Based on core Cα-RMSD, DSSP, and SASA, we reproduced AYEdes’s thermostability and AYEwt’s denaturation at 100°C as well as the intermediate stability of AYEwt-4mut and AYEwt-5mut. Using a contact analysis, we determined that mutations chosen for AYEwt-4mut and AYEwt-5mut did increase inter-residue interactions between secondary structure elements and maximize the amount of buried hydrophobic surface area, but not quite as predicted. For these designed variants and for AYEdes, we found that hydrophobic interactions were maximized, and solvent exposure of buried residues was minimized. We identified the most persistent interactions in each structure via a contact analysis, finding bulky, hydrophobic residues to be commonly involved in interactions in designed variants that were absent in AYEwt at high temperature. With these data, we designed additional variants, AYEwt-3mut, AYEwt-F58M, and AYEdes-M58F, to test mutations that we predicted would stabilize α1 and assess the effect of a seemingly destabilizing mutation in AYEdes, Phe58Met. The behavior of AYEwt-3mut showed intermediate thermostability and stabilization of α1, in agreement with our predictions, and it confirmed the ability of bulky hydrophobic residues to increase inter-secondary-structure contacts. While Met58 was involved in destabilizing interactions in AYEdes, Phe58 was only somewhat stabilizing in AYEdes, and Met58 was not particularly destabilizing in AYEwt.
Acknowledgements
The authors would like to thank Korin Wheeler for serving as the reader for Matthew Gill’s honors thesis and Amanda Jonsson for feedback on the manuscript. This work was supported by startup funds from the Santa Clara University College of Arts and Sciences, a DeNardo Research Award, and award number R15GM134439 from the National Institute of General Medical Sciences of the National Institutes of Health.
Conflict of interest
None.
Author contributions
MG and MEM designed the study, analyzed the data, and wrote the manuscript. MG performed the MD simulations and analysis calculations.
References
- Baker D. (2019) Prot. Sci., 28, 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck D.A.C., McCully M.E., Alonso D.O.V., Daggett V. (2000) In lucem molecular mechanics (in lucem molecular mechanics (ilmm) 2000–2020). University of Washington, Seattle. [Google Scholar]
- Becktel W.J. and Schellman J.A. (1987) Biopolymers, 26, 1859–1877. [DOI] [PubMed] [Google Scholar]
- Beglov D. and Roux B. (1998) J. Chem. Phys., 100, 9050. [Google Scholar]
- Best R.B., Zhu X., Shim J., Lopes P.E.M., Mittal J., Feig M., MacKerell A.D. (2012) J. Chem. Theory Comput., 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyken S.E., Chen Z., Groves B. et al. (2016) Science, 352, 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson J.W., Desjarlais J.R., Handel T.M., DeGrado W.F. (1998) Protein Sci., 7, 1404–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers M.C. and Daggett V. (2017) Mol. Syst. Des. Eng., 2, 9–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ C.D. and Gunsteren W.F. (2007) J. Chem. Phys., 126, 184110. [DOI] [PubMed] [Google Scholar]
- Dahiyat B.I. and Mayo S.L. (1997) Science, 278, 82–87. [DOI] [PubMed] [Google Scholar]
- Dantas G., Corrent C., Reichow S.L. et al. (2007) J. Mol. Biol., 366, 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas G., Kuhlman B., Callender D., Wong M., Baker D. (2003) J. Mol. Biol., 332, 449–460. [DOI] [PubMed] [Google Scholar]
- Dill K.A. (1990) Biochemistry, 29, 7133–7155. [DOI] [PubMed] [Google Scholar]
- Doreleijers J.F., Nederveen A.J., Vranken W., Lin J., Bonvin A.M.J.J., Kaptein R., Markley J.L., Ulrich E.L. (2005) J. Biomol. NMR, 32, 1–12. [DOI] [PubMed] [Google Scholar]
- Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., Sali A. (2006) Curr. Protoc. Bioinformatics, 15, 5.6.1–5.6.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrenberg A.M. and Swendsen R.H. (1988) Phys. Rev. Lett., 61, 2635–2638. [DOI] [PubMed] [Google Scholar]
- Fersht A. (1998) Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. W. H. Freeman, New York. [Google Scholar]
- Garcίa‐Sáez I., Reverter D., Vendrell J., Avilés F.X., Coll M. (1997) EMBO J., 16, 6906–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes R.L., Vilseck J.Z., Brooks C.L. (2018) Protein Sci., 27, 1910–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. (1996) J. Mol. Graph., 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Johnson L.B., Gintner L.P., Park S., Snow C.D. (2015) Protein Eng. Des. Sel., 28, 259–267. [DOI] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. (1998) J. Chem. Phys., 79, 926. [Google Scholar]
- Kabsch W. and Sander C. (1983) Biopolymers, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Koga N., Tatsumi-Koga R., Liu G., Xiao R., Acton T.B., Montelione G.T., Baker D. (2012) Nature, 491, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korendovych I.V. (2018) Protein engineering, Bornscheuer U.T. and Höhne M. (eds). Springer New York, New York, NY, pp. 15–23. [Google Scholar]
- Kuhlman B., Dantas G., Ireton G.C., Varani G., Stoddard B.L., Baker D. (2003) Science, 302, 1364–1368. [DOI] [PubMed] [Google Scholar]
- Lee B. and Richards F.M. (1971) J. Mol. Biol., 55, 379–380. [DOI] [PubMed] [Google Scholar]
- MacKerell A.D.J., Bashford D., Bellott et al. (1998) J. Phys. Chem. B, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- MacKerell A.D.J., Feig M., Brooks C.L. 3rd (2004) J. Comput. Chem., 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- Magliery T.J. and Regan L. (2004) Eur. J. Biochem., 271, 1595–1608. [DOI] [PubMed] [Google Scholar]
- Malakauskas S.M. and Mayo S.L. (1998) Nat. Struct. Mol. Biol., 5, 470–475. [DOI] [PubMed] [Google Scholar]
- McCully M.E. and Daggett V. (2012) Protein engineering handbook, Lutz S. and Bornscheuer U.T. (eds). Wiley-VCH, Weinheim, pp. 89–114. [Google Scholar]
- Nguyen C., Young J.T., Slade G.G., Oliveira R.J., McCully M.E. (2019) Biophys. J., 116, 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace C.N. (1992) J. Mol. Biol., 226, 29–35. [DOI] [PubMed] [Google Scholar]
- Pan Y. and Daggett V. (2001) Biochemistry, 40, 2723–2731. [DOI] [PubMed] [Google Scholar]
- Phillips J.C., Braun R., Wang W. et al. (2005) J. Comput. Chem., 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikkemaat M.G., Linssen A.B.M., Berendsen H.J.C., Janssen D.B. (2002) Protein Eng., Des. Sel., 15, 185–192. [DOI] [PubMed] [Google Scholar]
- Razvi A. and Scholtz J.M. (2006) Protein Sci., 15, 1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocklin G.J., Chidyausiku T.M., Goreshnik I. et al. (2017) Science, 357, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawle L., Huihui J., Ghosh K. (2017) J. Chem. Theory Comput., 13, 5065–5075. [DOI] [PubMed] [Google Scholar]
- Schellman J.A. (1987) Annu. Rev. Biophys. Biophys. Chem., 16, 115–137. [DOI] [PubMed] [Google Scholar]
- Shah P.S., Hom G.K., Ross S.A., Lassila J.K., Crowhurst K.A., Mayo S.L. (2007) J. Mol. Biol., 372, 1–6. [DOI] [PubMed] [Google Scholar]
- Shen M.Y. and Sali A. (2006) Protein Sci., 15, 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straatsma T.P. and Berendsen H.J.C. (1988) J. Chem. Phys., 89, 5876–5886. [Google Scholar]
- Wang J., Oliveira R.J., Chu X., Whitford P.C., Chahine J., Han W., Wang E., Onuchic J.N., Leite V.B. (2012) Proc. Natl. Acad. Sci., 109, 15763–15768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman M.I., Hart K.M., Sibbald C.A., Frederick T.E., Jimah J.R., Knoverek C.R., Tolia N.H., Bowman G.R. (2017) ACS Cent. Sci., 3, 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]