

Abstract
Enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase that suppresses gene expression. Previously, we developed a conditional null model where EZH2 is knocked out in uterus. Deletion of uterine EZH2 increased proliferation of luminal and glandular epithelial cells. Herein, we used RNA-Seq in wild-type (WT) and EZH2 conditional knockout (Ezh2cKO) uteri to obtain mechanistic insights into the gene expression changes that underpin the pathogenesis observed in these mice. Ovariectomized adult Ezh2cKO mice were treated with vehicle (V) or 17β-estradiol (E2; 1 ng/g). Uteri were collected at postnatal day (PND) 75 for RNA-Seq or immunostaining for epithelial proliferation. Weighted gene coexpression network analysis was used to link uterine gene expression patterns and epithelial proliferation. In V-treated mice, 88 transcripts were differentially expressed (DEG) in Ezh2cKO mice, and Bmp5, Crabp2, Lgr5, and Sprr2f were upregulated. E2 treatment resulted in 40 DEG with Krt5, Krt15, Olig3, Crabp1, and Serpinb7 upregulated in Ezh2cKO compared with control mice. Transcript analysis relative to proliferation rates revealed two module eigengenes correlated with epithelial proliferation in WT V vs. Ezh2cKO V and WT E2 vs. Ezh2cKO E2 mice, with a positive relationship in the former and inverse in the latter. Notably, the ESR1, Wnt, and Hippo signaling pathways were among those functionally enriched in Ezh2cKO females. Current results reveal unique gene expression patterns in Ezh2cKO uterus and provide insight into how loss of this critical epigenetic regulator assumingly contributes to uterine abnormalities.
Keywords: endometrial cancer, endometrium, epigenetics, estrogen, female reproduction, RNA-Seq, uterus
INTRODUCTION
Epigenetic changes in chromatin and DNA can result in altered gene expression without causing changes in DNA base sequences. Epigenetic mechanisms include posttranslational histone modification (e.g., methylation or acetylation), DNA methylation, and expression of both small and long noncoding RNAs (20). Epigenetic regulators, which include polycomb proteins and histone deacetylases, are essential for normal cell function and differentiation but when disrupted are associated with neoplastic and other pathologies (9).
Enhancer of zeste homolog 2 (EZH2) is the rate-limiting catalytic subunit of the polycomb repressive complex 2 (PRC2), a histone methyltransferase critical for histone modifications. The PRC2 complex regulates gene function by trimethylating histone 3 on lysine 27 (H3K27me3) (57). This methylation induces chromatin compaction and represses gene expression by obstructing transcriptional access to gene promoters. Deletion of epigenetic regulators such as EZH2 in the uterus will produce differential gene expression patterns identifiable through high throughput technology such as RNA-Seq, highlighting the importance of epigenetic mechanisms on transcriptional landscapes.
Upregulation of EZH2 is linked to pathologies, including endometrial and other cancers, and its expression has prognostic significance (15, 16, 47). However, loss-of-function mutations in Ezh2 also occur in conditions such as acute myeloid leukemia, as PRC2 inhibits expression of tumor suppressor genes as well as oncogenes (53). Global Ezh2 knockout is embryonic-lethal and results in abnormal mouse embryos with accumulated mesoderm cells (46). In uteri, altered EZH2 expression is associated with endometriosis and uterine fibroids (12, 65), in addition to endometrial cancer, suggestive of a critical uterine role for this epigenetic modifier.
Two groups recently created conditional knockout mice lacking uterine EZH2 (Ezh2cKO) to determine the consequences of EZH2 deletion (17, 44). Uterine EZH2 deletion induces aberrant epithelial proliferation, uterine hypertrophy, cystic endometrial hyperplasia, and altered E2 responsiveness (17, 44). In addition, Fang et al. (17) reported uterine epithelial stratification and increased expression of tumor markers. Predictably, uterine EZH2 deletion reduced H3K27me3 marks, suggesting transcriptomic effects (44). Analysis of Ezh2cKO mice underscores the importance of EZH2 for normal uterine growth and function. Uterine transcriptome analysis has been informative in understanding transcriptional effects of estrogen signaling through its main receptor ESR1 that induce uterine cell proliferation (63). We thus hypothesized that uterine EZH2 deletion would result in unique transcriptional changes associated with increased uterine epithelial cell proliferation.
In this study, we used RNA-Seq to compare gene expression in adult ovariectomized (OVX) wild-type (WT) and EzhcKO mice receiving either vehicle (V) or 17β-estradiol (E2) treatment. An ancillary goal was to examine E2 effects on EZH2-regulated mechanisms. Additionally, we used weighted gene coexpression network analysis (WGCNA) to link uterine transcriptomic changes to aberrant epithelial cell proliferation, the major pathology in Ezh2cKO mice (17, 44).
MATERIALS AND METHODS
Animals, animal care, and treatments.
Transgenic mice expressing Cre recombinase under control of the Pgr promoter on a mixed C57BL/6 and 129SvEv background were produced at the University of Florida, as previously described (44). Transgenic Pgr-Cre mice were bred with homozygous floxed Ezh2 mice. Male and female offspring expressing Pgr-Cre and heterozygous for floxed Ezh2 (Pgrwt/Cre, Ezh2+/flox) were then crossed to obtain females expressing Pgr-Cre and homozygous for floxed Ezh2 (Pgrwt/Cre, Ezh2flox/flox), whereas WT controls expressed floxed Ezh2 but no Cre (Pgrwt/wt, Ezh2flox/flox).
Female mice were OVX at postnatal day (PND) 60. Body weight increased in all groups by PND 74 after OVX (20.73 ± 0.76 vs. 23.03 ± 0.97 g). At this age, uterine weight of Ezh2cKO females is larger than that of WT mice (83.04 ± 20.41 vs. 186.03 ± 38.32 g) (44). Adult controls (Ezh2flox/flox) and Ezh2cKO (Ezh2flox/flox, PgrCre/+) mice (n = 4/group) were given either oil or 1 ng/g BW of E2 (E1024-1G, Sigma-Aldrich), and 24 h later, uteri were either fixed in 10% neutral buffered formalin for MKI67 immunostaining or frozen in liquid N2 and stored at −80°C for RNA extraction/RNA-Seq analysis. We selected a 1 ng/g E2 dose as this dose produces a submaximal epithelial proliferation response, whereas 10 ng/g produces maximal responses. The time of uterine collection was based on the maximal E2-induced proliferation response observed 24 h posttreatment (44). Body weights were WT V (20.31 ± 0.46 g); Ezh2cKO V (21.33 ± 1.51 g); WT E2, V (20.84 ± 0.73 g); and Ezh2cKO E2 (20.4 ± 0.71 g).
Mice were housed in standard mouse cages at 25°C, with 12 h:12 h light-dark cycles and ad libitum access to water and commercial rodent diet. All procedures and experiments were approved by the University of Florida Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Genotyping.
Genotyping for Pgr-Cre and floxed Ezh2 was performed at weaning by multiplex PCR on genomic DNA, as described (44). Briefly, specific primers for Pgr (primer 1, 5′-TATACCGATCTCCCTGGACG-3′; primer 2, 5′-ATGTTTAGCTGGCCCAAATG-3′; primer 3, 5′-CCCAAAGAGACACCAGGAAG-3′) and floxed Ezh2 (primer 1, 5′-CATGTGCAGCTTTCTGTTCA-3′; primer 2, 5′-CACAGCCTTTCTGCTCACTG-3′) (Jackson Laboratory, stock #022616) were used. PCR was performed by denaturing DNA at 94°C for 3 min, followed by 30 amplification cycles (94°C for 45 s, 60°C for 45 s, 72°C for 90 s) and a final extension step at 72°C for 5 min. The final PCR reaction mixture was visualized on a 1–2% agarose gel using ethidium bromide, and the expected PCR products for Pgr-Cre were at 500 bp and 300 bp for floxed Ezh2.
Immunohistochemistry for MKI67.
Immunohistochemistry for MKI67 was performed as described (44). Briefly, dissected uteri were formalin fixed and paraffin-embedded, sectioned at 5–6 μm, deparaffinized, and rehydrated. Following antigen retrieval, endogenous peroxidase activity was quenched with H2O2 in methanol. Slides were incubated with the primary antibody overnight (1:1,000 dilution) using rabbit monoclonal IgG for MKI67 (antibody ID AB 302459, catalog #ab16667, Abcam Inc.). Primary antibody binding was localized using the Vectastain ABC (kit catalog #PK-4000; Vector Laboratories, Burlingame, CA) and DAB Substrate Kit (Vector Laboratories); then sections were counterstained with Gill hematoxylin (catalog #245-654, Fisher Scientific).
Morphometric analysis of uterine MKI67 staining.
To quantify uterine MKI67 staining, images were captured with a ×25 objective and Olympus BH-2 microscope (Olympus, Center Valley, PA). Percentage of epithelial MKI67 staining was determined as described (44). Briefly, a box measuring 160 × 80 pixels was placed over 10 random luminal epithelial areas for each uterine section. Percentages of MKI67+ epithelial cells within each boxed region were determined by an investigator blinded to mouse genotype/treatment.
RNA isolation and RNA-Seq analysis.
Uterine tissue was frozen in liquid nitrogen, and stored at −80°C until used. Four animals per group were chosen based on our prior work and earlier RNA-Seq studies utilizing similar numbers of average reads per sample (5, 23, 26) that showed this replicate number should provide sufficient statistical power. RNA extraction was performed using the TRI-reagent (Thermo Fisher Scientific) and purified using Qiagen columns (Qiagen, Germantown, MD) following the manufacturers’ instructions. Total RNA concentration was determined with a Qubit 2.0 Fluorometer (Thermo Fisher), and RNA quality was assessed with Nanodrop (Agilent Technologies).
To produce RNA-Seq libraries, 200 ng of total RNA was used to isolate mRNA using a NEBNext Poly(A) mRNA magnetic isolation module (New England Biolabs, Ipswich, MA) and to perform RNA library construction with NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs) according to the manufacturer’s instructions. Briefly, extracted mRNA was fragmented in NEBNext first-strand synthesis buffer by heating at 94°C, followed by first-strand cDNA synthesis using reverse-transcriptase and random primers. Synthesis of double-stranded cDNA was performed using the 2nd-strand master mix provided with the kit. Resulting double-stranded cDNA was end-repaired, dA-tailing and ligated with NEBNext adaptors. Finally, libraries were enriched by 12 cycles of amplification and purified by Meg-Bind RxnPure Plus beads (Omega Biotek, Norcross, GA).
Barcoded libraries were sized on a bioanalyzer, and quantitated by Qubit and quantitative PCR (Kapa Biosystems Wilmington, MA). Eighteen individual libraries were pooled at equal molar concentrations (20 nM), and a total of four lanes of 2 × 100 bp were run on an Illumina HiSeq3000 (Illumina). RNA library construction and sequencing were performed at the Interdisciplinary Center for Biotechnology Research (ICBR), University of Florida.
Bioinformatics analyses of RNA-Seq data.
Reads acquired from the sequencing platform were processed with the Cutadapt program (39) to trim off sequencing adaptors, low-quality bases, and potential errors introduced during sequencing or library preparation. Reads with a quality phred-like score <20 and read length <40 bases were excluded from RNA-Seq analysis.
The 107,193 transcripts of GRCm38.p6 (also termed mm10 and released on January 9, 2012), were retrieved from the NCBI Refseq database and used as reference sequences for RNA-Seq analysis. All samples, as listed in Supplemental Table S1 (Supplemental Material available at URL: https://figshare.com/s/3b0de76388d1e1394790), were mapped independently to reference sequences by using the bowtie2 mapper (version 2.2.3) (32). Mapping results were processed with the same tools and scripts developed in-house at ICBR to remove potential PCR duplicates. Gene expression was assessed by counting number of mapped reads for each transcript (66). Significant up- and downregulated genes were selected using the q value [false discovery rate (FDR) ≤ 0.05] and fold-change for downstream analysis.
Principal component analyses (PCA) based on all identified genes in the various comparisons were done with the mixOmics package in R. Volcano plot analysis based on differentially expressed genes (DEG) for these comparisons were done with the OriginPro 2019 64 bit graphing program (OriginLab Corporation, Northampton, MA). Heatmap analysis based on DEG was done using the heatmapper site (http://www2.heatmapper.ca/expression/). Potential protein-protein interactions (PPI) based on DEG were determined with STRING (54) (https://string-db.org/). For functional enrichment analysis, DEG were imported in g:GOSt (https://biit.cs.ut.ee/gprofiler/gost), and the significance threshold used was Benjamini-Hochberg with an FDR ≤ 0.01. The output included all significant Gene Ontology (GO) terms and pathways (KEGG, Reactome, and WikiPathways). Pathway enrichment analysis using the overrepresentation analysis (ORA), and KEGG functional database was performed using WebGestalt (WEB-based GEne SeT AnaLysis Toolkit) (61) to obtain biological insights from DEG.
Key player analyses to identify coexpressed and hub genes were done as described previously (56). The key player package considers centrality score as determined based on eigenvector-based centrality.
WGCNA.
WGCNA describes correlation patterns among genes based on gene expression data. The WGCNA package in R (31) was used to find unsigned weighted gene coexpression modules. The blockwiseModules function was run with a soft thresholding power of 18 to indicate the gene cluster. To identify significant gene modules, correlation scores between genes and trait was used to rank the gene cluster, which utilized the eigengene network methodology by using Pearson Correlation/Bicor. Trait data used for this analysis were percentages of epithelial MKI67 staining, as detailed above. Genes within module eigengenes (ME) that were significant were further analyzed for potential PPI with STRING and for functional analysis or otherwise called ORA. For the latter, genes within significantly correlated modules were downloaded into g:GOSt (https://biit.cs.ut.ee/gprofiler/gost), and the significance threshold used was Benjamini-Hochberg with an FDR ≤ 0.01. Output included all significant GO terms and pathways (KEGG, Reactome, and WikiPathways). To examine for hub genes, PPI files generated with STRING were imported into the cytohubba app in Cytoscape (11, 51).
Statistical analysis for MKI67 immunostaining.
Uterine epithelial proliferation data were analyzed for normality by using the Wilk-Shapiro test (V9.4; SAS Analytics, Cary, NC) first. These data were then analyzed by the general linear model procedure in SAS with genotype, E2/V treatment, and their interaction as main effects, and individual mice were the experimental unit. A P value of ≤ 0.05 was considered significant. All data are presented as means ± SE.
RESULTS
Epithelial MKI67 staining.
As shown in Fig. 1, V-treated Ezh2cKO mice have a greater epithelial MKI67 staining than WT V mice (P ≤ 0.0001). However, E2 stimulated proliferation in both groups and abolished differences between Ezh2cKO and WT females (P = 0.63). WT E2 mice showed greater cell proliferation than WT V counterparts (P ≤ 0.0001). Ezh2cKO mice treated with E2 had further increases in epithelial proliferation relative to V-treated Ezh2cKO mice (P ≤ 0.0001). These results mirror results from our previous work testing three E2 concentrations in Ezh2cKO mice (44). We repeated this analysis in the current study as the integrative correlation analyses (detailed below) generally require results from the same animals to be tested across studies. For the few animals where this was not possible, RNA-Seq data were linked to the histological results for a comparably aged animal within the same treatment group.
Fig. 1.
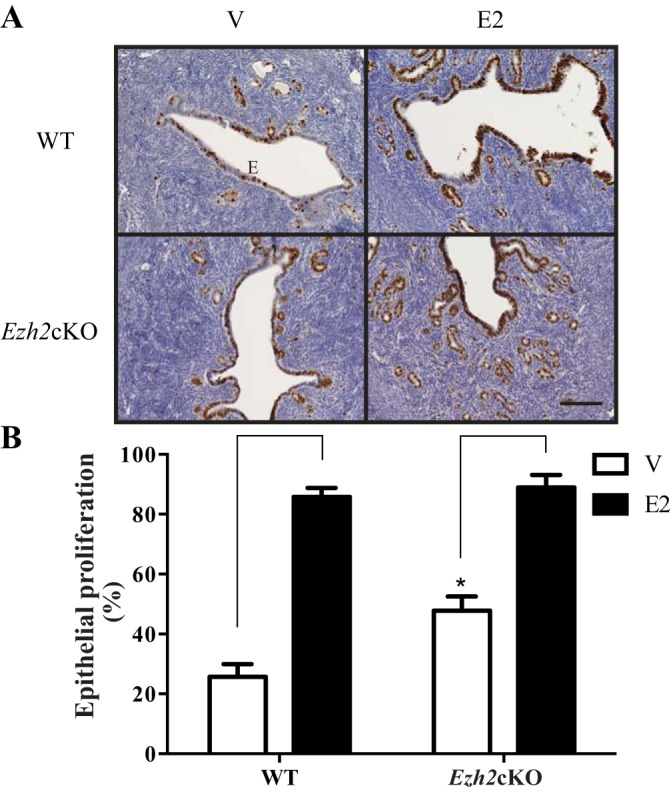
Epithelial (E) proliferation in EZH2 conditional knockout (Ezh2cKO) and wild-type (WT) uteri with or without 17β-estradiol (E2) treatment. The vehicle (V)-treated Ezh2cKO uteri show greater epithelial proliferation than the WT V group (P ≤ 0.0001), but proliferation was similar in both groups following E2 treatment. Brackets on bar indicate treatment effect within genotype. *Difference within same treatment. n = 6–9 for the various groups. All images are at the same magnification; magnification bar = 0.1 mm.
RNA-Seq.
Supplemental Table S1 summarizes alignment of RNA-Seq reads to the Mus musculus genome sequence. This number of reads is considered sufficient for eukaryotic transcriptome data (26). Supplemental File S1 (tab 1) includes genes identified in WT V vs. Ezh2cKO V comparisons, and information on fold change differences between the two groups and FDR for each gene. PCA based on all genes shows separation of these two groups and that most differences are attributed to the PC1 axis, which accounts for ~51% of the variation (Fig. 2A). Volcano plot analysis of DEG showed both up- and downregulated genes in Ezh2cKO V vs. WT V uteri (Fig. 2B). Hierarchical heatmap cluster analysis based only on DEG identified in WT V vs. EZH2cKO V comparisons revealed strong clustering within each of the two groups (Fig. 2C).
Fig. 2.

Gene expression profiles from vehicle-treated WT and Ezh2cKO uteri. A: a principal component analysis plot presenting the sample relationship based on PC1, 2, and 3, which accounted for 50.8, 22.9, and 12.6% of the variance, respectively. B: each detected gene as an individual dot, differentially expressed genes (DEG) are shown in red (those downregulated in Ezh2cKO V) or green (those upregulated in Ezh2cKO V); orange dots represent a trend for significance and non-significantly affected genes are shown in black. Gene expression is based on the log2 fold-change (x-axis) plotted against the log10(P value) for each gene. Based on the DEG, a heat map (C) was generated where each DEG is on a separate row and each column represents individual results. Both rows and columns are clustered using correlation distance and average linkage, as determined by the program http://www2.heatmapper.ca/.
There were 88 DEG in V-treated WT compared with Ezh2cKO mice (FDR < 0.05). Of these, 16 were downregulated and 72 upregulated in Ezh2cKO mice. Of the 88 genes, 42 had a −log2 fold-change >|1.5| with four downregulated and 38 upregulated. Some of the most relevant upregulated genes included cellular retinoic acid binding protein 2 (Crabp2), small proline-rich protein 2F (Sprr2f), leucine-rich repeat containing g protein-coupled receptor 5 (Lgr5), bone morphogenetic protein 5 (Bmp5), keratin 15 (Krt15), keratin 5 (Krt5), imprinted maternally expressed transcript (H19), cell division cycle associated 5 (Cdca5), oligodendrocyte transcription factor 3 (Olig3), serpin family b member 7 (Serpinb7), and aldehyde dehydrogenase 1 family member a3 (Aldh1a3) (Table 1), while glutathione peroxidase 3 (Gpx3), polymeric immunoglobulin receptor (Pigr), and sulfotransferase 1 family member d1 (Sult1d1) were notably downregulated. Table 1 lists the top significant DEG based on FDR ≤ 0.005.
Table 1.
Most significantly DEG in the uterus of WT and Ezh2cKO mice treated with V
| Gene ID | Fold Change | FDR | Expression in Ezh2cKO |
|---|---|---|---|
| Asb4 | 11.14817396 | 8.27E-20 | ↑ |
| Crabp2 | 3.848254091 | 1.14E-11 | ↑ |
| Gm38464 | 14.9607005 | 8.36E-11 | ↑ |
| Lgi3 | 3.979659533 | 2.18E-08 | ↑ |
| Cited4 | 0.345856692 | 3.07E-08 | ↓ |
| Sprr2f | 3.625020255 | 5.04E-08 | ↑ |
| Lgr5 | 2.369391701 | 9.79E-08 | ↑ |
| Gpx3 | 0.457093991 | 5.95E-07 | ↓ |
| Esco2 | 2.823937341 | 4.94E-06 | ↑ |
| Bmp5 | 2.833279186 | 9.72E-06 | ↑ |
| Krt15 | 9.578046978 | 1.79E-05 | ↑ |
| Sult1d1 | 0.466253774 | 2.96E-05 | ↓ |
| H19 | 3.042592602 | 3.51E-05 | ↑ |
| Olig3 | 3.533068202 | 3.99E-05 | ↑ |
| Tbx18 | 0.174968691 | 8.00E-05 | ↓ |
| Pclaf | 2.20718689 | 8.00E-05 | ↑ |
| Il17rb | 1.970889974 | 0.000214628 | ↑ |
| Kif11 | 2.496594524 | 0.000222291 | ↑ |
| Gm36834 | 5.804523659 | 0.000222291 | ↑ |
| Gm36317 | 3.023836965 | 0.000230323 | ↑ |
| Serpinb7 | 2.616756428 | 0.000251386 | ↑ |
| Aurka | 5.247556264 | 0.000251386 | ↑ |
| Aldh1a3 | 3.051121223 | 0.000389843 | ↑ |
| Ikzf3 | 22.96137032 | 0.000405102 | ↑ |
| Ppp1r1b | 2.821792684 | 0.000500617 | ↑ |
| Ugt2b34 | 0.379243567 | 0.000906668 | ↓ |
| Uox | 4.630955003 | 0.000926178 | ↑ |
| Pigr | 0.508582613 | 0.000979141 | ↓ |
| Arhgap11a | 2.183336895 | 0.000979141 | ↑ |
| Nasp | 3.706252837 | 0.001210914 | ↑ |
| Ren1 | 2.675262767 | 0.001580261 | ↑ |
| 4931428F04Rik | 3.170063922 | 0.001692858 | ↑ |
| Krt5 | 8.765587469 | 0.002053604 | ↑ |
| Mis18bp1 | 2.351257696 | 0.00231403 | ↑ |
| Prom2 | 7.170806062 | 0.002597374 | ↑ |
| Calml3 | 2.679000525 | 0.004045901 | ↑ |
| Btnl5-ps | 11.88243813 | 0.004399851 | ↑ |
| Cdca5 | 2.171016571 | 0.004825149 | ↑ |
| Pdlim3 | 0.53300295 | 0.004934818 | ↓ |
Underlined genes were identified to be key players. Boldfaced genes are discussed in results. DEG, differentially expressed gene; WT, wild type; Ezh2cKO, EZH2 conditional knockout; V, vehicle; FDR, false discovery rate.
Utilizing the STRING database (54), we evaluated for potential PPI. The program recognized 61 out of 88 inputted genes, creating a network with 66 nodes and 196 edges (Fig. 3). Key player analysis based on the strength and coexpression among DEG (3) revealed that Gpx3 and Sult1d1 were key player genes downregulated in Ezh2cKO V mice. Other key player genes circled in green (Fig. 3) include cell division cycle associated 5 (Cdca5) and tripartite motif containing 29 (TRIM29), which was upregulated. Downregulated genes included Sult1d1, and UDP glucuronosyltransferase 2 family, polypeptide B34 (Ugt2b34). Cdca5 showed extensive potential PPI of varying types with other protein products of DEG.
Fig. 3.

Cluster analysis of the protein-protein interaction (PPI) network based on DEG for the WT V vs. Ezh2cKO V comparison. A total of 61 DEGs were filtered into the DEGs’ PPI network complex that contained 66 nodes and 196 edges. List on the right denotes key player genes, which are also highlighted in the network.
STRING analysis of DEG in WT V vs. Ezh2cKO V (Table 2) yielded several significant GO terms in three categories (biological process, cellular component, and molecular function). These include cell cycle, cell division, mitotic chromosome condensation, and nuclear division in the biological process category, collectively suggesting that uterine Ezh2cKO deletion stimulates various processes involved in cell proliferation. KEGG pathway enrichment analyses were also performed based on DEG for this comparison. Several signaling pathways (Table 3), including estrogen (P = 0.007), hippo (P = 0.013), PI3K-AKT (P = 0.02), were enriched.
Table 2.
GO terms associated with DEG for WT V vs. Ezh2cKO V
| # Term ID | Term Description | FDR |
|---|---|---|
| Biological process GO terms | ||
| GO:0000278 | mitotic cell cycle | 0.00016 |
| GO:0007076 | mitotic chromosome condensation | 0.00016 |
| GO:0140014 | mitotic nuclear division | 0.00016 |
| GO:0007049 | cell cycle | 0.0002 |
| GO:0051301 | cell division | 0.00038 |
| Molecular function GO terms | ||
| GO:0001758 | retinal dehydrogenase activity | 0.0218 |
| GO:0004028 | 3-chloroallyl aldehyde dehydrogenase activity | 0.0218 |
| GO:0004748 | ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor | 0.0218 |
| Cellular component GO terms | ||
| GO:0000796 | condensin complex | 0.00068 |
| GO:0000793 | condensed chromosome | 0.0059 |
| GO:0000799 | nuclear condensin complex | 0.0065 |
| GO:0005971 | ribonucleoside-diphosphate reductase complex | 0.0065 |
| GO:0000794 | condensed nuclear chromosome | 0.012 |
Table 3.
Functional enrichment analysis of DEG based on WT V vs. Ezh2cKO V comparison
| Gene Set | Description | P Value |
|---|---|---|
| mmu04915 | estrogen signaling pathway | 0.0019 |
| mmu00232 | caffeine metabolism | 0.0125 |
| mmu05166 | human T cell leukemia virus 1 infection | 0.0144 |
| mmu04310 | Wnt signaling pathway | 0.0316 |
| mmu04390 | hippo signaling pathway | 0.0379 |
| mmu05020 | prion diseases | 0.0592 |
| mmu00410 | beta-alanine metabolism | 0.0631 |
| mmu04614 | renin-angiotensin system | 0.0651 |
| mmu04216 | ferroptosis | 0.0769 |
| mmu00330 | arginine and proline metabolism | 0.0866 |
Italicized pathways are mentioned in results.
We next compared genes differentially expressed in WT E2 vs Ezh2cKO E2 groups. PCA analysis based on all genes showed separation of these two groups and that most differences are attributed to the PC1 axis, which accounted for ~78% of the variation (Fig. 4A). Supplemental File S1 (tab 2) includes all genes identified in the WT E2 vs. Ezh2cKO E2 comparison and fold change differences between the two groups and FDR for each gene. Volcano plot analysis shows both up- and downregulated DEG in Ezh2cKO E2 vs. WT E2 uteri (Fig. 4B). Hierarchical heatmap cluster analysis based only on DEG in WT E2 vs. Ezh2cKO E2 reveals strong clustering within each of the two groups (Fig. 4C).
Fig. 4.

Gene expression profiles of WT and Ezh2cKO treated with E2. A principal component analysis plot (A) presenting the sample relationship based on PC1, 2, and 3, which accounted for 78.1, 12.3, and 4.5% of the variance, respectively. Volcano plot (B) represents each detected gene as an individual dot, DEG are shown in red (those downregulated in Ezh2cKO E2) or green (those upregulated in Ezh2cKO E2), and nonsignificantly affected genes are shown in black. Expression of genes is based on the log2 fold-change (x-axis) plotted against the log10(P value) for each gene. Based on the DEG, a heat map (C) was generated where each DEG is on a separate row and each column represents individual results. Both rows and columns are clustered using correlation distance and average linkage, as determined by the program http://www2.heatmapper.ca/.
Analysis of RNA-Seq data from WT E2 vs. Ezh2cKO E2 yielded 40 DEG (FDR < 0.05). Of these, seven were downregulated and 33 upregulated in Ezh2cKO mice (Fig. 4, B and C). Moreover, 26 had a −log2 fold-change > |1.5| with four downregulated and 22 upregulated. Some of the most significant (FDR ≤ 0.005) upregulated genes in Ezh2cKO E2 were Krt5, Krt15, Olig3 (cellular retinoic acid binding protein 2), Crabp1, and Serpinb7 (Table 4), whereas complement component 1, q subcomponent-like 1 (C1ql1) was downregulated in Ezh2cKO E2 relative to WT E2.
Table 4.
Most significantly DEG in the uterus of WT and Ezh2cKO mice treated with E2
| Gene ID | Fold Change | FDR | Expression in Ezh2cKO |
|---|---|---|---|
| Krt5 | 43.07656047 | 9.57E-31 | ↑ |
| Sult1d1 | 0.175423712 | 1.46E-11 | ↓ |
| Gm41284 | 6.17792266 | 3.21E-10 | ↑ |
| Krt15 | 15.49777885 | 3.89E-09 | ↑ |
| Olig3 | 6.848212907 | 4.05E-07 | ↑ |
| Crabp1 | 4.170093407 | 2.01E-06 | ↑ |
| Col17a1 | 50.45275038 | 3.74E-06 | ↑ |
| Serpinb7 | 6.851708108 | 6.97E-05 | ↑ |
| Nrk | 4.777750572 | 0.000338044 | ↑ |
| Obscn | 3.396918661 | 0.001332806 | ↑ |
| Epop | 0.339949928 | 0.001524958 | ↓ |
| Asprv1 | 0.541275388 | 0.001524958 | ↓ |
| Prnp | 3.02764155 | 0.001524958 | ↑ |
| Slco4c1 | 3.175247452 | 0.001524958 | ↑ |
| B4galnt4 | 1.780175145 | 0.00172243 | ↑ |
| Rbp1 | 2.331852965 | 0.00172243 | ↑ |
| C1ql1 | 0.179022409 | 0.004092399 | ↓ |
| Fam212a | 2.307705374 | 0.004092399 | ↑ |
| Gm9758 | 3.532058991 | 0.004092399 | ↑ |
Underlined genes were also identified to be key players; boldfaced genes are discussed in results.
Utilizing the STRING Database, we evaluated PPI. The software identified 30 of 40 DEG, detecting 40 nodes and 74 edges (Fig. 5). Representative key player genes for this comparison include Sult1d, which was downregulated in Ezh2cKO E2, but two other key players, insulin like growth factor 2 binding protein 1 (Igf2bp1) and Wnt family member 4 (Wnt4), were upregulated. Other key players are highlighted in green in Fig. 5. Surprisingly, many of the protein products for the key player genes did not demonstrate a considerable number of PPI. The few PPI for key players include S100A14 showing a PPI with KRT5, WNT4, was linked to prostaglandin E synthase (PTGES).
Fig. 5.

Cluster analysis of the PPI network for the WT E2 vs. Ezh2cKO E2 comparison. A total of 40 DEG were filtered into the DEG PPI network complex that contained 40 nodes and 74 edges. List on the right denotes key player genes, which are also highlighted in the network.
As with comparisons in V-treated groups, STRING analysis based on DEG identified in E2-treated WT vs. Ezh2cKO mice yielded enrichment for GO terms across the three categories (Table 5). Significant GO terms in the E2 group comparisons were related to DNA replication and metabolic processes, whereas those for vehicle-treated WT and Ezh2cKO mice related to mitotic processes. KEGG pathway enrichment analysis was performed based on DEG identified for this comparison (Table 6). Similar to WT V vs. Ezh2cKO V, DEG in these E2-treated groups showed functional enrichment for estrogen (P = 0.0019) and hippo (P = 0.038) signaling pathways. However, the Wnt pathway (P = 0.031) was also significantly enriched in this latter group comparison.
Table 5.
GO terms associated with DEG for WT E2 vs. Ezh2cKO E2
| # Term ID | Term Description | FDR |
|---|---|---|
| Biological process GO terms | ||
| GO:0006270 | DNA replication initiation | 4.04E-15 |
| GO:0006261 | DNA-dependent DNA replication | 1.94E-12 |
| GO:0006260 | DNA replication | 5.14E-12 |
| GO:0006259 | DNA metabolic process | 3.78E-07 |
| GO:0006268 | DNA unwinding involved in DNA replication | 1.23E-06 |
| Molecular function GO terms | ||
| GO:0004386 | helicase activity | 9.46E-07 |
| GO:0003678 | DNA helicase activity | 7.60E-05 |
| GO:0003688 | DNA replication origin binding | 0.00019 |
| GO:0004003 | ATP-dependent DNA helicase activity | 0.0012 |
| GO:0003697 | single-stranded DNA binding | 0.0013 |
| Cellular component GO terms | ||
| GO:0042555 | MCM complex | 3.76E-11 |
| GO:0005634 | nucleus | 0.00069 |
| GO:0005654 | nucleoplasm | 0.00069 |
| GO:0044428 | nuclear part | 0.00069 |
| GO:0070013 | Intracellular organelle lumen | 0.00069 |
Table 6.
Functional enrichment analysis of DEG based on WT E vs. Ezh2cKO E2 comparison
| Gene Set | Description | P Value |
|---|---|---|
| mmu04915 | estrogen signaling pathway | 0.0077 |
| mmu04728 | dopaminergic synapse | 0.0088 |
| mmu04390 | hippo signaling pathway | 0.013 |
| mmu00980 | metabolism of xenobiotics by cytochrome P450 | 0.0164 |
| mmu00982 | drug metabolism | 0.017 |
| mmu05031 | amphetamine addiction | 0.0192 |
| mmu00232 | caffeine metabolism | 0.02 |
| mmu04151 | PI3K-Akt signaling pathway | 0.0202 |
| mmu04924 | renin secretion | 0.0215 |
| mmu00830 | retinol metabolism | 0.0252 |
Italicized pathways are mentioned in results.
Key player genes and functional enrichment analysis of Ezh2cKO V vs. Ezh2cKO E2 comparison.
The full list of 252 DEG in V-treated Ezh2cKO vs. Ezh2cKO mice is provided in Supplemental File S1. To obtain further insights into EZH2 and E2 interaction, we examined 10 key player genes based on this comparison (Fig. 6). As shown, four of the key player genes are included in this diagram: farnesyl diphosphate synthase (Fdps), multiple EGF-like domains (Megf10), procollagen C-endopeptidase enhancer (Pcolce2), and carboxypeptidase Z (Cpz). Of these, Fdps and Pcolce2 showed the most interactions to other DEG based on this comparison. Fdps encodes an enzyme responsible for catalyzing farnesyl pyrophosphate production from isopentylpyrophosphate and dimethylallyl pyrophosphate, and the product, farnesyl pyrophosphate, serves as a key intermediate in cholesterol and sterol biosynthesis. GO terms associated with this gene include transferase activity and RNA binding. GO terms associated with Pcolce2 include heparin binding and metalloendopeptidase inhibitor activity. Additionally, we performed functional enrichment analysis based on the DEG identified in this comparison. Primary GO terms and pathways likely altered include those involved in reorganization of the extracellular matrix, cholesterol biosynthesis and metabolism, and steroid synthesis and metabolism.
Fig. 6.

Cluster analysis of the PPI network based on DEG for the Ezh2cKO V vs. Ezh2cKO E2 comparison. A total of 252 DEGs were filtered into the DEGs’ PPI network complex that contained 41 nodes and 55 edges. List on the right denotes key player genes, which are also highlighted in the network.
Integrative correlation analyses.
Two pairwise comparisons were used for these analyses: WT V vs. Ezh2cKO V (19,481 genes) and WT E2 vs. Ezh2cKO E2 (15,797 genes) were further analyzed with WGCNA (31). Individual modules are represented by different colors, and ME were then correlated with percent of epithelial MKI67 staining (detailed above), which resulted in 42 distinct color modules in this comparison.
Supplemental Fig. S1 shows the resulting dendrogram and heatmap for the WT V vs. Ezh2cKO V comparison. As shown in Fig. 7, two modules (Green and Sky Blue3) were positively associated with epithelial proliferation (Fig. 1). Genes within these two modules are listed in Supplemental File S2.
Fig. 7.

Relationship of weighted gene coexpression network analysis (WGCNA) results and percent epithelial proliferation for WT V vs. Ezh2cKO V comparison. Modules identified in Supplemental Fig. S1 were correlated with epithelial proliferation (Fig. 1). Each row corresponds to a module eigengene (ME), and colors represent the correlation coefficient between the ME and percentage of uterine epithelial cell proliferation. Numbers at the top of rows represents degree of correlation, and values with a negative integer indicate an inverse correlation with epithelial proliferation. Two color modules, Green and Sky Blue 3, positively correlated with epithelial proliferation (demarcated with boxes). A legend that ranges from blue to red shades (−1 to 1, respectively) is included to the right of the figure. These colors coincide with degree of correlation with darker blue representing strong negative correlation between an individual ME and proliferation and those ME more intensely red indicating a positive correlation. This is evident in the two ME, Green and Sky Blue 3 boxes, that are colored in dark red to indicate that the genes within these modules are positively associated with proliferation.
Extensive interactions for proteins encoded by genes within each of these two color modules were evident (Supplemental Figs. S2 and S3). Within the Green module, ESR1 is included in the center area of the PPI. Hub gene analysis further revealed that for this module, Esr1 was one of the top, hub genes (Supplemental Fig. S4). Other top hub genes for the Green module in this comparison included Mdm2, Nedd4, Smad3, Pik3ca, Pdgfra, and Huwe1. No significant hub genes were detected for Sky Blue3. We next examined genes within each of these modules for functional enrichment analyses; GO (19) and pathways functionally enriched in the Green and Sky Blue3 modules for the WT V vs. Ezh2cKO V are listed in Supplemental File S2. Example GO terms enriched include estrogen/ESR signaling, nuclear and intracellular hormone receptor binding, and histone binding, with those involved in steroid hormone signaling or histone proteins (Table 7).
Table 7.
Selective pathways associated with the Sky Blue3 and Green color modules for WT V vs. Ezh2cKO V comparison
| Module Color | Source | Pathway | FDR |
|---|---|---|---|
| Sky blue | GO:BP | metabolic process | 1.55E-06 |
| GO:BP | regulation of cellular metabolic process | 9.84E-06 | |
| GO:BP | regulation of metabolic process | 2.65E-05 | |
| GO:BP | apoptotic process | 0.000499702 | |
| GO:BP | regulation of MAPK cascade | 0.009607817 | |
| Green | GO:MF | steroid hormone receptor binding | 0.000040182 |
| GO:MF | nuclear hormone receptor binding | 5.12E-05 | |
| GO:MF | histone binding | 8.65E-05 | |
| GO:MF | hormone receptor binding | 0.000184918 | |
| GO:BP | histone acetylation | 0.00134373 | |
| GO:BP | intracellular steroid hormone receptor signaling pathway | 0.00141134 | |
| GO:BP | steroid hormone-mediated signaling pathway | 0.002336833 | |
| GO:BP | cellular hormone metabolic process | 0.002708761 | |
| KEGG | PI3K-Akt signaling pathway | 5.73E-05 | |
| KEGG | choline metabolism in cancer | 5.73E-05 | |
| KEGG | pathways in cancer | 0.000617863 | |
| KEGG | thyroid hormone signaling pathway | 0.001765468 | |
| KEGG | JAK-STAT signaling pathway | 0.002660921 | |
| KEGG | endocrine resistance | 0.003160081 | |
| KEGG | estrogen signaling pathway | 0.00363483 | |
| KEGG | prolactin signaling pathway | 0.005348187 | |
| KEGG | TNF signaling pathway | 0.005348187 | |
| KEGG | MAPK signaling pathway | 0.005348187 | |
| REAC | signaling by receptor tyrosine kinases | 1.24E-06 | |
| REAC | immune system | 0.000106784 | |
| REAC | PIP3 activates AKT signaling | 0.000106784 | |
| REAC | MAPK1/MAPK3 signaling | 0.000123755 | |
| REAC | PI5P, PP2A, and IER3 regulate PI3K/AKT signaling | 0.000397045 | |
| REAC | PI3K cascade | 0.002000441 | |
| REAC | PI3K/AKT activation | 0.002643746 | |
| REAC | IGF1R signaling cascade | 0.002643746 | |
| REAC | signaling by insulin receptor | 0.002643746 | |
| REAC | signaling by type 1 insulin-like growth factor 1 receptor (IGF1R) | 0.002828781 | |
| REAC | insulin receptor signaling cascade | 0.00302286 | |
| WP | EGFR1 signaling pathway | 0.001556579 | |
| WP | insulin signaling | 0.002021013 | |
| WP | IL-3 signaling pathway | 0.004400538 |
Italicized pathways are mentioned in results.
For the WT E2 vs. Ezh2cKO E2 comparison, 38 distinct color modules were identified (Supplemental Fig. S5), and two (Plum1 and Dark Green) were negatively associated with epithelial proliferation (Fig. 8). Genes within these two modules are listed in Supplemental File S3. PPI were then examined for proteins encoded by genes within these two color modules. Five clusters were evident for genes within the Plum1 pathway (Supplemental Fig. S6). Three of these only included two genes. The main one in the center includes BCL6 Transcription repressor (BCL6), CREB binding protein (CREBBP), MYB proto-oncogene, transcription factor (MYB), pleiotropic regulator 1 (PLRG1), heterogeneous nuclear ribonucleoprotein F (HNRNPF), BCAS2 pre-mRNA processing factor (BCAS2), and RNA binding motif protein 22 (RBM22). PPI for proteins encoded by genes in the Dark Green module included five main clusters. but of these, four clusters included only two genes. The one main cluster in the center contains ribosomal proteins and associated proteins (Supplemental Fig. S7). No significant hub genes were detected for either color module for this comparison.
Fig. 8.

Relationship of WGCNA results and percent epithelial proliferation for WT E2 vs. nomenclature Ezh2cKO E2 comparison. Modules identified in Supplemental Fig. S4 were then correlated with epithelial proliferation (Fig. 1). The same figure arrangement holds as listed in Fig. 6. As shown, two modules, Plum1 and Dark Green (demarcated with boxes), showed a significant inverse correlation with proliferation. A legend that ranges from blue to red shades (−1 to 1, respectively) is included to the right of the figure. These colors coincide with degree of correlation with darker blue representing strong negative correlation between an individual ME and proliferation and those ME more intensely red indicating a positive correlation. As shown, ME Plum 1 and Dark Green boxes are colored a dark blue, indicating a significant inverse correlation with proliferation.
Finally, we examined genes within each module by functional enrichment analyses. GO terms and pathways functionally enriched in the Plum1 and Dark Green modules for the WT E2 vs. Ezh2cKO E2 groups are listed in Supplemental File S3. Potential key pathways (Table 8) included epithelial development, and metabolism of RNA and lipids. Those involved in DNA or RNA processes are italicized.
Table 8.
Selective pathways associated with the Plum1 and Dark Green color modules for WT E2 vs. Ezh2cKO E2 comparison
| Module Color | Source | Pathway | FDR |
|---|---|---|---|
| Plum | GO:MF | organic cyclic compound binding | 0.000108362 |
| GO:MF | epithelium development | 0.002498768 | |
| GO:MF | RNA metabolic process | 0.004239583 | |
| GO:MF | cellular nitrogen compound biosynthetic process | 0.006146942 | |
| GO:BP | nucleic acid metabolic process | 0.006792618 | |
| GO:BP | aromatic compound biosynthetic process | 0.007047364 | |
| REAC | metabolism | 0.000679428 | |
| REAC | metabolism of RNA | 0.00862975 | |
| REAC | processing of capped intron-containing pre-mRNA | 0.00979419 | |
| REAC | metabolism of lipids | 0.009886223 | |
| WP | Mecp2 and associated Rett syndrome | 0.005864471 | |
| Dark green | GO:BP | cellular nitrogen compound metabolic process | 1.71E-07 |
| GO:BP | organic substance metabolic process | 2.31E-07 | |
| GO:BP | organic cyclic compound metabolic process | 4.64E-07 | |
| GO:BP | macromolecule metabolic process | 5.92E-07 | |
| GO:BP | nitrogen compound metabolic process | 5.92E-07 | |
| GO:BP | cellular aromatic compound metabolic process | 8.85E-07 | |
| GO:BP | nucleic acid metabolic process | 1.01E-06 | |
| GO:BP | regulation of metabolic process | 3.76E-05 | |
| GO:BP | cellular macromolecule metabolic process | 3.76E-05 | |
| GO:BP | negative regulation of DNA metabolic process | 0.000137343 | |
| GO:BP | RNA metabolic process | 0.000137343 | |
| GO:BP | cellular macromolecule biosynthetic process | 0.000281745 | |
| GO:BP | regulation of nitrogen compound metabolic process | 0.000491855 | |
| GO:BP | negative regulation of macromolecule metabolic process | 0.000561368 | |
| GO:BP | regulation of mRNA metabolic process | 0.000877929 | |
| GO:BP | regulation of macromolecule metabolic process | 0.000897712 | |
| GO:BP | mRNA metabolic process | 0.000897712 | |
| GO:BP | regulation of cellular macromolecule biosynthetic process | 0.002615055 | |
| GO:BP | organonitrogen compound metabolic process | 0.002907815 | |
| GO:BP | regulation of RNA metabolic process | 0.005351352 | |
| GO:CC | ribonucleoprotein complex | 0.002267432 |
Italicized pathways are mentioned in results.
DISCUSSION
Conditional deletion of Ezh2 in mouse uterus causes various phenotypic and functional changes, including increased uterine epithelial proliferation and cystic endometrial hyperplasia, as described here and in previous studies (17, 44). Uteri from non-OVX Ezh2cKO mice showed increased weight compared with WT controls, which was detectable by PND 22 and increased with age, with uteri from Ezh2cKO mice weighing almost four times as much as WT by PND 180 (44). Our main goal in this study was to delineate transcriptomic changes induced by loss of EZH2 to provide better understanding of uterine molecular pathways regulated by this protein. Integrative correlation analyses of RNA-Seq and cell proliferation data-derived MKI67 immunohistochemistry indicated changes in estrogen-related and other pathways that may be linked to phenotypic alterations caused by EZH2 deletion, allowing important insights into the relationship between EZH2 and epithelial proliferation and other critical pathways. Cre recombinase was under the control of the progesterone receptor (Pgr) promoter, resulting in transgenic mice that lack Ezh2 in all Pgr-expressing tissues, including uterus, mammary gland, cervix, vagina, oviduct, and select brain regions. In the mammary gland, loss of Ezh2 caused decreased numbers of terminal end buds and less extensive ductal branching (44). Conditional deletion of Ezh2 in the mammary gland thus led to effects opposite to those observed in the uterus where extensive epithelial proliferation was noted.
Signaling through the E2/ESR1 pathway regulates uterine development and normal uterine epithelial proliferation (13, 17). We and others previously reported high EZH2 expression in proliferative uterine epithelial cells (44), similar to what was seen in other proliferating cell types (60). Additionally, EZH2 is differentially regulated during the cell cycle, allowing establishment and maintenance of epigenetic marks in newly formed cells (41). Cell cycle-dependent EZH2 regulation occurs through posttranslational modifications, including phosphorylation of various amino acids within the protein. Phosphorylation of EZH2 on serine 21 by protein kinase B inhibits PRC2-mediated H3K27 trimethylation (8). In contrast, threonine residues 345 and 487 on EZH2 are also phosphorylation targets. This phosphorylation occurs during mitosis by cyclin-dependent kinases, which facilitate PRC2 interaction with noncoding RNAs, enabling recruitment to target genes (27). This EZH2 upregulation during the mitotic phase then catalyzes H3K27 trimethylation during chromatin replication.
The relationship between estrogen signaling and epigenetic regulation has been established, with estrogens modulating DNA methylation changes of estrogen-target genes (34), as well as changes in histone modifications (22). The EZH2/E2 relationship is unclear, with some reports suggesting E2 upregulates Ezh2 and others suggesting downregulation (14, 25, 49). Based on our RNA-Seq enriched pathways, EZH2 deletion enhances estrogen-regulated pathways, phenotypically reflected in increased uterine epithelial proliferation in Ezh2cKO mice. In addition, an EZH2 role in ESR1 coactivation was reported (52).
Loss of uterine EZH2 causes epithelial hyperplasia; this type of excessive proliferation is seen in precancerous endometrial lesions (18), which can progress to endometrial carcinoma (6). This uterine pathology could also impair fertility as cyclical regulation of epithelium proliferating is required for proper embryo implantation (29). Based on our observations, EZH2 is necessary to constrain uterine epithelial proliferation. Deregulation of H3K27 trimethylation and the machinery that maintains an adequate chromatin configuration disrupts normal transcription and is a landmark of cancer cells; mutations in these types of genes occur in various cancers. Particularly, gain-of-function mutations in EZH2 occur in endometrial (47) and other cancers (18, 62). Amounts of EZH2 frequently correlate with tumor growth, development, and prognosis (59). Conversely, loss-of-function mutations in EZH2 also can promote cancer initiation/progression. For example, 25% of T cell acute lymphoblastic leukemias show deletions of ezh2 (45), and EZH2 loss inhibits pancreatic regeneration and leads to pancreatic neoplasia (37).
Although high-grade endometrial carcinomas frequently overexpress EZH2, our model shows that loss of EZH2 leads to dysregulated and increased epithelial proliferation, a hallmark of precancerous lesions (18). Rats developmentally exposed to genistein have induction of phosphoinositide 3-kinase (PI3K)/AKT through membrane ESR signaling, which in turn phosphorylates and suppresses EZH2 leading to decreased levels of H3K27me3 in target genes. Genistein-induced reductions in EZH2 activity coincided with increased incidence and number of uterine leiomyomas (22). Thus, our uterine Ezh2cKO model represents an interesting approach to study uterine diseases and the consequences on pathology and fertility that are related to epigenetic changes. Additionally, studying epigenetic effects following EZH2 loss may facilitate understanding of agents that inhibit EZH2 as an approach to cancer therapy (24).
We observed that the majority of DEG were upregulated in Ezh2cKO mice. This result is consistent with the main role of EZH2 as a transcriptional inhibitor, although absolute effects on transcript expression are gene specific. If EZH2 normally represses a transcription factor, loss of EZH2 may upregulate that gene and genes it normally regulates. Thus, for tumor suppressor genes, EZH2 overexpression could lead to suppression of these genes and increased susceptibility to tumorigenic changes. Conversely, for proto-oncogenes, EZH2 loss may lead to unrestrained transcription of these genes with resulting neoplastic transformation. Thus, both under- and overexpression of EZH2 can lead to a greater risk of cancer. It is also important to consider that EZH2 has PRC2-independent roles that involve nonenzymatic and nonepigenetic mechanisms of gene regulation (28). An example of a nonepigenetic role of EZH2 is its ability to act as a coactivator of transcription factors, such as androgen receptor (64), and a transcriptional activator of genes that are ESR1 regulated in breast cancer cells (52). We will next consider specific gene expression changes identified in Ezh2cKO vs. WT mice.
In the V and E2 comparisons, Krt5 and Krt15 were significantly upregulated in Ezh2cKO mice. Previous studies also showed that KRT5, KRT6A, and KRT14 protein and mRNA expression were elevated in Ezh2cKO uteri relative to their WT counterparts (17). Based on these expression patterns and the enhanced uterine epithelial proliferation seen in these mice, it was postulated that EZH2 suppresses differentiation of basal-like cells and consequently restricts uncontrolled uterine epithelial proliferation that may culminate in tumorigenesis.
Another upregulated gene in Ezh2cKO V vs. WT V mice that was near significance (FDR = 0.06) was the maternally imprinted gene H19. E2 treatment increases H19 expression in uterus and mammary gland (1). Increased H19 expression in uterine stroma and myometrium is associated with increased severity of tumor invasion into the underlying uterine wall (36). Altered expression of genes involved in epithelial proliferation induced by loss of EZH2 is likely responsible for continued mitogenesis in Ezh2cKO V vs. WT V mice, but the exact nature of these changes remains to be established.
Leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5) was another gene upregulated in Ezh2cKO V mice relative to WT V, but this was not identified as a DEG in the E2 comparison. Previous work suggests that this gene is regulated by steroid hormones and may serve as an endometrial epithelial stem cell marker (7, 21, 55). Expression of Lgr5 is associated with deep-infiltrating endometriosis lesions (58). Changes in expression of H19, Lgr5, and other genes regulating epithelial proliferation may be involved in ovary-independent uterine epithelial proliferation in Ezh2cKO mice.
Key player analysis helped determine which DEG might be hub genes that drive expression of others. For the V comparison only, Cdca5, upregulated in Ezh2cKO uteri, was ranked as a key player. Moreover, STRING analysis revealed that CDCA5 had PPI with several other proteins encoded by DEG. Scant information is available on the uterine role of Cdca5, but it has been implicated in reproductive cancers (10, 33, 48).
Sult1d1 was downregulated in both V-and E2-treated Ezh2cKO uteri compared with WT and was considered a key player in both comparisons. In the V comparison, it was associated with the protein encoded by another key player DEG, UDP glucuronosyltransferase 2 family, polypeptide B34 (Ugt2b34), which was also downregulated in Ezh2cKO. No PPI were evident though for SULT1D1 in the E2 comparison. As a sulfotransferase, SULT1D1 is a detoxifying enzyme that inactivates endogenous dopamine-derived compounds, such as catecholamines (2), but no information is available on its uterine role.
Functional enrichment analysis revealed that estrogen and hippo signaling pathways were significant in both the V and E2 comparisons. The estrogen signaling pathway includes both genomic and nongenomic actions. Membrane ESR mediates nongenomic effects partly through activation of PI3K/AKT. This pathway induces AKT phosphorylation that, in turn, inhibits EZH2 action, resulting in reduced H3K27me3 epigenetic marks. In this way, membrane ESR signaling targets the epigenome (50). Differential binding and activation of xenoestrogens to membrane ESR may thus affect EZH2 inactivation, subsequent methylation of H3K27, and risk for uterine cancer (22). EZH2 may also directly interact with ESR1 and β-catenin and this epigenetic regulator link between estrogen and Wnt signaling pathways (52). Enhancement of both of these pathways promotes cell division. The Wnt pathway was enriched in the V comparison, and the PI3K/AKT signaling pathway was enriched in the E2 comparison. It is uncertain why there are differences between these two comparisons, but the collective findings support the existence of intertwining connections between estrogen receptor, PI3K-AKT, and Wnt pathways. Additionally, hippo signaling, enriched in both comparisons, may contribute to tumorigenesis due to disrupted EZH2 signaling (35).
The GO terms are consistent with increased cell proliferation, presumably due to activation of the signaling circuits above. In the V comparison, mitosis and cell cycle division are the top GO terms. DNA replication and associated processes are the top GO terms for the E2 comparison. Collectively, enrichment analysis and GO terms suggest that absence of uterine EZH2 stimulates pathways promoting cell proliferation, and administration of E2 to Ezh2cKO enhances effects through these and likely other pathways with the net effect of further heightening cell proliferation, consistent with the notion that estrogens and suppression of EZH2 act in tandem to stimulate tumorigenesis.
Another goal of this study was to examine genes and pathways enriched in Ezh2cKO mice treated with E2 or vehicle to examine potential E2 effects on EZH2-regulated mechanisms. The DEG identified in this comparison revealed that primary pathways altered in this group include reorganization of the extracellular matrix, cholesterol biosynthesis and metabolism, and steroid synthesis and metabolism.
To establish potential linkages between uterine genes identified based on the WT and Ezh2cKO comparisons and uterine epithelial proliferation, WGCNA was performed. In both comparisons, two modules significantly correlated with uterine proliferation, two positively and two inversely. This difference is presumably a function of the member genes and functionally enriched pathways in these different modules. Notably, in the Green module for the V comparison, Esr1 is a central hub gene with extensive PPI as shown in STRING analysis. Hub gene analysis further reveals that Esr1 is a top hub genes in this module. Correspondingly, functionally enriched pathways for this module include estrogen, PI3K/AKT, and other hormone/cell cycle signaling pathways. Additionally, histone protein modifications are functionally enriched in these two modules. The WGCNA with the V comparison support the functional enrichment analysis based on the DEG and further suggest that estrogen, PI3K/AKT, and histone protein modifications regulate uterine epithelial proliferation. This approach does not establish causation but lays the groundwork for future studies where antagonists to ESR1 and these other pathways can be administered to interrogate their role in uterine epithelial proliferation evident in ovariectomized Ezh2cKO mice.
Surprisingly, similar pathways were not altered in the E2 comparison. The Dark Green module, which was inversely related to proliferation in this comparison, consisted of many ribosomal protein-encoding genes. Stimulation of ribosomal protein mRNA and proteins by E2 has been shown in uteri (43), but it is unclear how these proteins may be negatively associated with epithelial proliferation.
Functional enrichment analyses for genes within both the Dark Green and Plum1 modules for the E2 comparison revealed enrichment for DNA and RNA metabolic processes and GO terms for apoptotic and regulation of cell differentiation. In the Dark Green module, one gene associated with such pathways is bone morphogenic protein 7 (Bmp7). In uteri, E2 and other xenoestrogens suppress epithelial apoptosis by downregulating this gene (30). Uterine deletion of Bmp7 results in extensive uterine epithelial proliferation (40). Homeobox A5 (Hoxa5), in the Plum1 module, was associated with several GO terms, including regulation of cell differentiation and epithelial development. Hoxa5 suppresses proliferation and induces apoptosis of cervical cancer cells (62). Taken together, functional enrichment on the significant modules associated with this comparison suggests that E2 augments epithelial proliferation in Ezh2cKO and initiates it in WT mice by suppressing apoptotic and cell differentiation genes.
While RNA-Seq provides insights into gene expression changes between Ezh2cKO and WT uteri, the cellular source of these transcriptomic changes cannot be discerned. For this reason, single cell RNA-Seq is now widely used in uterus (42) but requires identification of signature gene profiles that delineate certain cell populations. Tissue architecture is also destroyed by this approach. To overcome such limitations, tools such as the recently developed Visium Spatial Transcriptomics technology (4, 38) permit quantitative mRNA profiles of cells in intact tissue and may be useful to examine how gene expression changes in different uterine cell lineages relate to and possibly influence each other. In our previous work, we showed that there was overall downregulation of H3K27me3 marks (44). We will be using chromatin immunoprecipitation-Seq with an H3K27me3 antibody to identify specific uterine genes affected by this epigenetic modification in WT and Ezh2cKO mice.
In conclusion, conditional loss of EZH2 in uteri generally upregulates various genes associated with uterine diseases and has effects on estrogen, Wnt, and PI3K/AKT pathways. WGCNA confirmed that estrogen, PI3K/AKT, and other hormone/cell cycle signaling pathways and histone protein modifications are altered in Ezh2cKO mice and may be involved in uterine epithelial proliferation seen even after ovariectomy. Current findings reveal unique gene signature patterns in Ezh2cKO uteri and provide insights into overall EZH2 function.
GRANTS
These studies were supported by a grant from the University of Florida College of Veterinary Medicine and National Institutes of Health Grants HD-088006 and HD-087528 (to P. S. Cooke) and R01 ES-025547 (to C. S. Rosenfeld).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.M.M., M.K.N., C.S.R., and P.S.C. conceived and designed research; A.M.M., J.M., M.K.N., T.I.M., S.T., J.K., and C.S.R. performed experiments; A.M.M., J.M., F.Y., J.K., Z.L., Y.L., T.J., D.W., and C.S.R. analyzed data; A.M.M., J.M., Z.L., T.J., C.S.R., and P.S.C. interpreted results of experiments; A.M.M., J.M., and C.S.R. prepared figures; A.M.M., C.S.R., and P.S.C. drafted manuscript; A.M.M., J.M., T.I.M., S.T., T.J., C.S.R., and P.S.C. edited and revised manuscript; A.M.M., J.M., M.K.N., T.I.M., S.T., F.Y., Z.L., Y.L., T.J., D.W., C.S.R., and P.S.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Benjamin L. Graves for modifying the WGCNA r coding script for the current work and Drs. Franco DeMayo and John Lydon, who provided the original Pgr-Cre mice used for this work.
REFERENCES
- 1.Adriaenssens E, Lottin S, Dugimont T, Fauquette W, Coll J, Dupouy JP, Boilly B, Curgy JJ. Steroid hormones modulate H19 gene expression in both mammary gland and uterus. Oncogene 18: 4460–4473, 1999. doi: 10.1038/sj.onc.1202819. [DOI] [PubMed] [Google Scholar]
- 2.Alnouti Y, Klaassen CD. Tissue distribution and ontogeny of sulfotransferase enzymes in mice. Toxicol Sci 93: 242–255, 2006. doi: 10.1093/toxsci/kfl050. [DOI] [PubMed] [Google Scholar]
- 3.Azuaje FJ. Selecting biologically informative genes in co-expression networks with a centrality score. Biol Direct 9: 12, 2014. doi: 10.1186/1745-6150-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstråhle J, Tarish F, Tanoglidi A, Vickovic S, Larsson L, Salmén F, Ogris C, Wallenborg K, Lagergren J, Ståhl P, Sonnhammer E, Helleday T, Lundeberg J. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun 9: 2419, 2018. doi: 10.1038/s41467-018-04724-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bian F, Gao F, Kartashov AV, Jegga AG, Barski A, Das SK. Polycomb repressive complex 1 controls uterine decidualization. Sci Rep 6: 26061, 2016. doi: 10.1038/srep26061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boruban MC, Altundag K, Kilic GS, Blankstein J. From endometrial hyperplasia to endometrial cancer: insight into the biology and possible medical preventive measures. Eur J Cancer Prev 17: 133–138, 2008. doi: 10.1097/CEJ.0b013e32811080ce. [DOI] [PubMed] [Google Scholar]
- 7.Cervelló I, Gil-Sanchis C, Santamaría X, Faus A, Vallvé-Juanico J, Díaz-Gimeno P, Genolet O, Pellicer A, Simón C. Leucine-rich repeat-containing G-protein-coupled receptor 5-positive cells in the endometrial stem cell niche. Fertil Steril 107: 510–519.e3, 2017. doi: 10.1016/j.fertnstert.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 8.Cha T-L, Zhou BP, Xia W, Wu Y, Yang C-C, Chen C-T, Ping B, Otte AP, Hung M-C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 310: 306–310, 2005. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 9.Chase A, Cross NCP. Aberrations of EZH2 in cancer. Clin Cancer Res 17: 2613–2618, 2011. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Chen J, Zhao L, Song W, Xuan Z, Chen J, Li Z, Song G, Hong L, Song P, Zheng S. CDCA5, transcribed by E2F1, promotes oncogenesis by enhancing cell proliferation and inhibiting apoptosis via the AKT pathway in hepatocellular carcinoma. J Cancer 10: 1846–1854, 2019. doi: 10.7150/jca.28809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin C-H, Chen S-H, Wu H-H, Ho C-W, Ko M-T, Lin C-Y. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 8, Suppl 4: S11, 2014. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colón-Caraballo M, Monteiro JB, Flores I. H3K27me3 is an epigenetic mark of relevance in endometriosis. Reprod Sci 22: 1134–1142, 2015. doi: 10.1177/1933719115578924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunha GR, Lung B. The importance of stroma in morphogenesis and functional activity of urogenital epithelium. In Vitro 15: 50–71, 1979. doi: 10.1007/BF02627079. [DOI] [PubMed] [Google Scholar]
- 14.Doherty LF, Bromer JG, Zhou Y, Aldad TS, Taylor HS. In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: an epigenetic mechanism linking endocrine disruptors to breast cancer. Horm Cancer 1: 146–155, 2010. doi: 10.1007/s12672-010-0015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eskander RN, Ji T, Huynh B, Wardeh R, Randall LM, Hoang B. Inhibition of enhancer of zeste homolog 2 (EZH2) expression is associated with decreased tumor cell proliferation, migration, and invasion in endometrial cancer cell lines. Int J Gynecol Cancer 23: 997–1005, 2013. doi: 10.1097/IGC.0b013e318296a265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eskander RN, Tewari KS. Exploiting the therapeutic potential of the PI3K-AKT-mTOR pathway in enriched populations of gynecologic malignancies. Expert Rev Clin Pharmacol 7: 847–858, 2014. doi: 10.1586/17512433.2014.968554. [DOI] [PubMed] [Google Scholar]
- 17.Fang X, Ni N, Lydon JP, Ivanov I, Bayless KJ, Rijnkels M, Li Q. Enhancer of zeste 2 polycomb repressive complex 2 subunit Is required for uterine epithelial integrity. Am J Pathol 189: 1212–1225, 2019. doi: 10.1016/j.ajpath.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao Y, Li S, Li Q. Uterine epithelial cell proliferation and endometrial hyperplasia: evidence from a mouse model. Mol Hum Reprod 20: 776–786, 2014. doi: 10.1093/molehr/gau033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gene Ontology Consortium; Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32: D258–D261, 2004. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity 105: 4–13, 2010. doi: 10.1038/hdy.2010.54. [DOI] [PubMed] [Google Scholar]
- 21.Gil-Sanchis C, Cervelló I, Mas A, Faus A, Pellicer A, Simón C. Leucine-rich repeat-containing G-protein-coupled receptor 5 (Lgr5) as a putative human endometrial stem cell marker. Mol Hum Reprod 19: 407–414, 2013. doi: 10.1093/molehr/gat014. [DOI] [PubMed] [Google Scholar]
- 22.Greathouse KL, Bredfeldt T, Everitt JI, Lin K, Berry T, Kannan K, Mittelstadt ML, Ho SM, Walker CL. Environmental estrogens differentially engage the histone methyltransferase EZH2 to increase risk of uterine tumorigenesis. Mol Cancer Res 10: 546–557, 2012. doi: 10.1158/1541-7786.MCR-11-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He JP, Zhao M, Zhang WQ, Huang MY, Zhu C, Cheng HZ, Liu JL. Identification of gene expression changes associated with uterine receptivity in mice. Front Physiol 10: 125, 2019. doi: 10.3389/fphys.2019.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ihira K, Dong P, Xiong Y, Watari H, Konno Y, Hanley SJB, Noguchi M, Hirata N, Suizu F, Yamada T, Kudo M, Sakuragi N. EZH2 inhibition suppresses endometrial cancer progression via miR-361/Twist axis. Oncotarget 8: 13509–13520, 2017. doi: 10.18632/oncotarget.14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jefferson WN, Chevalier DM, Phelps JY, Cantor AM, Padilla-Banks E, Newbold RR, Archer TK, Kinyamu HK, Williams CJ. Persistently altered epigenetic marks in the mouse uterus after neonatal estrogen exposure. Mol Endocrinol 27: 1666–1677, 2013. doi: 10.1210/me.2013-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson SA, Spollen WG, Manshack LK, Bivens NJ, Givan SA, Rosenfeld CS. Hypothalamic transcriptomic alterations in male and female California mice (Peromyscus californicus) developmentally exposed to bisphenol A or ethinyl estradiol. Physiol Rep 5: e13133, 2017. doi: 10.14814/phy2.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaneko S, Li G, Son J, Xu C-F, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev 24: 2615–2620, 2010. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med 22: 128–134, 2016. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koot YEM, Teklenburg G, Salker MS, Brosens JJ, Macklon NS. Molecular aspects of implantation failure. Biochim Biophys Acta 1822: 1943–1950, 2012. doi: 10.1016/j.bbadis.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 30.Kusumegi T, Tanaka J, Kawano M, Yonemoto J, Tohyama C, Sone H. BMP7/ActRIIB regulates estrogen-dependent apoptosis: new biomarkers for environmental estrogens. J Biochem Mol Toxicol 18: 1–11, 2004. doi: 10.1002/jbt.20004. [DOI] [PubMed] [Google Scholar]
- 31.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559, 2008. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359, 2012. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li HY, Jin N, Han YP, Jin XF. Pathway crosstalk analysis in prostate cancer based on protein-protein network data. Neoplasma 64: 22–31, 2017. doi: 10.4149/neo_2017_103. [DOI] [PubMed] [Google Scholar]
- 34.Li S, Washburn KA, Moore R, Uno T, Teng C, Newbold RR, McLachlan JA, Negishi M. Developmental exposure to diethylstilbestrol elicits demethylation of estrogen-responsive lactoferrin gene in mouse uterus. Cancer Res 57: 4356–4359, 1997. [PubMed] [Google Scholar]
- 35.Li Y, Luo M, Shi X, Lu Z, Sun S, Huang J, Chen Z, He J. Integrated bioinformatics analysis of chromatin regulator EZH2 in regulating mRNA and lncRNA expression by ChIP sequencing and RNA sequencing. Oncotarget 7: 81715–81726, 2016. doi: 10.18632/oncotarget.13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lottin S, Adriaenssens E, Berteaux N, Leprêtre A, Vilain MO, Denhez E, Coll J, Dugimont T, Curgy JJ. The human H19 gene is frequently overexpressed in myometrium and stroma during pathological endometrial proliferative events. Eur J Cancer 41: 168–177, 2005. doi: 10.1016/j.ejca.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 37.Mallen-St Clair J, Soydaner-Azeloglu R, Lee KE, Taylor L, Livanos A, Pylayeva-Gupta Y, Miller G, Margueron R, Reinberg D, Bar-Sagi D. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev 26: 439–444, 2012. doi: 10.1101/gad.181800.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maniatis S, Äijö T, Vickovic S, Braine C, Kang K, Mollbrink A, Fagegaltier D, Andrusivová Ž, Saarenpää S, Saiz-Castro G, Cuevas M, Watters A, Lundeberg J, Bonneau R, Phatnani H. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 364: 89–93, 2019. doi: 10.1126/science.aav9776. [DOI] [PubMed] [Google Scholar]
- 39.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17: 10–12, 2011. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 40.Monsivais D, Clementi C, Peng J, Fullerton PT Jr, Prunskaite-Hyyryläinen R, Vainio SJ, Matzuk MM. BMP7 induces uterine receptivity and blastocyst attachment. Endocrinology 158: 979–992, 2017. doi: 10.1210/en.2016-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mu W, Starmer J, Yee D, Magnuson T. EZH2 variants differentially regulate polycomb repressive complex 2 in histone methylation and cell differentiation. Epigenetics Chromatin 11: 71, 2018. doi: 10.1186/s13072-018-0242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mucenski ML, Mahoney R, Adam M, Potter AS, Potter SS. Single cell RNA-seq study of wild type and Hox9,10,11 mutant developing uterus. Sci Rep 9: 4557, 2019. doi: 10.1038/s41598-019-40923-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muller RE, Knowler JT. The synthesis of ribosomal RNA and ribosomal protein and their incorporation into ribosomes in the uterus of the oestrogen-stimulated immature rat. FEBS Lett 174: 253–257, 1984. doi: 10.1016/0014-5793(84)81168-0. [DOI] [PubMed] [Google Scholar]
- 44.Nanjappa MK, Mesa AM, Medrano TI, Jefferson WN, DeMayo FJ, Williams CJ, Lydon JP, Levin ER, Cooke PS. The histone methyltransferase EZH2 is required for normal uterine development and function in mice. Biol Reprod 101: 306–317, 2019. doi: 10.1093/biolre/ioz097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, Asp P, Hadler M, Rigo I, De Keersmaecker K, Patel J, Huynh T, Utro F, Poglio S, Samon JB, Paietta E, Racevskis J, Rowe JM, Rabadan R, Levine RL, Brown S, Pflumio F, Dominguez M, Ferrando A, Aifantis I. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 18: 298–301, 2012. doi: 10.1038/nm.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol 21: 4330–4336, 2001. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oki S, Sone K, Oda K, Hamamoto R, Ikemura M, Maeda D, Takeuchi M, Tanikawa M, Mori-Uchino M, Nagasaka K, Miyasaka A, Kashiyama T, Ikeda Y, Arimoto T, Kuramoto H, Wada-Hiraike O, Kawana K, Fukayama M, Osuga Y, Fujii T. Oncogenic histone methyltransferase EZH2: A novel prognostic marker with therapeutic potential in endometrial cancer. Oncotarget 8: 40402–40411, 2017. doi: 10.18632/oncotarget.16316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phan NN, Wang CY, Li KL, Chen CF, Chiao CC, Yu HG, Huang PL, Lin YC. Distinct expression of CDCA3, CDCA5, and CDCA8 leads to shorter relapse free survival in breast cancer patient. Oncotarget 9: 6977–6992, 2018. doi: 10.18632/oncotarget.24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prusinski L, Al-Hendy A, Yang Q. Developmental exposure to endocrine disrupting chemicals alters the epigenome: Identification of reprogrammed targets. Gynecol Obstet Res 3: 1–6, 2016. doi: 10.17140/GOROJ-3-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenfeld CS, Cooke PS. Endocrine disruption through membrane estrogen receptors and novel pathways leading to rapid toxicological and epigenetic effects. J Steroid Biochem Mol Biol 187: 106–117, 2019. doi: 10.1016/j.jsbmb.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504, 2003. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, Sun L, Zhang Y, Chen Y, Li R, Zhang Y, Hong M, Shang Y. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol 27: 5105–5119, 2007. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skoda RC, Schwaller J. Dual roles of EZH2 in acute myeloid leukemia. J Exp Med 216: 725–727, 2019. doi: 10.1084/jem.20190250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D1: D447–D452, 2015. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tempest N, Baker AM, Wright NA, Hapangama DK. Does human endometrial LGR5 gene expression suggest the existence of another hormonally regulated epithelial stem cell niche? Hum Reprod 33: 1052–1062, 2018. doi: 10.1093/humrep/dey083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tesson BM, Breitling R, Jansen RC. DiffCoEx: a simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinformatics 11: 497, 2010. doi: 10.1186/1471-2105-11-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Treviño LS, Wang Q, Walker CL. Phosphorylation of epigenetic “readers, writers and erasers”: Implications for developmental reprogramming and the epigenetic basis for health and disease. Prog Biophys Mol Biol 118: 8–13, 2015. doi: 10.1016/j.pbiomolbio.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vallvé-Juanico J, Suárez-Salvador E, Castellví J, Ballesteros A, Taylor HS, Gil-Moreno A, Santamaria X. Aberrant expression of epithelial leucine-rich repeat containing G protein-coupled receptor 5-positive cells in the eutopic endometrium in endometriosis and implications in deep-infiltrating endometriosis. Fertil Steril 108: 858–867.e2, 2017. doi: 10.1016/j.fertnstert.2017.08.018. [DOI] [PubMed] [Google Scholar]
- 59.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RGAB, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419: 624–629, 2002. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 60.Visser HPJ, Gunster MJ, Kluin-Nelemans HC, Manders EMM, Raaphorst FM, Meijer CJLM, Willemze R, Otte AP. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol 112: 950–958, 2001. doi: 10.1046/j.1365-2141.2001.02641.x. [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 45, W1: W130–W137, 2017. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Yu C, Wang H. HOXA5 inhibits the proliferation and induces the apoptosis of cervical cancer cells via regulation of protein kinase B and p27. Oncol Rep 41: 1122–1130, 2019. doi: 10.3892/or.2018.6874. [DOI] [PubMed] [Google Scholar]
- 63.Winuthayanon W, Hewitt SC, Korach KS. Uterine epithelial cell estrogen receptor alpha-dependent and -independent genomic profiles that underlie estrogen responses in mice. Biol Reprod 91: 110, 2014. doi: 10.1095/biolreprod.114.120170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, Thornton JE, Gregory RI, Morrissey C, Vessella RL, Montironi R, Magi-Galluzzi C, Kantoff PW, Balk SP, Liu XS, Brown M. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338: 1465–1469, 2012. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang Q, Diamond MP, Al-Hendy A. Early life adverse environmental exposures increase the risk of uterine fibroid development: role of epigenetic regulation. Front Pharmacol 7: 40, 2016. doi: 10.3389/fphar.2016.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yao JQ, Yu F. DEB: A web interface for RNA-seq digital gene expression analysis. Bioinformation 7: 44–45, 2011. doi: 10.6026/97320630007044. [DOI] [PMC free article] [PubMed] [Google Scholar]