

Keywords: CD8+ T cells, lymphocytes, nonalcoholic steatohepatitis, stellate cells
Abstract
Nonalcoholic steatohepatitis (NASH) has increased in Western countries due to the prevalence of obesity. Current interests are aimed at identifying the type and function of immune cells that infiltrate the liver and key factors responsible for mediating their recruitment and activation in NASH. We investigated the function and phenotype of CD8+ T cells under obese and nonobese NASH conditions. We found an elevation in CD8 staining in livers from obese human subjects with NASH and cirrhosis that positively correlated with α-smooth muscle actin, a marker of hepatic stellate cell (HSC) activation. CD8+ T cells were elevated 3.5-fold in the livers of obese and hyperlipidemic NASH mice compared with obese hepatic steatosis mice. Isolated hepatic CD8+ T cells from these mice expressed a cytotoxic IL-10-expressing phenotype, and depletion of CD8+ T cells led to significant reductions in hepatic inflammation, HSC activation, and macrophage accumulation. Furthermore, hepatic CD8+ T cells from obese and hyperlipidemic NASH mice activated HSCs in vitro and in vivo. Interestingly, in the lean NASH mouse model, depletion and knockdown of CD8+ T cells did not impact liver inflammation or HSC activation. We demonstrated that under obese/hyperlipidemia conditions, CD8+ T cell are key regulators of the progression of NASH, while under nonobese conditions they play a minimal role in driving the disease. Thus, therapies targeting CD8+ T cells may be a novel approach for treatment of obesity-associated NASH.
NEW & NOTEWORTHY Our study demonstrates that CD8+ T cells are the primary hepatic T cell population, are elevated in obese models of NASH, and directly activate hepatic stellate cells. In contrast, we find CD8+ T cells from lean NASH models do not regulate NASH-associated inflammation or stellate cell activation. Thus, for the first time to our knowledge, we demonstrate that hepatic CD8+ T cells are key players in obesity-associated NASH.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) defines a series of liver pathologies, beginning with hepatic steatosis and progressing to liver fibrosis and even hepatic carcinoma (30, 31). Hepatic steatosis is characterized by the deposition of triglyceride (TG) in the cytoplasm of hepatocytes and is typically reversible. In contrast, the transition from hepatic steatosis to nonalcoholic steatohepatitis (NASH) is a key determinant in the development of irreversible liver fibrosis and/or hepatic carcinoma (2, 9, 14, 18, 43). Thus, understanding how hepatic steatosis transitions to NASH is important for preventing the more devastating forms of NAFLD. In addition to excess TG accumulation, immune cell infiltration into the liver is a key characteristic defining NASH. It is known that immune cells are fundamental contributors to both inflammation and to wound healing responses, which, under chronic conditions, can lead to fibrosis. However, limited attention has been placed on understanding the function of various immune cells during the transition from hepatic steatosis to NASH, especially under conditions that most commonly cause NASH—obesity and hyperlipidemia.
Hepatic stellate cells (HSCs) are considered the primary source of mediators that ultimately lead to hepatic fibrosis (17). Under homeostatic conditions, HSCs reside in the liver sinusoid in a quiescent state and serve as the major storage site for retinol lipid droplets (53). During liver injury, these cells release their lipid droplets, differentiate into myofibroblast-like cells and become activated, expressing α-smooth muscle actin (α-SMA) and secreting large quantities of extracellular matrix proteins.
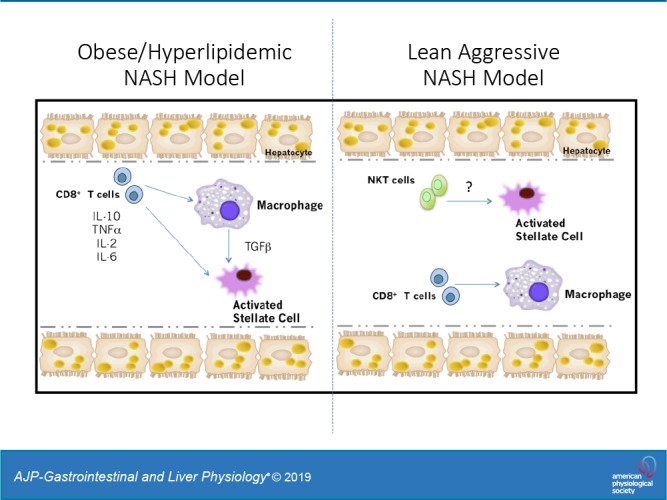
Innate and adaptive immune cells such as Kupffer cells (8, 24, 34), neutrophils (40), natural killer T cells (48, 56), and Th17 T cells (16, 27, 29, 41, 50) have been shown to be involved in hepatic steatosis, inflammation, and fibrosis associated with NAFLD onset. In models of acute liver injury, elevations in CD8+ T cells have been linked to liver fibrosis via activation of HSCs (12, 42). Currently, only two studies have investigated the role of CD8+ T cells in obese models of NASH (4, 56). Using a choline-deficient high-fat diet, or a high-fat high-carbohydrate diet (4, 56), investigators demonstrated that CD8+ T cells are key in driving hepatic inflammation and progression to NASH. However, what is unknown is the contribution of CD8+ T cells in other metabolic conditions linked to NASH, such as obesity/hyperlipidemia or nonobese conditions. Our findings indicate that CD8+ T cells are regulators of obesity and hyperlipidemia-associated NASH. This occurs through production of cytokines, such as IL-10 and TNFα, which drive the recruitment of macrophages and activation of HSCs. However, in the absence of obesity, CD8+ T cells play a limited role in driving NASH.
MATERIALS AND METHODS
Human Liver Tissue
Paraffin-embedded liver sections from bariatric surgery patients with normal, hepatic steatosis, NASH, and cirrhosis were identified through the pathology database using keyword searches. Inclusion criteria required bariatric surgery patients with normal, hepatic steatosis, NASH, and cirrhosis, and a body mass index (BMI) of 30 and above. Cases underwent analysis by a pathologist (M. C. Pacheco) for the various stages of NAFLD based on H&E and trichrome staining. Paraffin-embedded liver sections were prepared and stained with antibodies targeting CD8, α-SMA, and CD68. The Vanderbilt University Institutional Review Board granted approval for this study (IRB Protocol Number 150444). Whole-slide imaging and quantification of immunostaining were performed in the Digital Histology Shared Resource at Vanderbilt University Medical Center using Leica Digital Image Hub Software.
Mice
All animal care procedures were performed after approval from the Vanderbilt University Institutional Animal Care and Use Committee. Male 5-wk-old C57BL/6J, low-density lipoprotein receptor-deficient (LDLR−/−), CD8−/−, and green fluorescent protein (GFP) mice were originally purchased from Jackson Laboratories (Bar Harbor, ME) and crossed or further propagated in our colony.
Obese hepatic steatosis mouse model.
Male 10-wk C57BL/6J mice were ad libitum fed chow or Western diet (WD), which contains 42% of kcal from milk fat with 0.15% added cholesterol (Envigo Harlan-Teklad, Madison, WI) for 8 wk.
Obese/hyperlipidemic NASH mouse model.
Male 10-wk LDLR−/− or CD8−/−;LDLR−/− were ad libitum fed chow or WD for 8 wk.
Choline-deficient NASH model.
WT and CD8KO mice at 8–10 wk of age were ad libitum fed a high-fat choline-deficient diet 60% HF diet with 0.1% methionine and choline-deficient (HFCD) (cat. no. A06071302; Research Diets New Brunswick, NJ) for 10 wk. Carbohydrate and fatty acid composition for the WD and HFCD are detailed in Supplemental Table S1 (All supplemental material is available at https://doi.org/10.6084/m9.figshare.9965834).
Adoptive transfer studies.
For adoptive transfer studies, male 10-wk GFP+/+;LDLR−/− mice on WD for 8 wk were used as donors. Hepatic CD8+ T cells were isolated from donor mice by FACS sorting and pooled. Approximately 1.0 × 106 cells were injected retro-orbitally into WT chow-fed mice.
CD8 depletion studies.
Male 10-wk-old LDLR−/− or WT mice were injected intraperitoneally with rat anti-mouse CD8 antibody (Biolegend, San Diego, CA) or rat IgG (Biolegend) at the start of WD or HFCD feeding. Mice were injected weekly for 8–10 wk. Paraffin-embedded liver sections were stained and analyzed by a pathologist (S. A. Montgomery) for the various stages of NAFLD based on H&E and Sirius red staining (26).
Immune and Stellate Cell Isolation from Liver
Immune cell isolation.
Liver was excised and minced in 1 mg/ml collagenase in PBS. Minced liver was incubated at 37°C for 30 min. The cell suspension was filtered through a 100-μm filter and spun at 300 rpm for 3 min. The supernatant was collected and centrifuged at 1,500 rpm for 10 min. The pellet was resuspended in 40% Percoll and overlayed on top of 60% Percoll. The Percoll gradient was centrifuged at 2,000 g for 20 min. The two middle layers of the Percoll gradient were collected in 3% FBS in RPMI and centrifuged at 1,500 rpm for 10 min. The supernatant was discarded, and the pellet was resuspended in FACS buffer and stained for flow cytometry.
HSC isolation.
HSC methods were adopted from Mederacke et al. (33). Briefly, mice were anesthetized and perfused through the inferior vena cava with EGTA followed by protease and then 3.7 Mandl units per mg dry wt collagenase IV (Worthington Biochemical Corporation, Lakewood, NJ). The liver was removed and minced. The liver was further digested in protease/collagenase for 25 min at 40°C. Cells were centrifuged, and the cell pellet was washed with Grey’s balanced salt solution B. Cells were overlayed onto a Nycodenz gradient and centrifuged. HSCs were collected by removing the cell layer from the gradient, plated, and cultured in DMEM with 10% FBS.
Flow Cytometry
Cells isolated from various tissues were first incubated with Fc block for 5 min at RT, followed by incubation for 20 min at 4°C with fluorophore-conjugated antibodies in the following concentrations: T cell panel: CD4-Alexa 700 (1:100; BD Biosciences, San Jose, CA), TCRβ-APC-Cy7 (1:200; BD Biosciences), CD8-PE-Cy7 or FITC (1:200; BD Biosciences), CD44-PE (1:200; BD Biosciences), CD62L-APC (1:200; BD Biosciences), CD69-CF594 (1:200; BD Biosciences), and CD45-PercpCy5.5 (1:4,000; BD Biosciences). Macrophage panel: CD45-PercpCy5.5 or PE (1:4,000; BD Biosciences), F4/80-APC (1:100; eBioscience, San Diego, CA), CD11b-APC-Cy7 or FITC (1:200; BD Biosciences), and CD11c-A700 (1:200: BD Biosciences). Samples were processed on a 5-laser LSRII machine in the Vanderbilt Flow Cytometry Core, and data was analyzed using FlowJo software. Flow gating strategy for T cells and macrophages is provided in Supplemental Figure S1.
Immunofluorescence Analysis of Liver
Frozen liver sections were cut to 7-μm sections. Tissues were fixed in acetone for 10 min and then washed in PBS for 3 min. After fixation, tissue was washed with PBS and blocked with 5% goat serum. To stain for CD8+ T cells or SMA, a 1:100 dilution of anti-mouse CD8 or α-SMA (Abcam, Cambridge, MA) in goat serum was applied for 1 h at 25°C. Tissue was then washed with PBS and stained with a 1:1,000 dilution secondary-antibody conjugated to anti-mouse Alexa-488 (Cell Signaling Technology, Boston, MA). DAPI (1 μg/ml) was applied for nuclear staining, and tissue was mounted on glass slide with Immu-mount (Pittsburgh, PA). Images were acquired with a FV-1,000 Confocal Microscope.
RNA Isolation and Real-Time RT-PCR
RNA was isolated from ~30–50 mg of liver using the RNeasy mini kit from Qiagen (Valencia, CA). cDNA was synthesized using the iScript cDNA synthesis kit from BioRad (Hercules, CA). cDNA was diluted 1:2 or 1:10 and then used for real-time RT-PCR analysis on a BioRad iQ5 machine. Primer-probe sets were purchased from the “Assays-on-Demand” program at Applied Biosystems (Foster City, CA). Quantification of 18S was performed for each sample, and final relative concentration was determined by comparing each gene of interest to 18S using the ΔΔ CT method.
Western Analysis
Western blots were performed as previously described (3). Briefly, protein concentrations were determined using Pierce BCA Protein Assay (Rockford, IL). Membranes were blocked for 1 h in Odyssey blocking buffer (LI-COR, Lincoln, NE) at RT. Membranes were then probed with specific antibodies for α-SMA and β-actin. Blots were visualized using the Odyssey Infrared Imaging System (LI-COR). For the Odyssey, membranes were incubated with goat anti-rabbit IRDye 800CW secondary antibody at 1:10,000 dilution with 0.1% Tween-20 for 60 min protected from light. After washing in PBS+0.1% Tween-20, the membranes were scanned using an Odyssey Infrared Imaging System. Band intensity was quantified using Image Studio Lite version 3.1 software (Supplemental Figure S11).
T cells and HSCs Coculture
HSCs were isolated from chow-fed GFP+/+;LDLR−/− mice and cultured in DMEM with 10% FBS for 24 h. CD8+ and CD4+ T cells were collected from livers of LDLR−/− WD-fed mice by FACS sorting. Approximately 4.0×105 T cells were resuspended in DMEM with 10% FBS and cocultured with 2.0×105 plated HSCs.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 6.5 (GraphPad Software, San Diego, CA). For single time point measurements, statistical analyses were performed using two-tailed unpaired Student’s t-tests for two groups and one-way ANOVA for more than two groups followed by multiple comparisons. For genotype versus diet studies, two-way ANOVA tests followed by multiple comparisons test were performed.
RESULTS
CD8+ T Cells and SMA are Elevated During the Progression of NAFLD in Humans
Due to the detrimental outcomes of NASH, it is imperative to identify factors responsible for the development of the disease. We examined the types of immune cells as well as HSC activation in the liver of obese patients with various NAFLD pathologies. All subjects were obese and no differences in BMI were detected among the groups with different liver pathologies (Table 1). Histological staining revealed a significant increase in CD8 and SMA staining in cirrhotic livers compared with those with normal, steatosis, or NASH pathologies (Fig. 1). Interestingly, CD68 staining for macrophages was not different among groups.
Table 1.
Body mass index of obese patients
| Pathology | kg/m2 |
|---|---|
| Normal | 43.2 ± 3.9 |
| Steatosis | 48.3 ± 5.7 |
| NASH | 44.5 ± 9.0 |
| Cirrhosis | 51.9 ± 9.3 |
Data are means ± SE of n = 8–10 patients per group. Nonalcoholic steatohepatitis (NASH).
Fig. 1.

Data shown are of one-way ANOVA for CD8, smooth muscle action (SMA), and CD68 staining. CD8+ T cells and SMA expression are elevated during the progression of nonalcoholic fatty liver disease (NAFLD) in obese patients. Top: Human liver tissue sections were obtained from obese patients undergoing bariatric surgery that were diagnosed with normal, steatosis, nonalcoholic steatohepatitis (NASH), or cirrhosis. Bottom: representative images of trichrome staining, H&E, CD8, SMA, and CD68 staining (×20). Staining was quantified using Tissue Image Analysis for the Digital Image Hub. Data represent 8–10 patients per group. ****P < 0.0001.
We detected no significant increase in CD8 or SMA staining in patients with NASH compared with patients with steatosis or normal liver pathology. However, because the degree of NASH varied between patients based on the Brunt criteria (Table 2), we examined whether the degree of staining for CD8 correlated with the severity of NASH (10). Indeed, there was a positive correlation between the amount of CD8 and SMA staining and the severity of NASH (Supplemental Figure S2). However, CD68 and SMA staining in NASH patients were not correlated. In addition, the CD8 and SMA staining were visually localized to similar areas of the tissue (Supplemental Figure S3). These findings suggest that CD8+ T cells may influence HSCs activation under obese conditions of NAFLD.
Table 2.
Brunt score evaluation of obese patients
| Patient ID | Pathology | Zone of Fat Localization | Steatosis % | Steatosis Grade | Inflammation Grade | Ballooning | Fibrosis | Total NAS Score |
|---|---|---|---|---|---|---|---|---|
| 1AFA OV7 | NASH | 3 | 60 | 2 | 2 | 1 | 1a | 5 |
| 1AF7 OY9 | NASH | 3,2,1 | 70 | 3 | 2 | 2 | 2 | 7 |
| 1AEU OAT | NASH | 3,2 | 60 | 2 | 1 | 1 | 1a | 4 |
| 1AFK OKC | NASH | 3 | 25 | 1 | 2 | 0 | 2 | 3 |
| 1ADW O72 | NASH | 3,2,1; diffuse | 70 | 3 | 1 | 2 | 1a | 6 |
| 148X O9G | NASH | 3 | 20 | 1 | 2 | 2 | 1a | 5 |
| 149D OUV | NASH | 3,2 | 40 | 2 | 2 | 1 | 1a | 5 |
| 148T OD7 | NASH | 3 | 20 | 1 | 2 | 1 | 3 | 4 |
| 1492 O5E | NASH | 3,2,1 | 70 | 3 | 1 | 1 | 2 | 5 |
Brunt score evaluation of obese nonalcoholic steatohepatitis (NASH) patients based on zone of fat localization, % steatosis, steatosis grade, inflammation grade, ballooning, and fibrosis. NAS, NAFLD Activity Score.
Obese/Hyperlipidemic Mice Exhibit Characteristics of NASH
To further study the relationship between CD8+ T cells and activated stellate cells, two mouse models, one representing hepatic steatosis and the other representing NASH were used. Wild-type (WT) mice fed WD represent a model of obesity only, and LDLR−/− mice fed WD represent a model of obesity with hyperlipidemia (5, 51, 54). On a WD, LDLR−/− mouse have been reported to develop a number of characteristics seen in human NASH (6, 7, 28, 44). After 8 wk, WD increased adipose tissue mass as well as plasma insulin and glucose levels in both genotypes compared with chow-fed controls (Supplemental Figure S4, A–C). As expected, plasma cholesterol and TG levels were elevated in LDLR−/− mice on WD (Supplemental Figure S4, D and E).
WD increased liver mass and hepatic lipid accumulation in both genotypes (Fig. 2, A–C). Hepatic gene expression of Tnf and Il10 increased by 30-fold (Fig. 2, E and F) in both diet and genotype in the LDLR−/− mice on WD, while IFNγ protein levels in liver (Fig. 2G) were not impacted by diet or genotype. Regarding markers of fibrosis, Sirius red staining (Fig. 2H) and Timp1 gene expression (Fig. 2I) were significantly elevated in WD-fed LDLR−/− mice compared with WD-fed WT mice and chow-fed controls. Tgfb1 and Acta2, a gene that encodes SMA, a marker of HSC activation, was significantly increased in both WT and LDLR−/− fed WD- compared with chow-fed control mice (Fig. 2, J and K). LDLR−/− mice on WD scored higher for inflammation and displayed mild-to-moderate fibrosis compared with WT mice on WD (Table 3). Protein levels of SMA were slightly elevated in WD-fed LDLR−/− mice compared with WD-fed WT mice (Fig. 2K). These findings suggest the WD-fed WT mice represent a model of hepatic steatosis, while WD-fed LDLR−/− display characteristics of mild NASH.
Fig. 2.

LDLR−/− mice on Western diet (WD) represent a model of obesity/hyperlipidemia-induced nonalcoholic steatohepatitis (NASH). Wild-type (WT; open bars) and LDLR−/− (closed bars) mice were placed on chow or WD for 8 wks. Liver weights (A), hepatic lipid measured by Oil red O staining (B) (×10), and liver triglyceride (TG) concentration (C). D: hepatic H&E staining. Liver gene expression of Tnf (E) and Il10 (F) was measured by real-time RT-PCR, and protein levels of IFNγ were measured by ELISA (G). H: hepatic collagen deposition was measured by histological staining with Sirius red. Liver gene expression of Timp1 (I), and Tgfb1 (J) is shown. K: gene and protein expression of total liver alpha-smooth muscle actin (α-SMA). Data are means ± SE of n = 5–9 mice per group. Data analysis: Two-way ANOVA with Tukey correction for A, C, E, F, I, and J. *Genotype effects and +diet effects. * or +P < 0.05, ** or ++P < 0.01, +++P < 0.001, ++++P < 0.0001.
Table 3.
Histological scores of WD fed WT and LDLR−/− mice
| Mouse Genotype and Diet | Macrovesicular Steatosis | Microvesicular Steatosis | Hepatocellular Hypertrophy | NAFLD Score | Inflammation | Fibrosis |
|---|---|---|---|---|---|---|
| WT WD | 0 | 3 | 1 | 4 | 1 | absent |
| WT WD | 1 | 3 | 1 | 5 | 0 | absent |
| WT WD | 0 | 0 | 0 | 0 | 0 | absent |
| WT WD | 1 | 3 | 3 | 7 | 1 | mild |
| WT WD | 1 | 3 | 2 | 6 | 0 | absent |
| WT WD | 0 | 2 | 1 | 3 | 0 | mild |
| LDLR−/− WD | 1 | 2 | 1 | 4 | 0 | mild |
| LDLR−/− WD | 2 | 2 | 1 | 5 | 1 | mild |
| LDLR−/− WD | 1 | 2 | 1 | 4 | 3 | moderate |
| LDLR−/− WD | 2 | 3 | 2 | 7 | 2 | mild |
| LDLR−/− WD | 2 | 3 | 2 | 7 | 3 | mild |
| LDLR−/− WD | 3 | 2 | 2 | 7 | 3 | moderate |
| LDLR−/− WD | 2 | 2 | 1 | 5 | 3 | mild |
NAFLD, nonalcoholic fatty liver disease; WT, wild-type; LDLR−/−, low-density lipoprotein receptor-deficient; WD, Western diet.
Obese/Hyperlipidemic NASH Increases Hepatic CD8+ T Cells
Based on the increase in inflammatory and fibrotic gene expression in the NASH model, we investigated the immune cells present in liver under steatosis and NASH conditions. In the NASH model, the number of macrophages was increased with diet and genotype compared with WT (Supplemental Figure S4G). Likewise, there was a significant 3.5-fold increase in CD8+ T cells (P < 0.0001; Fig. 3A) along with a significant elevation in CD8 effector memory T cells (CD44+CD62L−) and activated CD8+ T cells (CD69+) (Fig. 3B). CD8+ T cells were dispersed throughout the liver and not localized to portal veins (Fig. 3C). Total CD4+ T cells were elevated under NASH conditions (Supplemental Figure S4H). However, no difference in activated CD4+ T cells or natural killer T cells was detected between groups (Supplemental Figure S4, I and J). We detected no difference in CD8+ T cells in spleen (Supplemental Figure S4K) or adipose tissue (Supplemental Figure S4L) between the steatosis and NASH models.
Fig. 3.

Hepatic CD8+ effector memory T cells are elevated in obesity/hyperlipidemia-induced NASH. Nonparenchymal cells were isolated from livers of wild-type (WT; open bars) and LDLR−/− (filled bars) mice on chow or Western diet (WD) for 8 wks. A: flow analysis of CD8+ T cells (CD8+TCRβ+). B: flow analysis of effector memory (CD44+CD62L-) and activated (CD69+) CD8+ T cells. C: immunofluorescence imaging of liver sections stained with CD8 (55) and DAPI (blue). Data are means ± SE of n = 5–6 mice per group. Representative images were captured at ×40. Data analysis: Two-way ANOVA with Tukey correction for A and B. *Genotype effects and +diet effects. ** or ++P < 0.01; ***P < 0.001.
Isolated hepatic CD8+ T Cells from Obese/Hyperlipidemic NASH Display a Cytotoxic IL-10 Phenotype
To examine whether macrophages or T cells were responsible for the elevation in cytokine gene expression detected in livers of the NASH model, hepatic macrophages and CD8+ T cells were isolated by magnetic sorting. Hepatic macrophages from the NASH model displayed a foam-like phenotype (Supplemental Figure S5A) and significant increases in inflammatory genes (Itgax and Tnf) and lipid metabolism genes (Cd36 and Lpl) (Supplemental Figure S5B, P < 0.01). Timp1 gene expression was elevated by 8-fold in hepatic macrophages of NASH model, while no difference in the Il10 gene was detected in macrophages among groups. Analysis of sorted hepatic CD8+ T cells yielded surprisingly different results. These CD8+ T cells were found to express a 25-fold increase in Il10 gene expression in the NASH compared with steatosis model (Supplemental Figure S5C, P < 0.005). Gene expression of Gzmb and Prf1 were also significantly increased (P < 0.05) in CD8+ T cells isolated from NASH model. There were no detectable differences in Tnf, Ifn, Il4, or Il6 genes.
To identify the cytokines secreted from hepatic CD8+ T cells, a cytokine array was performed on media collected from CD3/CD28 stimulated CD8+ T cells isolated from liver and spleen of the steatosis and NASH models. A significant increase in IL-10, TNFα, IL-6, and IL-2 secretion was detected in stimulated CD8+ T cells from livers of NASH compared with the steatosis model (Fig. 4). Interestingly, isolated CD8+ T cells from the spleens of the same mice displayed a completely different phenotype where IL-10 protein levels were considerably lower and not significantly different between the NASH and steatosis models (Supplemental Figure S6). These results demonstrated that under obese, hyperlipidemic, and NASH conditions, macrophages are a major source of TNFα and TIMP-1 expression, while CD8+ T cells are the primary source of IL-10. Furthermore, hepatic T cells from NASH mice have a cytokine profile distinct from splenic T cells.
Fig. 4.

Isolated hepatic CD8+ T cells express an IL-10 phenotype in obesity/hyperlipidemia-induced nonalcoholic steatohepatitis (NASH). Wild-type (WT) and LDLR−/− mice were placed on chow or WD for 8 wks. Mice were euthanized and livers collected for isolation of CD8+ T cells. Isolated CD8+ T cells were stimulated with IgG (control) or CD3/C28 for 72 h. Media was collected and secreted cytokine levels of IL-10, TNFα, Granzyme B, IL-6, IL-2, and IL-4 measured by Luminex. Data are means ± SE of n = 3 mice per group. Data analysis: two-way ANOVA with Tukey correction. *P < 0.05; ** or ++P < 0.01.
Hepatic CD8+ T Cells Regulate Inflammation, Macrophage Infiltration, and Stellate Cell Activation in the Obese/Hyperlipidemic NASH Model
CD8+ T cells regulate adipose tissue inflammation, macrophage recruitment, and insulin resistance under obese conditions (36). Conversely, in other models of inflammation and fibrosis, CD8 deficiency leads to a reduction in macrophage and lymphocyte populations (13). However, the extent to which this occurs in liver in an obesity and hyperlipidemia-associated model of NASH is unknown. To investigate the function of CD8+ T cells in the development of NASH in vivo, CD8 antibody depletion, global knockdown of CD8+ T cells in LDLR−/− mice, and CD8+ T cell adoptive transfer studies were conducted.
Using antibodies that target CD8+ T cells, acute (1-wk) CD8+ T cell depletion in the NASH model did not significantly reduce Cd8 gene expression but led to a significant reduction in Acta2, while Tnf, Il10, Tgfb1 and Timp1 were not impacted (Supplemental Figure S7A). Due to the mild reduction in CD8+ T cells in the acute study, a second chronic study was initiated in which CD8 antibody was administered weekly throughout the 8 wk of WD feeding. No differences in metabolic parameters were found among groups (Supplemental Figure S7B). Chronic CD8 antibody administration led to a significant 66% reduction of CD8+ T cells (Fig. 5A), which was also confirmed by measuring hepatic CD8 gene expression (data not shown). Similar to what has been demonstrated in adipose tissue (36), CD8 depletion also reduced macrophage levels in the liver (Fig. 5A). Likewise, there was a significant decrease in cytokine genes Il10, Tnf, Il2 (P < 0.05; Fig. 5B), as well as HSC-associated genes Tgfb1, Ch25h, and Acta2 (P < 0.05; Fig. 5C). By immunofluorescence we detected a reduction in SMA in livers of the CD8 depletion group compared with the IgG control (Fig. 5D). In comparison, global knockdown of CD8 in LDLR−/− mice decreased the number of hepatic macrophages and decreased SMA protein levels on WD compared with LDLR−/− mice on WD (Supplemental Figure S8).
Fig. 5.

Depletion of hepatic CD8+ T cells reduces liver macrophage populations and alpha-smooth muscle actin (α-SMA) expression in obesity/hyperlipidemia-induced nonalcoholic steatohepatitis (NASH) mice. LDLR−/− mice were placed on Western diet (WD) for 8 wks and injected weekly with IgG or anti-CD8 antibody throughout the duration of the diet. Mice were euthanized and livers isolated for subsequent analysis. A: flow analysis and quantification of hepatic CD8+TCRβ+ and CD11b+F4/80+ cells. B: Il10, Tnf, Il2, and Il6 gene expression in total liver. C: extracellular matrix markers, Tgfb1, Ch25h, Acta2, Timp1, and Col1a1 gene expression in total liver. D: immunofluorescence of liver sections stained with CD8 (55), α-SMA (11), and DAPI (blue) at ×40. Data are means ± SE of n = 10–12 mice per group. Data analysis: unpaired Student’s t-test. *P < 0.05 compared with IgG control group; **P < 0.01; ****P < 0.0001.
To further investigate the function of CD8+ T cells in development of NASH, an adoptive transfer study was initiated. To track the transfer of cells into the liver, GFP+/+;LDLR−/− mice were generated and fed a WD for 8 wk. Hepatic CD8+ T cells were isolated from these mice and injected into WT mice on chow diet. At 24-h postadoptive transfer, CD8+ T cells were detected in the liver and represented ~10% of the total hepatic CD8+ T cells (Fig. 6A). No change in inflammatory cytokines was detected (Fig. 6B), while a significant increase in Acta2 gene expression or SMA protein expression was found in mice receiving NASH CD8+ T cells (P < 0.001) (Fig. 6, C and D).
Fig. 6.

Adoptive transfer of hepatic CD8+ T cells from obesity/hyperlipidemia-induced nonalcoholic steatohepatitis (NASH) mice increases alpha-smooth muscle actin (α-SMA) expression. LDLR−/−;GFP+/+ mice were placed on Western diet (WD) for 8 wks. Mice were euthanized and hepatic CD8+ T cells were isolated from livers. Isolated CD8+ T cells were then injected into wild-type (WT) chow-fed mice and recipient mice were euthanized 24 h after injections. A: flow cytometry analysis of hepatic CD8+TCRβ+GFP+ cells. B: Tnf, Il10, Itgam, Itgax gene expression in total liver. C: Tgfb1, Timp1, and Acta2 gene expression in total liver. D: immunofluorescence of liver sections stained with α-SMA (11) and DAPI (blue) at ×40. Data are means ± SE of n = 5–6 mice per group. Data analysis: unpaired Student’s t-test. ****P < 0.0001.
Hepatic CD8+ T Cells from a Lean NASH Model Do Not Regulate Hepatic Inflammation and Stellate Cell Activation
Next, we sought to determine whether CD8+ T cells exert the same function on hepatic inflammation and HSC activation in a commonly used model of NASH. Diets deficient in methionine and choline are commonly used to induce severe liver disease in mice (32). However, one major caveat of this model is the loss in body weight and adipose tissue mass along with reduced insulin and glucose levels. To circumvent the weight loss, we used HFCD. WT mice on the HFCD diet maintained their body weight compared with mice on the HF diet, which gained weight (Supplemental Figure S9A). After 10 wk, HFCD-fed mice had reduced adipose tissue mass and fasting glucose levels compared with mice on HF diet (Supplemental Figure S9B). Comparable to published findings (32, 56) the HFCD increased liver mass compared with HF diet. Next, we examined hepatic immune cell populations. Similar to the obese/hyperlipidemia NASH model, CD8+ T cells and macrophages were elevated in the HFCD-fed mice (Supplemental Figure S9C). To investigate the function of these cells in NASH, CD8+ T cells were depleted using CD8 antibody. No differences in metabolic parameters were found among groups (Supplemental Figure S9, D and E). Chronic treatment of HFCD mice with CD8 antibody reduced CD8+ T cells with no impact on CD4+ T cells (Fig. 7A). However, CD8 antibody had no impact on gene expression of markers associated with hepatic inflammation, macrophages, or HSC activation in total liver or collagen deposition (Fig. 7, B and C).
Fig. 7.

CD8+ T cells do not regulate liver inflammation or activate hepatic stellate cells (HSCs) under nonobese nonalcoholic steatohepatitis (NASH) conditions. Wild-type (WT) mice were placed on high-fat diet with 0.1% methionine and deficient in choline (HFCD) diet for 10 wks and injected weekly with IgG or anti-CD8 antibody throughout the duration of the diet. Mice were euthanized, and livers were isolated for subsequent analysis. A: flow cytometric analysis and quantification of hepatic CD8+ and CD4+ T cells. B: gene expression in total liver. C: hepatic collagen deposition was measured by histological staining with Sirius red. Data are means ± SE of n = 10–12 mice per group. Data analysis: unpaired Student’s t-test. *P < 0.05.
Because CD8 antibody treatment reduced CD8+ T cells by ~50% and had no impact on hepatic inflammation or stellate cell activation, we examined whether complete knockdown of CD8+ T cells using CD8KO mice prevented HFCD-induced hepatic inflammation and HSC activation. Similar to WT mice on the HFCD, CD8KO mice maintained their body weight, remained glucose tolerant, and had reduced adipose tissue and liver mass compared with mice on HF diet (Supplemental Figure S10A). By flow analysis, HFCD induced CD8+ in WT, but total T cells, CD8+ effective memory, and activated T cells were eliminated in the CD8KO mice (Supplemental Figure S10B). In contrast to our findings in the obese NASH model, HFCD-induced macrophage infiltration was not impacted in CD8KO (Supplemental Figure S10C). Likewise, when we examined total liver gene expression, CD8 deficiency did not prevent HFCD-induced gene expression of HSC markers Acta2, Tgfb1, Timp1, and Col1a1, macrophage/monocyte markers Emr1, Itgax, and Itgam, or inflammatory markers Il10 and Tnf (Supplemental Figure S10D).
Hepatic CD8+ T Cells from Obese/Hyperlipidemic NASH Mice Directly Activate HSCs
We examined the gene expression of isolated hepatic CD8+ T cells from the lean NASH model by gene expression and found their gene expression profile was completely different compared with the CD8+ T cells isolated from the obese NASH model. Unlike the CD8+ T cells from the obese NASH model, these cells expressed low levels of Il10, Ifn, Gzmb and Tgfb1 genes (Fig. 8). Based on the cytokine expression profile and ability of CD8+ T cells to induce HSC activation in the obese and hyperlipidemia NASH model, we investigated the ability of CD8+ T cells from both the obese and lean NASH models to induce HSC activation directly. Coculture of isolated CD8+ T cells from the obese NASH model with isolated GFP+/+ HSCs significantly increased α-SMA staining compared with untreated HSCs (Fig. 9, A and B). Moreover, isolated CD4+ T cells from the obese NASH model did not activate HSCs. Similarly, CD8+ and CD4+ T cells from the nonobese NASH model did not activate HSCs (Fig. 9, C and D).
Fig. 8.

Phenotype of isolated CD8+ T cells from obese nonalcoholic steatohepatitis (NASH) and nonobese NASH model. Nonparenchymal cells were isolated from livers of Western diet (WD)-fed wild-type (WT), WD-fed LDLR−/−, high-fat (HF)-fed WT, and HF diet with 0.1% methionine and deficient in choline (HFCD)-fed WT on diet for 8 wks. FACS sorting and RNA isolation of CD8+ T cells were performed. Gene expression was measured by real-time PCR. Data are means ± SE of n = 3–5 mice per group. Data analysis: two-way ANOVA; *P < 0.05.
Fig. 9.

CD8+ T cells isolated from the obesity/hyperlipidemia-induced, but not lean nonalcoholic steatohepatitis (NASH), mice directly activate hepatic stellate cells (HSCs). HSCs were isolated from GFP+/+ chow-fed mice at 18 wks of age. HSCs were plated and incubated for 24 h. CD8+ T cells and CD4+ T cells were isolated from Western diet (WD)-fed LDLR−/− mice (ON) or high-fat diet with 0.1% methionine and deficient in choline (HFCD)-fed wild-type (WT) mice. Isolated T cells were cocultured with plated with HSCs for 3 days. Cells were fixed and stained for alpha-smooth muscle actin (α-SMA) (11). A and C: isolated HSC are fluorescent green and images were obtained by confocal microscopy at ×40. B and D: quantification of the activated HSCs using Image Xpress. Data are means ± SE of n = 3 mice per group. Data analysis: one-way ANOVA; ****P < 0.0001.
DISCUSSION
Our human data demonstrated a positive correlation between CD8+ T cells and stellate activation in obese patients with increasing degrees of NASH. Our data also demonstrated that during obesity, CD8+ T cells do not infiltrate the liver in large quantities until the onset of cirrhosis. We demonstrated that CD8+ T cells are the primary regulators of HSC activation in an obese/hyperlipidemic mouse model of NASH. Mechanistically, in the presence of obesity and hyperlipidemia, hepatic CD8+ T cells express an IL-10 phenotype, regulate hepatic inflammation, increase macrophage accumulation, and induce HSC activation. In contrast, in a lean choline-deficient NASH mouse model, hepatic CD8+ T cells have no impact on liver inflammation or HSC activation (Fig. 7 and Supplemental Fig. S10). We also find that isolated hepatic CD8+ T cells from the lean NASH model express a different phenotype to CD8+ T cells isolated from the obese hepatic steatosis mouse model (Fig. 8). Our data demonstrated that CD8+ T cells regulate HSC activation and liver injury in an obese/hyperlipidemic environment and played a minimal role in the development of NASH under nonobese conditions.
In humans, a strong relationship has been established between the progression of NAFLD and several metabolic conditions. Obesity, type 2 diabetes, elevated fasting blood glucose levels, hypertriglyceridemia, and hyperlipidemia have all been identified as risk factors for the development of NASH. We find in obese humans, CD8 staining positively correlated with HSC activation under NASH and cirrhotic conditions. To date, only two studies have identified positive correlations between elevations in CD8+ T cell infiltration in human liver and the degree of NASH (15, 56). However, correlations were not associated with hepatic stellate activation or body weight. Surprisingly, NAFLD in nonobese individuals is not well characterized. Of the limited clinical cases and epidemiology studies, nonobese NAFLD patients are typically nondiabetic, have a normal plasma lipid profile, and show no difference in severity of NASH or fibrosis compared with obese NAFLD patients (21). While our findings suggest obesity may contribute to the link between CD8+T cells and HSC activation, future studies are needed to assess the plasma lipid levels of obese and nonobese NASH patients and their impact on hepatic CD8+ T cell infiltration and fibrosis.
One caveat of our research findings is the use of the methionine/choline diet as a representative mouse model of lean NASH. Few patients with NAFLD are deficient for this amino acid and vitamin. Also, it is not known how either deficiency impacts immune cell function. NAFLD patients are primarily obese metabolic conditions, such as diabetes or dyslipidemia. Our study and others have reported that the commonly used methionine/choline-deficient (MCD) diet and WD yield two completely different pathologies of NASH. In mice, The MCD diet yields severe hepatic inflammation and fibrosis, due to impaired hepatic VLDL secretion. Where on the WD model, NASH is milder and recapitulates a number of the metabolic parameters associated with human NAFLD, such as obesity, hyperglycemia, and impaired glucose tolerance (20, 32).
It remains to be determined how obesity regulates hepatic CD8+ T cell function in NASH. Our data demonstrated obesity contributes to the function of hepatic CD8+ T cells in the progression of NASH. We also demonstrated that in the absence of obesity, hepatic CD8+ T cells do not regulate hepatic liver injury in NASH. Wolfe et al. (56) used an obese choline-deficient model, were the mice gained excessive adipose tissue mass and elevations in serum TG and cholesterol levels following 6 and 12 mo of diet. In their model CD8+ T cells did control stellate cell activation. Thus, crosstalk between the adipose tissue and liver could possibly drive T cell activation and liver injury under NASH conditions.
The activation of CD8+ T cells in hypercholesterolemic LDLR−/− mice fed WD is interesting, given that cholesterol metabolism is essential for CD8+ T cell activation (59). Following activation, CD8+ T cells use cholesterol biosynthesis and transport for producing granules and cytokines (59). Furthermore, cholesterol accumulation drives T cells toward a Th1 phenotype (40). In humans, statins have led to improvements in hepatic steatosis and inflammation (41). Similarly, in the absence of cholesterol, hepatic inflammation is reduced, suggesting CD8+ T cells infiltration or activation may be dependent on cholesterol (57). Cholesterol may also indirectly activate CD8+ T cells via antigen presenting cells. Dendritic cells, macrophages, and even stellate cells are capable of CD8+ T cell activation and proliferation. Dendritic cells and macrophages engulf oxidized LDL or free cholesterol (1, 19, 57). In humans and mice with NASH, Kupffer cells are found surrounding hepatocytes containing cholesterol crystals, and the Kupffer cells themselves can accumulate excess cholesterol leading to activation (19). In our study, we detected lipid-enriched macrophages in the obese and hyperlipidemic NASH model that were not present in the steatosis model. We also detected an elevation in the dendritic cell marker, CD11c+. Thus, macrophage and dendritic cell accumulation of cholesterol may contribute to CD8+ T cell activation. Given the function of CD8+ T cells in obesity-associated NAFLD, identification of factors responsible for activating these antigen-presenting macrophages and dendritic cells would shed light on specific antigens to target in NASH individuals.
Macrophages have been extensively studied in NAFLD and shown to increase as NAFLD progresses and regulate hepatic steatosis and inflammation (35, 52). Surprisingly, macrophages were not elevated in the livers of obese subjects with NASH or cirrhosis. Under normal and steatosis conditions, macrophages remained dispersed throughout the tissue but seem to accumulate closer to areas of collagen deposition in NASH and cirrhosis patients (Supplemental Figure S3). We demonstrated that depletion of CD8+ T cells regulates recruitment of hepatic CD11b+F4/80+ macrophages (Fig. 5A). In chronic liver injury, Kupffer cells act as the first responders to signals secreted from inflamed hepatocytes or infected cells. Kupffer cells can directly activate HSCs and both are capable of secreting inflammatory cytokines, such as CCL2, which recruit inflammatory Ly6highCD11b+ monocytes to the inflamed liver (49). As hepatic inflammation progresses, recruited monocytes also activate HSCs driving extracellular matrix deposition. It has been reported that CD8+ T cells control circulating Ly6ChighCD11b+ monocyte levels leading to reduced macrophage accumulation in atherosclerotic plaques and adipose tissue (13, 36). These monocytes are characterized by expression of CCR2, the receptor for CCL2. However, in our studies we did not detect differences in gene expression of Ccr2 or Itgam, the gene that encodes for CD11b. Bhattacharjee et al. (4) reported that depletion of CD8+ T cells reduces resident and recruited macrophages populations in an obese NASH mouse model by histological staining. It remains to be determined how hepatic CD8+ T cells regulate hepatic macrophage accumulation during NASH progression. These hepatic T cells may possibly release cytokines/factors to increase macrophage recruitment or proliferation.
Our findings suggest that under obese and hyperlipidemia conditions, hepatic CD8+ T cells have an IL-10 profile that regulates liver inflammation and stellate cell activation. While in the nonobese NASH model, CD8+ T cells do not contribute to liver injury or regulation of hepatic macrophage populations. IL-10 has also been reported to be elevated in patients with hepatic steatosis and NASH (39). IL-10 is well known to inhibit antigen-specific activation, proliferation, and production of proinflammatory cytokines by T cells and macrophages. Likewise, depletion or knockdown of IL-10 has led to reductions in liver injury in mouse models of NAFLD. While much emphasis is placed on the anti-inflammatory properties of IL-10, it also exhibits pro- and antifibrogenic properties. Studies have reported IL-10 induces TIMP-1 expression in macrophages (22, 23). IL-10 may also act to enhance survival of hepatic CD8+ T cells. In liver disease, such as hepatitis B virus infection (HBV), IL-10 secretion specifically from HBV-specific CD8+ T cells promotes liver disease. Thus IL-10 secretion from CD8+ T cells may act in an autocrine and paracrine fashion to promote cell survival allowing the progression of NASH.
Mechanistically, HSCs in the obese and hyperlipidemic model may be activated by a number of different factors, such as cytokines secreted from the CD8+ T cells, macrophages or hepatocytes. High concentrations of dietary factors, glucose, and cholesterol have been documented to activate HSC (45, 51, 60). In our studies, the cytokine profile of activated hepatic CD8+ T cells displayed higher levels of IL-10, TNFα, and IL-6 compared with hepatic CD8+ T cells from the steatosis model. TNFα has been reported to activate HSCs. We also detected an increase in TGFβ expression in hepatic CD8+ T cells, one of the major regulators of HSC activation. Furthermore, CD8 depletion studies reduced TGFβ gene expression (25, 46). The CD8+ T cells from the obese NASH model also expressed the cytotoxic proteins, perforin and granzyme. Cytotoxic T cells are also capable of destroying infected cells via the Fas ligand/Fas signaling pathway. Likewise, macrophages and natural killer T cells can secrete factors, such as TGFβ and proinflammatory cytokines, inducing HSC activation (37, 38, 47). Future studies are needed to determine, under NASH conditions, what cells types (Kupffer cells, hepatocytes, or HSC) are targeted by hepatic CD8+ T cells.
Collectively, our data demonstrated that metabolic conditions influence CD8+ T cells phenotype and function. In obesity- and hyperlipidemia-associated NASH, hepatic CD8+ T cells infiltrate the liver and contribute to macrophage accumulation and hepatic stellate activation. Therefore, CD8+ T cells are important drivers of hepatic inflammation and injury in obesity related NASH.
GRANTS
This work was supported by an American Diabetes Association Mentor-Based Postdoctoral Fellowship Grant 7-10-MI-05, UNCF-Merck Postdoctoral Science Research Fellowship, the Vanderbilt CTSA Grant UL1 TR000445 from National Center for Advancing Translational Sciences/National Institutes of Health (NCATS/NIH), and National Heart, Lung, and Blood Institute Grant K01HL121010 (to A. J. Kennedy). A. H. Hasty is supported by a Merit Award from the Veterans Affairs (Grant 5I01BX002195), an Established Investigator Award from the American Heart Association (Grant 12EIA827), and an Innovation Award from the American Diabetes Association (Grant 1-17-IBS-140). Whole-slide imaging and quantification of immunostaining were performed in the Digital Histology Shared Resource at Vanderbilt University Medical Center (VUMC). Confocal microscopy was performed using the VUMC Cell Imaging Shared Resource, supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637, and EY08126. Flow cytometry experiments were performed in the VMC Flow Cytometry Shared Resource, which is supported by the Vanderbilt Digestive Disease Research Center NIH Grant DK058404 from whom A. H. Hasty received scholarship funds. Animal histopathology and/or clinical services was performed by the Animal Histopathology & Laboratory Medicine Core at the University of North Carolina (UNC), which is supported in part by a National Cancer Institute Center Core Support (Grant 5P30CA016086-41) to the UNC Lineberger Comprehensive Cancer Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.H.H. and A.J.K. conceived and designed research; D.A.B., A.H.H., and A.J.K. performed experiments; D.A.B., M.C.P., M.K.W., S.A.M., A.H.H., and A.J.K. analyzed data; D.A.B., M.C.P., M.K.W., S.A.M., A.H.H., and A.J.K. interpreted results of experiments; D.A.B., A.H.H., and A.J.K. prepared figures; A.H.H. and A.J.K. drafted manuscript; D.A.B., M.C.P., M.K.W., S.A.M., A.H.H., and A.J.K. edited and revised manuscript; D.A.B., M.C.P., M.K.W., S.A.M., A.H.H., and A.J.K. approved final version of manuscript.
REFERENCES
- 1.Alderman CJ, Bunyard PR, Chain BM, Foreman JC, Leake DS, Katz DR. Effects of oxidised low density lipoprotein on dendritic cells: a possible immunoregulatory component of the atherogenic micro-environment? Cardiovasc Res 55: 806–819, 2002. doi: 10.1016/S0008-6363(02)00447-9. [DOI] [PubMed] [Google Scholar]
- 2.Alkhouri N, Tamimi TA, Yerian L, Lopez R, Zein NN, Feldstein AE. The inflamed liver and atherosclerosis: a link between histologic severity of nonalcoholic fatty liver disease and increased cardiovascular risk. Dig Dis Sci 55: 2644–2650, 2010. doi: 10.1007/s10620-009-1075-y. [DOI] [PubMed] [Google Scholar]
- 3.Anderson EK, Hill AA, Hasty AH. Stearic acid accumulation in macrophages induces toll-like receptor 4/2-independent inflammation leading to endoplasmic reticulum stress-mediated apoptosis. Arterioscler Thromb Vasc Biol 32: 1687–1695, 2012. doi: 10.1161/ATVBAHA.112.250142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar-Gonzalez RM, Shivakumar P, Kohli R. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol Commun 1: 299–310, 2017. doi: 10.1002/hep4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bieghs V, Van Gorp PJ, Wouters K, Hendrikx T, Gijbels MJ, van Bilsen M, Bakker J, Binder CJ, Lütjohann D, Staels B, Hofker MH, Shiri-Sverdlov R. LDL receptor knock-out mice are a physiological model particularly vulnerable to study the onset of inflammation in non-alcoholic fatty liver disease. PLoS One 7: e30668, 2012. doi: 10.1371/journal.pone.0030668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bieghs V, Verheyen F, van Gorp PJ, Hendrikx T, Wouters K, Lütjohann D, Gijbels MJ, Febbraio M, Binder CJ, Hofker MH, Shiri-Sverdlov R. Internalization of modified lipids by CD36 and SR-A leads to hepatic inflammation and lysosomal cholesterol storage in Kupffer cells. PLoS One 7: e34378, 2012. doi: 10.1371/journal.pone.0034378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bijnen M, Beelen N, Wetzels S, Gaar JV, Vroomen M, Wijnands E, Scheijen JL, van de Waarenburg MPH, Gijbels MJ, Cleutjens JP, Biessen EAL, Stehouwer CDA, Schalkwijk CG, Wouters K. RAGE deficiency does not affect non-alcoholic steatohepatitis and atherosclerosis in Western type diet-fed Ldlr-/- mice. Sci Rep 8: 15256, 2018. doi: 10.1038/s41598-018-33661-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int 26: 1175–1186, 2006. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 9.Bonora E, Targher G. Increased risk of cardiovascular disease and chronic kidney disease in NAFLD. Nat Rev Gastroenterol Hepatol 9: 372–381, 2012. doi: 10.1038/nrgastro.2012.79. [DOI] [PubMed] [Google Scholar]
- 10.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 94: 2467–2474, 1999. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 11.Caldwell SH, de Freitas LA, Park SH, Moreno ML, Redick JA, Davis CA, Sisson BJ, Patrie JT, Cotrim H, Argo CK, Al-Osaimi A. Intramitochondrial crystalline inclusions in nonalcoholic steatohepatitis. Hepatology 49: 1888–1895, 2009. doi: 10.1002/hep.22851. [DOI] [PubMed] [Google Scholar]
- 12.Casini A, Ricci OE, Paoletti F, Surrenti C. Immune mechanisms for hepatic fibrogenesis. T-lymphocyte-mediated stimulation of fibroblast collagen production in chronic active hepatitis. Liver 5: 134–141, 1985. doi: 10.1111/j.1600-0676.1985.tb00228.x. [DOI] [PubMed] [Google Scholar]
- 13.Cochain C, Koch M, Chaudhari SM, Busch M, Pelisek J, Boon L, Zernecke A. CD8+ T cells regulate monopoiesis and circulating Ly6C-high monocyte levels in atherosclerosis in mice. Circ Res 117: 244–253, 2015. doi: 10.1161/CIRCRESAHA.117.304611. [DOI] [PubMed] [Google Scholar]
- 14.Ekstedt M, Franzén LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 44: 865–873, 2006. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 15.Ghazarian M, Revelo XS, Nøhr MK, Luck H, Zeng K, Lei H, Tsai S, Schroer SA, Park YJ, Chng MHY, Shen L, D’Angelo JA, Horton P, Chapman WC, Brockmeier D, Woo M, Engleman EG, Adeyi O, Hirano N, Jin T, Gehring AJ, Winer S, Winer DA. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol 2: eaai7616, 2017. doi: 10.1126/sciimmunol.aai7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giles DA, Moreno-Fernandez ME, Stankiewicz TE, Cappelletti M, Huppert SS, Iwakura Y, Dong C, Shanmukhappa SK, Divanovic S. Regulation of inflammation by IL-17A and IL-17F modulates non-alcoholic fatty liver disease pathogenesis. PLoS One 11: e0149783, 2016. doi: 10.1371/journal.pone.0149783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol 6: 425–456, 2011. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, Bi Y, Xu M, Ma Z, Xu Y, Wang T, Li M, Liu Y, Lu J, Chen Y, Huang F, Xu B, Zhang J, Wang W, Li X, Ning G. Nonalcoholic fatty liver disease is associated with atherosclerosis in middle-aged and elderly Chinese. Arterioscler Thromb Vasc Biol 32: 2321–2326, 2012. doi: 10.1161/ATVBAHA.112.252957. [DOI] [PubMed] [Google Scholar]
- 19.Ioannou GN, Haigh WG, Thorning D, Savard C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J Lipid Res 54: 1326–1334, 2013. doi: 10.1194/jlr.M034876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanuri G, Bergheim I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int J Mol Sci 14: 11963–11980, 2013. doi: 10.3390/ijms140611963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim D, Kim WR. Nonobese fatty liver disease. Clin Gastroenterol Hepatol 15: 474–485, 2017. doi: 10.1016/j.cgh.2016.08.028. [DOI] [PubMed] [Google Scholar]
- 22.Kumada M, Kihara S, Ouchi N, Kobayashi H, Okamoto Y, Ohashi K, Maeda K, Nagaretani H, Kishida K, Maeda N, Nagasawa A, Funahashi T, Matsuzawa Y. Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation 109: 2046–2049, 2004. doi: 10.1161/01.CIR.0000127953.98131.ED. [DOI] [PubMed] [Google Scholar]
- 23.Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer JM. IL-10 inhibits metalloproteinase and stimulates TIMP-1 production in human mononuclear phagocytes. J Clin Invest 96: 2304–2310, 1995. doi: 10.1172/JCI118286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lanthier N, Molendi-Coste O, Cani PD, van Rooijen N, Horsmans Y, Leclercq IA. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. FASEB J 25: 4301–4311, 2011. doi: 10.1096/fj.11-189472. [DOI] [PubMed] [Google Scholar]
- 25.Li JT, Liao ZX, Ping J, Xu D, Wang H. Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J Gastroenterol 43: 419–428, 2008. doi: 10.1007/s00535-008-2180-y. [DOI] [PubMed] [Google Scholar]
- 26.Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, Havekes LM, Kleemann R, van den Hoek AM. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One 9: e115922, 2014. doi: 10.1371/journal.pone.0115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, She W, Wang F, Li J, Wang J, Jiang W. 3, 3′-Diindolylmethane alleviates steatosis and the progression of NASH partly through shifting the imbalance of Treg/Th17 cells to Treg dominance. Int Immunopharmacol 23: 489–498, 2014. doi: 10.1016/j.intimp.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 28.Lytle KA, Jump DB. Is Western diet-induced nonalcoholic steatohepatitis in Ldlr-/- mice reversible? PLoS One 11: e0146942, 2016. doi: 10.1371/journal.pone.0146942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma X, Hua J, Mohamood AR, Hamad AR, Ravi R, Li Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 46: 1519–1529, 2007. doi: 10.1002/hep.21823. [DOI] [PubMed] [Google Scholar]
- 30.Machado AP. Metabolic syndrome, atherosclerosis and thrombogenic risk. Rev Port Cardiol 25: 173–178, 2006. [PubMed] [Google Scholar]
- 31.Machado M, Cortez-Pinto H. Non-alcoholic steatohepatitis and metabolic syndrome. Curr Opin Clin Nutr Metab Care 9: 637–642, 2006. doi: 10.1097/01.mco.0000241677.40170.17. [DOI] [PubMed] [Google Scholar]
- 32.Machado MV, Michelotti GA, Xie G, Almeida Pereira T, Boursier J, Bohnic B, Guy CD, Diehl AM. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 10: e0127991, 2015. doi: 10.1371/journal.pone.0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mederacke I, Dapito DH, Affò S, Uchinami H, Schwabe RF. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc 10: 305–315, 2015. doi: 10.1038/nprot.2015.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139: 323–334.e7, 2010. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol 302: G1310–G1321, 2012. doi: 10.1152/ajpgi.00365.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15: 914–920, 2009. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 37.Park O, Jeong WI, Wang L, Wang H, Lian ZX, Gershwin ME, Gao B. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology 49: 1683–1694, 2009. doi: 10.1002/hep.22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman R, Dragomir AC, Aloman C, Schwabe RF. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58: 1461–1473, 2013. doi: 10.1002/hep.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rabelo F, Oliveira CP, Faintuch J, Mazo DF, Lima VM, Stefano JT, Barbeiro HV, Soriano FG, Alves VA, Carrilho FJ. Pro- and anti-inflammatory cytokines in steatosis and steatohepatitis. Obes Surg 20: 906–912, 2010. doi: 10.1007/s11695-010-0181-4. [DOI] [PubMed] [Google Scholar]
- 40.Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri-Sverdlov R, Hofker MH, Greve JW, Buurman WA. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS One 7: e52411, 2012. doi: 10.1371/journal.pone.0052411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rolla S, Alchera E, Imarisio C, Bardina V, Valente G, Cappello P, Mombello C, Follenzi A, Novelli F, Carini R. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci (Lond) 130: 193–203, 2016. doi: 10.1042/CS20150405. [DOI] [PubMed] [Google Scholar]
- 42.Safadi R, Ohta M, Alvarez CE, Fiel MI, Bansal M, Mehal WZ, Friedman SL. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology 127: 870–882, 2004. doi: 10.1053/j.gastro.2004.04.062. [DOI] [PubMed] [Google Scholar]
- 43.Santos RD, Agewall S. Non-alcoholic fatty liver disease and cardiovascular disease. Atherosclerosis 224: 324–325, 2012. doi: 10.1016/j.atherosclerosis.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 44.Subramanian S, Goodspeed L, Wang S, Kim J, Zeng L, Ioannou GN, Haigh WG, Yeh MM, Kowdley KV, O’Brien KD, Pennathur S, Chait A. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J Lipid Res 52: 1626–1635, 2011. doi: 10.1194/jlr.M016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugimoto R, Enjoji M, Kohjima M, Tsuruta S, Fukushima M, Iwao M, Sonta T, Kotoh K, Inoguchi T, Nakamuta M. High glucose stimulates hepatic stellate cells to proliferate and to produce collagen through free radical production and activation of mitogen-activated protein kinase. Liver Int 25: 1018–1026, 2005. doi: 10.1111/j.1478-3231.2005.01130.x. [DOI] [PubMed] [Google Scholar]
- 46.Sugimoto R, Enjoji M, Nakamuta M, Ohta S, Kohjima M, Fukushima M, Kuniyoshi M, Arimura E, Morizono S, Kotoh K, Nawata H. Effect of IL-4 and IL-13 on collagen production in cultured LI90 human hepatic stellate cells. Liver Int 25: 420–428, 2005. doi: 10.1111/j.1478-3231.2005.01087.x. [DOI] [PubMed] [Google Scholar]
- 47.Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, Xie G, Philips G, Chan IS, Karaca GF, Pereira TA, Chen Y, Mi Z, Kuo PC, Choi SS, Guy CD, Abdelmalek MF, Diehl AM. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 61: 1323–1329, 2012. doi: 10.1136/gutjnl-2011-301857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, Witek RP, Choi SS, Guy CD, Fearing CM, Teaberry V, Pereira FE, Adams DH, Diehl AM. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 51: 1998–2007, 2010. doi: 10.1002/hep.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol 60: 1090–1096, 2014. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 50.Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, Han X, Peng Y, Chen X, Shen L, Qiu D, Li Z, Ma X. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol 166: 281–290, 2011. doi: 10.1111/j.1365-2249.2011.04471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Okada Y, Kurihara C, Irie R, Yokoyama H, Shimamura K, Usui S, Ebinuma H, Saito H, Watanabe C, Komoto S, Kawaguchi A, Nagao S, Sugiyama K, Hokari R, Kanai T, Miura S, Hibi T. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 59: 154–169, 2014. doi: 10.1002/hep.26604. [DOI] [PubMed] [Google Scholar]
- 52.Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J Biol Chem 287: 40161–40172, 2012. doi: 10.1074/jbc.M112.417014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 14: 397–411, 2017. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 54.Walenbergh SM, Koek GH, Bieghs V, Shiri-Sverdlov R. Non-alcoholic steatohepatitis: the role of oxidized low-density lipoproteins. J Hepatol 58: 801–810, 2013. doi: 10.1016/j.jhep.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 55.Wilson CS, Chang AJ, Greene R, Machado S, Parsons MW, Takats TA, Zambetti LJ, Springer AL. Knockdown of inner arm protein IC138 in Trypanosoma brucei causes defective motility and flagellar detachment. PLoS One 10: e0139579, 2015. doi: 10.1371/journal.pone.0139579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, Lorentzen A, Einer C, Schulz S, Clavel T, Protzer U, Thiele C, Zischka H, Moch H, Tschöp M, Tumanov AV, Haller D, Unger K, Karin M, Kopf M, Knolle P, Weber A, Heikenwalder M. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26: 549–564, 2014. doi: 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 57.Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lütjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, van Bilsen M, Shiri-Sverdlov R, Hofker MH. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 48: 474–486, 2008. doi: 10.1002/hep.22363. [DOI] [PubMed] [Google Scholar]
- 59.Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C, Yan C, Wang L, Chang CC, Chang TY, Zhang T, Zhou P, Song BL, Liu W, Sun SC, Liu X, Li BL, Xu C. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 531: 651–655, 2016. doi: 10.1038/nature17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang F, Zhang Z, Kong D, Zhang X, Chen L, Zhu X, Lu Y, Zheng S. Tetramethylpyrazine reduces glucose and insulin-induced activation of hepatic stellate cells by inhibiting insulin receptor-mediated PI3K/AKT and ERK pathways. Mol Cell Endocrinol 382: 197–204, 2014. doi: 10.1016/j.mce.2013.09.020. [DOI] [PubMed] [Google Scholar]