

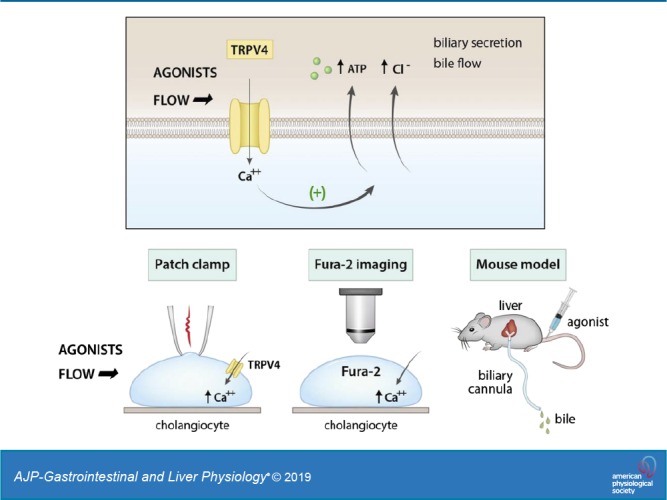
Keywords: biliary epithelium, channel, flow, liver, mechanosensitive
Abstract
Mechanosensitive signaling has emerged as a mechanism for the regulation of cholangiocyte transport and bile formation. The mechanical effect of fluid-flow, or shear, at the apical membrane of cholangiocytes regulates secretion through a process involving increases in [Ca2+]i and activation of Ca2+-activated Cl− channels. However, the initiating steps translating shear force to increases in intracellular calcium concentration ([Ca2+]i) are unknown. Transient receptor potential vanilloid member 4 (TRPV4), a nonselective cation channel present in the apical membrane of cholangiocytes, has been proposed as a potential mechanosensor. The aim of the present studies was to determine the potential role of TRPV4 in initiating mechanosensitive signaling in response to fluid-flow in cholangiocytes. TRPV4 expression was confirmed in both small and large mouse cholangiocytes. Exposure of cells to either fluid flow or specific TRPV4 pharmacological agonists rapidly increased both [Ca2+]i and membrane cation currents. Both flow- and agonist-stimulated currents displayed identical biophysical properties and were inhibited in the presence of TRPV4 antagonists or in cells after transfection with TRPV4 small interfering RNA. Transfection of mouse cholangiocytes with a TRPV4-enhanced green fluorescent protein construct increased the expression of TRPV4 and the magnitude of flow-stimulated currents. A specific TRPV4 agonist significantly increased the biliary concentration of ATP and bile flow in live mice when administered intravenously and increased ATP release from cholangiocyte monolayers when applied exogenously. The findings are consistent with a model in which activation of cholangiocyte TRPV4 translates shear force into an acute rise in membrane cation permeability, [Ca2+]i, ATP release, and bile flow. Understanding the role of mechanosensitive transport pathways may provide novel insights to modulate bile flow for the treatment of cholestatic liver disorders.
NEW & NOTEWORTHY These studies functionally characterize TRPV4 as a mechanosensitive channel in mouse cholangiocytes. By mediating a rapid rise in intracellular Ca2+, necessary for Ca2+-dependent secretion, TRPV4 represents a mechanosensor responsible for translating fluid flow into intracellular signaling and biliary secretion. Furthermore, intravenous infusion of a specific TRPV4 agonist increases bile flow in live mice. Understanding the role of TRPV4 in mechanosensitive transport pathways may provide novel insights to modulate bile flow during cholestasis.
INTRODUCTION
Cholangiocytes, also known as biliary epithelial cells, contribute to liver bile formation through the secretion of fluid and electrolytes (14). While the contribution of cholangiocytes to overall bile volume varies by species, it is felt that biliary epithelial secretion may account for up to 40% of bile volume in humans and even more in mice (3, 14, 23, 24). Cl− channels in the apical membrane of cholangiocytes provide the driving force for secretion (15), including those activated by increases in cAMP (26) and those activated by increases in intracellular Ca2+ (6). In single cholangiocytes and polarized biliary epithelial monolayers, Cl− efflux mediated by increases in intracellular Ca2+ are two- to threefold greater than that mediated by increases in cAMP (8), suggesting that Ca2+-dependent secretion is the predominant pathway contributing to biliary secretion and bile formation.
The cellular basis underlying regulated changes in cholangiocyte intracellular calcium concentration ([Ca2+]i) under physiological conditions is not entirely known. One stimulus which results in rapid increases in cholangiocyte [Ca2+]i is fluid flow, or shear force, at the apical plasma membrane (37). Moreover, the mechanosensitive increase in [Ca2+]i is linked to stimulation of Ca2+-activated Cl− channels in the apical membrane of both small and large cholangiocytes and an increase in biliary secretion (9). However, the mechanism by which fluid flow is translated to rapid increases in [Ca2+]i is unknown. While mechanical stimulation secondary to fluid flow is a stimulus for ATP release, P2 receptor activation, IP3 generation, and release of Ca2+ from intracellular stores, ATP release itself requires [Ca2+]i and is associated with rapid translocation of the Ca2+-dependent isoforms of PKC to the plasma membrane (7). Additionally, Ca2+ influx pathways exist in the apical membrane of cholangiocytes, allowing influx of Ca2+ from the extracellular space to replenish intracellular Ca2+ stores (5). Thus, several Ca2+ signaling pathways in cholangiocytes exist and represent specific targets which translate mechanical stimuli to biliary secretion and bile formation.
Interestingly, cholangiocytes express the transient receptor potential vanilloid member 4 (TRPV4) channel, a nonselective cation channel with high permeability to Ca2+ under physiological conditions, on the apical plasma membrane (20). In cholangiocytes, TRPV4 has been shown to be located on the primary cilium as well as nonciliary membrane locations (20, 21) and responds to changes in osmolality by regulating ATP release (20). In addition to activation by osmotic stress, TRPV4 is also activated by chemical and mechanical stress, including fluid flow, in other cell types (27). In endothelium (4) or renal collecting ducts (2), for example, fluid flow, or shear, results in rapid increases in TRPV4-mediated Ca2+ influx. If similar pathways are operative in cholangiocytes, TRPV4 may represent a potential candidate for the mechanosensor by which fluid flow is translated to intracellular signaling, including rapid increases in [Ca2+]i and stimulation of Ca2+-activated secretion. Targeting TRPV4 therefore may be a means to increase Ca2+-dependent secretion and promote bile flow.
The aim of the present studies, therefore, was to assess the potential role of TRPV4 in translating the mechanical effect of fluid flow at the apical membrane of mouse cholangiocytes into increases in [Ca2+]i and biliary secretion. Studies combining electrophysiology, fluorescent imaging, and direct measurements of bile flow were performed in individual cholangiocytes and intact murine models to functionally characterize the role of TRPV4 in mediating biliary secretion and bile formation.
METHODS
Cell models.
Studies were performed in mouse cholangiocytes isolated from the small or large bile ducts of normal mice (BALB/c) and were immortalized by transfection with the SV40 large-T antigen gene (18). These cells demonstrate identical properties to freshly isolated small and large mouse cholangiocytes (18, 35, 38). The cells were authenticated utilizing species-specific PCR primers and cell type-specific (cholangiocyte) primers (see Supplemental Fig. S1; at https://doi.org/10.6084/m9.figshare.10113281.v1 and https://doi.org/10.6084/m9.figshare.10250324.v1). No cell lines used in this study were found in the database of commonly misidentified cell lines that is maintained by ICLAC and NCBI Biosample, and all tested negative for mycoplasma. Both mouse large cholangiocytes (MLC) and mouse small cholangiocytes (MSC) were passaged at biweekly intervals and maintained in culture with minimum essential medium (GIBCO-BRL, Grand Island, NY) supplemented with 10% fetal bovine serum, penicillin (100 IU/ml) and streptomycin (100 μg/ml), and 2 mM l-glutamine at 37°C in a 5% CO2 incubator.
Immunofluorescence.
Localization of TRPV4 protein was performed in confluent MLC monolayers. MLC on collagen-coated tissue culture chamber slides (BD BioCoat) were fixed in 25% acetic acid-75% ethanol (vol/vol) for 10 min or 4% paraformaldehyde permeabilized with 1% Triton X-100 for 10 min, incubated with 5% normal donkey serum, and then incubated overnight at 4°C with rabbit anti-TRPV4 antibody (1:100, ACC-034; Alomone, Jerusalem, Israel) and then Dylight 488-conjugated donkey anti-rabbit (1:600, Jackson Immunoresearch) and counterlabeled with Alexa fluor 555 phalloidin to label actin. Control cells were prepared by omitting either primary or secondary antibodies from the incubation solution. The slides were coverslipped with Mowiol Æ 4-88 with 2.5% DABCO and left overnight in the dark at room temperature. The slides were imaged using a Leica TCS SP5 confocal microscope (Leica Micro-Systems, CMS) with custom software (Leica Micro-Systems LAS AF). Images were acquired using a frame size of 512 × 512 pixels and 4-line averaging was used to remove noise from the image. Images were then imported in ImageJ (https://rsb.info.nih.gov) using the LOCI Bio-formats plug-in (University of Wisconsin, Madison, WI).
Perfusion system.
Shear was applied to cells in a parallel plate chamber for Ca2+ imaging and to cells in an open top chamber for patch clamp recording (Warner Instruments, Hamden, CT). In each case, flow was applied with a dual syringe pump (Harvard Apparatus, Holliston, MA). The equation relating shear stress to volumetric flow rate through the chambers is given by τw = 6μQ/a2b, where μ is the viscosity of the solution (poise), Q is flow rate (mL/s), a is chamber height (cm), and b is chamber width (cm).
Measurement of TRPV4 currents.
Membrane TRPV4 currents were measured by whole cell patch-clamp techniques. Cells on a coverslip were mounted in a chamber (volume ~400 μL) and perfused at 2 mL/min with an extracellular solution containing (in mM): 150 NaCl, 6 CsCl, 1 MgCl2, 5 CaCl2, 10 HEPES, 10 glucose, and pH adjusted to 7.4 with 1 N NaOH. The concentration of CaCl2 varied between 0 and 15 mM, as indicated in the text. Nominally free Ca2+ was estimated at 10 µM. The osmolarity of the standard solution was 300 ± 10 as measured by a vapor pressure osmometer (Advanced Micro Osmometer). The internal pipette solution was composed of (in mM): 20 CsCl, 100 Cs-aspartate, 1 MgCl2, 4 Na2ATP, 10 BAPTA, 10 HEPES, and pH adjusted to 7.2 with CsOH. The [Ca2+]i was buffered in the presence of 10 mM BAPTA to 1 nM or to other intracellular concentrations as indicated. Patch pipettes were pulled from Corning 7052 glass and had a resistance of 3–5 MΩ. Recordings were made with an Axopatch ID amplifier (Axon Instruments, Foster City, CA), and were digitized (1 kHz) for storage on a computer and analyzed using pCLAMP v. 10.0 (Axon Instruments, Burlingame, CA) as previously described (11). The voltage protocol applied a holding potential –40 mV with a ramp from −100 mV to +100 mV over 450 ms at 5-s intervals (for real-time tracings). Pipette voltages (Vp) are referred to the bath. In the whole cell configuration, Vp corresponds to the membrane potential, and upward deflections of the current trace indicate outward membrane current. Results are compared with control studies measured on the same day to minimize any effects of day-to-day variability and are reported as current density (pA/pF) to normalize for differences in cell size. Capacitance and access resistance were monitored continuously.
Ca2+ imaging.
Cells were cultured for 24 h on 10-mm glass coverslips and then loaded with 2.5 μg/mL fura-2 AM (TEF Laboratories, Austin, TX) in isotonic extracellular buffer containing (in mM): 140 NaCl, 4 KCl, 2CaCl2, 1 MgCl2, 1 KH2PO4, 10 glucose, 10 HEPES (pH 7.4) supplemented with 0.01% pluronic F127 for 30 min at 22°C. In selected studies, Ca2+ was removed by inclusion of 0.2 mM EGTA in the solution. The coverslip was placed in the perfusion chamber on the stage of an inverted fluorescent microscope (Nikon TE2000). Changes of [Ca2+]i were measured at excitation wavelengths of 340 nm for calcium-bound fura-2 AM and 380 nm for calcium-free fura-2 AM and at emission wavelength of 510 nm. Experiments were performed at room temperature. In some experiments, patch recordings were combined with fluorometric recordings of [Ca2+]i. In these studies, the cells were loaded with 100 µM fura-2 Penta potassium salt via the patch pipette.
Total RNA isolation and RT-PCR analysis.
Total RNA was extracted using RNA STAT-60 Reagent (Tel-Test, Inc.) and Superscript. One-Step RT-PCR with Platinum Taq (Invitrogen) was used for RT-PCR. Mouse TRPV4 primers were forward 5′-CCA AGG ATG AGG GAG GCT-3′ and reverse 5′-GTC GGA TGA TGT GCT GAA AG-3′ [for a 351-bp fragment (NM_022017)]. One microgram of RNA was reverse transcribed in the presence of 100 pmol of oligo(dT) primer. Aliquots of 5% of the total cDNA were amplified with TaqDNA polymerase in a reaction mixture.
Western blot.
Total protein extracts were prepared from mouse cholangiocytes. Cells were washed twice with ice-cold PBS and lysed at 4°C with lysis buffer. Protein fractions were subjected to 7.5% SDS–polyacrylamide gel and transferred to nitrocellulose membranes. After blocking, immunoblots were incubated overnight with anti-mouse TRPV4 (1:200, Alomone Laboratories) This was followed by incubation with peroxidase-conjugated goat antirabbit antibody (dilution 1:10,000, Jackson Immuno Research Laboratories) and visualized with an ECL+ detection kit (GE Healthcare).
TRPV4 silencing.
TRPV4 was suppressed by specific mouse TRPV4 small interfering (si)RNA (NM_022017); 23-nucleotide siRNAs were designed and synthesized by IDT (5′-AGU UUG UCA CCA AGA UGU ACG ACC T-3′ and 3′-GUU CAA ACA GUG GUU CUA CAU GCU GGA-5′) and transfected with Lipofectamine RNAiMAX Reagent (Invitrogen). Noncoding Stealth RNAi (medium guanine cytosine duplex, Invitrogen) was utilized in control (mock) transfections. Block-it Fluorescent Oligo (Invitrogen) was used to optimize transfection conditions and for selection of transfected cells for whole cell patch-clamp current recording. Whole cell patch clamp experiments were done 48 h after transfection. Transfection efficiency and the degree of TRPV4 silencing were evaluated in all models by Western blot analysis.
Overexpression of TRPV4.
TRPV4/T/H6 in pCMV-Tag4 plasmid was a kind gift from Dr. Paul Blount (UT Southwestern Medical Center, Dallas, TX). In brief, TRPV4 was inserted into the PCMV-Tag4 vector after the addition of a new cut site, Kozac, N term H6 tag, and a thrombin site by PCR. The enhance green fluoresceent protein (EGFP)-TRPV4 construct was developed by inserting TRPV4 into pEGFP-C3 (Addgene, Watertown, MA). Transfections were done in MLC utilizing Lipofectamine 2000 (ThermoFisher Scientific, cat. no. 11668027) following the standard protocol. GFP fluorescence was visualized 24–48 h after transfection by fluorescent microscopy.
Measurement of ATP release.
Cellular ATP release was studied using the luciferin-luciferase (L-L) assay as previously described (12, 33). Cells on 35-mm tissue culture-treated dishes (Falcon, Becton Dickinson Labware, Franklin Lakes, NJ) were washed with PBS (600 μL × 2), 600 μL of Optimem (GIBCO) containing L-L (Fl-ATP Assay Mix; Sigma-Aldrich, St. Louis, MO) added, and then placed into a modified Turner TD 20/20 luminometer. After a 5-min equilibration period, a basal reading was obtained and then glycogen synthase kinase (GSK; 100 nM) or an equal volume of isotonic buffer (200 µL) was added (to account for ATP release due to mechanical stimulation). Readings were performed as cumulative bioluminescence over a 15-s interval and quantified as arbitrary light units (ALUs). Standard calibration curves were performed with known amounts of ATP added to the L-L-Optimem reagent during cell-free conditions (12). Lactate dehydrogenase (LDH) measurements were performed pre- and post-stimulus (GSK or isotonic exposure) using an enzymatic colorimetric cytotoxicity assay (CytoTox 96; Promega, Madison, WI), as previously described (37).
Measurement of bile flow in the bile duct-cannulated mouse model.
The bile duct was cannulated, and the relation between bile flow rate and ATP concentration in bile was measured in live mice. The bile duct was cannulated with a microfil tubing according to previously described techniques (30, 34). In brief, 3-mo-old C57BL/6 male mice weighing 20-30 g were anesthetized with Avertin 0.5 mg/g intraperitoneally (ip). The common bile duct was incised with a pair of fine iridectomy scissors ~6 mm below the hilum of the liver. A microfil tube (MF28G-5; WPI, Sarasota, FL) was passed through the incision and propelled toward the hilum for a distance of ~3 mm. Bile flow rate was recorded (µL·min−1·100 g body wt−1), and bile was collected in CryoTube vials (ThermoFisher Scientific) and immediately placed in liquid nitrogen. The ATP concentration in bile was measured with a standard L-L assay as described (13). Animal work described in this paper was approved and conducted under the oversight of the University of Pittsburgh Institutional Animal Care and Use Committee.
Reagents.
The 4α-phorbol 12,13-didecanoate (4α-PDD) compound was obtained from LC Laboratories (Woburn, MA) and GSK1016790A from Santa Cruz Biotechnology (Dallas, TX), and all other reagents were from Sigma-Aldrich (St. Louis, MO).
Statistics.
Results are presented as means ± SE, with n representing the number of culture plates or repetitions for each assay, as indicated. Statistical analysis included Fisher’s paired and unpaired t tests and ANOVA for multiple comparisons to assess statistical significance as indicated; P values < 0.05 were considered to be statistically significant.
RESULTS
Expression of TRPV4 mRNA and protein in biliary epithelium.
Utilizing selective primers for TRPV4, PCR products of the predicted size were detected in both MSC and MLC (Fig. 1A). Additionally, Western blot analysis, utilizing specific anti-TRPV4 antibody, detected a predicted protein band ∼91 kDa (Fig. 1B). Immunostaining of MLC monolayers revealed TRPV4 on the plasma membrane (Fig. 1C). Collectively, these results demonstrate the presence of TRPV4 in mouse biliary epithelium, with a predominant plasma membrane location.
Fig. 1.

Expression and localization of transient receptor potential vanilloid member 4 (TRPV4) in mouse biliary epithelial cells. A: RT-PCR. TRPV4 primers were used to detect TRPV4 expression in both small (MSC) and large (MLC) mouse cholangiocytes, represented by a band size at 351 (see methods). Representative of 5 trials. B: Western blot utilizing polyclonal anti-TRPV4 antibody (see methods). TRPV4 protein (2 bands, at ~85 and ~100 kDa) is present in whole cell lysates of MLC and MSC cells; loading control was β-actin. Representative blot of 5 trials. C: membrane localization of TRPV4 protein in polarized MLC monolayers. Staining with anti-TRPV4 antibody (green) demonstrates TRPV4 protein localized with actin (red) in the plasma membrane. Scale bar, 10 μm.
TRPV4 is a functional channel in cholangiocytes.
To evaluate functional expression of TRPV4 channels in cholangiocytes, we directly assessed the effects of TRPV4 agonists on channel activity, via whole cell patch clamp and [Ca2+]i, by fura-2 fluorescence. As shown in Fig. 2, exposure of MLC to the specific TRPV4 agonist 4α-PDD rapidly activated whole cell cation currents and simultaneously increased Ca2+ fluorescence as measured by fura-2 ratiometric imaging. Whole cell currents demonstrated outward rectification and a reversal near 0 mV (Ecat ∼0 mV). Channel activation was followed by rapid current decay when Ca2+ was present in the extracellular solution, consistent with the known biophysical properties of TRPV4 (27). Both the 4α-PDD-stimulated increase in whole cell currents and the increase in [Ca2+]i were inhibited by the TRPV4 antagonists ruthenium red and HC-067047 (Fig. 2C) (10). In separate studies, the compound GSK1016790A (GSK), which specifically and rapidly activates TRPV4, resulted in simultaneous activation of currents and increases in [Ca2+]i (Fig. 2B). Both ruthenium red and HC-067047 inhibited the whole cell currents and rise in [Ca2+]i in response to GSK (Fig. 2C). Last, transfection of MLC with TRPV4 siRNA significantly decreased expression of TRPV4 mRNA by 75 ± 4% and protein by 60 ± 10% (Fig. 3) and significantly inhibited both the increase in currents and increase [Ca2+]i in response to GSK compared with cells transfected with nontargeting siRNA (Fig. 2C). Similar results were observed with MSC [Supplemental Figs. S2 and S3; see https://doi.org/10.6084/m9.figshare.10113281.v1 and https://doi.org/10.6084/m9.figshare.10250324.v1 (captions)], and no differences were noted in the magnitude of agonist-stimulated currents or [Ca2+]i between cell types. Together, the results demonstrate that TRPV4 is functionally expressed in cholangiocytes and causes a rapid increase in [Ca2+]i upon direct, agonist-induced activation.
Fig. 2.

Specific agonists of transient receptor potential vanilloid member 4 (TRPV4) activate currents and increase intracellular calcium concentration ([Ca2+]i) in large mouse cholangiocytes (MLC). A, top: representative whole cell recording. The specific TRPV4 agonist 4α-phorbol-12,13-didecanoate (4αPDD; 1 µM) activates whole cell currents. Currents measured at +100 mV (○) and at −100 mV (●) are shown. Voltage protocol −100 mV to +100 mV ramp every 5 s. Bottom: cells on coverglass were loaded with fura-2 AM, washed with PBS, and exposed to 4αPDD (1 µM); y-axis values represent the ratio of fluorescence at 340 and at 380 nm. B, top: the specific TRPV4 agonist GSK (100 nM) activates whole cell currents. Currents measured at +100 mv (○) and at −100 mV (●) are shown. Bottom: fura-2-loaded MLC cells were exposed to glycogen synthase kinase (GSK; 100 nM). The y-axis values represent the ratio of fluorescence at 340 and at 380 nm. C, top: cumulative data demonstrating maximal current density (pA/pF) measured at +100 mV in response to 4αPDD or GSK. Currents were significantly inhibited by ruthenium red (RR, 10 µM, n = 9), HC-067047 (HC, 10 µM, n = 16), and after transfection with TRPV4 small interfering (si)RNA (n = 8). Bottom: cumulative data demonstrating the change in [Ca2+]i in response to 4αPDD or GSK in control cells (n = 78) and in the presence of RR (n = 46), HC (n = 54), or after transfection with TRPV4 siRNA (n = 27). Values represent relative change in fluorescence ratio 340/380. Maximal increase vs. basal **P ≤ 0.01, inhibitor vs. control, *P ≤ 0.05, vs. control **P ≤ 0.01, *P <0.05 vs. mock transfected.
Fig. 3.

Transfection of large mouse cholangiocytes (MLC) with plasmid for transient receptor potential vanilloid member 4 (TRPV4) small interfering (si)RNA decreases protein and RNA expression. A: representative Western blot demonstrating change in TRPV4 protein levels in cells transfected with nontargeting siRNA (scramble) and cells transfected with TRPV4 siRNA. β-Actin was used as loading control. B: cumulative data demonstrating change in TRPV4 protein (left) and mRNA (right) levels in control cells, cells transfected with nontargeting siRNA (scramble), and cells transfected with TRPV4 siRNA (*P < 0.05 vs. control or mock levels; n = 4 each).
Flow (shear) activates TRPV4-mediated membrane currents.
To characterize the biophysical and pharmacological properties of TRPV4-mediated currents in cholangiocytes in response to fluid flow, whole cell patch clamp studies were performed in single MLC in the presence or absence of defined flow exposures. A representative tracing is shown in Fig. 4. Under basal conditions, currents were small (5 ± 0.5 pA/pF at +100 mV). Exposure to flow (flow rate of 2 mL/min or shear of 0.64 dyne/cm2) resulted in rapid activation of currents within 10 ± 1.3 s, increasing current density to 110 ± 14.0 pA/pF at +100 mV (P < 0.001, n= 26). Currents were transient, exhibiting rapid inactivation even in the presence of continued flow exposure. Currents exhibited outward rectification, reversal potential of ∼0 mV, and rapid decay in Ca2+-containing extracellular buffer. Similar results were observed in MSC, and no differences in the magnitude or biophysical properties of flow-mediated currents were observed between cell types (Supplemental Figures S1 and S2; see https://doi.org/10.6084/m9.figshare.10113281.v1 and https://doi.org/10.6084/m9.figshare.10250324.v1). Thus, the flow-stimulated currents displayed identical biophysical properties to the currents directly activated by the specific TRPV4 agonists 4αPDD or GSK (Fig. 5). As with the agonist-stimulated currents, the flow-stimulated currents were inhibited by the TRPV4 antagonists ruthenium red or HC-067047 (Fig. 4). Likewise, transfection of MLC with TRPV4 siRNA decreased TRPV4 expression and abolished the flow-stimulated currents compared with cells transfected with nontargeting siRNA (Fig. 4). Together, the results demonstrate that fluid flow rapidly activates TRPV4 currents in mouse cholangiocytes.
Fig. 4.

Whole cell currents were measured during basal conditions and during exposure to flow of isotonic extracellular buffer (see methods). A: representative whole cell recording of large mouse cholangiocytes (MLC). Currents measured at +100 mV (○) and at −100 mV (●) are shown. Flow exposure (shear of 0.64 dyne/cm2) is indicated by the bar. Currents activated within 20 s of onset of flow and rapidly decayed. Ramp protocols (ramp from −100 mV to +100 mV over 450 ms; shown in inset) were obtained at (a*) and (b*), representing basal and maximal currents, respectively. Current-voltage (I-V) plot shown at the right was generated from these ramp protocols during basal and flow-stimulated conditions. B: representative whole cell current recordings of MLC after transfection with transient receptor potential vanilloid member 4 (TRPV4) small interfering (si)RNA (top trace) or in the presence of the TRPV4 channel inhibitor HC-067047 (100 µM, bottom trace) indicated by the bar. C: cumulative data demonstrating flow-stimulated currents in the presence or absence of the TRPV4 inhibitors ruthenium red (RR, 10 µM), HC-067047 (HC, 10 µM), or in cells after transfection with TRPV4 siRNA (n = 9). Values represent current density (pA/pF) measured at +100 mV (n = 7~9 each). Flow significantly increases current density vs. static (basal) conditions (#P < 0.01); flow-stimulated currents were significantly inhibited (*P < 0.05).
Fig. 5.

Comparison between flow- and agonist-stimulated transient receptor potential vanilloid member 4 (TRPV4) currents. Representative current traces (left) of large mouse cholangiocytes (MLC) in response to flow (shear of 0.64 dyne/cm2) (top) and after exposure to glycogen synthase kinase (GSK; 100 nM; bottom). Currents measured at +100 mV (○) and at −100 mV (●) are shown. Flow (shear of 0.64 dyne/cm2) or GSK (100 nM) exposure is indicated by the bar. Ramp protocols (inset), from holding potential of −100 mV to +100 mV over 450 ms, were obtained at (a*) and (b*), representing basal and maximal outward currents, respectively. I-V plots (right) were generated from these ramp protocols during basal and flow- (top) or GSK-stimulated (bottom) conditions as indicated.
Flow (shear) stimulates TRPV4-mediated increase in [Ca2+]i.
To determine the effects of fluid flow on TRPV4-mediated [Ca2+]i, MLC or MSC were loaded with fura-2, and fluorescence was measured under basal and flow-stimulated conditions. Exposure to flow (2 mL/min; shear = 0.64 dyne/cm2) rapidly increased Ca2+ fluorescence. The flow-stimulated increase in [Ca2+]i was inhibited by the TRPV4 antagonist HC-067047 (Fig. 6 and Supplemental Fig. S3; see https://doi.org/10.6084/m9.figshare.10113281.v1 and https://doi.org/10.6084/m9.figshare.10250324.v1). In separate studies, transfection of MLC with TRPV4 siRNA significantly inhibited the flow-mediated increase in [Ca2+]i compared with cells transfected with nontargeting siRNA (Fig. 6). Together, the results demonstrate that TRPV4 contributes to the flow-stimulated increase in [Ca2+]i in cholangiocytes.
Fig. 6.
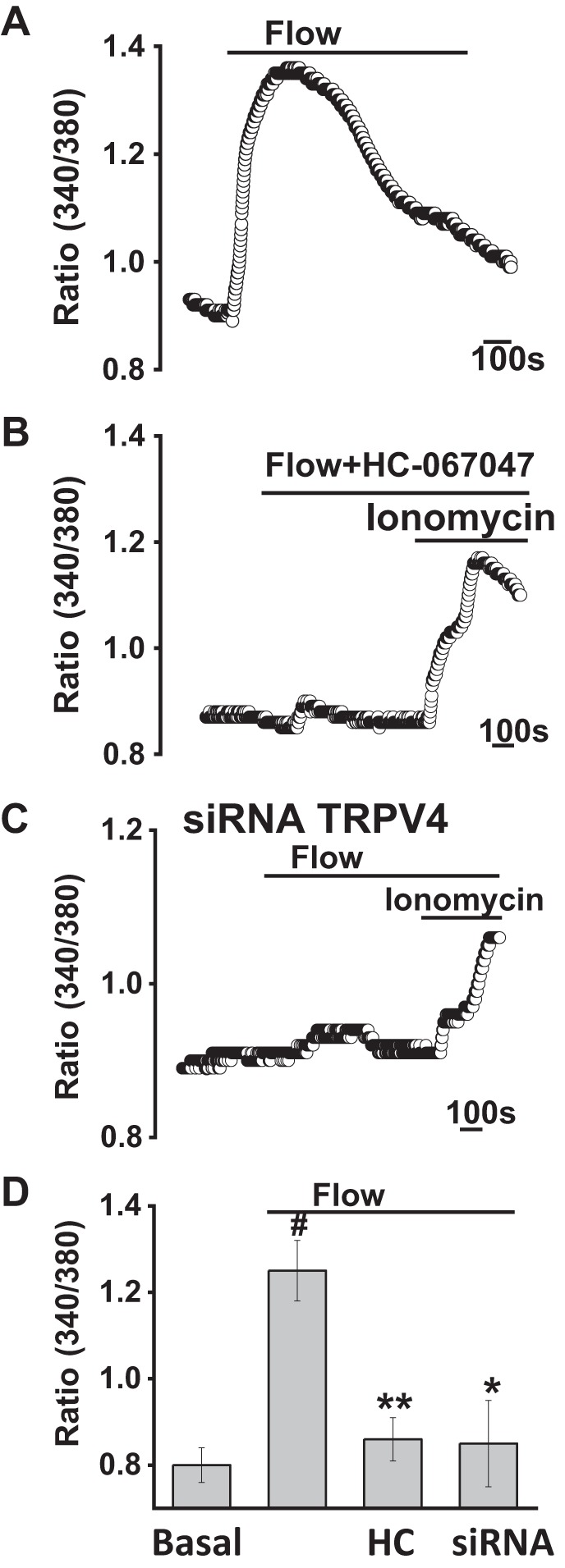
Flow-stimulated increases in intracellular calcium concentration ([Ca2+]i) in large mouse cholangiocytes (MLC). Representative Ca2+ fluorescence in response to fluid flow. MLC on coverglass were loaded with fura-2 AM and exposed to flow (2 mL/min; shear = 0.64 dyne/cm2); y-axis values represent the ratio of fluorescence at 340 and 380 nm; x-axis represents time (bar = 100 s). Control cells shown (A) vs. cells exposed to HC-067047 (HC, 10 µM; B) and after transfection with transient receptor potential vanilloid member 4 (TRPV4) small interfering (si)RNA (C). Cells were exposed to the Ca2+ ionophore ionomycin, as an internal control. D: cumulative data demonstrating the change in [Ca2+]i in response to fluid flow (shear = 0.64 dyne/cm2) in control cells (n = 78) and in the presence of HC-067047 (HC, n = 61), or after transfection with TRPV4 siRNA (n = 49). Values represent change in fluorescence ratio 340/380. Maximal increase vs. basal #P ≤ 0.01; inhibitor HC067047 vs. control **P ≤ 0.01; *P < 0.05 vs. mock transfected.
TRPV4 activity is modulated by extracellular Ca2+.
TRPV4 channel activity is modulated by extracellular Ca2+ through a mechanism that is not entirely understood. While the pore is primarily permeable to Ca2+ under physiological conditions, it is thought that selected amino acid residues in the pore region bind Ca2+ upon continued exposure, resulting in rapid channel inactivation (27). To determine whether similar modulation occurs in cholangiocytes, whole cell patch clamp studies were performed with defined concentrations of extracellular Ca2+. As shown in Fig. 7, when extracellular Ca2+ was lowered ([Ca2+]ext = 0) both the GSK- and flow-stimulated cation currents no longer demonstrated rapid inactivation but rather were sustained. These properties of sustained currents in nominal extracellular Ca2+ are consistent with the known biophysical properties of TRPV4-mediated currents in other cell types (28).
Fig. 7.

Extracellular Ca2+ modulates transient receptor potential vanilloid member 4 (TRPV4) channel activity. Representative whole cell patch clamp traces (top) and current-voltage (I-V) plots (bottom) in large mouse cholangiocytes (MLC) in response to flow (shear =0.64 dyne/cm2, left) or glycogen synthase kinase (GSK; 100 nM, right). When extracellular Ca2+ was removed (extracellular buffer [Ca2+] = 0), flow- and GSK-activated cation currents were sustained vs. when extracellular buffer contained 1.5 mM [Ca2+], as indicated by the bar (n = 7, P < 0.05).
Overexpression of TRPV4 increases channel activity in mouse cholangiocytes.
To further confirm and characterize the role of TRPV4 in cholangiocytes, we utilized a TRPV4-EGFP plasmid (see methods) to overexpress TRPV4 in cultured mouse cholangiocytes. The EGFP tag allowed selection of individual cells for patch clamp analysis. Transfection of cells with plasmid containing TRPV4-EGFP increased TRPV4 protein expression by 35 ± 3.4% compared with mock-transfected cells, based on Western blot analysis (Fig. 8). To determine whether this increase in expression was functionally significant, whole cell patch clamp studies were performed in MLC transfected with TRPV4-EGFP vs. controls (mock transfected). Fluid flow and GSK both stimulated TRPV4 currents in control and TRPV4-EGFP-transfected cells, although the magnitude of both flow- and GSK-stimulated currents was significantly greater in the TRPV4-EGFP-expressing cells (Fig. 8, B and C). In additional studies, TRPV4-EGFP-expressing cells also demonstrated significantly greater increases in [Ca2+]i compared with control cells (Fig. 8, D and E). These results demonstrate that, in response to both fluid flow and GSK, overexpression of TRPV4 in cholangiocytes results in significant increases in the magnitude of both whole cell currents and [Ca2+]i.
Fig. 8.

Overexpression of transient receptor potential vanilloid member 4 (TRPV4) in large mouse cholangiocytes (MLC) transfected with TRPV4-enhanced green fluorescent protein (EGFP). A: representative Western blot demonstrating TRPV4 protein levels in control cells (MLC) and after transfection with TRPV4-EGFP. Actin was used as loading control. In addition to increase of endogenous TRPV4 protein level, overexpressed EGFP-TRPV4 protein was present above. B: representative flow- and glycogen synthase kinase (GSK)-stimulated whole cell currents in control cells (open green and pink circles) and in cells after transfection with TRPV4-EGFP (open blue and black circles). C: cumulative data demonstrating maximal current density (pA/pF) in control cells, after mock transfected [EGFP(−)] and after transfection with TRPV4-EGFP [EGFP(+)] in response to flow (shear =0.64 dyne/cm2, n = 12) and GSK (100 nM, n = 15). Current density in TRPV4-EGFP transfected vs. mock transfected (**P < 0.01). D: representative calcium traces in fura-2-loaded control cells (pink triangles), mock-transfected (EGFP−, black circles) and in cells after transfection with TRPV4-EGFP (EGFP+, blue circles). The y-axis values represent the ratio of fluorescence at 340 and at 380 nm. E: cumulative data demonstrating the change in intracellular calcium concentration ([Ca2+]i) in response to fluid flow (shear = 0.64 dyne/cm2) or GSK in control cells (n = 45), cells after mock transfection (EGFP−, n = 20), and in cells after transfection with TRPV4-EGFP (n = 25). Values represent relative change in fluorescence ratio 340/380. Maximal increase fluorescence in TRPV4-EGFP transfected vs. mock transfected, **P ≤ 0.01.
Bile duct-cannulated murine model.
To assess the functional significance of direct TRPV4 activation on biliary secretion and bile flow, we directly measured bile flow in mice after bile duct cannulation. In response to intravenous administration of the TRPV4 agonist GSK (100 nM, 0.1 mL/g), bile flow increased significantly compared with infusion of normal saline (0.1 mL/g) (Table 1). The increase in GSK-stimulated bile flow was accompanied by an increase in the biliary concentration of ATP, consistent with previous reports of TRPV4-mediated ATP release in biliary epithelial cells (20). Based on standard calibration curves with known amounts of ATP (Supplemental Fig. S4; see https://doi.org/10.6084/m9.figshare.10113281.v1 and https://doi.org/10.6084/m9.figshare.10250324.v1), the amount of ATP released into bile in response to GSK was ∼25 nM, a concentration capable of activating cell surface purinergic receptors. Although systemic administration of GSK has been associated with cardiac collapse in animal models (36), we did not observe any acute cardiac side effects at the doses used in this study. Thus, intravenous administration of a direct TRPV4 agonist increases the concentration of ATP in bile and is associated with an increase in bile flow in a mouse model.
Table 1.
Bile flow and ATP concentration in bile duct-cannulated mice
| Basal | Saline | GSK | n | |
|---|---|---|---|---|
| Bile flow, µL·min−1·100 g−1 | 2.56 ± 0.45 | 2.67 ± 0.56 | 5.88 ± 1.12 | 6 P < 0.01 |
| Biliary ATP concentration, ALU (arbitrary light units) | 166.26 ± 10.1 | 171.2 ± 5.9 | 1,018.98 ± 24.3 | 6 P < 0.01 |
Values are expressed as means ± SE; n = number of mice. GSK, glycogen synthase kinase.
Measurement of ATP release from cholangiocyte monolayers.
We (25, 32, 37) have previously shown that mouse cholangiocytes release ATP into bile in response to fluid flow, cell swelling, or bile acids. Since the ATP concentration in mouse bile increased in response to the systemic administration of the TRPV4 agonist GSK, we sought to determine whether this agonist could directly cause ATP release from mouse cholangiocytes. As shown in Fig. 9, exposure of MLC to GSK (100 nM) resulted in a fourfold increase in ATP release compared with control or basal extracellular ATP levels. Of note, an equal volume of isotonic buffer resulted in a doubling of ATP release due to mechanical stimulation. No significant difference in pre- or post-stimulus LDH levels were observed, excluding cell lysis as the source of ATP release. Together, these results demonstrate that GSK is capable of directly stimulating ATP release from cholangiocytes and is consistent with previous studies where a similar increase in ATP release was observed from mouse cholangiocytes utilizing a different agonist of TRPV4 (20).
Fig. 9.
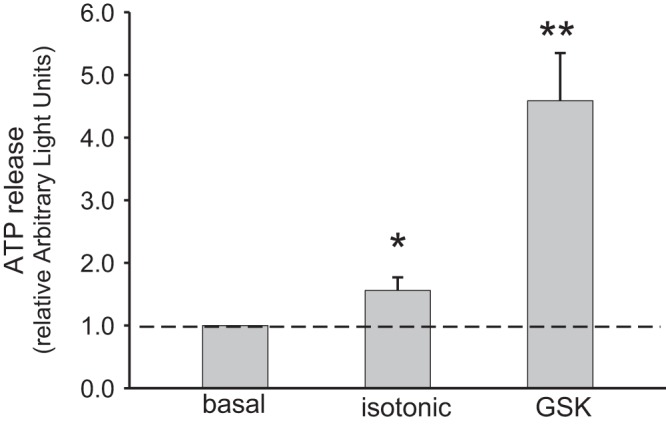
ATP release from large mouse cholangiocyte (MLC) monolayers. ATP release from MLC was detected using a luciferin-luciferase assay as described in methods. In response to the addition of transient receptor potential vanilloid member 4 (TRPV4) agonist glycogen synthase kinase (GSK; 100 nM), a large increase in ATP release was detected n = 6) compared with addition of an equal volume of isotonic buffer (n = 6) to account for mechanical stimulated ATP release. Results reported as relative change from basal in arbitrary light units (ALU). *P < 0.05, **P < 0.01.
DISCUSSION
These studies functionally characterize TRPV4 as a mechanosensitive channel in mouse biliary epithelial cells. By mediating a rapid rise in intracellular Ca2+, necessary for Ca2+-dependent secretion, TRPV4 represents a molecular candidate for the mechanosensor responsible for translating fluid flow into intracellular signaling and biliary secretion. Evidence for this is based on our studies and includes the following: 1) TRPV4 is expressed in both small and large mouse biliary epithelial models; 2) fluid flow leads to rapid onset of cation currents with identical biophysical properties to those activated by direct TRPV4 agonists; 3) fluid flow and application of pharmacological agonists to TRPV4 lead to a parallel increase in [Ca2+]i; 4) the increase in flow-stimulated currents and [Ca2+]i are both inhibited by pharmacological antagonists of TRPV4 or in cells transfected with specific TRPV4 siRNA; 5) overexpression of TRPV4 in mouse cholangiocytes increases both the magnitude of cation currents and [Ca2+]i in response to fluid flow; 6) exposure of MLC to the specific TRPV4 agonist GSK increases cellular ATP release; and 7) intravenous infusion of GSK increases bile flow in live mice. Together, these findings indicate that, by translating fluid flow into Ca2+-dependent secretion, TRPV4represents a critical component of ductular bile formation. If these studies performed in mouse biliary epithelial cells translate to in vivo conditions, several points, as well as uncertainties, deserve highlighting.
First, the mechanism by which fluid flow activates TRPV4 channels in biliary epithelial cells is unknown. Previously, in biliary epithelial cells, it was shown that TRPV4 localizes to the primary cilium, which may itself be a flow sensor (20). However, removal of the primary cilium from rat cholangiocytes decreases, but does not eliminate, TRPV4-mediated volume-dependent signaling through ATP release (20). Furthermore, TRPV4 has been shown to localize to nonciliary locations (21) or to the plasma membrane in epithelial models that do not express a primary cilium (22). Under the experimental conditions in our studies, individual cholangiocytes do not express a primary cilium. Therefore, the activation of TRPV4 appears to be independent of ciliary function. Moreover, in nonciliary-expressing cells, it has been suggested that direct mechanical effects on the plasma membrane, such as shear, stretch, or distention, may exert physical stresses directly on the TRPV4 protein, similar to other TRP channels (28, 29). Mechanical stimulation may also be transduced through cytoskeletal proteins such as actin or other scaffolding proteins (19). In keratinocytes, for example, colocalization and functional interactions between actin and TRPV4 have been demonstrated during mechanosensitive volume regulation (1). Last, it is not only possible but likely that fluid flow activates other signaling molecules that may modulate TRPV4 activity. In fact, we (7) have previously shown that fluid flow results in ATP release and rapid translocation of PKCα to the plasma membrane in cholangiocytes. Interestingly, TRPV4 contains several putative PKC phosphorylation sites, and it is therefore possible that flow-mediated kinase activity may have direct regulatory effects on channel function. The role of membrane stretch or distention, actin polymerization, or kinase phosphorylation in TRPV4 mechanosensitive functions requires further study.
Second, although membrane Cl− channels provide the driving force for biliary secretion and ductular bile flow, the molecular identification of the Cl− channel(s) involved in TRPV4-mediated Ca2+-dependent secretion is unknown. Two Cl− channels have been identified on a molecular basis in cholangiocytes: Ccystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-activated Cl− channel (15), and transmembrane member 16A (TMEM16A), a Ca2+-activated Cl− channel (6). CFTR is found on (14) the apical membrane of mouse cholangiocytes, forming the medium- and large-size bile ducts, but not small ducts, and is regulated via binding of the hormone secretin to basolateral receptors, increases in [cAMP]i and PKA-dependent channel phosphorylation (16, 17). In contrast, TMEM16A is found on the small, medium, and large-size bile ducts and is activated by increases in [Ca2+]i (6). Presumably, TRPV4, by mediating rapid Ca2+ influx, is linked to Ca2+-dependent TMEM16A activation. Interestingly, TMEM16A is also activated by fluid flow through a process that requires increases in [Ca2+]i and translocation of PKCα (9). Thus, TRPV4 may activate TMEM16A Cl− channels by mediating the initial and rapid increase in [Ca2+]i in response to fluid flow, although further studies are needed. Furthermore, by increasing Ca2+ influx from the external environment, TRPV4 may be a means to replete intracellular Ca2+ stores, such as those mediated by IP3 receptors (31), critical for continued Ca2+-dependent Cl− secretion (8). Thus, TRPV4 appears to be a critical member of the apical Ca2+-dependent secretory complex in biliary epithelium.
Third, the mechanism by which extracellular Ca2+ modulates TRPV4 activity in biliary epithelium is unknown. While TRPV4 is a nonselective cation channel with a high permeability to Ca2+ under physiological conditions, specific residues in the channel pore domain may directly bind Ca2+ ions resulting in rapid channel inactivation (27). Conversely, when the extracellular Ca2+ concentration is low, the channel remains open. We observed a similar phenomenon in our studies of biliary epithelial cells, where a decrease in the extracellular Ca2+ concentration led to an increase in channel open probability and sustained channel activity. TRPV4 may therefore be a means to fine-tune the extra- and intracellular concentrations of Ca2+. In this way, TRPV4 may not only contribute to the normal physiological regulation of biliary Ca2+ concentration, but also may have a potential role during pathological conditions in which the ductal concentration of Ca2+ may be altered, such as gallstone formation, polycystic disease, cholestasis, or other conditions.
Finally, the role of TRPV4 in liver diseases associated with altered Ca2+ signaling, such as polycystic kidney disease or cholestasis, is far from understood. Previously, it had been shown that TRPV4 was overexpressed in the liver of both rats and humans with polycystic kidney disease and was associated with cholangiocyte hyperproliferation (21). In these studies, application of TRPV4 agonists increased [Ca2+]i and reversed the hyperproliferative phenotype of biliary epithelial cells in culture, highlighting the potential use of TRPV4 as a therapeutic target in polycystic kidney disease. Additionally, abnormalities in mechanosensitive signaling along the bile duct may have important implications during cholestatic liver disease. A decrease in bile flow associated with cholestasis would be expected to inhibit mechanosensitive Ca2+ signaling and secretion. Our present studies, as well as those of others (20), demonstrate that systemic administration of a TRPV4 agonist to mice significantly increases bile flow, supporting the notion that targeting TRPV4 may be a means to increase choleresis. However, a note of caution is in order, as this compound has been associated with cardiopulmonary collapse in animal models when administered at higher doses (36). Thus, other TRPV4 agonists with more favorable side effect profiles will need to be developed before this strategy can be applied to the treatment of liver disease in humans. Overall, TRPV4 appears to contribute to normal Ca2+ homeostasis and may serve as a target for the treatment of cholestatic liver diseases.
Together, our studies provide evidence that TRPV4 is a functional mechanosensitive channel in biliary epithelium. By translating fluid flow to intracellular Ca2+ signaling, TRPV4 represents an important initiator of mechanosensitive signaling along the bile duct and an important member of the apical signaling complex necessary for ductular bile formation.
GRANTS
Research reported in this publication was supported by the Willis C. Maddrey M.D. endowment (UTSW), the Department of Pediatrics, University of Pittsburgh Medical Center (UPMC), and the National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health under award number P30 DK-120531 and award number R01 DK-078587 (A. Feranchak). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.F. conceived and designed research; Q.L., C.K., K.B., and J.S. performed experiments; Q.L., K.B., and A.F. analyzed data; Q.L., K.B., and A.F. interpreted results of experiments; Q.L., C.K., K.B., and A.F. prepared figures; Q.L. and A.F. drafted manuscript; Q.L. and A.F. edited and revised manuscript; A.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Paul Blount and Robin Wray, University of Texas Southwestern (UTSW), Dallas, TX, for kindly providing the constructs for TRPV4-EGFP.
REFERENCES
- 1.Becker D, Bereiter-Hahn J, Jendrach M. Functional interaction of the cation channel transient receptor potential vanilloid 4 (TRPV4) and actin in volume regulation. Eur J Cell Biol 88: 141–152, 2009. doi: 10.1016/j.ejcb.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Berrout J, Jin M, Mamenko M, Zaika O, Pochynyuk O, O’Neil RG. Function of transient receptor potential cation channel subfamily V member 4 (TRPV4) as a mechanical transducer in flow-sensitive segments of renal collecting duct system. J Biol Chem 287: 8782–8791, 2012. doi: 10.1074/jbc.M111.308411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyer JL. Bile formation and secretion. Compr Physiol 3: 1035–1078, 2013. doi: 10.1002/cphy.c120027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bubolz AH, Mendoza SA, Zheng X, Zinkevich NS, Li R, Gutterman DD, Zhang DX. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling. Am J Physiol Heart Circ Physiol 302: H634–H642, 2012. doi: 10.1152/ajpheart.00717.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doctor RB, Matzakos T, McWilliams R, Johnson S, Feranchak AP, Fitz JG. Purinergic regulation of cholangiocyte secretion: identification of a novel role for P2X receptors. Am J Physiol Gastrointest Liver Physiol 288: G779–G786, 2005. doi: 10.1152/ajpgi.00325.2004. [DOI] [PubMed] [Google Scholar]
- 6.Dutta AK, Khimji AK, Kresge C, Bugde A, Dougherty M, Esser V, Ueno Y, Glaser SS, Alpini G, Rockey DC, Feranchak AP. Identification and functional characterization of TMEM16A, a Ca2+-activated Cl- channel activated by extracellular nucleotides, in biliary epithelium. J Biol Chem 286: 766–776, 2011. doi: 10.1074/jbc.M110.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dutta AK, Khimji AK, Liu S, Karamysheva Z, Fujita A, Kresge C, Rockey DC, Feranchak AP. PKCα regulates TMEM16A-mediated Cl− secretion in human biliary cells. Am J Physiol Gastrointest Liver Physiol 310: G34–G42, 2016. doi: 10.1152/ajpgi.00146.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutta AK, Woo K, Doctor RB, Fitz JG, Feranchak AP. Extracellular nucleotides stimulate Cl− currents in biliary epithelia through receptor-mediated IP3 and Ca2+ release. Am J Physiol Gastrointest Liver Physiol 295: G1004–G1015, 2008. doi: 10.1152/ajpgi.90382.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dutta AK, Woo K, Khimji AK, Kresge C, Feranchak AP. Mechanosensitive Cl− secretion in biliary epithelium mediated through TMEM16A. Am J Physiol Gastrointest Liver Physiol 304: G87–G98, 2013. doi: 10.1152/ajpgi.00154.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, Hayward NJ, McNamara CR, Xue F, Moran MM, Strassmaier T, Uykal E, Owsianik G, Vennekens R, De Ridder D, Nilius B, Fanger CM, Voets T. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci USA 107: 19084–19089, 2010. doi: 10.1073/pnas.1005333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feranchak AP, Berl T, Capasso J, Wojtaszek PA, Han J, Fitz JG. p38 MAP kinase modulates liver cell volume through inhibition of membrane Na+ permeability. J Clin Invest 108: 1495–1504, 2001. doi: 10.1172/JCI200112190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feranchak AP, Fitz JG, Roman RM. Volume-sensitive purinergic signaling in human hepatocytes. J Hepatol 33: 174–182, 2000. doi: 10.1016/S0168-8278(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 13.Fiorotto R, Spirlì C, Fabris L, Cadamuro M, Okolicsanyi L, Strazzabosco M. Ursodeoxycholic acid stimulates cholangiocyte fluid secretion in mice via CFTR-dependent ATP secretion. Gastroenterology 133: 1603–1613, 2007. doi: 10.1053/j.gastro.2007.08.071. [DOI] [PubMed] [Google Scholar]
- 14.Fitz JG. Regulation of cholangiocyte secretion. Semin Liver Dis 22: 241–250, 2002. doi: 10.1055/s-2002-34502. [DOI] [PubMed] [Google Scholar]
- 15.Fitz JG, Basavappa S, McGill J, Melhus O, Cohn JA. Regulation of membrane chloride currents in rat bile duct epithelial cells. J Clin Invest 91: 319–328, 1993. doi: 10.1172/JCI116188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fitz JG, Cohn JA. Regulation of CFTR and other chloride channels in biliary epithelial cells. In: Biliary and Pancreatic Ductal Epithelia: Pathobiology and Pathophysiology, edited by Sirica A, Longnecker D. Philadelphia, PA: Marcel Decker, 1996. [Google Scholar]
- 17.Fitz JG, McGill J, Basavappa S, Cohn JA. Bile duct epithelial cells contain regulated chloride channels and CFTR. Clin Res 40: 319A, 1992. [Google Scholar]
- 18.Francis H, Glaser S, Demorrow S, Gaudio E, Ueno Y, Venter J, Dostal D, Onori P, Franchitto A, Marzioni M, Vaculin S, Vaculin B, Katki K, Stutes M, Savage J, Alpini G. Small mouse cholangiocytes proliferate in response to H1 histamine receptor stimulation by activation of the IP3/CaMK I/CREB pathway. Am J Physiol Cell Physiol 295: C499–C513, 2008. doi: 10.1152/ajpcell.00369.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goswami C, Kuhn J, Heppenstall PA, Hucho T. Importance of non-selective cation channel TRPV4 interaction with cytoskeleton and their reciprocal regulations in cultured cells. PLoS One 5: e11654, 2010. doi: 10.1371/journal.pone.0011654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gradilone SA, Masyuk AI, Splinter PL, Banales JM, Huang BQ, Tietz PS, Masyuk TV, Larusso NF. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci USA 104: 19138–19143, 2007. doi: 10.1073/pnas.0705964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gradilone SA, Masyuk TV, Huang BQ, Banales JM, Lehmann GL, Radtke BN, Stroope A, Masyuk AI, Splinter PL, LaRusso NF. Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology 139: 304–14.e2, 2010. doi: 10.1053/j.gastro.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knuth A, Gabbert H, Dippold W, Klein O, Sachsse W, Bitter-Suermann D, Prellwitz W, Meyer zum Büschenfelde KH. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J Hepatol 1: 579–596, 1985. doi: 10.1016/S0168-8278(85)80002-7. [DOI] [PubMed] [Google Scholar]
- 23.Lenzen R, Elster J, Behrend C, Hampel K-E, Bechstein W-O, Neuhaus P. Bile acid-independent bile flow is differently regulated by glucagon and secretin in humans after orthotopic liver transplantation. Hepatology 26: 1272–1281, 1997. doi: 10.1002/hep.510260527. [DOI] [PubMed] [Google Scholar]
- 24.Levine RA, Hall RC. Cyclic AMP in secretin choleresis. Evidence for a regulatory role in man and baboons but not in dogs. Gastroenterology 70: 537–544, 1976. doi: 10.1016/S0016-5085(76)80492-1. [DOI] [PubMed] [Google Scholar]
- 25.Li Q, Dutta A, Kresge C, Bugde A, Feranchak AP. Bile acids stimulate cholangiocyte fluid secretion by activation of transmembrane member 16A Cl- channels. Hepatology 68: 187–199, 2018. doi: 10.1002/hep.29804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGill JM, Basavappa S, Gettys TW, Fitz JG. Secretin activates Cl− channels in bile duct epithelial cells through a cAMP-dependent mechanism. Am J Physiol Gastrointest Liver Physiol 266: G731–G736, 1994. doi: 10.1152/ajpgi.1994.266.4.G731. [DOI] [PubMed] [Google Scholar]
- 27.Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol 286: C195–C205, 2004. doi: 10.1152/ajpcell.00365.2003. [DOI] [PubMed] [Google Scholar]
- 28.O’Neil RG, Heller S. The mechanosensitive nature of TRPV channels. Pflugers Arch 451: 193–203, 2005. doi: 10.1007/s00424-005-1424-4. [DOI] [PubMed] [Google Scholar]
- 29.Patel A, Sharif-Naeini R, Folgering JR, Bichet D, Duprat F, Honoré E. Canonical TRP channels and mechanotransduction: from physiology to disease states. Pflugers Arch 460: 571–581, 2010. doi: 10.1007/s00424-010-0847-8. [DOI] [PubMed] [Google Scholar]
- 30.Plaa GL, Becker BA. Demonstration of bile stasis in the mouse by a direct and an indirect method. J Appl Physiol 20: 534–537, 1965. doi: 10.1152/jappl.1965.20.3.534. [DOI] [PubMed] [Google Scholar]
- 31.Pusl T, Nathanson MH. The role of inositol 1,4,5-trisphosphate receptors in the regulation of bile secretion in health and disease. Biochem Biophys Res Commun 322: 1318–1325, 2004. doi: 10.1016/j.bbrc.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 32.Sathe MN, Woo K, Kresge C, Bugde A, Luby-Phelps K, Lewis MA, Feranchak AP. Regulation of purinergic signaling in biliary epithelial cells by exocytosis of SLC17A9-dependent ATP-enriched vesicles. J Biol Chem 286: 25363–25376, 2011. doi: 10.1074/jbc.M111.232868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor AL, Kudlow BA, Marrs KL, Gruenert DC, Guggino WB, Schwiebert EM. Bioluminescence detection of ATP release mechanisms in epithelia. Am J Physiol Cell Physiol 275: C1391–C1406, 1998. doi: 10.1152/ajpcell.1998.275.5.C1391. [DOI] [PubMed] [Google Scholar]
- 34.Tønsberg H, Holm R, Bjerregaard TG, Boll JB, Jacobsen J, Müllertz A. An updated and simplified method for bile duct cannulation of rats. Lab Anim 44: 373–376, 2010. doi: 10.1258/la.2010.010010. [DOI] [PubMed] [Google Scholar]
- 35.Ueno Y, Alpini G, Yahagi K, Kanno N, Moritoki Y, Fukushima K, Glaser S, LeSage G, Shimosegawa T. Evaluation of differential gene expression by microarray analysis in small and large cholangiocytes isolated from normal mice. Liver Int 23: 449–459, 2003. doi: 10.1111/j.1478-3231.2003.00876.x. [DOI] [PubMed] [Google Scholar]
- 36.Willette RN, Bao W, Nerurkar S, Yue TL, Doe CP, Stankus G, Turner GH, Ju H, Thomas H, Fishman CE, Sulpizio A, Behm DJ, Hoffman S, Lin Z, Lozinskaya I, Casillas LN, Lin M, Trout RE, Votta BJ, Thorneloe K, Lashinger ES, Figueroa DJ, Marquis R, Xu X. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J Pharmacol Exp Ther 326: 443–452, 2008. doi: 10.1124/jpet.107.134551. [DOI] [PubMed] [Google Scholar]
- 37.Woo K, Dutta AK, Patel V, Kresge C, Feranchak AP. Fluid flow induces mechanosensitive ATP release, calcium signalling and Cl- transport in biliary epithelial cells through a PKCzeta-dependent pathway. J Physiol 586: 2779–2798, 2008. doi: 10.1113/jphysiol.2008.153015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woo K, Sathe M, Kresge C, Esser V, Ueno Y, Venter J, Glaser SS, Alpini G, Feranchak AP. Adenosine triphosphate release and purinergic (P2) receptor-mediated secretion in small and large mouse cholangiocytes. Hepatology 52: 1819–1828, 2010. doi: 10.1002/hep.23883. [DOI] [PMC free article] [PubMed] [Google Scholar]