

Abstract
Preeclampsia (PE) is characterized by new-onset hypertension that usually occurs in the third trimester of pregnancy and is associated with oxidative stress and angiotensin II type 1 receptor agonistic autoantibodies (AT1-AAs). Inhibition of the AT1-AAs in the reduced uterine perfusion pressure (RUPP) rat, a model of PE, attenuates hypertension and many other characteristics of PE. We have previously shown that mitochondrial oxidative stress (mtROS) is a newly described PE characteristic exhibited by the RUPP rat that contributes to hypertension. However, the factors that cause mtROS in PE or RUPP are unknown. Thus, the objective of the current study is to use pharmacologic inhibition of AT1-AAs to examine their role in mtROS in the RUPP rat model of PE. AT1-AA inhibition in RUPP rats was achieved by administration of an epitope-binding peptide (′n7AAc′). Female Sprague-Dawley rats were divided into the following two groups: RUPP and RUPP + AT1-AA inhibition (RUPP + ′n7AAc′). On day 14 of gestation (GD), RUPP surgery was performed; ′n7AAc′ peptide (2 µg/μL) was administered by miniosmotic pumps in a subset of RUPP rats; and on GD19, sera, placentas, and kidneys were collected. mitochondrial respiration and mtROS were measured in isolated mitochondria using the Oxygraph 2K and fluorescent microplate reader, respectively. Placental and renal mitochondrial respiration and mtROS were improved in RUPP + ′n7AAc′ rats compared with RUPP controls. Moreover, endothelial cells (human umbilical vein endothelial cells) treated with RUPP + ′n7AAc′ sera exhibited less mtROS compared with those treated with RUPP sera. Overall, our findings suggest that AT1-AA signaling is one stimulus of mtROS during PE.
Keywords: AT1-AAs and preeclampsia, mitochondrial dysfunction, mitochondrial reactive oxygen species
INTRODUCTION
Preeclampsia (PE) affects 5–7% of all pregnancies in the United States (25, 27, 28). PE is the leading cause of maternal and fetal death and morbidity and is associated with maternal hemorrhage in underdeveloped countries. Although there is standard treatment for PE, it does not target a specific mechanism underlying the pathology of the disease. Importantly, there is no cure except for delivery of the fetus and placenta. The pathology underlying PE is not well understood. It is characterized by hypertension after the 20th wk of pregnancy and associated with immune activation, oxidative stress, endothelial dysfunction, and systemic vasoconstriction (2, 14, 15, 17, 26a). An important initiating event in the clinical manifestations of PE is thought to be placental ischemia, which is caused by reduced uteroplacental perfusion pressure because of abnormal spiral artery remodeling during placentation (1). Placental ischemia is associated with angiotensin II type 1 receptor agonistic autoantibodies (AT1-AAs), which are shown to be elevated in preeclamptic patients, and the reduced uterine perfusion pressure (RUPP) rat model of PE (9, 18, 31). AT1-AAs bind to a seven-amino-acid (7AA) sequence present on the second extracellular loop of the angiotensin II receptor type 1 (AT1R; see Ref. 31). AT1R signaling causes increased intracellular calcium levels and activation of the intracellular mitogen-activated protein kinase/extracellular signal regulated kinase pathway, which ultimately cause oxidative stress (ROS), vasoconstriction, and hypertension (10, 29, 32). Peptide competition experiments in our laboratory demonstrate that rat AT1-AAs interact with the same 7AA sequence of the AT1R. Based on this binding sequence (AFHYESQ), we have developed a capped inhibitory peptide (′n7AAc′) to bind to the AT1-AA and block AT1-AAs from binding to the AT1R. Protein “capping” of the NH2- and COOH-terminus of the peptide is a process commonly used to increase peptide half-life and to protect exogenous peptides from protein lysis and degradation when used in the whole animal. We have recently published that administration of ′n7AAc′ prevents hypertension and natural killer (NK) cell activation and lowers soluble fms-like tyrosine kinase-1 and preproendothelin-1 with improved nitrate/nitrite levels in RUPP rats, suggesting an important role of AT1-AAs in preeclamptic pathology (5). However, we did not know whether the ′n7AAc′ peptide improved mitochondrial function or ROS.
Studies from our laboratory and others have reported evidence for mitochondrial (mt) dysfunction and ROS in preeclamptic pathology (3, 6, 16, 21, 23, 30, 34). We have recently reported that RUPP rats exhibit placental and renal mtROS, and treatment with mitochondrial antioxidants (MitoQ/MitoTempo) attenuates high blood pressure, suggesting the importance of mtROS in PE (30). However, the studies exploring the factors that drive mtROS in PE are missing. Angiotensin II (ANG II) has been shown to cause mitochondrial dysfunction with increased mtROS in models of hypertension (11–13). Because AT1-AAs signal similarly to ANG II by binding to AT1R, it is possible that AT1-AAs might cause mtROS. We recently reported a role for AT1-AAs in causing renal and placental mtROS when infused in pregnant rats (6); however, it remains unknown if endogenous AT1-AAs are a stimulus for mitochondrial dysfunction and ROS in response to placental ischemia. Thus, we hypothesized that AT1-AA signaling is one possible mechanism by which mitochondrial dysfunction and mtROS are manifested in RUPP rats or in PE. Hence, the objective of the current study was to examine if inhibiting endogenous AT1-AAs by ′n7AAc′ will reduce mtROS in association with lowered blood pressures in RUPP rats.
METHODS
Animals
Twelve-week-old, 225-g, female Sprague-Dawley (SD) rats purchased from Envigo were used in the current study. Rats were housed in a temperature-controlled room (23°C) with a 12:12-h light-dark cycle with free access to standard rat chow and water. This study complied with guidelines of the University of Mississippi Medical Center, and the animals were handled according to the guiding principles published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee.
Materials and Reagents
Glutamate (49621)/malate (M1000), oligomycin (O4876), FCCP (C2920), rotenone (R8875), antimycin A (A8674), sucrose (84097), HEPES (H3375), EGTA (E3889), BSA (A9647), MOPS (M1254), KPi (795488), M199 (M4530), HRP (P8375), and SOD (S7571) were purchased from Sigma Aldrich (St. Louis, MO). DMEM (10566–016), FBS (16000044), antimycotic/antibiotic (15240062), MitoSOX red (M36008), and Amplex red UltraRed Reagent (A36006) were purchased from Thermo Fisher Scientific (Waltham, MA).
Effect of AT1-AA Inhibition on Placental Ischemia-Induced Mitochondrial-Mediated Oxidative Stress in RUPP Placenta and Kidney
RUPP surgery and AT1-AA inhibition.
Normal pregnant (NP) rats were divided into the following two groups: RUPP and RUPP + ′n7AAc′. The goal of the RUPP surgery was to create placental ischemia in pregnant rats that mimics ischemia in the preeclamptic placenta. Briefly, on gestation day (GD) 14, RUPP surgery was performed by placing surgical silver clips (0.203 mm diameter) on the abdominal aorta (1 clip) above the iliac bifurcation and on the ovarian arteries (1 clip each at 0.100 mm) on both sides under isoflurane anesthesia. This surgical procedure reduces blood flow to the uteroplacental unit by ~40%. The miniosmotic pumps containing ′n7AAc′ peptide (at a dose of 144 μg/day) were placed in the peritoneal cavity during the RUPP surgical procedure (5). On GD18, catheters were inserted in carotid arteries to measure blood pressure on GD19. Briefly, conscious arterial pressure was monitored with a pressure transducer (Cobe III transducer CDX Sema) and recorded continuously for 45 min after a 30-min stabilization period. Immediately after blood pressure measurements, placentas and kidneys were harvested and processed for mitochondrial experiments (30). Furthermore, serum samples were processed to be used in endothelial cell mtROS experiments (30). Differences in NP rats compared with RUPP rats have been published before and therefore were not compared in this study (5, 30).
Determination of AT1-AA activity.
Serum was analyzed for AT1-AA activity by cardiomyocyte assay. Antibodies were detected by the chronotropic responses to AT1 receptor-mediated stimulation of cultured neonatal rat cardiomyocytes coupled with receptor-specific antagonists as previously described (9, 29, 31). Chronotropic responses were measured and expressed in beats per minute.
Isolation of intact mitochondria.
Intact placental or renal mitochondria were isolated by differential centrifugation (6, 30). Briefly, placentas (2 placentas/rat) or kidneys (1 kidney/rat) were washed in ice-cold Mito I buffer (250 mM sucrose, 10 mM HEPES, 1 mM EGTA, and 0.1% BSA, pH 7.2), finely cut, and homogenized using a glass-glass Dounce homogenizer (4 mL buffer, 5 manual stokes). The homogenate was centrifuged at 4,000 rpm, 3 min at 4°C. The supernatant containing mitochondria was centrifuged at 10,000 rpm, 10 min at 4°C, to collect the mitochondrial pellet, which was washed with Mito I buffer (1 mL) one time, followed by Mito II (250 mM sucrose, 10 mM HEPES, and 0.1% BSA, pH 7.2; 1 mL one time) at 10,000 rpm, 10 min at 4°C. The final mitochondrial pellet was resuspended in 200 µL of Mito II buffer and used immediately for mitochondrial respiration and mtROS measurements.
Mitochondrial respiration.
Respiration measurements in the isolated placental or renal mitochondria were performed with an Oroboros Oxygraph-2K (30). State 3 (coupled respiratory state) and maximal respiration [electron transfer capacity (ETC)] rates were measured by adding ADP (5 mM) and FCCP (0.5 µM) to the isolated mitochondria in respiration buffer (100 mM KCl, 5 mM KPi, 1 mM EGTA, 1 mg/mL BSA, and 50 mM MOPS, pH 7.4) in the presence of the electron donors glutamate (10 mM)/malate (2 mM). Finally, nonmitochondrial oxygen consumption was measured by the addition of rotenone (0.5 µM) and antimycin A (2.5 µM). Nonmitochondrial respiration rate was subtracted from the state 3 or maximal rates to correct for nonmitochondrial respiration. The collected data were normalized for the amount of protein and expressed as picomoles of oxygen consumed per second per milligram mitochondrial protein.
Mitochondrial ROS production in isolated mitochondria.
Hydrogen peroxide (H2O2) production in placental or renal RUPP mitochondria was determined fluorometrically by using Amplex red reagent (6, 30). H2O2 oxidizes nonfluorescent Amplex red to the red fluorescent oxidation product, resorufin, in the presence of horseradish peroxidase (HRP). Briefly, mitochondria (0.4 mg/mL) were incubated in a 96-well plate with respiration buffer, superoxide dismutase (SOD, 40 U/mL), HRP (4 U/mL), and succinate (10 mM). Amplex red (10 µM) was added to the wells to start the reaction. The real-time production of H2O2 was recorded using a plate reader at 555/581 nm excitation/emission for 30 min at 25°C. Appropriate blanks without Amplex red or mitochondrial protein were included in the assay. The reagent concentrations in this assay were based on a microplate well final volume of 200 µL.
Role of AT1-AAs in Causing Vascular Endothelial Cell mtROS
Cell culture.
Human umbilical vein endothelial cells (HUVEC) were purchased from ATCC (catalog no. CRL-1730). Cells were cultured in 50:50 DMEM-M199 with 10% FBS and 1% antimycotic/antibiotic in a humidified atmosphere with 5% CO2, 20% O2, and 75% N2 at 37°C. The cells at passage 4 were cultured in six-well plates for the following experiments.
Mitochondrial-mediated ROS production in HUVECs.
Mitochondrial-specific ROS were measured using MitoSOX red, a novel fluorogenic dye specifically targeted to mitochondria in live cells. In brief, HUVECs were grown to 70% confluency in six-well culture plates (30). Cells were serum starved for 4 h before an overnight incubation with 10% experimental RUPP or RUPP + ′n7AAc′ serum. Experimental serum was washed off, and cells were incubated with MitoSOX red (5 µM) for 30 min at 37°C. Antimycin A (100 µM) was included in the experiment as a positive control. Serum-free medium was added after the cells were washed two times with DPBS, and the cells were incubated for an additional 4 h. Cells were collected and transferred to FACS tubes to run samples on the flow cytometer with 488 nm excitation to measure oxidized MitoSOX red in the FL2 and FL3 channels of a Gallios flow cytometer (Beckman Coulter, Brea, CA). At least 5,000 events for each sample were collected and analyzed. Significant differences between NP and RUPP sera to cause endothelial cell mtROS and mitochondrial dysfunction have been previously published and were not examined again in this study (30).
Statistical Analysis
All data are expressed in scatter plots as means ± SE. Statistical comparisons were made using Student’s t test. A value of P < 0.05 was considered statistically significant.
RESULTS
AT1-AA Inhibition Lowers AT1-AA Activity and Normalizes Blood Pressure in RUPP Rats
In vivo AT1-AA inhibition by ′n7AAc′ peptide significantly lowered circulating AT1-AA activity. Circulating AT1-AA activity is 17.91 ± 0.96 beats/min in RUPP rats, and RUPP + ′n7AAc′ circulating activity is 6.0 ± 0.20 beats/min (P < 0.05). Moreover, in vivo AT1-AA inhibition by ′n7AAc′ peptide attenuated hypertension in RUPP rats (n = 16) compared with RUPP controls (n = 6, 106 ± 2 vs. 120 ± 2 mmHg, P < 0.05; Fig. 1).
Fig. 1.

Angiotensin II type 1 receptor agonistic autoantibody (AT1-AA) inhibition normalizes blood pressure in reduced uterine perfusion pressure (RUPP) rats. RUPP surgery was performed on gestational day (GD) 14, and AT1-AA inhibitory peptide (′n7AAc′) was administered via osmotic minipumps. A: conscious blood pressure was measured on GD19, and sera were collected for determination of AT1-AA activation. AT1-AA inhibition reduced blood pressure in RUPPs treated with ′n7AAc′ (n = 16) vs. RUPPs (n = 6). B: n′7AAc′ reduced AT1-AA activity in the circulation, indicating binding of the antibody and inhibition of AT1 receptor activation. RUPPs treated with ′n7AAc′ (n = 7) vs. RUPPs (n = 8). MAP, mean arterial pressure; BPM, beats/min. Data are presented as means ± SE. *P < 0.05 vs. RUPP, Student’s t test.
AT1-AA Inhibition Attenuated ROS in RUPP Placenta
We have previously shown that mitochondrial respiration was reduced in RUPP placental mitochondria versus NP rats (30). In the current study, AT1-AA inhibition by ′n7AAc′ peptide caused a slight increase in state 3 respiration rate (Fig. 2A) and a trend toward higher maximal respiration rate (288.5 ± 37.01 pmol of O2·s−1·mg−1 vs. 174 ± 13 pmol·s−1·mg−1; Fig. 2B) in RUPP + ′n7AAc′ placental mitochondria (n = 9) versus RUPPs (n = 5). Furthermore, these improved respiration rates are associated with 60% fold decrease in mtROS production in placentas from RUPP rats treated with ′n7AAc′ peptide (n = 4; Fig. 2C) compared with RUPPs (n = 4).
Fig. 2.

Angiotensin II type 1 receptor agonistic autoantibody (AT1-AA) inhibition reduced mitochondrial oxidative stress (mtROS) production in reduced uterine perfusion pressure (RUPP) placenta. Respiration rates were measured in isolated mitochondria using Oxygraph 2K, and mtROS (H2O2) was measured using Amplex red assay with a fluorescent microplate reader. State 3 respiration (A) and maximal respiration (B) in RUPPs treated with AT1-AA inhibitory peptide [′n7AAc′; n = 11 (A) and 5 (B)] versus RUPPs [n = 4 (A) and 9 (B)]. Furthermore, AT1-AA inhibition significantly reduced mtROS production in RUPPs treated with ′n7AAc′ (n = 4) vs. RUPPs (n = 4) (C). Data are presented as means ± SE. *P < 0.05 vs. RUPP, Student’s t test.
AT1-AA Inhibition Attenuates ROS in RUPP Kidney
We have previously shown that mitochondrial respiration was reduced in RUPP renal mitochondria versus NP rats (30). In the current study, AT1-AA inhibition by ′n7AAc′ peptide shows a trend to increase the maximal respiration rate ( 651.2 ± 145.4 vs. 455.2 ± 95.63 pmol of O2·s−1·mg−1; Fig. 3B) in RUPP + ′n7AAc′ renal mitochondria (n = 6) vs. RUPPs (n = 6), which was associated with ~90% fold decrease in mtROS production in kidneys from RUPP rats treated with ′n7AAc′ peptide (n = 3; Fig. 3C) compared with RUPPs (n = 4).
Fig. 3.

Angiotensin II type 1 receptor agonistic autoantibody (AT1-AA) inhibition reduced mitochondrial oxidative stress (mtROS) production in reduced uterine perfusion pressure (RUPP) kidney. Respiration rates were measured in isolated mitochondria using the Oxygraph 2K, and mtROS (H2O2) was measured using Amplex red assay with a fluorescent microplate reader. State 3 respiration (A) and maximal respiration (B) in RUPPs treated with AT1-AA inhibitory peptide [′n7AAc′; n = 9 (A) and 6 (B)] vs. RUPPs [n = 6 (A) and 6 (B)]. Furthermore, AT1-AA inhibition significantly reduced mtROS production in RUPPs treated with ′n7AAc′ (n = 3) vs. RUPPs (n = 4) (C). Data are presented as means ± SE. *P < 0.05 vs. RUPP, Student’s t-test.
AT1-AA Inhibition Decreased HUVEC mtROS Caused by RUPP Sera
In our previously published study, we have shown that overnight incubation of HUVECs with 10% sera from RUPP rats shows significantly increased mtROS production compared with the cells that were incubated with NP sera (30). In this study, we report that sera from RUPPs treated with ′n7AAc′ (n = 4) attenuated mtROS compared with sera from RUPP control rats (n = 6, 1.14 ± 0.31 vs. 5.6 + 1.16% gated, P < 0.05; Fig. 4, A and B). These findings demonstrate a potentially important role of circulating AT1-AAs in causing mtROS in the systemic vasculature during PE.
Fig. 4.

Human umbilical vein endothelial cells (HUVECs) treated with serum from angiotensin II type 1 receptor agonistic autoantibodies (AT1-AA)-inhibited reduced uterine perfusion pressure (RUPP) rats show attenuated mitochondrial oxidative stress (mtROS) production. Serum (10%) from RUPPs (n = 4) treated with AT1-AA inhibitory peptide (′n7AAc′) significantly reduced mtROS in HUVECs compared with cells that were incubated with RUPP (n = 6) serum. A: dot plot and histogram showing the mitoSOX staining in HUVECs incubated with RUPP or RUPP + n7AAc. B: scatter plot showing quantified mitoSOX staining. SS, side scatter; FS, forward scatter; 7AA, seven amino acids. Data are presented as means ± SE. *P < 0.05 vs. RUPP, Student’s t test.
DISCUSSION
In the current study, we report novel findings on the role of AT1-AAs in causing mitochondrial dysfunction and mtROS production in the RUPP rat model of PE. AT1-AAs are agonistic antibodies that target the AT1R and are shown to be elevated in both preeclamptic women and RUPP rats (9, 18, 31). Moreover, we have recently published that placental ischemia induces mtROS that mediates hypertension in RUPP pregnant rats (30). In the current study, we replicated the data demonstrating that AT1-AA inhibition with the ′n7AAc′ attenuated hypertension in RUPP rats. Importantly, we show that this lower blood pressure is associated with reduced AT1-AA activity in the RUPP rats, confirming efficacy of the ′n7AAc′ AT1-AA-binding peptide to inhibit the AT1-AA from activating the AT1R. Moreover, the placental and renal respiration rates (maximal) were improved in RUPP rats treated with ′n7AAc′, however, not significantly, but could suggest that inhibiting AT1-AA activity improves electron transport chain (ETC) capacity in either the kidney or placenta. Interestingly, this was associated with significantly reduced renal and placental mtROS production. The improvement in mitochondrial function and attenuation of mtROS in the local placental tissue might have a beneficial effect on the mtROS-mediated damage in other organs such as the kidney or peripheral vasculature. In our previously published work, we reported that circulating factors in RUPP sera cause mtROS production in the cultured endothelial cells (30). Thus, we asked if inhibiting AT1-AAs in the sera of RUPP rats would improve endothelial cell mtROS. We show that the endothelial cells treated with sera from RUPP + ′n7AAc′ rats completely attenuated mtROS compared with cells that were treated with RUPP sera, suggesting the importance of circulating AT1-AAs in response to placental ischemia to affect the systemic vasculature.
A role for ANG II in causing oxidative stress has been well documented by a number of published studies. Specifically, ANG II has been shown to induce both NADPH oxidase (NOX) and mitochondrial-mediated ROS production (11, 12). Additionally, we have previously shown that AT1-AAs can cause placental ROS production by NOX activation and increase plasma isoprostanes (5, 24). More recently, we demonstrated that infusion of AT1-AA in pregnant rats is associated with activation of renal NK cells and increased renal mtROS. Furthermore, administration of a mixture of ′n7AAc′ and AT1-AAs in NP rats prevented elevated mtROS both in the placenta and kidney, indicating the importance of AT1-AA signaling in causing mtROS production in pregnant rats (6). Although we did not measure specific intracellular signaling involved in the production of mtROS, collectively these data demonstrate that AT1-AA-AT1R signaling results in mtROS and dysfunction in the kidney and placenta during pregnancy. However, what we do not know is, if AT1-AAs cause mitochondrial-mediated ROS similarly to ANG II. ANG II is not increased in the RUPP rat or in response to PE in patients; therefore, it is conceivable that direct inhibition of AT1-AA with the 7AA could improve both renal and placental mtROS independent of any action of ANG II. Moreover, it is possible that AT1-AAs cause mtROS via stimulating NK cell cytotoxicity toward AT1-AA-bound cells within the renal or placental tissues. To answer that question, future studies examining mtROS in the absence of NK cells during AT1-AA-induced hypertension during pregnancy may be performed.
Our previous results demonstrate that the mitochondrial targeted antioxidants, mitoQ and mitoTempo, attenuate hypertension in RUPP rats (30). Studies by Dikalova et al. showed that mitoTempo reduced mitochondrial superoxide levels, increased the state 3 respiration rate and respiratory coupled ratio, reduced NADPH oxidase activity, and restored nitric oxide levels in endothelial cells. Moreover, treatment of mice in vivo with mitoTempo attenuated hypertension in both ANG II and deoxycorticosterone acetate salt-sensitive models of hypertension (12). It has been shown that ANG II induces mitochondrial superoxide production by activation of reverse electron transfer (RET) within the ETC, revealing that the ultimate target of ANG II in causing mtROS is the ETC. Furthermore, as shown with mitoTempo, treatment of mice with malate (RET inhibitor) reduced blood pressure in ANG II-infused mice, suggesting that mtROS mediates the development of hypertension (11). Another study reported that ANG II causes reduced ETC activity and tricarboxylic acid cycle gene expression (19). Furthermore, a few studies that were focused on the role of ANG II in causing renal mitochondrial dysfunction revealed that ANG II in fact might play a role in mitochondrial damage in renal cell types (7, 8). In this study, we show for the first time that endogenous AT1-AAs, similarly to ANG II, cause mitochondrial impairment with elevated mtROS in the kidney in response to placental ischemia.
Doughan et al. showed that ANG II causes mitochondrial H2O2 production and reduces mitochondrial state 3 respiration in intact bovine aortic endothelial cells. Moreover, depletion of the p22phox subunit of NADPH oxidase or treatment with apocynin (NADPH oxidase inhibitor) reversed these effects on respiration and ROS production, suggesting that ANG II mediates mitochondrial dysfunction via NADPH oxidase in endothelial cells (13). Similar to ANG II, we demonstrate in this study that inhibiting circulating AT1-AAs in RUPP rats with ′n7AAc′ reduces endothelial cell mtROS.
Although in this study we demonstrate that AT1-AA play an important role in mtROS in response to placental ischemia, it is important to note that RUPP rats exhibit many PE pathological factors in addition to AT1-AAs. Factors such as soluble fms-like tyrosine kinase (sFlt-1, antiangiogenic factor involved in PE pathology) and TNF-α (inflammatory cytokine) are worth examining to better understand their contribution in causing mtROS in PE (Fig. 5). In fact, a study published by Brownfoot et al. demonstrated that sFlt-1 production is regulated by the ETC (4). Another recent study reported that sFlt-1 reduces mitochondrial respiration in endothelial and vascular smooth muscle cells in vitro (26). Moreover, TNF-α infusion increases superoxide and H2O2 production in cardiac mitochondria, which is associated with increased NADPH oxidase subunit expression and reduced ETC capacity. However, coadministration of losartan (AT1R blocker) prevented the mitochondrial damage, suggesting the TNF-α is upstream of ANG II-induced mitochondrial damage (22). However, it has been shown that AT1-AAs cause TNF-α or sFlt-1 production in RUPP rats (5, 33). Thus, further studies are needed to understand the molecular mechanisms underlying mtROS production in PE. Overall, findings from this and studies infusing the AT1-AA in rats indicate that AT1-AAs do play a role in causing mitochondrial dysfunction and mtROS in placentas, kidneys, and endothelial cells, which could in part be responsible for the placental, renal, or endothelial dysfunction associated with preeclamptic pathology. Our study highlights the role of AT1-AAs in causing mitochondrial dysfunction and mtROS production in the placenta and kidney of RUPP rats, and in cultured vascular endothelial cells. We believe that AT1-AA signaling may be one possible mechanism of mtROS in the PE pathology. Inhibiting AT1-AAs with ′n7AAc′ may serve as a potential therapeutic option to improve organ bioenergetics and treat hypertension during PE.
Fig. 5.
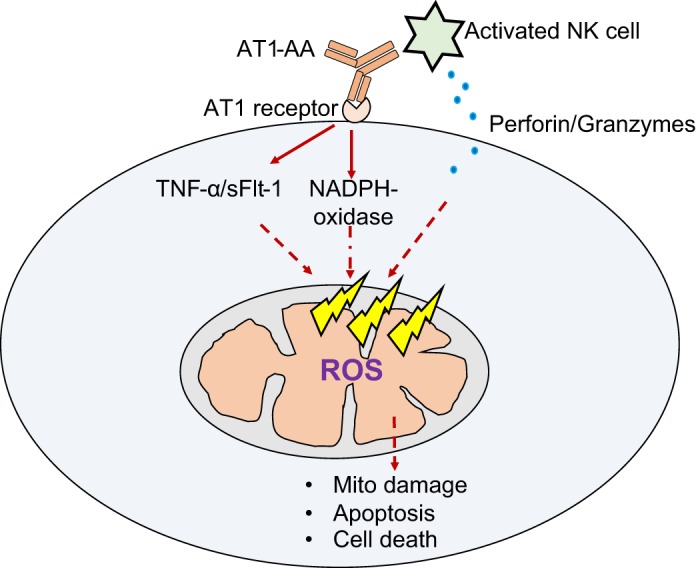
Cartoon showing series of events in angiotensin II type 1 receptor agonistic autoantibody (AT1-AA) induced mitochondrial dysfunction and mitochondrial oxidative stress (ROS) in a target cell (i.e., endothelial or placental cell). AT1-AA-induced mitochondrial oxidative stress is possibly mediated through a number of mechanisms such as activation of NADPH oxidases, production of soluble fms-like tyrosine kinase (sFlt-1) or TNF-α, and natural killer (NK) cell activation.
Perspectives and Significance
The role for oxidative stress in PE has been reported by a number of studies. We have previously documented mitochondrial dysfunction as a contributing source of oxidative stress in a rat model of PE. We have demonstrated that mtROS can contribute to the development of hypertension in a PE rat model. This was the first study to show the preclinical relevance of mtROS in PE. Whereas ours along with a few other studies demonstrate an important role of mtROS in PE, the molecular mechanisms underlying are still missing. Recently, we have reported important findings on the role of NK cell activation in causing mtROS in a PE rat model. With this study, we were able to show that another important factor that is involved in mtROS is AT1-AAs. However, the combined role of NK cell activation and AT1-AAs or role of additional PE pathological factors in causing mtROS are not known at this time. Further work on elucidating factors and signaling mechanisms may offer valuable knowledge on novel molecular targets that can be targeted to treat PE.
GRANTS
This research was supported by National Institutes of Health Grants RO1-HD-067541 and P20-GM-121334 (B. D. LaMarca) and R00-HL-103456 (D. C. Cornelius) and American Heart Association predoctoral fellowship 17PRE33660592/AHA (V. R. Vaka).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.R.V. and B.D.L. conceived and designed research; V.R.V., M.W.C., E.D., M.F., T.I., L.M.A., N.U., and D.C.C. performed experiments; V.R.V., M.W.C., E.D., M.F., T.I., L.M.A., N.U., R.D., G.W., and B.D.L. analyzed data; V.R.V., E.D., T.I., L.M.A., N.U., R.D., G.W., and B.D.L. interpreted results of experiments; V.R.V., M.W.C., E.D., M.F., L.M.A., and N.U. prepared figures; V.R.V., M.W.C., and B.D.L. drafted manuscript; V.R.V., M.W.C., E.D., L.M.A., N.U., R.D., G.W., and B.D.L. edited and revised manuscript; V.R.V., M.W.C., E.D., M.F., T.I., L.M.A., N.U., D.C.C., R.D., G.W., and B.D.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Robert Kramer, Dr. Jon Hosler, and Kristin Shirey for assistance with mitochondrial assays and analysis.
REFERENCES
- 1.Amaral LM, Wallace K, Owens M, LaMarca B. Pathophysiology and current clinical management of preeclampsia. Curr Hypertens Rep 19: 61, 2017. doi: 10.1007/s11906-017-0757-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aouache R, Biquard L, Vaiman D, Miralles F. Oxidative stress in preeclampsia and placental diseases. Int J Mol Sci 19: E1496, 2018. doi: 10.3390/ijms19051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beyramzadeh M, Dikmen ZG, Erturk NK, Tuncer ZS, Akbiyik F. Placental respiratory chain complex activities in high risk pregnancies. J Matern Fetal Neonatal Med 30: 2911–2917, 2017. doi: 10.1080/14767058.2016.1268594. [DOI] [PubMed] [Google Scholar]
- 4.Brownfoot FC, Hastie R, Hannan NJ, Cannon P, Tuohey L, Parry LJ, Senadheera S, Illanes SE, Kaitu’u-Lino TJ, Tong S. Metformin as a prevention and treatment for preeclampsia: effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am J Obstet Gynecol 214: 356.e1–356.e15, 2016. doi: 10.1016/j.ajog.2015.12.019. [DOI] [PubMed] [Google Scholar]
- 5.Cunningham MW Jr, Castillo J, Ibrahim T, Cornelius DC, Campbell N, Amaral L, Vaka VR, Usry N, Williams JM, LaMarca B. AT1-AA (angiotensin II type 1 receptor agonistic autoantibody) blockade prevents preeclamptic symptoms in placental ischemic rats. Hypertension 71: 886–893, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham MW Jr, Vaka VR, McMaster K, Ibrahim T, Cornelius DC, Amaral L, Campbell N, Wallukat G, McDuffy S, Usry N, Dechend R, LaMarca B. Renal natural killer cell activation and mitochondrial oxidative stress; new mechanisms in AT1-AA mediated hypertensive pregnancy. Pregnancy Hypertens 15: 72–77, 2019. doi: 10.1016/j.preghy.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Cavanagh EM, Piotrkowski B, Basso N, Stella I, Inserra F, Ferder L, Fraga CG. Enalapril and losartan attenuate mitochondrial dysfunction in aged rats. FASEB J 17: 1096–1098, 2003. doi: 10.1096/fj.02-0063fje. [DOI] [PubMed] [Google Scholar]
- 8.de Cavanagh EM, Toblli JE, Ferder L, Piotrkowski B, Stella I, Inserra F. Renal mitochondrial dysfunction in spontaneously hypertensive rats is attenuated by losartan but not by amlodipine. Am J Physiol Regul Integr Comp Physiol 290: R1616–R1625, 2006. doi: 10.1152/ajpregu.00615.2005. [DOI] [PubMed] [Google Scholar]
- 9.Dechend R, Gratze P, Wallukat G, Shagdarsuren E, Plehm R, Bräsen JH, Fiebeler A, Schneider W, Caluwaerts S, Vercruysse L, Pijnenborg R, Luft FC, Müller DN. Agonistic autoantibodies to the AT1 receptor in a transgenic rat model of preeclampsia. Hypertension 45: 742–746, 2005. doi: 10.1161/01.HYP.0000154785.50570.63. [DOI] [PubMed] [Google Scholar]
- 10.Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, Juepner A, Gulba DC, Mackman N, Haller H, Luft FC. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation 101: 2382–2387, 2000. doi: 10.1161/01.CIR.101.20.2382. [DOI] [PubMed] [Google Scholar]
- 11.Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassègue B, Griendling KK, Harrison DG, Dikalova AE. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid Redox Signal 20: 281–294, 2014. doi: 10.1089/ars.2012.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116, 2010. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 102: 488–496, 2008. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 14.Granger JP. Inflammatory cytokines, vascular function, and hypertension. Am J Physiol Regul Integr Comp Physiol 286: R989–R990, 2004. doi: 10.1152/ajpregu.00157.2004. [DOI] [PubMed] [Google Scholar]
- 15.Harmon AC, Cornelius DC, Amaral LM, Faulkner JL, Cunningham MW Jr, Wallace K, LaMarca B. The role of inflammation in the pathology of preeclampsia. Clin Sci (Lond) 130: 409–419, 2016. doi: 10.1042/CS20150702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Illsinger S, Janzen N, Sander S, Schmidt KH, Bednarczyk J, Mallunat L, Bode J, Hagebölling F, Hoy L, Lücke T, Hass R, Das AM. Preeclampsia and HELLP syndrome: impaired mitochondrial function in umbilical endothelial cells. Reprod Sci 17: 219–226, 2010. doi: 10.1177/1933719109351597. [DOI] [PubMed] [Google Scholar]
- 17.LaMarca B, Cornelius DC, Harmon AC, Amaral LM, Cunningham MW, Faulkner JL, Wallace K. Identifying immune mechanisms mediating the hypertension during preeclampsia. Am J Physiol Regul Integr Comp Physiol 311: R1–R9, 2016. doi: 10.1152/ajpregu.00052.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LaMarca B, Wallace K, Herse F, Wallukat G, Martin JN Jr, Weimer A, Dechend R. Hypertension in response to placental ischemia during pregnancy: role of B lymphocytes. Hypertension 57: 865–871, 2011. doi: 10.1161/HYPERTENSIONAHA.110.167569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larkin JE, Frank BC, Gaspard RM, Duka I, Gavras H, Quackenbush J. Cardiac transcriptional response to acute and chronic angiotensin II treatments. Physiol Genomics 18: 152–166, 2004. doi: 10.1152/physiolgenomics.00057.2004. [DOI] [PubMed] [Google Scholar]
- 21.Mando’ C, Marino MA, Miriam F, Palma CD, Borelli M, Trabattoni D, Stampalija T, Ferrazzi E, Clementi E, Cetin I. OS048 mitochondrial content and function in placental cells and tissuesof preeclampsia and IUGR. Pregnancy Hypertens 2: 203, 2012. doi: 10.1016/j.preghy.2012.04.049. [DOI] [PubMed] [Google Scholar]
- 22.Mariappan N, Elks CM, Haque M, Francis J. Interaction of TNF with angiotensin II contributes to mitochondrial oxidative stress and cardiac damage in rats. PLoS One 7: e46568, 2012. doi: 10.1371/journal.pone.0046568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsubara S, Minakami H, Sato I, Saito T. Decrease in cytochrome c oxidase activity detected cytochemically in the placental trophoblast of patients with pre-eclampsia. Placenta 18: 255–259, 1997. doi: 10.1016/S0143-4004(97)80059-8. [DOI] [PubMed] [Google Scholar]
- 24.Parrish MR, Wallace K, Tam Tam KB, Herse F, Weimer A, Wenzel K, Wallukat G, Ray LF, Arany M, Cockrell K, Martin JN, Dechend R, LaMarca B. Hypertension in response to AT1-AA: role of reactive oxygen species in pregnancy-induced hypertension. Am J Hypertens 24: 835–840, 2011. doi: 10.1038/ajh.2011.62. [DOI] [PubMed] [Google Scholar]
- 25.Roberts JM, Pearson G, Cutler J, Lindheimer M; NHLBI Working Group on Research on Hypertension During Pregnancy . Summary of the NHLBI working group on research on hypertension during pregnancy. Hypertension 41: 437–445, 2003. doi: 10.1161/01.HYP.0000054981.03589.E9. [DOI] [PubMed] [Google Scholar]
- 26.Sánchez-Aranguren LC, Espinosa-González CT, González-Ortiz LM, Sanabria-Barrera SM, Riaño-Medina CE, Nuñez AF, Ahmed A, Vasquez-Vivar J, López M. Soluble Fms-like tyrosine kinase-1 alters cellular metabolism and mitochondrial bioenergetics in preeclampsia. Front Physiol 9: 83, 2018. doi: 10.3389/fphys.2018.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26a.Sánchez-Aranguren LC, Prada CE, Riaño-Medina CE, Lopez M. Endothelial dysfunction and preeclampsia: role of oxidative stress. Front Physiol 5: 372, 2014. doi: 10.3389/fphys.2014.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet 365: 785–799, 2005. doi: 10.1016/S0140-6736(05)17987-2. [DOI] [PubMed] [Google Scholar]
- 28.Sibai BM, Caritis S, Hauth J; National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network . What we have learned about preeclampsia. Semin Perinatol 27: 239–246, 2003. doi: 10.1016/S0146-0005(03)00022-3. [DOI] [PubMed] [Google Scholar]
- 29.Thway TM, Shlykov SG, Day MC, Sanborn BM, Gilstrap LC III, Xia Y, Kellems RE. Antibodies from preeclamptic patients stimulate increased intracellular Ca2+ mobilization through angiotensin receptor activation. Circulation 110: 1612–1619, 2004. doi: 10.1161/01.CIR.0000142855.68398.3A. [DOI] [PubMed] [Google Scholar]
- 30.Vaka VR, McMaster KM, Cunningham MW Jr, Ibrahim T, Hazlewood R, Usry N, Cornelius DC, Amaral LM, LaMarca B. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertension 72: 703–711, 2018. doi: 10.1161/HYPERTENSIONAHA.118.11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jüpner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest 103: 945–952, 1999. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang S, Zhong Q, Qiu Z, Chen X, Chen F, Mustafa K, Ding D, Zhou Y, Lin J, Yan S, Deng Y, Wang M, Zhou Y, Liao Y, Zhou Z. Angiotensin II receptor type 1 autoantibodies promote endothelial microparticles formation through activating p38 MAPK pathway. J Hypertens 32: 762–770, 2014. doi: 10.1097/HJH.0000000000000083. [DOI] [PubMed] [Google Scholar]
- 33.Zhou CC, Irani RA, Zhang Y, Blackwell SC, Mi T, Wen J, Shelat H, Geng YJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody-mediated tumor necrosis factor-alpha induction contributes to increased soluble endoglin production in preeclampsia. Circulation 121: 436–444, 2010. doi: 10.1161/CIRCULATIONAHA.109.902890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zsengellér ZK, Rajakumar A, Hunter JT, Salahuddin S, Rana S, Stillman IE, Ananth Karumanchi S. Trophoblast mitochondrial function is impaired in preeclampsia and correlates negatively with the expression of soluble fms-like tyrosine kinase 1. Pregnancy Hypertens 6: 313–319, 2016. doi: 10.1016/j.preghy.2016.06.004. [DOI] [PubMed] [Google Scholar]