

Abstract
Endothelial dysfunction, characterized by reduced bioavailability of nitric oxide and increased oxidative stress, is a hallmark characteristic in diabetes and diabetic nephropathy (DN). High levels of asymmetric dimethylarginine (ADMA) are observed in several diseases including DN and are a strong prognostic marker for cardiovascular events in patients with diabetes and end-stage renal disease. ADMA, an endogenous endothelial nitric oxide synthase (NOS3) inhibitor, is selectively metabolized by dimethylarginine dimethylaminohydrolase (DDAH). Low DDAH levels have been associated with cardiac and renal dysfunction, but its effects on DN are unknown. We hypothesized that enhanced renal DDAH-1 expression would improve DN by reducing ADMA and restoring NOS3 levels. DBA/2J mice injected with multiple low doses of vehicle or streptozotocin were subsequently injected intrarenally with adenovirus expressing DDAH-1 (Ad-h-DDAH-1) or vector control [Ad-green fluorescent protein (GFP)], and mice were followed for 6 wk. Diabetes was associated with increased kidney ADMA and reduced kidney DDAH activity and DDAH-1 expression but had no effect on kidney DDAH-2 expression. Ad-GFP-treated diabetic mice showed significant increases in albuminuria, histological changes, glomerular macrophage recruitment, inflammatory cytokine and fibrotic markers, kidney ADMA levels, and urinary thiobarbituric acid reactive substances excretion as an indicator of oxidative stress, along with a significant reduction in kidney DDAH activity and kidney NOS3 mRNA compared with normal mice. In contrast, Ad-h-DDAH-1 treatment of diabetic mice reversed these effects. These data indicate, for the first time, that DDAH-1 mediates renal tissue protection in DN via the ADMA-NOS3-interaction. Enhanced renal DDAH-1 activity could be a novel therapeutic tool for treating patients with diabetes.
Keywords: asymmetric dimethylarginine, diabetic nephropathy, dimethylarginine dimethylaminohydrolase, nitric oxide
INTRODUCTION
Diabetes mellitus is a serious medical condition that leads to a variety of microvascular and macrovascular complications in addition to hyperglycemia. One of the most serious complications is diabetic nephropathy (DN) leading to end-stage renal disease (ESRD), affecting 20–40% of patients with type 1 and 2 diabetes (3, 14, 44, 48). Early histological changes in DN include glomerular hyperfiltration and hypertrophy, followed by glomerular basement membrane thickening, fibrosis, type I collagen deposition, macrophage infiltration, endothelial dysfunction, and mesangial matrix accumulation (5, 6, 9, 20, 70), leading to glomerular sclerosis and ESRD (5, 48). Current therapies, including blood pressure and glucose control and other lifestyle changes, have been only modestly successful in delaying the progression of renal failure in diabetes (2).
Endothelial dysfunction, characterized by reduced bioavailability of nitric oxide (NO) and increased oxidative stress, is a hallmark characteristic of diabetes (21) and DN (23). Under conditions of low arginine levels or hyperglycemia, endothelial NO synthase (eNOS; NOS3) is uncoupled, producing reactive oxygen species and oxidative stress in lieu of NO (16, 65). Low or absent NOS3 has been shown to exacerbate DN (63, 72). Thus, elucidating the basis for vascular dysfunction in DN is critical (11). Asymmetric dimethylarginine (ADMA) is a strong endogenous inhibitor of NO synthesis (19, 39, 58, 61). High levels of ADMA are observed in hypertension (24, 38), hypercholesterolemia (13, 34), diabetes (32), and chronic kidney disease (CKD) (61), and are a strong prognostic marker of cardiovascular events in patients with diabetes (1, 49, 54) and ESRD (61, 75). ADMA is highly elevated in adults with type 1 diabetes, leading to increased risk of ESRD and cardiac dysfunction (19). Clinical studies have shown that elevated ADMA is associated with increased mortality in patients with type 2 diabetes (74) and is an accurate indicator of albuminuria, impaired glomerular filtration rate, and hypertension (57). Elevated ADMA is also associated with insulin resistance in patients with type 1 diabetes (18), and vitamin E treatment decreased circulating ADMA levels while increasing NO levels (45). However, the precise mechanism underlying ADMA elevation in these diseases is not fully understood.
ADMA is metabolized by dimethylarginine dimethylaminohydrolase (DDAH) into dimethylamine and citrulline (28, 40, 59), which are not inhibitors of NOS. Two isoforms of DDAH (DDAH-1 and DDAH-2) are encoded by two different genes, with distinct tissue distribution (29, 41). Although both are expressed in the kidney, mainly in glomerular endothelial cells, macula densa, and tubular cells (55, 56), DDAH-1 is the critical isozyme to degrade ADMA (25, 42). Low DDAH-1 levels have been associated with CKD, high systolic blood pressure, and increased urinary and plasma ADMA levels (36, 46). Introduction of DDAH in animal models with high ADMA reduced tubular fibrosis and improved systolic blood pressure (36, 37, 73). DDAH-1-deficient mice had increased plasma and kidney ADMA levels, reduced urine and plasma NO levels, and decreased endothelial cell proliferation (25), an effect completely reversed by DDAH-1 overexpression (71). However, a direct effect of DDAH on diabetic kidney disease had not previously been determined.
In the present study, we showed that diabetes is associated with increased kidney ADMA and reduced kidney DDAH activity and DDAH-1 expression but has no effect on kidney DDAH-2 expression. Intrarenal DDAH-1 overexpression using an adenoviral vector significantly reduced kidney injury, as evidenced by reduced albuminuria, glomerular histology score, glomerular macrophage recruitment, inflammatory cytokine and fibrotic markers, kidney ADMA levels, and urinary thiobarbituric acid-reactive substances (TBARS) excretion, along with a significant increase in kidney DDAH activity and kidney NOS3 mRNA expression. Taken together, our data suggest that enhancing renal DDAH-1 activity might be a promising therapeutic modality for DN.
MATERIALS AND METHODS
Diabetic mouse model.
All animal experiments were performed in 6-wk-old male DBA/2J background mice (strain no. 000671, The Jackson Laboratory, Bar Harbor, ME). Diabetes was induced by intraperitoneal injection of 50 mg/kg streptozotocin (STZ) for 5 days as recommended by the Animal Models of Diabetes Complications Consortium as an optimal model of DN (15, 17). Adenovirus expressing DDAH-1 (Ad-h-DDAH-1; 1 × 109 infectious units) or control vector [adenovirus expressing green fluorescent protein (Ad-GFP)] were directly and slowly injected into the superficial cortex in six locations (20 µL each) bilaterally as previously described (30, 31). Adenovirus was injected only into mice that had been confirmed to be diabetic at 2 wk following the final STZ injection. After 6 wk, plasma and 24-h urine samples were collected, mice were euthanized, and kidneys were removed for further experiments. All animal experiments were approved by the University of Texas Health Science Center at San Antonio and Penn State Hershey Institutional Animal Care and Use Committees.
Adenovirus vectors.
Adenovirus vectors with expression of human DDAH-1 (Ad-h-DDAH-1; catalog no. ADV-206691) or green fluorescent protein (Ad-GFP; catalog no. 1060) driven by the cytomegalovirus promoter were obtained from Vector BioLabs (Malvern, PA).
Analytical methodology.
Urine albumin excretion was measured by ELISA using an Albuwell M kit (Exocell, Philadelphia, PA) as previously described (5, 6, 64). Urine creatinine was determined using a Creatinine Liquid Reagents Assay Kit (Diazyme Laboratories, Poway, CA). Urinary TBARS Assay was performed according to the instruction provided by Animal Models of Diabetes Complications Consortium as previously described (68, 69). Blood glucose was measured using Contour Next glucose meter (Ascensia Diabetes Care, Parsippany, NJ) as previously described (5, 6, 64). Systolic blood pressure was determined using CODA Non-Invasive Blood Pressure System (Kent Scientific, Torrington, CT) as previously described (5, 6, 64).
Western blot analysis.
As previously described (69), kidney lysates were prepared by homogenization in lysis buffer (0.1% Triton X-100) supplemented with protease inhibitor cocktail tablets (Roche, Cambridge, MA). The BCA protein assay (ThermoFisher, Waltham, MA) was used to determine protein concentration. Thirty micrograms of kidney lysates were separated on a NuPAGE 4~12% bis-Tris Gel (Invitrogen, Carlsbad, CA), and the separated proteins were transferred onto a polyvinylidene difluoride membrane (Invitrogen). After being blocked with 5% nonfat dry milk, membranes were incubated with DDAH-1 (Abcam, Cambridge, MA, 1:3,000 dilution), DDAH-2 (Abcam, 1:2,000 dilution), or GAPDH (Cell Signaling Technology, Danvers, MA, 1:1,000 dilution) primary antibody overnight at 4°C followed by incubation with anti-rabbit IgG-horseradish peroxidase secondary antibody (Cell Signaling Technology, 1:3,000 dilution) for 1 h at room temperature. Membranes were developed using enhanced chemiluminescence solutions (ThermoFisher Scientific) after a wash followed by exposure to X-ray film. Densitometry was performed using ImageJ (NIH; https://imagej.nih.gov/ij/index.html).
Immunohistochemistry.
Mouse kidney tissues were fixed in 10% formalin and embedded in paraffin, and 3-μm sections were cut. Immunohistochemistry was performed on paraffin-embedded sections with anti-mouse Mac-2 antibody (clone M3/38, Cedarlane, Burlington, NC) as previously described (64, 69). Forty glomeruli per mouse were examined with a ×40 objective in a blinded manner, and the number of macrophages in the section were divided by 40 glomeruli to obtain the average macrophage/glomerulus value for each mouse. Images were taken with an Olympus BX51 microscope and DP71 digital camera using Microsuite Basic 2.6 image software. Images were obtained with a ×100 (oil) objective with a total magnification of ×1,000.
Renal histopathology.
Kidneys were fixed in 4% paraformaldehyde and embedded in paraffin, and 5-μm sections were cut. Sections were stained with periodic acid-Schiff and examined at ×400 in a blinded manner, and scores were averaged. Semiquantitative scores (0–4+) were assigned based on the masked reading, as previously described (72). In brief, 12 sections/mouse were graded from 0 to 4+, where 0 represents no lesion and 1, 2, 3, and 4+ represent mesangial matrix expansion or sclerosis, involving ≤25, 25–50, 50–75, or >75% of the glomerular tuft area, respectively. Images were taken with an Olympus BX51 microscope and DP71 digital camera using Microsuite Basic 2.6 image software. Images were taken at ×40 objective magnification. For ×100 images, pictures were taken on a Nikon Eclipse E600 scope using a Nikon Digital Camera DYM1200.
RT-PCR.
RNA was extracted from kidneys using TRIzol (Invitrogen, Carlsbad, CA), and cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Real-time PCR was performed on a CFX384 real-time system (Bio-Rad, Hercules, CA) using Taqman primers for mouse NOS3 (Mm00435217_m1), IL-1β (Mm00434228_m1), fibronectin (Mm01256744_m1), arginase-2 (Mm00477592_m1), and 18S rRNA (Mm03928990_g1) (all from ThermoFisher). The reaction was performed using a protocol of 50°C for 2 min, 95°C for 10 min, and then 39 cycles of 95°C for 15 s and 60°C for 1 min. Threshold cycle (Ct) values were normalized to 18S rRNA, and ΔCt values were compared with normal samples.
DDAH activity assay.
DDAH activity was determined using the procedure described by Tain and Baylis (52). Briefly, kidneys were homogenized in lysis buffer containing 0.1% Triton X-100, incubated with urease (1:10 ratio), mixed with 1 mM ADMA, incubated at 37°C for 45 min, and treated with 4% sulfosalicylic acid before centrifugation at 3,000 g for 10 min. The supernatant (300 µl) was mixed with an equal amount of color mixture (1 part of oxime reagent:2 parts of antipyrine/H2SO4) and incubated for 110 min at 60°C in the dark. The reaction was stopped by cooling samples at −20°C for 10 min, and 200-µl samples were then loaded into a 96-well plate. Citrulline was measured at 466 nm.
ADMA quantification.
ADMA levels were determined using a Mouse Asymmetrical Dimethylarginine ELISA kit (MBS1601018, MyBiosource, San Diego, CA) using the manufacturer’s instructions. Briefly, 20–45 mg of kidney tissue were homogenized in 300 µL of 1× PBS and then measured for protein concentration using Pierce BCA protein assay kit (ThermoFisher). Lysate (50 µL) was loaded in duplicate into strips and incubated with 10 µL of anti-ADMA antibody and 50 µL of streptavidin-horseradish peroxidase for 60 min at 37°C. Wells were then washed and developed for 10 min, and optical density was read at 450 nm. Data are presented as nanomoles per grams of tissue used.
Statistical analysis.
Comparisons between groups were conducted using GraphPad Prism software (version 7.04, San Diego, CA). Results are expressed as means ± SE. An unpaired t test was used for comparison between two groups. One-way ANOVA using Fisher’s least significant difference test as a post hoc test was used to compare significance between more than two groups. P < 0.05 represented a significant difference.
RESULTS
Diabetes regulates kidney ADMA and DDAH-1 levels.
Diabetic mice had elevated kidney ADMA levels (Fig. 1A), reduced kidney DDAH activity (Fig. 1B), and reduced DDAH-1 expression (Fig. 1C), but kidney DDAH-2 expression was unaffected (Fig. 1D). Since only DDAH-1 was significantly reduced in DN, we decided to use an adenovirus vector to overexpress DDAH-1 specifically in the kidney in all subsequent experiments.
Fig. 1.

Diabetes increases kidney asymmetric dimethylarginine (ADMA) and reduces kidney dimethylarginine dimethylaminohydrolase (DDAH) activity and DDAH-1 expression. A: kidney ADMA levels were determined by ELISA. B: DDAH activity was determined as described in materials and methods. C and D: levels of DDAH-1 and DDAH-2 protein in normal and diabetic mouse kidneys were determined by Western blot analysis and normalized to GAPDH. Western blots are representative of 5 mice/group. Data are presented as means ± SE. *P < 0.05 compared with normal.
Kidney DDAH-1 delivery reduced albuminuria in diabetic mice.
Previous studies have routinely demonstrated that diabetic mice have elevated 24-h urine albumin excretion and the urine albumin-to-creatinine ratio compared with normal mice (67, 69). Similarly, Ad-GFP-treated diabetic mice showed a significantly higher urine albumin-to-creatinine ratio (Fig. 2) compared with normal mice after 6 wk of diabetes. Intrarenal Ad-h-DDAH-1-treated diabetic mice had a significantly reduced urine albumin-to-creatinine ratio compared with Ad-GFP-treated diabetic mice. Importantly, there were no significant differences between Ad-GFP-treated and Ad-h-DDAH-1-treated diabetic mice in blood glucose levels (499 ± 1 vs. 485 ± 7 mg/dL) or systolic blood pressure (118 ± 3 vs. 123 ± 4 mmHg), respectively.
Fig. 2.
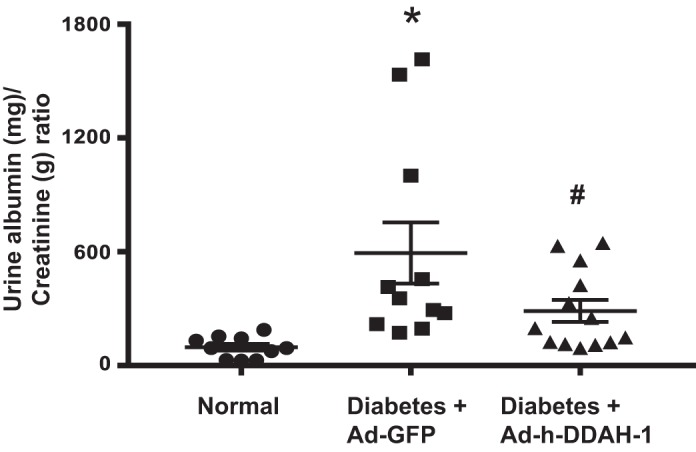
Administration of adenovirus expressing dimethylarginine dimethylaminohydrolase-1 (Ad-h-DDAH-1) reduces albuminuria in diabetic mice. Urine was collected from mice and analyzed for the urine albumin-to-creatinine ratio. Data are presented as means ± SE; n = 10–13 mice/group. *P < 0.005 compared with normal; #P < 0.05 compared with adenovirus expressing green fluorescent protein (Ad-GFP)-treated diabetic mice.
Kidney DDAH-1 delivery restored kidney DDAH-1 activity and ADMA levels in diabetic mice.
Since diabetes was associated with reduced kidney DDAH activity along with increased kidney ADMA levels (Fig. 1), we next determined whether intrarenal Ad-h-DDAH-1 delivery could restore these parameters. As shown in Fig. 3, Ad-GFP-treated diabetic mice showed significantly lower kidney DDAH activity (Fig. 3A) and increased kidney ADMA levels (Fig. 3B) compared with normal mice after 6 wk of diabetes. Intrarenal Ad-h-DDAH-1-treated diabetic mice restored kidney DDAH activity and kidney ADMA levels to their normal levels.
Fig. 3.
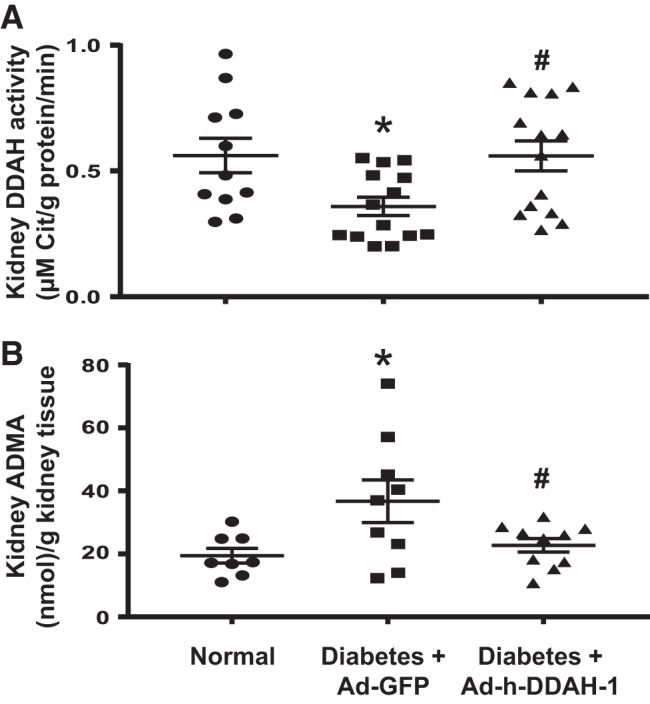
Administration of adenovirus expressing dimethylarginine dimethylaminohydrolase-1 (Ad-h-DDAH-1) improves mouse kidney DDAH activity and reduces renal asymmetric dimethylarginine (ADMA) in diabetic mice. A: kidneys were analyzed for DDAH activity as described in materials and methods. B: ADMA levels were measured in mouse kidney samples by ELISA. Data are presented as means ± SE; n = 8–14 mice/group. *P < 0.05 compared with normal; #P < 0.05 compared with adenovirus expressing green fluorescent protein (Ad-GFP)-treated diabetic mice.
Kidney DDAH-1 delivery restored kidney NOS3 mRNA expression and urinary TBARS levels in diabetic mice.
Since ADMA is a strong endogenous inhibitor of NO synthesis (19, 39, 58, 61) and plays a role in NOS uncoupling in endothelial cells, resulting in the production of reactive oxygen species and oxidative stress in lieu of NO (4, 50), we examined the effect of intrarenal Ad-h-DDAH-1 delivery on NOS3 expression and oxidative stress as measured by TBARS. Whereas kidney NOS3 mRNA expression was reduced (Fig. 4A) and levels of urinary TBARS greatly increased (Fig. 4B) in Ad-GFP-treated diabetic mice compared with normal mice, kidney NOS3 mRNA expression and urinary TBARS excretion were restored to normal levels in Ad-h-DDAH-1-treated diabetic mice.
Fig. 4.
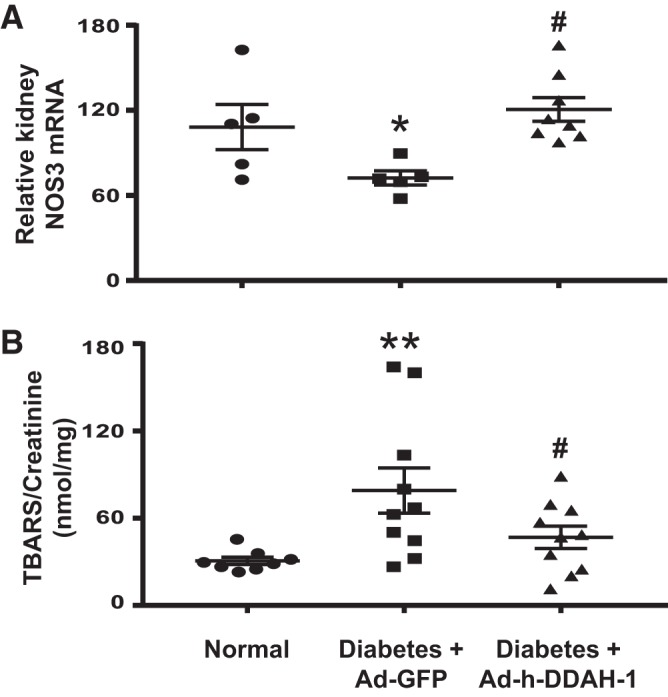
Administration of adenovirus expressing dimethylarginine dimethylaminohydrolase-1 (Ad-h-DDAH-1) increases kidney endothelial nitric oxide synthase (NOS3) mRNA and reduces urinary thiobarbituric acid-reactive substances (TBARS) in diabetic mice. A: total RNA was isolated from mouse kidneys, and RT-PCR was performed to determine NOS3 mRNA levels normalized to 18S rRNA. B: mouse 24-h urine was collected for the measurement of TBARS. Data are presented as means ± SE; n = 5–10 mice/group. *P < 0.05 and **P < 0.001 compared with normal; #P < 0.005 compared with adenovirus expressing green fluorescent protein (Ad-GFP)-treated diabetic mice.
Kidney DDAH-1 delivery decreased renal histological score in diabetic mice.
Periodic acid-Schiff staining showed significantly increased glomerular cellularity and mesangial expansion after 6 wk of diabetes in Ad-GFP-treated diabetic mice compared with normal mice (Fig. 5). In contrast, Ad-h-DDAH-1-treated diabetic mice had significantly reduced glomerular scores compared with Ad-GFP-treated diabetic mice (Fig. 5).
Fig. 5.

Administration of adenovirus expressing dimethylarginine dimethylaminohydrolase-1 (Ad-h-DDAH-1) reduces kidney pathology in diabetic mice. Paraffin-embedded mouse kidney sections were subjected to periodic acid-Schiff staining. Scale bar = 50 µm (×40) and 20 µm (×100). Data are presented as means ± SE; n = 8–11 mice/group. *P < 0.005 compared with normal; #P < 0.005 compared with adenovirus expressing green fluorescent protein (Ad-GFP)-treated diabetic mice.
Kidney DDAH-1 delivery decreased glomerular macrophage infiltration in diabetic mice.
To determine whether kidney DDAH is critical for kidney macrophage infiltration in DN, we examined the distribution and number of Mac-2-positive macrophages in the glomeruli by immunohistochemistry (Fig. 6). The number of glomerular macrophages in normal mice was low but increased significantly in Ad-GFP-treated diabetic mice after 6 wk of diabetes (Fig. 6). Intrarenal Ad-h-DDAH-1-treated diabetic mice had significantly reduced glomerular macrophage recruitment compared with Ad-GFP-treated diabetic mice.
Fig. 6.

Administration of adenovirus expressing dimethylarginine dimethylaminohydrolase-1 (Ad-h-DDAH-1) reduces glomerular macrophage infiltration in diabetic mice. A: glomerular macrophage recruitment was visualized by immunohistochemical staining using Mac-2 antibody, and pictures were taken at ×100 magnification. Red arrows indicate macrophages. Scale bar = 20 µm. B: macrophage numbers per glomerulus were counted in a blinded manner. The number of glomerular macrophages was counted in 40 glomeruli/section (number of macrophages in glomeruli divided by the number of glomeruli) under ×40 magnification and averaged. Data are presented as means ± SE; n = 8–12 mice/group. *P < 0.001 compared with normal; #P < 0.005 compared with adenovirus expressing green fluorescent protein (Ad-GFP)-treated diabetic mice.
Kidney DDAH-1 delivery decreased inflammatory cytokines and fibrotic markers in diabetic mice.
Increased inflammatory cytokines and fibrotic markers are major features of and important predictors of DN (6, 9, 67). Therefore, we further assessed the anti-inflammatory and antifibrotic effects of intrarenal DDAH-1 delivery in diabetic mice (Fig. 7). Kidney arginase-2, fibronectin, and IL-1β mRNA expression was significantly increased in Ad-GFP-treated diabetic mice compared with normal mice. Intrarenal Ad-h-DDAH-1-treated diabetic mice resulted in significantly reduced kidney arginase-2, fibronectin, and IL-1β mRNA expression after 6 wk of diabetes.
Fig. 7.

Administration of adenovirus expressing dimethylarginine dimethylaminohydrolase-1 (Ad-h-DDAH-1) reduces inflammatory cytokines and fibrotic markers in diabetic mice. RT-PCR was performed on RNA extracted from kidney and analyzed for the indicated genes using indicated Taqman primers. Threshold cycle (Ct) values were normalized to 18S rRNA values and then compared with normal mice. Data are presented as means ± SE; n = 6–7 mice/group. *P < 0.01 and **P < 0.001 compared with normal; #P < 0.05 compared with adenovirus expressing green fluorescent protein (Ad-GFP)-treated diabetic mice.
DISCUSSION
DDAH-1 plays a pivotal role to maintain endothelial function under physiological and pathological conditions, yet its role in diabetic kidney injury had not previously been determined. This study shows that intrarenal DDAH-1 delivery mediates renal tissue protection as proven by a reduction in albuminuria, histopathological changes, kidney macrophage recruitment, and inflammatory cytokine and fibrotic markers during diabetes. The renal tissue-protective effect of DDAH-1 is likely mediated by a reduction of ADMA and restoration of kidney NOS3 expression and activity, thus reducing oxidative stress. These findings reveal an important role for DDAH-1 in the pathogenesis of DN and provide evidence for enhancing renal DDAH-1 activity as a potential therapeutic modality for treating patients with diabetes.
Although both DDAH-1 and DDAH-2 are expressed in the kidney (29), albeit in different renal cell types, we show that expression DDAH-1, but not of DDAH-2, is altered in diabetes. Zhu et al. (73) showed that treatment of diabetic rats with DDAH-2 improved myocardial function and lower blood glucose levels. Unlike DDAH-2, DDAH-1 exerts its effects mainly in the kidneys (41, 51). DDAH-1 is a critical enzyme to degrade ADMA (25, 42). Low DDAH-1 levels have been associated with CKD and hypertension and increased cardiovascular risk (36, 46, 60). DDAH-1-deficient mice had increased ADMA levels, reduced NO production, and decreased cell proliferation; an effect completely reversed by DDAH-1 overexpression (71). Although previous reports have shown that diabetes is associated with reduced renal DDAH activity and increased renal and urinary ADMA (27, 33, 47), a direct effect of DDAH on diabetic kidney disease remained unknown.
DDAH-1 knockdown reduced proximal tubule fluid reabsorption and induced adverse kidney pathology without affecting systolic blood pressure (12). Our results show that increased renal expression of DDAH-1 did not affect systolic blood pressure in diabetic mice despite restoring ADMA levels and renal NOS3 mRNA expression.
DDAH-1 converts ADMA to dimethylamine and citrulline, and increased levels of ADMA are widely used as an indicator of diabetes complications (36, 46). Polymorphisms in both DDAH isoforms have been linked with type 1 and 2 diabetes as well as elevated serum ADMA (22, 35) but not albuminuria. Only one DDAH polymorphism (single-nucleotide polymorphism rs805304) is correlated with increased risk of glomerular filtration rate decline (53). Our data indicate that DDAH-1 activity is critical for regulating ADMA levels in diabetes.
Although the role of NO in DN is well established, the expression and regulation of NO and NOS in DN is controversial (26). We have previously shown that the production of NO is diminished in diabetes and restored to normal levels by insulin (8). Lai et al. (27) showed an association between elevated ADMA and the reduction in NO and DDAH activity in diabetic rats. ADMA, equipotent with NG-monomethyl-l-arginine, inhibits NOS3, which becomes uncoupled and generates superoxide in lieu of NO (43). Our data show increased NOS3 expression as a result of increased kidney DDAH-1, which could explain the protective effect of DDAH-1 in DN via reducing ADMA levels. These results are in agreement with a previous report showing that ADMA infusion resulted in a reduction of NOS3 expression (mRNA, protein, and phospho-eNOS) and NO levels, along with increased superoxide production, in the porcine coronary artery (66). The mechanism by which DDAH-1 overexpression increases NOS3 mRNA expression is not known. However, as DDAH-1 overexpression likely promotes ADMA conversion to citrulline and dimethylamine, decreases in ADMA levels result in reduced inhibition of NOS3 activity and expression, NOS3 uncoupling, and generation of superoxide in lieu of NO (43). Furthermore, reduced ADMA levels may promote NO synthesis because ADMA can competitively inhibit cellular uptake of arginine, the nitrogenous substrate for NOS3. Our study is only focused on ADMA/NOS3 interaction and therefore is limited for not examining other NOS isoforms and measuring NOS protein expression, NOS uncoupling, or activity, which also could play a role in the protective effect of DDAH-1 overexpression in DN. Additional studies are needed to elucidate the role of ADMA in regulating NOS3 expression.
DDAH-1 resulted in less mesangial expansion and glomerular hypercellularity in diabetes, indicating a possible contribution of DDAH-1 deficiency to the initiation and/or progression of diabetic renal fibrosis. This is also confirmed by reduced fibronectin in DDAH-1-overexpressing mice in our study. The renal protective effect of DDAH-1 also correlates with a significant reduction of kidney macrophage infiltration and inflammatory cytokines. Whether the reduction in macrophage recruitment is mediated directly by DDAH-1 or indirectly by reducing diabetic renal injury is not clear at this time. Additional studies are required to elucidate the role of DDAH-1 in kidney macrophage infiltration. However, we previously showed that arginase-2 mediates renal tissue injury and pharmacological blockade or genetic deficiency of arginase-2 protects both the development and progression of DN via a NOS3-dependent pathway (39a, 67a, 68). Whether DDAH-1 and arginase-2 interact directly or indirectly is not clear at this time.
In conclusion, we have shown that intrarenal adenoviral delivery of DDAH-1 reduces DN by preventing the increase in ADMA and restoring expression of NOS3. Thus, strategies to enhance renal DDAH-1 activity may represent promising therapeutic modalities for treatment of DN.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-094930 and DK-094930S1.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.M.M. and A.S.A. conceived and designed research; M.D.W., T.G., and K.S. performed experiments; M.D.W., K.S., T.K.C., and A.S.A. analyzed data; M.D.W., S.M.M., and A.S.A. interpreted results of experiments; M.D.W. and A.S.A. prepared figures; M.D.W. and A.S.A. drafted manuscript; M.D.W., S.M.M., and A.S.A. edited and revised manuscript; M.D.W., T.G., K.S., T.K.C., S.M.M., and A.S.A. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank R. Padmanabhan for technical assistance.
REFERENCES
- 1.Abbasi F, Asagmi T, Cooke JP, Lamendola C, McLaughlin T, Reaven GM, Stuehlinger M, Tsao PS. Plasma concentrations of asymmetric dimethylarginine are increased in patients with type 2 diabetes mellitus. Am J Cardiol 88: 1201–1203, 2001. doi: 10.1016/S0002-9149(01)02063-X. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Rahman EM, Saadulla L, Reeves WB, Awad AS. Therapeutic modalities in diabetic nephropathy: standard and emerging approaches. J Gen Intern Med 27: 458–468, 2012. doi: 10.1007/s11606-011-1912-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alhaider AA, Korashy HM, Sayed-Ahmed MM, Mobark M, Kfoury H, Mansour MA. Metformin attenuates streptozotocin-induced diabetic nephropathy in rats through modulation of oxidative stress genes expression. Chem Biol Interact 192: 233–242, 2011. doi: 10.1016/j.cbi.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Antoniades C, Shirodaria C, Leeson P, Antonopoulos A, Warrick N, Van-Assche T, Cunnington C, Tousoulis D, Pillai R, Ratnatunga C, Stefanadis C, Channon KM. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J 30: 1142–1150, 2009. doi: 10.1093/eurheartj/ehp061. [DOI] [PubMed] [Google Scholar]
- 5.Awad AS, Gao T, Gvritishvili A, You H, Liu Y, Cooper TK, Reeves WB, Tombran-Tink J. Protective role of small pigment epithelium-derived factor (PEDF) peptide in diabetic renal injury. Am J Physiol Renal Physiol 305: F891–F900, 2013. doi: 10.1152/ajprenal.00149.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awad AS, Kinsey GR, Khutsishvili K, Gao T, Bolton WK, Okusa MD. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am J Physiol Renal Physiol 301: F1358–F1366, 2011. doi: 10.1152/ajprenal.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Awad AS, Webb RL, Carey RM, Siragy HM. Renal nitric oxide production is decreased in diabetic rats and improved by AT1 receptor blockade. J Hypertens 22: 1571–1577, 2004. doi: 10.1097/01.hjh.0000133718.86451.6a. [DOI] [PubMed] [Google Scholar]
- 9.Awad AS, You H, Gao T, Cooper TK, Nedospasov SA, Vacher J, Wilkinson PF, Farrell FX, Brian Reeves W. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int 88: 722–733, 2015. doi: 10.1038/ki.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balakumar P, Chakkarwar VA, Krishan P, Singh M. Vascular endothelial dysfunction: a tug of war in diabetic nephropathy? Biomed Pharmacother 63: 171–179, 2009. doi: 10.1016/j.biopha.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 12.Bell T, Araujo M, Luo Z, Tomlinson J, Leiper J, Welch WJ, Wilcox CS. Regulation of fluid reabsorption in rat or mouse proximal renal tubules by asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase 1. Am J Physiol Renal Physiol 315: F74–F78, 2018. doi: 10.1152/ajprenal.00560.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Böger RH, Bode-Böger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, Cooke JP. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 98: 1842–1847, 1998. doi: 10.1161/01.CIR.98.18.1842. [DOI] [PubMed] [Google Scholar]
- 14.Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 8: 29, 2010. doi: 10.1186/1478-7954-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breyer MD, Böttinger E, Brosius FC 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K; AMDCC . Mouse models of diabetic nephropathy. J Am Soc Nephrol 16: 27–45, 2005. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 16.Brodsky SV, Gao S, Li H, Goligorsky MS. Hyperglycemic switch from mitochondrial nitric oxide to superoxide production in endothelial cells. Am J Physiol Heart Circ Physiol 283: H2130–H2139, 2002. doi: 10.1152/ajpheart.00196.2002. [DOI] [PubMed] [Google Scholar]
- 17.Brosius FC 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T; Animal Models of Diabetic Complications Consortium . Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caglar K, Yilmaz MI, Sonmez A, Cakir E, Kaya A, Acikel C, Eyileten T, Yenicesu M, Oguz Y, Bilgi C, Oktenli C, Vural A, Zoccali C. ADMA, proteinuria, and insulin resistance in non-diabetic stage I chronic kidney disease. Kidney Int 70: 781–787, 2006. doi: 10.1038/sj.ki.5001632. [DOI] [PubMed] [Google Scholar]
- 19.Carmann C, Lilienthal E, Weigt-Usinger K, Schmidt-Choudhury A, Hörster I, Kayacelebi AA, Beckmann B, Chobanyan-Jürgens K, Tsikas D, Lücke T. The L-arginine/NO pathway, homoarginine, and nitrite-dependent renal carbonic anhydrase activity in young people with type 1 diabetes mellitus. Amino Acids 47: 1865–1874, 2015. doi: 10.1007/s00726-015-2027-9. [DOI] [PubMed] [Google Scholar]
- 20.Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int 75: 1145–1152, 2009. doi: 10.1038/ki.2009.86. [DOI] [PubMed] [Google Scholar]
- 21.Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 108: 1527–1532, 2003. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 22.Fogarty RD, Abhary S, Javadiyan S, Kasmeridis N, Petrovsky N, Whiting MJ, Craig JE, Burdon KP. Relationship between DDAH gene variants and serum ADMA level in individuals with type 1 diabetes. J Diabetes Complications 26: 195–198, 2012. doi: 10.1016/j.jdiacomp.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 23.Goligorsky MS, Chen J, Brodsky S. Workshop: endothelial cell dysfunction leading to diabetic nephropathy: focus on nitric oxide. Hypertension 37: 744–748, 2001. doi: 10.1161/01.HYP.37.2.744. [DOI] [PubMed] [Google Scholar]
- 24.Goonasekera CD, Shah V, Rees DD, Dillon MJ. Vascular endothelial cell activation associated with increased plasma asymmetric dimethyl arginine in children and young adults with hypertension: a basis for atheroma? Blood Press 9: 16–21, 2000. doi: 10.1080/080370500439371. [DOI] [PubMed] [Google Scholar]
- 25.Hu X, Atzler D, Xu X, Zhang P, Guo H, Lu Z, Fassett J, Schwedhelm E, Böger RH, Bache RJ, Chen Y. Dimethylarginine dimethylaminohydrolase-1 is the critical enzyme for degrading the cardiovascular risk factor asymmetrical dimethylarginine. Arterioscler Thromb Vasc Biol 31: 1540–1546, 2011. doi: 10.1161/ATVBAHA.110.222638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komers R, Anderson S. Paradoxes of nitric oxide in the diabetic kidney. Am J Physiol Renal Physiol 284: F1121–F1137, 2003. doi: 10.1152/ajprenal.00265.2002. [DOI] [PubMed] [Google Scholar]
- 27.Lai YL, Aoyama S, Ohata M, Otsuka N, Shiokawa H, Tomono S, Fujiwara Y, Kanazawa H, Miyoshi N, Ohshima H. Dysregulation of dimethylargininedimethylaminohydrolase/asymmetric dimethylarginine pathway in rat type II diabetic nephropathy. J Clin Biochem Nutr 51: 143–149, 2012. doi: 10.3164/jcbn.11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leiper J, Vallance P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc Res 43: 542–548, 1999. doi: 10.1016/S0008-6363(99)00162-5. [DOI] [PubMed] [Google Scholar]
- 29.Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, Whitley GS, Vallance P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem J 343: 209–214, 1999. doi: 10.1042/bj3430209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li XC, Cook JL, Rubera I, Tauc M, Zhang F, Zhuo JL. Intrarenal transfer of an intracellular fluorescent fusion of angiotensin II selectively in proximal tubules increases blood pressure in rats and mice. Am J Physiol Renal Physiol 300: F1076–F1088, 2011. doi: 10.1152/ajprenal.00329.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li XC, Zhuo JL. Proximal tubule-dominant transfer of AT(1a) receptors induces blood pressure responses to intracellular angiotensin II in AT(1a) receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol 304: R588–R598, 2013. doi: 10.1152/ajpregu.00338.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, Kimoto M, Tsuji H, Reaven GM, Cooke JP. Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation 106: 987–992, 2002. doi: 10.1161/01.CIR.0000027109.14149.67. [DOI] [PubMed] [Google Scholar]
- 33.Liu X, Xu X, Shang R, Chen Y. Asymmetric dimethylarginine (ADMA) as an important risk factor for the increased cardiovascular diseases and heart failure in chronic kidney disease. Nitric Oxide 78: 113–120, 2018. doi: 10.1016/j.niox.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundman P, Eriksson MJ, Stühlinger M, Cooke JP, Hamsten A, Tornvall P. Mild-to-moderate hypertriglyceridemia in young men is associated with endothelial dysfunction and increased plasma concentrations of asymmetric dimethylarginine. J Am Coll Cardiol 38: 111–116, 2001. doi: 10.1016/S0735-1097(01)01318-3. [DOI] [PubMed] [Google Scholar]
- 35.Marra M, Marchegiani F, Ceriello A, Sirolla C, Boemi M, Franceschi C, Spazzafumo L, Testa I, Bonfigli AR, Cucchi M, Testa R. Chronic renal impairment and DDAH2-1151 A/C polymorphism determine ADMA levels in type 2 diabetic subjects. Nephrol Dial Transplant 28: 964–971, 2013. doi: 10.1093/ndt/gfs516. [DOI] [PubMed] [Google Scholar]
- 36.Matsuguma K, Ueda S, Yamagishi S, Matsumoto Y, Kaneyuki U, Shibata R, Fujimura T, Matsuoka H, Kimoto M, Kato S, Imaizumi T, Okuda S. Molecular mechanism for elevation of asymmetric dimethylarginine and its role for hypertension in chronic kidney disease. J Am Soc Nephrol 17: 2176–2183, 2006. doi: 10.1681/ASN.2005121379. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto Y, Ueda S, Yamagishi S, Matsuguma K, Shibata R, Fukami K, Matsuoka H, Imaizumi T, Okuda S. Dimethylarginine dimethylaminohydrolase prevents progression of renal dysfunction by inhibiting loss of peritubular capillaries and tubulointerstitial fibrosis in a rat model of chronic kidney disease. J Am Soc Nephrol 18: 1525–1533, 2007. doi: 10.1681/ASN.2006070696. [DOI] [PubMed] [Google Scholar]
- 38.Matsuoka H, Itoh S, Kimoto M, Kohno K, Tamai O, Wada Y, Yasukawa H, Iwami G, Okuda S, Imaizumi T. Asymmetrical dimethylarginine, an endogenous nitric oxide synthase inhibitor, in experimental hypertension. Hypertension 29: 242–247, 1997. doi: 10.1161/01.HYP.29.1.242. [DOI] [PubMed] [Google Scholar]
- 39.Morris SM., Jr Arginine metabolism revisited. J Nutr 146: 2579S–2586S, 2016. doi: 10.3945/jn.115.226621. [DOI] [PubMed] [Google Scholar]
- 39a.Morris SM Jr, Gao T, Cooper TK, Kepka-Lenhart D, Awad AS. Arginase-2 mediates diabetic renal injury. Diabetes 60: 3015–3022, 2011. doi: 10.2337/db11-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogawa T, Kimoto M, Sasaoka K. Purification and properties of a new enzyme, NG,NG-dimethylarginine dimethylaminohydrolase, from rat kidney. J Biol Chem 264: 10205–10209, 1989. [PubMed] [Google Scholar]
- 41.Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol 293: H3227–H3245, 2007. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- 42.Pope AJ, Karrupiah K, Kearns PN, Xia Y, Cardounel AJ. Role of dimethylarginine dimethylaminohydrolases in the regulation of endothelial nitric oxide production. J Biol Chem 284: 35338–35347, 2009. doi: 10.1074/jbc.M109.037036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pou S, Keaton L, Surichamorn W, Rosen GM. Mechanism of superoxide generation by neuronal nitric-oxide synthase. J Biol Chem 274: 9573–9580, 1999. doi: 10.1074/jbc.274.14.9573. [DOI] [PubMed] [Google Scholar]
- 44.Raptis AE, Viberti G. Pathogenesis of diabetic nephropathy. Exp Clin Endocrinol Diabetes 109, Suppl 2: S424–S437, 2001. doi: 10.1055/s-2001-18600. [DOI] [PubMed] [Google Scholar]
- 45.Saran R, Novak JE, Desai A, Abdulhayoglu E, Warren JS, Bustami R, Handelman GJ, Barbato D, Weitzel W, D’Alecy LG, Rajagopalan S. Impact of vitamin E on plasma asymmetric dimethylarginine (ADMA) in chronic kidney disease (CKD): a pilot study. Nephrol Dial Transplant 18: 2415–2420, 2003. doi: 10.1093/ndt/gfg406. [DOI] [PubMed] [Google Scholar]
- 46.Shi L, Zhao C, Wang H, Lei T, Liu S, Cao J, Lu Z. Dimethylarginine dimethylaminohydrolase 1 deficiency induces the epithelial to mesenchymal transition in renal proximal tubular epithelial cells and exacerbates kidney damage in aged and diabetic mice. Antioxid Redox Signal 27: 1347–1360, 2017. doi: 10.1089/ars.2017.7022. [DOI] [PubMed] [Google Scholar]
- 47.Shibata R, Ueda S, Yamagishi S, Kaida Y, Matsumoto Y, Fukami K, Hayashida A, Matsuoka H, Kato S, Kimoto M, Okuda S. Involvement of asymmetric dimethylarginine (ADMA) in tubulointerstitial ischaemia in the early phase of diabetic nephropathy. Nephrol Dial Transplant 24: 1162–1169, 2009. doi: 10.1093/ndt/gfn630. [DOI] [PubMed] [Google Scholar]
- 48.Shumway JT, Gambert SR. Diabetic nephropathy-pathophysiology and management. Int Urol Nephrol 34: 257–264, 2002. doi: 10.1023/A:1023244829975. [DOI] [PubMed] [Google Scholar]
- 49.Stühlinger MC, Abbasi F, Chu JW, Lamendola C, McLaughlin TL, Cooke JP, Reaven GM, Tsao PS. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA 287: 1420–1426, 2002. doi: 10.1001/jama.287.11.1420. [DOI] [PubMed] [Google Scholar]
- 50.Sud N, Wells SM, Sharma S, Wiseman DA, Wilham J, Black SM. Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterial endothelial cells: role of mitochondrial dysfunction. Am J Physiol Cell Physiol 294: C1407–C1418, 2008. doi: 10.1152/ajpcell.00384.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sydow K, Schmitz C, von Leitner EC, von Leitner R, Klinke A, Atzler D, Krebs C, Wieboldt H, Ehmke H, Schwedhelm E, Meinertz T, Blankenberg S, Böger RH, Magnus T, Baldus S, Wenzel U. Dimethylarginine dimethylaminohydrolase1 is an organ-specific mediator of end organ damage in a murine model of hypertension. PLoS One 7: e48150, 2012. doi: 10.1371/journal.pone.0048150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tain YL, Baylis C. Determination of dimethylarginine dimethylaminohydrolase activity in the kidney. Kidney Int 72: 886–889, 2007. doi: 10.1038/sj.ki.5002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanhäuserová V, Tomandl J, Pácal L, Klepárník M, Malúšková D, Bartáková V, Kuricová K, Rehořová J, Stěpánková S, Svojanovský J, Olšovský J, Bělobrádková J, Krusová D, Jurajda M, Mužík J, Pavlík T, Kaňková K. ADMA, SDMA and L-arginine/ADMA ratio but not DDAH genetic polymorphisms are reliable predictors of diabetic nephropathy progression as identified by competing risk analysis. Kidney Blood Press Res 36: 200–208, 2012. doi: 10.1159/000343409. [DOI] [PubMed] [Google Scholar]
- 54.Tarnow L, Hovind P, Teerlink T, Stehouwer CD, Parving HH. Elevated plasma asymmetric dimethylarginine as a marker of cardiovascular morbidity in early diabetic nephropathy in type 1 diabetes. Diabetes Care 27: 765–769, 2004. doi: 10.2337/diacare.27.3.765. [DOI] [PubMed] [Google Scholar]
- 55.Tojo A, Kimoto M, Wilcox CS. Renal expression of constitutive NOS and DDAH: separate effects of salt intake and angiotensin. Kidney Int 58: 2075–2083, 2000. doi: 10.1111/j.1523-1755.2000.00380.x. [DOI] [PubMed] [Google Scholar]
- 56.Tojo A, Welch WJ, Bremer V, Kimoto M, Kimura K, Omata M, Ogawa T, Vallance P, Wilcox CS. Colocalization of demethylating enzymes and NOS and functional effects of methylarginines in rat kidney. Kidney Int 52: 1593–1601, 1997. doi: 10.1038/ki.1997.490. [DOI] [PubMed] [Google Scholar]
- 57.Triches CB, Quinto M, Mayer S, Batista M, Zanella MT. Relation of asymmetrical dimethylarginine levels with renal outcomes in hypertensive patients with and without type 2 diabetes mellitus. J Diabetes Complications 32: 316–320, 2018. doi: 10.1016/j.jdiacomp.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 58.Tsikas D, Bollenbach A, Hanff E, Kayacelebi AA. Asymmetric dimethylarginine (ADMA), symmetric dimethylarginine (SDMA) and homoarginine (hArg): the ADMA, SDMA and hArg paradoxes. Cardiovasc Diabetol 17: 1, 2018. doi: 10.1186/s12933-017-0656-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ueda S, Kato S, Matsuoka H, Kimoto M, Okuda S, Morimatsu M, Imaizumi T. Regulation of cytokine-induced nitric oxide synthesis by asymmetric dimethylarginine: role of dimethylarginine dimethylaminohydrolase. Circ Res 92: 226–233, 2003. doi: 10.1161/01.RES.0000052990.68216.EF. [DOI] [PubMed] [Google Scholar]
- 60.Valkonen VP, Tuomainen TP, Laaksonen R. DDAH gene and cardiovascular risk. Vasc Med 10, Suppl 1: S45–S48, 2005. doi: 10.1177/1358836X0501000107. [DOI] [PubMed] [Google Scholar]
- 61.Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 339: 572–575, 1992. doi: 10.1016/0140-6736(92)90865-Z. [DOI] [PubMed] [Google Scholar]
- 63.Wang CH, Li F, Hiller S, Kim HS, Maeda N, Smithies O, Takahashi N. A modest decrease in endothelial NOS in mice comparable to that associated with human NOS3 variants exacerbates diabetic nephropathy. Proc Natl Acad Sci USA 108: 2070–2075, 2011. doi: 10.1073/pnas.1018766108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wetzel MD, Gao T, Venkatachalam M, Morris SM Jr, Awad AS. l-Homoarginine supplementation prevents diabetic kidney damage. Physiol Rep 7: e14235, 2019. doi: 10.14814/phy2.14235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci USA 93: 6770–6774, 1996. doi: 10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xuan C, Lun LM, Zhao JX, Wang HW, Wang J, Ning CP, Liu Z, Zhang BB, He GW. L-citrulline for protection of endothelial function from ADMA-induced injury in porcine coronary artery. Sci Rep 5: 10987, 2015. doi: 10.1038/srep10987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.You H, Gao T, Cooper TK, Reeves WB, Awad AS. Macrophages directly mediate diabetic renal injury. Am J Physiol Renal Physiol 305: F1719–F1727, 2013. doi: 10.1152/ajprenal.00141.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67a.You H, Gao T, Cooper TK, Morris SM Jr, Awad AS. Arginase inhibition: a new treatment for preventing progression of established diabetic nephropathy. Am J Physiol Renal Physiol 309: F447–F455, 2015. doi: 10.1152/ajprenal.00137.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.You H, Gao T, Cooper TK, Morris SM Jr, Awad AS. Arginase inhibition mediates renal tissue protection in diabetic nephropathy by a nitric oxide synthase 3-dependent mechanism. Kidney Int 84: 1189–1197, 2013. doi: 10.1038/ki.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.You H, Gao T, Cooper TK, Morris SM Jr, Awad AS. Diabetic nephropathy is resistant to oral l-arginine or l-citrulline supplementation. Am J Physiol Renal Physiol 307: F1292–F1301, 2014. doi: 10.1152/ajprenal.00176.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.You H, Gao T, Raup-Konsavage WM, Cooper TK, Bronson SK, Reeves WB, Awad AS. Podocyte-specific chemokine (C-C motif) receptor 2 overexpression mediates diabetic renal injury in mice. Kidney Int 91: 671–682, 2017. doi: 10.1016/j.kint.2016.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang P, Xu X, Hu X, Wang H, Fassett J, Huo Y, Chen Y, Bache RJ. DDAH1 deficiency attenuates endothelial cell cycle progression and angiogenesis. PLoS One 8: e79444, 2013. doi: 10.1371/journal.pone.0079444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, Breyer MD, Harris RC. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol 17: 2664–2669, 2006. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu ZD, Ye JM, Fu XM, Wang XC, Ye JY, Wu XR, Hua P, Liao YQ, Xuan W, Duan JL, Li WY, Fu H, Xia ZH, Zhang X. DDAH2 alleviates myocardial fibrosis in diabetic cardiomyopathy through activation of the DDAH/ADMA/NOS/NO pathway in rats. Int J Mol Med 43: 749–760, 2019. doi: 10.3892/ijmm.2018.4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zobel EH, von Scholten BJ, Reinhard H, Persson F, Teerlink T, Hansen TW, Parving HH, Jacobsen PK, Rossing P. Symmetric and asymmetric dimethylarginine as risk markers of cardiovascular disease, all-cause mortality and deterioration in kidney function in persons with type 2 diabetes and microalbuminuria. Cardiovasc Diabetol 16: 88, 2017. doi: 10.1186/s12933-017-0569-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zoccali C, Bode-Böger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, Cataliotti A, Bellanuova I, Fermo I, Frölich J, Böger R. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet 358: 2113–2117, 2001. doi: 10.1016/S0140-6736(01)07217-8. [DOI] [PubMed] [Google Scholar]