

Abstract
Carbamoylphosphate synthetase 1 (CPS1) deficiency is a rare inborn error of metabolism leading often to neonatal onset hyperammonemia with coma and high mortality. The biochemical features of the disease are nonspecific and cannot distinguish this condition from other defects of the urea cycle, namely N‐acetylglutamate synthase deficiency. Therefore, molecular genetic investigation is required for confirmation of the disease, and nowadays this is done with increasing frequency applying next‐generation sequencing (NGS) techniques. Our laboratory has a long‐standing interest in CPS1 molecular genetic investigation and receives samples from centers in Europe and many other countries. We perform RNA‐based CPS1 molecular genetic investigation as first line investigation and wanted in this study to evaluate our experience with this approach as compared to NGS. In the past 15 years, 297 samples were analyzed, which were referred from 37 countries. CPS1 deficiency could be confirmed in 155 patients carrying 136 different genotypes with only a single mutation recurring more than two times. About 10% of the total 172 variants comprised complex changes (eg, intronic changes possibly affecting splicing, deletions, insertions, or deletions_insertions), which would have been partly missed if only NGS was done. Likewise, RNA analysis was crucial for correct interpretation of at least half of the complex mutations. This study gives highest sensitivity to RNA‐based CPS1 molecular genetic investigation and underlines that NGS should be done together with copy number variation analysis. We propose that unclear cases should be investigated by RNA sequencing in addition, if this method is not used as the initial diagnostic procedure.
Keywords: carbamoylphosphate synthetase 1, CPS1, next‐generation sequencing, RNA analysis, urea cycle defects
1. INTRODUCTION
Carbamoylphosphate synthetase 1 (CPS1, E.C. 6.3.4.16) catalyzes, as first and rate‐limiting reaction of the urea cycle, the entry of ammonia into the cycle. This enzyme is encoded by CPS1 (MIM *608307), located on chromosome 2q35 and composed of 38 exons leading to a 1500 amino acid protein.1, 2, 3 Mutations in CPS1 can result in reduced or absent enzyme function leading to hyperammonemia and other features of CPS1 deficiency such as neonatal or late onset encephalopathy with vomiting, seizures, and coma if left untreated (CPS1D, MIM #237300).4 Diagnosis of CPS1D is based on a biochemical profile with increased plasma ammonia, decreased plasma citrulline, and a normal or low orotic acid in urine. Confirmation of the diagnosis requires either enzyme analysis in liver or small intestinal tissue or, recommended as method of choice, molecular genetic investigation.5, 6
More than 230 CPS1 mutations are currently reported.7, 8, 9, 10, 11, 12, 13, 14, 15 Molecular genetic investigation for CPS1D can use different methods: exon per exon sequencing (direct Sanger sequencing), next generation sequencing (NGS) as part of a (often custom‐made) gene panel or whole exome or whole genome sequencing with or without the analysis of copy number variation (CNV), and RNA analysis.16, 17, 18
While automated sequencing applying NGS became more widely available in recent years and showed improved detection rates if compared to direct Sanger sequencing,19 our laboratory performed in most cases RNA analysis for the confirmation of CPS1D.6, 16 Source of RNA can be liver tissue but also skin fibroblasts17 or, hereby improving the turn‐around time, peripheral lymphocytes that were stimulated with phytohemagglutinin hereby applying a straightforward and less invasive protocol.16 Advantage of the latter approach is its simplicity and an improved sensitivity since, for instance, deletions or insertions caused by intronic changes are picked up in addition as their effect on RNA is instantly observable.
In this study, we wanted to evaluate our experience in performing RNA‐based CPS1 molecular genetic investigation over the past 15 years. To do so, we investigated all found CPS1 variants in this period, identified with varying methods, and analyzed the sensitivity of the applied approaches. We hypothesized that RNA analysis would reduce the number of missed mutations if compared to direct Sanger sequencing, and would even be better than NGS techniques.
2. MATERIALS AND METHODS
From 2004 to 2018, a total of 297 samples were referred with a suspicion of CPS1D (analyses were done at the University Children's Hospital Münster, Germany, until 2008, and at the University Children's Hospital Zurich since 2008). From (index) patients, our laboratory received mainly cultured fibroblasts or heparin blood, but also EDTA blood for DNA isolation, DNA, dried blood spots, or shock‐frozen liver tissue. Parents' samples were used in case the index patient was deceased and no material was available, and/or for confirmation of obligate heterozygosity. From the referring letter, we tried to collect information on origin of patients, onset of disease, and severity of the clinical course. As molecular genetic investigation was part of clinical diagnostics, there was no requirement for an approval by the respective ethics committees.
Assuming that single nucleotide changes in exons or flanking intronic sequences would not pose a problem for neither of the sequencing methods, we focused our analysis on complex mutations including intronic changes possibly affecting splicing as well as deletions, insertions, and INDELs. Four different sequencing methods were considered: direct Sanger sequencing of DNA isolated from EDTA blood or dried blood spots, NGS without or with CNV, and RNA sequencing using liver, fibroblasts, or lymphocytes. We arbitrarily defined ±50 bp flanking the exons as limit for an in general “probably detected” change with however maybe uncertain interpretation. For analyzing splicing variants within these limits, softwares Human Splicing Finder and MutationTaster were used.20, 21
3. RESULTS
During the study period, 297 samples were sent from 37 countries in Europe, North and South America, Australia, Asia, and Africa. CPS1D could be confirmed in 155 patients (52 diagnosed in Münster and 103 in Zurich) from 33 countries; patients referred from France (n = 32) and Turkey (n = 27) comprised the largest groups of positive samples. For the confirmed cases, we received mainly fibroblasts (n = 79) or heparin blood (n = 57) for index patient testing, but also two liver biopsies. In 17 families, there was no sufficient material available from the index patient. Therefore, mutations were initially searched for in parents (16 heparin blood samples and 1 fibroblast cell line), and were, whenever possible, confirmed in DNA (often derived from dried blood spots) of the patient.
Clinical information was not always provided, but the majority of the patients showed a neonatal onset. No strict correlation was found between type of mutation and onset of disease,11 except for a deletion in exon 25 (c.3037_3039delGTG) that was always associated with a neonatal onset of severe disease.22 Of the total 155 patients, 83 were homozygous but clinical information was too scarce to further correlate this. The remaining 72 patients were compound heterozygous.
Underlining genetic heterogeneity at the CPS1 locus, we identified a total of 136 different genotypes with 172 different variants, of which 56% were missense (n = 97), 17% deletions (n = 29), 15% splice‐site (n = 26), 6% nonsense (n = 10), 4% insertions (n = 7), and 2% INDELs (n = 3; Figure 1 and Table 1). Of the total 172 different variants, 31 missense, 2 nonsense, 13 splice‐errors, and 16 Del/Ins/Dup were not yet reported in literature (summarized in Tables S1 and S2).
Figure 1.
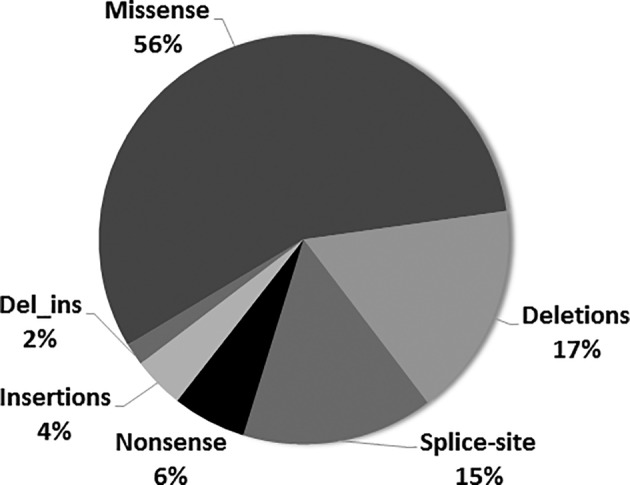
Graph showing the distribution of all CPS1 mutations in this study
Table 1.
Summary of CPS1 mutations of the study cohort
| Item | # |
|---|---|
| Index patients | 155 |
| Genotypes | |
| Homozygous | 64 |
| Heterozygous‐compound | 72 |
| Total | 136 |
| Recurrent | 1 |
| Mutations | |
| Missense | 97 |
| Nonsense | 10 |
| Splice‐errors | 26 |
| Deletions | 29 |
| Insertions/duplications | 7 |
| Del_Ins | 3 |
| Total mutations | 172 |
| Recurrent | 9 |
Note: Recurrence of genotypes or mutations in this study was defined as occurrence >2 times.
The most frequent mutation in this study, present in 17 Turkish patients, was the homozygous deletion in exon 25 c.3037_3039delGTG (p.Val1013del). Next, the missense mutation c.2339G>A (p.Arg780His) in exon 19 was found in seven patients (of varying ethnic background and nationality), of which two are homozygous for this change. The most frequent splice‐site mutation was c.3558+1G>C (p.Glu1161_Arg1186del) in exon 29, identified in three patients.
As reported before,11 distribution of CPS1 mutations shows predominance of the catalytic domains (116 of 172 mutations): the bicarbonate phosphorylation domain (exons 13‐20) was affected by 61 mutations and the carbamate phosphorylation domain (exons 25‐34) by 55 mutations (Figure 2).
Figure 2.
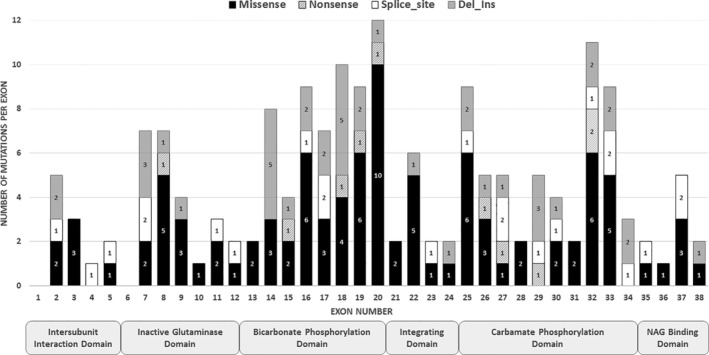
Distribution of mutations over the CPS1 gene and its domains
Parental DNA could be investigated in 98 families confirming obligate carriership in 93 parents. In five families, only one of the parents was found to be a carrier, namely in three parents only the mother and in two parents only the father.
We identified and further analyzed 14 complex mutations (ie, intronic changes possibly affecting splicing, deletions, insertions, and INDELs), which were all found in single patients only and contributed to 14 different genotypes; within this subcohort, only three patients were homozygous. Details to the complex mutations in this study are summarized in Table 2. From these 14 complex mutations, using exon‐wise sequencing or NGS without CNV, five mutations would have been missed; using NGS even with CNV, two mutations would have been missed. RNA analysis did in fact identify all these mutations, and was necessary for a correct interpretation in half of the cases (Table 2).
Table 2.
Summary of 14 complex mutations of the CPS1 gene and their identification by different methods
| Exon | Nucleotide | Protein | RNA | State in this study | Detection only with RNA analysis | Detection with NGS ± CNV | Possible misalignment error with NGS ± CNV | Correct interpretation only with RNA analysis | Human splicing finder | Mutation taster |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | c.127‐26_127‐24delinsCAG | p.(Ala43Aspfs*22) | r.127_128ins(23) | Heterozygous | x | x | Affecting splicing | Benign | ||
| 7 | c.622‐24A>G | p.(Asp208_Lys237del) | r.622_711del (Exon 7) | Homozygous | x | x | Benign | Benign | ||
| 7 | c.622‐52_c.711+1416del | p.(Asp208_Lys237del) | r.622_711del (Exon 7) | Heterozygous | x with CNV | |||||
| 7 | c.622‐7A>G | p.(Lys207_Asp208insTrpGln) | r.621_622insTGGCAG | Homozygous | x | x | Affecting splicing | Disease causing | ||
| 8 | c.712‐430_766del | p.(Arg238Metfs*5) | r.712_840del (Exon 8) | Heterozygous | x with CNV | |||||
| 9 | c.947G>T | p.(Arg316Metfs*2) | r.946_947insTGTGA | Heterozygous | x | x | Affecting splicing | Disease causing | ||
| 15 | c.1549+124_2391+800del | p.(Val518Hisfs*8) | r.1550_2391del (Exon 15‐19) | Homozygous | xa | |||||
| 17 | c.1837‐8A>G | p.(Ala613Phefs*25) | r.1836_1837insTTTCTAG | Heterozygous | x | x | Affecting splicing | Disease causing | ||
| 18 |
c.2079_2080ins CATTCATTCATTCATT |
p.(Val694Hisfs*8) |
r.2079_2080ins CATTCATTCATTCATT |
Heterozygous | x | |||||
| 24 | c.2895+429_c.2960‐281del | p.(Glu966Alafs*27) | r.2896_2959del (Exon 24) | Heterozygous | x with CNV | |||||
| 25 | c.2960‐18A>G | p.(Gly987Valfs*33) |
r.2959_2960ins TCTCATTGTCTCTGCAG |
Heterozygous | x | x | Affecting splicing | Benign | ||
| 30 | c.3559‐745A>G |
p.(Arg1186_Val1187ins LysProArgLeuSerLys*) |
r.3558_3559ins(94) | Heterozygous | x | |||||
| 34 | c.4102‐239A>G | p.(Gln1368Serfs*15) | r.4101_4102ins(89) | Heterozygous | x | |||||
| 38 | c.4405‐9T>G | p.(Val1469Ilefs*4) | r.4404_4405insATTTTCAG | Heterozygous | x | x | Affecting splicing | Disease causing |
4. DISCUSSION
CPS1D is a rare metabolic condition with a nonspecific biochemical profile thus requiring additional tests.5 Nowadays, confirmation of the disease is usually done by molecular genetic investigation that led to the reporting of more than 230 CPS1 mutations underlining the genetic heterogeneity at this locus.7, 8, 9, 10, 11, 12, 13, 14 In recent years, NGS became the preferred method for CPS1 molecular genetic investigation either as part of (often custom‐made) gene panels or of whole exome or genome sequencing. In our laboratory, RNA sequencing was the preferred method in the past 15 years.11, 16 In this study, we investigated retrospectively the theoretical sensitivity if different sequencing approaches would have been used for detecting CPS1 mutations, and, based on our findings, suggest a diagnostic algorithm for molecular genetic testing of CPS1D.
We based our analysis on 155 genetically confirmed cases comprising 172 different variants and 136 different genotypes. Main finding was that in CPS1D, private mutations are the rule, and in about 10% of the genotypes a complex mutation is present. Underlining the relevance of RNA sequencing, 7/14 complex mutations required for correct interpretation RNA analysis, and 5/14 complex mutations would have been missed if NGS was done without CNV analysis. In contrast, RNA sequencing had identified all mutations.
Almost all of the potentially missed variants are intronic substitutions, deletions, or insertions affecting splicing. For instance, the deep intronic change c.3559‐745A>G creates a novel donor splice site leading to an insertion of a 94 bp pseudoexon with a premature termination codon (p.[Arg1186_Val1187insLysProArgLeuSerLys*]). The same occurred in the case of c.4102‐239A>G (p.[Gln1368Serfs*15]). As both these mutations lie in deep intronic sequences, likely neither direct Sanger sequencing nor NGS (with or without CNV) would have detected them. In contrast, RNA analysis using phytohemagglutinin stimulated lymphocytes or fibroblasts did in fact identify these mutations. Other examples illustrating the same principle are summarized in Table 2. Some of these additional examples comprise deletions (c.622‐52_711+1416del, c.712‐430_766del, c.2895+429_2960‐281del), which would have only been detected if CNV analysis was added to NGS. RNA analysis is however also important in case of changes close to the exonic sequences. As shown for mutation c.622‐24A>G, while this change would have likely been identified by all sequencing methods (apart possibly from whole exome sequencing), correct interpretation as a splicing mutation was not offered by in silico prediction (Table 2).
We had previously shown that RNA analysis can substantially shorten the time to diagnosis in CPS1D.16 We add here an improved diagnostic yield as another benefit of performing RNA analysis. With these two advantages in mind, we propose to consider adding RNA sequencing in so far mutation‐negative patients with a strong suspicion for CPS1D. In addition, it needs to be remembered that RNA sequencing is a straightforward and easy to perform analysis requiring only basic sequencing facilities but not high‐throughput techniques rendering establishing and performing this method feasible in many places. Based on these findings and considerations, we propose the following diagnostic algorithm: if available, RNA sequencing can be the first line genetic test in suspected CPS1D; in all other cases, NGS together with CNV should be performed with the option of adding RNA sequencing in mutation‐negative patients with a strong suspicion for CPS1D or in order to correctly interpret unclear intronic alterations that could possibly affect splicing.
In summary, molecular genetic investigation for CPS1D remains challenging as the specific locus shows a high variability with most mutations being private. In addition, many intronic sequences of CPS1 are likewise prone to changes that may be missed with NGS, even if CNV analysis is added. Unclear cases may therefore need to be investigated by RNA sequencing in addition, if this method is not used as the initial diagnostic procedure.
Supporting information
Table S1 Novel missense mutations (n = 31) in the CPS1 gene
Table S2 Novel nonsense mutations (n = 2), deletions (n = 12), duplications (n = 2), insertions (n = 1), delins (n = 1), and splice errors (n = 13) of the CPS1 gene
ACKNOWLEDGMENTS
The authors are grateful to all colleagues from the many centers who provided samples of their patients for CPS1 mutation studies. This study was supported by the Swiss National Science Foundation (grant 320030_176088 to J.H.). Molecular genetic investigation for CPS1 deficiency performed at University Children's Hospital Zurich is supported by Recordati Rare Diseases. This company however was not involved in planning or performing of this study, or in writing this manuscript.
Isler J, Rüfenacht V, Gemperle C, Allegri G, Häberle J. Improvement of diagnostic yield in carbamoylphosphate synthetase 1 (CPS1) molecular genetic investigation by RNA sequencing. JIMD Reports. 2020;52:28–34. 10.1002/jmd2.12091
Communicating Editor: Manuel Schiff
Funding information Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung, Grant/Award Number: 320030_176088
REFERENCES
- 1. Häberle J, Schmidt E, Pauli S, et al. Gene structure of human carbamylphosphate synthetase 1 and novel mutations in patients with neonatal onset. Hum Mutat. 2003;21:444. [DOI] [PubMed] [Google Scholar]
- 2. Hoshide R, Soejima H, Ohta T, et al. Assignment of the human carbamyl phosphate synthetase I gene (CPS1) to 2q35 by fluorescence in situ hybridization. Genomics. 1995;28:124‐125. [DOI] [PubMed] [Google Scholar]
- 3. Summar ML, Hall LD, Eeds AM, et al. Characterization of genomic structure and polymorphisms in the human carbamyl phosphate synthetase I gene. Gene. 2003;311:51‐57. [DOI] [PubMed] [Google Scholar]
- 4. Häberle J, Rubio V. Disorders of the urea cycle and related enzymes In: Saudubray JM, Baumgartner M, Walter JH, eds. Inborn Metabolic Diseases. 6th ed. Heidelberg: Springer; 2016:295‐308. [Google Scholar]
- 5. Häberle J, Burlina A, Chakrapani A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: first revision. J Inherit Metab Dis. 2019;42:1192‐1230. [DOI] [PubMed] [Google Scholar]
- 6. Rüfenacht V, Häberle J. Mutation analysis of urea cycle disorders. J Pediatr Biochem. 2014;4:33‐44. [Google Scholar]
- 7. Aoshima T, Kajita M, Sekido Y, et al. Novel mutations (H337R and 238‐362del) in the CPS1 gene cause carbamoyl phosphate synthetase I deficiency. Hum Hered. 2001a;52:99‐101. [DOI] [PubMed] [Google Scholar]
- 8. Aoshima T, Kajita M, Sekido Y, et al. Carbamoyl phosphate synthetase I deficiency: molecular genetic findings and prenatal diagnosis. Prenat Diagn. 2001b;21:634‐637. [DOI] [PubMed] [Google Scholar]
- 9. Finckh U, Kohlschutter A, Schafer H, Sperhake K, Colombo JP, Gal A. Prenatal diagnosis of carbamoyl phosphate synthetase I deficiency by identification of a missense mutation in CPS1 . Hum Mutat. 1998;12:206‐211. [DOI] [PubMed] [Google Scholar]
- 10. Funghini S, Thusberg J, Spada M, et al. Carbamoyl phosphate synthetase 1 deficiency in Italy: clinical and genetic findings in a heterogeneous cohort. Gene. 2012;493:228‐234. [DOI] [PubMed] [Google Scholar]
- 11. Häberle J, Shchelochkov OA, Wang J, et al. Molecular defects in human carbamoyl phosphate synthetase I: mutational spectrum, diagnostic and protein structure considerations. Hum Mutat. 2011;32:579‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ihara K, Nakayama H, Hikino S, Hara T. Mutation in CPS1 . Hum Genet. 1999;105:375. [Google Scholar]
- 13. Khayat M. Novel human pathological mutations. Gene symbol: CPS1. Disease: carbamoyl phosphate synthetase I deficiency. Hum Genet. 2009;125:336. [PubMed] [Google Scholar]
- 14. Kurokawa K, Yorifuji T, Kawai M, et al. Molecular and clinical analyses of Japanese patients with carbamoylphosphate synthetase 1 (CPS1) deficiency. J Hum Genet. 2007;52:349‐354. [DOI] [PubMed] [Google Scholar]
- 15. Mitchell S, Ellingson C, Coyne T, et al. Genetic variation in the urea cycle: a model resource for investigating key candidate genes for common diseases. Hum Mutat. 2009;30:56‐60. [DOI] [PubMed] [Google Scholar]
- 16. Kretz R, Hu L, Wettstein V, Leiteritz D, Häberle J. Phytohemagglutinin stimulation of lymphocytes improves mutation analysis of carbamoylphosphate synthetase 1. Mol Genet Metab. 2012;106:375‐378. [DOI] [PubMed] [Google Scholar]
- 17. Rapp B, Häberle J, Linnebank M, et al. Genetic analysis of carbamoylphosphate synthetase I and ornithine transcarbamylase deficiency using fibroblasts. Eur J Pediatr. 2001;160:283‐287. [DOI] [PubMed] [Google Scholar]
- 18. Wang J, Shchelochkov OA, Zhan H, et al. Molecular characterization of CPS1 deletions by array CGH. Mol Genet Metab. 2011;102:103‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Djemie T, Weckhuysen S, von Spiczak S, et al. Pitfalls in genetic testing: the story of missed SCN1A mutations. Mol Genet Genomic Med. 2016;4:457‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Desmet FO, Hamroun D, Lalande M, Collod‐Beroud G, Claustres M, Beroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11:361‐362. [DOI] [PubMed] [Google Scholar]
- 22. Hu L, Diez‐Fernandez C, Rüfenacht V, et al. Recurrence of carbamoyl phosphate synthetase 1 (CPS1) deficiency in Turkish patients: characterization of a founder mutation by use of recombinant CPS1 from insect cells expression. Mol Genet Metab. 2014;113:267‐273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Novel missense mutations (n = 31) in the CPS1 gene
Table S2 Novel nonsense mutations (n = 2), deletions (n = 12), duplications (n = 2), insertions (n = 1), delins (n = 1), and splice errors (n = 13) of the CPS1 gene