

Abstract
Introduction
The prevalence of Wilson disease (WD) in Costa Rica is among the highest reported in the world, 4.9:100 000. Previous investigators have also described a burden of autosomal recessive conditions in this country. Genetic testing for WD began in 2010 as a strategy for earlier detection due to the country's high prevalence. Here we describe what we have learned about the genotype and phenotype of the Costa Rican pediatric population with WD.
Methods
We completed a retrospective review of medical records from pediatric individuals (<18 years of age) with molecular testing for ATP7B between 2010 and 2015. We documented phenotype and genotype for cases with WD as defined by the international scoring system.
Results
Thirty‐four WD cases from 28 families were included, 15 female and 19 male patients. The most frequent pathogenic variant in ATP7B was NM_000053:c.3809A>G, p.Asn1270Ser, with 58.8% of affected individuals homozygous for this variant. Age of diagnosis ranged from 1 to 17 years, with an average of 8.8 ± 3.6 years. All individuals who presented with acute liver failure (n = 6) were homozygous for the p.Asn1270Ser variant (Chi‐squared, P < .05).
Discussion
Molecular testing has facilitated the detection of presymptomatic patients with WD in Costa Rica. We hope that ongoing efforts in the delivery of clinical services lead to optimized molecular screening for WD and other genetic conditions in Costa Rica.
Keywords: acute liver failure, ATP7B, genotype and phenotype, pediatric, Wilson disease
SYNOPSIS.
Here we describe what we have learned about the genotype and phenotype of the Costa Rican pediatric population with WD.
1. INTRODUCTION
Wilson disease (WD) is an autosomal recessive disorder in which copper's metabolism is altered due to pathogenic variants in the ATP7B gene. The condition's phenotype can vary widely from acute liver failure (ALF) to neuropsychiatric manifestations, and occurs as a result of the toxic effects of excess copper in tissues.1, 2, 3 WD is a treatable condition, and early detection through molecular testing can decrease morbidity and mortality.4, 5 Costa Rica has the highest prevalence of WD in the world: 4.9:100 000, as per previous reports, as well as a burden of other autosomal recessive conditions.6, 7 Previous studies have reported a high rate of acute liver failure among WD patients in Costa Rica, approximately 17% in children, in comparison to other reports where only 5% was observed.8, 9, 10, 11 To address this, the Medical Genetics and Metabolism Department at the National Children's Hospital (HNN) now provides molecular diagnoses for a group of conditions including Wilson disease (OMIM #277900). Molecular testing for early detection was implemented as a strategic measure to reduce morbidity and mortality.12, 13, 14 The project's goal was to optimize the benefits of molecular testing through extended familial cascade screening, genetic counseling, and reproductive planning.15
In this study, we characterize the phenotype and genotype of the Costa Rican pediatric population with WD who had molecular testing between 2010 and 2015 and compare the results with a previous report from our center to demonstrate the impact of strategic measures in the delivery of genetics services.
2. METHODS
We completed a retrospective review of medical records from pediatric individuals (<18 years of age) with molecular testing for ATP7B between 2010 and 2015 (n = 140). We included patients with WD defined as a score ≥ 4 points in the Leipzig or EASL scoring system (n = 34).5
2.1. Phenotype
Phenotypic data was collected from medical records. Phenotype was defined by clinical presentation at time of diagnosis as: presymptomatic, hepatic, acute liver failure (ALF), neuropsychiatric, or coombs‐negative acute hemolytic anaemia.5 ALF was defined as onset of clinical symptoms <8 weeks with evidence of impaired hepatic function, INR > 2, and any degree of encephalopathy.16
2.2. Genotype
Molecular testing for ATP7B began in 2010 with DNA PCR amplification and restriction enzyme fragment length polymorphism testing.17 An initial 4‐exon panel included exons 6, 7, 8, and 18, with variants p.Met645Arg, p.Met665Ile, p.Leu708Pro, and p.Asn1270Ser (GRCh37/hg19, NM_000053). This strategy was later expanded to a 7‐exon panel that included exons 6, 7, 8, 14, 17, 18, and 21. This selection was based on previous Costa Rican population estimates on variant frequencies.17 By 2013, the HNN testing center had the ability to sequence by the Sanger method all 21 exons and splicing regions in ATP7B.
2.3. Statistical analysis
We transferred the information gathered into a collection sheet using the Epi Info Software, where we compiled Leipzig scores to determine patient inclusion.5 Statistical analyses were performed using the STATA version 14 and R Software version 3.4.4.
3. RESULTS
A total of 140 pediatric individuals had molecular testing for ATP7B variants between 2010 and 2015, due to liver disease of unknown etiology or suspicion of WD due to family history. A total of 34 pediatric patients, from 28 families, were confirmed to have WD by a score ≥ 4 points in the Leipzig scoring system. From the 34 diagnosed patients, 23 were new and 11 had been previously reported.18, 19 Previously reported patients had prior clinical diagnosis and were later confirmed molecularly (Table 1). There were 15 female and 19 male patients diagnosed with no significant age difference between sexes (females = 7.9 ± 4.5 years, males = 9.4 ± 2.6 years, Welch ANOVA P = .2654). Age of diagnosis ranged from 1‐17 years, with an average of 8.8 ± 3.6 years. Those presenting with symptoms upon initial evaluation had an average age of 10.1 ± 2.1 years.
Table 1.
Characteristics of pediatric patients with WD in Costa Rica at presentation (n = 34)
| Family‐case | Province | Age (years) | Sex | Kinship | Genotype | Phenotype | Previously reported | Leipzig score | KF ring | 24‐hour urinary copper (μmol/24 hours) | Ceruloplasmin (g/L)/ceruloplasmin ferroxidase activity (IU/L) | Hemolytic anemia coombs‐neg | INR | AST/ALT (IU/L) | Total bilirubin (mg/dL) | Liver copper (μg/g dry wt) | Modified Nazer score (if ALF) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1‐1* | San José | 6.4 | Female | Sibling | p.N1270S/p.N1270S | Hepatic | + | 8 | – | 14.25 | 0.04 g/L | – | 1.00 | 38/62 | 0.7 | – | – |
| 1‐2 | San José | 10.3 | Female | Sibling | p.N1270S/p.N1270S | Presymptomatic | + | 8 | – | 6.9 | 1 IU/L | – | 1.00 | 85/121 | 0.6 | – | – |
| 2‐1* | Alajuela | 8.1 | Male | – | p.N1270S/p.N1270S | Hepatic | – | 8 | – | 1.41 | 0.00 g/L | – | 1.00 | 184/406 | 0.4 | 11.2 | – |
| 3‐1* | Puntarenas | 7.5 | Male | Sibling | p.N1270S/p.N1270S | Hepatic | + | 8 | – | 4.8 | 7 IU/L | – | 1.00 | 193/341 | 0.5 | – | – |
| 3‐2 | Puntarenas | 4.5 | Male | Sibling | p.N1270S/p.N1270S | Presymptomatic | + | 8 | – | 5.12 | 13 IU/L | – | – | 127/174 | 0.8 | – | – |
| 4‐1* | San José | 11.1 | Male | Sibling | p.N1270S/p.N1270S | Acute liver failure | – | 5 | – | – | – | + | 2.64 | 263/45 | 12.4 | – | 10 |
| 4‐2 | San José | 2.6 | Female | Sibling | p.N1270S/p.N1270S | Presymptomatic | – | 6 | – | 0.6 | 0.00 g/L | – | 1.00 | 47/35 | 0.4 | – | – |
| 5‐1* | Limón | 12.4 | Male | – | p.N1270S/p.L708P | Hepatic | – | 9 | + | 3 | 0.00 g/L | – | 1.40 | 98/30 | 1.0 | – | – |
| 6‐1* | Heredia | 9.5 | Male | Sibling | p.N1270S/p.L708P | Hepatic | – | 11 | – | 22.96 | 0.09 g/L | + | 1.46 | 216/123 | 12.1 | 4.66 | – |
| 6‐2 | Heredia | 15.7 | Male | Sibling | p.N1270S/p.L708P | Presymptomatic | – | 8 | – | 1.19 | 0.03 g/L | – | – | – | – | 4.75 | – |
| 7‐1* | Puntarenas | 12.1 | Female | – | p.N1270S/p.N1270S | Acute liver failure | – | 13 | – | 43.39 | 0.00 g/L | + | 3.15 | 236/101 | 30.1 | – | 11 |
| 8‐1* | San José | 8.3 | Female | – | p.N1270S/p.N1270S | Hepatic | – | 9 | – | 10 | 0.04 g/L | – | 1.77 | 102/60 | 0.9 | 2.1 | – |
| 9‐1*± | San José | 9.8 | Female | – | p.N1270S/p.N1270S | Acute liver failure | – | 13 | – | 26.45 | 0.00 g/L | + | 2.90 | 161/22 | 46.7 | – | 10 |
| 10‐1*± | Cartago | 13.7 | Female | 3rd Cousin | p.N1270S/p.N1270S | Acute liver failure | – | 15 | + | 162.14 | 9 IU/L | + | 3.23 | 127/19 | 47.7 | – | 9 |
| 10‐2 | San José | 17.9 | Female | 3rd Cousin | p.N1270S/p.T1434M | Presymptomatic | – | 9 | – | 16.1 | 0.17 g/L | – | 1.06 | 22/16 | 0.8 | 14.8 | – |
| 11‐1 | San José | 4.6 | Female | – | p.N1270S/p.N1270S | Presymptomatic | + | 6 | – | 1.07 | 6 IU/L | – | 1.10 | 52/56 | 0.2 | – | – |
| 12‐1 | San José | 13.1 | Female | – | p.N1270S/p.M645R | Presymptomatic | – | 8 | – | 17.6 | 0.00 g/L | – | 1.04 | 237/104 | 0.4 | – | – |
| 13‐1* | San José | 11.8 | Male | – | p.N1270S/p.N1270S | Coombsneg hemolytic anemia | + | 8 | – | 2.5 | 11 IU/L | + | 1.40 | 124/16 | 8.1 | – | – |
| 14‐1*† | Cartago | 9.8 | Male | – | p.N1270S/p.N1270S | Acute liver failure | – | 10 | – | – | 0.10 g/L | + | 2.46 | 147/21 | 35.9 | – | 9 |
| 15‐1 | Guanacaste | 6.3 | Male | – | p.N1270S/– | Presymptomatic | + | 4 | – | 1.59 | 35 IU/L | – | 1.00 | 127/206 | 0.6 | 25.3 | – |
| 16‐1 | San José | 1.7 | Female | – | p.N1270S/p.L708P | Presymptomatic | + | 8 | – | 6.8 | 0.00 g/L | – | 1.10 | 43/25 | 0.4 | – | – |
| 17‐1 | San José | 10.1 | Male | – | p.N1270S/p.N1270S | Presymptomatic | – | 10 | – | 25.5 | 0.03 g/L | – | 1.15 | 144/186 | 0.8 | 6.8 | – |
| 18‐1* | Puntarenas | 5.3 | Female | Sibling | p.M645R/– | Presymptomatic | – | 7 | – | 11.3 | 0.00 g/L | – | 1.06 | 87/111 | 0.4 | 23.77 | – |
| 18‐2 | Puntarenas | 5.9 | Female | Sibling | p.M645R/– | Presymptomatic | – | 5 | – | 7.84 | 0.00 g/L | – | – | – | – | – | – |
| 19‐1* | San José | 8.1 | Female | – | p.N1270S/p.N1270S | Acute liver failure | – | 13 | + | 71.65 | 6 IU/L | + | 3.15 | 219/36 | 50.8 | 5.59 | 11 |
| 20‐1* | San José | 10.5 | Male | – | p.N1270S/– | Hepatic | + | 9 | + | 7.16 | 11 IU/L | – | 1.18 | 152/158 | 2.2 | 6.78 | – |
| 21‐1 | Puntarenas | 6.8 | Male | – | p.N1270S/p.M645R | Presymptomatic | + | 8 | – | 5.04 | 7 IU/L | – | – | 143/208 | 0.7 | – | – |
| 22‐1 | San José | 6.5 | Female | – | p.N1270S/p.N1270S | Presymptomatic | – | 8 | – | 3.52 | 0.03 g/L | – | 1.09 | 100/197 | 1.1 | – | – |
| 23‐1 | San José | 9.9 | Male | – | p.N1270S/p.N1270S | Presymptomatic | – | 7 | – | 2.86 | 0.00 g/L | – | 1.10 | 262/231 | 0.8 | – | – |
| 24‐1* | Cartago | 11.8 | Male | – | p.N1270S/p.N1270S | Hepatic | – | 7 | – | – | 0.03 g/L | + | 3.90 | 230/100 | 9.0 | – | – |
| 25‐1* | San José | 7.3 | Male | – | p.N1270S/– | Hepatic | + | 6 | – | 8.26 | 0.02 g/L | – | – | 130/54 | 2.0 | 3.77 | – |
| 26‐1* | San José | 11.4 | Male | – | p.N1270S/p.N1270S | Hepatic | – | 8 | – | 21.07 | 0.00 g/L | – | 3.10 | 191/86 | 2.4 | – | – |
| 27‐1 | Alajuela | 12.3 | Male | – | –/– | Presymptomatic | – | 5 | – | 9.46 | 0.19 g/L | – | 1.10 | 25/12 | 1.4 | 13.7 | – |
| 28‐1* | San José | 11.6 | Male | – | p.N1270S/– | Hepatic | – | 4 | – | 35.43 | 0.00 g/L | – | 1.76 | 177/169 | 1.7 | – | – |
Abbreviations: ALF, acute liver failure; KF, Kayser–Fleischer; ULN, upper limit of normal; * = index case; ± = liver transplant; − = no; + = yes; † deceased.
We estimated pediatric prevalence as the proportion of affected individuals over the population of individuals <18 years of age per region, according to the Costa Rican National Population Census data.20 The national prevalence of pediatric WD was estimated to be 2.2:100000. The proportion of affected individuals varied markedly depending on the region of the country from 0.6 to 3.9:100000 (Figure 1). The central province of San José was the most affected with a prevalence of 3.9:100 000, followed by the province of Puntarenas with 3.6:100 000. The difference in prevalence with previous adult population studies may be attributable to the natural history of the condition and its onset later in adulthood.6
Figure 1.
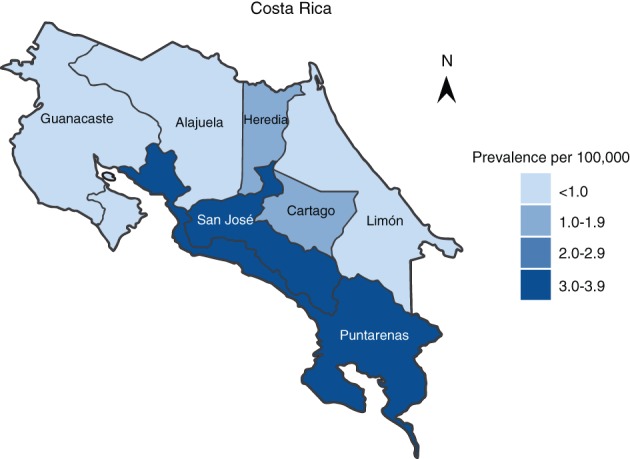
Prevalence of pediatric patients with Wilson Disease in Costa Rica by province
Biallelic pathogenic variants were identified in 27 of the 34 individuals who met Leipzig criteria for WD. The most frequent pathogenic variant detected in our population in ATP7B (hg19) was NM_000053:c.3809A>G, p.Asn1270Ser (Table 2). The majority of pediatric WD cases (58.8%) were homozygous for this missense variant. Five different pathogenic variants were detected in WD cases: p.Asn1270Ser, p.Met645Arg, p.Leu708Pro, p.Thr1434Met, and p.Met665Ile. Individuals with unidentified variants had four of the 21 exons sequenced. Efforts are underway to identify variants in individuals with WD and ≤1 variant identified. From 106 individuals without WD, 23 were heterozygous for one pathogenic variant in ATP7B (19 p.Asn1270Ser, 2 p.Met645Arg, 1 p.Thr1434Met and 1 p.Met665Ile).
Table 2.
Genotype frequency in pediatric patients with Wilson disease in Costa Rica
| Genotype | Number of patients (n = 34) |
|---|---|
| Homozygous | |
| p.N1270S/p.N1270S | 20 (58.8%) |
| Compound heterozygous | |
| p.N1270S/p.M645R | 2 (5.9%) |
| p.N1270S/p.L708P | 4 (11.7%) |
| p.N1270S/p.T1434M | 1 (3.0%) |
| One variant detected | |
| p.N1270S/− | 4 (11.7%) |
| p.M645R/− | 2 (5.9%) |
| No variants detected | |
| −/− | 1 (3.0%) |
We documented the phenotype at onset with biochemical parameters. The most frequent clinical presentation was hepatic in 50% of cases (Table 3). The hepatic phenotype included: acute hepatitis, chronic hepatitis, hepatomegaly, or ALF. No patients presented as neuropsychiatric WD. All patients had decreased ceruloplasmin, most cases (32 of 34) in levels considerably below 50% of the lower normal limit (defined as 0.1 g/L or 28 IU/L for ceruloplasmin ferroxidase activity). Average urinary copper was 18.9 ± 30.7 μmol/ 24 hours, including patients under penicillamine challenge. The international scoring system for WD (EASL or Leipzig criteria) previously validated in pediatric patients, was highly sensitive capturing all patients in this cohort.5, 21
Table 3.
Phenotypic presentations at diagnosis in pediatric patients with Wilson disease in Costa Rica
| Phenotype | Number of patients (n = 34) | % | Jimenez et al. (n = 35)a , b | % |
|---|---|---|---|---|
| Hepatic | 11 | 32% | 13 | 37% |
| Acute liver failure (deceased) | 6 (1) | 18% | 11 (6) | 31% |
| Presymptomatic | 16 | 47% | 6 | 17% |
| Coombs negative hemolytic anemia | 1 | 3% | 4 | 11% |
| Neurologic | 0 | 0% | 1 | 3% |
Jiménez et al.19
Ten patients between the two studies (six with presymptomatic and four with hepatic phenotype) overlap due to a four‐year study intersection (2002‐2006).
ALF occurred mainly in the context of chronic liver disease. Nonetheless, acute presentation was distinguished from chronic disease by symptom duration before diagnosis, clinical signs of chronic liver disease or portal hypertension. Most cases of ALF (4 of 6) were in more advanced stages of encephalopathy at time of presentation. Of the six cases presenting with ALF, four were females and two were male patients. Two patients with ALF were transplanted, three were not transplanted and survived, and one male patient died at age 9 years and 4 months due to fulminant liver failure (Tables 1 and 3).22 In the pediatric population with WD, all individuals with acute liver failure (n = 6) were homozygous for the p.Asn1270Ser variant (Figure 2). We found no significant difference in age of presentation between homozygous and heterozygous phenotypes. Sixteen of the 34 patients (47%) were detected in a presymptomatic stage through molecular cascade screening due to family history.
Figure 2.
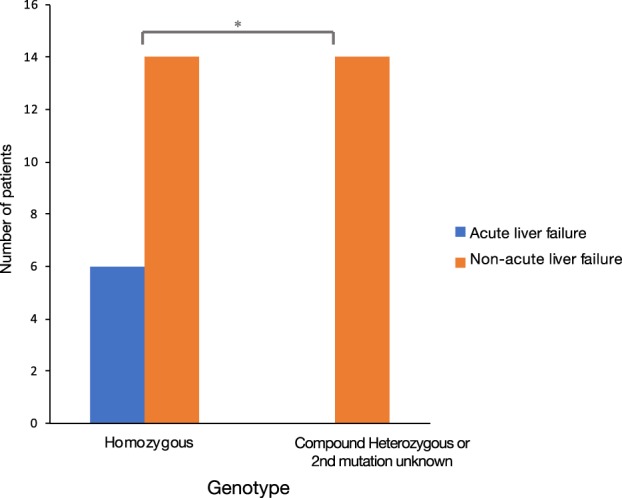
Genotype‐phenotype correlation. Comparison of homozygous p.Asn1270Ser phenotypes (n = 20) to heterozygous phenotypes (n = 14) in pediatric patients with Wilson disease (Chi‐squared, P < .05)
Jiménez et al.,19 characterized the pediatric population with WD at the same center before the advent of molecular testing, between 1992 and 2006 (Table 3). We documented a total of 34 patients diagnosed in a time span of 13 years and previously Jimenez et al. had documented a total of 35 cases diagnosed in a span of 14 years.19 There is a similar number of cases diagnosed per year between the two studies and a reduction in age of diagnosis from 10 ± 2 to 8.8 ± 3.4 years. Jimenez et al. documented six presymptomatic cases, captured due to family history of WD. We observed a total of sixteen presymptomatic cases. Although six presymptomatic patients from our cohort overlap with Jimenez et al. due to prior clinical diagnosis between 2002 and 2006, there is an increase of at least 30% of patients being captured presymptomatically.19 Mortality decreased from six cases to one case between the two studies.19
4. DISCUSSION
In this study, we describe what we have learned about the genotype and phenotype of the Costa Rican pediatric population with WD. Since our Institution serves as the national reference center for Clinical Genetics, Pediatric Gastroenterology, and Hepatology, we speculate to have captured nearly all pediatric WD cases in the nation. We compared data with a previous report from our center to demonstrate the impact of strategic measures in the delivery of genetics services.
First, we observed an increase in presymptomatic detection attributable to familial cascade genetic screening.23 Expanded cascade screening identified 16 presymptomatic patients with family history of WD. Earlier detection was possible with molecular testing for ATP7B genetic variants. We hypothesize early diagnosis and treatment prevented complications from chronic disease. In some instances, testing may have spared individuals from serial clinical evaluations or invasive testing. Since individuals with pathogenic variants in one allele can have slightly altered biochemical parameters, molecular testing may be helpful to distinguish between affected and carrier status. Future efforts to include the adult population could contribute to further characterize of the Costa Rican WD population.
Second, the number of pediatric cases identified over time was similar between Jimenez et al. (2009) and our cohort. Interestingly, a higher prevalence of cases in the central province of San José is consistent with prior reports and cases described here are moderately genetically homogenous.6, 24 Explanations for the higher incidence of WD in our population combined with a higher central region prevalence and a more genetically homogenous population, include consanguinity and a possible founder effect.11 Interestingly, the first reports of the p.Asn1270SSer variant were documented in the Mediterranean region (Sicily and Turkey) and later in Costa Rica.11, 12, 25 This missense variant has since been identified in various populations across the globe, specifically in Latino, Asian, African, and European populations.26 Rare recessive conditions, like WD, tend to be enriched in smaller, more genetically isolated populations.27 Founder effects are somewhat common among humans and have been identified before in Costa Rica.7, 27 However, the haplotype from a single common ancestor must be identified to determine if there is a founder effect for WD.11
Third, we observed that all pediatric cases with ALF had homozygous p.Asn1270Ser pathogenic variants. We find this observation noteworthy since the p.Asn1270Ser missense variant is located in the ATP hinge domain, which may be essential for protein function. Previous functional studies in Saccharomyces cerevisiae documented that wild type and other missense variants rescue delta ccc2, the yeast homologue of ATP7B,8 yet, the p.Asn1270Ser mutant could not rescue functionality of delta ccc2.8 Therefore, we hypothesize that the p.Asn1270Ser variant may have a highly deleterious effect, however, mutation status has not been demonstrated to relate to more severe phenotypes.28, 29
As genetic services in Costa Rica continue to grow, genetic knowledge continues to expand, and clinical applications increase, strategies to deliver services can be optimized for our population. Delivery of clinical services in the future may include widespread molecular screening in regions with high prevalence and copy number variant detection to improve sensitivity. Genetic sequencing offers the potential to be diagnostic, especially in cases where biochemical parameter interpretation is challenging. Molecular testing for more common recessive conditions in our nation has contributed to population characterization and earlier detection. We hope that our ongoing efforts lead to optimized molecular and biochemical screening for WD in Costa Rica.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
ETHICS APPROVAL AND INFORMED CONSENT
This investigation was approved by the Scientific Ethical Committee at the National Children's Hospital of Costa Rica (CEC‐HNN‐019‐2017) and followed Good Clinical Practice standards as well as local and international human research regulations. Data was de‐identified for analyses. The provision of individual consent forms is not required for review of medical records. The CENDEISSS Bioethics Committee approved the publication of this study.
ANIMAL RIGHTS
This article does not contain studies with animal subjects performed by any of the authors.
ACKNOWLEDGMENTS
We gratefully acknowledge the teams at Programa Nacional de Tamizaje Neonatal, Hospital Nacional de Niños, “Dr. Carlos Sáenz Herrera”; Unidad de Investigación, Hospital San Juan de Dios, Caja Costarricense de Seguro Social and Centro de Desarrollo Estratégico e Información en Salud y Seguridad Social (CENDEISSS). We would also like to thank Colleen Carlston, Jiyoo Chang, Joseph T. Shieh, and all pediatric program clinical staff at the University of Costa Rica.
Penon‐Portmann M, Lotz‐Esquivel S, Chavez Carrera A, et al. Wilson disease in Costa Rica: Pediatric phenotype and genotype characterization. JIMD Reports. 2020;52:55–62. 10.1002/jmd2.12098
Communicating Editor: Johannes Häberle
REFERENCES
- 1. Bandmann O, Weiss KH, Kaler SG. Wilson's disease and other neurological copper disorders. Lancet Neurol. 2015;14(1):103‐113. 10.1016/S1474-4422(14)70190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nat Rev. 2018;4(21):1‐20. 10.1038/s41572-018-0018-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanzi R, Petrukhin K, Chernov I, et al. The Wilson disease gene is a putative copper transporting P‐type ATPase similar to the menkes gene. Nat Genet. 1993;5(4):327‐337. 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 4. Durand F, Bernuau J, Giostra E, et al. Wilson's disease with severe hepatic insufficiency: beneficial effects of early administration of D‐penicillamine. Gut. 2001;48:849‐852. 10.1136/gut.48.6.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. The European Association for the Study of Liver . EASL clinical practice guidelines: Wilson's disease. J Hepatol. 2012;56(3):671‐685. 10.1016/j.jhep.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 6. Hevia‐Urrutia F, Alvarado‐Echeverria I, Sanabria‐Castro A, et al. National Alliance for Wilsons disease: health policy in Costa Rica. Hepatol Med Policy. 2017;2:1‐6. 10.1186/s41124-016-0012-x. [DOI] [Google Scholar]
- 7. Kurtz CL, Karolyi L, Seyberth HW, et al. A common NKCC2 mutation in costa Rican Bartter's syndrome patients: evidence for a founder effect. J Am Soc Nephrol: JASN. 1997;8(11):1706‐1711. [DOI] [PubMed] [Google Scholar]
- 8. Iida M, Terada K, Sambongi Y, et al. Analysis of functional domains of Wilson disease protein (ATP7B) in Saccharomyces cerevisiae . FEBS Lett. 1998;428(3):281‐285. https://doi.org/S0014-5793(98)00546-8. [DOI] [PubMed] [Google Scholar]
- 9. Mainardi V, Rando AEK, Valverde M, et al. Acute liver failure due to Wilson disease: eight years of the national liver transplant program in Uruguay. Ann Hepatol. 2019;18(1):187‐192. 10.5604/01.3001.0012.7911. [DOI] [PubMed] [Google Scholar]
- 10. Roberts EA, Socha P. Wilson disease in children. Handb Clin Neurol. 2017;142:141‐156. 10.1016/B978-0-444-63625-6.00012-4. [DOI] [PubMed] [Google Scholar]
- 11. Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype‐phenotype correlation, and functional analyses. Am J Hum Genet. 2007a;61:317‐328. 10.1086/514864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet. 2006;120(2):151‐159. 10.1007/s00439-006-0202-5. [DOI] [PubMed] [Google Scholar]
- 13. Gaffney D, Walker JL, O'Donnell JG, et al. DNA‐based presymptomatic diagnosis of Wilson disease. J Inherit Metab Dis. 1992;15(2):161‐170. 10.1007/BF01799625. [DOI] [PubMed] [Google Scholar]
- 14. Ohura T, Abukawa D, Shiraishi H, et al. Pilot study of screening for Wilson disease using dried blood spots obtained from children seen at outpatient clinics. J Inherit Metab Dis. 1999;22(1):74‐80. 10.1023/A:1005455401076. [DOI] [PubMed] [Google Scholar]
- 15. ACMG Board of Directors . Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2015;17(6):505‐507. 10.1038/gim.2015.41. [DOI] [PubMed] [Google Scholar]
- 16. Mccullough AJ, Fleming CR, Thistle JL, et al. Diagnosis of Wilson's disease presenting as fulminant hepatic failure. Gastroenterology. 1983;84(1):161‐167. 10.1016/S0016-5085(83)80181-4. [DOI] [PubMed] [Google Scholar]
- 17. Centeno Cerdas C. Descripción mutacional del gen ATP7B en pacientes pediátricos con enfermedad de Wilson en la población pediátrica Costarricense. San José: Universidad de Costa Rica; 2009. [Google Scholar]
- 18. Henao JA, Valverde K, Ávila ML. Anemia hemolítica como presentación inicial de enfermedad de Wilson: un caso pediátrico. Arch Argent Pediatr. 2016;114(6):e436‐e439. 10.5546/aap.2016.e436. [DOI] [PubMed] [Google Scholar]
- 19. Jiménez G, Cambronero V, Morales C, Mora A, Guzmán C, Jiménez‐Rivera C. Enfermedad de Wilson: experiencia pediátrica en Costa Rica. Gastroenterol Hepatol. 2009;32(4):274‐278. 10.1016/j.gastrohep.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 20. INEC Costa Rica, I. N. de E. y C . (2008). Estimaciones y proyecciones de población por sexo y edad, 1950–2100 Retrieved from http://inec.go.cr.
- 21. Dhawan A. Evaluation of the scoring system for the diagnosis of Wilson's disease in children. Liver Int. 2005;25(3):680‐681. 10.1111/j.1478-3231.2005.01072.x. [DOI] [PubMed] [Google Scholar]
- 22. Dhawan A, Taylor RM, Cheeseman P, De Silva P, Katsiyiannakis L, Mieli‐Vergani G. Wilson's disease in children: 37‐year experience and revised King's for liver transplantation. Liver Transpl. 2005;11(4):441‐448. 10.1002/lt.20352. [DOI] [PubMed] [Google Scholar]
- 23. Saborio‐Rocafort M. Experiencia en la prestacion de servicios de genética en Costa Rica. Bol Sanit Panam. 1993;115(1):25‐31. [Google Scholar]
- 24. Hevia F, Miranda M. The special problem of Wilson's disease in Costa Rica—an unexpected high prevalence. Gastroenterol Int. 1989;2(4):1‐943. 10.1016/j.neurol.2013.05.002. [DOI] [Google Scholar]
- 25. Rodriguez‐Castro KI, Hevia‐Urrutia FJ, Sturniolo GC. Wilson's disease: a review of what we have learned. World J Hepatol. 2015;7(29):2859‐2870. 10.4254/wjh.v7.i29.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. BioRxiv. 2019;531210 10.1101/531210. [DOI] [Google Scholar]
- 27. Venegas PB, Novak JM, Oscar CA, et al. Cystic fibrosis mutations in Costa Rica. Hum Biol. 2013;75(2):179‐188. 10.1353/hub.2003.0039. [DOI] [PubMed] [Google Scholar]
- 28. Ferenci P, Stremmel W, Członkowska A, et al. Age and sex but not ATP7B genotype effectively influence the clinical phenotype of Wilson disease. Hepatology. 2019;69(4):1464‐1476. 10.1002/hep.30280. [DOI] [PubMed] [Google Scholar]
- 29. Sandahl TD, Laursen TL, Munk DE, Vilstrup H, Weiss KH, Ott P. The prevalence of Wilson disease. An update. Hepatology. 2019;0‐3. 10.1002/hep.30911. [DOI] [PubMed] [Google Scholar]