

Abstract
Background
Because of the high number of people with schizophrenia not responding adequately to monotherapy with antipsychotic agents, the evidence regarding the efficacy and safety of additional medication was examined in a number of clinical trials. One approach to this research question was the use of benzodiazepines, as monotherapy as well as in combination with antipsychotics.
Objectives
To determine the efficacy, acceptability, and tolerability of benzodiazepines in people with schizophrenia and schizophrenia‐like psychoses.
Search methods
In February 2011, we updated the literature search of the previous version of this systematic review (last search March 2005). We searched the trial register of the Cochrane Schizophrenia Group (containing methodical searches of BIOSIS, CINAHL, Dissertation abstracts, EMBASE, LILACS, MEDLINE, PSYNDEX, PsycINFO, RUSSMED, Sociofile, supplemented with hand searching of relevant journals and numerous conference proceedings). Additionally, we inspected references of all identified studies for further relevant studies and contacted authors of relevant publications in order to obtain missing data from existing trials. We applied no language restrictions.
Selection criteria
We included all randomised controlled trials comparing benzodiazepines (as monotherapy or as adjunctive agent) with antipsychotic drugs or placebo for the pharmacological management of schizophrenia and/or schizophrenia‐like psychoses.
Data collection and analysis
Review authors (MD and CL) analysed independently the new references of the update‐search referring to the inclusion criteria. MD and CL extracted all data from the included trials. For dichotomous outcomes we calculated risk ratios (RR) and their 95% confidence intervals (CI). We analysed continuous data by using mean differences (MD) and their 95% CI. We assessed each pre‐selected outcome from the included trials with the risk of bias tool.
Main results
The 2011 update search yielded three further randomised controlled trials. The review currently includes 34 studies with 2657 participants. Most studies were characterised by a small sample size, short duration, and incomplete outcome data reporting.
Benzodiazepine monotherapy is compared with placebo in eight trials. The proportion of participants with no clinically important response did not significantly differ between those given benzodiazepines or placebo (N = 382, 6 RCTs, RR 0.67 CI 0.44 to 1.02). The results from the various rating scales applied to assess global and mental state were inconsistent.
Fourteen studies examined benzodiazepine monotherapy in comparison with antipsychotic monotherapy. Clinically important treatment response assessment revealed no statistically significant difference between the study groups (30 minutes: N = 44, 1 RCT, RR 0.91 CI 0.58 to 1.43; 60 minutes: N = 44,1 RCT, RR 0.61 CI 0.20 to 1.86; 12 hours: N = 66, 1 RCT, RR 0.75 CI 0.44 to 1.30; pooled short‐term studies: N = 112, 2 RCTs, RR 1.48 CI 0.64 to 3.46). Desired sedation occurred significantly more often among participants in the benzodiazepine group than in the antipsychotic group at 20 and 40 minutes. No significant between‐group differences could be identified for global and mental state or occurrence of adverse effects.
Twenty trials compared benzodiazepine augmentation of antipsychotics with antipsychotic monotherapy. Referring to clinically important response, statistically significant improvement could be demonstrated only for the first 30 minutes of augmentation treatment (30 minutes: 1 RCT, N = 45, RR 0.38 CI 0.18 to 0.80; 60 minutes: N = 45,1 RCT, RR 0.07 CI 0.00 to 1.13; 12 hour: N = 67,1 RCT, RR 0.85 CI 0.51 to 1.41; pooled short‐term studies: N = 511, 6 RCTs, RR 0.87 CI 0.49 to 1.54). Analyses of the global and mental state yielded no between‐group differences except for desired sedation at 30 as well as 60 minutes (30 minutes: N = 45, 1 RCT, RR 2.25 CI 1.18 to 4.30; 60 minutes: N = 45, 1 RCT, RR 1.39 CI 1.06 to 1.83).
Authors' conclusions
There is currently no convincing evidence to confirm or refute the practise of administering benzodiazepines as monotherapy or in combination with antipsychotics for the pharmacological treatment of schizophrenia and schizophrenia‐like psychosis. Low‐quality evidence suggests that benzodiazepines are effective for very short‐term sedation and could be considered for calming acutely agitated people with schizophrenia. Measured by the overall attrition rate, the acceptability of benzodiazepine treatment appears to be adequate. Adverse effects were generally poorly reported. High‐quality future research projects with large sample sizes are required to clarify the evidence of benzodiazepine treatment in schizophrenia, especially regarding long‐term augmentation strategies.
Keywords: Humans, Antipsychotic Agents, Antipsychotic Agents/adverse effects, Antipsychotic Agents/therapeutic use, Benzodiazepines, Benzodiazepines/adverse effects, Benzodiazepines/therapeutic use, Randomized Controlled Trials as Topic, Schizophrenia, Schizophrenia/drug therapy
Plain language summary
Benzodiazepines for schizophrenia
Antipsychotic drugs are the primary method of treatment for people suffering from mental illness. However, many people with mental health problems do not respond well to antipsychotics which often are very good at treating positive symptoms (e.g. hearing voices or seeing things), but not so good for negative symptoms (e.g. loss of emotions, inactivity). In addition, antipsychotics can sometimes cause debilitating side effects such as movement disorders, weight gain, sleepiness and dizziness. If someone does not respond well to traditional antipsychotic drugs, psychiatrists are faced with the choice of switching to a different type of drug that may work better on its own; or adding a new drug or drugs to supplement the original antipsychotic drug treatment.
Benzodiazepines can be taken alone or in combination with more traditional antipsychotic drugs. They cause sedation, calmness and relax the muscles, so are helpful in calming down agitated people with anxiety, sleep problems, seizures, alcohol withdrawal and acute mental health problems.
This review found 34 studies with 2657 people. It compared benzodiazepines when used alone as the only medication or when used in combination with another drug for people with schizophrenia. Information from the 34 studies was generally poor, incomplete and badly reported. The 34 studies were of short duration and were small in size. The review suggests that there is little evidence to support the use of benzodiazepines either alone or in combination. However, benzodiazepines do have sedative properties that can calm people down and help them become less agitated for short periods of time. More research, particularly involving benzodiazepines as add‐on treatment used in combination with traditional antipsychotic drugs, is required.
This plain language summary has been written by Benjamin Gray, Service User and Service User Expert, Rethink Mental Illness, Email: ben.gray@rethink.org.
Summary of findings
Summary of findings for the main comparison. BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT for schizophrenia.
| Benzodiazepines as sole treatment versus placebo as sole treatment for schizophrenia | ||||||
|
Patient or population: people with schizophrenia Setting: inpatients and outpatients Intervention: benzodiazepines as sole treatment versus placebo as sole treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Benzodiazepines as sole treatment versus placebo as sole treatment | |||||
| No clinically important response to treatment ‐ short term | 667 per 1000 | 447 per 1000 (293 to 680) | RR 0.67 (0.44 to 1.02) | 382 (6 studies) | ⊕⊝⊝⊝ very low1,2,3 | |
| Leaving the study early due to any reason (overall acceptability of treatment) | 0 per 1000 | 0 per 1000 (0 to 0) | RR 0.89 (0.57 to 1.38) | 440 (8 studies) | ⊕⊝⊝⊝ very low4,5 | |
| Leaving the study early due to adverse effects (overall tolerability of treatment) | See comment | See comment | Not estimable | 161 (4 studies) | ⊕⊝⊝⊝ very low6,7 | |
| Number of participants tranquillised at endpoint of the study (Desired sedation/tranquillisation) | See comment | See comment | See comment | See comment | See comment | No included trial provided data for that outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes8. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Serious study limitations: High risk of bias regarding outcome data reporting in Merlis 1962 and attrition in two trials (Gundlach 1966; Hankoff 1962). 2 Substancial level of heterogeneity. 3 Serious imprecision: The 95% confidence interval around the pooled risk ratio includes both "no effect" and "appreciable benefit". Downgraded by 1. 4 High risk of bias regarding outcome data reporting in Merlis 1962 and attrition in two trials (Gundlach 1966; Hankoff 1962). Nishikawa 1982 used a cross‐over design. 5 Serious imprecision: The 95% confidence interval around the pooled risk ratio and the best estimate of effect includes both "no effect" and "appreciable benefit". Downgraded by 1. 6Merlis 1962 was characterised by incomplete outcome data reporting and Nishikawa used a cross‐over study design. 7 There were only few studies included that were characterised by a small sample size; one of these used a cross‐over design (Nishikawa 1982) and another one reported outcome data incompletely (Merlis 1962).
8 The basis for the assumed risk was the risk in the pooled control group of the relevant studies.
Summary of findings 2. BENZODIAZEPINES versus ANTIPSYCHOTICS for schizophrenia.
| Benzodiazepines versus antipsychotics for schizophrenia | ||||||
|
Patient or population: people with schizophrenia Setting: inpatients and outpatients Intervention: benzodiazepine monotherapy versus antipsychotic monotherapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Benzodiazepines versus antipsychotics | |||||
| No clinically important response to treatment ‐ ultra short term (at 60 minutes) | 286 per 1000 | 174 per 1000 (57 to 532) | RR 0.61 (0.2 to 1.86) | 44 (1 study) | ⊕⊝⊝⊝ very low1,2 | |
| No clinically important response to treatment ‐ ultra short term (at 12 hours) | 514 per 1000 | 386 per 1000 (226 to 668) | RR 0.75 (0.44 to 1.3) | 66 (1 study) | ⊕⊕⊝⊝ low2 | |
| No clinically important response to treatment ‐ short term | 150 per 1000 | 222 per 1000 (96 to 519) | RR 1.48 (0.64 to 3.46) | 112 (2 studies) | ⊕⊝⊝⊝ very low3,4 | |
| Leaving the study early due to any reason (overall acceptability of treatment) | 0 per 1000 | 0 per 1000 (0 to 0) | RR 0.73 (0.45 to 1.18) | 738 (13 studies) | ⊕⊝⊝⊝ very low5,6 | |
| Leaving the study early due to adverse effects (overall tolerability of treatment) | 0 per 1000 | 0 per 1000 (0 to 0) | RR 13 (0.78 to 216.39) | 444 (5 studies) | ⊕⊝⊝⊝ very low7,8 | |
| Desired sedation ‐ ultra short term ‐ asleep at 12 hours | 514 per 1000 | 386 per 1000 (226 to 668) | RR 0.75 (0.44 to 1.3) | 66 (1 study) | ⊕⊕⊝⊝ low8 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes10. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 The trial was not blinded. 2 Serious imprecision: The 95% confidence interval around the risk ratio includes both "no effect" and "appreciable benefit". Downgraded by 1. 3 Both trials were characterised by incomplete outcome data reporting. 4 Serious imprecision: The 95% confidence interval around the pooled risk ratio and the best estimate of effect includes both "no effect" and "appreciable harm". Downgraded by 1. 5 Two trials were non‐blinded (Garza‐Trevino 1989; TREC‐Rio 2003); Lerner 1979,Merlis 1962 and Hankoff 1962 reported outcome data incomplete and Nishikawa used a cross‐over study design. 6 Serious imprecision: The 95% confidence interval around the best estimate of effect includes both "no effect" and "appreciable benefit". Downgraded by 1. 7TREC‐Rio 2003 was not blinded, Lerner 1979 and Merlis 1962 reported outcome data incompletely and Nishikawa 1982 used a cross‐over trial design. 8 Serious imprecision: The 95% confidence interval of the risk ratio includes both "no effect" and appreciable harm". Downgraded by 1. 9 Both included studies were conducted as open trials.
10 The basis for the assumed risk was the risk in the pooled control group of the relevant studies.
Summary of findings 3. ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS for schizophrenia.
| Adjunctive benzodiazepines + antipsychotics versus placebo/no adjunctive treatment + antipsychotics for schizophrenia | ||||||
|
Patient or population: people with schizophrenia Setting: inpatients and outpatients Intervention: adjunctive benzodiazepines + antipsychotics versus placebo/no adjunctive treatment + antipsychotics | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Adjunctive benzodiazepines + antipsychotics versus placebo/no adjunctive treatment + antipsychotics | |||||
| No clinically important response to treatment ‐ ultra short term (at 60 minutes) | 286 per 1000 | 20 per 1000 (0 to 323) | RR 0.07 (0 to 1.13) | 45 (1 study) | ⊕⊝⊝⊝ very low1,2 | |
| No clinically important response to treatment ‐ ultra short term (at 12 hours) | 514 per 1000 | 437 per 1000 (262 to 725) | RR 0.85 (0.51 to 1.41) | 67 (1 study) | ⊕⊕⊝⊝ low2 | |
| No clinically important response to treatment ‐ short term (3 weeks or longer) | 307 per 1000 | 267 per 1000 (150 to 473) | RR 0.87 (0.49 to 1.54) | 511 (6 studies) | ⊕⊝⊝⊝ very low3,4 | |
| Leaving the study early due to any reason (overall acceptability of treatment) | 0 per 1000 | 0 per 1000 (0 to 0) | RR 1.36 (0.81 to 2.3) | 1185 (19 studies) | ⊕⊝⊝⊝ very low5,6 | |
| Leaving the study early due to adverse effects (overall tolerability of treatment) ‐ short term | 0 per 1000 | 0 per 1000 (0 to 0) | RR 3.24 (0.68 to 15.45) | 415 (6 studies) | ⊕⊝⊝⊝ very low7,8 | |
| Desired sedation ‐ ultra short term ‐ tranquillised at 60 minutes | 714 per 1000 | 992 per 1000 (757 to 1000) | RR 1.39 (1.06 to 1.83) | 45 (1 study) | ⊕⊕⊝⊝ low1 | |
| Desired sedation ‐ ultra short term ‐ asleep at 12 hours | 514 per 1000 | 437 per 1000 (262 to 725) | RR 0.85 (0.51 to 1.41) | 67 (1 study) | ⊕⊕⊝⊝ low9 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes10. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 The study was not double‐blinded. 2 Serious imprecision: The 95% confidence interval of the risk ratio includes both "no effect" and "appreciable benefit". Downgraded by 1. 3Xuan 2007 was not blinded and Lingjaerde 1982 used a cross‐over study design. 4 Serious imprecision: The 95% confidence interval of the pooled risk ratio includes both "no effect" and "appreciable benefit". Downgraded by 1. 5 Five studies were not blinded und three used a cross‐over design. 6 Serious imprecision: The 95% confidence interval of the pooled risk ratio includes both "no effect" and "appreciable harm". Downgraded by 1. 7 One study used a cross‐over‐design and three were characterised by selective reporting. 8 Serious imprecision: The 95% confidence interval of the pooled risk ratio and best estimate of effect includes both "no effect" and "appreciable harm". Downgraded by 1. 9 Serious imprecision: The 95% confidence interval of the risk ratio includes both "no effect" and "appreciable harm". Downgraded by 1.
10 The basis for the assumed risk was the risk in the pooled control group of the relevant studies.
Background
Description of the condition
Schizophrenia is a chronic and disabling psychiatric disorder. It afflicts approximately one per cent of the population worldwide with little gender differences. Schizophrenia ranks among the seven most frequent causes listed by the WHO for loss of years of life due to disability (WHO 2001). Its typical manifestations are "positive" symptoms such as fixed, false beliefs (delusions) and perceptions without cause (hallucinations), "negative" symptoms such as apathy and lack of drive, disorganisation of behaviour and thought, and catatonic symptoms such as mannerisms and bizarre posturing (Carpenter 1994). The degree of suffering and disability is considerable with 80% to 90% of those affected not working (Marvaha 2004) and up to 10% dying by suicide (Tsuang 1978).
Description of the intervention
Benzodiazepines are mainly characterised by their sedative and muscle‐relaxing properties. Therefore, these compounds are traditionally used for the pharmacotherapy of anxiety disorders, sleep disorders, seizures, and alcohol withdrawal. In schizophrenia, the administration of benzodiazepines can be considered to sedate and calm agitated people with acute schizophrenic episodes (Leucht 2011). The adverse effects that are experienced most frequently are drowsiness, dizziness, problems with concentration, and "paradoxical effects" including irritability, impulsivity as well as seizures. Rare, but very severe adverse effects of benzodiazepines are respiratory depression/arrest in short‐term treatment and, more commonly, the risk of dependence in case of long‐term medication.
Benzodiazepines are often classified according to their elimination half‐live (e.g. short‐acting with less than six hours and long‐acting with more than 24 hours).
How the intervention might work
The mechanism of action of benzodiazepines is characterised by enhanced activity of the inhibiting neurotransmitter GABA (gamma‐aminobutyric acid). The GABA‐A receptor in the synapsis of neurons has specific receptors for benzodiazepines. Interaction of benzodiazepines with these receptors results in a hyperpolarisation through increased opening of chloride ion channels (increase of the membrane potential). These molecular alterations result in an enhancement of GABA‐A receptor activity followed by decline of the excitability of neurons. All in all, these mechanisms cause the calming effects of the benzodiazepines due to reduced synaptic communication between neurons (Benkert 2011).
Why it is important to do this review
Antipsychotic drugs can be regarded as core treatment for both, acute and long‐term treatment of schizophrenia (Falkai 2005; Falkai 2006). The effectiveness of antipsychotics in schizophrenia could be demonstrated in a wide range of randomised controlled trials (Leucht 2009). But many people with schizophrenia do not achieve full remission of symptoms despite adequate antipsychotic drug treatment. At the present time, clozapine is considered as gold standard in the case of treatment‐resistance (Essali 2009), but due to its risk profile (especially in terms of agranulocytosis), most guidelines recommend clozapine administration only after non‐response to at least two adequate trials with different other antipsychotic compounds (Leucht 2011). However, many individuals with psychoses fail to respond satisfactorily to conventional treatment with first‐ and second‐generation antipsychotics, and clinicians are often faced with the choice of switching to alternate types of medication, or augmenting existing antipsychotics with other drugs or treatment options.
Therefore, the evaluation of additive therapeutic strategies in the presence of remaining relevant schizophrenic symptoms has high clinical relevance. Over the last forty years a variety of adjunctive treatment options have been evaluated concerning effectiveness in the treatment of schizophrenia (Christison 1991). In this context, the administration of various compounds such as carbamazepine (Leucht 2007a), lithium (Leucht 2007b), benzodiazepines, beta‐blockers (Cheine 2001) and valproate (Schwarz 2008) were examined in people with psychosis refractory to antipsychotic monotherapy. Additionally to the pharmacological approaches, other somatic therapy strategies such as electroconvulsive therapy have also been evaluated for people with schizophrenia (Tharyan 2005).
In this systematic review we examine the role of benzodiazepines in the treatment of schizophrenia. Companion reviews of our work‐group have examined the anticonvulsant carbamazepine (Leucht 2007a), lithium (Leucht 2007b) and valproate (Schwarz 2008) as sole or adjunctive treatment for schizophrenia. In contrast to this systematic review, another Cochrane review examined the effectiveness of benzodiazepines in acute psychoses irrespective of the underlying diagnosis (Gillies 2005), while we only included studies on schizophrenia and related disorders.
Objectives
To review the effectiveness of benzodiazepines for the treatment of schizophrenia and schizophrenia‐like psychoses, including examining whether:
benzodiazepines monotherapy is an effective treatment option for schizophrenia and schizophrenia‐like psychoses; and
benzodiazepine augmentation of antipsychotic medication is an effective treatment for the same illnesses.
Methods
Criteria for considering studies for this review
Types of studies
We only included studies that randomly assigned participants with schizophrenia or related disorders. We excluded quasi‐randomised studies such as those using allocation by day of the week, date of birth, alternate allocation. This decision is based on the evidence of a strong relationship between allocation concealment and direction of effect (Schulz 1995).
If a trial was described as "double‐blind", but randomisation was not explicitly mentioned, we implied that the study was randomised and assessed the inclusion of the trial in a sensitivity analysis. If there was no substantive difference within primary outcomes (see Types of outcome measures) when these "implied randomisation" studies were added, then we included these in the final analysis. If there was a substantive difference, we only analysed clearly randomised trials and described the results of the sensitivity analysis in the text.
Where people were given additional medications within the treatment group receiving benzodiazepines, we only included data if the adjunct treatment was evenly distributed between groups and it was only the medication with benzodiazepines that was randomised.
Randomised cross‐over studies were eligible, but only data up to the point of first cross‐over were used to avoid biases due to carry‐over effects of the treatments (Elbourne 2002).
Types of participants
We included people with the main diagnosis of schizophrenia and other types of schizophrenia‐like psychoses (e.g. schizophreniform, schizoaffective, or delusional disorders), irrespective of the diagnostic system applied. There is no clear evidence that the schizophrenia‐like psychoses are caused by fundamentally different disease processes or require different treatment approaches (Carpenter 1994). In accordance with the general strategy of the Cochrane Schizophrenia Group (Adams 2011), we also included studies that had used other diagnostic criteria than those of the International Statistical Classification of Diseases and Related Health Problems (ICD) and the Diagnostic and Statistical Manual of Mental Disorders (DSM). These diagnostic criteria are not meticulously used in clinical routine either, so broader inclusion criteria will enhance generalisability and representativeness. Trials randomising people with schizophrenia were included without restrictions concerning age, gender, and comorbidities.
To be included, at least 50% of the participants within a trial had to have a schizophrenic syndrome, or the results exclusively regarding the participants with schizophrenia were provided by the authors.
Types of interventions
Benzodiazepine alone.
Placebo (or no intervention).
Benzodiazepine in combination with any antipsychotic treatment: any dose.
Antipsychotic treatment (any dose).
Antipsychotic treatment (any dose) in combination with placebo (or no intervention).
Benzodiazepines could be applied in any dose and any route of administration (e.g. oral tablets, oral liquids, intramuscular injections, or intravenous injections).
Types of outcome measures
We grouped all outcomes by time ‐ ultra short term (up to 24 hours), short term (up to 12 weeks), medium term (13 to 26 weeks) and long term (over 26 weeks).
Primary outcomes
1. No clinically important response to treatment.
We defined this as less than 50% reduction from the baseline value of a rating scale such as the "Positive and Negative Syndrome Scale" (PANSS; Kay 1987) or the "Brief Psychiatric Rating Scale" (BPRS; Overall 1962) because validation studies have shown that this definition is clinically meaningful (Leucht 2005a; Leucht 2005b; Leucht 2006). If these data were not available, we used the definition provided in the included studies.
Secondary outcomes
1. Leaving the study early
1.1 Leaving the study early due to any reason (as a measure of overall acceptability) 1.2 Leaving the study early due to adverse effects (as a measure of overall tolerability) 1.3 Leaving the study early due to inefficacy of treatment (as a measure of overall efficacy)
2. Global state
2.1 Average score/change of the global state 2.2 Relapse ‐ as defined by each of the studies
3. Mental state
3.1 No clinically significant improvement of the general mental state ‐ as defined by each of the studies 3.2 Average score/change of the general mental state 3.3 No clinically significant response in terms of anxiety ‐ as defined by each of the studies 3.4 Average score/change of anxiety 3.5 No clinically significant response in terms of positive symptoms ‐ as defined by each of the studies 3.6 Average score/change of positive symptoms 3.7 No clinically significant response in terms of negative symptoms ‐ as defined by each of the studies 3.8 Average score/change of negative symptoms 3.9 No clinically significant response in terms of depressive symptoms ‐ as defined by each of the studies 3.10 Average score/change of depressive symptoms 3.11 No clinically significant response in terms of manic symptoms ‐ as defined by each of the studies 3.12 Average score/change of manic symptoms 3.13 Number of participants with clinically desired sedation 3.14 Average score/change of vigilance
4. Behaviour
4.1 General behaviour 4.2 Specific behaviours 4.2.1 Social functioning 4.2.2 Aggression against self, others, or property
5. Service utilisation
5.1 Days in hospital 5.2 Change in hospital status
6. Adverse effects
6.1 General adverse effects ‐ total number of patients with adverse effects 6.2 Specific adverse effects
7. Economic outcomes
7.1. Average change in total cost of medical and mental health care 7.2. Total indirect and direct costs
Search methods for identification of studies
No language restriction was applied to avoid the problem of "language bias" (Egger 1997b).
Electronic searches
The search methods for this 2011 update are documented below, for previous searches see Appendix 1.
Cochrane Schizophrenia Group Trials Register (February 2011)
We searched the register using the phrases:
[*abecarnil* OR *adinazolam* OR *AHN 086* OR *alpidem* OR *alprazolam* OR *anthramycin* OR *arfendazam* OR *azepam* OR *bentazepam* OR *benzodiazepine* OR *bretazenil* OR *bromazepam* OR *brotizolam* OR *CGS 20625* OR *CGS 8216* OR *CGS 9895* OR *CGS 9896* OR *chlordiazepoxide* OR *ciclotizolam* OR *ciprazafone* OR *clobazam* OR *clonazepa* OR *clotiazepam* OR *CP 32961* OR *DBCE* OR *devazepide* OR *dextofisopam* OR *diazepam* OR *dikaliumclorazepat* OR *divaplon* OR *dulozafone* OR *ELB 139* OR *emapunil* OR *endixaprine* OR *eszopiclone* OR *etizolam* OR *FG 7142* OR *flumazenil* OR *flunitrazepam* OR *flurazepam* OR *flutazoram* OR *GABA* OR *gedocarnil* OR *girisopam* OR *imidazenil* OR *JL 13* OR *KC 5944* OR *L 365260* OR *lorazepam* OR *lormetazepam* OR *lorzafone* OR *magnesium* OR *MCC* OR *medazepam* OR *metaclazepam* OR *midazolam* OR *nerisopam* OR *nitrazepam* OR *nordazepam* OR *oxazepam* OR *pazinaclone* OR *PBCC* OR *perlapine* OR *pinasepam* OR *pipequaline* OR *Pirenzepine* OR *PK 11195* OR *prazepam* OR *premazepam* OR *quazepam* OR *radequinil* OR *reclazepam* OR *ricasetron* OR *rilmazafone* OR *Ro 15‐4513* OR *Ro 19‐5686* OR *Ro 5‐4864* OR *sarmazenil* OR *SL 75102* OR *SR 95195* OR *suriclone* OR *temazepam* OR *tiagabine* OR *timelotem* OR *tofisopam* OR *tracazolate* OR *trepipam* OR *triazolam* OR *triflubazam* OR *Y 23684* OR *zaleplon* OR *zalospirone* OR *ZAPA* OR *ZK 90798* OR *ZK 91296* OR *ZK 93423* OR *zolam* OR *zolpidem* OR *zopiclone* in interventions of STUDY]
This register is compiled by systematic searches of major databases, hand searches and conference proceedings (http://szg.cochrane.org/cochrane‐schizophrenia‐group‐specialised‐register).
Searching other resources
1. Reference searching
We inspected the references of all identified studies for further relevant studies.
2. Personal contact
We contacted the first/corresponding author of each included study for information regarding unpublished trials and for missing information.
Data collection and analysis
Methods used in data collection and analysis for this update are documented below, for previous methods please see Appendix 2.
Selection of studies
Review authors MD and CL inspected all abstracts of studies identified as above and identified potentially relevant reports. In addition, to ensure reliability, MT and SL inspected a random sample of these abstracts, comprising 10% of the total. Where disagreement occurred this was resolved by discussion, or if there was still doubt, the full article was acquired for further inspection. The full articles of relevant reports were acquired for reassessment and carefully inspected for a final decision on inclusion (see Criteria for considering studies for this review). Once the full articles were obtained, in turn MD and CL inspected all full reports and independently decided whether they met the inclusion criteria. MD and CL were not blinded to the names of the authors, institutions, or journal of publication. Where difficulties or disputes arose, we asked author SL for help and if it was impossible to decide, we added these studies to those awaiting assessment and contacted the authors of the papers for clarification.
Data extraction and management
1. Extraction
Review authors MD and CL extracted data from all included studies. In addition, to ensure reliability, MT independently extracted data from a random sample of these studies, comprising 10% of the total. Again, any disagreement was discussed, decisions documented and, if necessary, we contacted the authors of studies for clarification. With remaining problems, SL helped to clarify issues and these final decisions were documented. We extracted data presented only in graphs and figures whenever possible but included the data only if two review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary. If studies were multicentre, wherever possible, we extracted data relevant to each component centre separately.
2. Management
2.1 Forms
We extracted data on standard simple forms.
2.2 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint) which can be difficult to conduct in unstable conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis as we used mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011).
2.3 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to all data before inclusion: (a) data from studies of at least 200 participants were entered in the analysis irrespective of the following rules, because skewed data pose less of a problem in large studies; (b) endpoint data: when a scale starts from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation. If this value was lower than one, it strongly suggested a skew and the study was excluded. If this ratio was higher than one but below two, there is suggestion of skew. We entered the study and tested whether its inclusion or exclusion substantially changed the results. If the ratio was larger than two the study was included, because skew is less likely (Altman 1996; Higgins 2011). (c) change data: when continuous data are presented on a scale which includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We entered the study, because change data tend to be less skewed and because excluding studies would also lead to bias, because not all the available information was used.
In case of skewness we displayed the data in an 'other data table' and did not calculate effect sizes. These tables can be found within the section "data analysis".
2.4 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.5 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into "clinically improved" or "not clinically improved". It can be generally assumed that if there is a reduction less than 50% in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986), this could be considered as "No clinically significant response" (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.6 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and used GRADE profiler (GRADE Profiler) to import data from RevMan 5 (Review Manager (RevMan)) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision making. We selected the following main outcomes for inclusion in the summary of findings table.
1. No clinically important response to treatment
2. Acceptability of treatment
leaving the study early due to any reason
leaving the study early due to adverse effects (tolerability of treatment)
3. Desired sedation/tranquillisation
number of participants tranquillised at endpoint of the study (ultra short term)
Assessment of risk of bias in included studies
Three review authors (MD, CL, MT) worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to measure trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data, selective reporting, and other biases. We did not include studies in this systematic review if sequence generation was inadequate.
If the raters disagreed, the final rating was made by consensus, with the involvement of another member of the review group (SL). Where inadequate details of randomisation and other characteristics of trials were provided, we contacted the authors of the studies in order to obtain further information. Non‐concurrence in quality assessment was reported, but if disputes arose as to which category a trial was to be allocated, again, resolution was made by discussion.
The level of risk of bias was noted in both, the text of the review and in the Table 1; Table 2; Table 3.
Measures of treatment effect
1. Dichotomous data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000).
2. Continuous data
For continuous outcomes, we estimated mean differences (MDs) between groups with their 95% confidence intervals (CIs).
Unit of analysis issues
1. Cluster trials
Studies increasingly employ "cluster randomisation" (such as randomisation by clinician or practice) but analysis and pooling of clustered data pose problems. Firstly, authors often fail to account for intra‐class correlation (ICC) in clustered studies, leading to a "unit of analysis" error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If clustering had not been accounted for in primary studies, we planned to present data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. If in subsequent versions of this review we include cluster trials, we will seek to contact first authors of studies to obtain ICCs for their clustered data and will adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will present these data as if from a non‐cluster randomised study but adjust for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a "design effect". This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC is not reported it will be assumed to be 0.1 (Ukoumunne 1999).
If cluster studies had been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involves more than two treatment arms, if relevant, the additional treatment arms were presented in comparisons. If data are binary these were simply added and combined within the two‐by‐two table. If data were continuous, we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions. Where the additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
1. Overall loss of credibility
At some degree loss of follow‐up, data must lose credibility (Xia 2009). The loss to follow‐up in randomised schizophrenia trials is often considerable calling the validity of the results into question. Nevertheless, it is unclear what degree of attrition leads to a high degree of bias. We did not exclude data from trials on the basis of the percentage of participants completing them. We, however, used the 'Risk of bias' tool described above to indicate potential bias when more than 25% of the participants left the studies prematurely, when the reasons for attrition differed between the intervention and the control group, and when no appropriate imputation strategies were applied.
2. Dichotomous data
Data were presented on a "once‐randomised‐always‐analyse" basis, assuming an intention‐to‐treat (ITT) analysis. If the authors applied such a strategy, we used their results. If the original authors presented only the results of the per‐protocol or completer population, we assumed that those participants lost to follow‐up would have had the same percentage of events as those who remained in the study.
3. Continuous data
3.1 General
Intention‐to‐treat was used when available. We anticipated that in some studies, in order to perform an ITT analysis, the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leon 2006). Therefore, where LOCF data were used in the analysis, it was indicated in the review.
3.2 Standard deviations
If standard deviations(SDs) were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals (CIs) available for group means, and either 'P' value or 't' values available for differences in mean, we can calculate them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): When only the SE is reported, SDs are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, t or F values, CIs, ranges or other statistics. If these formulae did not apply, we calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations. When such situations or participant groups arose, these were fully discussed.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods. When such methodological outliers arise these were fully discussed.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
Heterogeneity between studies was investigated by considering the I2 method alongside the Chi2 P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2011). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a confidence interval for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, was interpreted as evidence of substantial levels of heterogeneity (Higgins 2011). When substantial levels of heterogeneity were found in the primary outcomes, we explored reasons for heterogeneity (see Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997a). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In other cases, where funnel plots are possible, we sought statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage concerning the random‐effects model. It puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose random‐effects model for all analyses but additionally we investigated the use of a fixed‐model approach in sensitivity analyses for the primary outcomes.
Subgroup analysis and investigation of heterogeneity
Investigation of heterogeneity
If inconsistency was high, this was reported. First, we investigated whether data had been entered correctly. Second, if data were correct, the graph was visually inspected and outlying studies were successively removed to see if heterogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, data were presented. If not, data were not pooled and issues discussed. We know of no supporting research for this 10% cut off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity was obvious, we simply stated hypotheses regarding these for future reviews or versions of this review. We did not perform further analyses.
Sensitivity analysis
All sensitivity analyses were only applied for the primary outcome.
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as implied randomisation. For the primary outcomes, we included these studies in sensitivity analyses and if there were no substantive differences when the implied randomised studies were added to those with better description of randomisation, then all data were employed from these studies.
2. Implication of non double‐blind trials
We aimed to assess the exclusion of trials that were not double‐blind in a sensitivity analysis.
3. Fixed versus random‐effects models
All data were synthesised using a random‐effects model, however, we also calculated data for the primary outcome using a fixed‐effect model to evaluate whether the greater weights assigned to larger trials with greater event rates, altered the significance of the results compared with the more evenly distributed weights in the random‐effects model.
Results
Description of studies
Please also see Characteristics of included studies, Characteristics of excluded studies, and Characteristics of studies awaiting classification.
Results of the search
The initial search strategy identified more than 180 citations, of which about 100 were clearly unrelated to the topic of this review. We obtained and inspected 82 articles. Forty‐one studies were excluded and a further three studies are awaiting assessment. Overall 31 studies were included.
The update search in February 2011 yielded 80 further references of which 34 were potentially relevant and were regarded more closely. Three of these reports were included (Wang 2000; Ma 2006; Xuan 2007) and 24 studies were excluded and the reasons for exclusion documented in the Characteristics of included studies table. One citation could be a further relevant randomised trial (Davis 2008), but due to insufficient information it had to be classified as awaiting assessment. We have written to the corresponding author. Six references retrieved in the update search were reports of studies that had been already included in the first version of this review (a flowchart of the systematic literature search is provided in Figure 1). This systematic review now includes 34 studies while 66 reports are listed in the excluded studies table and four studies are still awaiting assessment.
1.
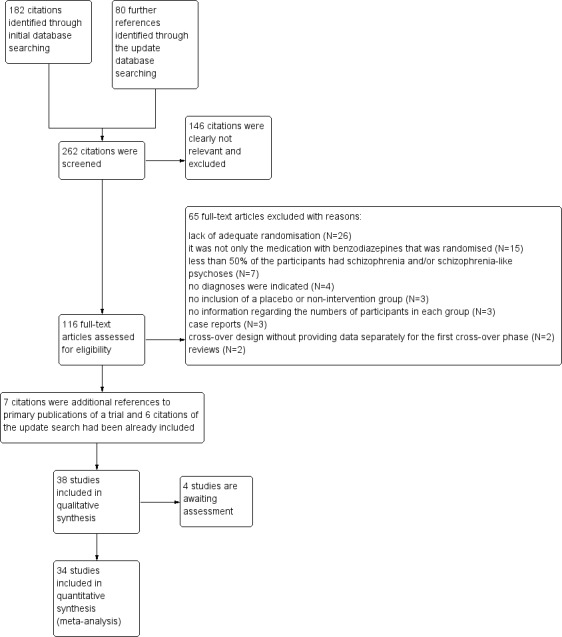
Study flow diagram.
Included studies
The 34 included studies were published between 1962 and 2007. Most of the studies were published in English, four in Chinese (Wang 2000; Wang 2003; Ma 2006; Xuan 2007) and one study in German (Marneros 1979).
2.1 Methods
All studies were randomised ("implied randomisation" in case of Marneros 1979) and most used double‐blind‐blind methodology. For further details please see sections below on allocation and blinding.
2.2 Study design
Thirty studies had a parallel group design, whereas four were designed as cross‐over studies (Holden 1968; Lingjaerde 1979; Lingjaerde 1982; Nishikawa 1982). No trial employed a cluster randomisation. Regarding dealing with missing data, many studies did not clearly indicate which analysis method was applied. Some stated the use of a completers‐only analysis, whereas, no trial report mentioned a last observation carried forward (LOCF) approach.
2.3 Duration
In 25 studies the benzodiazepines were administered over a short‐term period from one to 10 weeks (N = 25), in seven studies over an ultra short‐term period ‐ up to 24 hours, and in two studies (Cheung 1981; Nishikawa 1982) over a long‐term period (from 18 months to three years).
2.4 Participants
The 34 studies included a total of 2657 people. The number of people included in each study ranged from 12 (Nestoros 1982) to 301 (TREC‐Rio 2003). Nineteen studies included only persons with schizophrenia or schizophrenia‐like psychosis. The applied diagnostic criteria varied to a considerable degree because the studies were carried out at different times during a period of 45 years. Fifteen studies additionally included people with other diagnoses. They were included because at least 50% of the trial sample had a diagnosis of schizophrenia or schizophrenia‐like psychosis. There was one exception to this rule. Azima 1962 included 184 persons of which only 22 were diagnosed with schizophrenia or schizophrenia‐like psychosis, but the data used in this analysis exclusively refer to the 22 people with schizophrenia or schizophrenia‐like psychosis.
2.5 Setting
Eighteen trials included exclusively inpatients, five trials randomised outpatient participants. One study included both, inpatients and outpatients (Lingjaerde 1979), and another trial was conducted in an emergency room (TREC‐Rio 2003). There was no information regarding the setting available for the remaining nine trials examined in this review.
2.6 Interventions
Seven studies compared benzodiazepine as a sole agent with placebo and 13 trials compared benzodiazepines with antipsychotic drugs both given as monotherapy. Twenty included studies examined the effects of benzodiazepines as add‐on medication to antipsychotic compounds. Diazepam (N = 7) and chlordiazepoxide (N=7) were the most frequently administered benzodiazepines followed by clonazepam (N = 6) and lorazepam (N = 5). Other benzodiazepines such as alpidem, alrazolam, camazepam, estazolam, flunitrazepam, and midazolam were given in only a few cases. Dose of medication varied due to the duration of the studies and due to the mode of administration (see Characteristics of included studies).
The most often administered antipsychotic drug was haloperidol (N = 13) followed by chlorpromazine (N = 4). Occasionally fluphenazine, thioridazine or risperidone were given. Some studies did not indicate the antipsychotic used, but it was stated that the dose of the antipsychotic agents was kept constant.
2.7 Outcomes
2.7.1 No clinically important response to treatment
The primary outcome criteria "no clinically important response to treatment" was reported only by a limited number of the included studies (16 trials).
2.7.2 Rating scales
Different rating scales were used to assess clinical response and adverse effects. Details of scales that provided usable data are shown below. Reasons for exclusion of data from other instruments are given under "outcomes" in the Characteristics of included studies table.
2.7.2.1 Global state
2.7.2.1.1 Clinical Global Impression ‐ CGI (Guy 1976) A rating instrument commonly used in studies on schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement during therapy. A seven‐point scoring system is usually used with low scores indicating decreased severity and/or greater recovery. Chouinard 1993 and Foster 1997 reported data from this scale.
2.7.2.2 Mental state
2.7.2.2.1 Agitated Behaviour Scale ‐ ABS (Corrigan 1989) This is a 14‐item scale used for monitoring agitation levels. This scale has been applied in emergency department settings. The means and standard deviations for the total score and subscale scores are based on samples of persons with traumatic brain injury treated during the acute phases of recovery on an inpatient rehabilitation unit. A prospective sample of all participants with brain injuries, regardless of whether they were demonstrating agitation, revealed an overall mean ABS score of 21.01 and a standard deviation of 7.35 (Corrigan 1989). Battaglia 1997 used this scale.
2.7.2.2.2 Brief Psychiatric Rating Scale ‐ BPRS (Overall 1962) A brief rating scale used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. The scale has 18 items, and each item can be defined on a seven‐point scale varying from "not present" (1) to "extremely severe" (7). Scoring goes from 18 ‐126. Barbee 1992; Minervini 1990; Foster 1997; Wang 2000; Ma 2006 and Xuan 2007 reported data from the BPRS scale. Battaglia 1997 selected eleven psychosis/anxiety items (hostility, suspiciousness, uncooperativeness, unusual thought content, disorganised conceptualisation, hallucinatory behaviour, grandiosity, anxiety, excitement, tension and mannerisms/posturing) and termed this modified version the MBPRS.
2.7.2.2.3 Inpatient Multidimensional Psychiatric Scale ‐ IMPS (Lorr 1962) A rating scale used to assess the severity of a range of psychiatric symptoms. Higher scores indicate more symptoms. Further details could not be obtained. Chouinard 1993 reported data from this scale. 2.7.2.2.4 Malamud‐Sands Scale ‐ MMS (Malamud 1947) The scale consists of 19 items which can be divided into three major groups. The first seven comprise behavior items that can be directly observed at the time when the patient is interviewed. The next four are functions which are also objectively observable but which are based on the continuous observations by ward personnel during 24 hours which are reported to the rating psychiatrist. The third group comprises eight functions which can be evaluated only on the basis of an interview and on communication with the participants themselves. Only Merlis 1962 provided data from this scale.
2.7.2.2.5 Positive and Negative Symptom Scale ‐ PANSS (Kay 1987) A 30‐item scale used to assess the severity and range of general, positive and negative symptoms in schizophrenia. Each item is defined on a seven‐point severity scale from absent (1) to extreme (7). Scoring goes from 30 ‐210. Wang 2003 used this scale.
2.7.2.2.6 Scale for the Assessment of Negative Symptoms ‐ SANS (Andreasen 1989) This six‐point scale contains a global rating of the following negative symptoms: alogia, affective blunting, avolition‐apathy, anhedonia‐associality and attention impairment. Higher scores indicate more symptoms. Barbee 1992 reported data from this scale.
2.7.2.3 Behaviour
2.7.2.3.1 Nurses Observation Scale for Inpatient Evaluation ‐ NOSIE (Honigfeld 1965) An 80‐item scale with items rated on a five‐point scale from zero (not present) to four (always present). Ratings are based on behaviour over the previous three days. The seven headings are social competence, social interest, personal neatness, cooperation, irritability, manifest psychosis and psychotic depression. The total score ranges from zero to 320 with high scores indicating a poor outcome. Chouinard 1993 reported data from this scale.
2.7.2.4 Adverse effects
2.7.2.4.1 Simpson Angus Scale ‐ SAS (Simpson 1970) This 10‐item scale, with a scoring system of zero to four for each item, measures drug‐induced parkinsonism, a short term drug‐induced movement disorder. A low score indicates low levels of parkinsonism. Foster 1997 used this scale.
2.7.2.4.2 Treatment Emergent Symptom Scale/Form ‐ TESS/F (Guy 1976) This checklist assesses a variety of characteristics for each adverse effects, including severity, relationship to the drug, temporal characteristics (timing after a dose, duration and pattern during the day), contributing factors, course, and action taken to counteract the effect. Symptoms can be listed a priori or can be recorded as observed by the investigator. Ma 2006 reported data from this scale.
2.7.3 Missing outcomes
The primary outcome criteria "No clinically important response to treatment" was incompletely reported by the included studies. Special aspects, as for example, aggression and manic symptoms, were evaluated only by very few studies. Again, behaviour and adverse effects were incompletely reported. No data were available for economic consequences of treatment.
Excluded studies
Sixty‐five studies were excluded and displayed in the Characteristics of excluded studies table. The reasons for exclusion were that the participants were not randomised (N = 22), the randomisation procedure was inappropriate (N = 4), or in case of adjunct treatment it was not only the medication with benzodiazepines that was randomised (N = 15). Further reasons were that the studies did not include a placebo or non‐intervention group (N = 3), they were designed as cross‐over studies with unusable data of the first cross‐over phase (N = 2), case reports (N = 3), reviews (N = 2), did not report numbers in each group (N = 3), or the participants with schizophrenia and/or schizophrenia‐like psychosis were less than 50% of the trial sample and data were not provided separately for people with schizophrenia (N = 7), or no diagnosis was indicated (N = 4).
Studies awaiting assessment
We contacted the authors of two cross‐over studies (Maculans 1964; Ungvari 1999) to obtain data from the first cross‐over phase. We also contacted a further first author (Salzman 1991) to clarify initial group size and missing standard deviation. We have not yet received feedback from these authors. Another trial is currently awaiting assessment after the update search (Davis 2008)
Ongoing studies
We found no ongoing studies.
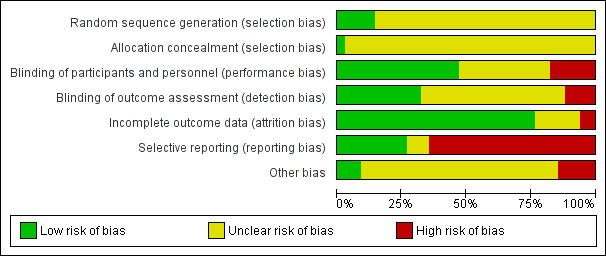
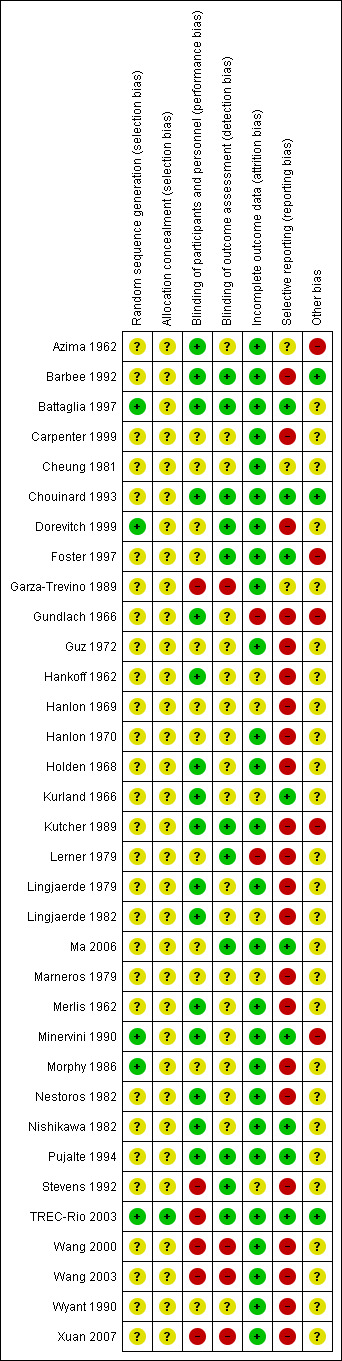
Risk of bias in included studies
For graphical representations of our judgements of risk of bias please refer to Figure 2 and Figure 3. Full details of judgements are provided in the risk of bias tables of the included studies.
2.
3.
Allocation
The participants of three studies (Battaglia 1997; Dorevitch 1999; TREC‐Rio 2003) were assigned by a table of random numbers and two trials were balanced for each group of six participants (Minervini 1990; Morphy 1986). These trials were classified as having a "Low risk of bias" in the quality score ranking. No further details on the randomisation method were available in 24 studies. Three trials stated at least to have used a "stratified randomisation procedure" (Hanlon 1969; Hanlon 1970; Carpenter 1999,) and in one study the randomisation procedure was based on a lottery (Wang 2000). In one publication, no randomisation was mentioned (Marneros 1979). Because the trial was described as "double‐blind" we implied that the study was randomised. All these 31 studies were given the quality score "Unclear risk of bias".
Regarding concealment of allocation, only one study provided enough information to permit judgement of "Low risk of bias" in the quality score (TREC‐Rio 2003).
Blinding
Performance‐Bias: Most of the included studies were declared as "double‐blind". In 10 of these, the trial authors provided no further information concerning the mechanism of blinding ("Unclear risk of bias"), while 16 provided information to allow judgement with "Low risk of bias" in the quality tool. Most of these trials used identical capsules for blinding. Two studies (Wyant 1990; Ma 2006 ) were described as single‐blind without further information ("Unclear risk of bias") and six studies (Garza‐Trevino 1989; Stevens 1992; Wang 2000; Wang 2003; TREC‐Rio 2003; Xuan 2007) were not blinded (classified as "High risk of bias").
Detection‐Bias: 19 publications did not provide enough information to allow classification of high or low risk of bias. In 11 trials, we assessed a "Low risk" for a detection‐bias, while four studies appeared to have a "High risk" regarding this bias (Garza‐Trevino 1989; Wang 2000; Wang 2003; Xuan 2007). The open studies of Stevens 1992 and TREC‐Rio 2003 were blinded for the assessment of the main outcome and therefore classified as "Low risk of bias".
Incomplete outcome data
The overall‐attrition (participants who left the trials early for any indication) was low (< 10%) in 27 trials (rated as "Low risk of bias" with the exception of the study by Kurland 1966 that was characterised by highly different attrition rates in the intervention‐ and control group and therefore classified as "Unclear risk of bias") and moderate (10% to 25%) in five studies (Hankoff 1962; Hanlon 1969; Marneros 1979; Lingjaerde 1982; Stevens 1992). Moderate attrition was judged as "Unclear risk of bias" because the trial authors of these studies did not provide sufficient information to judge if the analysis methods were appropriate to deal with the missing data. In two trials the attrition could be considered as high (> 25%) (Gundlach 1966; Lerner 1979) (rated as "High risk of bias"). Thirteen studies reported the number of participants leaving the study early due to adverse effects and 11 studies reported the number of participants leaving the study early due to inefficacy of treatment. In most of the publications the trial authors used a completers‐analysis.
Selective reporting
The outcome‐data reporting was incomplete in 22 studies (rated as "High risk of bias"). In several instances the data had to be estimated from figures which led to imprecision. Ten trials appeared to be free of selective reporting and were rated with "Low risk of bias" in the quality score, although in many of these publications the reporting of adverse effects was incomplete.
Other potential sources of bias
Only three studies seemed to be free of other potential sources of bias and were assigned a "Low risk of bias" (Cheung 1981; Barbee 1992; TREC‐Rio 2003). In 25 studies the risk of bias was considered as "unclear" due to a lack of available information in the publications. Thus, there was insufficient information to assess whether an important risk of bias exists. Two trials were characterised through extreme baseline imbalances regarding the different study groups (Azima 1962; Foster 1997). Other reasons for rating "High risk of bias" are provided in the Characteristics of included studies table.
Effects of interventions
See: Table 1; Table 2; Table 3
1. COMPARISON 1: BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT
1.1 Primary outcome: No clinically important response to treatment
Six short‐term studies (Azima 1962, Gundlach 1966, Hankoff 1962, Merlis 1962, Minervini 1990, Nestoros 1982) reported the number of participants without a clinically significant response to treatment. Combining the data of these trials demonstrated no significant difference in favour of any treatment modality (N = 382, 6 RCTs, RR 0.67 CI 0.44 to 1.02). The I² value of 62% and the significant heterogeneity test (P = 0.02) indicated a substantial level of heterogeneity. The included long‐term studies did not provide data for the primary outcome.
1.2 Secondary outcomes
1.2.1 Leaving the study early
1.2.1.1 Leaving the study early due to any reason
Eight studies contributed data to this outcome. Seven studies were short term and one was long term. In the short‐term studies there was no statistically significant difference between participants treated with benzodiazepines and placebo (N = 417, 7 RCTs, RR 0.89 CI 0.57 to 1.38) and in the long‐term trial, no participant left the trial early (Nishikawa 1982).
1.2.1.2 Leaving the study early due to adverse effects
Three short‐term studies (Merlis 1962; Minervini 1990; Nestoros 1982) indicated the reason for premature discontinuation was due to adverse effects. As there no one left early in either group, the risk ratio could not be estimated. The same holds true for the only long‐term study (Nishikawa 1982).
1.2.1.3 Leaving the study early due to inefficacy of treatment
Again, four trials (Merlis 1962; Minervini 1990; Nestoros 1982; Nishikawa 1982) stated the number of participants leaving the study early was due to inefficacy of treatment. No attrition was observed.
1.2.2 Global state
1.2.2.1 Relapse
Two trials reported relapse rates after one year, one short‐term study (Carpenter 1999) and one long‐term study (Nishikawa 1982). We found no significant difference between the treatment and placebo groups in both trials (short term: N = 35, 1 RCT, RR 0.67 CI 0.36 to 1.23; long term: N = 23, 1 RCT, RR 1.00 CI 0.85 to 1.18).
1.2.3 Mental state
1.2.3.1 General: BPRS and MMS scale
Merlis 1962 analysed the number of participants unimproved or worse according to the BPRS and the MMS total scores. There were no significant differences between groups, neither in terms of the BPRS (N = 60,1 RCT, RR 1.00 CI 0.39 to 2.53), nor in terms of the MMS (N = 60,1 RCT, RR 0.90 CI 0.52 to 1.57). In contrast to the dichotomous data, we found that the mean BPRS scores at three weeks favoured benzodiazepines compared with placebo on a statistically significant level (Minervini 1990, N = 66,1 RCT, MD ‐17.60 CI ‐22.61 to ‐12.59).
1.2.3.2 Specific: anxiety symptoms (TSRS and HAM‐A scale) Two studies rated anxiety (Gundlach 1966; Minervini 1990) with special scales. Gundlach 1966 used the TSRS at six weeks and found no evidence for significant group difference (N = 39,1 RCT, RR 1.16 CI 0.63 to 2.15). Minervini 1990 employed the HAM‐A rating scale at three weeks and reported a significant advantage in favour of the treatment group. However, data were skewed and could therefore only be displayed in an "other data table".
1.2.4 Adverse effects
1.2.4.1 General: total number of participants with adverse effects
Only Gundlach 1966 analysed how many participants experienced at least one adverse effect. The difference between the two groups was statistically significant (N = 100,1 RCT, RR 1.44 CI 1.02 to 2.04, number needed to harm (NNTH) 5 CI 3 to 50) with more participants in the benzodiazepine group suffering from adverse effects than in the placebo group.
1.2.4.2 Specific adverse effects
1.2.4.2.1 Anorexia Hankoff 1962 reported on anorexia. No participant experienced this adverse effect (N = 122,1 RCT, RR not estimable).
1.2.4.2.2 Autonomic reactions Gundlach 1966 observed 13 participants in the benzodiazepine group and seven participants in the placebo group with autonomic reactions such as flushing or dry mouth. The difference did not reach statistical significance (N = 100,1 RCT, RR 1.71 CI 0.75 to 3.93).
1.2.4.2.3 Cardiovascular reactions, depression and dryness of mouth Hankoff 1962 reported on cardiovascular reactions, depression and dryness of mouth, but no participant in neither group experienced these adverse effects (N = 122,1 RCT, RR not estimable).
1.2.4.2.4 Energy level Two studies (Hankoff 1962; Gundlach 1966) examined changes in energy level. We found no significant difference regarding an increase in energy level between groups (N = 222, 2 RCTs, RR 0.62 CI 0.02 to 22.78). An I² value of 69% was assessed and the Chi2 test indicated a P value of P = 0.07. For the pooled results of both trials, significantly more participants with a decrease in energy level in the benzodiazepine group than in the control group were identified (N = 222, 2 RCTs, RR 2.18 CI 1.38 to 3.43, NNTH 5 CI 3 to 25).
1.2.4.2.5 Gastrointestinal reactions Three studies (Gundlach 1966; Hankoff 1962; Minervini 1990) indicated the numbers of participants experiencing gastrointestinal reactions on the prescribed study medication, but no significant difference between the groups could be identified (3 RCTs, N = 288, RR 0.99 CI 0.29 to 3.37).
1.2.4.2.6 Headache Regarding headache, there was no significant difference between benzodiazepines and placebo (Gundlach 1966; Hankoff 1962) (N = 222, 2 RCTs, RR 0.75 CI 0.20 to 2.81).
1.2.4.2.7 Insomnia In Hankoff 1962 only one participant in the placebo group complained about insomnia (N = 122,1 RCT, RR 1.14 CI 0.05 to 27.28).
1.2.4.2.9 Movement disorder Two studies (Azima 1962; Hankoff 1962) investigated the occurrence of movement disorders and we found participants in the benzodiazepine group experienced significantly more ataxia compared to the placebo group (N = 144, 2 RCTs, RR 8.18 CI 1.35 to 49.74, NNTH not significant). Hankoff 1962 also reported on EPS in general, but no participant in either group experienced this adverse effect (N = 122,1 RCT, RR not estimable).
1.2.4.2.10 Sedation Altogether, in two studies (Minervini 1990; Nestoros 1982) only one participant in the pooled benzodiazepine group suffered from extreme sedation (N = 78, 2 RCTs, RR 3.00 CI 0.15 to 61.74).
1.3 Sensitivity analyses
1.3.1 Implication of randomisation
We aimed to conduct a sensitivity analysis in terms of the primary outcome when a trial was not clearly stated to be randomised but described as "double‐blind". This affects no study which contributed data to this comparison. Therefore, the sensitivity analysis was not undertaken.
1.3.2 Implication of non double‐blind trials
No non‐double blind trial was included in the pooled data‐analysis of the primary outcome.Therefore, no sensitivity analysis was conducted.
1.4 Fixed and random effects
When using a fixed‐effect model to analyse the data for the primary outcome, we assessed a statistically significant superiority of benzodiazepines compared with placebo (N = 382, 6 RCTs, RR 0.66 CI 0.5 to 0.8) accompanied by a substantial level of heterogeneity (I² = 62%, heterogeneity test: P = 0.02).
1.5 'Summary of findings' table
The results of three outcomes ‐ no clinically important response to treatment, leaving the study early due to any reason, and leaving the study early due to adverse effects ‐ were considered more closely in a 'Summary of findings' table (see Table 1). The judgements derived from this instrument were used for the discussion section of the review (see Discussion ‐ Summary of main results
2. COMPARISON 2: BENZODIAZEPINES versus ANTIPSYCHOTICS
2.1 Primary outcome: No clinically important response to treatment
Focussing on global clinical response only four studies (Battaglia 1997; Garza‐Trevino 1989; Hankoff 1962; Merlis 1962) evaluated the number of participants who experienced no clinically significant response to treatment. Garza‐Trevino 1989 investigated the participants after 30 and 60 minutes, Battaglia 1997 after 12 hours, Hankoff 1962 after two weeks, and Merlis 1962 after four weeks. We found no significant differences between groups (30 minutes: N = 44,1 RCT, RR 0.91 CI 0.58 to 1.43; 60 minutes: N = 44,1 RCT, RR 0.61 CI 0.20 to 1.86; 12 hours: N = 66,1 RCT, RR 0.75 CI 0.44 to 1.30; pooled short‐term studies: N = 112, 2 RCTs, RR 1.48 CI 0.64 to 3.46). In summary, no significant evidence was found to suggest that one agent was superior to the other.
2.2 Secondary outcomes
2.2.1 Leaving the study early
2.2.1.1 Leaving the study early due to any reason
Thirteen studies indicated the number of participants leaving the study early due to any reason. Seven studies were ultra short term, four were short term, and two were long‐term studies. In neither group was the difference between the medication with benzodiazepines and antipsychotics significant (ultra short: N = 514, 7 RCTs, RR 0.67 CI 0.33 to 1.36; short term: N = 161, 4 RCTs, RR 0.65 CI 0.27 to 1.56; long term: N = 63, 2 RCTs, 2 RCTs, RR 5.00 CI 0.26 to 96.13).
2.2.1.2 Leaving the study early due to adverse effects
Regarding attrition due to adverse terms in three studies of the ultra short‐term category (Lerner 1979; TREC‐Rio 2003; Wyant 1990), no statistically significant difference was found between benzodiazepines and antipsychotics (N = 351, 3 RCTs, RR 13.00 CI 0.78 to 216.39). Only Merlis 1962 and Nishikawa 1982 reported on leaving early due to adverse effects in the short and long term, respectively, but again, no significant differences could be detected between the pooled results of the intervention and control groups (short term: N = 60,1 RCT, RR not estimable; long term: N = 33,1 RCT, RR not estimable).
2.2.1.3 Leaving the study early due to inefficacy of treatment
Additionally, there were no significant differences in terms of premature discontinuation due to inefficacy of treatment in either category (ultra short term: N = 311, 2 RCTs, RR not estimable; short term: N = 76, 2 RCTs, RR 0.33 CI 0.02 to 7.14; long term: N = 33,1 RCT, RR not estimable).
2.2.2 Global state
2.2.2.1 CGI severity score
Three studies (Chouinard 1993; Foster 1997; Lerner 1979) reported on the mean CGI severity score at endpoint as a measure of the participants' global state. Foster 1997 assessed one and four hours, Chouinard 1993 two hours, and Lerner 1979 four, and 24 hours. Foster 1997 revealed a statistically significant superiority of benzodiazepines at one hour (N = 37, 1 RCT, MD ‐0.67 CI ‐1.09 to ‐0.25) but not at four hours (N = 37, 1 RCT, MD ‐0.62 CI ‐1.36 to 0.12). The data at two hours (Chouinard 1993), four hours (Lerner 1979), and 24 hours (Lerner 1979) were skewed and could only be displayed in an "other data table".
2.2.2.2 Relapse
Relapse was reported by Carpenter 1999 at four weeks, and by Cheung 1981 and Nishikawa 1982 at one year or longer. There was no significant between‐group difference in the short‐term study (N = 33, 1 RCT, RR 0.84 CI 0.43 to 1.66) as well as in the pooled overall result of the two long‐term studies (N = 63, 2 RCTs, RR 2.02 CI 0.37 to 11.04). With an I² value of 83% and a significant heterogeneity test (P = 0.01), the heterogeneity between the two long‐term studies (Cheung 1981; Nishikawa 1982) was substantial.
2.2.3 Mental state
2.2.3.1 General: BPRS, MBPRS and MMS scale
Merlis 1962 reported on the number of participants without an improvement in general mental state using the BPRS and the MMS scale, but the data from both scales were not significantly different between groups (BPRS: N = 60,1 RCT, RR 2.5 CI 0.6 to 10.3; MMS: N = 60,1 RCT, RR 1.3 CI 0.6 to 2.1). Foster 1997 and Lerner 1979 evaluated the general mental state by mean BPRS total score at one hour (Foster 1997), four hours (Lerner 1979; Foster 1997), and 12 to 24 hours (Lerner 1979). Because of skewness the 12 to 24‐hour data by Lerner 1979 were displayed in an 'other data table' (see Analysis 2.11). Foster 1997 showed wide confidence intervals without a statistically significant difference between groups (N = 37, 1 RCT: one hour MD ‐3.26 CI ‐10.7 to 4.1; four hours MD ‐1.73 CI ‐9.6 to 6.1). Battaglia 1997 rated the mean MBPRS scores at one and 12 hours. One‐hour data were statistically significant favouring the control group (N = 66,1 RCT, MD 6.00 CI 0.68 to 11.32). Twelve‐hour data were not significantly different (N = 66,1 RCT, MD 1.00 CI ‐4.32 to 6.32).
2.11. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 11 Mental state: 2b . General ‐ BPRS ‐ ultra short term (skewed data).
| Mental state: 2b . General ‐ BPRS ‐ ultra short term (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Mean BPRS score at 4 hours | ||||
| Lerner 1979 | Benzodiazepines | 11.40 | 4.20 | 11 |
| Lerner 1979 | Antipsychotics | 3.80 | 5.20 | 9 |
| Mean BPRS score at 12‐24 hours | ||||
| Lerner 1979 | Benzodiazepines | 6.30 | 6.80 | 20 |
| Lerner 1979 | Antipsychotics | 8.0 | 8.30 | 20 |
2.2.3.2 Specific: manic symptoms (IMPS subscore)
Because of skewness data by Chouinard 1993 could only be displayed in an "other data table".
2.2.3.3 Specific: sedation
Three studies (Battaglia 1997; Garza‐Trevino 1989; TREC‐Rio 2003) evaluated sedation at 20 minutes (TREC‐Rio 2003), 30 minutes (Garza‐Trevino 1989), 40 minutes (TREC‐Rio 2003), 60 minutes (Garza‐Trevino 1989; TREC‐Rio 2003), 120 minutes (TREC‐Rio 2003), and 12 hours (Battaglia 1997). The results at 20 minutes showed a statistically significant superiority of benzodiazepines (N = 301, 1 RCT, RR 1.32 CI 1.16 to 1.49, number needed to benefit (NNTB) 5 CI 3 to 8). The results of Garza‐Trevino 1989 at 30 minutes were not significant but showed a trend similar to the results of TREC‐Rio 2003 at 20 minutes (N = 44,1 RCT, RR 1.17 CI 0.53 to 2.59). At 40 minutes, we again found a significant superiority of benzodiazepines in terms of sedation compared to antipsychotic drugs (N = 301, 1 RCT, RR 1.13 CI 1.04 to 1.23, NNTB 9 CI 6 to 33). However, sedation at 60 minutes was not significantly different (N = 345, 2 RCTs, RR 1.07 CI 1.00 to 1.15). Also, no significant difference was found at 120 minutes in TREC‐Rio 2003 (N = 301, 1 RCT, RR 1.04 CI 0.98 to 1.10). The 12‐hour data of the study by Battaglia 1997 were equivocal (N = 66,1 RCT, RR 0.75 CI 0.44 to 1.30). Chouinard 1993 examined sedation with the mean CGI sedation score at two hours. We could not reveal any significant between‐group difference (N = 16, 1 RCT, MD 0.10 ‐0.98 to 1.18).
2.2.4 Behaviour
2.2.4.1 General: modified NOSIE score
Chouinard 1993 reported on general behaviour at 2 hours using the NOSIE scale. The data were skewed and could therefore only be displayed in the "other data table".
2.2.4.2 Specific: aggression
Aggression data were reported (TREC‐Rio 2003) at different times with three criteria: Needing restraints within two hours, needing additional tranquillising drugs at two hours, and any episode of aggression at 24 hours. None of these items revealed any significant differences between groups (needing restraints, N = 301, 1 RCT, RR 0.82 CI 0.55 to 1.22); (any episode of aggression, N = 301, 1 RCT, RR 1.12 CI 0.78 to 1.62); (needing additional drugs, N = 301, 1 RCT, RR 0.28 CI 0.06 to 1.34). Battaglia 1997 reported on aggression using the mean ABS score at one and at 12 hours. There were no significant differences between groups (mean ABS at one hour, N = 66, 1 RCT, MD 4.00 CI ‐1.32 to 9.32; mean ABS at 12 hours, N = 66, 1 RCT, MD 3.00 CI ‐2.32 to 8.32).
2.2.5 Service utilisation
TREC‐Rio 2003 documented how many participants were still in hospital after two weeks of pharmacotherapy. There was no statistically significant between‐group difference between benzodiazepine and antipsychotic treated participants (N = 301, 1 RCT, RR 0.96 CI 0.77 to 1.18).
2.2.6 Adverse effects
2.2.6.1 General: total number of patients with adverse effects
Two studies (Battaglia 1997; Hankoff 1962) analysed how many participants experienced at least one adverse effects without verifying any statistically significant difference between the study groups (N = 118, 2 RCTs, RR 0.73 CI 0.45 to 1.17).
2.2.6.2 Specific adverse effects
2.2.6.2.1 Anorexia In Hankoff 1962, two participants in the antipsychotic drug group suffered from anorexia. The between‐group difference was not significant (N = 52, 1 RCT, RR 0.19 CI 0.01 to 3.69).
2.2.6.2.2 Cardiovascular reactions Only Hankoff 1962 reported on the rate of cardiovascular reactions in the two treatment groups, but no statistically significant difference was found (N = 52, 1 RCT, RR 0.19 CI 0.01 to 3.69).
2.2.6.2.3 Energy level Hankoff 1962 observed six participants in the benzodiazepine group and 10 participants in the antipsychotic group with a decrease in energy level (N = 52, 1 RCT, RR 0.56 CI 0.24 to 1.30). Also the outcome of "increase in energy level" was equivocal (N = 52, 1 RCT, RR 0.93 CI 0.06 to 14.03).
2.2.6.2.4 Depression Only Hankoff 1962 reported on depression without revealing any significant difference between the two study groups ( N = 52, 1 RCT, RR 0.19 CI 0.01 to 3.69).
2.2.6.2.5 Dizziness In Battaglia 1997, benzodiazepines did not produce more dizziness than antipsychotics (N = 66, 1 RCT, RR 1.13 CI 0.25 to 5.19).
2.2.6.2.6 Dryness of mouth Two studies (Hankoff 1962; Battaglia 1997) examined dryness of mouth. We could not identify any significant difference between groups (N = 118, 2 RCTs, RR 1.36 CI 0.35 to 5.33).
2.2.6.2.7 Gastric reactions, headache and insomnia In Hankoff 1962, no participant suffered from gastric reactions or insomnia (N = 52, 1 RCT, RR not estimable). Two participants in the antipsychotic group experienced a headache, but data were not statistically significant (N = 52, 1 RCT, RR 0.19, CI 0.01 to 3.69).
2.2.6.2.8 Movement disorder Altogether, three studies provided information regarding movement disorders (Battaglia 1997; Foster 1997; Hankoff 1962). These included reports referring to the movement disorders ataxia (Hankoff 1962; Battaglia 1997), dystonia (Battaglia 1997), parkinsonism (Battaglia 1997), slight dysarthria (Battaglia 1997), tremor (Battaglia 1997), and extrapyramidal adverse effects (EPS) in general (Foster 1997; Hankoff 1962) without revealing any statistically significant between‐group differences: ataxia (ultra short term: N = 66, 1 RCT, RR 2.26 CI 0.22 to 23.71; short term: N = 52, 1 RCT, RR 0.31 CI 0.030 to 2.78), dystonia (N = 66, 1 RCT, RR 0.16, CI 0.02 to 1.24), parkinsonism (N = 66, 1 RCT, RR 0.16 CI 0.01 to 2.99), slight dysarthria (N = 66, 1 RCT, RR 0.56 CI 0.11 to 2.87), tremor (N = 66,1 RCT, RR 0.56 CI 0.05 to 5.93), and EPS in general (ultra short term: N = 37,1 RCT, RR not estimable; short term: N = 52, 1 RCT, RR 0.19 CI 0.01 to 3.69). Chouinard 1993 evaluated movement disorders with the mean "Parkinsonism Total Score" and the mean "Tardive Dyskinesia Score" at two hours. The data were skewed and could therefore only be displayed in the "other data table". Battaglia 1997 and Foster 1997 reported on the use of antiparkinson medication, but we found no significant difference between groups (N = 103, 2 RCTs, RR 0.50 CI 0.17 to 1.47).
2.2.6.2.9 Respiratory depression In the relatively large trial by TREC‐Rio 2003 only one participant with benzodiazepine medication suffered from respiratory depression (N = 301, 1 RCT, RR 2.98 CI 0.12 to 72.58).
2.2.6.2.10 Sedation Extreme sedation (Foster 1997) was equivocal between groups (N = 37, 1 RCT, RR 1.76 CI 0.33 to 9.364).
2.2.6.2.11 Seizure In TREC‐Rio 2003, one participant in the antipsychotic group experienced a seizure, but data did not reach a statistically significant level (N = 301, 1 RCT, RR 0.33 CI 0.01 to 8.06).
2.3 Sensitivity analyses
2.3.1 Implication of randomisation
Because all trials that contributed data to the primary outcome were clearly stated to be randomised, the reason for performing the preplanned sensitivity analysis was not given. Therefore, sensitivity analysis was not undertaken.
2.3.2 Implication of non double‐blind trials
No non‐double blind trials were included in the pooled data‐analysis of the primary outcome.Therefore, no sensitivity analysis was conducted.
2.4 Fixed and random effects
Using a fixed‐effect model to analyse the data for the primary outcome did not alter the main findings (statistically significant between‐group differences) in the relevant comparisons (fixed‐effect model: 30 minutes: N = 44, 1 RCT, RR 0.91 CI 0.58 to 1.43; 60 minutes: N = 44, 1 RCT, RR 0.61 CI 0.20 to 1.86; 12 hours: N = 66, 1 RCT, RR 0.75 CI 0.44 to 1.30; pooled short‐term studies: N = 112, 2 RCTs, RR 1.58 CI 0.68 to 3.67).
2.5 'Summary of findings' table
The results of four outcomes ‐ no clinically important response to treatment, leaving the study early due to any reason, leaving the study early due to adverse effects, and desired sedation ‐ were considered more closely in a 'Summary of findings' table (see Table 2). The judgements derived from this instrument were used for the discussion section of the review (see Discussion ‐ Summary of main results).
3. COMPARISON 3: ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS
3.1 Primary outcome: No clinically important response to treatment
Data concerning the primary outcome were reported by eight studies at different time points. The number of participants failing to achieve clinically important response to treatment was provided for the study duration of 30 minutes (Garza‐Trevino 1989), 60 minutes (Garza‐Trevino 1989), 12 hours (Battaglia 1997), and three weeks or longer (Guz 1972; Hanlon 1969; Hanlon 1970; Lingjaerde 1982; Morphy 1986; Xuan 2007). Only the results at 30 minutes showed a statistically significant superiority of the benzodiazepine augmentation (30 minutes: N = 45, 1 RCT, RR 0.38 CI 0.18 to 0.80, NNTH 2, CI 2 to 7. Neither 60 minutes data (N = 45, 1 RCT, RR 0.07 CI 0.00 to 1.13), 12 hour data (N = 67, 1 RCT, RR 0.85 CI 0.51 to 1.41) nor three weeks or longer data were statistically significant (N = 511, 6 RCTs, RR 0.87 CI 0.49 to 1.54). Regarding the pooled results of the six short‐term studies substantial heterogeneity ‐ represented through a significant heterogeneity test (P = 0.008) and an I² value of 68% ‐ must be considered.
3.2 Secondary outcomes
3.2.1 Leaving the study early
3.2.1.1 Leaving the study early due to any reason
Nineteen studies contributed to the outcome "number of participants leaving the study early" due to any reason. Three studies fell in the ultra short term and 16 studies fell in the short‐term category. In the ultra short‐term studies, no participant in either group left the study early (N = 140, 3 RCTs, RR not estimable). Short‐term studies revealed a slight trend towards higher attrition rates in the intervention group (antipsychotics plus benzodiazepines), but the difference was not statistically significant (N = 1045, 16 RCTs, RR 1.36 CI 0.81 to 2.30).
3.2.2.2 Leaving the study early due to adverse effects
Assessing the number of participants with premature termination of treatment due to adverse effects did not reveal any significant difference between the benzodiazepine augmentation group and the control group (N = 415, 6 RCTs, RR 3.24 CI 0.68 to 15.45).
3.2.2.3 Leaving the study early due to inefficacy of treatment
The between‐group difference was not statistically significant concerning the number of participants leaving the study early due to inefficacy of treatment (N = 347, 6 RCTs, RR 0.76 CI 0.17 to 3.42).
3.2.3 Global state
3.3.1 CGI severity score
Lingjaerde 1982 applied the mean CGI severity score for the evaluation of the global state, but the data were skewed and could therefore only be displayed in an "other data" table.
3.2.4 Mental state
3.2.4.1 General (BPRS, MBPRS and PANSS scale)
Five studies examined the participants' general state at different time points and using different scales. In Battaglia 1997 there were no statistically significant between‐group differences in mean MBPRS scores at one hour (N = 67,1 RCT, MD ‐4.00 CI ‐9.51 to 1.51) and at 12 hours (N = 67, 1 RCT, MD 2.00 CI ‐3.27 to 7.27). In Barbee 1992 mean BPRS scores at three days favoured significantly the control group (N = 28, 1 RCT, MD 6.81 CI 0.32 to 13.30) but not at one hour ( N = 28, 1 RCT, MD ‐3.11 CI ‐8.86 to 2.64), 24 hours (N = 28, 1 RCT, MD 0.01 CI ‐7.28 to 7.28), and two days (N = 28, 1 RCT, MD 2.00 CI ‐3.86 to 7.86). In Wang 2003, we found no significant difference in terms of PANSS total scores at two weeks (N = 80, 1 RCT, MD ‐6.20 CI ‐12.55 to 0.15) and in Stevens 1992 there was no significant difference in terms of percentage BPRS reduction at 28 days (N = 61, 1 RCT, MD ‐4.50 CI ‐14.93 to 5.93). In the same way the difference between the mean BPRS total score of the two study groups were statistically not significant in Xuan 2007 at eight weeks (N = 97, 1 RCT, MD ‐3.20 Cl ‐6.54 to 0.14).
3.2.4.2.1 Specific: anxiety symptoms (HAM‐A score)
Morphy 1986 analysed anxiety with the mean HAM‐A score at endpoint and Guz 1972 with the mean HAM‐A change score from baseline to endpoint. The data of both studies were skewed and could therefore only be presented in an "other data" table.
3.2.4.2.2 Specific: negative symptoms (SANS score)
Barbee 1992 assessed negative symptoms using the mean SANS score at endpoint and found a slight superiority of the control group. Again, the data were skewed and could only be displayed in an "other data" table.
3.2.4.2.3 Specific: sedation
Sedation data were reported by Garza‐Trevino 1989 and Battaglia 1997. In the benzodiazepine augmentation group of Garza‐Trevino 1989, significantly more participants were tranquillized at 30 minutes (N = 45, 1 RCT, RR 2.25 CI 1.18 to 4.30, NNTB 2 CI 2 to 7) and at 60 minutes (N = 45, 1 RCT, RR 1.39 CI 1.06 to 1.83, NNTB 3 CI 2 to 11) in comparison to the control group. In Battaglia 1997, we found no significant difference in terms of participants who were asleep at 12 hours (N = 67, 1 RCT, RR 0.85 CI 0.51 to 1.41).
3.2.5 Behaviour
3.2.5.1 Specific: aggression
Aggression data were reported in two studies (Battaglia 1997; Dorevitch 1999). Dorevitch 1999 documented the number of participants needing restraints within 120 minutes but nobody needed sanctions (N = 28, 1 RCT, RR not estimable). Battaglia 1997 evaluated aggression with the mean ABS score at one and at 12 hours, but there was no statistically significant between‐group difference measurable (one hour: N = 67, 1 RCT, MD ‐3.00 CI ‐8.27 to 2.27; 12 hours: N = 67, 1 RCT, MD 0.00 CI ‐5.27 to 5.27).
3.2.6 Service use
Only one study examined service utilisation (Barbee 1992) and reported on the number of participants who were still in hospital after three days without identifying any significant difference (N = 28, 1 RCT, RR 0.90 CI 0.54 to 1.50).
3.2.7 Adverse effects
3.2.7.1 General: total number of participants with adverse effects, TESS score
Two studies (Battaglia 1997; Kurland 1966) indicated the total number of participants experiencing at least one adverse effect. We found no statistically significant differences between groups (N = 151, 2 RCTs, RR 0.91 Cl 0.54 to 1.53). In the same way, we found no significant difference regarding the TESS score which was reported in Ma 2006 (N = 120,1 RCT, MD 0.61 Cl ‐0.42 to 1.64).
3.2.7.2 Spectfic adverse effects
3.2.7.2.1 Anorexia Only one participant in the control group of Guz 1972 suffered from anorexia, a non‐significant difference (N = 60,1 RCT, RR 0.33 CI 0.01 to 7.87).
3.2.7.2.2 Allergic reaction Kurland 1966 reported the occurrence of allergic reactions. There was no significant difference between both study arms detectable (N = 84,1 RCT, RR 1.36 CI 0.24 to 7.75).
3.2.7.2.3 Blurred vision Guz 1972 and Kurland 1966 evaluated the number of participants in each group that experienced blurred vision. In Guz 1972, one participant in the combination group experienced blurred vision and in Kurland 1966, one participant in the control group. Therefore, no statistically significant difference could be assessed (N = 144, 2 RCTs, RR 0.96 CI 0.10 to 9.04).
3.2.7.2.4 Cardiovascular reaction Combining the results of Guz 1972 and Kurland 1966 regarding cardiovascular reactions, no significant difference between groups could be identified (N = 144, 2 RCTs, RR 1.53 CI 0.19 to 12.11).
3.2.7.2.5 Chills One trial (Kurland 1966), described that chills did not occur in either group (N = 84,1 RCT, RR not estimable).
3.2.7.2.6 Confusion, depression and diarrhoea Guz 1972 found no significant difference between the intervention and control groups concerning confusion (N = 60, 1 RCT, RR 0.33 CI 0.01 to 7.87), depression (N = 60,1 RCT, RR 1.00 CI 0.07 to 15.26), and diarrhoea (N = 60, 1 RCT, RR 3.00 CI 0.13 to 70.83).
3.2.7.2.7 Dizziness Dizziness was reported in four trials (Battaglia 1997; Lingjaerde 1982; Ma 2006; Pujalte 1994) occurring in 18 participants in the pooled intervention group and nine participants in the pooled control group. This difference did not reach statistical significance for all four studies (N = 257, 4 RCTs, RR 1.96 CI 0.88 to 4.37), but only investigating the three short‐term studies revealed a significant between‐group difference (N = 190, 3 RCTs, RR 2.58 CI 1.1 to 6.2; NNTH not significant).
3.2.7.2.8 Drowsiness In Battaglia 1997 and Kurland 1966 the pooled between‐group difference was not statistically significant in terms of drowsiness (N = 151, 2 RCTs, RR 0.52 CI 0.13 to 2.05).
3.2.7.2.9 Dryness of mouth Four studies (Battaglia 1997, Guz 1972, Kurland 1966, Lingjaerde 1982) reported on dryness of mouth and we found no significant difference between groups (N = 269, 4 RCTs, RR 1.63 CI 0.38 to 6.97).
3.2.7.2.10 Excitation Guz 1972 documented one participant with excitation in the control group (N = 60, 1 RCT, RR 0.33, CI 0.01 to 7.87).
3.2.7.2.11 Gastrointestinal reaction In Kurland 1966 there was no significant difference between groups concerning gastrointestinal reaction (N = 84, 1 RCT, RR 0.30 CI 0.01 to 7.25).
3.2.7.2.12 Headache Combining the results of Kurland 1966 and Lingjaerde 1982 did not reveal any significant between‐group difference (N = 142, 2 RCTs, RR 0.78 CI 0.04 to 14.93).
3.2.7.2.13 Increased salivation and insomnia Guz 1972 and Kurland 1966 evaluated the occurrence of increased salivation (N = 144, 2 RCTs, RR 1.98 CI 0.26 to 14.91) and insomnia (N = 144, 2 RCTs, RR 1.50 CI 0.41 to 5.44), without revealing any statistically significant between‐group difference.
3.2.7.2.14 Lactation In Kurland 1966 the between‐group difference was not statistically significant in terms of lactation (N = 84, 1 RCT, RR 0.30 CI 0.01 to 7.25).
3.2.7.2.15 Movement disorder We found no significant difference in terms of specific movement disorders such as ataxia (Battaglia 1997; Guz 1972), dystonia (Battaglia 1997; Kurland 1966), parkinsonism (Battaglia 1997; Guz 1972; Kurland 1966) slight dysarthria (Battaglia 1997; Lingjaerde 1982), or tremor (Battaglia 1997; Guz 1972). Summarizing the three short‐term studies (Barbee 1992; Hanlon 1969; Stevens 1992), significantly fewer participants in the benzodiazepine augmentation group received antiparkinson medication at least once (N = 215, 3 RCTs, RR 0.74 CI 0.61 to 0.90; NNTH not significant).
3.2.7.2.16 Restlessness In Guz 1972 and Lingjaerde 1982,, six participants in the benzodiazepine augmentation group and five participants in the control group suffered from restlessness, a non‐significant difference (N = 118, 2 RCTs, RR 1.16 CI 0.40 to 3.36).
3.2.7.2.17 Sensory disturbances Sensory disturbances were reported by Kurland 1966. The difference between the groups was not statistically significant (N = 84, 1 RCT, RR 0.91 CI 0.06 to 14.06).
3.2.7.2.18 Sleep disorder There was no statistically significant between‐group difference in the one trial which reported on sleep disorders (Lingjaerde 1982, N = 58, 1 RCT, RR 0.41 CI 0.02 to 9.60).
3.2.7.2.19 Somnolence We found reports of somnolence (Guz 1972; Lingjaerde 1982) which was significantly higher in the benzodiazepine augmentation group than in the control group (N = 118, 2 RCTs, RR 3.30 CI 1.04 to 10.40, NNTH 8 CI 5 to 50).
3.2.7.2.20 Vomiting Guz 1972 reported the numbers of participants experiencing vomiting, but no significant difference between the study groups could be detected (N = 60, 1 RCT, RR 3.00 CI 0.13 to 70.83).
3.3 Sensitivity analysis
3.3.1 Implication of randomisation
We aimed to conduct a sensitivity analysis in terms of the primary outcome when a trial was not clearly stated to be randomised but described as "double‐blind". In this comparison, this affects the study of Marneros 1979. Because this trial did not provide any data that were included in the meta‐analytic calculations measuring the primary outcome; this sensitivity analysis was not undertaken.
3.3.2 Implication of non double‐blind trials
In terms of the evaluation of an augmentation of antipsychotics with benzodiazepines there was one open trial included in the pooled data‐analysis of the primary outcome (Xuan 2007). Removing this trial did not convert the results in terms of statistically significant between‐group differences.
3.4 Fixed and random effects
Using a fixed‐effect model to analyse the data for the primary outcome did not alter the main findings (statistically significant between‐group differences) in the relevant comparisons (fixed‐effect model: 30 minutes: N = 45, 1 RCT, RR 0.37 CI 0.18 to 0.80; 60 minutes: N = 45, 1 RCT, RR 0.07 CI 0.00 to 1.13; 12 hours: N = 67, 1 RCT, RR 0.85 CI 0.51 to 1.41; pooled short‐term studies: N = 511, 6 RCTs, RR 0.96 CI 0.72 to 1.27).
3.5 'Summary of findings' table
The results of four outcomes ‐ no clinically important response to treatment, leaving the study early due to any reason, leaving the study early due to adverse effects, and desired sedation ‐ were considered more closely in a 'Summary of findings' table (see Table 3). The judgements derived from this instrument were used for the discussion section of the review (see Discussion ‐ Summary of main results).
4. Publication bias
Due to the small number of included trials providing usable data concerning the primary outcome, we did not perform a funnel plot analysis.
5. Investigation of heterogeneity
Statistical tests revealed substantial levels of heterogeneity (defined by an I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 test) in the outcomes "No clinically important response to treatment" (Comparison 1. and 3.) and relapse rates (Comparison 2.), but we did not identify clear reasons explaining this heterogeneity. As the number of trials and participants is still low for the individual outcomes, some of this heterogeneity may be due to chance alone. Regarding the outcome "relapse", the different duration of the trials could be a possible reason for the high heterogeneity.
Discussion
Summary of main results
General
It is common practise to use benzodiazepines for a variety of purposes in schizophrenia including anxiety, agitation, disruptive behaviour, motor disturbances, and sometimes for the augmentation of antipsychotic drugs to alleviate positive symptoms. Analysing 34 randomised controlled trials with a total number of 2657 participants, we found no supportive evidence for this widespread clinical practice, except for short‐term sedation of agitated schizophrenic persons.
1. COMPARISON 1: BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT
1.1 No clinically important response to treatment
Based on the data of six short‐term studies, we found no evidence supporting the general use of benzodiazepines as monotherapy to improve symptomatology in people with schizophrenia. Study participants randomised to benzodiazepines alone did not differ significantly from those given placebo in achieving clinically important response to treatment. Statistically significant heterogeneity in the pooled results led to an overestimate of treatment effect with the fixed‐effect model that was not evident when the (more appropriate) random‐effects model was applied to pool data for this outcome.
The fact that a statistically significant difference in favour of benzodiazepine treatment occurred in the sensitivity analysis when applying a fixed‐effect model can be attributed to the substantial level of heterogeneity between the single trials contributing data to this comparison.
1.2 Leaving the study early
About 15% of participants (64 out of 440) left the study early due to any reason. Since there was no statistically significant between‐group difference, monotherapy with benzodiazepines may be generally acceptable for people with schizophrenia. Analysing specific reasons for leaving the studies early, adverse effects or inefficacy of treatment, we also observed no significant differences between benzodiazepines and placebo.
1.3 Relapse rate
Because two small trials (Carpenter 1999; Nishikawa 1982) revealed no significant difference in relapse rates between the study groups, it can be concluded that for relapse prevention benzodiazepines are not an appropriate alternative to antipsychotic drugs.
1.4 Mental state
Results regarding the participants' mental state were rarely reported and difficult to interpret because different scales were used. Whereas one small study (Merlis 1962), found no significant difference in terms of the number of participants with an improvement of general mental state, Minervini 1990 (who analysed the mean BPRS after three weeks) found benzodiazepines to be significantly superior to placebo. Although Minervini 1990 claimed that all participants were in remission and their psychotic symptoms "controlled" and that they were all not receiving antipsychotics, the mean BPRS score was quite high for such a condition (mean BPRS about 67). In addition, the data were derived from only one study and thus need replication. Therfore, we advise caution in the interpretation of this result. Again, very limited data were available for specific aspects of the mental state and no statistically significant outcome data were revealed.
1.5 Adverse effects
The incomplete reporting of adverse effects considerably limits the interpretation of these outcomes. There were significantly more participants treated with benzodiazepines who suffered from any adverse effects, but this result was based on only one study (Gundlach 1966). Again, based on very limited data, benzodiazepines decreased the participants' energy level more often than placebo. This finding can be explained by the sedating properties of benzodiazepines. Benzodiazepines were also associated with more ataxia than placebo.
2. COMPARISON 2: BENZODIAZEPINES versus ANTIPSYCHOTICS
2.1 No clinically important response to treatment
Only four studies including 222 participants reported on the number of participants with a failure to achieve clinically important response to treatment, and no significant difference was found. Compared with placebo (Comparison 1.), the results of this examination (benzodiazepine versus antipsychotic monotherapy) do not support the general use of benzodiazepines as sole agents in people with schizophrenia and related disorders.
2.2. Leaving the study early
Overall 44 out of 738 participants (6 %) terminated the studies prematurely with no statistical significant difference between benzodiazepines and antipsychotics, both given as sole agents. This result was consistent irrespective of the trial duration (ultra short, short, or long term). When specific reasons for leaving the studies early ‐ adverse effects or inefficacy of treatment ‐ were indicated, again there was no significant difference between groups. This result was quite surprising as it could be assumed that more participants in the antipsychotic group discontinued due to adverse effects and more participants in the benzodiazepine group due to inefficacy of treatment, at least in longer‐term studies. Because the total number of participants discontinuing the studies was generally few, there may have been a lack of statistical power and larger trials are needed to assess these outcomes.
2.3 Global state
One study found a superiority of benzodiazepines compared to placebo analysing the mean CGI severity score at one hour (Foster 1997). This difference may be caused by the more sedating effects of antipsychotic drugs, but it requires a replication, especially since the superiority vanished in the study at four hours.
2.4 Relapse rate
There was no difference in terms of relapse rates both for the short‐term and long‐ter studies, but the occurrence of high heterogeneity limits the interpretation of the results.
2.5 Mental state
Only four studies analysed the general mental state measured by different scales and only one trial could verify a significant difference in favour of antipsychotics at one hour. Thus, the results were not consistent and further studies comparing benzodiazepines with antipsychotics are needed. Sedation as a desired treatment effect was more prevalent among the participants receiving benzodiazepines than antipsychotics. Nevertheless, a statistically significant difference was only found at 20 minutes and at 40 minutes but not at 30 minutes, 60 minutes, 120 minutes, and 12 hours. Again, more studies are required to replicate this finding.
2.6 Behaviour
Neither general nor specific aspects of behaviour such as aggression showed any significant superiority between any group. Again, the analysis was limited by the fact that only two trials reported usable data on behaviour.
2.7 Service utilisation
The only outcome on service utilisation analysed was the number of participants who were still in hospital after two weeks. There was no significant difference in the only study that contributed to this comparison.
2.8 Adverse effects
There was no significant difference between benzodiazepines and antipsychotic drugs neither in terms of "any adverse effect" nor in terms of the specific adverse effects anorexia, cardiovascular reactions, change in energy level, depression, dizziness, dryness of mouth, gastric reactions, headaches, insomnia, movement disorders, respiratory depression, extreme sedation, and seizure. Again, since only four studies (Battaglia 1997; Foster 1997; Hankoff 1962; TREC‐Rio 2003) provided data for these outcomes, no firm conclusion can be drawn.
3. COMPARISON 3: ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS
3.1 No clinically important response to treatment
The addition of benzodiazepines to antipsychotics may improve the global state of people with schizophrenia for a very short time frame.This finding was statistically significant at 30 minutes, but the difference diminished over time. For the results at one and 12 hours as well as for the short‐term trials, the analyses failed to identify any significant between‐group differences. Again, the sedating and anxiolytic properties of benzodiazepines may contribute to this short‐term effect. The results of the short‐term studies are limited by substantial heterogeneity between the included trials.
3.2 Leaving the study early
In this comparison 56 out of 1185 participants (5%) discontinued the regular study phase early. There was no significant difference between groups, neither in terms of leaving early for any reason nor for inefficacy of treatment or for adverse effects, suggesting an equal acceptability of benzodiazepine augmentation compared with that of taking antipsychotics alone or combined with placebo.
3.3 Mental state
Few studies presented data on the mental state. Various scales and various assessment times were used which may in part explain why the results were very inconsistent with some studies showing a superiority of antipsychotic monotherapy whereas, others showed a reversed trend. No firm conclusion can be drawn on the basis of the available data. The very little data on anxiety and on negative symptoms were skewed and thus, could not be used in analysis. Sedation as a desired treatment effect was reported by two studies at different time points. At 30 and 60 minutes significantly more participants in the benzodiazepine augmentation group were sedated than in the control group. At 12 hours this effect was not reproducible. Again, the sedating effect of benzodiazepines is the only one that could be found relatively consistently in this systematic review, but due to the small sample size more evidence is necessary.
3.4 Behaviour
Only two ultra short‐term studies reported on aggression using different criteria. Neither measure showed any difference between groups, but it is surprising how few studies evaluated this important outcome.
3.5 Service utilisation
Only one study (Barbee 1992), reported on how many participants were still in hospital after three days and did not find a significant difference between groups. The interpretation of this outcome is again hampered by the small number of participants included.
3.6 Adverse effects
The reporting of adverse effects was very sparse. Only two studies (N = 151) reported on the number of participants with at least one adverse effect and found no significant difference between the benzodiazepine plus antipsychotic group and the control group. In the same way the TESS score, assessed in only one study (N = 120), revealed no significant between‐group difference. There was also no significant difference in terms of anorexia, allergic reaction, blurred vision, cardiovascular reaction, chills, confusion, depression, diarrhoea, drowsiness, dryness of mouth, excitation, gastrointestinal reaction, headache, increased salivation, insomnia, lactation, movement disorders, restlessness, sensory disturbances, sleep disorder and vomiting. Significantly more participants in the benzodiazepine augmentation group experienced somnolence and dizziness when analysing only the short‐term studies. Nevertheless, if there are any consistent effects of benzodiazepines that could be detected by this review, then it was the sedating characteristics of benzodiazepines. These properties can be used therapeutically, but benzodiazepines must be carefully dosed to avoid respiratory depression. In this context, it should be considered that sedation in agitated schizophrenic people can be also achieved by combining high‐potency with low‐potency antipsychotics (Leucht 2011). Fewer participants in the benzodiazepine augmentation group received antiparkinson medication. It is possible that benzodiazepines alleviate extrapyramidal adverse effects of antipsychotic drugs. As adverse effects were generally poorly reported, future research projects are needed to assess this aspect of treatment.
Overall completeness and applicability of evidence
Some limitations regarding this systematic review must be considered, e.g. the trials were usually characterised by small sample sizes and the outcomes were often incompletely reported thus not allowing their inclusion in the meta‐analytic calculations. As a result of variability in the trial designs and different outcome scales being applied, pooling the results was often impossible. One reason for this diversity is that the trials were carried out during a very long period from 1962 to 2007. There is an important distinction between no evidence for an effect (as is the case in this review) and evidence that there is no effect. Thus, no firm conclusions on the efficacy and tolerability of benzodiazepines for schizophrenia can be drawn. Further adequate trials are required to clarify this research question.
Quality of the evidence
All included trials were randomised and most of them were double‐blind, but details were often not presented. Therefore, it appears unclear whether the studies were adequately randomised, whether treatment allocation was really concealed, and whether sufficient blinding could be assured over the whole trial period. In 27 out of 34 studies the overall attrition was classified as low (<10%). Within this context, it should be considered that many of these trials were characterised by a short trial duration. Some studies evaluated only a time frame fewer than 24 hours. Only 12 trials were judged to be free of selective reporting, and there were studies with other problems such as extreme baseline imbalances etc. Based on the GRADE approach the quality of the evidence can be regarded as low or very low (see Table 1; Table 2; Table 3).
Potential biases in the review process
A priori we decided to pool all benzodiazepines in this systematic review. This could be probably a potential bias because benzodiazepines differ in terms of potency and speed of metabolism and elimination. Additionally, the route of administration (oral or parenteral) could probably limit the comparability of the trials results.
The decision to pool all studies irrespective of the administered antipsychotic drug (Comparison 2. and 3.) is justified for efficacy related outcomes because most antipsychotics are characterised by comparable effectiveness (Leucht 2009). The pooling of the antipsychotic compounds seems to be more problematic for adverse effects because antipsychotic drugs differ to a large extent in this regard. Thus, any differences in Comparison 2. and 3. in terms of adverse effects cannot be generalised to all antipsychotic compounds.
The study search was largely based on the trial register of the Cochrane Schizophrenia Group (Adams 2011). This is largely made up of searches of published literature. It is possible that there are unpublished studies we are not aware of and the possibility of a publication bias must be regarded.
We have chosen to use the random‐effects model for our meta‐analytic calculations to consider variability between the included studies. This technique does emphasise the results from smaller trials and it is these studies that are likely to be most prone to bias. Within this context, it must be accounted that applying a fixed‐effect model instead of a random‐effects approach in a sensitivity analysis of the primary outcome converted non‐significant results to significant between‐group difference in Comparison 1. (benzodiazepine monotherapy versus placebo).
Agreements and disagreements with other studies or reviews
We are aware of one Cochrane review investigating the utilisation of benzodiazepines in acute psychosis irrespective of the underlying diagnosis (Gillies 2005). Thus, this review differs from the present one which evaluated only trials that included people with the main diagnosis of a schizophrenia and other types of schizophrenia(‐like) psychoses (e.g. schizophreniform, schizoaffective, or delusional disorders). As in this review, Gillies 2005 concluded that the findings were insufficient to derive evidence‐based treatment recommendations.
Our meta‐analytic findings support the advice of international treatment guidelines which recommend explicitly antipsychotic monotherapy as first‐line medication for the management of schizophrenia (e.g. Falkai 2005; Falkai 2006; Lehman 2004; NICE 2009). Due to their well established sedating properties, the administration of benzodiazepines should be considered primarily for the short‐term management of acutely highly agitated patients with schizophrenia but not for the medium and long term pharmacological treatment of schizophrenia and schizophrenia‐like psychosis.
Authors' conclusions
Implications for practice.
1. For clinicians
Although benzodiazepines are very frequently used in clinical practise, we found, apart from short‐term sedation, no empirical evidence to support their widespread use. At best, benzodiazepines may effectively be used to calm or sedate people with schizophrenia in the ultra short term with an acute episode of a schizophrenic disorder. This systematic review is unable to quantify the effects of benzodiazepines for other outcomes that benzodiazepines are commonly used for in clinical practise such as reducing anxiety or improving positive symptoms of schizophrenia and related disorders.
2. For people with schizophrenia
People with schizophrenia should know that benzodiazepines may help them to calm down during exacerbations of the disorder and that their use for this indication is not associated with a high number of adverse effects.
3. For managers and policy makers
This systematic review provides evidence that benzodiazepines main indication of benzodiazepines in schizophrenia (sedation of acute agitated people with schizophrenia) is supported by this systematic review. No evidence is available for the effectiveness of the general prescription of benzodiazepines in schizophrenia.
Implications for research.
1. General
Any future trials should respect standards of measuring outcomes and of reporting trial data in order to enhance the comparability of study results (Begg 1996). Therefore, the updated version of the CONSORT Statement should be considered (Schulz 2010).
2. Specific
Benzodiazepines are very frequently prescribed in the treatment of schizophrenia. It is therefore sobering how poorly their efficacy and safety have been examined up to date in randomised controlled research projects. We suggest to conduct further trials on benzodiazepines, especially as "add‐on" medication to antipsychotic drugs, to evaluate a potential alleviation of the positive symptoms of schizophrenia.
What's new
| Date | Event | Description |
|---|---|---|
| 27 June 2012 | New search has been performed | Three further studies are included (Ma 2006, Wang 2000, Xuan 2007) and 24 studies have been added to the "excluded studies table". The text has been rewritten and every included trial assessed with the 'Risk of bias' tool. The main findings did not change substantially. |
| 27 June 2012 | New citation required but conclusions have not changed | Substantive update: conclusions not changed |
Acknowledgements
We would like to thank Profs. and Drs. Carpenter, Cheung, Huf, Lingjaerde, Mauri, Minervini, and Ungvari for their reply to our letters or for sending us their original patient data. We would like to acknowledge the contribution of Anja Volz who helped with data extraction and writing of the report in previous versions of the review. We also wish to thank Samantha Roberts, Mark Fenton and Judy Wright for the trial searching and the editorial base of the Cochrane Schizophrenia Group for their continuous help.
Appendices
Appendix 1. Previous search methods
1. Electronic searching 1.1 We searched Cochrane Schizophrenia Group's Register of Trials (August 1998**) using the phrase:
[and (benzodiazepine* or alprazolam* or bromazepam* or neo* or brotizolam* or chlordiazepoxid* or clobazam* or clotiazepam* or diazepam* or dikaliumclorazepat* or flunitrazepam* or flurazepam* or loprazolam* or lorazepam* or lormetazepam* or medazepam* or metaclazepam* or midazolam* or nitrazepam* or nordazepam* or oxazepam* or prazepam* or temazepam* or triazolam*]
1.2 We searched Biological Abstracts (1980 ‐ August 1998) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (benzodiazepine* or alprazolam* or bromazepam* or neo* or brotizolam* or chlordiazepoxid* or clobazam* or clotiazepam* or diazepam* or dikaliumclorazepat* or flunitrazepam* or flurazepam* or loprazolam* or lorazepam* or lormetazepam* or medazepam* or metaclazepam* or midazolam* or nitrazepam* or nordazepam* or oxazepam* or prazepam* or temazepam* or triazolam*]
1.3 We searched the Cochrane Library (issue 3, 1998) using the Phrase:
[and (benzodiazepine* or alprazolam* or bromazepam* or neo* or brotizolam* or chlordiazepoxid* or clobazam* or clotiazepam* or diazepam* or dikaliumclorazepat* or flunitrazepam* or flurazepam* or loprazolam* or lorazepam* or lormetazepam* or medazepam* or metaclazepam* or midazolam* or nitrazepam* or nordazepam* or oxazepam* or prazepam* or temazepam* or triazolam*] 1.4 We searched EMBASE (1980 ‐ August 1998) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (benzodiazepine* or alprazolam* or bromazepam* or neo* or brotizolam* or chlordiazepoxid* or clobazam* or clotiazepam* or diazepam* or dikaliumclorazepat* or flunitrazepam* or flurazepam* or loprazolam* or lorazepam* or lormetazepam* or medazepam* or metaclazepam* or midazolam* or nitrazepam* or nordazepam* or oxazepam* or prazepam* or temazepam* or triazolam*]
1.5 We searched MEDLINE (1966 ‐ August 1998) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (benzodiazepine* or alprazolam* or bromazepam* or neo* or brotizolam* or chlordiazepoxid* or clobazam* or clotiazepam* or diazepam* or dikaliumclorazepat* or flunitrazepam* or flurazepam* or loprazolam* or lorazepam* or lormetazepam* or medazepam* or metaclazepam* or midazolam* or nitrazepam* or nordazepam* or oxazepam* or prazepam* or temazepam* or triazolam*]
1.6 We searched PsycLIT (1886 ‐ August 1998) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (benzodiazepine* or alprazolam* or bromazepam* or neo* or brotizolam* or chlordiazepoxid* or clobazam* or clotiazepam* or diazepam* or dikaliumclorazepat* or flunitrazepam* or flurazepam* or loprazolam* or lorazepam* or lormetazepam* or medazepam* or metaclazepam* or midazolam* or nitrazepam* or nordazepam* or oxazepam* or prazepam* or temazepam* or triazolam*]
1.7 We searched PSYNDEX (1974 ‐ August 1998) using the Cochrane Schizophrenia Group's phrase for randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (benzodiazepine* or alprazolam* or bromazepam* or neo* or brotizolam* or chlordiazepoxid* or clobazam* or clotiazepam* or diazepam* or dikaliumclorazepat* or flunitrazepam* or flurazepam* or loprazolam* or lorazepam* or lormetazepam* or medazepam* or metaclazepam* or midazolam* or nitrazepam* or nordazepam* or oxazepam* or prazepam* or temazepam* or triazolam*]
**The search was repeated twice ‐ for the last time in March 2005 ‐ due to a delay in summarising the results. For these updates we only searched the Cochrane Schizophrenia Group's Register of Trials.
2. Reference lists We search all references of articles selected for inclusion for further relevant trials.
3. Personal contact We contacted the first authors of included studies identified from the first search for further relevant studies.
Appendix 2. Previous methods: data collection and analyses
[For definitions of terms used in this, and other sections, please refer to the Glossary]
1. Study selection We inspected all study citations identified by the searches independently, and the full reports of the studies of agreed relevance were obtained. Where agreement could not be reached, we acquired the full report for more detailed scrutiny. If the disagreement could not be resolved from published information, we added the article to those awaiting assessment and the authors of the study were contacted for clarification.
2. Quality assessment The methodological quality of the trials included in this review was assessed using the criteria described in the Cochrane Handbook. These criteria are based on the evidence of a strong relationship between allocation concealment and the potential for bias in the results (Schulz 1995) and are defined as below:
A. Low risk of bias (adequate allocation concealment) B. Moderate risk of bias (some doubt about the results) C. High risk of bias (inadequate allocation concealment)
For the purpose of the analysis in this review, trials were included if they met the criteria A or B.
3. Data extraction Anja Volz and/or Vesal Khors and/or Donna Gillies independently extracted the data from included studies and the data were checked by Stefan Leucht. Again, any disagreements were discussed, the decisions documented and, if necessary, the authors of the studies were contacted for clarification. Justification for excluding references from the review were documented.
4. Data management 4.1 Intention‐to‐treat For studies that did not specify the reasons for people leaving the study early ('dropped out'), the reviewers assumed that these people had no change in the clinical outcome variables. Wahlbeck and colleagues highlighted the problem of high drop‐out rates in randomised controlled antipsychotic drug trials (Wahlbeck 2001). As there is no consensus on the level of drop‐out that makes results meaningless, all trials were included in the main analysis. We performed a sensitivity analysis excluding those trials with a greater than 50% drop‐out rate to test whether this procedure significantly changed the results of the primary outcomes. When insufficient data were provided to identify the original group size (prior to drop‐outs), the authors were contacted and the trials were allocated to the 'awaiting assessment' list.
4.2 Cross‐over design We expected that some trials would use a crossover design. In order to exclude the potential additive effect in the second or later stages on these trials, only data from the first stage were analysed.
4.3 Cluster trials Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) ‐ whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated ‐ causing type I errors (Bland 1997, Gulliford 1999). Secondly, RevMan does not currently support meta‐analytic pooling of clustered dichotomous data, even when these are correctly analysed by the authors of primary studies, since the 'design effect' (a statistical correction for clustering) cannot be incorporated.
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol ‐ to indicate the presence of a probable unit of analysis error. Subsequent versions of this review will seek to contact first authors of studies to seek intra‐class correlation co‐efficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, then we also presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect. We have sought statistical advice from the MRC Biostatistics Unit, Cambridge, UK. Dr Julian Higgins advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intraclass correlation co‐efficient (ICC) [Design effect = 1+(m‐1)*ICC]. Should the ICC not be reported it was assumed to be 0.1 (Ukoumunne 1999). Where cluster studies were appropriately analysed taking into account intra‐class correlation coefficients and relevant data documented in the report, synthesis with other studies was possible using the generic inverse variance technique.
4.4 Data types Outcomes are assessed using continuous (for example changes on a behaviour scale), categorical (for example, one of three categories on a behaviour scale, such as 'little change', 'moderate change' or 'much change') or dichotomous measures (for example, either 'no important changes' or 'important changes' in a person's behaviour). Currently RevMan does not support categorical data so they could not be analysed as such.
4.4.1 Dichotomous data: Where possible efforts were made to convert outcome measures to dichotomous data. This may be done by identifying cut off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. If authors of studies used predefined cut off points for determining clinical effectiveness then, where appropriate, we used these.
For dichotomous outcomes, we calculated the relative risk (RR) with a 95% confidence interval (CI) based on a random effects model. If the overall results were statistically significant, the Number Needed to Treat (NNT) or the Number Needed to Harm (NNH) were calculated as the inverse of the risk reduction. It has been shown that RR is more intuitive than odds ratios (Boissel 1999) and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect. We therefore chose the relative risk as an effect size measure.
4.4.2 Continuous data 4.4.2.1 Normal distribution: Continuous data on outcomes in trials relevant to mental health issues are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data the following standards were applied. When a scale starts from zero, the standard deviation, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution ‐ Altman 1996). Endpoint scores on scales often have a finite start and end point and this rule can be applied. When continuous data were presented on a scale which includes a possibility of negative values (such as change on a scale) it is impossible to tell whether data are non‐normally distributed (skewed) or not. It is thus preferable to use scale endpoint data, which typically cannot have negative values. If endpoint data were not available, we chose to use change data, because the statistics used in Metaview are rather robust towards skewness. If a scale starts from a positive value (such as PANSS, which can have values from 30‐210) the calculation described above should be modified to take the scale starting point into account. In these cases skewness is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score.
4.4.2.2 Intention‐to‐treat versus completer analyses: in the case of continuous data it was supposed that in many cases an intention‐to‐treat analysis would not be available, so that the data had to be analysed as they were presented in the original publications.
4.4.2.3 Summary statistic: for continuous outcomes, a weighted mean difference (MD) between groups was estimated using a random effects model. Whenever possible we took the opportunity to make direct comparisons between trials that used the same measurement instrument to quantify specific outcomes. Where continuous data was presented from different scales rating the same effect, both sets of data were presented and the general direction of effect inspected.
4.4.2.4 Rating scales: a wide range of instruments are available to measure mental health outcomes. These instruments vary in quality and many are not valid, or even ad hoc. For outcome instruments some minimum standards have to be set. Continuous data from rating scales were included only if the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000), the instrument was either a self report or completed by an independent rater or relative (not the therapist), and the instrument could be considered a global assessment of an area of functioning. However, as it was expected that therapists would frequently also be the rater, such data were commented on with 'prone to bias'.
4.5 Data display Whenever possible data were entered into RevMan in such a way that the area to the left of the line of no effect indicated a favourable outcome for benzodiazepine alone or benzodiazepine augmentation.
5. Heterogeneity We assessed heterogeneity by visual inspection of the graphs and supplemented this procedure using the I‐squared statistic. This measure provides an estimate of the percentage of variability due to heterogeneity rather than chance alone. Where the I‐squared estimate was greater than or equal to 75%, we interpreted this as indicating the presence of high levels of heterogeneity. In such cases we investigated the reasons for heterogeneity. 6. Publication bias Data from all included trials were entered into a funnel graph (trial effect versus trial size or 'precision') in an attempt to investigate the likelihood of overt publication bias. A formal test of funnel plot asymmetry (suggesting potential publication bias) was not undertaken (Egger 1997a).
Data and analyses
Comparison 1. BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 No clinically important response to treatment ‐ short term | 6 | 382 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.44, 1.02] |
| 2 Leaving the study early ‐ any reason | 8 | 440 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.57, 1.38] |
| 2.1 short term | 7 | 417 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.57, 1.38] |
| 2.2 long term | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Leaving the study early due to adverse effects | 4 | 161 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.1 short term | 3 | 138 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 long term | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Leaving the study early due to inefficacy of treatment | 4 | 161 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.1 short term | 3 | 138 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 long term | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Global state: 1. Relapse | 2 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.41, 1.74] |
| 5.1 short term | 1 | 35 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.36, 1.23] |
| 5.2 long term | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.85, 1.18] |
| 6 Mental state: 1. General ‐ various scales (unimproved or worse) ‐ short term | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 BPRS | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.39, 2.53] |
| 6.2 MMS | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.9 [0.52, 1.57] |
| 7 Mental state: 2. General mean BPRS at 3 weeks (high=poor) ‐ short term | 1 | 66 | Mean Difference (IV, Random, 95% CI) | ‐17.60 [‐22.61, ‐12.59] |
| 8 Mental state: 3a. Specific ‐ anxiety TSRS at 6 weeks (unimproved or worse) ‐ short term | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.63, 2.15] |
| 9 Mental state: 3b. Specific ‐ anxiety mean HAM‐A at 3 weeks (skewed data) ‐ short term | Other data | No numeric data | ||
| 10 Adverse effects: 1. total number of participants with adverse effects ‐ short term | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 1.44 [1.02, 2.04] |
| 11 Adverse effects: 2. Anorexia ‐ short term | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 12 Adverse effects: 3. Autonomic reaction (flushing, dryness of mouth etc) ‐ short term | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.75, 3.93] |
| 13 Adverse effects: 4. Cardiovascular reactions ‐ short term | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 14 Adverse effects: 5. Depression ‐ short term | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Adverse effects: 6. Dryness of mouth ‐ short term | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 16 Adverse effects: 7. Energy level change ‐ short term | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 Increased energy level (Insomnia, tension etc.) | 2 | 222 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.02, 22.78] |
| 16.2 Decreased energy level (sleepiness, motor inhibition) | 2 | 222 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [1.38, 3.43] |
| 17 Adverse effects: 8. Gastrointestinal reactions ‐ short term | 3 | 288 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.29, 3.37] |
| 18 Adverse effects: 9. Headache ‐ short term | 2 | 222 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.20, 2.81] |
| 19 Adverse effects: 10. Insomnia ‐ short term | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.05, 27.28] |
| 20 Adverse effects: 11. Movement disorders ‐ short term | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 20.1 Ataxia | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 8.18 [1.35, 49.74] |
| 20.2 EPS | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 21 Adverse effects: 12. Sedation (extreme) ‐ short term | 2 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.15, 61.74] |
| 22 Sensitivity analysis (no clinically important response to treatment) ‐ use of fixed‐effects model | 6 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.52, 0.83] |
1.1. Analysis.

Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 1 No clinically important response to treatment ‐ short term.
1.2. Analysis.
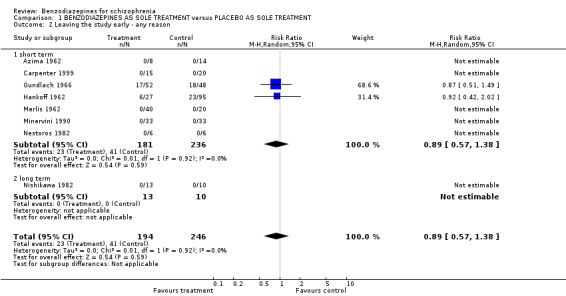
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 2 Leaving the study early ‐ any reason.
1.3. Analysis.
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 3 Leaving the study early due to adverse effects.
1.4. Analysis.
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 4 Leaving the study early due to inefficacy of treatment.
1.5. Analysis.
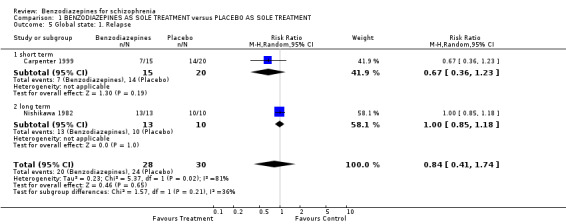
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 5 Global state: 1. Relapse.
1.6. Analysis.
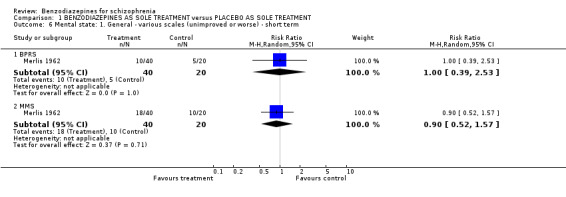
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 6 Mental state: 1. General ‐ various scales (unimproved or worse) ‐ short term.
1.7. Analysis.
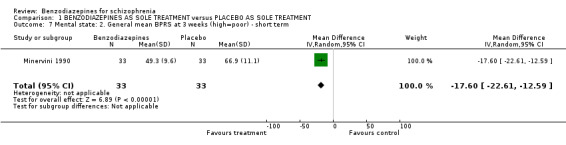
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 7 Mental state: 2. General mean BPRS at 3 weeks (high=poor) ‐ short term.
1.8. Analysis.
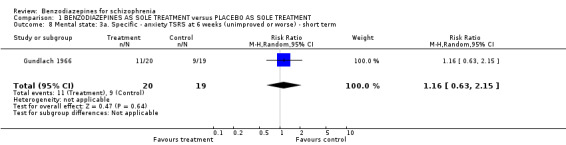
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 8 Mental state: 3a. Specific ‐ anxiety TSRS at 6 weeks (unimproved or worse) ‐ short term.
1.9. Analysis.
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 9 Mental state: 3b. Specific ‐ anxiety mean HAM‐A at 3 weeks (skewed data) ‐ short term.
| Mental state: 3b. Specific ‐ anxiety mean HAM‐A at 3 weeks (skewed data) ‐ short term | ||||
|---|---|---|---|---|
| Study | Invervention | Mean | SD | N |
| Minervini 1990 | Benzodiazepine | 7.60 | 6.90 | 33 |
| Minervini 1990 | Placebo | 31.40 | 8.50 | 33 |
1.10. Analysis.
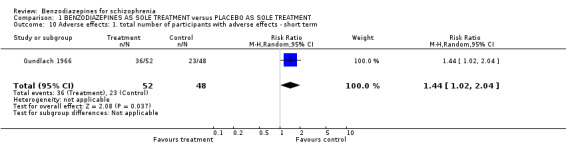
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 10 Adverse effects: 1. total number of participants with adverse effects ‐ short term.
1.11. Analysis.
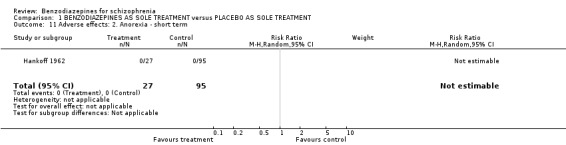
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 11 Adverse effects: 2. Anorexia ‐ short term.
1.12. Analysis.

Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 12 Adverse effects: 3. Autonomic reaction (flushing, dryness of mouth etc) ‐ short term.
1.13. Analysis.
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 13 Adverse effects: 4. Cardiovascular reactions ‐ short term.
1.14. Analysis.
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 14 Adverse effects: 5. Depression ‐ short term.
1.15. Analysis.
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 15 Adverse effects: 6. Dryness of mouth ‐ short term.
1.16. Analysis.
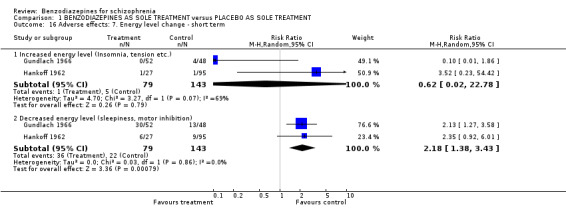
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 16 Adverse effects: 7. Energy level change ‐ short term.
1.17. Analysis.
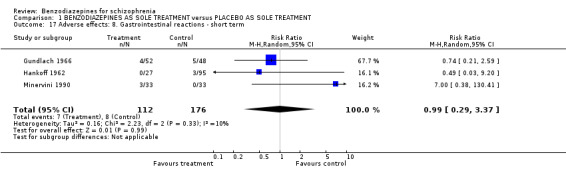
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 17 Adverse effects: 8. Gastrointestinal reactions ‐ short term.
1.18. Analysis.
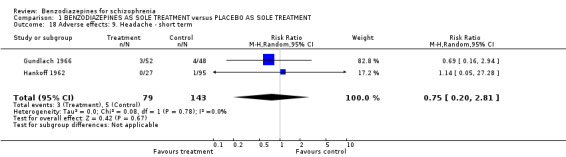
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 18 Adverse effects: 9. Headache ‐ short term.
1.19. Analysis.
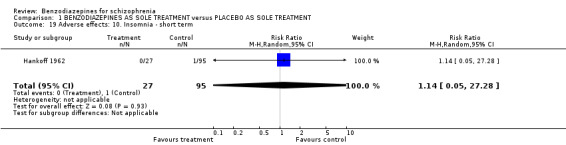
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 19 Adverse effects: 10. Insomnia ‐ short term.
1.20. Analysis.
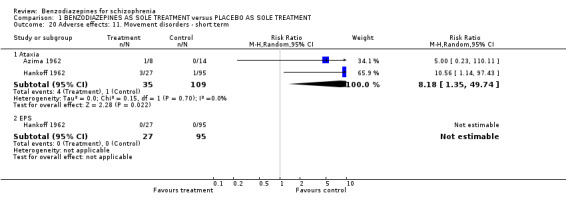
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 20 Adverse effects: 11. Movement disorders ‐ short term.
1.21. Analysis.
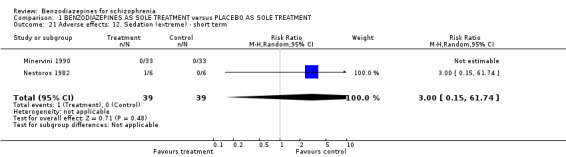
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 21 Adverse effects: 12. Sedation (extreme) ‐ short term.
1.22. Analysis.
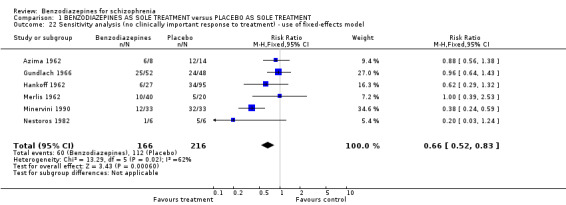
Comparison 1 BENZODIAZEPINES AS SOLE TREATMENT versus PLACEBO AS SOLE TREATMENT, Outcome 22 Sensitivity analysis (no clinically important response to treatment) ‐ use of fixed‐effects model.
Comparison 2. BENZODIAZEPINES versus ANTIPSYCHOTICS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 No clinically important response to treatment ‐ ultra short term | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Unimproved or worse at 30 minutes | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.58, 1.43] |
| 1.2 Unimproved or worse at 60 minutes | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.20, 1.86] |
| 1.3 Unimproved or worse at 12 hours | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.44, 1.30] |
| 2 No clinically important response to treatment ‐ short term | 2 | 112 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [0.64, 3.46] |
| 3 Leaving the study early ‐ any reason | 13 | 738 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.45, 1.18] |
| 3.1 ultra short term | 7 | 514 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.33, 1.36] |
| 3.2 short term | 4 | 161 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.27, 1.56] |
| 3.3 long term | 2 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.26, 96.13] |
| 4 Leaving the study early due to adverse effects | 5 | 444 | Risk Ratio (M‐H, Random, 95% CI) | 13.0 [0.78, 216.39] |
| 4.1 ultra short term | 3 | 351 | Risk Ratio (M‐H, Random, 95% CI) | 13.0 [0.78, 216.39] |
| 4.2 short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.3 long term | 1 | 33 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Leaving the study early due to inefficacy of treatment | 5 | 420 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.14] |
| 5.1 ultra short term | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 short term | 2 | 76 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.14] |
| 5.3 long term | 1 | 33 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Global state: 2a. Mean CGI severity score ‐ ultra short term (high=poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Mean CGI severity score at 1 hour | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.09, ‐0.25] |
| 6.2 Mean CGI severity score at 4 hours | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐0.62 [‐1.36, 0.12] |
| 7 Global state: 2b. Mean CGI severity score ‐ ultra short term (skewed data) | Other data | No numeric data | ||
| 7.1 Mean CGI severity score at 2 hours | Other data | No numeric data | ||
| 7.2 Mean CGI severity score at 4 hours | Other data | No numeric data | ||
| 7.3 Mean CGI severity score at 24 hours | Other data | No numeric data | ||
| 8 Global state: 3. Relapse | 3 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.72, 2.22] |
| 8.1 short term (by 4 weeks) | 1 | 33 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.43, 1.66] |
| 8.2 long term (by 1 year or longer) | 2 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 2.02 [0.37, 11.04] |
| 9 Mental state: 1. General (various scales) ‐ short term | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 BPRS (unimproved or worse) | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.5 [0.60, 10.34] |
| 9.2 MMS (unimproved or worse) | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.60, 2.13] |
| 10 Mental state: 2a. General ‐ BPRS ‐ ultra short term (high=poor) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10.1 Mean BPRS score at 1hour | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐3.26 [‐10.65, 4.13] |
| 10.2 Mean BPRS score at 4 hours | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐1.73 [‐9.60, 6.14] |
| 10.3 Mean MBPRS at 1 hour | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 6.0 [0.68, 11.32] |
| 10.4 Mean MBPRS at 12 hours | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 1.0 [‐4.32, 6.32] |
| 11 Mental state: 2b . General ‐ BPRS ‐ ultra short term (skewed data) | Other data | No numeric data | ||
| 11.1 Mean BPRS score at 4 hours | Other data | No numeric data | ||
| 11.2 Mean BPRS score at 12‐24 hours | Other data | No numeric data | ||
| 12 Mental state: 3a. Specific ‐mania ‐ ultra short term (skewed data) | Other data | No numeric data | ||
| 12.1 Mean IMPS subscore | Other data | No numeric data | ||
| 13 Mental state: 3b. Specific ‐ sedation ‐ ultra short term (high=better) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 Tranquilized or asleep at 20 minutes | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [1.16, 1.49] |
| 13.2 Tranquilized or asleep at 30 minutes | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.53, 2.59] |
| 13.3 Tranquilized or asleep at 40 minutes | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [1.04, 1.23] |
| 13.4 Tranquilized or asleep at 60 minutes | 2 | 345 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [1.00, 1.15] |
| 13.5 Tranquilized or asleep at 120 minutes | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.98, 1.10] |
| 13.6 Asleep at 12 hours | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.44, 1.30] |
| 14 Mental state: 3c. Specific ‐ sedation, mean CGI sedation score ‐ ultra short term (at 2 hours) (high=poor) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.98, 1.18] |
| 15 Behaviour: 1. General ‐ mean modified NOSIE score ‐ ultra short term (at 2 hours)(skewed data) | Other data | No numeric data | ||
| 16 Behaviour: 2a. Specific ‐ aggression ‐ ultra short term | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 Needing restraints within 120 min. | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.55, 1.22] |
| 16.2 Any episode of aggression at 24 hours | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.78, 1.62] |
| 16.3 Use of additional tranquilising drugs at 120 minutes | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 0.28 [0.06, 1.34] |
| 17 Behaviour: 2b. Specific ‐ aggression ‐ ultra short term | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 17.1 Mean ABS after 1 hour | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 4.0 [‐1.32, 9.32] |
| 17.2 Mean ABS after 12 hours | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 3.0 [‐2.32, 8.32] |
| 18 Service use: 1. Still in hospital ‐ short term (after 2 weeks) | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.77, 1.18] |
| 19 Adverse effects: 1. total number of participants with adverse‐effects | 2 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.45, 1.17] |
| 19.1 ultra short term | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.48, 1.66] |
| 19.2 short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.25, 1.15] |
| 20 Adverse effects: 2. Anorexia ‐ short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.69] |
| 21 Adverse effects: 3.Cardiovascular reactions ‐ short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.69] |
| 22 Adverse effects: 4. Change in energy level ‐ short term | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 22.1 Decreased energy level | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.24, 1.30] |
| 22.2 Increased energy level | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.06, 14.03] |
| 23 Adverse effects: 5. Depression ‐ short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.69] |
| 24 Adverse effects: 6. Dizziness ‐ ultra short term | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.25, 5.19] |
| 25 Adverse effects: 7. Dryness of mouth | 2 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.35, 5.33] |
| 25.1 ultra short term | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.88 [0.49, 7.24] |
| 25.2 short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 7.26] |
| 26 Adverse effects: 8. Gastric reactions ‐ short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 27 Adverse effects: 9. Headaches ‐ short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.69] |
| 28 Adverse effects: 10. Insomnia ‐ short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 29 Adverse effects: 11a. Movement disorders | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 29.1 Ataxia ‐ ultra short term | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 2.26 [0.22, 23.71] |
| 29.2 Ataxia ‐ short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.03, 2.78] |
| 29.3 Dystonia ‐ ultra short term | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 0.16 [0.02, 1.24] |
| 29.4 Parkinsonism ‐ ultra short term | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 0.16 [0.01, 2.99] |
| 29.5 Slight dysarthria ‐ ultra short term | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.11, 2.87] |
| 29.6 Tremor ‐ ultra short term | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.05, 5.93] |
| 29.7 EPS ‐ ultra short term | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 29.8 EPS ‐ short term | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.69] |
| 29.9 Use of antiparkinson medication ‐ ultra short term | 2 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.17, 1.47] |
| 30 Adverse effects: 11b. Movement disorders ‐ ultra short term (skewed data) | Other data | No numeric data | ||
| 30.1 Mean Parkinson Total Score at 2 hours | Other data | No numeric data | ||
| 30.2 Mean Tardive Dyskinesia Total Score | Other data | No numeric data | ||
| 31 Adverse effects: 12. Respiratory depression ‐ ultra short term | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 2.98 [0.12, 72.58] |
| 32 Adverse effects: 13. Sedation (extreme) ‐ ultra short term | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.76 [0.33, 9.36] |
| 33 Adverse effects: 14. Seizure ‐ ultra short term | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.06] |
| 34 Sensitivity analysis (no clinically important response to treatment, ultra short term) ‐ use of a fixed‐effects model | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 34.1 Unimproved or worse at 30 minutes | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.58, 1.43] |
| 34.2 Unimproved or worse at 60 minutes | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.20, 1.86] |
| 34.3 Unimproved or worse at 12 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.44, 1.30] |
| 35 Sensitivity analysis (no clinically important response to treatment, short term) ‐ use of a fixed‐effects model | 2 | 112 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.68, 3.67] |
2.1. Analysis.
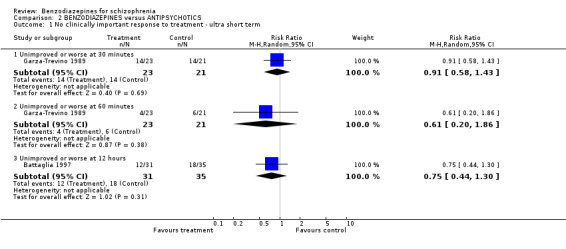
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 1 No clinically important response to treatment ‐ ultra short term.
2.2. Analysis.
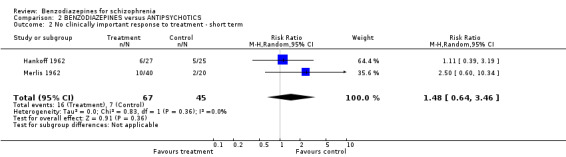
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 2 No clinically important response to treatment ‐ short term.
2.3. Analysis.
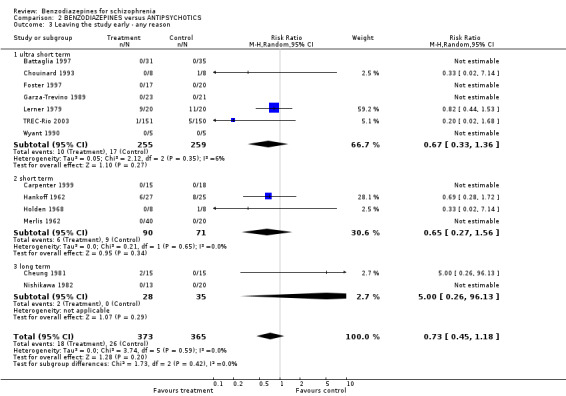
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 3 Leaving the study early ‐ any reason.
2.4. Analysis.
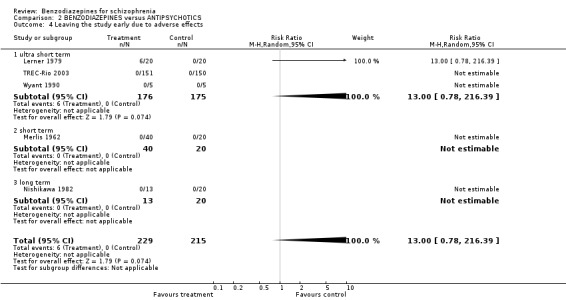
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 4 Leaving the study early due to adverse effects.
2.5. Analysis.
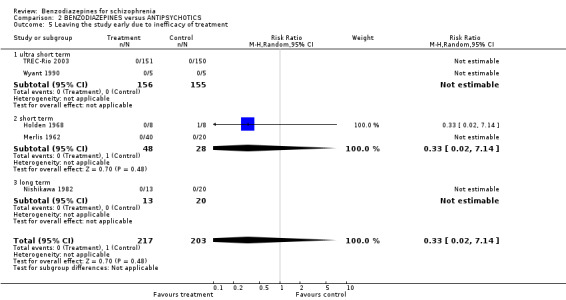
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 5 Leaving the study early due to inefficacy of treatment.
2.6. Analysis.
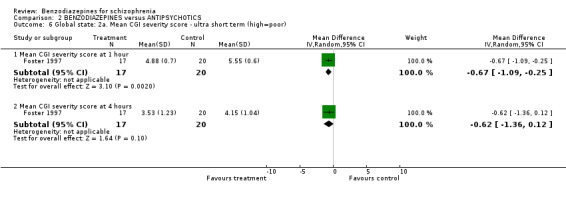
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 6 Global state: 2a. Mean CGI severity score ‐ ultra short term (high=poor).
2.7. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 7 Global state: 2b. Mean CGI severity score ‐ ultra short term (skewed data).
| Global state: 2b. Mean CGI severity score ‐ ultra short term (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Mean CGI severity score at 2 hours | ||||
| Chouinard 1993 | Benzodiazepines | 2.20 | 1.30 | 8 |
| Chouinard 1993 | Antipsychotics | 2.60 | 1.30 | 8 |
| Mean CGI severity score at 4 hours | ||||
| Lerner 1979 | Benzodiazepines | 0.90 | 0.67 | 11 |
| Lerner 1979 | Antipsychotics | 0.50 | 0.05 | 9 |
| Mean CGI severity score at 24 hours | ||||
| Lerner 1979 | Benzodiazepines | 0.40 | 0.83 | 20 |
| Lerner 1979 | Antipsychotics | 1.0 | 0.83 | 8 |
2.8. Analysis.
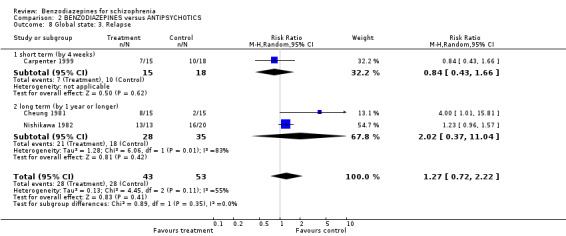
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 8 Global state: 3. Relapse.
2.9. Analysis.
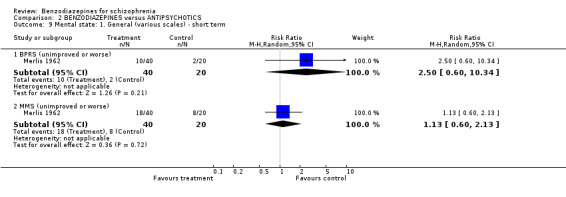
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 9 Mental state: 1. General (various scales) ‐ short term.
2.10. Analysis.
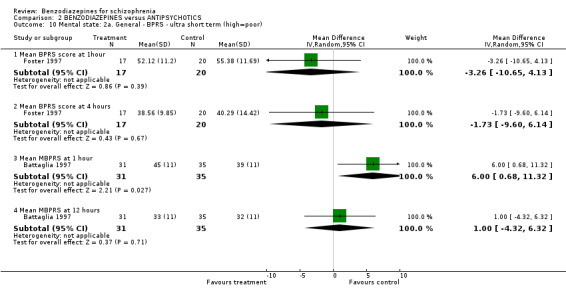
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 10 Mental state: 2a. General ‐ BPRS ‐ ultra short term (high=poor).
2.12. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 12 Mental state: 3a. Specific ‐mania ‐ ultra short term (skewed data).
| Mental state: 3a. Specific ‐mania ‐ ultra short term (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Mean IMPS subscore | ||||
| Chouinard 1993 | Benzodiazepines | 10.0 | 7.20 | 8 |
| Chouinard 1993 | Antipsychotics | 7.40 | 3.80 | 8 |
2.13. Analysis.

Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 13 Mental state: 3b. Specific ‐ sedation ‐ ultra short term (high=better).
2.14. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 14 Mental state: 3c. Specific ‐ sedation, mean CGI sedation score ‐ ultra short term (at 2 hours) (high=poor).
2.15. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 15 Behaviour: 1. General ‐ mean modified NOSIE score ‐ ultra short term (at 2 hours)(skewed data).
| Behaviour: 1. General ‐ mean modified NOSIE score ‐ ultra short term (at 2 hours)(skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Chouinard 1993 | Benzodiazepines | 1.30 | 1.60 | 8 |
| Chouinard 1993 | Antipsychotics | 1.70 | 1.60 | 8 |
2.16. Analysis.
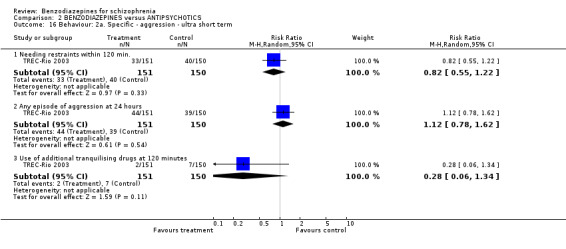
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 16 Behaviour: 2a. Specific ‐ aggression ‐ ultra short term.
2.17. Analysis.
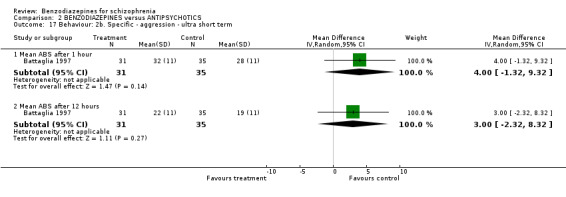
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 17 Behaviour: 2b. Specific ‐ aggression ‐ ultra short term.
2.18. Analysis.

Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 18 Service use: 1. Still in hospital ‐ short term (after 2 weeks).
2.19. Analysis.
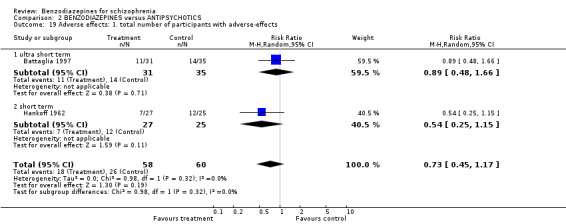
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 19 Adverse effects: 1. total number of participants with adverse‐effects.
2.20. Analysis.
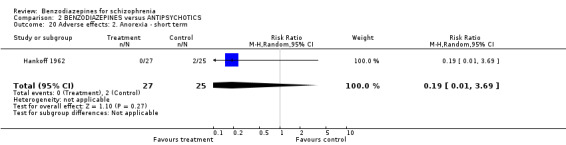
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 20 Adverse effects: 2. Anorexia ‐ short term.
2.21. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 21 Adverse effects: 3.Cardiovascular reactions ‐ short term.
2.22. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 22 Adverse effects: 4. Change in energy level ‐ short term.
2.23. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 23 Adverse effects: 5. Depression ‐ short term.
2.24. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 24 Adverse effects: 6. Dizziness ‐ ultra short term.
2.25. Analysis.
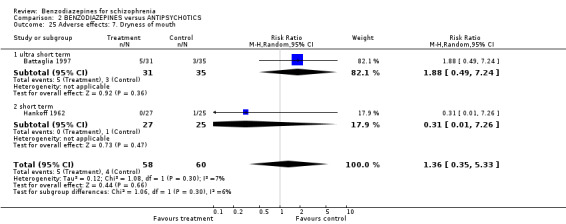
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 25 Adverse effects: 7. Dryness of mouth.
2.26. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 26 Adverse effects: 8. Gastric reactions ‐ short term.
2.27. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 27 Adverse effects: 9. Headaches ‐ short term.
2.28. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 28 Adverse effects: 10. Insomnia ‐ short term.
2.29. Analysis.
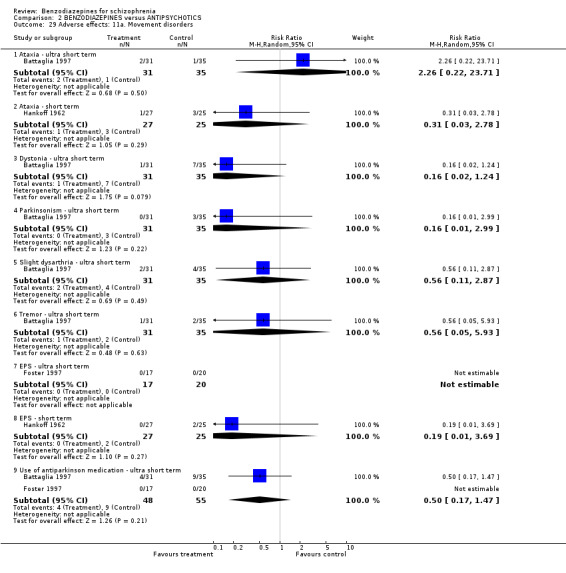
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 29 Adverse effects: 11a. Movement disorders.
2.30. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 30 Adverse effects: 11b. Movement disorders ‐ ultra short term (skewed data).
| Adverse effects: 11b. Movement disorders ‐ ultra short term (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Mean Parkinson Total Score at 2 hours | ||||
| Chouinard 1993 | Benzodiazepines | 6.50 | 4.90 | 7 |
| Chouinard 1993 | Antipsychotics | 8.50 | 4.90 | 8 |
| Mean Tardive Dyskinesia Total Score | ||||
| Chouinard 1993 | Benzodiazepines | 3.0 | 2.0 | 7 |
| Chouinard 1993 | Antipsychotics | 0.90 | 2.0 | 8 |
2.31. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 31 Adverse effects: 12. Respiratory depression ‐ ultra short term.
2.32. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 32 Adverse effects: 13. Sedation (extreme) ‐ ultra short term.
2.33. Analysis.
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 33 Adverse effects: 14. Seizure ‐ ultra short term.
2.34. Analysis.
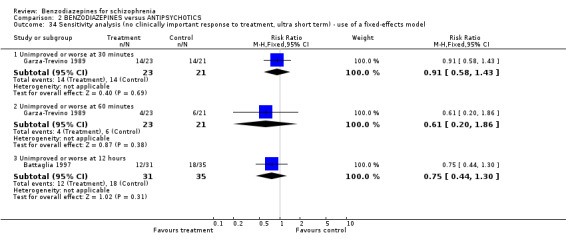
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 34 Sensitivity analysis (no clinically important response to treatment, ultra short term) ‐ use of a fixed‐effects model.
2.35. Analysis.
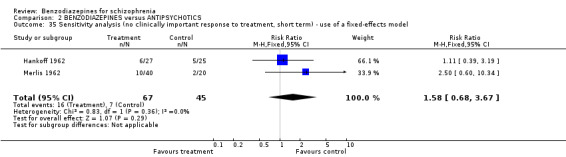
Comparison 2 BENZODIAZEPINES versus ANTIPSYCHOTICS, Outcome 35 Sensitivity analysis (no clinically important response to treatment, short term) ‐ use of a fixed‐effects model.
Comparison 3. ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 No clinically important response to treatment ‐ ultra short term | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Unimproved or worse at 30 minutes | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.18, 0.80] |
| 1.2 Unimproved or worse at 60 minutes | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.00, 1.13] |
| 1.3 Unimproved or worse at 12 hours | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.51, 1.41] |
| 2 No clinically important response to treatment ‐ short term | 6 | 511 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.49, 1.54] |
| 3 Leaving the study early any reason | 19 | 1185 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.81, 2.30] |
| 3.1 ultra short term | 3 | 140 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 short term | 16 | 1045 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.81, 2.30] |
| 4 Leaving the study early due to adverse effects ‐ short term | 6 | 415 | Risk Ratio (M‐H, Random, 95% CI) | 3.24 [0.68, 15.45] |
| 5 Leaving the study early due to inefficacy of treatment ‐ short term | 6 | 347 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.17, 3.42] |
| 6 Global state: 2. Mean CGI severity score ‐ short term (high=poor) | Other data | No numeric data | ||
| 7 Mental state: 1. General ‐ ultra short term (various scales) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 Mean BPRS score after 1 hour (high = poor) | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐3.11 [‐8.86, 2.64] |
| 7.2 Mean MBPRS after 1 hour (high = poor) | 1 | 67 | Mean Difference (IV, Random, 95% CI) | ‐4.0 [‐9.51, 1.51] |
| 7.3 Mean MBPRS after 12 hours (high = poor) | 1 | 67 | Mean Difference (IV, Random, 95% CI) | 2.0 [‐3.27, 7.27] |
| 7.4 Mean BPRS score after 24 hours (high = poor) | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐7.26, 7.28] |
| 8 Mental state: 1. General ‐ short term (various scales) | 4 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 Mean BPRS score at 2 days (high = poor) | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 2.0 [‐3.86, 7.86] |
| 8.2 Mean BPRS score at 3 days (high = poor) | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 6.81 [0.32, 13.30] |
| 8.3 Mean PANSS at 2 weeks (high = poor) | 1 | 80 | Mean Difference (IV, Random, 95% CI) | ‐6.20 [‐12.55, 0.15] |
| 8.4 Mean percentage BPRS reduction at 28 days (low = poor) | 1 | 61 | Mean Difference (IV, Random, 95% CI) | ‐4.5 [‐14.93, 5.93] |
| 8.5 Mean BPRS at 8 weeks (high = poor) | 1 | 97 | Mean Difference (IV, Random, 95% CI) | ‐3.20 [‐6.54, 0.14] |
| 9 Mental state: 2a. Specific ‐ anxiety ‐ short term (skewed data) | Other data | No numeric data | ||
| 9.1 Summary of mean HAM‐A score at 4 weeks | Other data | No numeric data | ||
| 9.2 Hamilton Anxiety Score (total difference from baseline to 4 weeks) | Other data | No numeric data | ||
| 10 Mental state: 2b. Specific ‐ negative symptoms (SANS score at endpoint) ‐ short term (skewed data) | Other data | No numeric data | ||
| 11 Mental state: 3. Specific ‐ sedation ‐ ultra short term | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 Tranquilized at 30 minutes | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 2.25 [1.18, 4.30] |
| 11.2 Tranquilized at 60 minutes | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [1.06, 1.83] |
| 11.3 Asleep at 12 hours | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.51, 1.41] |
| 12 Behaviour: 1. Specific ‐aggression ‐ ultra short term (needing restraints within 120 minutes) | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13 Behaviour: 2. Specific ‐ aggression ‐ ultra short term | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 13.1 Mean ABS at 1 hour (high = poor) | 1 | 67 | Mean Difference (IV, Random, 95% CI) | ‐3.0 [‐8.27, 2.27] |
| 13.2 Mean ABS at 12 hours (high = poor) | 1 | 67 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐5.27, 5.27] |
| 14 Service use: 1. Still in hospital ‐ short term (at 3 days) | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.9 [0.54, 1.50] |
| 15 Adverse effects: 1. total number of participants with adverse‐effects | 2 | 151 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.54, 1.53] |
| 15.1 ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.46, 1.61] |
| 15.2 short term | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.41, 2.61] |
| 16 Adverse effects: 2. TESS score ‐ short term | 1 | 120 | Mean Difference (IV, Random, 95% CI) | 0.61 [‐0.42, 1.64] |
| 17 Adverse effects: 3. Anorexia ‐ short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 18 Adverse effects: 4. Allergic reaction ‐ short term | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.24, 7.75] |
| 19 Adverse effects: 5. Blurred vision ‐ short term | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.10, 9.04] |
| 20 Adverse effects: 6. Cardiovascular reactions ‐ short term | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.19, 12.11] |
| 21 Adverse effects: 7. Chills ‐ short term | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 22 Adverse effects: 8. Confusion ‐ short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 23 Adverse effects: 9. Depression ‐ short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.26] |
| 24 Adverse effects: 10. Diarrhoea ‐ short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 25 Adverse effects: 11. Dizziness | 4 | 257 | Risk Ratio (M‐H, Random, 95% CI) | 1.96 [0.88, 4.37] |
| 25.1 ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.13, 4.09] |
| 25.2 short term | 3 | 190 | Risk Ratio (M‐H, Random, 95% CI) | 2.58 [1.08, 6.15] |
| 26 Adverse effects: 12. Drowsiness | 2 | 151 | Risk Ratio (M‐H, Random, 95% CI) | 0.52 [0.13, 2.05] |
| 26.1 ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.13, 4.09] |
| 26.2 short term | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.03, 2.80] |
| 27 Adverse effects: 13. Dryness of mouth | 4 | 269 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [0.38, 6.97] |
| 27.1 ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 3.28 [0.36, 29.97] |
| 27.2 short term | 3 | 202 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.14, 6.58] |
| 28 Adverse effects: 14. Excitation ‐ short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 29 Adverse effects: 15. Gastrointestinal reactions ‐ short term | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.01, 7.25] |
| 30 Adverse effects: 16. Headache ‐ short term | 2 | 142 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.04, 14.93] |
| 31 Adverse effects: 17. Increased salivation ‐ short term | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [0.26, 14.91] |
| 32 Adverse effects: 18. Insomnia ‐ short term | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [0.41, 5.44] |
| 33 Adverse effects: 19. Lactation ‐ short term | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.01, 7.25] |
| 34 Adverse effects: 20. Movement disorders | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 34.1 Ataxia ‐ ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 3.28 [0.36, 29.97] |
| 34.2 Ataxia ‐ short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.12, 3.71] |
| 34.3 Dystonia ‐ ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.07, 1.40] |
| 34.4 Dystonia ‐ short term | 1 | 88 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.97] |
| 34.5 Parkinsonism ‐ ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.03, 2.32] |
| 34.6 Parkinsonism ‐ short term | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 0.26 [0.04, 1.60] |
| 34.7 Slight dysarthria ‐ ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.20, 3.39] |
| 34.8 Slight dysarthria ‐ short term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 34.9 Tremor ‐ ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.05, 5.75] |
| 34.10 Tremor ‐ short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.2 [0.02, 1.61] |
| 34.11 Use of antiparkinson medication ‐ ultra short term | 1 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.36 [0.11, 1.23] |
| 34.12 Use of antiparkinson medication ‐ short term | 3 | 215 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.61, 0.90] |
| 35 Adverse effects: 21. Restlessness ‐ short term | 2 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.40, 3.36] |
| 36 Adverse effects: 22. Sensory disturbances ‐ short term | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.06, 14.06] |
| 37 Adverse effects: 23. Sleep disorder ‐ short term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.02, 9.60] |
| 38 Adverse effects: 24. Somnolence ‐ short term | 2 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 3.30 [1.04, 10.40] |
| 39 Adverse effects: 25. Vomitting ‐ short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 40 Sensitivity analysis (no clinically important response to treatment, ultra short term) ‐ exclusion of studies which were not double‐blind‐effects model | 5 | 414 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.41, 1.62] |
| 41 Sensitivity analysis (no clinically important response to treatment, ultra short term) ‐ use of a fixed‐effects model | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 41.1 Unimproved or worse at 30 minutes | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.18, 0.80] |
| 41.2 Unimproved or worse at 60 minutes | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.07 [0.00, 1.13] |
| 41.3 Unimproved or worse at 12 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.51, 1.41] |
| 42 Sensitivity analysis (no clinically important response to treatment, short term) ‐ use of a fixed‐effects model | 6 | 511 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.72, 1.27] |
3.1. Analysis.
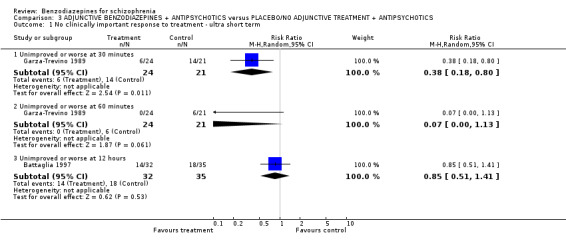
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 1 No clinically important response to treatment ‐ ultra short term.
3.2. Analysis.
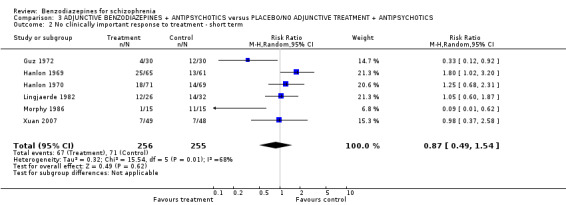
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 2 No clinically important response to treatment ‐ short term.
3.3. Analysis.
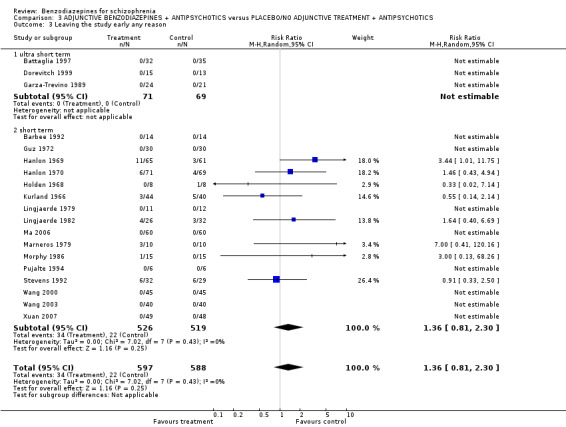
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 3 Leaving the study early any reason.
3.4. Analysis.
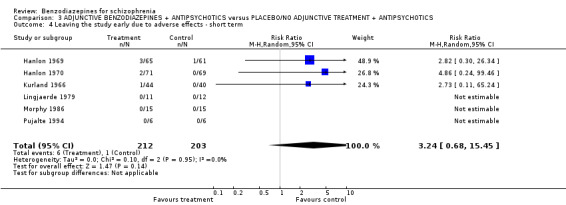
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 4 Leaving the study early due to adverse effects ‐ short term.
3.5. Analysis.
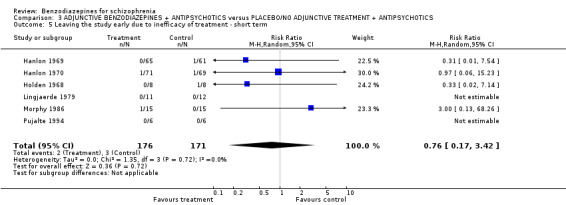
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 5 Leaving the study early due to inefficacy of treatment ‐ short term.
3.6. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 6 Global state: 2. Mean CGI severity score ‐ short term (high=poor).
| Global state: 2. Mean CGI severity score ‐ short term (high=poor) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Lingjaerde 1982 | Adjunctive benzodiazepines + antipsychotics | 4.60 | 5.16 | 22 |
| Lingjaerde 1982 | Placebo/no adjunctive treatment | 4.55 | 4.30 | 29 |
3.7. Analysis.
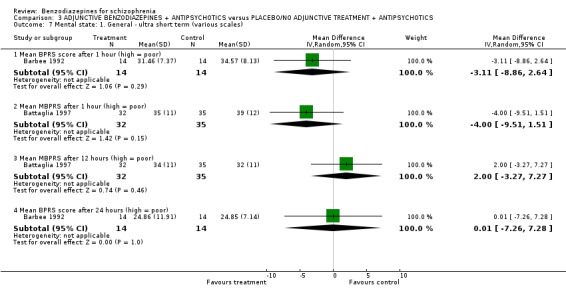
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 7 Mental state: 1. General ‐ ultra short term (various scales).
3.8. Analysis.
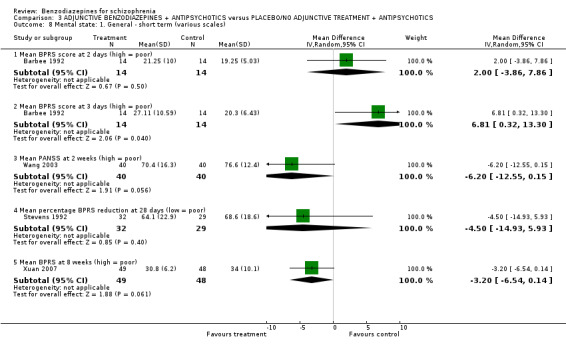
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 8 Mental state: 1. General ‐ short term (various scales).
3.9. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 9 Mental state: 2a. Specific ‐ anxiety ‐ short term (skewed data).
| Mental state: 2a. Specific ‐ anxiety ‐ short term (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Summary of mean HAM‐A score at 4 weeks | ||||
| Morphy 1986 | Adjunctive benzodiazepines + antipsychotics | 0.96 | 0.65 | 13 |
| Morphy 1986 | Placebo/no adjunctive treatment + antipsychotics | 1.29 | 0.38 | 15 |
| Hamilton Anxiety Score (total difference from baseline to 4 weeks) | ||||
| Guz 1972 | Adjunctive benzodiazepines + antipsychotics | 15.47 | 11.23 | 30 |
| Guz 1972 | Placebo/no adjunctive treatment + antipsychotics | 12.5 | 11.23 | 30 |
3.10. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 10 Mental state: 2b. Specific ‐ negative symptoms (SANS score at endpoint) ‐ short term (skewed data).
| Mental state: 2b. Specific ‐ negative symptoms (SANS score at endpoint) ‐ short term (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Barbee 1992 | Benzodiazepines + antipsychotics | 23.31 | 11.66 | 14 |
| Barbee 1992 | Placebo + antipsychotics | 20.92 | 11.47 | 14 |
3.11. Analysis.
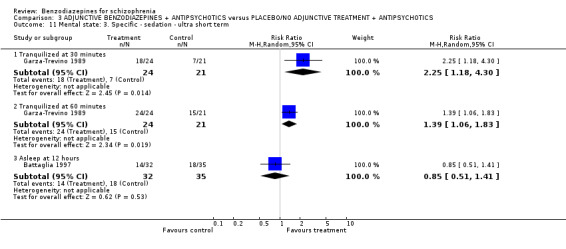
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 11 Mental state: 3. Specific ‐ sedation ‐ ultra short term.
3.12. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 12 Behaviour: 1. Specific ‐aggression ‐ ultra short term (needing restraints within 120 minutes).
3.13. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 13 Behaviour: 2. Specific ‐ aggression ‐ ultra short term.
3.14. Analysis.
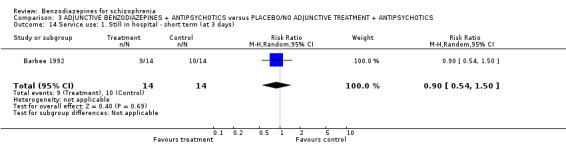
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 14 Service use: 1. Still in hospital ‐ short term (at 3 days).
3.15. Analysis.
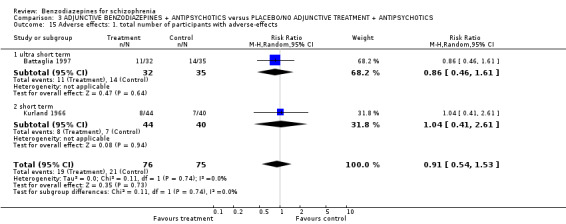
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 15 Adverse effects: 1. total number of participants with adverse‐effects.
3.16. Analysis.
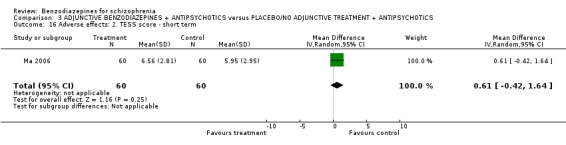
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 16 Adverse effects: 2. TESS score ‐ short term.
3.17. Analysis.
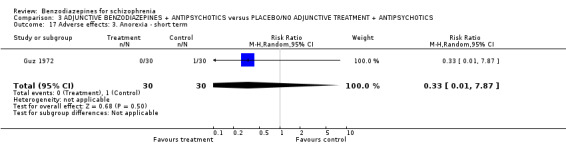
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 17 Adverse effects: 3. Anorexia ‐ short term.
3.18. Analysis.
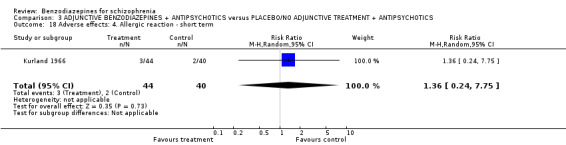
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 18 Adverse effects: 4. Allergic reaction ‐ short term.
3.19. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 19 Adverse effects: 5. Blurred vision ‐ short term.
3.20. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 20 Adverse effects: 6. Cardiovascular reactions ‐ short term.
3.21. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 21 Adverse effects: 7. Chills ‐ short term.
3.22. Analysis.
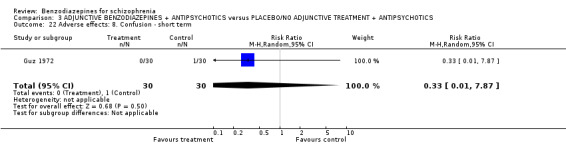
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 22 Adverse effects: 8. Confusion ‐ short term.
3.23. Analysis.
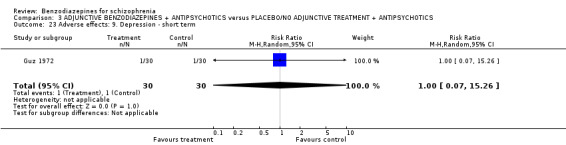
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 23 Adverse effects: 9. Depression ‐ short term.
3.24. Analysis.
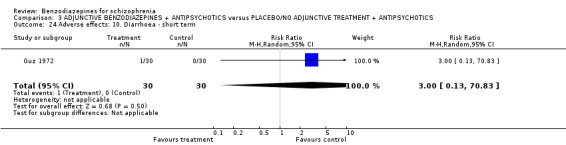
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 24 Adverse effects: 10. Diarrhoea ‐ short term.
3.25. Analysis.
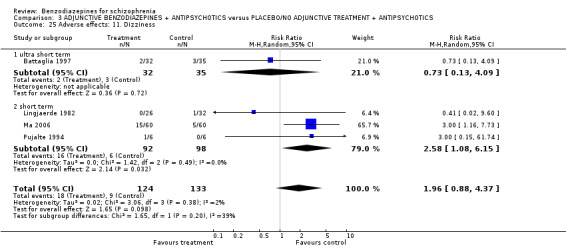
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 25 Adverse effects: 11. Dizziness.
3.26. Analysis.
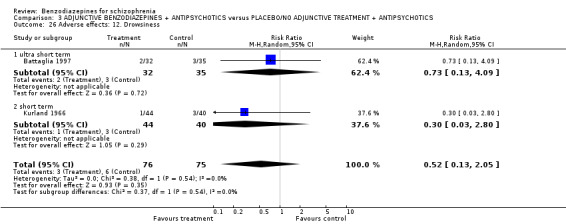
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 26 Adverse effects: 12. Drowsiness.
3.27. Analysis.
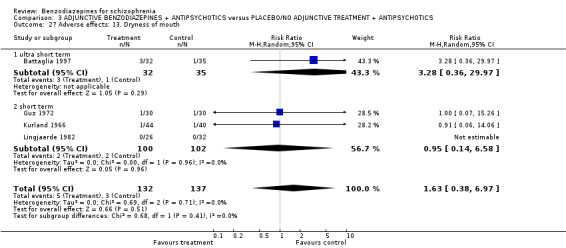
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 27 Adverse effects: 13. Dryness of mouth.
3.28. Analysis.
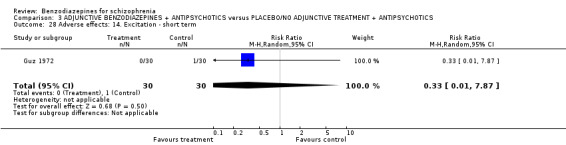
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 28 Adverse effects: 14. Excitation ‐ short term.
3.29. Analysis.

Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 29 Adverse effects: 15. Gastrointestinal reactions ‐ short term.
3.30. Analysis.
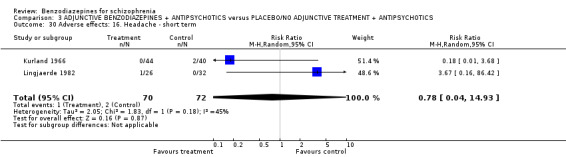
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 30 Adverse effects: 16. Headache ‐ short term.
3.31. Analysis.
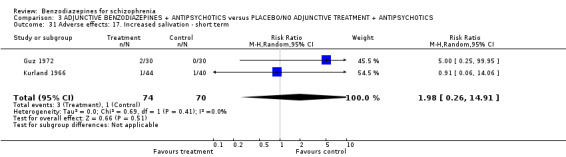
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 31 Adverse effects: 17. Increased salivation ‐ short term.
3.32. Analysis.
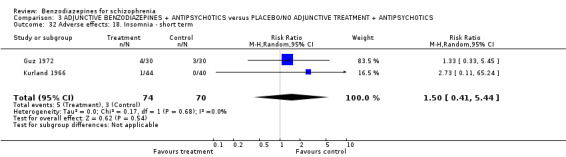
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 32 Adverse effects: 18. Insomnia ‐ short term.
3.33. Analysis.
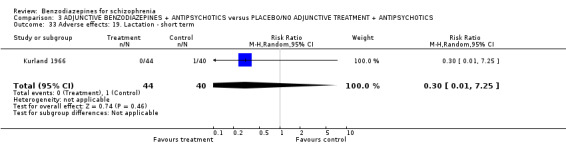
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 33 Adverse effects: 19. Lactation ‐ short term.
3.34. Analysis.
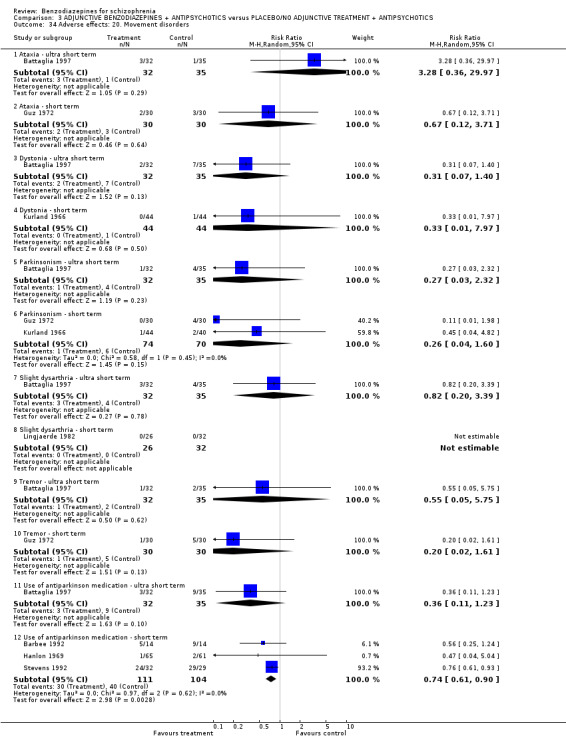
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 34 Adverse effects: 20. Movement disorders.
3.35. Analysis.
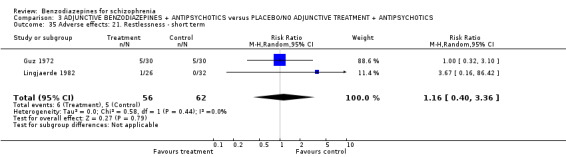
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 35 Adverse effects: 21. Restlessness ‐ short term.
3.36. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 36 Adverse effects: 22. Sensory disturbances ‐ short term.
3.37. Analysis.
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 37 Adverse effects: 23. Sleep disorder ‐ short term.
3.38. Analysis.
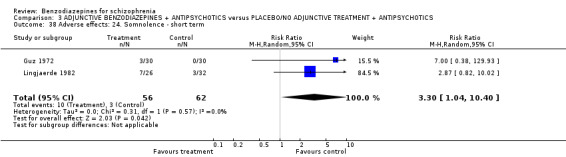
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 38 Adverse effects: 24. Somnolence ‐ short term.
3.39. Analysis.
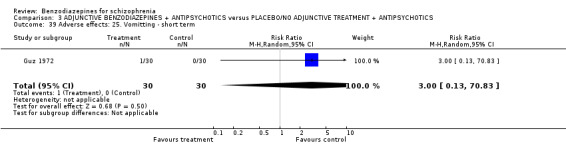
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 39 Adverse effects: 25. Vomitting ‐ short term.
3.40. Analysis.
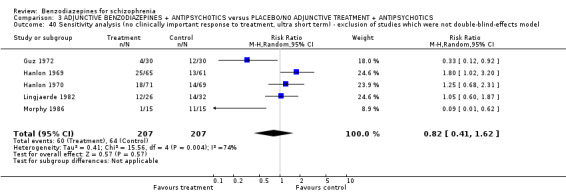
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 40 Sensitivity analysis (no clinically important response to treatment, ultra short term) ‐ exclusion of studies which were not double‐blind‐effects model.
3.41. Analysis.
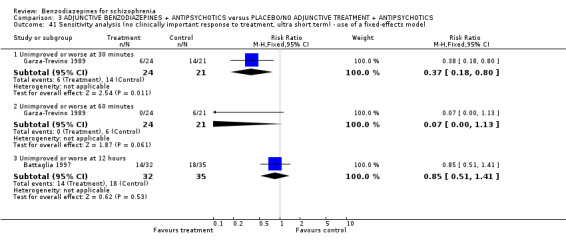
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 41 Sensitivity analysis (no clinically important response to treatment, ultra short term) ‐ use of a fixed‐effects model.
3.42. Analysis.
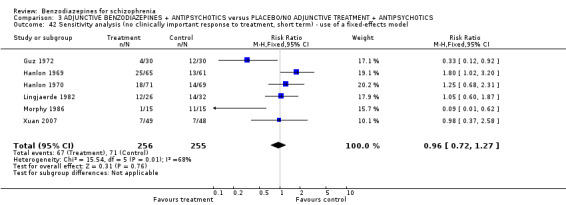
Comparison 3 ADJUNCTIVE BENZODIAZEPINES + ANTIPSYCHOTICS versus PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 42 Sensitivity analysis (no clinically important response to treatment, short term) ‐ use of a fixed‐effects model.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Azima 1962.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: mean 3 weeks (no fixed endpoint) (short‐term trial duration). Location: Allan Memorial Hospital, McGill University, Montreal, Canada. Setting: not indicated. |
|
| Participants | Diagnosis: neurotic reactions (N = 150), schizophrenia (N = 22)*, psychotic depression (N = 12); diagnostic criteria not indicated. History: all with anxiety as target symptom. N = 184. Age: mean 41 years. Sex: 82 M, 102 F. | |
| Interventions | 1. Chlordiazepoxide: mean dose: 70 mg/d. N = 92 (only 8 participants with schizophrenia). 2. Placebo. N = 92 (only 14 participants with schizophrenia). | |
| Outcomes | Leaving the study early.
Global state. Unable to use: Adverse effects (no numbers). |
|
| Notes | *only the 22 schizophrenic patients were included in the meta‐analytic calculations. Main results/conclusion: Benzodiazepine was superior to placebo in terms of anxiety. Chlordiazepoxide: 23.4% appreciable improvement Placebo: 11.7% appreciable improvement. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Multi‐blind”; “identical placebos”. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Multi‐blind”; “evaluation of drug effects is made by different members of the research team and the final conclusion is the result of pooling of the multiple observations”. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | Unclear risk | The outcomes that are of interest in the review have been reported in the pre‐specific way. The adverse effects were not fully addressed. |
| Other bias | High risk | “In the present study, in conjunction with Librium, 3 tranquillizers, 2 anti‐depressants, 4 placebos, were investigated in the unit.” No fixed endpoint for the study (mean three weeks; range: 5 days to 3 months). The majority of participants were treatment‐resistant. Regarding this review the main focus on the anti‐anxiety effect of the medication to judge improvement could be a possible risk of bias. |
Barbee 1992.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 72 hours (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (DSM‐III‐R), acutely psychotic. History: not indicated. N = 28. Age: mean 33.4 years. Sex: not indicated. | |
| Interventions | 1. Alprazolam: dose 1 mg + haloperidol 5 mg (starting dose). N = 14.
2. Placebo + haloperidol: dose 5 mg (starting dose). N = 14. Medication dose was repeated after each rating depending on psychopathology. |
|
| Outcomes | Leaving the study early.
Mental state: BPRS, SANS.
Still in hospital at the end of the study.
Use of antiparkinson medication. Unable to use: Adverse effects: SAS (no numbers). |
|
| Notes | Main results/conclusion:
For both groups as a whole the tranquillizing effect was found to be significant for SAPS, the SANS, the BPRS, and BPRS psychotic subscale. There were no significant differences between the two groups on any of these measures, at any of the time points, suggesting that the two treatments had similar overall effects. The trial was sponsored "in part" by a grant from the manufacturers of alprazolam. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”; “identically appearing placebo tablet” |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Two investigators administered the scales and were blind to each other’s ratings and the drug assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (Simpson and Angus Scale). The adverse effects were not fully addressed (only the five most frequent). |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Battaglia 1997.
| Methods | Allocation: randomised; computer‐generated table of random numbers.
Blindness: double.
Design: parallel.
Duration: 24 hours (ultra short‐term trial duration). Location: five sites in the USA: University of Texas Southwestern Medical Center, Dallas; Harbor‐UCLA Medical Center, Los Angelas; University of Nebraska Medical Center, Omaha; Albert Einstein Medical Center, Phiadelphia; and Harbor‐view Medical Center, Seattle. Setting: inpatients (multicentre). |
|
| Participants | Diagnosis*: mania (N = 13), psychoactive substance use (N = 16), psychosis NOS (N = 27), schizophrenia (N = 47), schizophreniform disorder (N = 1). N = 98. Age: mean 34.2 years. Sex: 73 M, 25 F. | |
| Interventions | 1. Lorazepam: dose 2 mg + haloperidol 5 mg. N = 32.
2. Lorazepam: dose 2 mg. N = 31.
3. Haloperidol: dose 5 mg. N = 35. Medication was applied intramuscular; 1‐6 injections within 12 hours. |
|
| Outcomes | Leaving the study early. Mental state: ABS, CGI, MBPRS. Asleep after 12 hours. Adverse effects. dryness of mouth, dizziness, movement disorder, total number of adverse effects, use of antiparkinson medication. | |
| Notes | *6 of 98 participants received more than one diagnosis.
Main results/conclusion:
Significant mean differences on the ABS (hour 1) and MBPRS (hour 2 and 3) suggest that tranquillization was most rapid in patients receiving the combination treatment. The trial was sponsored "in part" by a grant from the manufacturers of lorazepam. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were "sequentially assigned on the basis of a computer‐generated table of random numbers." |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Double‐blind". "The emergency department psychiatrist who treated and rated the patient remained blinded to the identity of the patient’s medication throughout treatment." The non‐blinding of others is unlikely to introduce bias. The "syringes containing the study drug were prepared by a non‐blinded nurse or hospital pharmacist and delivered to the emergency department as needed." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Double‐blind". "The emergency department psychiatrist who treated and rated the patient remained blinded to the identity of the patient’s medication throughout treatment." The non‐blinding of others is unlikely to introduce bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. "Data for two patients were excluded from the efficacy analysis because of receiving antipsychotic medication shortly before entering the study." Due to the low attrition the risk of bias might be rather low. |
| Selective reporting (reporting bias) | Low risk | The outcomes that are of interest in the review have been reported in the pre‐specific way. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Carpenter 1999.
| Methods | Allocation: stratified randomisation.
Blindness: double.
Design: parallel.
Duration: 4 weeks (followed by a 2‐week period of drug taper ‐ single blind) (short‐term trial duration). Location: Maryland Psychiatric Research Center, Department of Psychiatry, University of Maryland, Baltimore, USA. Setting: outpatients. |
|
| Participants | Diagnosis: schizophrenia and schizoaffective disorder (DSM‐III‐R). N = 53. History: mean duration of illness: 12.6 years. Age: mean 35.6 years. Sex: 38 M, 15 F. | |
| Interventions | 1. Diazepam: dose 10 mg tid. N = 15. 2. Placebo. N = 20. 3. Fluphenazine: dose 5 mg tid. N = 18. | |
| Outcomes | Leaving the study early. Global state: Relapse. | |
| Notes | At the end of the 4‐week treatment period, 30% of the placebo‐treated and 53% of diazepam‐treated participants had not advanced to the next stage of exacerbation. There was no statistically significant difference between fluphenazine and diazepam treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “A stratified randomization procedure was used to assign drug treatment to balance study groups on gender, prior social function, and past duration of hospital care.” No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed. The trial authors did not provide raw data of the BPRS total score or the CGI. Reporting on adverse effects was incomplete. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Cheung 1981.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 18 months (long‐term trial duration). Location: South Kwai Chung Psychiatric Centre, Hong Kong. Setting: outpatients. |
|
| Participants | Diagnosis: schizophrenia (according to criteria of Parkes). History: mean duration of illness: 10.1 years; relapse: 36‐60 months prior to study. N = 30. Age: mean 40.1 years. Sex: 12 M, 18 F. | |
| Interventions | 1. Benzodiazepines (no further details). N = 15. 2. Constant dose of antipsychotics: dose (chlorpromazine equivalent =139 +/‐ 78 mg/d). N = 15. | |
| Outcomes | Leaving the study early. Global state: Relapse. | |
| Notes | Females and late‐onset patients have a significantly better prognosis than male and early‐onset cases. The benzodiazepine group had a significantly higher relapse rate than the antipsychotic group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Two participants left the trial early in the benzodiazepine group, the total number of patients leaving the trial early is rather low (6.7%). The analysis was based on completer data, but due to the low attrition the risk of bias might be rather low. Additionally, the trial authors provided a worst‐case‐scenario analysis. |
| Selective reporting (reporting bias) | Unclear risk | The outcomes that are of interest in the review have been reported in the pre‐specific way. The adverse effects were not fully addressed. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Chouinard 1993.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 2 hours (ultra short‐term trial duration). Location: 2 psychiatric hospitals in Montreal, Canada. Setting: not indicated. |
|
| Participants | Diagnosis: bipolar disorder (N = 7), chronic schizophrenia (N = 5), schizoaffective disorder (N = 3), brief reactive psychosis (N = 1), all acutely agitated (DSM‐III). N = 16. Age: mean 34.2 years. Sex: 10 M, 6 F. | |
| Interventions | 1. Clonazepam: dose 1‐2 mg i.m. at 0, 0.5 and 1 hour + placebo. N = 8. 2. Haloperidol: dose 5‐10 mg i.m. at 0, 0.5 and 1 hour + procyclidine (antiparkinsonian medication). N = 8. | |
| Outcomes | Global state: CGI Severity Scale, CGI Sedation Scale. Mental state: Target Manic Symptoms, IMPS Mania Subscale. Behaviour: NOSIE subscale. Adverse effects: Parkinsonism Total Score*, Tardive Dyskinesia Total Score*. |
|
| Notes | *The haloperidol group received antiparkinson medication during the study.
Both medications produced significant reduction of manic symptoms within two hours of initial treatment; however, haloperidol produced beneficial results more rapidly than clonazepam. The trial was supported "in part" by a grant from the manufacturers of clonazepam. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”. “To ensure double‐blind conditions, the clonazepam group received procyclidine placebo with each dose.” The nurse administering the drugs i.m. injections “was not blind to the treatment allocations”, but this was unlikely to introduce bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “Double‐blind”. The evaluating psychiatrist was blind to treatment allocations. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | One participant left the trial early in the benzodiazepine group; the total number of patients leaving the trial early is rather low (6.25%). The trial authors did not provide any information how they considered with the drop‐out in the analysis, but due to the low attrition the risk of bias might be rather low. |
| Selective reporting (reporting bias) | Low risk | The outcomes that are of interest in the review have been reported in the pre‐specific way with the exception of adverse effects, which were not fully addressed. |
| Other bias | Low risk | “Of the 16 patients, 12 (6 in each group) were receiving neuroleptics at study entry.” |
Dorevitch 1999.
| Methods | Allocation: randomised; table of random numbers.
Blindness: double.
Design: parallel.
Duration: 2 hours (ultra short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 19), schizoaffective (N = 7) and bipolar disorder (N = 2), acute exacerbation (DSM‐IV). N = 28. Age: 20‐60 years. Sex: 13 M, 15 F. | |
| Interventions | 1. Flunitrazepam: dose 1 mg i.m. + antipsychotics. N = 15. 2. Haloperidol: dose 5 mg i.m. + antipsychotics. N = 13. | |
| Outcomes | Leaving the study early. Mental state: OAS. | |
| Notes | Significantly more rapid onset of sedation in flunitrazepam group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “Patients were assigned by a table of random numbers.” No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind“; no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “Double‐blind”; ”All ratings were completed by the same rater, who was blind to the study medications.” Comment: Probably done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (e.g. SDs are missing). Adverse effects were not fully addressed. |
| Other bias | Unclear risk | Antipsychotic drug treatment in both study groups additionally to flunitrazepam and haloperidol. Four patients were taking additional medications including mood stabilizers and lorazepam. |
Foster 1997.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 4 hours (ultra short‐term trial duration). Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: schizophrenia (N = 13), bipolar disorder (N = 13), schizoaffective (N = 4), psychotic disorder NOS (N = 7), acute exacerbation (DSM‐III‐R). N = 37. Age: mean 41.8 years. Sex: 26 M, 11 F. | |
| Interventions | 1. Lorazepam: dose 2 mg every 30 min for 4 hours. N = 17. 2. Haloperidol: dose 5 mg every 30 min for 4 hours. N = 20. | |
| Outcomes | Leaving the study early. Global state: CGI. Mental state: BPRS. Adverse effects: EPS, extreme sedation, use of antiparkinson medication. |
|
| Notes | Rapid tranquillisation. No differences were found either in the number of doses administered or in the administration route selected. Lorazepam may provide an excellent alternative for rapid tranquillisation of the acutely agitated psychotic patient in the emergency room setting. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind”; no further details |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “Double‐blind”. “All raters were unaware of the specific medication received by the patient.” Raters were trained by the first author of the trial, and “periodic calibration training sessions were conducted to control for rater drift. Initial training was continued until raters agreed on item scores at least 90% of the time.” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | Low risk | The outcomes that are of interest in the review have been reported in the pre‐specific way with the exception of adverse effects, which were not fully addressed. |
| Other bias | High risk | Baseline‐imbalance regarding the diagnosis randomised to the both different treatment groups. |
Garza‐Trevino 1989.
| Methods | Allocation: randomised; no further details.
Blindness: non‐blind.
Design: parallel.
Duration: 60 minutes (ultra short‐term trial duration). Location: General psychiatric hospital operated by the Department of Psychiatry of the University of Texas Medical School, Houston, USA. Setting: not indicated. |
|
| Participants | Diagnosis: schizophrenia (N = 16), mania (N = 22), atypical psychotic disorder (N = 16), miscellaneous diagnosis (N = 14), severely agitated. N = 68. Age: mean 31 years. Sex: 41 M, 27 F. | |
| Interventions | 1. Lorazepam: dose 4 mg i.m. every 30 min + haloperidol 5 mg i.m. every 30 min. N = 24. 2. Haloperidol: dose 5 mg i.m. every 30 min. N = 21. 3. Lorazepam: dose 4 mg i.m. every 30 min. N = 23. | |
| Outcomes | Leaving the study early. Global state: Global impression. Mental state: Tranquillized after 30 minutes and 60 minutes. |
|
| Notes | These analyses confirmed that the combination treatment was significantly superior to each of the components in producing rapid tranquillization after statistical correction for differences in baseline severity of agitation. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Non‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Non‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | Unclear risk | The outcomes that are of interest in the review have been reported in the pre‐specific way. No information regarding adverse effects available. |
| Other bias | Unclear risk | Baseline‐imbalance concerning sex‐distribution. |
Gundlach 1966.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 6 weeks (short‐term trial duration). Location: Kings County Psychiatric Hospital Center, Brooklyn, New York, USA. Setting: not indicated. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 22), other schizophrenic types (N = 41), involutions (N = 9), anxiety reactions (N = 18), character disorders (N = 8), other diagnosis (N = 2). N = 100. Age: mean 40 years. Sex: 39 M, 61 F. | |
| Interventions | 1. Diazepam: dose 15‐40 mg/d. N = 52. 2. Placebo. N = 48. | |
| Outcomes | Leaving the study early.
Mental state: anxiety according to TSRS*.
Adverse effects: autonomic reactions (perspiration, flushing, dryness of mouth etc.), change in energy level, gastric reactions (nausea/vomiting), headache. Unable to use: Mental state: MMPI, MAS (incomplete numbers). |
|
| Notes | *Results referred to 39 participants with schizophrenia, who were rated with TSRS. 63% on diazepam were improved in anxiety as contrasted to 55% of the placebo cases. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”. “Placebo was matched in appearance.” “The psychiatrists, social workers and other personnel dealing with the patients did not have access the code indicating which patients were treated with diazepam and which with placebo.” |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition was high (32.7% in the benzodiazepine group and 37.5% in the placebo group). |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed. |
| Other bias | High risk | The drop‐out rate mentioned in the text differs from the drop‐out rate presented in the tables of the publication. |
Guz 1972.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 4 weeks (short‐term trial duration). Location: University of Sao Paulo Psychiatric Clinic, Brazil. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 49), dementia (N = 3), maniac‐depressive (N = 3), paranoia (N = 2), psychosis with epilepsy (N = 1), phobic neurosis (N = 1), drug dependency (N = 1), acute exacerbation. N = 60. Age: mean 27 years. Sex: not indicated. | |
| Interventions | 1. Lorazepam 6 mg/d + 3 mg/d haloperidol. N = 30.
2. Placebo + 3 mg/d haloperidol. N = 30. Haloperidol dose could be adjusted weekly. |
|
| Outcomes | Leaving the study early.
Mental state: Global impression,
Adverse effects: anorexia, akathisia, blurring of vision, confusion, depression, diarrhoea, dizziness, dry mouth, excitation, increased salivation, insomnia, parkinsonism, restlessness, somnolence, cardiovascular reactions (tachycardia), tremor, gastric reactions (vomiting). Unable to use: Mental state: HAM‐A, TSRS (incomplete numbers). |
|
| Notes | The benzodiazepine derivative did have a beneficial effect on some individual symptoms. Adverse effects were no more frequent and no more severe in the lorazepam than in the placebo group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind”, no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”, no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (e.g. SDs are missing). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Hankoff 1962.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 2 weeks (short‐term trial duration). Location: not indicated. Setting: outpatients. |
|
| Participants | Diagnosis*: schizophrenia (N = 90), non schizophrenic (N = 44). N = 174. Age*: 20‐71 years. Sex*: 70 M, 64 F. | |
| Interventions | 1. Chlordiazepoxide: dose 10 mg tid. N = 27. 2. Placebo. N = 95. 3. Chlorpromazine: dose 50 mg tid. N = 25. 4. Meprobamate: dose 200 mg tid. N = 27. | |
| Outcomes | Leaving the study early.
Global state: Global impression.
Adverse effects: anorexia, ataxia, cardiovascular reactions (palpitations), decreased energy level (retardation), depression, dry mouth, EPS, gastric reactions (nausea), headaches, increased energy level (irritability/nervousness), insomnia. Unable to use: Mental state: Manifest Anxiety Rating Scale, AACL (incomplete numbers). |
|
| Notes | *Demographic data is indicated only for completer. A decrease in anxiety was indicated. A significant difference was noted for only one of the treatment groups, chlorpromazine. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”; “All the drugs and their placebos were administered in pink No. 2 capsules.” |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | With 23.2% the attrition was rather high. The authors provided a completer analysis. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed. |
| Other bias | Unclear risk | “Certain variations in the composition of the groups emerged.” |
Hanlon 1969.
| Methods | Allocation: stratified randomisation.
Blindness: double.
Design: parallel.
Duration: 32 days (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 135), not indicated (N = 55) (research psychiatrists´ diagnoses). History: 75% of the patients were severely ill with at least one previous hospitalisation. N = 190. Age: mean 36 years. Sex: 60 M, 130 F. | |
| Interventions | 1. Chlordiazepoxide: mean dose: 29 mg/d + thioridazine 468 mg/d. N = 65. 2. Placebo + thioridazine: mean dose: 470 mg. N = 61. 3. Imipramine: mean dose: 75 mg/d + thioridazine 429 mg/d. N = 64. | |
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects.
Leaving the study early due to inefficacy of treatment.
Global state: Global impression.
Adverse effects: blurred vision, constipation, drowsiness, dry mouth, headache, insomnia, nasal congestion, palpitation, parkinsonism, vertigo. Unable to use: Global state: CGI (incomplete numbers). Mental state: IMPS, SGPG, Group Behavior Scale, MMPI, Depression Index (incomplete numbers). Behaviour: NOSIE (incomplete numbers). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned “within sex”; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind”, no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”, no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The attrition was rather low in the thioridazine plus placebo group (4.9%) but moderate in the thioridazine plus chlordiazepoxide group (16.9%), overall 11.1%. The trial authors provided a modified completer‐analysis. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (e.g. SDs are missing). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Hanlon 1970.
| Methods | Allocation: stratified randomisation.
Blindness: double.
Design: parallel.
Duration: 32 days (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (DSM‐III‐R). History: "chronic", acute exacerbation. N = 211. Age: mean 36.5 years. Sex: 57 M, 154 F. | |
| Interventions | 1. Chlordiazepoxide: mean dose: 37.4 mg/d + fluphenazine 6.6 mg/d. N = 71. 2. Placebo + fluphenazine: mean dose: 6.6 mg/d. N = 69. 3. Imipramine: mean dose: 91.8 mg/d + fluphenazine 6.6 mg/d. N = 71. | |
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects.
Leaving the study early due to inefficacy of treatment.
Global state: CGI. Unable to use: Mental state: IMPS, SGPG, Group Behavior Scale, MMPI, WAIS (incomplete numbers). Behaviour: NOSIE. Adverse effects: (incomplete numbers). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned “within sex”; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind”, no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”, no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | With 7.1% the overall attrition was rather low (8.5% in the fluphenazine plus chlordiazepoxide group and 5.8% in the fluphenazine plus placebo group). The trial authors provided a modified completer‐analysis, but due to the low attrition the risk of bias might be rather low. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (e.g. SDs are missing). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Holden 1968.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: cross‐over.
Duration: 8 weeks (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 24); subtypes: hebephrenic schizophrenia (N = 7), paranoid schizophrenia (N = 6), catatonic schizophrenia (N = 1), chronic undifferentiated (N = 8), not indicated (N = 2). History: "chronic". N = 24. Age: mean 33 years. Sex: 24 M. | |
| Interventions | 1. Chlordiazepoxide: mean dose: 1 mg/kg/d. N = 8. 2. Chlordiazepoxide + thioridazine: mean dose: 0.5 mg/kg/d and 2.5 mg/kg/d. N = 8. 2. Thioridazine: mean dose: 5 mg/kg/d. N = 8. | |
| Outcomes | Leaving the study early.
Leaving the study early due to inefficacy of treatment. Unable to use: Global state: CGI, Psychopathologic Cluster Rating (incomplete numbers). Adverse effects: (incomplete numbers). |
|
| Notes | In global rating, both thioridazine and the combined drug treatments were effective in 20 of 22 participants who completed the study. The trial was sponsored by the manufacturers of chlordiazepoxide. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”. “The drugs were prepared in identical capsules.” Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The attrition was rather low (8.3%). The analysis was based on completer data, but due to the low attrition the risk of bias might be rather low. |
| Selective reporting (reporting bias) | High risk | The outcomes of interest for this review were not fully addressed (e.g. no raw data regarding the BPRS were provided; SDs were missing). |
| Other bias | Unclear risk | Cross‐over study design. |
Kurland 1966.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 28 days (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N =˜163), not indicated (N =˜18). History: acute exacerbation. N = 181. Age: mean 39 years. Sex: 64 M, 117 F. | |
| Interventions | 1. Chlordiazepoxide: dose 30 mg/d + chlorpromazine: mean dose: 2.70 mg/lb/d. N = 44. 2. Placebo + chlorpromazine: mean dose: 2.70 mg/lb/d. N = 40. 3. Chlorpromazine: mean dose: 2.70 mg/lb/d, + imipramine 75 mg/d. N = 35. | |
| Outcomes | Leaving the study early. Leaving the study early due to adverse effects. Adverse effects: allergic reactions (skin rash), cardiovascular reactions (vasomotor disturbances), drowsiness, dry mouth, gastrointestinal reactions, increased salivation, insomnia, lactation, movement disorder, sensory disturbances. | |
| Notes | Most common adverse effects for combined drug treatment versus chlorpromazine only. The trial was sponsored by the manufacturers of chlordiazepoxide. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”; “Chlordiazepoxide, imipramine, and placebo were dispensed from the pharmacy in identical opaque white capsules, so that neither patients nor research personnel knew which of theses medications was being administered.” Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”, no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The attrition was rather low in the chlordiazepoxide plus chlorpromazine group (6.8%) and moderate in the chlorpromazine plus placebo group (12.5%), overall 9.5%. |
| Selective reporting (reporting bias) | Low risk | The outcomes that are of interest in the review have been reported in the pre‐specific way. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Kutcher 1989.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 7 days (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 5), schizoaffective disorder (N = 4), bipolar manic disorder (N = 4), psychotic depression (N = 1), not indicated (N = 1). History: acute phase of psychotic illness and neuroleptic‐induced akathisia. N = 15. Age: mean 18 years. Sex: 9 M, 6 F. | |
| Interventions | 1. Clonazepam: dose 1 mg/d + antipsychotics*. N = 7. 2. Placebo + antipsychotics*. N = 8. | |
| Outcomes | Leaving the study early. Unable to use: Adverse effects: akathisia scale (no numbers). |
|
| Notes | *Medication was administered in addition to antiparkinsonian agents and lithium. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double blind”; tablets were “identical in appearance” Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “Double‐blind”, “Akathisia ratings were repeated blind to the patient’s medication status.” Comment: Probably done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | One of 15 participants left the trial early (7.1%). The analysis was based on completer data, but due to the low attrition the risk of bias might be rather low. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (akathisia scale). |
| Other bias | High risk | “All patients continued on their routine medications (neuroleptics and others) over the duration of the trial.” |
Lerner 1979.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 24 hours (ultra short‐term trial duration). Location: Eitanim Psychiatric Hospital, Jerusalem, Israel. Setting: not indicated. |
|
| Participants | Diagnosis*: paranoid schizophrenia (N = 9), schizoaffective schizophrenia (N = 9), subtypes of schizophrenia (N = 5), paranoid states (N = 2) and mania (N = 5). History: acute exacerbation. N = 40. Age: mean 33 years. Sex: not indicated. | |
| Interventions | 1. Diazepam: dose 30‐40 mg/24 hours. N = 20.
2. Haloperidol: dose 35 mg/24 hours. N = 20. Medication was applied intravenous; 3 injection within 24 hours. |
|
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects. Global state: CGI** . Mental state: BPRS**. |
|
| Notes | *Diagnosis of 10 participants was not indicated. **Scores of 20 participants were missing. Only the results of 20 participants were analysed (11 in the benzodiazepine group and 9 in the antipsychotic group). No significant differences in the clinical response of actively psychotic patients to diazepam or to haloperidol within the first 24 hours of drug treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “Double‐blind”; clinical ratings “were performed by consensus of two physicians unaware of the treatment administered.” Comment: Probably done. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition was very high (50%). 4 patients were not rated “because the raters were unavailable.” |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (SDs were missing). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Lingjaerde 1979.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: cross‐over.
Duration: 3 weeks (short‐term trial duration). Location: 3 psychiatric hospitals in Norway. Setting: inpatients. |
|
| Participants | Diagnosis: hebephrenic schizophrenia (N = 9), paranoid schizophrenia (N = 7), residual schizophrenia (N = 2), undifferentiated schizophrenia (N = 1), reactive paranoid psychosis (N = 3), chronic epileptic psychosis (N = 1). History: "chronic". N = 23. Age: mean 47 years. Sex: 19 M, 4 F. | |
| Interventions | 1. Diazepam: dose 15 mg/d + constant dose of antipsychotics. N = 11*. 2. Placebo + constant dose of antipsychotics. N = 12*. | |
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects.
Leaving the study early due to inefficacy of treatment. Unable to use: Mental state: BPRS (incomplete numbers). Behaviour: NOSIE (incomplete numbers). Adverse effects: incomplete numbers. |
|
| Notes | * More than half of all patients received antiparkinsonian medication.
This study indicates that diazepam, in a dosage of 15 mg daily, has a beneficial effect when given as an adjunct to neuroleptics in chronic psychotic patients. The trial was sponsored by the manufacturers of diazepam. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”. “Tablets of identical appearance.” Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No drop‐outs; but the data for one participant regarding the BPRS and for two participants regarding the NOSIE scale were missing. Due to the low attrition the risk of bias might be rather low. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (e.g. the SDs were missing). |
| Other bias | Unclear risk | Additionally to the study medication the participants received further their previous medication which contains two different antipsychotic agents in 11 participants. Cross‐over study design. |
Lingjaerde 1982.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: cross‐over.
Duration: 3 weeks (short‐term trial duration). Location: 10 psychiatric departments in Denmark and Norway. Setting: inpatients and outpatients. |
|
| Participants | Diagnosis: hebephrenic or simple schizophrenia (N = 24), paranoid schizophrenia (N = 20), unspecified schizophrenia (N = 8), manic‐depressive psychosis (N = 3), reactive, paranoid psychosis (N = 3); (ICD‐8 diagnosis). History: subchronic or chronic. N = 58. Age: mean 34.7 years (M), 44 years (F). Sex: 28 M, 30 F. | |
| Interventions | 1. Estazolam: dose 6 mg/d + constant dose of antipsychotics*. N = 26. 2. Placebo + constant dose of antipsychotics*. N = 32. | |
| Outcomes | Leaving the study early.
Global state: CGI severity scale.
Adverse effects: dizziness, dryness of mouth, headache, restlessness, somnolence, slight dysarthria, sleep disorder (difficulties going to sleep). Unable to use: Mental state: BPRS, CPRS (no usable data). |
|
| Notes | *Patients received additionally antiparkinsonian agents (N = 22), tricyclic antidepressants (N = 5), antiepileptic (N = 3), and lithium (N = 1).
Both treatment groups there was a significant improvement during the first 3 weeks period with regard to both, global clinical state and auditory hallucinations.
It was concluded that estazolam (in addition to antipsychotic compounds) was significantly superior to placebo in terms of the global clinical state. The trial was sponsored by one of the manufacturers of estazolam. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”. “Similar‐looking placebo tablets” Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The attrition was moderate (12.1%). It was not clearly indicated which analysis‐method was applied. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (incomplete numbers). |
| Other bias | Unclear risk | The difference in mean age at baseline was statistically significant. Some patients received no antipsychotic agent, whereas other participants received one or more antipsychotics during the trial. Cross‐over study design. |
Ma 2006.
| Methods | Allocation: randomised; no further details. Blindness: single‐blind, the raters did not involved in the study. Design: parallel. Duration: 4 weeks (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (CCMD‐3) with auditory hallucination. History: mean duration of illness: 25.2 months (SD = 13.86). N = 120. Age: mean 25.55 years (SD = 7.385). Sex: 76 M, 44 F. |
|
| Interventions | 1. Injection of clonazepam into Ting‐gong (acupuncture point): dose: 2mg/d (1mg at each acupuncture point, total 14 times) + antipsychotic: dose amount to chlorpromazine dose was 595.35 ±185.60mg/d. N = 60. 2. Antipsychotic: dose: amount to chlorpromazine dose was 583.85 ± 190.80mg/d. N = 60. |
|
| Outcomes | Leaving the study early. Mental state: 12th item score of the BPRS (hallucination item). Adverse effects: TESS. |
|
| Notes | Conclusion: "Injection of clonazepam into Tinggong is safe and effective therapy for auditory hallucination of schizophrenia." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Single‐blind; no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Single‐blinded, the raters did not involved in the study. Comment: Probably done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | Low risk | The outcomes that are of interest in the review have been reported in the pre‐specific way. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Marneros 1979.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 21 days (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (criteria of K. Schneider). History: acute exacerbation. N = 20. Age: 20‐55 years. Sex: not indicated. | |
| Interventions | 1. Camazepam: dose 40 mg/d + haloperidol 12 mg/d. N = 10. 2. Placebo + haloperidol: dose 12 mg/d. N = 10. | |
| Outcomes | Leaving the study early. Unable to use: Global state: CGI (incomplete numbers). |
|
| Notes | Adjunctive treatment with benzodiazepine was significantly better in affective symptoms, like aggression etc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind”, no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”, no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Three patients in the camazepame group left the trial early (30%) and no participant in the placebo group; the total number of patients leaving the trial early is moderate (15%). The trial authors provided the individual patient data. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed. The adverse effects were not fully addressed. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Merlis 1962.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 4 weeks (proceeded by a 2 week wash‐out period) (short‐term trial duration). Location: Center Islip State Hospital, Central Islip, New York, USA. Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 32), hebephrenic schizophrenia (N = 12), catatonic schizophrenia (N = 12), other subtypes (N = 14), psychosis and mental deficiency (N = 5), psychosis and psychopathic personality (N = 5). History: "chronic". N = 80. Age: mean 38 years. Sex: 40 M, 40 F. | |
| Interventions | 1. Chlordiazepoxide: dose 25 mg/d. N = 20. 2. Diazepam: dose 10 mg/d. N = 20. 3. Chlorpromazine: dose 50 mg/d. N = 20. 4. Placebo. N = 20. | |
| Outcomes | Leaving the study early. Leaving the study early due to adverse effects. Leaving the study early due to inefficacy of treatment. Mental state: BPRS ‐ not improved (physicians´ rating), MMS (nurses´rating). | |
| Notes | No significant difference between the groups. The trial was supported "in part" by the manufacturers of chlordiazepoxide and diazepam. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”. “Capsules of identical appearance, identified on the ward only by code letter.” Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed. The trial authors did not provide raw data of the BPRS score or the MMS. Reporting on adverse effects was incomplete. |
| Other bias | Unclear risk | “Neither of the two psychiatrists had had previous experience with the BPRS‐Scale.” |
Minervini 1990.
| Methods | Allocation: randomised; balanced for each group of six patients.
Blindness: double.
Design: parallel.
Duration: 3 weeks (short‐term trial duration). Location: Psychiatric Department of the "Opera Don Uva" Hospital, Bisceglie, Italy. Setting: inpatients. |
|
| Participants | Diagnosis: disorganized‐type schizophrenia (N = 11), catatonic‐type schizophrenia (N = 3), paranoid‐type schizophrenia (N = 43), undifferentiated‐type schizophrenia (N = 9). (DSM‐III). History: "chronic" without antipsychotic medication and anxiety not related to schizophrenic process. N = 66. Age: mean 46.5 years. Sex: 66 F. | |
| Interventions | 1. Alpidem: dose 100 mg/d. N = 33. 2. Placebo. N = 33. | |
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects.
Leaving the study early due to inefficacy of treatment. Global state: CGI. Mental state: BPRS, HAM‐A. Adverse effects: Gastrointestinal reactions (vomiting), sedation (extreme). |
|
| Notes | Alpidem was significantly more effective than placebo in improving HRSA score. Considering the results of CGI at day 21, more patients were moderately to markedly improved on alpidem than on placebo. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned; “Randomisation was balanced for each group of six patients.” No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Alpidem tablets of 50 mg and placebo tablets were identical in size and appearance.” Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | Low risk | The outcomes have been reported in the pre‐specific way. |
| Other bias | High risk | The main focus of this investigated trial was on the anti‐anxiety effect of the additional benzodiazepine medication. This can be judge as a possible risk of bias. “The patients were all chronic institutionalised schizophrenics whose anxiety was not directly linked to the schizophrenic process.” |
Morphy 1986.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 4 weeks (short‐term trial duration). Location: Buffalo Veterans Administration Medical Center Mental Health Clinic, Buffalo, USA. Setting: outpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 30) (DSM‐III). History: chronic schizophrenic with moderate‐to‐severe symptoms of anxiety. N = 30. Age: mean 47 years. Sex: 29 M, 1 F. | |
| Interventions | 1. Alprazolam: dose maximum 3 mg/d + constant dose of antipsychotics. N = 15. 2. Placebo + constant dose of antipsychotics. N = 15. | |
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects.
Leaving the study early due to inefficacy of treatment. Global state: CGI. Mental state: HAM‐A. Unable to use: Mental state: Wittenborn Psychiatric Rating Scale, Raskin‐Covi Scales (no numbers). |
|
| Notes | Alprazolam was shown to be more effective than placebo in the relieving symptoms of anxiety. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned; “randomization was forced so that in each consecutive group of six patients, three patients received alprazolam and three patients received placebo.” No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | One alprazolam‐treated patient (6.7%) was dropped from the study early in the protocol and the data were not available for the analyses. Due to the low attrition the risk of bias might be rather low. |
| Selective reporting (reporting bias) | High risk | The outcomes of interest for this review were not fully addressed (e.g. no raw data regarding the BPRS were provided). The adverse effects were not fully addressed. |
| Other bias | Unclear risk | The main focus of this investigated trial was on the anti‐anxiety effect of the additional benzodiazepine medication. This can be judged as a possible risk of bias. |
Nestoros 1982.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 1 week (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 12) (RDC, DSM‐III). History: "chronic", acute exacerbation. N = 12. Age: not indicated. Sex: not indicated. | |
| Interventions | 1. Diazepam: dose 70‐400 mg/d. N = 6. 2. Placebo. N = 6. | |
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects.
Leaving the study early due to inefficacy of treatment.
Global state: CGI. Unable to use: Mental state: BPRS, SS‐PSE, SARS, SCL‐90 (incomplete numbers). Behaviour: NOSIE (incomplete numbers). |
|
| Notes | Within a few hours to a few days from the onset of diazepam treatment both, positive and negative schizophrenic symptoms were dramatically eliminated in 5 out of 6 participants. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”; “matched tablets”. Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed. The trial authors did not provide raw data of the BPRS score, the SCL‐90, the SARS and the SS‐PSE. Reporting on adverse effects was incomplete. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Nishikawa 1982.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: cross‐over.
Duration: 3 years (long‐term trial duration). Location: Seiwakai Nishikawa Hospital, Hamada, Japan. Setting: outpatients. |
|
| Participants | Diagnosis: schizophrenia (DSM‐III‐R). History: "chronic". N = 55. Age: mean 33.7 years. Sex: 37 M, 18 F. | |
| Interventions | 1. Diazepam: dose 15 mg/d. N = 13. 2. Haloperidol: dose 3 mg/d. N = 10. 3. Placebo. N = 10. 4. Chlorpromazine: dose 75 mg/d. N = 10. 5. Imipramine: dose 50 mg/d. N = 12. | |
| Outcomes | Leaving the study early. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Global state: relapse. | |
| Notes | The drug initially assigned was administered until relapse signs appeared. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”. “Drug appearance, with respect to powder colour, taste and volume, was made identical by adding a kind of stomachics.” Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Double‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Regarding the outcomes of interest for this review in the case of cross‐over‐studies (only data up to the point of the first cross‐over were used), there was no risk of bias due to the attrition: “the treatments with the first allocated drugs were performed in all patients.” |
| Selective reporting (reporting bias) | Low risk | The outcomes that are of interest in the review have been reported in the pre‐specific way with the exception of adverse effects, which were not fully addressed. |
| Other bias | Unclear risk | Cross‐over study design. |
Pujalte 1994.
| Methods | Allocation: randomised; no further details.
Blindness: double.
Design: parallel.
Duration: 2 weeks (short‐term trial duration). Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: schizophrenia (N = 10), schizoaffective disorder (N = 2) (DSM‐III‐R). History: neuroleptic induced akathisia. N = 12. Age: not indicated. Sex: 8 M, 4 F. | |
| Interventions | 1. Clonazepam: dose 0.5‐2.5 mg/d + constant antipsychotics*. N = 6. 2. Placebo + constant antipsychotics*. N = 6. | |
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects.
Leaving the study early due to inefficacy of treatment.
Adverse effects: drowsiness. Unable to use: Adverse effects: Barnes Rating Scale for Drug Induced Akathisia (skewed data) |
|
| Notes | *Patients received anticholinergic agents in a constant dose. The trial was supported by the manufacturers of clonazepam. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “Double‐blind”; “identical appearance” of the study medication. Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “Double‐blind”. “The patients were all examined by the same independent psychiatrist, who was unaware of the medication dose.” Comment. Probably done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | Low risk | The outcomes that are of interest in the review have been reported in the pre‐specific way. Reporting on adverse effects was incomplete. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Stevens 1992.
| Methods | Allocation: randomised; no further details.
Blindness: non blind.
Design: parallel.
Duration: 28 days (short‐term trial duration). Location: Department of Psychiatry, University of Tübingen, Germany. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (DSM‐III‐R). History: "chronic". N = 61. Age: mean ˜ 40 years. Sex: 33 M, 28 F. | |
| Interventions | 1. Lorazepam: dose 0.05 mg/kg/d + haloperidol 0.5 mg/kg/d. N = 32. 2. Placebo + haloperidol 0.5 mg/kg/d. N = 29. | |
| Outcomes | Leaving the study early.
Adverse effects: use of antiparkinson medication at least once. Unable to use: Mental state: BPRS (no usable data). Adverse effects: SAS (no usable data). |
|
| Notes | None of the patient treated with lorazepam and haloperidol achieved better BPRS score or subscores, nor did their condition improve faster than in patient treated with haloperidol alone. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | “The study design was open.” |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The mean outcome (BPRS) was rated “by raters ignorant of the patient’s current medication.” Thus, the study was single‐blinded concerning the main‐outcome. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The attrition was moderate in the haloperidol plus lorazepam group (18.75%) and in the haloperidol plus placebo group (20.7%), overall 19.7%. The trial authors provided a completer‐analysis. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed. The trial authors did not provide raw data of the BPRS score (only relative scores). Reporting on adverse effects was incomplete. |
| Other bias | Unclear risk | “to avoid bias introduced by group differences in entry scores that did not reach significance or violated homogeneity of variance, the comparison was based on improvement calculated as the quotient of actual score/initial score.” |
TREC‐Rio 2003.
| Methods | Allocation: randomised; table of random numbers.
Blindness: rater‐blinded in terms of the primary outcome.
Design: parallel.
Duration: 2 weeks (short‐term trial duration). Location: 3 public psychiatric hospitals in Rio de Janeiro, Brazil. Setting: emergency room. |
|
| Participants | Diagnosis*: psychosis (N = 219), substance misuse (N = 51), other (N = 30). History: acute aggressive or agitated behaviour. N = 301. Age: mean 38 years. Sex: 136 M, 155 F. | |
| Interventions | 1. Midazolam: dose 15 mg i.m. (N = 124) or midazolam 7.5 mg i.m. (N = 26). 2. Antipsychotic + promethazine: dose haloperidol 10 mg i.m. (N = 71) or haloperidol 5 mg i.m. (N = 77) + promethazine 25‐50 mg i.m. | |
| Outcomes | Leaving the study early. Leaving the study early due to adverse effects. Leaving the study early due to inefficacy of treatment. Mental state: tranquillized or asleep after 20, 40, 60 and 120 min., another episode of aggression/agitation after 24 hours, not needing restraints after 120 min., needing a doctor within 24 hours, still in hospital at the end of the study. Adverse effects: grand‐mal seizure, respiratory depression. | |
| Notes | *Diagnosis of 1 participant is not indicated. According to the authors 150 persons were in the category F20 of ICD‐10. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned; “In Britain one collaborator used Microsoft Excel to generate even random numbers less than 10. These block sizes were than applied to a table of random numbers.” Comment: Probably done. |
| Allocation concealment (selection bias) | Low risk | “To help ensure concealment of allocation, [one collaborator] produced a table of allocation sequence independent of block size. He sent these tables to a Brazilian colleague independent of the TREC team, who ensured that the correct drug was in the consecutively numbered local pack before it was sealed. These packs were constructed of cardboard, were identical, and were sealed firmly with tape, across which the consecutive number was written.” Comment: Probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | “Open treatment.” |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The ratings of the primary outcome were performed by raters “blind to the allocated treatment, and unknown to the clinicians looking after the patient.” Comment: Probably done. Thus, concerning the main‐outcome (tranquillised or asleep by 20 minutes), the study was single‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | With 2% the overall attrition was rather low (0.7% in the midazolam group and 3.3% in the haloperidol plus promethazine group). Due to the low attrition the risk of bias might be rather low. |
| Selective reporting (reporting bias) | Low risk | The outcomes that are of interest in the review have been reported in the pre‐specific way with the exception of adverse effects, which were not fully addressed. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Wang 2000.
| Methods | Allocation: randomised according to lottery; no further details. Blindness: non‐blind. Design: parallel. Duration: 2 weeks (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (CCMD‐2‐R), refractory auditory hallucinations. History: mean duration of illness: 12.1 years (SD = 8.31). N = 90. Age: mean 38.9 years (SD = 10.355). Sex: 60 M, 31 F. |
|
| Interventions | 1. Clonazepam (injected into Ting‐gong [acupuncture point]): dose: 2 mg/d (1mg at each acupuncture point) + antipsychotics. N = 45. 2. Antipsychotics. N = 45. |
|
| Outcomes | Leaving the study early. Mental state: 12th item score of the BPRS (hallucination item). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | randomised according to lottery; allocated by acupuncturist; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Non‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Non‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed. PANSS total score was assessed but no data were provided in the publication. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Wang 2003.
| Methods | Allocation: randomised; no further details.
Blindness: non blind.
Design: parallel.
Duration: 8 weeks (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia. History: first episode. N = 80. Age: unclear. Sex: unclear. | |
| Interventions | 1. Risperidone: dose 2‐5 mg/d + clonazepam i.m. 2‐4 mg/d. N = 40 2. Risperidone: dose 2‐5 mg/d. N = 40 | |
| Outcomes | Leaving the study early. Mental state: PANSS* Adverse effects: TESS. | |
| Notes | *usable data only for 2‐week treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not double‐blind; no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not double‐blind; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (usable data only for 2‐week treatment). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Wyant 1990.
| Methods | Allocation: randomised; no further details.
Blindness: single.
Design: parallel.
Duration: 2 hours (ultra short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: chronic paranoid schizophrenia (N = 5), chronic undifferentiated schizophrenia (N = 10). History: "chronic". N = 15. Age: benzodiazepine group mean age ˜35 years, haloperidol group mean age ˜43 years. Sex: 15 M. | |
| Interventions | 1. Midazolam: dose 5 mg i.m. N = 5. 2. Haloperidol: dose 10 mg i.m. N = 5. 3. Sodium amytal: dose 250 mg i.m. N = 5. | |
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects.
Leaving the study early due to inefficacy of treatment. Unable to use: Global state: CGI (incomplete numbers). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Single‐blind”; no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Single‐blind”; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (CGI). The adverse effects were not addressed. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Xuan 2007.
| Methods | Allocation: randomised; no further details. Blindness: non‐blind. Design: parallel. Duration: 8 weeks (short‐term trial duration). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (CCMD‐3); History: first episode 62 patients; recurrence 35 patients. N = 97. Age: mean 30.05 (SD = 9). Sex: 51 M, 46 F. |
|
| Interventions | 1. Risperidone: dose 3.9 ± 1.0 mg/d (2‐6 mg/d) [initial dose 0.5‐1mg/d, increased to treatment dose within two weeks] + Benzodiazepines (clonazepam or lorazepam or alprazolam). Benzodiazepines dose was not described, it was used in 7‐10 days, not beyond 2 weeks. N = 49. 2. Risperidone: dose 3.6 ± 0.9 mg/d (2‐6 mg/d) [initial dose 0.5‐1 mg/d, increased to treatment dose within two weeks]. N = 48. |
|
| Outcomes | Leaving the study early. Mental state: BPRS. Unable to use: Adverse effects: TESS. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned; no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Non‐blind; no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Non‐blind; no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcomes that are of interest in the review were not fully addressed (TESS score). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
General abbreviations: F ‐ females. i.m. ‐ intramuscular. M ‐ males. mg ‐ milligram. min ‐ minutes. N ‐ number of participants. SD ‐ standard deviation. tid ‐ three times daily.
Diagnostic criteria: CCMD‐3 ‐ Chinese Classification of Mental Disorders, third revision. DSM‐II ‐ Diagnostic and Statistical Manual of Mental disorders, second edition. DSM‐III ‐ Diagnostic and Statistical Manual of Mental disorders, third edition. DSM‐III‐R ‐ Diagnostic and Statistical Manual of Mental disorders, third edition, revised. ICD‐8/9/10 ‐ International Classification of Diseases, eighth/ninth/tenth revision. RDC ‐ Research Diagnostic Criteria.
Global state scales: CGI ‐ Clinical Global Impression. OCR ‐ Overall Clinical Rating.
Mental state/Behaviour scales: AACL ‐ Affect Adjective Check List. ABS ‐ Agitated Behavior Scale. BDI ‐ Beck Depression Inventory. BPRS ‐ Brief Psychiatric Rating Scale. BRMAS ‐ Bech‐Rafaelsen Scale for Mania. CPRS ‐ Comprehensive Psychopathology Rating Scale. HAM‐A ‐ Hamilton Rating Scale of Anxiety. IMPS ‐ Inpatient Multidimensional Rating Scale. MAS ‐ Manifest Anxiety Scale. MBPRS ‐ Modified Brief Psychiatric Rating Scale. MMPI ‐ Minnesota Multiphasic Personality Inventory. MMS ‐ Malamud‐Sands Scale. MMSE ‐ Mini Mental State Examination. MS ‐ Manchester Scale. MSM ‐ Murphy Scale for Mania. MSRS ‐ Manic State Rating Scale. NH ‐ (Modified) New Haven Schizophrenia Index. NOSIE ‐ Nurses Observation Scale for Inpatient Evaluation. OAS ‐ Overt Aggression Scale. PANSS ‐ Positive and Negative Symptoms Scale. PIP ‐ Psychotic Inpatient Profile. SANS ‐ Scale for Assessment of Negative Symptoms. SAPS ‐ Scale for Assessment of Positive Symptoms. SARS ‐ Simpson Angus Rating Scale. SCI ‐ Structured Clinical Interview. SCL‐90 ‐ 90 Item Self‐Assessment Questionnaire. SGPG ‐ Spring Grove Psychiatric Rating Scale. SRSS ‐ Self‐Rating Symptom Scale. SS‐PSE ‐ Schizophrenia Subscale of the Present State Examination. TSRS ‐ Target Symptom Rating Scale. VAS ‐ Visual Analogue Scale. WAIS ‐ Wechsler Adult Intelligence Scale.
Adverse effect scales: AIMS ‐ Abnormal Involuntary Movement Scale. BFCRS ‐ Bush‐Francis Catatonia Rating Scale. BARS ‐ Barnes Akathisia Rating Scale. CGI ‐ Clinical Global Impression, side effects. EPS ‐ Extrapyramidal side effects. EPSE ‐ Extrapyramidal side effects (modified Simpson & Angus Scale). FSUCL ‐ Fischers Somatische Symptome or Unerwünschte Effekte Check List. MRS ‐ Modified Rogers Scale. SAFTEE ‐ Scale for Assessment of Treatment Emergent Events. SAS ‐ Simpson and Angus Scale. TD ‐ tardive dyskinesia. TESS ‐ Treatment Emergent Symptom Scale. UKU ‐ UKU Side effect Scale. VPAS ‐ Van Putten´s Akinesia Scale.
Diagnosis: NOS ‐ not otherwise specified.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adler 1985 | Allocation: randomised, double‐blind. Participants: people with schizophrenia. Interventions: antipsychotics + lorazepam vs. antipsychotics + beta‐blockers. Outcomes: no usable data in terms of any outcome. |
| Alexander 2004 | Allocation: randomised. Participants: most participants were diagnosed to have mania; only 18.5 % of the patients had schizophrenia. |
| Altamura 1987 | Allocation: randomised. Participants: people with schizophrenia. Interventions: clonazepam + haloperidol versus haloperidol + placebo. Outcomes: no usable data. |
| Ananth 1979 | Allocation: randomised, double‐blind. Participants: chronic geriatric patients with psychiatric symptomatology; unclear how many had schizophrenia‐like psychoses. |
| Bao 2007 | Allocation: randomised. Participants: people with schizophrenia. Interventions: aripiprazole + clonazepam versus clozapine. |
| Bienek 1997 | Allocation: randomised, double‐blind. Participants: people with bipolar disorder, psychosis not otherwise specified, schizophrenia, brief reactive psychosis, substance‐induced psychosis, undifferentiated schizophrenia. Interventions: lorazepam versus antipsychotics + lorazepam. |
| Bobruff 1981 | Allocation: randomised. Participants: patients with tardive dyskinesia, no diagnosis given. |
| Cohen 1987 | Allocation: not randomised. |
| Crosse 1974 | Allocation: not randomised. |
| Csernansky 1988 | Allocation: randomised. Participants: people with schizophrenia. Interventions: alprazolam + antipsychotics versus placebo + antipsychotics. Outcomes: no usable data. |
| Dubin 1988 | Allocation: not randomised, review. |
| Ge Qianrong 2004 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone + clonazepam versus haloperidol. |
| Greenfeld 1987 | Allocation: not randomised, case report. |
| Haas1982 | Allocation: not randomised. |
| Hanus 1973 | Allocation: not randomised, controlled clinical trial. |
| Hartelius 1978 | Allocation: randomised. Participants: only 39 of 143 participants had schizophrenia; their data were not presented separately. |
| Harvey 2004 | Allocation: randomised, partially blinded. Participants: people with schizophrenia (mildly symptomatic). Interventions: controlled cross‐over trial with haloperidol versus lorazepam. Outcomes: no usable data. |
| He 2007 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone + clonazepam versus clozapine. |
| Hekimian 1967 | Allocation: not adequately randomised, alternate allocation. |
| Hesso 1975 | Allocation: not randomised. |
| Horiguchi 1992 | Allocation: randomised. Participants: people with chronic schizophrenia. Interventions: antipsychotics + clonazepam versus antipsychotics + antiparkinson medication. |
| Hovens 2005 | Allocation: not randomised. |
| Hu 2004 | Allocation: not randomised. |
| Itil 1972 | Allocation: not randomised. |
| Jibiki 1994 | Allocation: not randomised. |
| Jirmeson 1982 | Allocation: not randomised. |
| Jungkunz 1984 | Allocation: randomised. Participants: people with schizophrenia. Interventions: methionine versus diazepam, no appropriate comparison group. |
| Kang 2006 | Allocation: randomised. Participants: people with schizophrenia. Interventions: quetiapine + clonazepam versus chlorpromazine versus clozapine. |
| Karson 1982 | Allocation: not randomised. |
| Kellner 1975 | Allocation: randomised. Participants: people with schizophrenia. Interventions: thioridazine or trifluoperazine versus chlordiazepoxide versus placebo. Outcomes: no usable data. |
| Knott 2006 | Allocation: randomised, double‐blind. Participants: unclear how many patients had schizophrenia or related disorders; no diagnosis given. |
| Li 2004 | Allocation: randomised. Participants: people with schizophrenia. Interventions: loxapine versus risperidone. |
| Li 2007 | Allocation: randomised according to hospital admission order/number. |
| Maar 1983 | Allocation: randomised, double‐blind. Participants: people with schizophrenia were excluded. |
| Mei 2006 | Allocation: randomised. Participants: people with schizophrenia. Interventions: quetiapine + clonazepam versus haloperidol. |
| Monroe 1967 | Allocation: not randomised. |
| Nestros 1983 | Allocation: not randomised. |
| Nobay 2004 | Allocation: randomised, double‐bind. Participants: unclear how many patients had schizophrenia or related disorders; no diagnosis given. |
| Pato 1989 | Allocation: not randomised. |
| Pecknold 1993 | Allocation: not randomised, retrospective survey. |
| Qin 2005 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone + alprazolam versus clozapine. |
| Remington 1993 | Allocation: not randomised, case report. |
| Richards 1998 | Allocation: randomised, double‐blind. Participants: only 10 % with schizophrenia‐like psychoses. |
| Ruskin 1978 | Allocation: not randomised. |
| Salam 1988 | Allocation: not randomised, case series. |
| Saletu 1971 | Allocation: not randomised. Participants: healthy individuals. |
| Shafti 2005 | Allocation: unclear whether randomised. Participants: people with schizophrenia. Interventions: alprazolam versus clomipramine versus citalopram versus placebo. |
| Soldatos 1986 | Allocation: not randomised. |
| Sterlin 1971 | Allocation: not randomised. |
| Stonehill 1966 | Allocation: unclear whether randomised, double‐blind, cross‐over design. Participants: people with psychosis. Interventions: diazepam, chlordiazepoxide, LA XIV, LA XVII. Outcomes: no usable data. |
| Subramaney 1997 | Allocation: randomised, double‐blind. Participants: organic hallucinations (psychoactive substance abuse) (N = 24), schizophrenia (N = 16), bipolar disorders (N = 14). |
| Tang 2007 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone + clonazepam versus haloperidol. |
| TREC‐Vellore 2004 | Allocation: randomised. Participants: schizophrenia (N = 37), acute psychosis (N = 22), mania (N = 97), depression (N = 19), substance abuse (N = 10), other (N = 15). |
| Wang 2004a | Allocation: randomised according to hospital admission serial number. |
| Wang 2004b | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone + clonazepam versus clozapine versus haloperidol. |
| Wang 2005 | Allocation: randomised. Participants: people with schizophrenia. Interventions: quetiapine + clonazepam versus haloperidol. |
| Wang Jian 1995 | Allocation: randomised according to hospital admission order/number, stratified randomisation. |
| Weber 1983 | Allocation: randomised, cross‐over design. Participants: people with schizophrenia or organic brain syndrome and tardive dyskinesia. Interventions: diazepam versus no treatment. Outcomes: no usable data. |
| Weckowicz 1960 | Allocation: study group assignment on the basis of rating. |
| Wolkowitz 1986 | Allocation: not randomised. |
| Wolkowitz 1988 | Allocation: not randomised, review. |
| Wu 2006 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone + clonazepam versus haloperidol. |
| Yang 2003 | Allocation: randomised. Participants: schizophrenia. Interventions: risperidone + clonazepam versus haloperidol versus clozapine (no adequate comparison group). |
| Yu 2006 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone + clonazepam versus haloperidol. |
| Zhang 2007 | Allocation: randomised. Participants: people with schizophrenia. Interventions: olanzapine + clonazepam versus haloperidol. |
| Zhang YingHui 2008 | Allocation: randomised. Participants: people with schizophrenia. Interventions: quetiapine + clonazepam versus haloperidol. |
Regarding abbreviations see footnotes under the table Characteristics of included studies
Characteristics of studies awaiting assessment [ordered by study ID]
Davis 2008.
| Methods | Allocation: randomisation. Blindness: not indicated. Design: not indicated. Duration: not indicated. Setting: not indicated. |
| Participants | acutely agitated schizophrenic patients |
| Interventions | 1. haloperidol 5‐15 mg/d. N = 21. 2. clonazepam 2‐6 mg/d. N = 20. 3. haloperidol 5‐15 mg/d + clonazepam 2‐6 mg/d. N = 21. |
| Outcomes | BPRS total score, BPRS agitation (primary outcome) and psychosis subscales, CGI at 2, 4, 6 and 24 hours. |
| Notes | Regarding the BPRS agitation subscale the clonazepam augmentation and haloperidol monotherapy produced equal response rate at 6 and 24 hours, but a better response compared to a monotherapy with clonazepam. When performing this systematic review, the full publication of the trial results was not yet available. |
Maculans 1964.
| Methods | Allocation: randomisation. Blindness: double. Design: cross‐over. Duration: 3 weeks medication with each drug and 1‐week placebo interval between the active drug treatment. Setting: inpatient. |
| Participants | Diagnosis: psychotic patients, chronically ill, with histories of long‐term hospitalisations and known refractoriness to all therapeutic modalities. paranoid schizophrenia (N = 9), chronic undifferentiated schizophrenia (N = 26), childhood schizophrenia (N = 1), chronic brain syndrome combined with alcohol intoxication (N = 1). Age: average 31.5 years. Sex: not indicated. |
| Interventions | 1. Chlorprothixene 100 mg.
2. Chlorpromazine 100 mg. 3. Diazepam 10 mg. |
| Outcomes | Leaving the study early.
Global state: Clinical assessment of patient response to each course of therapy ("good", "fair", "poor"). Adverse effects. |
| Notes | There is no information available regarding the results of the first trial phase up to the point of first cross‐over. |
Salzman 1991.
| Methods | Allocation: implied randomisation. Blindness: double. Design: parallel. Duration: 48 hours. Setting: inpatient. |
| Participants | Diagnosis: "acute psychotic disruptive behavior" in all participants; schizophrenia (N = 26), bipolar disorder (N = 11), schizoaffective disorder (N = 4), organic mental disorder (N = 6), other including psychotic depression, personality disorder (N = 13). Age: mean 30.5 years in the lorazepam group and 37.9 in the haloperidol group. Sex: not indicated. |
| Interventions | 1. Lorazepam i.m. 2 mg. 2. Haloperidol i.m. 5 mg. |
| Outcomes | Leaving the study early.
Global state: CGI. Mental state: BPRS. |
| Notes | Lorazepam as well as haloperidol were added to ongoing antipsychotic treatment. When performing this systematic review, the full publication of the trial results was not yet available. |
Ungvari 1999.
| Methods | Allocation: randomisation. Blindness: double. Design: cross‐over. Duration: 12 weeks (6 weeks each for lorazepam and placebo with a 4‐week wash‐out period in‐between). Setting: inpatient. |
| Participants | Diagnosis: schizophrenia according to DSM‐IV. "All subjects were severely disabled chronic patients (duration of illness > 5 years)" Age: mean 44.89 years in the lorazepam group and 43.5 in the placebo group. Sex: 12 M, 5 F. |
| Interventions | 1. Lorazepam 6 mg. N = 9 (first phase of the study). 2. Placebo. N = 9 (first phase of the study). |
| Outcomes | Leaving the study early.
Leaving the study early due to adverse effects. Global state: CGI. Mental state: BPRS, HDRS. Behaviour: NOSIE. Adverse effects: AIMS, SAS, BARS, VPAS, BFCRS, MRS. |
| Notes | There is no information available regarding the results of the first trial phase up to the point of first cross‐over. |
Regarding abbreviations see footnotes under the table "Characteristics of included studies".
Differences between protocol and review
In difference to the previous version of this systematic review we did not intend to include continuous data from rating scales only if a.) the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b.) the measuring instrument has not been written or modified for that particular trial.
In this updated systematic review we calculated the number needed to treat to provide benefit /to induce harm statistic (NNTB/H), and its 95% confidence interval (CI) as the inverse of the risk differences for statistically significant results.
We did not aim to calculate the standardised mean differences (SMDs) as effect size measures.
Contrary to our statements in the previous version of this systematic review, we did not exclude trials because of unclear allocation concealment.
In this updated systematic review we decided to display skewed data in an 'other data table' and did not calculate effect sizes. These tables can be found within the section "data analysis".
Contributions of authors
Markus Dold: study selection, data extraction, statistical analyses, writing of the report (updated version of the review). Magdolna Tardy: study selection, data extraction (updated version of the review). Chunbo Li: study selection, data extraction (updated version of the review). Vesal Khorsand: protocol development, data extraction (previous version of the review). Donna Gillies: data extraction (previous version of the review). Stefan Leucht: protocol development, checking study selection and data extraction, writing of the report (previous and updated version of the review).
Sources of support
Internal sources
Freistaat Bayern, Germany.
External sources
German Ministry of Health, German Research Network on Schizophrenia, funded by the German Federal Ministry of Education and Research (grant 01GI933x), Germany.
Declarations of interest
Markus Dold: received travel payment from Janssen‐Cilag. Magdolna Tardy: none to declare. Chunbo Li: none to declare. Vesal Khorsand: none to declare. Donna Gillies: none to declare. Stefan Leucht: has received honoraria for consulting/advisory boards from Alkermes, BristolMyersSquibb, EliLilly, Janssen, Johnson&Johnson, Medavante, Roche, lecture honoraria from AstraZeneca, BristolMyersSquibb, EliLilly, EssexPharma, Janssen, Johnson&Johnson, Lundbeck Institute, Pfizer, SanofiAventis, and EliLilly has provided medication for a trial with SL as the primary investigator.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Azima 1962 {published data only}
- Azima H, Arthurs D, Silver A. The effects of chlordiazepoxide (librium) in anxiety states: A multi‐blind study. Canadian Psychiatric Journal 1962;7:44. [DOI] [PubMed] [Google Scholar]
Barbee 1992 {published data only}
- Barbee J, Mancuso D, Freed C, Todorov AA. Alprazolam as a neuroleptic adjunct in the emergency treatment of schizophrenia [published erratum appears in American Journal of Psychiatry 1992 ;149:1129] [see comments]. American Journal of Psychiatry 1992;149:506‐10. [DOI] [PubMed] [Google Scholar]
Battaglia 1997 {published data only}
- Battaglia J, Moss S, Rush J, Kang J, Mendoza R, Leedom L, et al. Haloperidol, lorazepam, or both for psychotic agitation? A multicenter, prospective, double‐blind, emergency department study. American Journal of Emergency Medicine 1997;15:335‐40. [DOI] [PubMed] [Google Scholar]
Carpenter 1999 {published data only}
- Carpenter T, Buchanan W, Kirkpatrick B, Breier F. Diazepam treatment of early signs of exacerbation in schizophrenia. American Journal of Psychiatry 1999;156:299‐303. [DOI] [PubMed] [Google Scholar]
Cheung 1981 {published data only}
- Cheung HK. Schizophrenics fully remitted on neuroleptics for 3‐5 years‐To stop or continue drugs?. British Journal of Psychiatry 1981;138:490‐4. [DOI] [PubMed] [Google Scholar]
Chouinard 1993 {published data only}
- Chouinard G, Annable L, Turnier L, Holobow N, Szkrumelak N. A double‐blind randomized clinical trial of rapid tranquilization with i.m. clonazepam and i.m. haloperidol in agitated psychotic patients with manic symptoms. Canadian Journal of Psychiatry 1993;38(Suppl 4):114‐21. [PubMed] [Google Scholar]
Dorevitch 1999 {published data only}
- Dorevitch A, Kalian M, Shlafman M, Lerner V. Treatment of long‐term tardive dyskinesia with Vitamin E. Biological Psychiatry 1997;41:114‐6. [DOI] [PubMed] [Google Scholar]
- Dorevith A, Katz N, Zemishlany Z, Aizenberg D, Weizman A. Intramuscular flunitrazepam versus intramuscular haloperidol in the emergency treatment of aggressive psychotic behavior. American Journal of Psychiatry 1999;156:142‐4. [DOI] [PubMed] [Google Scholar]
Foster 1997 {published data only}
- Foster S, Kessel J, Berman ME, Simpson GM. Efficacy of lorazepam and haloperidol for rapid tranquilization in a psychiatric emergency room setting. International Clinical Psychopharmacology 1997;12:175‐9. [DOI] [PubMed] [Google Scholar]
Garza‐Trevino 1989 {published data only}
- Garza‐Trevino ES, Hollister LE, Overall JE, Alexander WF. Efficacy of combinations of intramuscular antipsychotics and sedative‐hypnotics for control of psychotic agitation. American Journal of Psychiatry 1989;146:1598‐601. [DOI] [PubMed] [Google Scholar]
Gundlach 1966 {published data only}
- Gundlach R, Engelhardt DM, Hankoff L, Paley H, Rudorfer L, Bird E. A Double‐blind outpatient study of diazepam (Valium) and placebo. Psychopharmacologia 1966;9:81‐92. [DOI] [PubMed] [Google Scholar]
Guz 1972 {published data only}
- Guz I. Lorazepam and haloperidol in the treatment of schizophrenic patients [Lorazepam e haloperidol em esquizofrenia]. Jornal Brasileiro de Psiquiatria 1988;37:205‐7. [Google Scholar]
- Guz I, Moraes R, Sartoretto JN. The therapeutic effects of lorazepam in psychotic patients treated with haloperidol ‐ A double blind study. Current Therapeutic Research 1972;14:767‐74. [PubMed] [Google Scholar]
Hankoff 1962 {published data only}
- Hankoff L, Rudorfer L, Paly HM. A reference study of ataraxics. A two‐week double blind outpatient evaluation. The Journal of New Drugs 1962;179:173‐8. [DOI] [PubMed] [Google Scholar]
Hanlon 1969 {published data only}
- Hanlon TE, Kay Y, Agallianos D, Berman A, Bethon C, Kolber F, et al. Combined drug treatment of newly hospitalized, acutely ill psychiatric patients. Diseases of the Nervous System 1969;30:104‐16. [PubMed] [Google Scholar]
Hanlon 1970 {published data only}
- Hanlon TE, Ota KY, Kurland AA. Comparative effects of fluphenazine, fluphenazine‐chlordiazepoxide and fluphenazine‐imipramine. Diseases of the Nervous System 1970;31:171‐7. [PubMed] [Google Scholar]
- Hanlon TE, Robert JB, Kurland AA. Effects od control techniques on therapeutic outcome in a controlled trial. International Pharmacopsychiatry 1975;10:169‐76. [DOI] [PubMed] [Google Scholar]
Holden 1968 {published data only}
- Holden JMC, Holden UP. Weight changes with schizophrenic psychosis and psychotropic drug therapy. Psychosomatics 1970;11:551‐61. [DOI] [PubMed] [Google Scholar]
- Holden JMC, Itil TM. Laboratory changes with chlordiazepoxide and thioridazine, alone and combined. Canadian Psychiatric Association Journal 1969;14:299‐301. [DOI] [PubMed] [Google Scholar]
- Holden JMC, Itil TM, Keskiner A, Fink M. Thioridazine and chlordiazepoxide, alone and combined, in the treatment of chronic schizophrenia. Comprehensive Psychiatry 1968;9:633‐43. [DOI] [PubMed] [Google Scholar]
Kurland 1966 {published data only}
- Kurland AA, Bethon GD, Michaux MH, Agallianos DD. Chlorpromazine‐chlordiazepoxide and chlorpromazine‐imipramine treatment: side effects and clinical laboratory findings. Journal of New Drugs 1966;6:80‐95. [PubMed] [Google Scholar]
- Michaux MH, Kurland AA, Agallianos DD. Chlorpromazine‐chlordiazepoxide and chlorpromazine‐imipramine treatment of newly hospitalized, acutely ill psychiatric patients. Current Therapeutic Research Clininical and Experimental 1966;8(Suppl):117‐52. [PubMed] [Google Scholar]
Kutcher 1989 {published data only}
- Kutcher S, Williamson P, MacKenzie S, Marton P, Ehrlich M. Successful clonazepam treatment of neuroleptic‐induced akathisia in older adolescents and young adults: a double‐blind, placebo‐controlled study. Journal of Clinical Psychopharmacology 1989;9:403‐6. [PubMed] [Google Scholar]
Lerner 1979 {published data only}
- Lerner Y, Lwow E, Levitin A, Belmaker RH. Acute high‐dose parenteral haloperidol treatment of psychosis. American Journal of Psychiatry 1979;136:1061‐4. [DOI] [PubMed] [Google Scholar]
Lingjaerde 1979 {published data only}
- Lingjaerde O, Engstrand E, Ellingsen P, Robak OH. Antipsychotic effect of diazepam when given in addition to neuroleptics in chronic psychotic patients ‐ A double‐blind clinical trial. Current Therapeutic Research 1979;26:505‐14. [Google Scholar]
Lingjaerde 1982 {published data only}
- Lingjaerde O. Effect of the benzodiazepine derivative estazolam in patients with auditory hallucinations. A multicentre double‐blind, cross‐over study. Acta Psychiatrica Scandinavia 1982;65:339‐54. [DOI] [PubMed] [Google Scholar]
Ma 2006 {published data only}
- Ma C. The control study of injection of clonazepam into tinggong for auditory hallucination of schizophrenia. Journal of Clinical Psychosomatic Diseases 2006;12:338‐9. [Google Scholar]
Marneros 1979 {published data only}
- Marneros A. Anxiolytische Zusatzbehandlung bei den affektbetonten Schizophrenien. Ein Doppelblindversuch. Therapiewoche 29 1979;44:7533‐8. [Google Scholar]
Merlis 1962 {published data only}
- Merlis S, Turner WJ, Krumholz W. A double‐blind comparison of diazepam, chlordiazepoxide and chlorpromazine in psychotic patients. Journal of Neuropsychiatry 1962;3:133‐8. [PubMed] [Google Scholar]
Minervini 1990 {published data only}
- Minervini MG, Priore P, Farolfi A, Cesana B, Morselli PL. Double blind, controlled study of the efficacy and safety of alpidem in the treatment of anxiety in schizophrenic in‐patients. Pharmacopsychiatry 1990;23:102‐6. [DOI] [PubMed] [Google Scholar]
Morphy 1986 {published data only}
- Morphy M. A double‐blind comparison of alprazolam and placebo in the treatment of anxious schizophrenic outpatients. Current Therapeutic Research 1986;40:551‐60. [Google Scholar]
Nestoros 1982 {published data only}
- Nestoros JN, Suranyi‐Cadotte BE, Spees RC, Schwartz G, Vasavan Nair NP. Diazepam in high doses is effective in schizophrenia. Progress in Neuro‐ Psychopharmacolgy and Biological Psychiatry 1982;6:513‐6. [DOI] [PubMed] [Google Scholar]
Nishikawa 1982 {published data only}
- Nishikawa T, Tsuda A, Tanaka M, Koga I, Uchida Y. Prophylactic effect of neuroleptics in symptom‐free schizophrenics. Psychopharmacology 1982;77:301‐4. [DOI] [PubMed] [Google Scholar]
Pujalte 1994 {published data only}
- Pujalte D, Battai T, Hue B, Alric R, Pouget R, Blayac JP, et al. A double‐blind comparison of clonazepam and placebo in the treatment of neuroleptic‐induced akathisia. Clinical Neuropharmacology 1994;17:236‐42. [DOI] [PubMed] [Google Scholar]
Stevens 1992 {published data only}
- Stevens A, Stevens I, Mahal A, Gaertner HJ. Haloperidol and lorazepam combined: clinical effects and drug plasma levels in the treatment of acute schizophrenic psychosis. Pharmacopsychiatry 1992;25:273‐7. [DOI] [PubMed] [Google Scholar]
TREC‐Rio 2003 {published data only}
- TREC Collaborative Group. Rapid tranquillisation for agitated patients in emergency psychiatric rooms: a randomised trial of midazolam versus haloperidol plus promethiazine. BMJ 2003;327:708‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2000 {published data only}
- Wang G, Ji R, Pei G. the control study of injection of clonazepam into t´ingkung for refractory auditory hallucination of schizophrenics. Journal of Clinical Psychosomatic Diseases 2000;6:143‐4. [Google Scholar]
Wang 2003 {published data only}
- Wang X, Wang X, Zhou H. An efficacy study of risperidone combined with clonazepam injection in the treatment of paranoid schizophrenia. Journal of Clinical Psychologic Medicine 2003;13:217‐8. [Google Scholar]
Wyant 1990 {published data only}
- Wyant M, Diamond BI, O'Neal E, Sloan A, Borison RL. The use of midazolam in acutely agitated psychiatric patients. Psychopharmacology Bulletin 1990;26:126‐9. [PubMed] [Google Scholar]
Xuan 2007 {published data only}
- Xuan GH, Chu Y. Clinical observation on the effect of risperidone combined with BZD on schizophrenia. China Tropical Medicine 2007;7:244. [Google Scholar]
References to studies excluded from this review
Adler 1985 {published data only}
- Adler L, Angrist B, Peselow E, Corwin J, Rotrosen J. Efficacy of propranolol in neuroleptic‐induced akathisia. Journal of Clinical Psychopharmacology 1985;5:164‐6. [PubMed] [Google Scholar]
Alexander 2004 {published data only}
- Alexander J, Tharyan P, Adams C, John T, Mol C, Philip J. Rapid tranquillisation of violent or agitated patients in a psychiatric emergency setting. Pragmatic randomised trial of intramuscular lorazepam v. haloperidol plus promethazine. British Journal of Psychiatry 2004;186:63‐9. [DOI] [PubMed] [Google Scholar]
Altamura 1987 {published data only}
- Altamura AC, Mauri MC, Mantero M, Brunetti M. Clonazepam/haloperidol combination therapy in schizophrenia: A double blind study. Acta Psychiatrica Scandinavica 1987;76:702‐6. [DOI] [PubMed] [Google Scholar]
Ananth 1979 {published data only}
- Ananth JV, Sohn JH, Ban TA, Lehmann HE. Doxepin in geriatric patients. Current Therapeutic Research 1979;25:133‐8. [Google Scholar]
Bao 2007 {published data only}
- Bao LY, Wang KM. The effectiveness of aripiprazole combined with clonazepam treated with agitation in schizophrenia. Medical Journal of Chinese People's Health 2007;5:ni. [Google Scholar]
Bienek 1997 {published data only}
- Bienek SA, Ownby RL, Dominguez RA. A double‐blind study of lorazepam versus the combination of haloperidol and lorazepam in managing agitation. Pharmacotherapy 1998;18:57‐62. [PubMed] [Google Scholar]
Bobruff 1981 {published data only}
- Bobruff A, Gardos G, Tarsy D, Rapkin R, Cole J, Moore P. Clonazepam and phenobarbital in tardive dyskinesia. American Journal of Psychiatry 1981;138:189‐93. [DOI] [PubMed] [Google Scholar]
Cohen 1987 {published data only}
- Cohen S, Khan A. Adjunctive benzodiazepines in acute schizophrenia. Neuropsychobiology 1987;18:9‐12. [DOI] [PubMed] [Google Scholar]
Crosse 1974 {published data only}
- Crosse B. Clinical trials of sulpiride (1403 R.D.‐‐Dogmatil): Its advantages prescribed alone or in combination [Essais cliniques du sulpiride (1403 R. D. ‐ Dogmatil): Son Intérêt en prescription isolée et en association]. Psychologie Médicale 1974;6:1623‐30. [Google Scholar]
Csernansky 1988 {published data only}
- Csernansky JG, Riney SJ, Lombrozo L, Overall JE, Hollister LE. Double‐blind comparison of alprazolam, diazepam, and placebo for the treatment of negative symptoms. Archive of General Psychiatry 1988;45:655‐9. [DOI] [PubMed] [Google Scholar]
- Csernansky JG, Tacke U, Rusen D, Hollister LE. The effect of benzodiazepines on tardive dyskinesia symptoms. Journal of Clinical Psychopharmacology 1988;8:154. [DOI] [PubMed] [Google Scholar]
Dubin 1988 {published data only}
- Dubin WR. Rapid tranquilization: antipsychotics or benzodiazepines. Journal of Clinical Psychiatry 1988;49:5‐11. [PubMed] [Google Scholar]
Ge Qianrong 2004 {published data only}
- Ge Q, Liang X, Wang X. A comparative study of risperidone combining clonazepam and haloperidol in the treatment of patients with schizophrenia. Shandong Archives of Psychiatry 2004;17:134‐6. [Google Scholar]
Greenfeld 1987 {published data only}
- Greenfeld D, Conrad C, Kincare P, Bowers MB. Treatment of catatonia with low‐dose lorazepam. American Journal of Psychiatry 1987;144:1224‐5. [DOI] [PubMed] [Google Scholar]
Haas1982 {published data only}
- Haas S, Emrich HM, Beckmann H. Analgesic and euphoric effects of high dose diazepam in schizophrenia. Neuropsychobiology 1982;8:123‐8. [DOI] [PubMed] [Google Scholar]
Hanus 1973 {published data only}
- Hanus M, Guelfi J. A new tranquilizer: lorazepam (158 cases) [Un nouveau tranquillisant: le Lorazepam (158 observations)]. Semaine des Hopitaux (Therapeutique) 1973;49:349‐54. [PubMed] [Google Scholar]
Hartelius 1978 {published data only}
- Hartelius H, Larsson AK, Lepp M, Malm U, Arvidsson A, Dahlstrom H. A controlled long‐term study of flunitrazepam, nitrazepam and placebo, with special regard to withdrawal effects. Acta Psychiatrica Scandinavica 1978;58:1‐15. [DOI] [PubMed] [Google Scholar]
Harvey 2004 {published data only}
- Harvey AT, Flockhart D, Gorski JC, Greenblatt DJ, Burke M, Werder S, et al. Intramuscular haloperidol or lorazepam and QT intervals in schizophrenia. Journal of Clinical Pharmacology 2004;44:1173‐84. [DOI] [PubMed] [Google Scholar]
He 2007 {published data only}
- He W. [Title only available in Chinese characters]. Modern Medicine and Health 2007;23:527. [Google Scholar]
Hekimian 1967 {published data only}
- Hekimian LJ, Friedhoff AJ. A controlled study of placebo, chlordiazepoxide and chlorpromazine with thirty male schizophrenic patients. Diseases of Nervous System 1967;28:675‐8. [PubMed] [Google Scholar]
Hesso 1975 {published data only}
- Hesso R, Retterstol N, Torp H. Clinical trial with a new substance (PLP 100‐127) in order to asses therapeutic efficacy and dependence creating properties. Behavioral Neuropsychiatry 1975;7:13‐7. [PubMed] [Google Scholar]
Horiguchi 1992 {published data only}
- Horiguchi J, Nishimatsu O. Usefulness of antiparkinsonian drugs during neuroleptic treatment and the effect of clonazepam on akathisia and parkinsonism occurred after antiparkinsonian drug withdrawal: a double‐blind study. Japanese Journal of Psychiatry and Neurology 1992;46:733‐9. [DOI] [PubMed] [Google Scholar]
Hovens 2005 {published data only}
- Hovens JE, Dries PJT, Melman CTM, Wapenaar RJC, Loonen AJM. Oral risperidone with lorazepam versus oral zuclopenthixol with lorazepam in the treatment of acute psychosis in emergency psychiatry: a prospective, comparative, open‐label study. Journal of Psychopharmacology 2005;19:51‐7. [DOI] [PubMed] [Google Scholar]
Hu 2004 {published data only}
- Hu W. Investigation of administering BZD medicament in outpatients with psychosis. Heath Psychology Journal 2004;12:43. [Google Scholar]
Itil 1972 {published data only}
- Itil T, Gannon P, Cora R, Polvan N, Akpinar S, Elveris F, et al. SCH‐12,041, a new anti‐anxiety agent (quantitative pharmaco‐electroencephalography and clinical trials). Behavioral Neuropsychiatry 1972;4:15‐24. [PubMed] [Google Scholar]
Jibiki 1994 {published data only}
- Jibiki I, Yamaguchi N, Momonoi F. Beneficial effect of high‐dose clotiazepam on intractable auditory hallucinations in chronic schizophrenic patients. An open trial. European Journal of Clinical Pharmacology 1994;46:367‐9. [DOI] [PubMed] [Google Scholar]
Jirmeson 1982 {published data only}
- Jimerson DC, Kammen DP, Post RM, Docherty JP, Bunney WE. Diazepam in schizophrenia: a preliminary double‐blind trial. American Journal of Psychiatry 1982;139:489‐91. [DOI] [PubMed] [Google Scholar]
Jungkunz 1984 {published data only}
- Jungkunz G, Nedopil N, Ruther E. Acute effects of the synthetic analogue of methionine enkephalin FK 33‐824 in schizophrenic patients. A double blind trial. Pharmacopsychiatry 1984;17:76‐8. [DOI] [PubMed] [Google Scholar]
Kang 2006 {published data only}
- Kang M. Clinical study on efficacy of quetiapine combined with clonazepam treated with acute agitation in schizophrenia patients. Linchuang Jingshen Yixue Zazhi 2006;16:221‐2. [Google Scholar]
Karson 1982 {published data only}
- Karson C, Weinberger D, Bigelow L, Wyatt RJ. Clonazepam treatment of chronic schizophrenia: negative results in a double‐blind, placebo‐controlled trial. American Journal of Psychiatry 1982;139:1627‐8. [DOI] [PubMed] [Google Scholar]
Kellner 1975 {published data only}
- Kellner R, Wilson RM, Muldawer MD, Pathak D. Anxiety in schizophrenia. The responses to chlordiazepoxide in an intensive design study. Archives of General Psychiatry 1975;32:1246‐54. [DOI] [PubMed] [Google Scholar]
Knott 2006 {published data only}
- Knott JC, Taylor DM, Castle DJ. Randomized clinical trial comparing intravenous midazolam and droperidol for sedation of the acutely agitated patient in the emergency department. Annals of Emergency Medicine 2006;47:61‐7. [DOI] [PubMed] [Google Scholar]
Li 2004 {published data only}
- Li AF. [Title only available in Chinese characters]. Chinese Journal of Clinical Medicine 2004;3:33‐4. [Google Scholar]
Li 2007 {published data only}
- Li C, Zhu S, Wang H, Chen H, Yu Y, Liu D, et al. Safety and efficacy of clonazepam, haloperidol and haloperidol combined with clonazepam in the treatment of schizophrenia with excitement and agitation. Shanghai Archives of Psychiatry 2007;19:150‐2. [Google Scholar]
Maar 1983 {published data only}
- Maar S, Schuback G. Comparison of two benzodiazepines in general psychosomatic disturbances [Wirkungsvergleich zweier Benzodiazepine bei psychovegetativen Allgemeinstörungen]. Therapiewoche 1983;33:1251‐8. [Google Scholar]
Mei 2006 {published data only}
- Mei Y, Pan J, Ma P. Clinical observation on acute excitation of schizophrenia treated with QIWEI combined with clonazepam. Chinese Nursing Research 2006;20:1176‐7. [Google Scholar]
Monroe 1967 {published data only}
- Monroe RR, Dale R. Chlordiazepoxide in the treatment of patients with "activated EEG's". Diseases of Nervous System 1967;28:390‐6. [PubMed] [Google Scholar]
Nestros 1983 {published data only}
- Nestros JN, Nair NPV, Pulman JR, Schwartz G, Bloom D. High doses of diazepam improve neuroleptic‐resistant chronic schizophrenic patients. Psychopharmacology 1983;81:42‐7. [DOI] [PubMed] [Google Scholar]
Nobay 2004 {published data only}
- Nobay F, Simon BC, Levitt MA, Dresden GM. A prospective, double‐blind, randomized trial of midazolam versus haloperidol versus lorazepam in the chemical restraint of violent and severely agitated patients. Academic Emergency Medicine 2004;11:744‐9. [DOI] [PubMed] [Google Scholar]
Pato 1989 {published data only}
- Pato CN, Wolkowitz OM, Rapaport M, Schulz SC, Pickar D. Benzodiazepine augmentation of neuroleptic treatment in patients with schizophrenia. Psychopharmacology Bulletin 1989;25:263‐6. [PubMed] [Google Scholar]
Pecknold 1993 {published data only}
- Pecknold JC. Survey of the adjuvant use of benzodiazepines for treating outpatients with schizophrenia. The Journal of Psychiatric Neurosciences 1993;18:82‐4. [PMC free article] [PubMed] [Google Scholar]
Qin 2005 {published data only}
- Qin Y, Kang W, Song L. A comparative study of risperidone combining alprazolam and clozapine in the treatment of patients with schizophrenia. Heath Psychology Journal 2005;13:444‐5. [Google Scholar]
Remington 1993 {published data only}
- Remington G, Fornazzari L, Setha R. Placebo response in refractory tardive akathisia. Canadian Journal of Psychiatry 1993;38:248‐9. [DOI] [PubMed] [Google Scholar]
Richards 1998 {published data only}
- Richards JR, Derlet RW, Duncan DR. Chemical restraint for the agitated patient in the emergency department: lorazepam versus droperidol. The Journal of Emergency Medicine 1998;16:567‐73. [DOI] [PubMed] [Google Scholar]
Ruskin 1978 {published data only}
- Ruskin P, Averbush I, Belmaker RH, Dasberg H. Benzodiazepind in chronic schizophrenia. Biological Psychiatry 1979;14:557‐8. [PubMed] [Google Scholar]
Salam 1988 {published data only}
- Salam SA, Kilzieh N. Lorazepam treatment of psychogenic catatonia: an update. Journal of Clinical Psychiatry 1988;49:16‐21. [PubMed] [Google Scholar]
Saletu 1971 {published data only}
- Saletu B, Saletu M, Itil T. Effect of minor and major tranquilizers on somatosensory evoked potentials. Psychopharmacologia 1971;24:349‐58. [DOI] [PubMed] [Google Scholar]
Shafti 2005 {published data only}
- Shafti SS. Rehabilitation of schizophrenia: adjunctive therapy of negative symptoms. Iranian Rehabilitation Journal 2004;2:ni. [Google Scholar]
- Shafti SS, Rey S, Abad A. Drug‐specific responsiveness of negative symptoms. International Journal of Psychosocial Rehabilitation 2005;10:43‐51. [Google Scholar]
Soldatos 1986 {published data only}
- Soldatos CR, Sakkas PN, Bergiannaki JD, Stefanis CN. Behavioral side effects of triazolam in psychiatric inpatients: report of five cases. Drug Intelligence and Clinical Pharmacy 1986;20:294‐7. [DOI] [PubMed] [Google Scholar]
Sterlin 1971 {published data only}
- Sterlin C, Augustin E, Ban TA, Jarrold L. Doxepin as adjuvant medication in the treatment of chronic schizophrenic patients: a comparative study. Current Therapy Research Clinical and Experimental 1971;13:50‐2. [PubMed] [Google Scholar]
Stonehill 1966 {published data only}
- Stonehill E, Lee H, Ban TA. A comparative study with Benzodiazepines in chronic psychotic patients. Diseases of the Nervous System 1966;27:411‐3. [PubMed] [Google Scholar]
Subramaney 1997 {published data only}
- Subramaney U, Brook S, Berk M. A prospective randomised double blind controlled study of the efficacy of Lorazepam versus Clothiapine in the control of acutely behaviourally disturbed patients. 10th ECNP (European College of Neuropsychopharmacology) Congress, Vienna, Austria Sep 13 ‐ 17, 1997.
Tang 2007 {published data only}
- Tang Q, Yang L, Lai G, Zhand J. A clinical study of risperidone oral solution combined with clonazepam injection in the treatment of acute excitement phase schizophrenia patient. Shanghai Archives of Psychiatry 2007;19:153‐5. [Google Scholar]
TREC‐Vellore 2004 {published data only}
- Alexander J, Tharyan P, Adams CE, John T, Mol C, Philip J. Rapid tranquilisation of violent or agitated patients in a psychiatric emergency setting: a pragmatic randomised trial of intramuscular lorazepam versus haloperidol plus promethiazine. British Journal of Psychiatry 2004;185:63‐9. [DOI] [PubMed] [Google Scholar]
Wang 2004a {published data only}
- Wang L, Li B, Zhao Z. A controlled study of compound diazepam in adjunctive treatment for schizophrenia. Journal of Binzhou Medical College 2004;27:111‐2. [Google Scholar]
Wang 2004b {published data only}
- Wang G, Cai ZJ, Wang LF. A multicenter study of risperidone treatment for acute agitation in patients with schizophrenia. Chinese Journal of Psychiatry 2004;37:88‐91. [Google Scholar]
Wang 2005 {published data only}
- Wang DB, Xu SQ, Tang QP, Ying YF, Mei YT, He FX. Treatment of 80 cases of schizophrenia manifesting acute erethitic symptoms with quetiapine combined with clonazepam. Herald of Medicine 2005;24:687‐8. [Google Scholar]
Wang Jian 1995 {published data only}
- Wang J, Wang x, Chen H. Clinical controlled study of perphenazine combined with alprazolam in treatment of negative symptoms. Chinese Journal of Pharmaco Epidemiology 1995;4:137‐9. [Google Scholar]
Weber 1983 {published data only}
- Weber SS, Dufresne RL, Becker RE, Mastrati P. Diazepam in tardive dyskinesia. Drug Intelligence and Clinical Pharmacy 1983;17:523‐7. [DOI] [PubMed] [Google Scholar]
Weckowicz 1960 {published data only}
- Weckowicz TE, Ward T. Clinical trial of RO 5‐0690 and chlorpromazine on disturbed chronic schizophrenic patients. Diseases of the Nervous System 1960;21:527‐8. [PubMed] [Google Scholar]
Wolkowitz 1986 {published data only}
- Wolkowitz OM, Pickar D, Doran AR, Breier A, Tarell J, Paul SM. Combination alprazolam‐neuroleptic treatment of the positive and negative symptoms of schizophrenia. American Journal of Psychiatry 1986;143:85‐7. [DOI] [PubMed] [Google Scholar]
Wolkowitz 1988 {published data only}
- Wolkowitz OM, Breier A, Doran A, Kelsoe J, et al. Alprazolam augmentation of the antipsychotic effects of fluphenazine in schizophrenic patients ‐ Preliminary results. Archives of General Psychiatry 1988;45:664‐71. [DOI] [PubMed] [Google Scholar]
Wu 2006 {published data only}
- Wu Z. [Title only available in Chinese characters]. Journal of Qiqihar Medical College 2006;27:536‐7. [Google Scholar]
Yang 2003 {published data only}
- Yang X, Wang Z, Ling Z, et al. A randomly controlled comparison of risperidone added with intramuscular clonazepam in the treatment of excitement of schizophrenia. Shanghai Archives of Psychiatry 2003;15:98‐104. [Google Scholar]
Yu 2006 {published data only}
- Yu J. [Title only available in Chinese characters]. Nervous Diseases and Mental Hygiene 2006;6:129‐30. [Google Scholar]
Zhang 2007 {published data only}
- Zhang HS. Study of olanzapine combined with clonazepam in treatment of schizophrenia with acute psychomotor excitation. Linchuang Jingshen Yixue Zazhi 2007;17:239‐40. [Google Scholar]
Zhang YingHui 2008 {published data only}
- Zhang YingHui. [Title only available in Chinese characters]. Shandong Archives of Psychiatry 2008;21:59‐60. [Google Scholar]
References to studies awaiting assessment
Davis 2008 {published data only}
- Davis JM, Wang B, He Y, Jin H, Hu Q, Zhang M. The emergency treatment of acutely agitated psychotic schizophrenia patient. International Journal of Neuropsychopharmacology 2008;11(Suppl 1):163. [Google Scholar]
Maculans 1964 {published data only}
- Maculans GA. Comparison of diazepam, chlorprothixine and chlorpromazine in chronic schizophrenic patients. Diseases of the Nervous System 1964;25:164‐8. [PubMed] [Google Scholar]
Salzman 1991 {published data only}
- Salzman C, Solomon D, Miyawaki E, Glassman R, Rood L, Flowers E, et al. Parenteral lorazepam versus parenteral haloperidol for the control of psychotic disruptive behavior. Journal of Clinical Psychiatry 1991;52:177‐80. [PubMed] [Google Scholar]
Ungvari 1999 {published data only}
- Ungvari GS, Chiu HFK, Chow LY, Lau BST, Tang WK. Lorazepam for chronic catatonia: a randomized, double‐blind, placebo‐controlled cross‐over study. Psychopharmacology 1999;142:393‐8. [DOI] [PubMed] [Google Scholar]
Additional references
Adams 2011
- Adams CE, Coutinho E, Duggan L, Leucht S, Tharyan P, Davis JM, et al. Cochrane Schizophrenia Group. The Cochrane Library Chichester, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreasen 1989
- Andreasen NC. Scale for the assessment of negative symptoms. British Journal of Psychiatry 1989;155:53‐8. [PubMed] [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials: the CONSORT statement. Journal of American Medicale Assosiation 1996;276:637‐9. [DOI] [PubMed] [Google Scholar]
Benkert 2011
- Benkert O, Hippius H. Kompendium der Psychiatrischen Pharmakotherapie. 8. Berlin‐Heidelberg‐New York: Springer, 2011. [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54:405‐11. [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Cheine 2001
- Cheine M, Ahonen J, Wahlbeck K. Beta‐blocker supplementation of standard drug treatment for schizophrenia. Cochrane Database of Systematic Reviews 2001, Issue 3. [DOI: 10.1002/14651858.CD000234] [DOI] [PubMed] [Google Scholar]
Christison 1991
- Christison GW, Kirch DG, Wyatt RJ. When symptoms persist: choosing among alternative somatic treatments for schizophrenia. Schizophrenia Bulletin 1991;17:217‐45. [DOI] [PubMed] [Google Scholar]
Corrigan 1989
- Corrigan JD. Development of a scale for assessment of agitation following traumatic brain injury. Journal of Clinical and Experimental Neuropsychology 1989;11:261‐77. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Abstracts of 8th International Cochrane Colloquium. Cape Town, South Africa, 2000 Oct 25‐28th.
Divine 1992
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7:623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997a
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997b
- Egger M, Zellweger‐Zähner T, Schneider M, Junker C, Lengeler C, Antes G. Language bias in randomised controlled trials published in English and German. Lancet 1997;350:326‐9. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31:140‐9. [DOI] [PubMed] [Google Scholar]
Essali 2009
- Essali A, Al‐Haj Haasan N, Li C, Rathbone J. Clozapine versus typical neuroleptic medication for schizophrenia. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD000059.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Falkai 2005
- Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, Möller HJ, WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, Part 1: acute treatment of schizophrenia. World Journal of Biological Psychiatry 2005;6:132‐91. [DOI] [PubMed] [Google Scholar]
Falkai 2006
- Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, Möller HJ, WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: long‐term treatment of schizophrenia. World Journal of Biological Psychiatry 2005;7:5‐40. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59:7‐10. [DOI] [PubMed] [Google Scholar]
Gillies 2005
- Gillies D, Beck A, McCloud A, Rathbone J. Benzodiazepines alone or in combination with antipsychotic drugs for acute psychosis. Cochrane Database of Systematic Reviews 2005, Issue 4. [DOI: 10.1002/14651858.CD003079.pub2] [DOI] [PubMed] [Google Scholar]
GRADE Profiler [Computer program]
- GRADE Working Group. GRADE Profiler. Version 3.2. GRADE Working Group, 2004.
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology (DOTES: Dosage Record and Treatment Emergent Symptom Scale). Rockville: National Institute of Mental Health, 1976. [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Honigfeld 1965
- Honigfeld G, Klett CJ. The nurses' observation scale for inpatient evaluation. A new scale for measuring improvement in chronic schizophrenia. Journal of Clinical Psychology 1965;21:65‐71. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and negative syndrome scale (PANSS) manual. Positive and negative syndrome scale (PANSS) manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A. The positive and negative symptom scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13:261‐75. [DOI] [PubMed] [Google Scholar]
Lehman 2004
- Lehman AF, Lieberman JA, Dixon LB, McGlashan TH, Miller AL, Perkins DO, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. American Journal of Psychiatry 2004;161:1‐56. [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59:1001‐5. [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79:231‐8. [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [DOI] [PubMed] [Google Scholar]
Leucht 2006
- Leucht S, Kane JM, Etschel E, Kissling W, Hamann J, Engel RR. Linking the PANSS, BPRS, and CGI: clinical implications. Neuropsychopharmacology 2006;31:2318‐25. [DOI] [PubMed] [Google Scholar]
Leucht 2007a
- Leucht S, Kissling W, McGrath J, White P. Carbamazepine for schizophrenia. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD001258.pub2] [DOI] [PubMed] [Google Scholar]
Leucht 2007b
- Leucht S, McGrath J, Kissling W. Lithium for schizophrenia. Cochrane Database of Systematic Reviews 2003, Issue 3. [DOI: 10.1002/14651858.CD003834.pub2] [DOI] [PubMed] [Google Scholar]
Leucht 2009
- Leucht S, Arbter D, Engel RR, Kissling W, Davis JM. How effective are second‐generation antipsychotic drugs? A meta‐analysis of placebo‐controlled trials. Molecular Psychiatry 2009;14:429‐47. [DOI] [PubMed] [Google Scholar]
Leucht 2011
- Leucht S, Heres S, Kissling W, Davis JM. Evidence‐based pharmacotherapy of schizophrenia. International Journal of Neuropsychopharmacology 2011;14:269‐84. [DOI] [PubMed] [Google Scholar]
Lorr 1962
- Lorr M, McNair DM, Klett CJ, Lasky JJ. Evidence of ten psychotic symptoms. Journal of Consulting Psychology 1962;26:185. [DOI] [PubMed] [Google Scholar]
Malamud 1947
- Malamud W, Sands SL. A revision of the psychiatric rating scale. American Journal of Psychiatry 1947;104:231‐7. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Marvaha 2004
- Marvaha S, Johnson S. Schizophrenia and employment ? a review. Social Psychiatry and Psychiatric Epidemiology 2004;39:337‐49. [DOI] [PubMed] [Google Scholar]
NICE 2009
- National Institute for Clinical Excellence (NICE). Core interventions in the treatment and management of schizophrenia in primary and secondary care (update). NICE, 2009. [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:790‐812. [Google Scholar]
Review Manager (RevMan) [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Schulz 2010
- Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. Trials 2010;11:32. [Google Scholar]
Schwarz 2008
- Schwarz C, Volz A, Li C, Leucht S. Valproate for schizophrenia. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.CD004028.pub3] [DOI] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Simpson 1970
- Simpson M, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica 1970;212:11‐9. [DOI] [PubMed] [Google Scholar]
Tharyan 2005
- Tharyan P. Electroconvulsive therapy for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD000076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tsuang 1978
- Tsuang MT. Suicide in schizophrenics, manics, depressives, and surgical controls: a comparison with general population suicide mortality. Archives of General Psychiatry 1978;35:153‐55. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3:1‐75. [PubMed] [Google Scholar]
Wahlbeck 2001
- Wahlbeck K, Tuunainen A, Ahokas A, Leucht S. Drop‐out rates in randomised antipsychotic drug trials. Psychopharmacology 2001;155:230‐3. [DOI] [PubMed] [Google Scholar]
WHO 2001
- World Health Organization. World Health Report 2001. Mental health ‐ new understanding, new hope. Genf: World Health Organization, 2001. [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33:254‐7. [Google Scholar]
References to other published versions of this review
Volz 2007
- Volz A, Khorsand V, Gillies D, Leucht S. Benzodiazepines for schizophrenia. Cochrane Database of Systematic Reviews 2007, Issue 1. [DOI: 10.1002/14651858.CD006391] [DOI] [PubMed] [Google Scholar]