

Abstract
Synapses are well known as the main structures responsible for transmitting information through the release and recognition of neurotransmitters by pre- and post-synaptic neurons. These structures are widely formed and eliminated throughout the whole lifespan via processes termed synaptogenesis and synaptic pruning, respectively. Whilst the first pro-cess is needed for ensuring proper connectivity between brain regions and also with the periphery, the second phenomenon is important for their refinement by eliminating weaker and unnecessary synapses and, at the same time, maintaining and fa-voring the stronger ones, thus ensuring proper synaptic transmission. It is well-known that synaptic elimination is modulated by neuronal activity. However, only recently the role of the classical complement cascade in promoting this phenomenon has been demonstrated. Specifically, microglial cells recognize activated complement component 3 (C3) bound to synapses tar-geted for elimination, triggering their engulfment. As this is a highly relevant process for adequate neuronal functioning, dis-ruptions or exacerbations in synaptic pruning could lead to severe circuitry alterations that could underlie neuropathological alterations typical of neurological and neuropsychiatric disorders. In this review, we focus on discussing the possible in-volvement of excessive synaptic elimination in Alzheimer’s disease, as it has already been reported dendritic spine loss in post-synaptic neurons, increased association of complement proteins with its synapses and, hence, augmented microglia-mediated pruning in animal models of this disorder. In addition, we briefly discuss how this phenomenon could be related to other neurological disorders, including multiple sclerosis and schizophrenia.
Keywords: Synaptic elimination, synaptic plasticity, microglia, complement cascade, alzheimer’s disease, multiple sclerosis, schizophrenia
1. INTRODUCTION
Synapse maintenance and elimination are essential mechanisms underlying synaptic plasticity in the central nervous system (CNS), as both processes are likely involved in learning and lifelong memory processing and storage [1-4]. Not surprisingly, these phenomena have been long known to rely on neuronal activity. In fact, studies from the 1970s demonstrated that monocular deprivation affected the decision regarding which synapses are kept or pruned [5-9]. Sensory deprivation significantly disrupts eye-specific segregation, with the intact eye occupying a much greater thalamocortical projection compared to the deprived one [8]. Meanwhile, pharmacological experiments clearly show that inhibiting one eye activity by tetrodoxin injection impaired eye-specific segregation in fetal cat dorsolateral geniculate nucleus (dLGN) [9].
Equally remarkable are the differences in the spatiotemporal regulation at which these events occur. Indeed, all brain regions experience a sharp increase in synapse formation at early post-natal stages, followed by a period of synaptic pruning [10, 11]. The pruning of overly produced synapses is highly important to maintain the stronger connections, while eliminating the weaker ones, ensuring adequate propagation of information. Interestingly, it has been shown that brain regions involved in sensory information, such as the thalamus, auditory and visual cortices, mature earlier than those responsible for higher cognitive processing, such as the hippocampus and the pre-frontal cortex [10-18]. While the first ones reach their adult levels at late childhood, with their pruning period spanning from mid-to-late infancy, the latter regions only do so at early adulthood, since synaptic elimination only surpasses synaptogenesis rates from puberty onwards [10, 11, 14].
Until the last two decades, little was known regarding the main mechanisms related to synaptic elimination. Perhaps, the most notable discovery in that matter has been the involvement of glial cells and immune molecules in its regulation [19-22]. Given the importance of this event throughout life, it is not surprising that interference in synaptic pruning regulation could lead to neuropathological alterations. This review aims to provide an updated view of the principal molecular mechanisms encompassing synaptic pruning, focusing later on its possible involvement in relevant neurological disorders.
1.1. Molecular Mechanisms of Synaptic Elimination
Synaptic pruning is a phenomenon that heavily relies on activity, with both long-term potentiation (LTP) and long-term depression (LTD) inducing morphological changes at the synaptic level [13, 23, 24]. While LTP promotes spine turnover and synapse stabilization [13, 25], LTD, on the other hand, leads to spine shrinkage and loss, enhanced varicosities turnover and their separation from dendritic spines Fig. (1) [13, 24, 26, 27]. It is worth mentioning that LTD can be induced by two distinct molecular mechanisms: N-methyl-D-aspartate receptor (NMDAR)-dependent and metabotropic glutamate receptors (mGluR)-dependent LTD [28]. In the NMDAR-dependent LTD (NMDAR-LTD), after glutamate activation of the receptor, there is a modest Ca2+ influx that triggers a yet-unknown intracellular kinase activation. This kinase phosphorylates the GluR2 subunit of α-amino-3-hydroxy-5-methylisoxazole propionic acid receptor (AMPAR) at Ser-880, leading to its endocytosis, thus reducing the amount of AMPAR available in the spine surface and leading to LTD. In addition, this Ca2+ influx activates calcineurin, which, in turn, activates the protein phosphatase 1 (PP1). PP1 dephosphorylates the AMPAR subunit GluR1 at Ser-845, strongly reducing AMPAR conductance [28]. On the other hand, mGluR-dependent LTD (mGluR-LTD) requires the activation of group I mGluRs (mGluRI), namely mGluR1 or mGluR5, members of the G-protein coupled receptor (GPCR) superfamily. Upon their activation, Gq and Gα11 proteins trigger a series of events ultimately leading to protein kinase C (PKC) activation. Then, PKC phosphorylates GluR2 also at Ser-880, prompting AMPAR internalization and LTD [28].
Fig. (1).
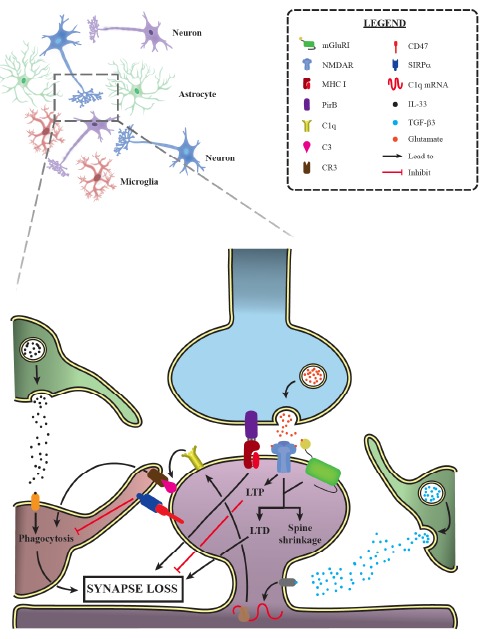
Main mechanisms of synaptic elimination during development. Recent data have indicated a relevant role of immune molecules in developmental synaptic pruning. As it is shown here, astrocytes secrete TGF-β3, leading to the downstream upregulation of the complement component C1q by neurons. Once in the plasma membrane, C1q triggers the activation of the classical complement cascade later culminating in C3 accumulation, which is recognized by microglia via CR3, thus prompting the phagocytosis of target synapses. IL-33, another astrocytic-secreted factor, is essential for synaptic engulfment by microglial cells. In addition, MHC I and its binding partner PirB have been demonstrated to promote synaptic elimination, while pre-synaptic CD47 signals via SIRPα, inhibiting synaptic uptake by microglial cells. Finally, LTD induction, following NMDAR and group I mGluRs activation, is also capable of promoting synapse loss, while LTP inhibits this phenomenon. mGluRI: group I metabotropic glutamate receptor; NMDAR: N-methyl-D-aspartate receptor; MHC I: major histocompatibility complex class I; PirB: paired immunoglobulin-like receptor B; CR3: complement receptor 3; CD47: cluster of differentiation 47; SIRPα: signal-regulatory protein α; IL-33: interleukin 33; TGF-β3: transforming growth factor β3. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
Interestingly, LTD-triggered spine shrinkage is reversed by the NMDAR antagonist APV and independent of AMPAR endocytosis [24,29]. Moreover, genetic deletion of GluN1 and GluN2B, two NMDAR subunits, promotes reduced spine density and impaired LTD in the cortex and hippocampus [30]. Notwithstanding, while NMDAR is involved in both LTD and spine shrinkage, biochemical evidence suggests these events are triggered by distinct signaling pathways Fig. (1) [24, 31]. More recently, Henson and colleagues [32] have demonstrated an augmentation in unpaired synapses upon NMDA application to mixed hippocampal and cortical cultures, providing extra evidence for the NMDAR role in synaptic elimination Fig. (1) [32]. On the other hand, an important role of mGluRI in LTD-mediated synapse elimination has been identified [33-35]. Surprisingly, it has been found that in mice lacking mGluR subtype 1 (mGluR1) not only is LTD impaired, but also these animals display deficits in surplus pruning of climbing fibers (CFs) synapses onto Purkinje cells (PCs) in the cerebellum [33, 34]. Moreover, mGluR1-/- and mGluR5-/- animals show higher synapse number than their wild-type (WT) littermates in the dLGN and layer IV of the cortical cortex, respectively [36, 37]. Pharmacological evidence also indicates that chemical LTD induction by mGluRI agonist application leads to an overall reduction in synaptic strength, followed by persistent synapse elimination Fig. (1) [38, 39]. Equally remarkable, this effect only happens when LTD is induced repeatedly.
Despite the relevance of these two types of LTD in mediating synaptic morphological changes, little is known about whether they act in concert to drive synaptic loss. Nonetheless, recent evidence shows that LTD weakens synapses with the ones with lower glutamatergic release probability being selectively eliminated in the following 7 days of stimulation, an effect that is spread to their closest neighbors [40]. In addition, NMDAR-dependent synaptic weakening and spine shrinkage occurs after 1 h of LTD induction. Notably, mGluRI activation is only required when this phenomenon occurs in large spines [41].
Even though the literature presents more evidence for mGluR- and NMDAR-LTD involvement in spine shrinkage and synaptic elimination, other molecular circuits take part in this process as well. For instance, semaphorins 7A and 5B are known to promote synaptic pruning while semaphorin 3A is thought to prevent it [42, 43]. Moreover, γ-aminobutyric acid (GABA)ergic inhibition has been proposed to be involved in synaptic loss during CFs elimination in the cerebellum and at puberty in the female hippocampus [44, 45]. Also, glutamatergic receptors δ1 and δ2 interact with neurexins in a process mediated via Cbln2 and Cbln1, respectively, promoting the formation and maintenance of synapses in the CNS [46, 47]. It is also worth citing the novel role played by the β-catenin/N-cadherin complex in the synapses. Throughout development, those spines that accumulate higher amounts of β-catenin/N-cadherin are preferably maintained and form stable spines, while those neighbor spines with lower levels of this protein complex are eliminated in an activity-dependent fashion [48]. These two proteins are also found pre-synaptically, functioning in the neocortex by stabilizing synapses and reducing their excessive turnover [49]. Despite having mentioned the main known mechanisms underlying synaptic pruning, additional mechanisms could also prove relevant (for more information on this topic, please refer to [1]).
1.2. Immune Molecules Play a Central Role in Glial-mediated Synaptic Pruning
The most notable recent finding concerning synaptic pruning is the discovery that immune molecules mediate synaptic elimination in the brain. Although erstwhile, the brain was considered an immune-privileged organ, at the dawn of the 21st century, it was shown that the immune system is involved in synaptic pruning. In a pioneer work, Huh and colleagues [50] argued that Class I Major histocompatibility complex (MHC-I) is expressed in neurons throughout the development in an activity-dependent manner, being necessary for adequate dLGN eye-specific segregation and sustained NMDAR-LTD. Later studies, aiming at understanding this phenomenon, have demonstrated that MHC-I molecules colocalize with dendritic spines and that MHC-I knockout (KO) animals display increased frequency of excitatory post-synaptic currents (mEPSCs) in both hippocampal
and visual cortical neuronal cultures [51]. This phenomenon is accompanied by an increase in pre-synaptic terminal buttons size and vesicular number, which, according to the authors, could reflect altered synaptic scaling in MHC-I KO animals. In addition, mice lacking two MHC-I molecules, H2-Db and H2-Kb, has been demonstrated to display impaired visual cortex and dLGN synaptic plasticity and eye-specific segregation [52-54]. Strikingly, this phenotype is accompanied by disrupted synaptic elimination and impaired LTD, which is rescued by inducing H2-Db expression Fig. (1) [53]. Accordingly, blockade or complete deletion of PirB, an MHC-I receptor, phenocopies the elevated spine density and LTD impairments observed in the visual cortex and hippocampus of MHC-I KO animals Fig. (1) [55-58]. Albeit these findings are observed in younger animals during specific periods, evidence suggests that these MHC molecules and PirB are later upregulated in microglia and neurons, especially during aging [59]. Thus, these MHC molecules appear to play a role in synaptic plasticity during learning processes, memory formation and aging, events closely associated with synapse turnover [4]. Furthermore, it is worth mentioning that MHC-I molecules are found to colocalize with the complement component C1q, another immune factor widely involved in synaptic elimination and expressed at the same developmental periods throughout mice lifespan [54, 60].
The direct involvement of the classical complement cascade in synaptic pruning has been elegantly demonstrated firstly in the dLGN [19]. Throughout the development, immature astrocytes secrete transforming growth factor β3 (TGF-β3) [20], leading to upregulation of all C1q subunits in retinal ganglion cells (RGCs) Fig. (1), with C1q localizing in close proximity with dendritic spines during the peaking pruning period [19]. When C1q is knocked out, mice display deficient dLGN eye-specific segregation due to impaired synaptic elimination [19]. This phenotype has been replicated in KO animals for C3, a protein downstream of C1q in the classical complement cascade and also found to colocalize with synapses Fig. (1). Since activated C3 is recognized by the complement receptor 3 (CR3), which is solely expressed in microglia in the brain parenchyma, and that microglia makes contact with synaptic structures [21], it has been hypothesized that surveillant microglia might be implicated in synaptic pruning [22] Fig. (1). In fact, microglial cells actively engulf C3-tagged synapses, a phenomenon dependent on neuronal activity, given that intraocular injection of TTX elevated microglia-mediated pruning [61]. Interestingly, microglial CR3 activation has also been associated with LTD induction, further adding evidence for the complement involvement in synaptic plasticity [62]. In addition, biochemical investigations suggest that complement-mediated pruning is probably associated with apoptotic-like mechanisms [63-66].
These studies opened up an avenue of investigation aiming at demonstrating that microglia is a key player in this synaptic plasticity mechanism. For instance, mice bearing homozygous deletion for the CX3CL1 receptor (CX3CR1), exclusively found in microglia, display delayed synaptic elimination in the hippocampus, which is accompanied by long-lasting electrophysiological and behavioral deficits [67, 68]. This observation could be in part explained by the reduced number of microglial cells multiplying or migrating into the CA1 region during the synaptic pruning period, although it is also possible that disrupted apoptotic-like mechanisms play a role as well [67, 69]. Moreover, loss of progranulin (GRN) in microglia, a protein involved in lysosomal and autophagy cargo degradation [70, 71], leads to increased synaptic pruning in thalamocortical circuits [70]. Interestingly, microglial cells are more activated in GRN KO mice, leading to increased C1q and C3 secretion, which could account for the excessive synaptic elimination observed. On the other hand, elevated GRN levels block synaptic pruning, with mice displaying higher synaptic density in the hippocampus [72]. Similar findings are observed in mice deficient for Atg7, a protein required for autophagy, suggesting that this process is also needed for efficient microglia-mediated synaptic pruning [73]. Also, it is relevant to take in consideration that microglial activities are dependent on astrocyte-secreted interleukin 33 (IL-33), as animals deficient in this protein have disrupted synaptic elimination Fig. (1) [74]. Furthermore, microglial cells also play important roles in synapse formation throughout development, since animals depleted of this cell type display significant electrophysiological and synaptogenic impairments [75].
In addition, other studies have also fostered the investigation of new immunological signaling pathways in this phenomenon. Recently, CD47, a transmembrane immunoglobulin superfamily protein, has been shown to act as a protective signal preventing synaptic engulfment by microglia Fig. (1) [76]. This protein localizes preferentially at pre-synaptic terminals of more active synapses, signaling to microglial cells via SIRPα Fig. (1). Meanwhile, Bjartmar and colleagues [77] have provided the first evidence for neuronal pentraxins (NPs) participation in synaptic elimination. Deletion of NP-1, NP-2 and NP receptor (NPR) leads to increased synaptic numbers in the dLGN [77]. Notwithstanding, according to Koch and Ullian [78] these unpruned structures are comprised mostly of silent synapses, which do not contain AMPAR receptors, being unable to display AMPAR-mediated transmission and, hence, exhibit defective synaptic maturation. Another group of molecules thought to render synapses more mature in this context are the C1q-like proteins (C1qls) [79, 80]. These proteins signal via BAI3 in the CNS [80] and promote synapse formation and maintenance in the medial pre-frontal cortex (mPFC), since knocking down their expression levels leads to significant synaptic puncta reduction [79]. Surprisingly, a deeper inspection of the transcriptomic data produced by Chen et al. [81] indicates that NPs are upregulated in the CA1-CA3 hippocampal regions during LTP, while C1ql1 show significant downregulation. At first, this data seems odd, as LTP is implicated in synapse stabilization [25]; nevertheless, it is also possible that NPs and C1ql1 are involved in the elevated synaptic turnover observed in this phenomenon [24].
Perhaps one of the most curious findings concerning microglial participation in synaptic plasticity mechanisms has come from the work developed by Weinhard and coworkers [82]. By analyzing the CA1 region of the hippocampus in newborn mice at post-natal day 15 (P15), these authors have identified that microglial cells do not phagocytose the whole synapse. Instead, these cells only engulf and uptake pre-synaptic material in a process of partial phagocytosis, so-called trogocytosis, in a complement-independent fashion. Additionally, microglia promote the protrusion of new filopodia from mature dendritic spine contacts [83]. Despite the novelty of this work, it should be taken with caution. First of all, the lack of complement-mediated synapse removal might be an exclusive feature of the CA1 region in the hippocampus. Indeed, this seems to be the only region protected from age-dependent synaptic density decline in the whole hippocampus [84]. Moreover, for all other hippocampal regions, deletion of C3 rescues this phenotype, suggesting a prominent role of the classical complement cascade in developmentally regulated synaptic elimination. Finally, other mechanisms could replace microglia-mediated spine engulfment in CA1. For example, astrocytes have been shown to prune synapses via MERTK- and MEGF10-mediated phagocytosis in the dLGN [85] and it is still unclear whether this phenomenon takes place in the hippocampus as well.
Since disruptions in synaptic pruning have been associated with an increased burden in neurological disorders, in the next sections, we are going to concentrate our discussion in the evidences for its involvement in the neuropathological alterations observed in Alzheimer’s Disease (AD), Multiple Sclerosis (MS) and Schizophrenia (SCZ). Our major focus will be given to findings implicating immunological factors and glial cells as driving forces for altered synaptic elimination in each one of these diseases.
2. SYNAPTIC PRUNING IN NEUROLOGICAL DISORDERS
2.1. Alzheimer’s Disease
Dementia is a syndrome that affects approximately 50 million people worldwide and its most common cause is AD, which responds for 60-70% of all cases [86-88]. The first case of AD was described by the German psychiatrist Alois Alzheimer, who identified the symptoms as progressive cognitive impairment, memory loss, language disability, spatial and temporal disorientation, hallucinations and delusions [89]. This disease is also characterized by evident cerebral atrophy and neurodegeneration, affecting cortical and subcortical regions, with greater synapse than neuronal loss [90-96]. Additionally, other hallmarks of AD found in the brain of affected patients are extracellular senile plaques, intraneuronal neurofibrillary tangles, gliosis, microglial activation and neuroinflammation [97-105].
These senile plaques are the result of oligomeric amyloid β (Aβ) peptide aggregation. Cleavage of amyloid precursor protein (APP), a cell surface receptor [106], occurs naturally in the brain, generating soluble APPα when APP is processed by α-secretase [107]. However, cleavage of APP by β-secretase and γ-secretase leads to the production of pathogenic Aβ forms of 40 and 42 amino acid residues, termed Aβ1-40 and Aβ1-42, respectively [108-110]. The presence of a qualitatively altered allele of γ-secretase results in higher production of Aβ1-42, which is more susceptible to aggregation and toxicity [111] and is associated with higher pathogenicity compared to Aβ1-40, which is the most abundant isoform produced in healthy brains [112].
Interestingly, the load of soluble oligomeric Aβ has been implicated in the synaptic loss observed in AD patients, even enabling the distinction between them and subjects with profuse plaques and tangles but with no symptoms of dementia [113]. Studies employing AD mouse models have shown that increased levels of soluble Aβ correlate with the decrease in synaptic density [114]. Notably, soluble Aβ represents the main factor contributing to the synaptotoxic and neurotoxic effects observed in AD [115, 116], leading to severe impairments in synaptic plasticity [117-120]. These oligomers are responsible for blocking hippocampal and cortical LTP [119-123], while also inducing LTD [119, 120, 124, 125], spine and neuronal loss [120, 125, 126]. The underlying mechanism appears to involve changes in glutamatergic neurotransmission mediated mainly by mGluRI and NMDAR Fig. (2). Further studies have demonstrated that the activation of NMDAR localized extrasynaptically (eNMDAR) is an important event involved in Aβ synaptic toxicity [117, 120, 124, 126-128]. This activation of eNMDAR triggers nitric oxide (NO) production in neuronal cultures exposed to Aβ oligomers [129]. In turn, NO promotes Cdk5 activation via S-Nitrosylation and its downstream signaling cascade leads to spine loss Fig. (2) [130]. Injection of these forms of Aβ in rodents has been proven as a useful model of mimicking synaptic pathology and by extension, the cognitive deficits found in AD [131-134]. These findings further reassert the role of soluble Aβ forms in the synaptic changes key to the etiology of AD.
Fig. (2).
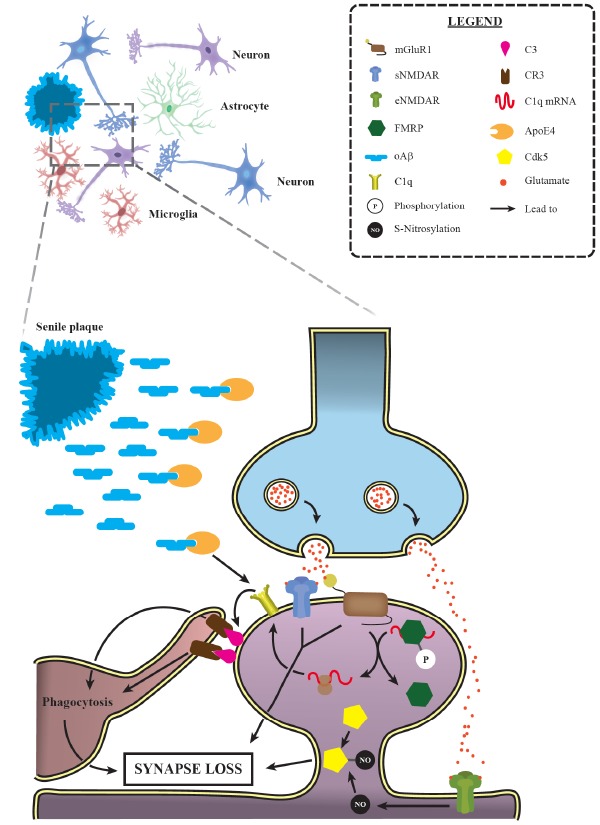
Activation of synaptic pruning mechanisms is relevant for AD progression. Progressive synaptic loss is one of the main morphological features of Alzheimer’s disease, observed even earlier than senile plaque deposition. Several studies have demonstrated the prominent role of the classical complement cascade in such event. Indeed, it has been shown that ApoE4, a major AD risk factor, leads to C1q accumulation in animal models. Additionally, mGluR1 activation allows a greater C1q mRNA translation by promoting the dephosphorylation of FMRP, an RNA-binding protein that represses translation. These data provide a suitable explanation for the increased levels of C1q and its downstream complement factor C3 in AD, which, in turn, trigger targeted synapses phagocytosis by microglial cells. Moreover, NMDAR and group I mGluRs activation have also been involved in the excessive synaptic loss observed during AD. Interestingly, the activation of extrasynaptic NMDAR (eNMDAR) induces the production of the S-nitrosylation of Cdk5, another factor implicated in AD excessive synaptic elimination. mGluR1: metabotropic glutamate receptor 1; sNMDAR: synaptic N-methyl-D-aspartate receptor; eNMDAR: extrasynaptic N-methyl-D-aspartate receptor; FMRP: fragile X mental retardation protein; oAβ: oligomeric amyloid β; CR3: complement receptor 3; ApoE4: Apolipoprotein E ε4; Cdk5: cyclin-dependent kinase 5. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
Early synaptic loss is well established as a key feature of AD. For instance, studies have shown 38%, 14% and 44-55% reduction in the number of synapses in biopsies of AD patients at the temporal and frontal cortices and the hippocampus, respectively [94, 135-138]. Since then, other studies using distinct approaches endorsed the central role of synaptic loss in the pathology of AD. Further analysis using post-mortem tissues from AD patients confirms a decline in the synaptic number in the temporal, parietal and frontal lobes [139-141]. Also, a reduction of synaptic contacts have been noticed in the outer molecular layer of the hippocampal dentate gyrus, the CA1 region and the precuneus [142]. Moreover, presynaptic protein levels can also be used to estimate synaptic terminal loss [143]. The expressions of proteins such as synaptophysin, syntaxin, secretoneurin, PSD95 and glutamatergic vesicular transporters and receptors are shown to be significantly diminished in AD patients and in animal models [113, 144-150]. Notably, this PSD95 reduction has been shown to be driven by degradation via proteasome, a process mediated by NMDAR activity and that takes place exclusively at glutamatergic synapses [151]. A correlation between the cognitive shortcoming of AD patients and reduced expression level of synaptic proteins such as synaptobrevin, synaptotagmin and Rab3a has also been described [152], although results obtained in different studies may vary according to brain region and protein analyzed [143]. Furthermore, it has been suggested that synaptic loss is the strongest correlate with the cognitive deficits found in AD and might be a process restricted to excitatory synapses [147, 148, 150, 151, 153-155].
Neurofibrillary tangles (NFT), another key feature of AD, are mainly made of paired helical filaments of the
phosphorylated microtubule-associated protein tau (MAPT). This MAPT causes the expansion of senile plaques and exacerbates the formation of plaque-associated dystrophic neurites containing its misfolded form [156]. In addition, NFT located in pyramidal neurons correlates with changes in morphology and decreased the density of dendritic spines [157]. Nonetheless, previous reports indicate that accumulation of MAPT at spines, rather than NFTs, are linked to the onset of tau-related pathologies in AD [101, 102, 158-162]. Accordingly, oligomeric Tau promotes the reduction of synaptic markers and impair mitochondrial function [162]. These effects are mediated via Tau relocation from axons to dendrites upon its phosphorylation [159]. Indeed, Hoover et al. [159], using a transgenic model mimicking Tau phosphorylation in vivo, have shown that this post-translational modification leads to LTP and memory formation impairments. Moreover, they have observed that hyperphosphorylated Tau dissociates from microtubules and is mislocalized to dendritic spines. This leads to reduced AMPAR and NMDAR surface levels in these structures, impairing appropriate glutamatergic transmission, which could account for the observed phenotypes and synapse loss often found in AD models even before NFT deposition [159]. Interestingly, this role of Tau in modulating synaptic plasticity is required for Aβ-induced synaptic defects, with both proteins being found at the same post-synaptic compartment [158, 160, 163]. Intriguingly, reducing Tau levels in a dose-dependent manner in a mouse model of AD has been shown to be neuroprotective, significantly dampening Aβ-excitotoxic effects by preventing NMDAR and PSD95 uncoupling, thus securing proper LTP induction, preventing neuronal loss and improving memory formation and locomotor coordination in behavioral tests [161, 164].
2.1.1. Overheating the Developmental Engine in AD: When Complement-mediated Synaptic Pruning Becomes Harmful
It is well known that AD etiology shares environmental and genetic risk factors [165-166]. The majority of the cases of AD are sporadic and affect elder individuals (Late Onset AD, abbreviated as LOAD), while the remaining, which is about 5% of the cases, have an autosomal dominant inheritance pattern, occurring before 65 years of age (Early Onset AD, shortened as EOAD) [167]. It has been shown that in several EOAD patients, there is greater expression and release of Aβ1-42 peptide, which aggregates more rapidly in insoluble amyloid fibrils [168-171], frequently giving rise to the brain plaques that characterize the disease [172]. In addition, the risk of developing AD is highly associated with the expression pattern of distinct isoforms of the apolipoprotein E (ApoE) gene in both EOAD and LOAD patients, whose protein product binds with high avidity to Aβ Fig. (2) [173-175]. Indeed, while the presence of ApoE2 allele is protective [176, 177], the number of ApoE4 allele copies is proportional to the risk of developing LOAD and inversely correlated with the age of onset and years of survival [174-176, 178]. Moreover, recent research has also demonstrated that the ApoE4 isoform stimulates greater APP transcription compared to other isoforms, thereby increasing Aβ levels [179]. Also, homozygous ApoE4 carriers display elevated production of Aβ oligomers, which might be implicated in dendritic spine dysfunction and severe memory impairment [180]. Several lines of evidence indicate that ApoE is highly relevant to AD development, making this gene a suitable candidate to study synaptic loss in this disorder. Using transgenic mouse models expressing distinct human ApoE alleles, authors have shown that the ApoE4 isoform produces the greatest reduction in both pre- and post-synaptic markers density, dendritic spine number and dendritic arbor complexity in the cortex, amygdala and hippocampus [181-186]. Furthermore, transcriptomic data obtained from human induced pluripotent stem cells (hiPSCs)-derived neurons bearing the ApoE4 allele indicates that genes involved in synaptic regulation are among those most differently expressed compared to those of ApoE3 neurons [187]. Amongst them, C1ql3 and neuronal pentraxin 2 are downregulated in ApoE4 neurons, which is very interesting as these genes might be involved in synaptic elimination. Likewise, distinct ApoE alleles have been found to interfere with astrocytic synaptic pruning capacity [188]. Interestingly, murine astrocytes expressing the human ApoE2 isoform show greater synapse phagocytosis compared to those cells expressing ApoE3 and ApoE4, with the latter displaying the lowest engulfment index [188]. This compromised ability of astrocytes in eliminating senescent synapses by ApoE4 mice leads to the accumulation of C1q in the hippocampus, especially in aged animals Fig. (2). If this augmented C1q targets senescent synapses, this could trigger higher synaptic engulfment by microglia. However, more studies will be needed to clarify this issue. It is also important to mention that early reports indicate enhanced microglial activation in an ApoE4 dose-dependent manner, with AD homozygous individuals displaying greater numbers of activated microglia in the frontal and temporal cortices compared to homozygous ApoE3 patients [189]. Taking in consideration that Aβ comprises an inflammatory stimulus to the brain [190, 191] and that ApoE4 microglia exhibit increased activation [189] and augmented levels of proinflammatory transcripts [187], these data argue in favor of a prominent role for ApoE4 in mediating neuroinflammation in AD.
Neuroinflammatory mediators have also been associated with the pathology of AD. The presence of the membrane attack complex (MAC) and the immune activation of microglia have been shown to be better correlated with the number of synapses than the load of neurofibrillary tangles and Aβ deposition [192]. Another finding worth mentioning is that the decreased level of complement defense 59, a protein that prevents MAC assembly, in the cortex and hippocampus of AD patients correlates with the reduction in synaptophysin [193]. These data further highlight the association of the complement system and AD development. In addition, previous reports indicate that C1q and C3, two components of the classical complement cascade, display age-dependent upregulation in the hippocampus and cerebral cortex [194]. This rise in C1q and C3 levels is even greater in AD models, suggesting their involvement in disease progression Fig. (2). Indeed, genetic deletion of C1q and C3 in AD mouse models promotes reduced neurodegeneration, plaque deposition, gliosis, and microglia activation, as well as increased expression of synaptic markers [195, 196].
Later studies helped to clarify the association between synaptic loss and reactivation of the synaptic pruning process during the course of AD [197]. For instance, Hong and colleagues [198] have demonstrated the detrimental role of the complement system and microglia in AD early synaptic loss. Increased deposition of C1q and C3 at synapses of hippocampus and cortex is observed in the human APP (hAPP) transgenic mouse model of AD, leading to accentuated synaptic engulfment by microglial cells before the onset of plaque deposition Fig. (2). This effect is mediated by oligomeric Aβ, since its intracerebroventricular injection prompts the deposition of C1q and C3 in the hippocampus of WT mice [198]. In contrast, ablation of the complement receptor CR3 reduces the synaptic loss and the number of phagocytic microglia [198]. Similar results are observed in Tau-P301S transgenic mice [199]. In these animals, neurodegeneration and hippocampal atrophy are present only after 12 months of age, while gliosis and synaptic dysfunction are detected in 6-month-old animals. Proteomic data indicate progressive accumulation of C1q at synapses, especially at the post-synaptic compartment, at the same age it is possible to identify excessive hyperphosphorylated Tau in these structures [199]. This evidence led the authors to hypothesize that the reduction in synaptic markers in the hippocampus is related to exaggerated pruning by microglial cells. In fact, this has been shown to be the case, as treatment of cultures with anti-C1q antibody inhibited microglia-mediated synaptic elimination. In contrast, other proteomics studies failed to identify the enrichment of complement components in the hAPP mouse model and in AD patient post-mortem brain samples [200, 201]. These contradictory data indicate that more studies should be conducted to fully clarify the role of the complement cascade in AD, aiming at distinguishing whether the progressive C1q accumulation at synapses, found by Dejanovic et al [199], is a sole consequence of the disorder or is also affected by the aging process.
Remarkably, Litvinchuk et al [202] have provided the first evidence for the participation of C3a, one of the two C3 cleavage products, and its cognate receptor C3aR in AD synapse loss, using the Tau-P301S mouse model. While C3b acts as an opsonin and signals via CR3 in microglia, C3a is usually implicated in triggering inflammatory signaling (for more information about AD neuroinflammation and the classical complement cascade, refer to [203]). In this study, genetic deletion of C3aR has led to a significant reduction in astrogliosis and microgliosis, while also rescuing LTP deficits, enhancing synaptophysin and PSD-95 levels and blocking neurodegeneration in the hippocampus. In addition, authors have also shown that C3a rendered microglia more activated, as indicated by enhanced synaptic markers uptake and CD68 staining, an effect attenuated in the double mutant (C3a KO/Tau-P301S) mouse model. It is also important to mention that C3a/C3aR pathway disruption in the Tau-P301S mice reduces tumor necrosis factor α (TNF-α), IL-1β and IL-6 back to WT levels [202]. Moreover, in the same study, RNA-seq analysis revealed that both microglia and astrocytes display neurotoxic features, a phenotype partly rescued by C3aR deletion. Interestingly, this microglial transcriptomic profile corroborates the data found by Karen-Shaul et al [204], indicating that during AD progression microglial cells assume a more proinflammatory and activated profile, thus contributing to the neuroinflammation observed in this disorder. On the other hand, the astrocytic transcriptomic profile partially resembles the features of A1 reactive astrocytes characterized by Liddelow and collaborators [202, 205]. A1 astrocytes are shown to impair synaptic formation and neuronal electrophysiological properties, while also upregulating C3 mRNA levels [205]. Given that microglial cells are the major source of C1q in the brain [206], that this protein is accumulated in AD mouse models and the proinflammatory profile displayed by microglia in AD, one could suppose that these activated microglia could lead to the formation of reactive A1 astrocytes [205]. These two neurotoxic glial cells could be, at least in part, responsible for both the synaptic defects and neurodegeneration observed in AD. Finally, recent data indicate a prominent role for mGluR1 activation in increasing C1q expression at dendritic spines, favoring their uptake by microglial cells in the hippocampus of mice injected with Aβ1-40 oligomers [207]. This phenomenon relies on fragile X mental retardation protein (FMRP) dephosphorylation, allowing local translation of FMRP-silenced mRNAs Fig. (2). Since mGluR1 activation is also involved in LTD induction, this data provides important evidence connecting LTD and complement-dependent synaptic elimination. Altogether, these results suggest a prominent role of the complement cascade in mediating early synaptic loss in AD.
2.1.2. Other Synaptic Pruning-related Molecules also Contribute to Exaggerated Synaptic Loss in AD
Other synaptic pruning-related molecules have also been established as contributors to AD development. For instance, caspase-3, a classic proapoptotic factor, is elevated in AD brains, being specially enriched at the post-synaptic density [208]. In line with this evidence, D’amelio et al [209] have demonstrated that caspase-3 triggers AMPAR dephosphorylation via calcineurin in an AD mouse model, without any obvious cell death. This reduction of AMPAR at spines leads to increased LTD and diminished hippocampal apical dendritic spine levels in 3-month old mice, triggering subsequent learning and memory deficits [209]. These abnormalities are rescued by intrahippocampal administration of the caspase-3 inhibitor, z-DEVD-fmk, suggesting the participation of apoptotic-like mechanisms in AD early synaptic elimination and making this pathway an interesting target for future therapeutic interventions. On the other hand, the human ortholog of PirB, LilrB2, comprises a high affinity Aβ receptor [210]. This MHC-I receptor also mediates altered synaptic plasticity and memory deficits often observed in AD models. Indeed, its genetic deletion in mouse hippocampal slices rescues LTP induction by oligomeric Aβ. Moreover, transgenic AD animals with PirB ablation perform better in memory tasks, compared to diseased animals bearing intact PirB copies. In contrast, IL-33 is reduced in AD patients’ brains [211]. Interestingly, when this cytokine is administered to hAPP mice, it rescues LTP induction, while also improving Aβ clearance by microglia, making IL-33 an interesting therapeutic target [212]. Despite IL-33 importance in inducing microglia-mediated synaptic pruning, its effects might not be specific to this phenomenon, as IL-33 may simply activate microglial phagocytic capacity, which could explain why this cytokine is able to enhance Aβ uptake and degradation.
Neuronal pentraxin 1 (NP-1), progranulin (GRN) and the CX3CL1/CX3CR1 also take part in AD neuropathological events. NP-1 levels, for example, are elevated in the hippocampus and plasma of AD individuals [213]. Likewise, NP-1 is upregulated in neuronal cultures treated with Aβ, where it mediates cell death and reduction of synaptophysin levels [214]. Intriguingly, progranulin levels in the blood and cerebrospinal fluid (CSF) increase with disease progression, correlating with cognitive decline and memory disturbance [215, 216]. Despite this, genetic deletion of GRN in hAPP mouse models worsens cognitive behavior, also leading to elevated numbers of activated microglia in the hippocampus, higher TNF-α and IL-1α mRNA levels and increased deposition of C1q at dendritic spines [217, 218]. This effect seems to be mediated by microglial cells, as specific deletion of GRN in this cell type leads to increased plaque burden and decreased phagocytic index [217]. In contrast, overexpression of this molecule prevents Aβ-mediated cell toxicity, diminishes plaque deposition and rescues neurodegeneration and memory deficits [217]. Therefore, these data may indicate that increased progranulin levels throughout the course of AD might reflect an attempt to reestablish the proper homeostatic process by increasing microglial clearance of Aβ, thus diminishing plaque accumulation. On the other hand, the role of CX3CL1/CX3CR1 is less well defined. While CX3CL1 is downregulated in the CSF and brain of AD patients, no change has been reported for its receptor level compared to healthy individuals [219, 220]. Moreover, deficiency in either membrane-anchored CX3CL1 or in its receptor increases Aβ phagocytosis, reducing plaque deposition [221, 222]. However, the lack of CX3CR1 has been shown to increase Tau pathological effects via elevated p-Tau levels [220, 221]. In addition, literature data are controversial regarding the activation status of CX3CR1-/- microglia in AD. While Lee and colleagues [222] report reduced microglial activation and TNF-α mRNA levels in APP/PS1;CX3CR1-/- mouse model, Cho et al [220] demonstrate the opposite in hAPP;CX3CR1-/- mice. Therefore, what is the precise role of CX3CL1/CX3CR1 in AD neuroinflammation is still a matter of debate, while its role in AD synaptic elimination remains insofar unknown. Taken together, these data support the involvement of several modulators of synaptic elimination in AD progression.
Despite the vast majority of studies aiming at understanding synapse loss in AD has focused on investigating elevated synaptic elimination, an interesting alternative hypothesis that might explain the reduced synapse numbers observed is disrupted synaptogenesis. Interestingly, researchers have found that soluble Aβ leads to increased pre-synaptic Ca2+ levels, promoting higher phosphorylation levels of Ca2+/calmodulin-dependent protein kinase IV (CaMKIV) and, ultimately, impairing activity-dependent synaptogenesis [223]. In addition, Levi et al. [224] demonstrated that human ApoE4 transgenic mice display deficient environmental-stimulated synaptogenesis, resulting in learning and memory formation deficits, compared to human ApoE3 transgenic mice. These evidences imply that impaired synaptogenesis could also play an important role in AD development and progression. However, whether the reduction in synapse number observed in this disease is due to diminished synapse formation, increased synaptic elimination or both is a matter that remains to be investigated.
Given the importance of synapse loss in AD development, therapies aiming at restoring normal synaptic levels or preventing their loss could benefit individuals affected by this disorder. For example, using a gene delivery system, Kampen & Kay [225] overexpressed progranulin in an AD transgenic mouse model. In this study, it is possible to observe that GRN expression leads to reduced Aβ plaque burden, astrogliosis and microgliosis, as well as increased synaptic density in the hippocampus. In another report, daily injections of the Heat-shock protein 90 (HSP90) inhibitor 17-AAG prevent Aβ-driven synapse loss and memory deficits in vivo, likely by pre- and post-synaptic proteins upregulation, hence strengthening the existing synapses [226]. Interestingly, Resveratrol, a Sirtuin 1 activator, protects rats exposed to oligomeric Aβ from displaying disturbed memory acquisition and LTP induction [227]. These effects are probably mediated by neurogenesis and blockade of microglial and astrocytic activation, substantially reducing neuroinflammation [228, 229]. These two Resveratrol effects combined could contribute to increase neuronal and synaptic number in AD patients’ brains, while also dampening the neurotoxic effects mediated by reactive glial cells. Thus, this drug comprises one of the most promising candidates for AD treatment in the near future. Finally, clinical trials using intranasal insulin applications have been successful in ameliorating cognitive functions of patients exhibiting mild to moderate AD [230]. Insulin works by preventing Aβ-derived diffusible ligands binding to neuronal surface, resulting in significant diminished spine loss [231]. It is worth mentioning that this strategy is currently under Phase II/III clinical trials (ClinicalTrials.gov Identifier: NCT01767909).
2.2. Multiple Sclerosis
Multiple Sclerosis (MS) is one of the most frequent demyelinating CNS disorders, affecting around 3 million people worldwide [232]. Patients bearing this neurological disorder often present motor, cognitive, visual and sensory deficits throughout the course of the disease, which worsens with aging [233-237]. These symptoms are widely viewed as the result of extensive white matter demyelination, associated with blood-brain barrier disruption, leading to massive inflammatory infiltration, followed by astrogliosis and microgliosis, neuronal injury, extensive excitotoxicity and neuroinflammation [233, 234, 238-242].
Even though most researchers have focused on the inflammatory aspects concerning white matter demyelination and its associated lesions, gray matter alterations have been regarded as an important aspect in MS, especially concerning the development of cognitive deficits [241]. For instance, it has been shown that decreased cognitive processing is associated with cortical lesions and atrophy in several brain regions [243], including the basal ganglia [244], thalamus [244, 245], sensorimotor, entorhinal and cingulate cortices [246], cerebellum [247], amygdala [248] and hippocampus [249, 250]. One of the mechanisms responsible for this phenotype is the extensive neuronal and glial cell loss present in the MS lesions and the extensive demyelination of cortical structures [250-253]. As such, several studies have identified that proinflammatory cascades, induced by both peripheric and activated CNS resident immune cells, are associated with augmented apoptosis in MS patients and in the Experimental Autoimmune Encephalomyelitis (EAE) mouse model [252-256]. Thus, TNF-α has been found to induce oligodendrocytes programmed cell death in a caspase-independent fashion [254]. Moreover, Interferon γ (IFN-γ) promotes the cell death of oligodendrocytes and also of microglial cells, contributing to the reduction in myelination and gray matter volume observed in MS patients [255, 256]. In addition, IL-1β and glutamatergic excitotoxicity have also been demonstrated to activate apoptosis, triggering neuronal cell death via the p53 cascade [257], while augmented oxidative stress, due to increased reactive oxygen species production, is also implied to take part in the cell loss that takes place in MS [258]. Furthermore, the classical complement cascade plays a significant role in the induction of apoptosis of neuronal cells in MS. In fact, early reports have provided evidence that animals lacking the complement component C3 in an EAE model have milder disease phenotypes compared to their WT littermates, including decreased demyelination and diminished number of infiltrated macrophages and T cells [259]. Subsequent studies not only corroborate this data [260, 261], but also indicate the involvement of C5 in mediating cell death in an acute EAE model [262]. Additionally, recent findings obtained by Watkins et al [263] have shown increased C1q, C3b and C9 expression in the gray matter lesions in the cortex. These authors have also found elevated levels of microglial cells positive for the complement receptors CR3, C3aR and C5aR. Altogether, these data strongly suggest the formation of the membrane attack complex in the CNS cells of MS patients, leading to their death by apoptosis.
2.2.1. Synaptic Elimination might be Central for the Cognitive Decline During MS Progression
Synaptic elimination is an important neurodegenerative process that appears to take place in the CNS parenchyma of MS individuals and to lead to extensive the cognitive decline. As such, post-mortem analysis of MS patients and EAE mouse models have provided evidence for decreased synapse number in the hippocampus, insular, frontotemporal and occipital cortices, and striatum [250, 264-267]. In addition, the staining intensities for synapsin I, synaptophysin and PSD-95 are also reduced in the hippocampus of MS patients and in EAE mice [266, 267]. Even though these results and the involvement of the classical complement cascade in MS could suggest increased synaptic pruning, direct evidence for such a phenomenon in MS is sparse. Indeed, Machalidou and colleagues [268] have found increased C1q and C3d immunoreactivity in all hippocampal regions in the brain of MS patients. Also, they have demonstrated that these two complement factors colocalize with synaptophysin, both in neuronal synapses and inside activated microglia, indicating these cells are engulfing complement targeted synapses. These data suggest that synaptic pruning contributes to the diminished synapse density observed in MS patients. Conversely, CD47, which acts as a “don’t-eat-me” signal, preventing phagocytosis of synapses by microglia, is downregulated in chronic MS lesions [269, 270]. Intriguingly, when CD47 is deleted in an EAE mouse model, the disease fails to develop to its full extent [270]. In contrast, when this molecule is blocked during the disease course, it worsens EAE development in treated animals, on which it is possible to observe increased myelin phagocytosis by murine macrophages [270]. Although these studies did not access CD47 role in synaptic pruning, its downregulation during MS progression could prompt both activated microglia and infiltrating macrophages to excessively prune synapses due to inefficient recognition of this protective signal.
On the other hand, Freria et al. [271], upon analyzing spinal cord synapses, found that increased MHC-I expression in both astrocytes and microglia correlates with decreased immunostaining for synaptophysin, suggesting that MHC-I levels are inversely related to synaptic density. Genetic studies indicated that a cohort of MS patients possesses higher frequency of two progranulin alleles (rs9897526 A and rs5848 T) that are associated with increased disease severity and decreased progranulin expression levels [272]. In marked contrast, Vercellino et al [273] have provided evidence for strong expression of progranulin in activated microglia, macrophages and neurons around cortical lesions in MS patients. Moreover, they also show higher CSF progranulin levels in patients displaying the progressive form of the disease, compared to those in clinical remission. Adding more confusion to the role played by progranulin in MS, De Riz and colleagues [274] have argued that this factor does not play a significant role in this disease since they have found its CSF levels to be more correlated with aging than with MS per se. Despite progranulin importance in synaptic elimination, there is a lack of substantial evidence for the participation of progranulin in MS synapse loss, especially because only a few studies have aimed at investigating its role in this disease. In addition, previous reports indicate increased brain, CSF and serum levels of CX3CL1 in MS patients compared to healthy individuals [275, 276]. Since CX3CL1 expression in astrocytes does not differ between MS patients and controls [277], it is possible that increased CSF levels of this chemokine could come, at least in part, from neuronal origin or increased release of soluble CX3CL1 from the astrocytic plasma membrane. Furthermore, data points out that CX3CL1 expression in the brain is regulated by adenosine signaling and that blocking CX3CL1 impairs EAE development, especially dampening lymphocytes migration into the brain parenchyma [278]. Also, CX3CR1 deletion markedly decreases Natural Killer cell influx into the brain in EAE [278]. It is also worth mentioning that neuronal pentraxin receptor (NPR) is upregulated in MS patients, which might indicate that neuronal pentraxins are somehow involved in this disease progression [279]. However, whether these immune molecules are implicated in the enhanced synaptic elimination observed in MS patients is still unknown. Even though these data do not conclusively point to a direct association between increased synaptic elimination and disease progression, the altered levels of the aforementioned molecules in MS might indicate this could be the case.
Regarding deficient synaptogenesis, a phenomenon that could also explain the observed reduction in synaptic staining intensities in MS patients and EAE mouse models, little is known. Indeed, one of the fewest evidences pointing in this direction indicates a negative correlation between synapse number and paralysis severity in an EAE mouse model [280]. In this study, proinflammatory cytokines released by T cells have been shown to either augment the production of secreted protein acidic and rich in cysteine (SPARC) or inhibit SPARC-like 1 (SPARCL1) synthesis. While SPARCL1 promotes the formation of excitatory synapses, SPARC blocks it [280]. Albeit this data provides support for disrupted synaptogenesis, the lack of evidence limits any further conclusion concerning the importance of this phenomenon in MS progression. Altogether, future research should be conducted to understand better to what extent increased synaptic pruning or decreased synaptogenesis could influence MS development.
Despite the scarcity of data regarding the molecular mechanisms involved in spine loss in MS, it is well known that glutamatergic and GABAergic transmissions are altered in this disease, especially in the case of the first system, which is implicated in triggering excitotoxic neuronal damage [239, 266, 281-283]. Elevated TNF-α level is observed during MS progression, leading to substantial increase in spontaneous excitatory post-synaptic currents (sEPSCs) and subsequent neuronal damage due to augmented glutamate release [240]. This elevated level of glutamate seems to be largely due to increased TNF-α release by astrocytes and microglia, ultimately leading to impaired memory in EAE and increased neurotoxicity [283, 284]. In addition, this cytokine also mediates increased synaptic turnover, even before microglial activation and lymphocyte infiltration in EAE mouse brains, suggesting that this proinflammatory molecule might also play an important role in modulating synapse density in MS [285]. Interestingly, treating EAE mice with the cannabinoid receptors 1 and 2 (CB1 and CB2, respectively) agonist WIN55,512-2 significantly ameliorates disease progression, even reducing cellular infiltrates in the spinal cord [286]. This effect is largely mediated by CB1, triggering subsequent downregulation of TNF-α levels, which, in turn, diminishes the glutamatergic excitotoxicity [286, 287]. Similar results are observed when microglial cells are treated with either the gap junction blocker carbenoxolone or the glutaminase inhibitor 6-diazo-5-oxo-L-norleucine, with both drugs preventing the excessive glutamate release [288]. Thereby, therapeutic strategies aiming at reducing either TNF-α or released glutamate levels comprise an attractive approach to ameliorate MS progression.
2.3. Schizophrenia
SCZ is a neurodevelopmental disorder that is characterized by high level of synapse elimination. This disorder is estimated to affect approximately 1% of the entire world population and its symptoms begin to manifest at late adolescence and early adulthood [289-290]. SCZ symptoms are often divided into three categories: positive symptoms (hallucinations, delusions, thought disorder and psychosis), negative symptoms (avolition, depression, anhedonia and loss of social drive) and cognitive deficits (disorganized speech, thought and attention deficits) [290, 291]. Anatomical studies using imaging techniques, such as magnetic resonance imaging (MRI), have identified several brain alterations in schizophrenic individuals. Thus, researchers have found substantial enlargements of the third and lateral ventricles, which are correlated with augmented cognitive deficits [292-295]. In addition, it is also perceived a 3% and 5.5-6.5% reduction in the whole cerebrum and hippocampal volume, respectively, as well as overall reduced cortical thickness [294-296]. Interestingly, this reduction in brain volume is not accompanied by the corresponding decrease in neuronal numbers. In fact, studies suggest that not only are neuronal numbers similar between schizophrenic and healthy individuals, but they might also be increased in the former group, especially in the pre-frontal cortex, a region implicated in SCZ [297-299].
Based on these findings, researchers hypothesize that SCZ brain volume and cortical thickness reductions are caused by altered morphological features in the neuropil [300]. Post-mortem tissue analyses have shown substantially decreased number of dendritic spines in several cortical structures, including the striatum, the subiculum and the temporal, auditory and pre-frontal cortices [300-305]. These evidences provide strong support for the hypothesis firstly proposed by Feinberg in 1983, that SCZ could arise from excessive synaptic pruning during adolescence. This phenomenon could lead to significant cortical connectivity alterations in the adulthood, and, ultimately, to the characteristic symptoms observed throughout disease progression [306]. Despite almost four decades after being proposed, the mechanisms whereby elevated synaptic elimination could account for the onset of SCZ are still poorly understood.
Even though schizophrenia etiology is still mostly unknown, several genetic and environmental risk factors are thought to trigger this neurodevelopmental disorder [289, 307]. Notably, genetic studies have provided the majority of evidences linking SCZ and abnormal synaptic pruning. For instance, Sekar and colleagues [308] have demonstrated that schizophrenic individuals possess genetic structural variants favoring the greater expression of C4A mRNA, one of the C4 isoforms found in the human genome. C4 activates C3 in the classical complement cascade and these authors have demonstrated that mice lacking C4 show abnormal eye-specific segregation in the dLGN as well as reduced C3-puncta colocalization at synapses [308]. Corroborating C4 involvement in SCZ, Prasad et al [309] have identified that both C4 isoforms (C4A and C4B) expression correlates with neuropil contraction in SCZ patients. Furthermore, chronic schizophrenic patients display higher blood levels of C4, while those at high-risk of developing psychosis present increased levels of both C3 and C4 compared to healthy individuals [310]. On the other hand, SIRPα, the CD47 receptor, is downregulated in the dorsolateral pre-frontal cortex of schizophrenic individuals [311]. These findings are particularly remarkable, as C4 is a pruning-promoting molecule, while SIRPα takes part in the protective signaling preventing synaptic elimination. Additionally, if brain complement component levels are actually elevated in SCZ, it is possible to speculate the involvement of mGluR1 in this phenomenon, similarly to what has been observed in Alzheimer’s disease [207]. As such, augmented levels of mGluR1 has been observed in the pre-frontal cortex of SCZ patients [312], while non-synonymous mutations in the mGluR1 gene (GRM1) has been found in a cohort of schizophrenic individuals [313]. Moreover, the downstream target of mGluR1 activation, FMRP, is decreased in the blood of SCZ individuals [314]. Since C1q mRNA is repressed upon FMRP binding, these data might suggest a connection between mGluR1 activation, which could also lead to LTD induction, and increased complement-dependent synaptic pruning in SCZ. In remarkable contrast, other genetic studies, using distinct schizophrenic patient cohorts, failed to detect the association between the classical complement cascade and schizophrenia [315-317]. This contradiction might reflect the high heterogenicity found between schizophrenic patients and the polygenic nature of this disorder. Therefore, further functional studies, using novel strategies, should be carried out to elucidate the actual contribution of the complement cascade to SCZ development.
Bergon and colleagues [318] have performed a large meta-analysis based on gene expression alterations found in schizophrenic individuals compared to healthy ones in order to find other suitable candidates important for synaptic elimination. Intriguingly, it has been shown that CX3CR1 is downregulated in both the brain and blood in SCZ patients. Moreover, another study identified that the genetic variant coding for CX3CR1A55T is associated with SCZ [319]. The functional validation of this mutated CX3CR1 form in vitro indicated that it leads to impairments in downstream signaling upon CX3CL1 binding, which might affect the microglial phenotype in this disorder. Additionally, it is important to mention that common genetic variant analyses indicate that polymorphisms in the MHC locus are significantly associated with SCZ [320, 321]. Another genetic study has identified progranulin genetic variants conferring higher risk for schizophrenia [322], whereas others found that glypicans, which are responsible for neuronal pentraxin release regulation [323], are also related to this disease [324].
Post-mortem analyses and imaging techniques point to the activation of microglial cells in SCZ [324-326]. This microglial phenotype suggests the occurrence of neuroinflammation and enhanced levels of proinflammatory molecules in the brain of schizophrenic individuals [327-328]. For instance, evidences indicate elevated TNF-α and IL-6 in the brain, blood and CSF of SCZ patients [328-331]. On the other hand, increased IL-1β levels, a classic proinflammatory cytokine, have been implicated in SCZ, although these results are disputable [330-332]. Nonetheless, whether these cytokines contribute to promote microglia-mediated synaptic pruning in schizophrenia is still unknown. In an attempt to identify whether this is the case, studies using animals to model environmental factors have been used. Indeed, using the maternal immune activation model, which mimics pre-natal infection as a risk factor in SCZ [333], Mattei and colleagues [334] have verified that the microglia of the offspring display an activated phenotype with transcriptomic profile and cytokine expression partially matching those found in SCZ patients. Surprisingly, synaptic density in these animals is also reduced [335]. Unfortunately, the behavioral alterations observed in these animals recapitulate those associated with autism and not schizophrenia [336]. In addition, another major drawback of using this kind of model is the absence of the genetic component of SCZ, which does not allow the execution of studies aiming at analyzing the interaction between genetic and environmental risk factor.
The use of iPSCs derived from schizophrenic individuals could partially solve this issue, at least for studies attempting to investigate major changes at the molecular and cellular levels. Indeed, Brennand and colleagues [337] have found that iPSC-differentiated neurons derived from SCZ patients exhibit reduced neuronal connectivity and neurite numbers and decreased expression of PSD95 and glutamate ionotropic receptor kainate 1 expression. Additionally, Sellgren et al [338] have shown that microglial-like cells derived from schizophrenic individuals display a higher phagocytic index of synaptosomes obtained from hiPSC-derived neuronal preparations. These data indicate increased synaptic engulfment capacity by microglia in SCZ, which could underpin the elevated synapse loss observed in this condition. Interestingly, this higher phagocytic capacity of SCZ microglial-cells is inhibited by minocycline treatment, a drug recently suggested for schizophrenia treatment [338, 339].
CONCLUSION
In recent years, several lines of evidence have indicated a prominent role of synaptic elimination as a key component in modulating neuroplasticity in the CNS. Nonetheless, disturbances in this process are likely underpinning the cognitive deficits often observed in many of the most relevant neurological disorders afflicting humankind. In this review, we summarized the most up-to-date information available in the scientific literature, regarding what is currently known about synaptic pruning in AD, MS and SCZ, three of the most relevant neurological disorders. Nevertheless, there is still much to be learned about this process, especially concerning the involvement of immune molecules and glial cells in mediating synapse maintenance/elimination in these disorders.
ACKNOWLEDGEMENTS
P.L.C., I.B.Q.L, E.M.A.M., N.C.S., T.D. and F.M.R. were responsible for the manuscript writing, proofreading and editing.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by CNPq (grant number 429049/2018-8) and FAPEMIG (grant number PPM-00212-18) grants to F.M.R and by the grant from the Ministry of Education, Science, Research and Sport of the Slovak Republic to T.D., DB Biotech, Slovakia. P.L.C. and I.B.Q.L. are recipients of CAPES PhD and Masters scholarships, respectively.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Riccomagno M.M., Kolodkin A.L. Sculpting neural circuits by axon and dendrite pruning. Annu. Rev. Cell Dev. Biol. 2015;31:779–805. doi: 10.1146/annurev-cellbio-100913-013038. [http://dx.doi.org/10.1146/annurev-cellbio-100913-013038]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lichtman J.W., Colman H. Synapse elimination and indelible memory. Neuron. 2000;25(2):269–278. doi: 10.1016/s0896-6273(00)80893-4. [http://dx.doi.org/10.1016/S0896-6273(00)80893-4]. [PMID: 10719884]. [DOI] [PubMed] [Google Scholar]
- 3.Kano M., Hashimoto K. Synapse elimination in the central nervous system. Curr. Opin. Neurobiol. 2009;19(2):154–161. doi: 10.1016/j.conb.2009.05.002. [http://dx.doi.org/10.1016/j.conb.2009.05.002]. [PMID: 19481442]. [DOI] [PubMed] [Google Scholar]
- 4.Yang G., Pan F., Gan W.B. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462(7275):920–924. doi: 10.1038/nature08577. [http://dx.doi.org/10.1038/nature08577]. [PMID: 19946265]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fukui Y., Bedi K.S. Quantitative study of the development of neurons and synapses in rats reared in the dark during early postnatal life. 1. Superior colliculus. J. Anat. 1991;174:49–60. [PMID: 2032942]. [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C., Regehr W.G. Developmental remodeling of the retinogeniculate synapse. Neuron. 2000;28(3):955–966. doi: 10.1016/s0896-6273(00)00166-5. [http://dx.doi.org/10.1016/S0896-6273(00)00166-5]. [PMID: 11163279]. [DOI] [PubMed] [Google Scholar]
- 7.Cragg B.G. The development of synapses in kitten visual cortex during visual deprivation. Exp. Neurol. 1975;46(3):445–451. doi: 10.1016/0014-4886(75)90118-1. [http://dx.doi.org/10.1016/0014-4886(75)90118-1]. [PMID: 1112285]. [DOI] [PubMed] [Google Scholar]
- 8.Shatz C.J., Stryker M.P. Ocular dominance in layer IV of the cat’s visual cortex and the effects of monocular deprivation. J. Physiol. 1978;281:267–283. doi: 10.1113/jphysiol.1978.sp012421. [http://dx.doi.org/10.1113/jphysiol.1978.sp012421]. [PMID: 702379]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shatz C.J., Stryker M.P. Prenatal tetrodotoxin infusion blocks segregation of retinogeniculate afferents. Science. 1988;242(4875):87–89. doi: 10.1126/science.3175636. [http://dx.doi.org/10.1126/science.3175636]. [PMID: 3175636]. [DOI] [PubMed] [Google Scholar]
- 10.Huttenlocher P.R., Dabholkar A.S. Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 1997;387(2):167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [http://dx.doi.org/10.1002/(SICI)1096-9861(19971020)387:2<167:AID-CNE1>3.0.CO;2-Z]. [PMID: 9336221]. [DOI] [PubMed] [Google Scholar]
- 11.Petanjek Z., Judaš M., Šimic G., Rasin M.R., Uylings H.B., Rakic P., Kostovic I. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc. Natl. Acad. Sci. USA. 2011;108(32):13281–13286. doi: 10.1073/pnas.1105108108. [http://dx.doi.org/10.1073/pnas.1105108108]. [PMID: 21788513]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoshiko M., Arnoux I., Avignone E., Yamamoto N., Audinat E. Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 2012;32(43):15106–15111. doi: 10.1523/JNEUROSCI.1167-12.2012. [http://dx.doi.org/10.1523/JNEUROSCI.1167-12.2012]. [PMID: 23100431]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nägerl U.V., Eberhorn N., Cambridge S.B., Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44(5):759–767. doi: 10.1016/j.neuron.2004.11.016. [http://dx.doi.org/10.1016/j.neuron.2004.11.016]. [PMID: 15572108]. [DOI] [PubMed] [Google Scholar]
- 14.Yildirim M., Mapp O.M., Janssen W.G., Yin W., Morrison J.H., Gore A.C. Postpubertal decrease in hippocampal dendritic spines of female rats. Exp. Neurol. 2008;210(2):339–348. doi: 10.1016/j.expneurol.2007.11.003. [http://dx.doi.org/10.1016/j.expneurol.2007.11.003]. [PMID: 18096161]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koss W.A., Belden C.E., Hristov A.D., Juraska J.M. Dendritic remodeling in the adolescent medial prefrontal cortex and the basolateral amygdala of male and female rats. Synapse. 2014;68(2):61–72. doi: 10.1002/syn.21716. [http://dx.doi.org/10.1002/syn.21716]. [PMID: 24105875]. [DOI] [PubMed] [Google Scholar]
- 16.Huttenlocher P.R. Synaptic density in human frontal cortex - developmental changes and effects of aging. Brain Res. 1979;163(2):195–205. doi: 10.1016/0006-8993(79)90349-4. [http://dx.doi.org/10.1016/0006-8993(79)90349-4]. [PMID: 427544]. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto K., Kano M. Synapse elimination in the devel-oping cerebellum. Cellular and molecular life sciences. Cell. Mol. Life Sci. 2013;70(24):4667–4680. doi: 10.1007/s00018-013-1405-2. [http://dx.doi.org/10.1007/s00018-013-1405-2]. [PMID: 23811844]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elston G.N., Oga T., Fujita I. Spinogenesis and pruning scales across functional hierarchies. J. Neurosci. 2009;29(10):3271–3275. doi: 10.1523/JNEUROSCI.5216-08.2009. [http://dx.doi.org/10.1523/JNEUROSCI.5216-08.2009]. [PMID: 19279264]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevens B., Allen N.J., Vazquez L.E., Howell G.R., Christopherson K.S., Nouri N., Micheva K.D., Mehalow A.K., Huberman A.D., Stafford B., Sher A., Litke A.M., Lambris J.D., Smith S.J., John S.W., Barres B.A. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [http://dx.doi.org/10.1016/j.cell.2007.10.036]. [PMID: 18083105]. [DOI] [PubMed] [Google Scholar]
- 20.Bialas A.R., Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 2013;16(12):1773–1782. doi: 10.1038/nn.3560. [http://dx.doi.org/10.1038/nn.3560]. [PMID: 24162655]. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Tremblay M.E., Lowery R.L., Majewska A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8(11):e1000527. doi: 10.1371/journal.pbio.1000527. [http://dx.doi.org/10.1371/journal.pbio.1000527]. [PMID: 21072242]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nimmerjahn A., Kirchhoff F., Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [http://dx.doi.org/10.1126/science.1110647]. [PMID: 15831717]. [DOI] [PubMed] [Google Scholar]
- 23.Coleman J.E., Nahmani M., Gavornik J.P., Haslinger R., Heynen A.J., Erisir A., Bear M.F. Rapid structural remodeling of thalamocortical synapses parallels experience-dependent functional plasticity in mouse primary visual cortex. J. Neurosci. 2010;30(29):9670–9682. doi: 10.1523/JNEUROSCI.1248-10.2010. [http://dx.doi.org/10.1523/JNEUROSCI.1248-10.2010]. [PMID: 20660250]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Q., Homma K.J., Poo M.M. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44(5):749–757. doi: 10.1016/j.neuron.2004.11.011. [http://dx.doi.org/10.1016/j.neuron.2004.11.011]. [PMID: 15572107]. [DOI] [PubMed] [Google Scholar]
- 25.De Roo M., Klauser P., Muller D. LTP promotes a selective long-term stabilization and clustering of dendritic spines. PLoS Biol. 2008;6(9):e219. doi: 10.1371/journal.pbio.0060219. [http://dx.doi.org/10.1371/journal.pbio.0060219]. [PMID: 18788894]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bastrikova N., Gardner G.A., Reece J.M., Jeromin A., Dudek S.M. Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc. Natl. Acad. Sci. USA. 2008;105(8):3123–3127. doi: 10.1073/pnas.0800027105. [http://dx.doi.org/10.1073/pnas.0800027105]. [PMID: 18287055]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becker N., Wierenga C.J., Fonseca R., Bonhoeffer T., Nägerl U.V. LTD induction causes morphological changes of presynaptic boutons and reduces their contacts with spines. Neuron. 2008;60(4):590–597. doi: 10.1016/j.neuron.2008.09.018. [http://dx.doi.org/10.1016/j.neuron.2008.09.018]. [PMID: 19038217]. [DOI] [PubMed] [Google Scholar]
- 28.Gladding C.M., Fitzjohn S.M., Molnár E. Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacol. Rev. 2009;61(4):395–412. doi: 10.1124/pr.109.001735. [http://dx.doi.org/10.1124/pr.109.001735]. [PMID: 19926678]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He K., Lee A., Song L., Kanold P.O., Lee H.K. AMPA receptor subunit GluR1 (GluA1) serine-845 site is involved in synaptic depression but not in spine shrinkage associated with chemical long-term depression. J. Neurophysiol. 2011;105(4):1897–1907. doi: 10.1152/jn.00913.2010. [http://dx.doi.org/10.1152/jn.00913.2010]. [PMID: 21307330]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brigman J.L., Wright T., Talani G., Prasad-Mulcare S., Jinde S., Seabold G.K., Mathur P., Davis M.I., Bock R., Gustin R.M., Colbran R.J., Alvarez V.A., Nakazawa K., Delpire E., Lovinger D.M., Holmes A. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J. Neurosci. 2010;30(13):4590–4600. doi: 10.1523/JNEUROSCI.0640-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X.B., Yang Y., Zhou Q. Independent expression of synaptic and morphological plasticity associated with long-term depression. J. Neurosci. 2007;27(45):12419–12429. doi: 10.1523/JNEUROSCI.2015-07.2007. [http://dx.doi.org/10.1523/JNEUROSCI.2015-07.2007]. [PMID: 17989307]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henson M.A., Tucker C.J., Zhao M., Dudek S.M. Long-term depression-associated signaling is required for an in vitro model of NMDA receptor-dependent synapse pruning. Neurobiol. Learn. Mem. 2017;138:39–53. doi: 10.1016/j.nlm.2016.10.013. [http://dx.doi.org/10.1016/j.nlm.2016.10.013]. [PMID: 27794462]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kano M., Hashimoto K., Kurihara H., Watanabe M., Inoue Y., Aiba A., Tonegawa S. Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron. 1997;18(1):71–79. doi: 10.1016/s0896-6273(01)80047-7. [http://dx.doi.org/10.1016/S0896-6273(01)80047-7]. [PMID: 9010206]. [DOI] [PubMed] [Google Scholar]
- 34.Ichise T., Kano M., Hashimoto K., Yanagihara D., Nakao K., Shigemoto R., Katsuki M., Aiba A. mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science. 2000;288(5472):1832–1835. doi: 10.1126/science.288.5472.1832. [http://dx.doi.org/10.1126/science.288.5472.1832]. [PMID: 10846166]. [DOI] [PubMed] [Google Scholar]
- 35.Huber K.M., Kayser M.S., Bear M.F. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288(5469):1254–1257. doi: 10.1126/science.288.5469.1254. [http://dx.doi.org/10.1126/science.288.5469.1254]. [PMID: 10818003]. [DOI] [PubMed] [Google Scholar]
- 36.Narushima M., Uchigashima M., Yagasaki Y., Harada T., Nagumo Y., Uesaka N., Hashimoto K., Aiba A., Watanabe M., Miyata M., Kano M. The Metabotropic glutamate receptor subtype 1 Mediates experience-dependent maintenance of mature synaptic connectivity in the visual thalamus. Neuron. 2016;91(5):1097–1109. doi: 10.1016/j.neuron.2016.07.035. [http://dx.doi.org/10.1016/j.neuron.2016.07.035]. [PMID: 27545713]. [DOI] [PubMed] [Google Scholar]
- 37.Chen C.C., Lu H.C., Brumberg J.C. mGluR5 knockout mice display increased dendritic spine densities. Neurosci. Lett. 2012;524(1):65–68. doi: 10.1016/j.neulet.2012.07.014. [http://dx.doi.org/10.1016/j.neulet.2012.07.014]. [PMID: 22819970]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shinoda Y., Tanaka T., Tominaga-Yoshino K., Ogura A. Persistent synapse loss induced by repetitive LTD in developing rat hippocampal neurons. PLoS One. 2010;5(4):e10390. doi: 10.1371/journal.pone.0010390. [http://dx.doi.org/10.1371/journal.pone.0010390]. [PMID: 20436928]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shinoda Y., Kamikubo Y., Egashira Y., Tominaga-Yoshino K., Ogura A. Repetition of mGluR-dependent LTD causes slowly developing persistent reduction in synaptic strength accompanied by synapse elimination. Brain Res. 2005;1042(1):99–107. doi: 10.1016/j.brainres.2005.02.028. [http://dx.doi.org/10.1016/j.brainres.2005.02.028]. [PMID: 15823258]. [DOI] [PubMed] [Google Scholar]
- 40.Wiegert J.S., Oertner T.G. Long-term depression triggers the selective elimination of weakly integrated synapses. Proc. Natl. Acad. Sci. USA. 2013;110(47):E4510–E4519. doi: 10.1073/pnas.1315926110. [http://dx.doi.org/10.1073/pnas.1315926110]. [PMID: 24191047]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oh W.C., Hill T.C., Zito K. Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc. Natl. Acad. Sci. USA. 2013;110(4):E305–E312. doi: 10.1073/pnas.1214705110. [http://dx.doi.org/10.1073/pnas.1214705110]. [PMID: 23269840]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uesaka N., Uchigashima M., Mikuni T., Nakazawa T., Nakao H., Hirai H., Aiba A., Watanabe M., Kano M. Retrograde semaphorin signaling regulates synapse elimination in the developing mouse brain. Science. 2014;344(6187):1020–1023. doi: 10.1126/science.1252514. [http://dx.doi.org/10.1126/science.1252514]. [PMID: 24831527]. [DOI] [PubMed] [Google Scholar]
- 43.O’Connor T.P., Cockburn K., Wang W., Tapia L., Currie E., Bamji S.X. Semaphorin 5B mediates synapse elimination in hippocampal neurons. Neural Dev. 2009;4:18. doi: 10.1186/1749-8104-4-18. [http://dx.doi.org/10.1186/1749-8104-4-18]. [PMID: 19463192]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakayama H., Abe M., Morimoto C., Iida T., Okabe S., Sakimura K., Hashimoto K. Microglia permit climbing fiber elimination by promoting GABAergic inhibition in the developing cerebellum. Nat. Commun. 2018;9(1):2830. doi: 10.1038/s41467-018-05100-z. [http://dx.doi.org/10.1038/s41467-018-05100-z]. [PMID: 30026565]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Afroz S., Parato J., Shen H., Smith S.S. Synaptic pruning in the female hippocampus is triggered at puberty by extrasynaptic GABAA receptors on dendritic spines. eLife. 2016;5:e15106. doi: 10.7554/eLife.15106. [http://dx.doi.org/10.7554/eLife.15106]. [PMID: 27136678]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tao W., Díaz-Alonso J., Sheng N., Nicoll R.A. Postsynaptic δ1 glutamate receptor assembles and maintains hippocampal synapses via Cbln2 and neurexin. Proc. Natl. Acad. Sci. USA. 2018;115(23):E5373–E5381. doi: 10.1073/pnas.1802737115. [http://dx.doi.org/10.1073/pnas.1802737115]. [PMID: 29784783]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uemura T., Lee S.J., Yasumura M., Takeuchi T., Yoshida T., Ra M., Taguchi R., Sakimura K., Mishina M. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141(6):1068–1079. doi: 10.1016/j.cell.2010.04.035. [http://dx.doi.org/10.1016/j.cell.2010.04.035]. [PMID: 20537373]. [DOI] [PubMed] [Google Scholar]
- 48.Bian W.J., Miao W.Y., He S.J., Qiu Z., Yu X. Coordinated spine pruning and maturation mediated by inter-spine competition for cadherin/catenin complexes. Cell. 2015;162(4):808–822. doi: 10.1016/j.cell.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 49.Li M. Y., Miao W. Y., Wu Q. Z., He S. J., Yan G., Yang Y., Liu J. J., Taketo M. M., Yu X. A critical role of presynaptic cadherin/ catenin/p140Cap complexes in stabilizing spines and functional synapses in the neocortex. Neuron. 2017;94(6):1155–1172. doi: 10.1016/j.neuron.2017.05.022. e8. [DOI] [PubMed] [Google Scholar]
- 50.Huh G.S., Boulanger L.M., Du H., Riquelme P.A., Brotz T.M., Shatz C.J. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290(5499):2155–2159. doi: 10.1126/science.290.5499.2155. [http://dx.doi.org/10.1126/science.290.5499.2155]. [PMID: 11118151]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goddard C.A., Butts D.A., Shatz C.J. Regulation of CNS synapses by neuronal MHC class I. Proc. Natl. Acad. Sci. USA. 2007;104(16):6828–6833. doi: 10.1073/pnas.0702023104. [http://dx.doi.org/10.1073/pnas.0702023104]. [PMID: 17420446]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee H., Brott B.K., Kirkby L.A., Adelson J.D., Cheng S., Feller M.B., Datwani A., Shatz C.J. Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature. 2014;509(7499):195–200. doi: 10.1038/nature13154. [http://dx.doi.org/10.1038/nature13154]. [PMID: 24695230]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adelson J.D., Sapp R.W., Brott B.K., Lee H., Miyamichi K., Luo L., Cheng S., Djurisic M., Shatz C.J. Developmental sculpting of intracortical circuits by MHC class I H2-Db and H2-Kb. Cereb. Cortex. 2016;26(4):1453–1463. doi: 10.1093/cercor/bhu243. [http://dx.doi.org/10.1093/cercor/bhu243]. [PMID: 25316337]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Datwani A., McConnell M.J., Kanold P.O., Micheva K.D., Busse B., Shamloo M., Smith S.J., Shatz C.J. Classical MHCI molecules regulate retinogeniculate refinement and limit ocular dominance plasticity. Neuron. 2009;64(4):463–470. doi: 10.1016/j.neuron.2009.10.015. [http://dx.doi.org/10.1016/j.neuron.2009.10.015]. [PMID: 19945389]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vidal G. S., Djurisic M., Brown K., Sapp R. W., Shatz C. J. Developmental sculpting of intracortical circuits by MHC class I H2-Db and H2-Kb. Cereb. Cortex. 26(4):1453–1463. doi: 10.1093/cercor/bhu243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Syken J., Grandpre T., Kanold P.O., Shatz C.J. PirB restricts ocular-dominance plasticity in visual cortex. Science. 2006;313(5794):1795–1800. doi: 10.1126/science.1128232. [http://dx.doi.org/10.1126/science.1128232]. [PMID: 16917027]. [DOI] [PubMed] [Google Scholar]
- 57.Bochner D.N., Sapp R.W., Adelson J.D., Zhang S., Lee H., Djurisic M., Syken J., Dan Y., Shatz C.J. Blocking PirB up-regulates spines and functional synapses to unlock visual cortical plasticity and facilitate recovery from amblyopia. Sci. Transl. Med. 2014;6(258):258ra140. doi: 10.1126/scitranslmed.3010157. [http://dx.doi.org/10.1126/scitranslmed.3010157]. [PMID: 25320232]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Djurisic M., Brott B.K., Saw N.L., Shamloo M., Shatz C.J. Activity-dependent modulation of hippocampal synaptic plasticity via PirB and endocannabinoids. Mol. Psychiatry. 2018;24(8):1206–1219. doi: 10.1038/s41380-018-0034-4. [PMID: 29670176]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.VanGuilder Starkey H.D., Van Kirk C.A., Bixler G.V., Imperio C.G., Kale V.P., Serfass J.M., Farley J.A., Yan H., Warrington J.P., Han S., Mitschelen M., Sonntag W.E., Freeman W.M. Neuroglial expression of the MHCI pathway and PirB receptor is upregulated in the hippocampus with advanced aging. J. Mol. Neurosci. 2012;48(1):111–126. doi: 10.1007/s12031-012-9783-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stephan A.H., Madison D.V., Mateos J.M., Fraser D.A., Lovelett E.A., Coutellier L., Kim L., Tsai H.H., Huang E.J., Rowitch D.H., Berns D.S., Tenner A.J., Shamloo M., Barres B.A. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 2013;33(33):13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013. [http://dx.doi.org/10.1523/JNEUROSCI.1333-13.2013]. [PMID: 23946404]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schafer D.P., Lehrman E.K., Kautzman A.G., Koyama R., Mardinly A.R., Yamasaki R., Ransohoff R.M., Greenberg M.E., Barres B.A., Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [http://dx.doi.org/10.1016/j.neuron.2012.03.026]. [PMID: 22632727]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang J., Malik A., Choi H.B., Ko R.W., Dissing-Olesen L., MacVicar B.A. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron. 2014;82(1):195–207. doi: 10.1016/j.neuron.2014.01.043. [http://dx.doi.org/10.1016/j.neuron.2014.01.043]. [PMID: 24631344]. [DOI] [PubMed] [Google Scholar]
- 63.Ertürk A., Wang Y., Sheng M. Local pruning of dendrites and spines by caspase-3-dependent and proteasome-limited mechanisms. J. Neurosci. 2014;34(5):1672–1688. doi: 10.1523/JNEUROSCI.3121-13.2014. [http://dx.doi.org/10.1523/JNEUROSCI.3121-13.2014]. [PMID: 24478350]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Györffy B.A., Kun J., Török G., Bulyáki É., Borhegyi Z., Gulyássy P., Kis V., Szocsics P., Micsonai A., Matkó J., Drahos L., Juhász G., Kékesi K.A., Kardos J. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc. Natl. Acad. Sci. USA. 2018;115(24):6303–6308. doi: 10.1073/pnas.1722613115. [http://dx.doi.org/10.1073/pnas.1722613115]. [PMID: 29844190]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fraser D.A., Pisalyaput K., Tenner A.J. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J. Neurochem. 2010;112(3):733–743. doi: 10.1111/j.1471-4159.2009.06494.x. [http://dx.doi.org/10.1111/j.1471-4159.2009.06494.x]. [PMID: 19919576]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meng L., Mulcahy B., Cook S.J., Neubauer M., Wan A., Jin Y., Yan D. The cell death pathway regulates synapse elimination through cleavage of gelsolin in caenorhabditis elegans neurons. Cell Rep. 2015;11(11):1737–1748. doi: 10.1016/j.celrep.2015.05.031. [http://dx.doi.org/10.1016/j.celrep.2015.05.031]. [PMID: 26074078]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paolicelli R.C., Bolasco G., Pagani F., Maggi L., Scianni M., Panzanelli P., Giustetto M., Ferreira T.A., Guiducci E., Dumas L., Ragozzino D., Gross C.T. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [http://dx.doi.org/10.1126/science.1202529]. [PMID: 21778362]. [DOI] [PubMed] [Google Scholar]
- 68.Zhan Y., Paolicelli R.C., Sforazzini F., Weinhard L., Bolasco G., Pagani F., Vyssotski A.L., Bifone A., Gozzi A., Ragozzino D., Gross C.T. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 2014;17(3):400–406. doi: 10.1038/nn.3641. [http://dx.doi.org/10.1038/nn.3641]. [PMID: 24487234]. [DOI] [PubMed] [Google Scholar]
- 69.Sokolowski J.D., Chabanon-Hicks C.N., Han C.Z., Heffron D.S., Mandell J.W. Fractalkine is a “find-me” signal released by neurons undergoing ethanol-induced apoptosis. Front. Cell. Neurosci. 2014;8:360. doi: 10.3389/fncel.2014.00360. [http://dx.doi.org/10.3389/fncel.2014.00360]. [PMID: 25426022]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lui H., Zhang J., Makinson S.R., Cahill M.K., Kelley K.W., Huang H.Y., Shang Y., Oldham M.C., Martens L.H., Gao F., Coppola G., Sloan S.A., Hsieh C.L., Kim C.C., Bigio E.H., Weintraub S., Mesulam M.M., Rademakers R., Mackenzie I.R., Seeley W.W., Karydas A., Miller B.L., Borroni B., Ghidoni R., Farese R.V., Jr, Paz J.T., Barres B.A., Huang E.J. Progranulin deficiency promotes circuit-Specific synaptic pruning by microglia via complement activation. Cell. 2016;165(4):921–935. doi: 10.1016/j.cell.2016.04.001. [http://dx.doi.org/10.1016/j.cell.2016.04.001]. [PMID: 27114033]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang M.C., Srinivasan K., Friedman B.A., Suto E., Modrusan Z., Lee W.P., Kaminker J.S., Hansen D.V., Sheng M. Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J. Exp. Med. 2017;214(9):2611–2628. doi: 10.1084/jem.20160999. [http://dx.doi.org/10.1084/jem.20160999]. [PMID: 28778989]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang K., Li Y.J., Guo Y., Zheng K.Y., Yang Q., Yang L., Wang X.S., Song Q., Chen T., Zhuo M., Zhao M.G. Elevated progranulin contributes to synaptic and learning deficit due to loss of fragile X mental retardation protein. Brain. 2017;140(12):3215–3232. doi: 10.1093/brain/awx265. [http://dx.doi.org/10.1093/brain/awx265]. [PMID: 29096020]. [DOI] [PubMed] [Google Scholar]
- 73.Kim H.J., Cho M.H., Shim W.H., Kim J.K., Jeon E.Y., Kim D.H., Yoon S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry. 2017;22(11):1576–1584. doi: 10.1038/mp.2016.103. [http://dx.doi.org/10.1038/mp.2016.103]. [PMID: 27400854]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vainchtein I.D., Chin G., Cho F.S., Kelley K.W., Miller J.G., Chien E.C., Liddelow S.A., Nguyen P.T., Nakao-Inoue H., Dorman L.C., Akil O., Joshita S., Barres B.A., Paz J.T., Molofsky A.B., Molofsky A.V. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359(6381):1269–1273. doi: 10.1126/science.aal3589. [http://dx.doi.org/10.1126/science.aal3589]. [PMID: 29420261]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parkhurst C.N., Yang G., Ninan I., Savas J.N., Yates J.R., III, Lafaille J.J., Hempstead B.L., Littman D.R., Gan W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155(7):1596–1609. doi: 10.1016/j.cell.2013.11.030. [http://dx.doi.org/10.1016/j.cell.2013.11.030]. [PMID: 24360280]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lehrman E. K., Wilton D. K., Litvina E. Y., Welsh C. A., Chang S. T., Frouin A., Walker A. J., Heller M. D., Umemori H., Chen C., Stevens B. CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Develop-ment. Neuron, 2018;100(1):120–134. doi: 10.1016/j.neuron.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bjartmar L., Huberman A.D., Ullian E.M., Rentería R.C., Liu X., Xu W., Prezioso J., Susman M.W., Stellwagen D., Stokes C.C., Cho R., Worley P., Malenka R.C., Ball S., Peachey N.S., Copenhagen D., Chapman B., Nakamoto M., Barres B.A., Perin M.S. Neuronal pentraxins mediate synaptic refinement in the developing visual system. J. Neurosci. 2006;26(23):6269–6281. doi: 10.1523/JNEUROSCI.4212-05.2006. [http://dx.doi.org/10.1523/JNEUROSCI.4212-05.2006]. [PMID: 16763034]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koch S.M., Ullian E.M. Neuronal pentraxins mediate silent synapse conversion in the developing visual system. J. Neurosci. 2010;30(15):5404–5414. doi: 10.1523/JNEUROSCI.4893-09.2010. [http://dx.doi.org/10.1523/JNEUROSCI.4893-09.2010]. [PMID: 20392962]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martinelli D.C., Chew K.S., Rohlmann A., Lum M.Y., Ressl S., Hattar S., Brunger A.T., Missler M., Südhof T.C. Expression of C1ql3 in discrete neuronal populations controls efferent synapse numbers and diverse behaviors. Neuron. 2016;91(5):1034–1051. doi: 10.1016/j.neuron.2016.07.002. [http://dx.doi.org/10.1016/j.neuron.2016.07.002]. [PMID: 27478018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bolliger M.F., Martinelli D.C., Südhof T.C. The cell-adhesion G protein-coupled receptor BAI3 is a high-affinity receptor for C1q-like proteins. Proc. Natl. Acad. Sci. USA. 2011;108(6):2534–2539. doi: 10.1073/pnas.1019577108. [http://dx.doi.org/10.1073/pnas.1019577108]. [PMID: 21262840]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen P.B., Kawaguchi R., Blum C., Achiro J.M., Coppola G., O’Dell T.J., Martin K.C. Mapping Gene Expression in Excitatory Neurons during Hippocampal Late-Phase Long-Term Potentiation. Front. Mol. Neurosci. 2017;10:39. doi: 10.3389/fnmol.2017.00039. [http://dx.doi.org/10.3389/fnmol.2017.00039]. [PMID: 28275336]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weinhard L., di Bartolomei G., Bolasco G., Machado P., Schieber N.L., Neniskyte U., Exiga M., Vadisiute A., Raggioli A., Schertel A., Schwab Y., Gross C.T. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018;9(1):1228. doi: 10.1038/s41467-018-03566-5. [http://dx.doi.org/10.1038/s41467-018-03566-5]. [PMID: 29581545]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miyamoto A., Wake H., Ishikawa A.W., Eto K., Shibata K., Murakoshi H., Koizumi S., Moorhouse A.J., Yoshimura Y., Nabekura J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016;7:12540. doi: 10.1038/ncomms12540. [http://dx.doi.org/10.1038/ncomms12540]. [PMID: 27558646]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shi Q., Colodner K.J., Matousek S.B., Merry K., Hong S., Kenison J.E., Frost J.L., Le K.X., Li S., Dodart J.C., Caldarone B.J., Stevens B., Lemere C.A. Complement C3-Deficient mice fail to display age-related hippocampal decline. J. Neurosci. 2015;35(38):13029–13042. doi: 10.1523/JNEUROSCI.1698-15.2015. [http://dx.doi.org/10.1523/JNEUROSCI.1698-15.2015]. [PMID: 26400934]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chung W.S., Clarke L.E., Wang G.X., Stafford B.K., Sher A., Chakraborty C., Joung J., Foo L.C., Thompson A., Chen C., Smith S.J., Barres B.A. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504(7480):394–400. doi: 10.1038/nature12776. [http://dx.doi.org/10.1038/nature12776]. [PMID: 24270812]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patterson C. World Alzheimer Report 2018; Alzheimer's Disease International (ADI): London, September. 2018, 2018:1–48. [Google Scholar]
- 87.S. Freel, K. Seeher, N. Chowdhary, S. Sivananthan, A. M. Pot., editors. Towards a dementia plan: a WHO guide. World Health Organization (WHO):; Geneva: 2018. (WHO), W. H. O. p. 78.http://apps.who.int/iris/handle/10665/272642(accessed January 6th, 2019) [Google Scholar]
- 88.Alzheimer A., Stelzmann R.A., Schnitzlein H.N., Murtagh F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde. Clin. Anat. 1995;8(6):429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 89.Colon E.J. The cerebral cortex in presenile dementia. A quantitative analysis. Acta Neuropathol. 1973;23(4):281–290. doi: 10.1007/BF00687457. [http://dx.doi.org/10.1007/BF00687457]. [PMID: 4718195]. [DOI] [PubMed] [Google Scholar]
- 90.Shefer V.F. Absolute number of neurons and thickness of the cerebral cortex during aging, senile and vascular dementia, and Pick’s and Alzheimer’s diseases. Neurosci. Behav. Physiol. 1973;6(4):319–324. doi: 10.1007/BF01182672. [http://dx.doi.org/10.1007/BF01182672]. [PMID: 4781784]. [DOI] [PubMed] [Google Scholar]
- 91.Najlerahim A., Bowen D.M. Regional weight loss of the cerebral cortex and some subcortical nuclei in senile dementia of the Alzheimer type. Acta Neuropathol. 1988;75(5):509–512. doi: 10.1007/BF00687139. [http://dx.doi.org/10.1007/BF00687139]. [PMID: 3376753]. [DOI] [PubMed] [Google Scholar]
- 92.Mann D.M. Alzheimer’s disease and Down’s syndrome. Histopathology. 1988;13(2):125–137. doi: 10.1111/j.1365-2559.1988.tb02018.x. [http://dx.doi.org/10.1111/j.1365-2559.1988.tb02018.x]. [PMID: 2971602]. [DOI] [PubMed] [Google Scholar]
- 93.Hubbard B.M., Anderson J.M. A quantitative study of cerebral atrophy in old age and senile dementia. J. Neurol. Sci. 1981;50(1):135–145. doi: 10.1016/0022-510x(81)90048-4. [http://dx.doi.org/10.1016/0022-510X(81)90048-4]. [PMID: 7229656]. [DOI] [PubMed] [Google Scholar]
- 94.Davies C.A., Mann D.M., Sumpter P.Q., Yates P.O. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J. Neurol. Sci. 1987;78(2):151–164. doi: 10.1016/0022-510x(87)90057-8. [http://dx.doi.org/10.1016/0022-510X(87)90057-8]. [PMID: 3572454]. [DOI] [PubMed] [Google Scholar]
- 95.Whitwell J.L., Przybelski S.A., Weigand S.D., Knopman D.S., Boeve B.F., Petersen R.C., Jack C.R. Jr 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer’s disease. Brain. 2007;130(Pt 7):1777–1786. doi: 10.1093/brain/awm112. [http://dx.doi.org/10.1093/brain/awm112]. [PMID: 17533169]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Spires T.L., Meyer-Luehmann M., Stern E.A., McLean P.J., Skoch J., Nguyen P.T., Bacskai B.J., Hyman B.T. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 2005;25(31):7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [http://dx.doi.org/10.1523/JNEUROSCI.1879-05.2005]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Glenner G.G., Wong C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120(3):885–890. doi: 10.1016/s0006-291x(84)80190-4. [http://dx.doi.org/10.1016/S0006-291X(84)80190-4]. [PMID: 6375662]. [DOI] [PubMed] [Google Scholar]
- 98.Masters C.L., Multhaup G., Simms G., Pottgiesser J., Martins R.N., Beyreuther K. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985;4(11):2757–2763. doi: 10.1002/j.1460-2075.1985.tb04000.x. [http://dx.doi.org/10.1002/j.1460-2075.1985.tb04000.x]. [PMID: 4065091]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Masters C.L., Simms G., Weinman N.A., Multhaup G., McDonald B.L., Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA. 1985;82(12):4245–4249. doi: 10.1073/pnas.82.12.4245. [http://dx.doi.org/10.1073/pnas.82.12.4245]. [PMID: 3159021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knafo S., Alonso-Nanclares L., Gonzalez-Soriano J., Merino-Serrais P., Fernaud-Espinosa I., Ferrer I., DeFelipe J. Widespread changes in dendritic spines in a model of Alzheimer’s disease. Cereb. Cortex. 2009;19(3):586–592. doi: 10.1093/cercor/bhn111. [http://dx.doi.org/10.1093/cercor/bhn111]. [PMID: 18632740]. [DOI] [PubMed] [Google Scholar]
- 101.Yoshiyama Y., Higuchi M., Zhang B., Huang S.M., Iwata N., Saido T.C., Maeda J., Suhara T., Trojanowski J.Q., Lee V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351. doi: 10.1016/j.neuron.2007.01.010. [http://dx.doi.org/10.1016/j.neuron.2007.01.010]. [PMID: 17270732]. [DOI] [PubMed] [Google Scholar]
- 102.Kandimalla R., Manczak M., Yin X., Wang R., Reddy P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018;27(1):30–40. doi: 10.1093/hmg/ddx381. [http://dx.doi.org/10.1093/hmg/ddx381]. [PMID: 29040533]. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 103.Hamelin L., Lagarde J., Dorothée G., Potier M.C., Corlier F., Kuhnast B., Caillé F., Dubois B., Fillon L., Chupin M., Bottlaender M., Sarazin M. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer’s disease. Brain. 2018;141(6):1855–1870. doi: 10.1093/brain/awy079. [http://dx.doi.org/10.1093/brain/awy079]. [PMID: 29608645]. [DOI] [PubMed] [Google Scholar]
- 104.Dani M., Wood M., Mizoguchi R., Fan Z., Walker Z., Morgan R., Hinz R., Biju M., Kuruvilla T., Brooks D.J., Edison P. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain. 2018;141(9):2740–2754. doi: 10.1093/brain/awy188. [http://dx.doi.org/10.1093/brain/awy188]. [PMID: 30052812]. [DOI] [PubMed] [Google Scholar]
- 105.Xiang Z., Haroutunian V., Ho L., Purohit D., Pasinetti G.M. Microglia activation in the brain as inflammatory biomarker of Alzheimer’s disease neuropathology and clinical dementia. Dis. Markers. 2006;22(1-2):95–102. doi: 10.1155/2006/276239. [http://dx.doi.org/10.1155/2006/276239]. [PMID: 16410654]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kang J., Lemaire H.G., Unterbeck A., Salbaum J.M., Masters C.L., Grzeschik K.H., Multhaup G., Beyreuther K., Müller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325(6106):733–736. doi: 10.1038/325733a0. [http://dx.doi.org/10.1038/325733a0]. [PMID: 2881207]. [DOI] [PubMed] [Google Scholar]
- 107.Shoji M., Golde T.E., Ghiso J., Cheung T.T., Estus S., Shaffer L.M., Cai X.D., McKay D.M., Tintner R., Frangione B. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258(5079):126–129. doi: 10.1126/science.1439760. [http://dx.doi.org/10.1126/science.1439760]. [PMID: 1439760]. [DOI] [PubMed] [Google Scholar]
- 108.Kimberly W.T., Esler W.P., Ye W., Ostaszewski B.L., Gao J., Diehl T., Selkoe D.J., Wolfe M.S. Notch and the amyloid precursor protein are cleaved by similar gamma-secretase(s). Biochemistry. 2003;42(1):137–144. doi: 10.1021/bi026888g. [http://dx.doi.org/10.1021/bi026888g]. [PMID: 12515548]. [DOI] [PubMed] [Google Scholar]
- 109.Kimberly W.T., Wolfe M.S. Identity and function of gamma-secretase. J. Neurosci. Res. 2003;74(3):353–360. doi: 10.1002/jnr.10736. [http://dx.doi.org/10.1002/jnr.10736]. [PMID: 14598311]. [DOI] [PubMed] [Google Scholar]
- 110.Vassar R., Bennett B.D., Babu-Khan S., Kahn S., Mendiaz E.A., Denis P., Teplow D.B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M.A., Biere A.L., Curran E., Burgess T., Louis J.C., Collins F., Treanor J., Rogers G., Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [http://dx.doi.org/10.1126/science.286.5440.735]. [PMID: 10531052]. [DOI] [PubMed] [Google Scholar]
- 111.Szaruga M., Veugelen S., Benurwar M., Lismont S., Sepulveda-Falla D., Lleo A., Ryan N.S., Lashley T., Fox N.C., Murayama S., Gijsen H., De Strooper B., Chávez-Gutiérrez L. Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. J. Exp. Med. 2015;212(12):2003–2013. doi: 10.1084/jem.20150892. [http://dx.doi.org/10.1084/jem.20150892]. [PMID: 26481686]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fu L., Sun Y., Guo Y., Chen Y., Yu B., Zhang H., Wu J. [Google Scholar]; Yu X., Kong W., Wu H. Comparison of neurotoxicity of different aggregated forms of Abeta40, Abeta42 and Abeta43 in cell cultures. Journal of peptide science. Eur. Peptide Soc. 2017;23(3):245–251. doi: 10.1002/psc.2975. [DOI] [PubMed] [Google Scholar]
- 113.Lue L.F., Kuo Y.M., Roher A.E., Brachova L., Shen Y., Sue L., Beach T., Kurth J.H., Rydel R.E., Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999;155(3):853–862. doi: 10.1016/s0002-9440(10)65184-x. [http://dx.doi.org/10.1016/S0002-9440(10)65184-X]. [PMID: 10487842]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tomiyama T., Matsuyama S., Iso H., Umeda T., Takuma H., Ohnishi K., Ishibashi K., Teraoka R., Sakama N., Yamashita T., Nishitsuji K., Ito K., Shimada H., Lambert M.P., Klein W.L., Mori H. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 2010;30(14):4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [http://dx.doi.org/10.1523/JNEUROSCI.5825-09.2010]. [PMID: 20371804]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lambert M.P., Barlow A.K., Chromy B.A., Edwards C., Freed R., Liosatos M., Morgan T.E., Rozovsky I., Trommer B., Viola K.L., Wals P., Zhang C., Finch C.E., Krafft G.A., Klein W.L. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 1998;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [http://dx.doi.org/10.1073/pnas.95.11.6448]. [PMID: 9600986]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shankar G.M., Bloodgood B.L., Townsend M., Walsh D.M., Selkoe D.J., Sabatini B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007;27(11):2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [http://dx.doi.org/10.1523/JNEUROSCI.4970-06.2007]. [PMID: 17360908]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wei W., Nguyen L.N., Kessels H.W., Hagiwara H., Sisodia S., Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010;13(2):190–196. doi: 10.1038/nn.2476. [http://dx.doi.org/10.1038/nn.2476]. [PMID: 20037574]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stéphan A., Laroche S., Davis S. Generation of aggregated beta-amyloid in the rat hippocampus impairs synaptic transmission and plasticity and causes memory deficits. J. Neurosci. 2001;21(15):5703–5714. doi: 10.1523/JNEUROSCI.21-15-05703.2001. [http://dx.doi.org/10.1523/JNEUROSCI.21-15-05703.2001]. [PMID: 11466442]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang H.W., Pasternak J.F., Kuo H., Ristic H., Lambert M.P., Chromy B., Viola K.L., Klein W.L., Stine W.B., Krafft G.A., Trommer B.L. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924(2):133–140. doi: 10.1016/s0006-8993(01)03058-x. [http://dx.doi.org/10.1016/S0006-8993(01)03058-X]. [PMID: 11750898]. [DOI] [PubMed] [Google Scholar]
- 120.Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E., Smith I., Brett F.M., Farrell M.A., Rowan M.J., Lemere C.A., Regan C.M., Walsh D.M., Sabatini B.L., Selkoe D.J. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008;14(8):837–842. doi: 10.1038/nm1782. [http://dx.doi.org/10.1038/nm1782]. [PMID: 18568035]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang Z., Jackson R.J., Hong W., Taylor W.M., Corbett G.T., Moreno A., Liu W., Li S., Frosch M.P., Slutsky I., Young-Pearse T.L., Spires-Jones T.L., Walsh D.M. Human Brain-Derived Aβ Oligomers Bind to Synapses and Disrupt Synaptic Activity in a Manner That Requires APP. J. Neurosci. 2017;37(49):11947–11966. doi: 10.1523/JNEUROSCI.2009-17.2017. [http://dx.doi.org/10.1523/JNEUROSCI.2009-17.2017]. [PMID: 29101243]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen Q.S., Kagan B.L., Hirakura Y., Xie C.W. Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J. Neurosci. Res. 2000;60(1):65–72. doi: 10.1002/(SICI)1097-4547(20000401)60:1<65::AID-JNR7>3.0.CO;2-Q. [http://dx.doi.org/10.1002/(SICI)10974547(20000401)60:1<65:AID-JNR7>3.0.CO;2-Q]. [PMID: 10723069]. [DOI] [PubMed] [Google Scholar]
- 123.Walsh D.M., Klyubin I., Fadeeva J.V., Cullen W.K., Anwyl R., Wolfe M.S., Rowan M.J., Selkoe D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [http://dx.doi.org/10.1038/416535a]. [PMID: 11932745]. [DOI] [PubMed] [Google Scholar]
- 124.Li S., Hong S., Shepardson N.E., Walsh D.M., Shankar G.M., Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62(6):788–801. doi: 10.1016/j.neuron.2009.05.012. [http://dx.doi.org/10.1016/j.neuron.2009.05.012]. [PMID: 19555648]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hsieh H., Boehm J., Sato C., Iwatsubo T., Tomita T., Sisodia S., Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52(5):831–843. doi: 10.1016/j.neuron.2006.10.035. [http://dx.doi.org/10.1016/j.neuron.2006.10.035]. [PMID: 17145504]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Talantova M., Sanz-Blasco S., Zhang X., Xia P., Akhtar M.W., Okamoto S., Dziewczapolski G., Nakamura T., Cao G., Pratt A.E., Kang Y.J., Tu S., Molokanova E., McKercher S.R., Hires S.A., Sason H., Stouffer D.G., Buczynski M.W., Solomon J.P., Michael S., Powers E.T., Kelly J.W., Roberts A., Tong G., Fang-Newmeyer T., Parker J., Holland E.A., Zhang D., Nakanishi N., Chen H.S., Wolosker H., Wang Y., Parsons L.H., Ambasudhan R., Masliah E., Heinemann S.F., Piña-Crespo J.C., Lipton S.A. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA. 2013;110(27):E2518–E2527. doi: 10.1073/pnas.1306832110. [http://dx.doi.org/10.1073/pnas.1306832110]. [PMID: 23776240]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lei M., Xu H., Li Z., Wang Z., O’Malley T.T., Zhang D., Walsh D.M., Xu P., Selkoe D.J., Li S. Soluble Aβ oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis. 2016;85:111–121. doi: 10.1016/j.nbd.2015.10.019. [http://dx.doi.org/10.1016/j.nbd.2015.10.019]. [PMID: 26525100]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao J., Li A., Rajsombath M., Dang Y., Selkoe D. J., Li S. Soluble Abeta Oligomers Impair Dipolar Heterodendritic Plasticity by Activation of mGluR in the Hippocampal CA1 Region. iScience. 2018;6:138–150. doi: 10.1016/j.isci.2018.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Molokanova E., Akhtar M.W., Sanz-Blasco S., Tu S., Piña-Crespo J.C., McKercher S.R., Lipton S.A. Differential effects of synaptic and extrasynaptic NMDA receptors on Aβ-induced nitric oxide production in cerebrocortical neurons. J. Neurosci. 2014;34(14):5023–5028. doi: 10.1523/JNEUROSCI.2907-13.2014. [http://dx.doi.org/10.1523/JNEUROSCI.2907-13.2014]. [PMID: 24695719]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Qu J., Nakamura T., Cao G., Holland E.A., McKercher S.R., Lipton S.A. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc. Natl. Acad. Sci. USA. 2011;108(34):14330–14335. doi: 10.1073/pnas.1105172108. [http://dx.doi.org/10.1073/pnas.1105172108]. [PMID: 21844361]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cleary J.P., Walsh D.M., Hofmeister J.J., Shankar G.M., Kuskowski M.A., Selkoe D.J., Ashe K.H. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 2005;8(1):79–84. doi: 10.1038/nn1372. [http://dx.doi.org/10.1038/nn1372]. [PMID: 15608634]. [DOI] [PubMed] [Google Scholar]
- 132.Figueiredo C.P., Clarke J.R., Ledo J.H., Ribeiro F.C., Costa C.V., Melo H.M., Mota-Sales A.P., Saraiva L.M., Klein W.L., Sebollela A., De Felice F.G., Ferreira S.T. Memantine rescues transient cognitive impairment caused by high-molecular-weight aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 2013;33(23):9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013. [http://dx.doi.org/10.1523/JNEUROSCI.0482-13.2013]. [PMID: 23739959]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ledo J.H., Azevedo E.P., Clarke J.R., Ribeiro F.C., Figueiredo C.P., Foguel D., De Felice F.G., Ferreira S.T. Amyloid-β oligomers link depressive-like behavior and cognitive deficits in mice. Mol. Psychiatry. 2013;18(10):1053–1054. doi: 10.1038/mp.2012.168. [http://dx.doi.org/10.1038/mp.2012.168]. [PMID: 23183490]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Morroni F., Sita G., Tarozzi A., Rimondini R., Hrelia P. Early effects of Aβ1-42 oligomers injection in mice: Involvement of PI3K/Akt/GSK3 and MAPK/ERK1/2 pathways. Behav. Brain Res. 2016;314:106–115. doi: 10.1016/j.bbr.2016.08.002. [http://dx.doi.org/10.1016/j.bbr.2016.08.002]. [PMID: 27498145]. [DOI] [PubMed] [Google Scholar]
- 135.King R.D., Brown B., Hwang M., Jeon T., George A.T. Alzheimer’s Disease neuroimaging initiative. Fractal dimension analysis of the cortical ribbon in mild Alzheimer’s disease. Neuroimage. 2010;53(2):471–479. doi: 10.1016/j.neuroimage.2010.06.050. [http://dx.doi.org/10.1016/j.neuroimage.2010.06.050]. [PMID: 20600974]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Clare R., King V.G., Wirenfeldt M., Vinters H.V. Synapse loss in dementias. J. Neurosci. Res. 2010;88(10):2083–2090. doi: 10.1002/jnr.22392. [http://dx.doi.org/10.1002/jnr.22392]. [PMID: 20533377]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Scheff S.W., Price D.A., Schmitt F.A., Mufson E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging. 2006;27(10):1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [http://dx.doi.org/10.1016/j.neurobiolaging.2005.09.012]. [PMID: 16289476]. [DOI] [PubMed] [Google Scholar]
- 138.Scheff S.W., Price D.A., Schmitt F.A., DeKosky S.T., Mufson E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68(18):1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [http://dx.doi.org/10.1212/01.wnl.0000260698.46517.8f]. [PMID: 17470753]. [DOI] [PubMed] [Google Scholar]
- 139.Scheff S.W., Price D.A. Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol. 1993;33(2):190–199. doi: 10.1002/ana.410330209. [http://dx.doi.org/10.1002/ana.410330209]. [PMID: 8434881]. [DOI] [PubMed] [Google Scholar]
- 140.Scheff S.W., DeKosky S.T., Price D.A. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol. Aging. 1990;11(1):29–37. doi: 10.1016/0197-4580(90)90059-9. [http://dx.doi.org/10.1016/0197-4580(90)90059-9]. [PMID: 2325814]. [DOI] [PubMed] [Google Scholar]
- 141.Reddy P.H., Mani G., Park B.S., Jacques J., Murdoch G., Whetsell W., Jr, Kaye J., Manczak M. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J. Alzheimers Dis. 2005;7(2):103–117. doi: 10.3233/jad-2005-7203. [http://dx.doi.org/10.3233/JAD-2005-7203]. [PMID: 15851848]. [DOI] [PubMed] [Google Scholar]
- 142.Scheff S.W., Price D.A., Schmitt F.A., Roberts K.N., Ikonomovic M.D., Mufson E.J. Synapse stability in the precuneus early in the progression of Alzheimer’s disease. J. Alzheimers Dis. 2013;35(3):599–609. doi: 10.3233/JAD-122353. [http://dx.doi.org/10.3233/JAD-122353]. [PMID: 23478309]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Honer W.G. Pathology of presynaptic proteins in Alzheimer’s disease: more than simple loss of terminals. Neurobiol. Aging. 2003;24(8):1047–1062. doi: 10.1016/j.neurobiolaging.2003.04.005. [http://dx.doi.org/10.1016/j.neurobiolaging.2003.04.005]. [PMID: 14643376]. [DOI] [PubMed] [Google Scholar]
- 144.Masliah E., Mallory M., Alford M., DeTeresa R., Hansen L.A., McKeel D.W., Jr, Morris J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56(1):127–129. doi: 10.1212/wnl.56.1.127. [http://dx.doi.org/10.1212/WNL.56.1.127]. [PMID: 11148253]. [DOI] [PubMed] [Google Scholar]
- 145.Wakabayashi K., Honer W.G., Masliah E. Synapse alterations in the hippocampal-entorhinal formation in Alzheimer’s disease with and without Lewy body disease. Brain Res. 1994;667(1):24–32. doi: 10.1016/0006-8993(94)91709-4. [http://dx.doi.org/10.1016/0006-8993(94)91709-4]. [PMID: 7895080]. [DOI] [PubMed] [Google Scholar]
- 146.Marksteiner J., Kaufmann W.A., Gurka P., Humpel C. Synaptic proteins in Alzheimer’s disease. Journal of Molecular Neuroscience: MN. 2002;18(1-2):53–63. doi: 10.1385/JMN:18:1-2:53. [http://dx.doi.org/10.1385/JMN:18:1-2:53]. [DOI] [PubMed] [Google Scholar]
- 147.Almeida C.G., Tampellini D., Takahashi R.H., Greengard P., Lin M.T., Snyder E.M., Gouras G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005;20(2):187–198. doi: 10.1016/j.nbd.2005.02.008. [http://dx.doi.org/10.1016/j.nbd.2005.02.008]. [PMID: 16242627]. [DOI] [PubMed] [Google Scholar]
- 148.Canas P. M., Simoes A. P., Rodrigues R. J., Cunha R. A. Predominant loss of glutamatergic terminal markers in a beta-amyloid peptide model of Alzheimer's disease. Neuropharmacology, 2014;76(Pt A)):51–56. doi: 10.1016/j.neuropharm.2013.08.026. [DOI] [PubMed] [Google Scholar]
- 149.Berchtold N.C., Coleman P.D., Cribbs D.H., Rogers J., Gillen D.L., Cotman C.W. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol. Aging. 2013;34(6):1653–1661. doi: 10.1016/j.neurobiolaging.2012.11.024. [http://dx.doi.org/10.1016/j.neurobiolaging.2012.11.024]. [PMID: 23273601]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mitew S., Kirkcaldie M.T., Dickson T.C., Vickers J.C. Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging. 2013;34(10):2341–2351. doi: 10.1016/j.neurobiolaging.2013.04.010. [http://dx.doi.org/10.1016/j.neurobiolaging.2013.04.010]. [PMID: 23643146]. [DOI] [PubMed] [Google Scholar]
- 151.Roselli F., Tirard M., Lu J., Hutzler P., Lamberti P., Livrea P., Morabito M., Almeida O.F. Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J. Neurosci. 2005;25(48):11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005. [http://dx.doi.org/10.1523/JNEUROSCI.3034-05.2005]. [PMID: 16319306]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sze C.I., Bi H., Kleinschmidt-DeMasters B.K., Filley C.M., Martin L.J. Selective regional loss of exocytotic presynaptic vesicle proteins in Alzheimer’s disease brains. J. Neurol. Sci. 2000;175(2):81–90. doi: 10.1016/s0022-510x(00)00285-9. [http://dx.doi.org/10.1016/S0022-510X(00)00285-9]. [PMID: 10831767]. [DOI] [PubMed] [Google Scholar]
- 153.DeKosky S.T., Scheff S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 1990;27(5):457–464. doi: 10.1002/ana.410270502. [http://dx.doi.org/10.1002/ana.410270502]. [PMID: 2360787]. [DOI] [PubMed] [Google Scholar]
- 154.Terry R.D., Masliah E., Salmon D.P., Butters N., DeTeresa R., Hill R., Hansen L.A., Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [http://dx.doi.org/10.1002/ana.410300410]. [PMID: 1789684]. [DOI] [PubMed] [Google Scholar]
- 155.Chen M.K., Mecca A.P., Naganawa M., Finnema S.J., Toyonaga T., Lin S.F., Najafzadeh S., Ropchan J., Lu Y., McDonald J.W., Michalak H.R., Nabulsi N.B., Arnsten A.F.T., Huang Y., Carson R.E., van Dyck C.H. Assessing Synaptic Density in Alzheimer Disease With Synaptic Vesicle Glycoprotein 2A Positron Emission Tomographic Imaging. JAMA Neurol. 2018;75(10):1215–1224. doi: 10.1001/jamaneurol.2018.1836. [http://dx.doi.org/10.1001/jamaneurol.2018.1836]. [PMID: 30014145]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jackson R.J., Rudinskiy N., Herrmann A.G., Croft S., Kim J.M., Petrova V., Ramos-Rodriguez J.J., Pitstick R., Wegmann S., Garcia-Alloza M., Carlson G.A., Hyman B.T., Spires-Jones T.L. Human tau increases amyloid β plaque size but not amyloid β-mediated synapse loss in a novel mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2016;44(12):3056–3066. doi: 10.1111/ejn.13442. [http://dx.doi.org/10.1111/ejn.13442]. [PMID: 27748574]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Merino-Serrais P., Benavides-Piccione R., Blazquez-Llorca L., Kastanauskaite A., Rábano A., Avila J., DeFelipe J. The influence of phospho-τ on dendritic spines of cortical pyramidal neurons in patients with Alzheimer’s disease. Brain. 2013;136(Pt 6):1913–1928. doi: 10.1093/brain/awt088. [http://dx.doi.org/10.1093/brain/awt088]. [PMID: 23715095]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fein J.A., Sokolow S., Miller C.A., Vinters H.V., Yang F., Cole G.M., Gylys K.H. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am. J. Pathol. 2008;172(6):1683–1692. doi: 10.2353/ajpath.2008.070829. [http://dx.doi.org/10.2353/ajpath.2008.070829]. [PMID: 18467692]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hoover B.R., Reed M.N., Su J., Penrod R.D., Kotilinek L.A., Grant M.K., Pitstick R., Carlson G.A., Lanier L.M., Yuan L.L., Ashe K.H., Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68(6):1067–1081. doi: 10.1016/j.neuron.2010.11.030. [http://dx.doi.org/10.1016/j.neuron.2010.11.030]. [PMID: 21172610]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Takahashi R.H., Capetillo-Zarate E., Lin M.T., Milner T.A., Gouras G.K. Co-occurrence of Alzheimer’s disease ß-amyloid and τ pathologies at synapses. Neurobiol. Aging. 2010;31(7):1145–1152. doi: 10.1016/j.neurobiolaging.2008.07.021. [http://dx.doi.org/10.1016/j.neurobiolaging.2008.07.021]. [PMID: 18771816]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ittner L.M., Ke Y.D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B.C., Christie M.J., Napier I.A., Eckert A., Staufenbiel M., Hardeman E., Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142(3):387–397. doi: 10.1016/j.cell.2010.06.036. [http://dx.doi.org/10.1016/j.cell.2010.06.036]. [PMID: 20655099]. [DOI] [PubMed] [Google Scholar]
- 162.Lasagna-Reeves C.A., Castillo-Carranza D.L., Sengupta U., Clos A.L., Jackson G.R., Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [http://dx.doi.org/10.1186/1750-1326-6-39]. [PMID: 21645391]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shipton O.A., Leitz J.R., Dworzak J., Acton C.E., Tunbridge E.M., Denk F., Dawson H.N., Vitek M.P., Wade-Martins R., Paulsen O., Vargas-Caballero M. Tau protein is required for amyloid beta-induced impairment of hippocampal long-term potentiation. J. Neurosci. 2011;31(5):1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [http://dx.doi.org/10.1523/JNEUROSCI.2610-10.2011]. [PMID: 21289177]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Roberson E.D., Scearce-Levie K., Palop J.J., Yan F., Cheng I.H., Wu T., Gerstein H., Yu G.Q., Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [http://dx.doi.org/10.1126/science.1141736]. [PMID: 17478722]. [DOI] [PubMed] [Google Scholar]
- 165.Gatz M., Reynolds C.A., Fratiglioni L., Johansson B., Mortimer J.A., Berg S., Fiske A., Pedersen N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry. 2006;63(2):168–174. doi: 10.1001/archpsyc.63.2.168. [http://dx.doi.org/10.1001/archpsyc.63.2.168]. [PMID: 16461860]. [DOI] [PubMed] [Google Scholar]
- 166.Emahazion T., Feuk L., Jobs M., Sawyer S.L., Fredman D., St Clair D., Prince J.A., Brookes A.J. SNP association studies in Alzheimer’s disease highlight problems for complex disease analysis. Trends Genet. 2001;17(7):407–413. doi: 10.1016/s0168-9525(01)02342-3. [http://dx.doi.org/10.1016/S0168-9525(01)02342-3]. [PMID: 11418222]. [DOI] [PubMed] [Google Scholar]
- 167.Zhu X.C., Tan L., Wang H.F., Jiang T., Cao L., Wang C., Wang J., Tan C.C., Meng X.F., Yu J.T. Rate of early onset Alzheimer’s disease: a systematic review and meta-analysis. Ann. Transl. Med. 2015;3(3):38. doi: 10.3978/j.issn.2305-5839.2015.01.19. [PMID: 25815299]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Goate A., Chartier-Harlin M.C., Mullan M., Brown J., Crawford F., Fidani L., Giuffra L., Haynes A., Irving N., James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. doi: 10.1038/349704a0. [http://dx.doi.org/10.1038/349704a0]. [PMID: 1671712]. [DOI] [PubMed] [Google Scholar]
- 169.Jarrett J.T., Berger E.P., Lansbury P.T., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32(18):4693–4697. doi: 10.1021/bi00069a001. [http://dx.doi.org/10.1021/bi00069a001]. [PMID: 8490014]. [DOI] [PubMed] [Google Scholar]
- 170.Jarrett J.T., Berger E.P., Lansbury P.T., Jr The C-terminus of the beta protein is critical in amyloidogenesis. Ann. N. Y. Acad. Sci. 1993;695:144–148. doi: 10.1111/j.1749-6632.1993.tb23043.x. [http://dx.doi.org/10.1111/j.1749-6632.1993.tb23043.x]. [PMID: 8239273]. [DOI] [PubMed] [Google Scholar]
- 171.Suzuki N., Cheung T.T., Cai X.D., Odaka A., Otvos L., Jr, Eckman C., Golde T.E., Younkin S.G. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264(5163):1336–1340. doi: 10.1126/science.8191290. [http://dx.doi.org/10.1126/science.8191290]. [PMID: 8191290]. [DOI] [PubMed] [Google Scholar]
- 172.Roher A.E., Lowenson J.D., Clarke S., Woods A.S., Cotter R.J., Gowing E., Ball M.J. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1993;90(22):10836–10840. doi: 10.1073/pnas.90.22.10836. [http://dx.doi.org/10.1073/pnas.90.22.10836]. [PMID: 8248178]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.van Duijn C.M., de Knijff P., Cruts M., Wehnert A., Havekes L.M., Hofman A., Van Broeckhoven C. Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer’s disease. Nat. Genet. 1994;7(1):74–78. doi: 10.1038/ng0594-74. [http://dx.doi.org/10.1038/ng0594-74]. [PMID: 8075646]. [DOI] [PubMed] [Google Scholar]
- 174.Strittmatter W.J., Weisgraber K.H., Huang D.Y., Dong L.M., Salvesen G.S., Pericak-Vance M., Schmechel D., Saunders A.M., Goldgaber D., Roses A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1993;90(17):8098–8102. doi: 10.1073/pnas.90.17.8098. [http://dx.doi.org/10.1073/pnas.90.17.8098]. [PMID: 8367470]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Strittmatter W.J., Saunders A.M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G.S., Roses A.D., Apolipoprotein E. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1993;90(5):1977–1981. doi: 10.1073/pnas.90.5.1977. [http://dx.doi.org/10.1073/pnas.90.5.1977]. [PMID: 8446617]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chartier-Harlin M.C., Parfitt M., Legrain S., Pérez-Tur J., Brousseau T., Evans A., Berr C., Vidal O., Roques P., Gourlet V. Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: analysis of the 19q13.2 chromosomal region. Hum. Mol. Genet. 1994;3(4):569–574. doi: 10.1093/hmg/3.4.569. [http://dx.doi.org/10.1093/hmg/3.4.569]. [PMID: 8069300]. [DOI] [PubMed] [Google Scholar]
- 177.Talbot C., Lendon C., Craddock N., Shears S., Morris J.C., Goate A. Protection against Alzheimer’s disease with apoE epsilon 2. Lancet. 1994;343(8910):1432–1433. doi: 10.1016/s0140-6736(94)92557-7. [http://dx.doi.org/10.1016/S0140-6736(94)92557-7]. [PMID: 7910910]. [DOI] [PubMed] [Google Scholar]
- 178.Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W., Roses A.D., Haines J.L., Pericak-Vance M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [http://dx.doi.org/10.1126/science.8346443]. [PMID: 8346443]. [DOI] [PubMed] [Google Scholar]
- 179.Huang Y.A., Zhou B., Wernig M., Sudhof T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell. 168(3):427–441. doi: 10.1016/j.cell.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hashimoto T., Serrano-Pozo A., Hori Y., Adams K.W., Takeda S., Banerji A.O., Mitani A., Joyner D., Thyssen D.H., Bacskai B.J., Frosch M.P., Spires-Jones T.L., Finn M.B., Holtzman D.M., Hyman B.T., Apolipoprotein E. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012;32(43):15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [http://dx.doi.org/10.1523/JNEUROSCI.1542-12.2012]. [PMID: 23100439]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dumanis S.B., Tesoriero J.A., Babus L.W., Nguyen M.T., Trotter J.H., Ladu M.J., Weeber E.J., Turner R.S., Xu B., Rebeck G.W., Hoe H.S. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci. 2009;29(48):15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009. [http://dx.doi.org/10.1523/JNEUROSCI.4026-09.2009]. [PMID: 19955384]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Klein R.C., Mace B.E., Moore S.D., Sullivan P.M. Progressive loss of synaptic integrity in human apolipoprotein E4 targeted replacement mice and attenuation by apolipoprotein E2. Neuroscience. 2010;171(4):1265–1272. doi: 10.1016/j.neuroscience.2010.10.027. [http://dx.doi.org/10.1016/j.neuroscience.2010.10.027]. [PMID: 20951774]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nwabuisi-Heath E., Rebeck G.W., Ladu M.J., Yu C. ApoE4 delays dendritic spine formation during neuron development and accelerates loss of mature spines in vitro. ASN Neuro. 2014;6(1):e00134. doi: 10.1042/AN20130043. [PMID: 24328732]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhu Y., Nwabuisi-Heath E., Dumanis S.B., Tai L.M., Yu C., Rebeck G.W., LaDu M.J. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60(4):559–569. doi: 10.1002/glia.22289. [http://dx.doi.org/10.1002/glia.22289]. [PMID: 22228589]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Andrews-Zwilling Y., Bien-Ly N., Xu Q., Li G., Bernardo A., Yoon S.Y., Zwilling D., Yan T.X., Chen L., Huang Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 2010;30(41):13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Koffie R.M., Hashimoto T., Tai H.C., Kay K.R., Serrano-Pozo A., Joyner D., Hou S., Kopeikina K.J., Frosch M.P., Lee V.M., Holtzman D.M., Hyman B.T., Spires-Jones T.L. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain. 2012;135(Pt 7):2155–2168. doi: 10.1093/brain/aws127. [http://dx.doi.org/10.1093/brain/aws127]. [PMID: 22637583]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lin Y.T., Seo J., Gao F., Feldman H.M., Wen H.L., Pen-ney J., Cam H.P., Gjoneska E., Raja W.K., Cheng J., Rueda R., Kritskiy O., Abdurrob F., Peng Z., Milo B., Yu C.J., Elmsaouri S., Dey D., Ko T., Yankner B.A., Tsai L.H. APOE4 Causes Widespread Molecular and Cellular Altera-tions Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018;98(6):1141–1154. doi: 10.1016/j.neuron.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chung W.S., Verghese P.B., Chakraborty C., Joung J., Hyman B.T., Ulrich J.D., Holtzman D.M., Barres B.A. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. USA. 2016;113(36):10186–10191. doi: 10.1073/pnas.1609896113. [http://dx.doi.org/10.1073/pnas.1609896113]. [PMID: 27559087]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Egensperger R., Kösel S., von Eitzen U., Graeber M.B. Microglial activation in Alzheimer disease: Association with APOE genotype. Brain Pathol. 1998;8(3):439–447. doi: 10.1111/j.1750-3639.1998.tb00166.x. [http://dx.doi.org/10.1111/j.1750-3639.1998.tb00166.x]. [PMID: 9669695]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Currais A., Quehenberger O.A. M. A.; Daugherty, D.; Ma-her, P.; Schubert, D. Amyloid proteotoxicity initiates an in-flammatory response blocked by cannabinoids. NPJ Aging Mech. Dis. 2016;2:16012. doi: 10.1038/npjamd.2016.12. [http://dx.doi.org/10.1038/npjamd.2016.12]. [PMID: 28721267]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bornemann K.D., Wiederhold K.H., Pauli C., Ermini F., Stalder M., Schnell L., Sommer B., Jucker M., Staufenbiel M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am. J. Pathol. 2001;158(1):63–73. doi: 10.1016/s0002-9440(10)63945-4. [http://dx.doi.org/10.1016/S0002-9440(10)63945-4]. [PMID: 11141480]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lue L.F., Brachova L., Civin W.H. Rogers, J. Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J. Neuropathol. Exp. Neurol. 1996;55(10):1083–1088. [http://dx.doi.org/10.1097/00005072-199655100-00008]. [PMID: 8858005]. [PubMed] [Google Scholar]
- 193.Yang L.B., Li R., Meri S., Rogers J., Shen Y. Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer’s disease. J. Neurosci. 2000;20(20):7505–7509. doi: 10.1523/JNEUROSCI.20-20-07505.2000. [http://dx.doi.org/10.1523/JNEUROSCI.20-20-07505.2000]. [PMID: 11027207]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Reichwald J., Danner S., Wiederhold K.H., Staufenbiel M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J. Neuroinflammation. 2009;6:35. doi: 10.1186/1742-2094-6-35. [http://dx.doi.org/10.1186/1742-2094-6-35]. [PMID: 19917141]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Fonseca M.I., Zhou J., Botto M., Tenner A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2004;24(29):6457–6465. doi: 10.1523/JNEUROSCI.0901-04.2004. [http://dx.doi.org/10.1523/JNEUROSCI.0901-04.2004]. [PMID: 15269255]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shi Q., Chowdhury S., Ma R., Le K.X., Hong S., Caldarone B.J., Stevens B., Lemere C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017;9(392):eaaf6295. doi: 10.1126/scitranslmed.aaf6295. [http://dx.doi.org/10.1126/scitranslmed.aaf6295]. [PMID: 28566429]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shen Y., Meri S. Yin and Yang: complement activation and regulation in Alzheimer’s disease. Prog. Neurobiol. 2003;70(6):463–472. doi: 10.1016/j.pneurobio.2003.08.001. [http://dx.doi.org/10.1016/j.pneurobio.2003.08.001]. [PMID: 14568360]. [DOI] [PubMed] [Google Scholar]
- 198.Hong S., Beja-Glasser V.F., Nfonoyim B.M., Frouin A., Li S., Ramakrishnan S., Merry K.M., Shi Q., Rosenthal A., Barres B.A., Lemere C.A., Selkoe D.J., Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. doi: 10.1126/science.aad8373. [http://dx.doi.org/10.1126/science.aad8373]. [PMID: 27033548]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dejanovic B., Huntley M.A., De Maziere A., Meilandt W.J., Wu T., Srinivasan K., Jiang Z., Gandham V., Friedman B.A., Ngu H., Foreman O., Carano R.A.D., Chih B., Klumperman J., Bakalarski C., Hanson J.E., Sheng M. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron. 2018;100(6):1322–1336. doi: 10.1016/j.neuron.2018.10.014. [http://dx.doi.org/10.1016/j.neuron.2018.10.014]. [DOI] [PubMed] [Google Scholar]
- 200.Savas J.N., Wang Y.Z., DeNardo L.A., Martinez-Bartolome S., McClatchy D.B., Hark T.J., Shanks N.F., Cozzolino K.A., Lavallée-Adam M., Smukowski S.N., Park S.K., Kelly J.W., Koo E.H., Nakagawa T., Masliah E., Ghosh A., Yates J.R. III Amyloid Accumulation Drives Proteome-wide Alterations in Mouse Models of Alzheimer’s Disease-like Pathology. Cell Rep. 2017;21(9):2614–2627. doi: 10.1016/j.celrep.2017.11.009. [http://dx.doi.org/10.1016/j.celrep.2017.11.009]. [PMID: 29186695]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Seyfried N.T., Dammer E.B., Swarup V., Nandakumar D., Duong D.M., Yin L., Deng Q., Nguyen T., Hales C.M., Wingo T., Glass J., Gearing M., Thambisetty M., Troncoso J.C., Geschwind D.H., Lah J.J., Levey A.I. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 2017;4(1):60–72. doi: 10.1016/j.cels.2016.11.006. [http://dx.doi.org/10.1016/j.cels.2016.11.006]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Litvinchuk A., Wan Y.W., Swartzlander D.B., Chen F., Cole A., Propson N.E., Wang Q., Zhang B., Liu Z., Zheng H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron. 2018;100(6):1337–1353. doi: 10.1016/j.neuron.2018.10.031. [http://dx.doi.org/10.1016/j.neuron.2018.10.031]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shen Y., Yang L., Li R. What does complement do in Alzheimer’s disease? Old molecules with new insights. Transl. Neurodegener. 2013;2(1):21. doi: 10.1186/2047-9158-2-21. [http://dx.doi.org/10.1186/2047-9158-2-21]. [PMID: 24119446]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., David E., Baruch K., Lara-Astaiso D., Toth B., Itzkovitz S., Colonna M., Schwartz M., Amit I. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169(7):1276–1290. doi: 10.1016/j.cell.2017.05.018. [http://dx.doi.org/10.1016/j.cell.2017.05.018]. [DOI] [PubMed] [Google Scholar]
- 205.Liddelow S.A., Guttenplan K.A., Clarke L.E., Bennett F.C., Bohlen C.J., Schirmer L., Bennett M.L., Münch A.E., Chung W.S., Peterson T.C., Wilton D.K., Frouin A., Napier B.A., Panicker N., Kumar M., Buckwalter M.S., Rowitch D.H., Dawson V.L., Dawson T.M., Stevens B., Barres B.A. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi: 10.1038/nature21029. [http://dx.doi.org/10.1038/nature21029]. [PMID: 28099414]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Fonseca M.I., Chu S.H., Hernandez M.X., Fang M.J., Modarresi L., Selvan P., MacGregor G.R., Tenner A.J. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflammation. 2017;14(1):48. doi: 10.1186/s12974-017-0814-9. [http://dx.doi.org/10.1186/s12974-017-0814-9]. [PMID: 28264694]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bie B., Wu J., Foss J.F., Naguib M. Activation of mGluR1 mediates C1q-dependent microglial phagocytosis of glutamatergic synapses in Alzheimer’s Rodent models. Mol. Neurobiol. 2019;56(8):5568–5585. doi: 10.1007/s12035-019-1467-8. [http://dx.doi.org/10.1007/s12035-019-1467-8]. [PMID: 30652266]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Louneva N., Cohen J.W., Han L.Y., Talbot K., Wilson R.S., Bennett D.A., Trojanowski J.Q., Arnold S.E. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am. J. Pathol. 2008;173(5):1488–1495. doi: 10.2353/ajpath.2008.080434. [http://dx.doi.org/10.2353/ajpath.2008.080434]. [PMID: 18818379]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.D’Amelio M., Cavallucci V., Middei S., Marchetti C., Pacioni S., Ferri A., Diamantini A., De Zio D., Carrara P., Battistini L., Moreno S., Bacci A., Ammassari-Teule M., Marie H., Cecconi F. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2011;14(1):69–76. doi: 10.1038/nn.2709. [http://dx.doi.org/10.1038/nn.2709]. [PMID: 21151119]. [DOI] [PubMed] [Google Scholar]
- 210.Kim T., Vidal G.S., Djurisic M., William C.M., Birnbaum M.E., Garcia K.C., Hyman B.T., Shatz C.J. Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science. 2013;341(6152):1399–1404. doi: 10.1126/science.1242077. [http://dx.doi.org/10.1126/science.1242077]. [PMID: 24052308]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Chapuis J., Hot D., Hansmannel F., Kerdraon O., Ferreira S., Hubans C., Maurage C.A., Huot L., Bensemain F., Laumet G., Ayral A.M., Fievet N., Hauw J.J., DeKosky S.T., Lemoine Y., Iwatsubo T., Wavrant-Devrièze F., Dartigues J.F., Tzourio C., Buée L., Pasquier F., Berr C., Mann D., Lendon C., Alpérovitch A., Kamboh M.I., Amouyel P., Lambert J.C. Transcriptomic and genetic studies identify IL-33 as a candidate gene for Alzheimer’s disease. Mol. Psychiatry. 2009;14(11):1004–1016. doi: 10.1038/mp.2009.10. [http://dx.doi.org/10.1038/mp.2009.10]. [PMID: 19204726]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Fu A.K., Hung K.W., Yuen M.Y., Zhou X., Mak D.S., Chan I.C., Cheung T.H., Zhang B., Fu W.Y., Liew F.Y., Ip N.Y. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc. Natl. Acad. Sci. USA. 2016;113(19):E2705–E2713. doi: 10.1073/pnas.1604032113. [http://dx.doi.org/10.1073/pnas.1604032113]. [PMID: 27091974]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ma Q.L., Teng E., Zuo X., Jones M., Teter B., Zhao E.Y., Zhu C., Bilousova T., Gylys K.H., Apostolova L.G., LaDu M.J., Hossain M.A., Frautschy S.A., Cole G.M. Neuronal pentraxin 1: A synaptic-derived plasma biomarker in Alzheimer’s disease. Neurobiol. Dis. 2018;114:120–128. doi: 10.1016/j.nbd.2018.02.014. [http://dx.doi.org/10.1016/j.nbd.2018.02.014]. [PMID: 29501530]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Abad M.A., Enguita M., DeGregorio-Rocasolano N., Ferrer I., Trullas R. Neuronal pentraxin 1 contributes to the neuronal damage evoked by amyloid-beta and is overexpressed in dystrophic neurites in Alzheimer’s brain. J. Neurosci. 2006;26(49):12735–12747. doi: 10.1523/JNEUROSCI.0575-06.2006. [http://dx.doi.org/10.1523/JNEUROSCI.0575-06.2006]. [PMID: 17151277]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Suarez-Calvet M., Capell A., Araque Caballero M.A., Morenas-Rodriguez E., Fellerer K., Franzmeier N., Klein-berger G., Eren E., Deming Y., Piccio L., Karch C.M., Cruchaga C., Paumier K., Bateman R.J., Fagan A.M., Mor-ris J.C., Levin J., Danek A., Jucker M., Masters C.L., Rossor M.N., Ringman J.M., Shaw L.M., Trojanowski J.Q., Weiner M., Ewers M., Haass C., Dominantly Inherited Alzheimer N. Alzheimer’s Disease Neuroimaging, I., CSF progranulin increases in the course of Alzheimer’s disease and is associated with sTREM2, neurodegeneration and cog-nitive decline. EMBO Mol. Med. 2018;10(12) doi: 10.15252/emmm.201809712. [http://dx.doi.org/10.15252/emmm.201809712]. [PMID: 30482868]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cooper Y.A., Nachun D., Dokuru D., Yang Z., Karydas A.M., Serrero G., Yue B. Alzheimer’s Disease Neuroimag-ing, I.; Boxer, A. L.; Miller, B. L.; Coppola, G., Progranulin levels in blood in Alzheimer’s disease and mild cognitive im-pairment. Ann. Clin. Transl. Neurol. 2018;5(5):616–629. doi: 10.1002/acn3.560. [http://dx.doi.org/10.1002/acn3.560]. [PMID: 29761124]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Minami S.S., Min S.W., Krabbe G., Wang C., Zhou Y., Asgarov R., Li Y., Martens L.H., Elia L.P., Ward M.E., Mucke L., Farese R.V., Jr, Gan L. Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 2014;20(10):1157–1164. doi: 10.1038/nm.3672. [http://dx.doi.org/10.1038/nm.3672]. [PMID: 25261995]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Takahashi H., Klein Z.A., Bhagat S.M., Kaufman A.C., Kostylev M.A., Ikezu T., Strittmatter S.M. Alzheimer’s Disease Neuroimaging, I. Opposing effects of progranulin de-ficiency on amyloid and tau pathologies via microglial TYROBP network. Acta Neuropathol. 2017;133(5):785–807. doi: 10.1007/s00401-017-1668-z. [http://dx.doi.org/10.1007/s00401-017-1668-z]. [PMID: 28070672]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Perea J.R., Lleó A., Alcolea D., Fortea J., Ávila J., Bolós M. Decreased CX3CL1 levels in the Cerebrospinal fluid of patients With Alzheimer’s Disease. Front. Neurosci. 2018;12:609. doi: 10.3389/fnins.2018.00609. [http://dx.doi.org/10.3389/fnins.2018.00609]. [PMID: 30245615]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Cho S.H., Sun B., Zhou Y., Kauppinen T.M., Halabisky B., Wes P., Ransohoff R.M., Gan L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2011;286(37):32713–32722. doi: 10.1074/jbc.M111.254268. [http://dx.doi.org/10.1074/jbc.M111.254268]. [PMID: 21771791]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lee S., Xu G., Jay T.R., Bhatta S., Kim K.W., Jung S., Landreth G.E., Ransohoff R.M., Lamb B.T. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J. Neurosci. 2014;34(37):12538–12546. doi: 10.1523/JNEUROSCI.0853-14.2014. [http://dx.doi.org/10.1523/JNEUROSCI.0853-14.2014]. [PMID: 25209291]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lee S., Varvel N.H., Konerth M.E., Xu G., Cardona A.E., Ransohoff R.M., Lamb B.T. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010;177(5):2549–2562. doi: 10.2353/ajpath.2010.100265. [http://dx.doi.org/10.2353/ajpath.2010.100265]. [PMID: 20864679]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Park D., Na M., Kim J.A., Lee U., Cho E., Jang M., Chang S. Activation of CaMKIV by soluble amyloid-β1-42 impedes trafficking of axonal vesicles and impairs activity-dependent synaptogenesis. Sci. Signal. 2017;10(487):eaam8661. doi: 10.1126/scisignal.aam8661. [PMID: 28698220]. [DOI] [PubMed] [Google Scholar]
- 224.Levi O., Jongen-Relo A.L., Feldon J., Roses A.D., Michaelson D.M. ApoE4 impairs hippocampal plasticity isoform-specifically and blocks the environmental stimulation of synaptogenesis and memory. Neurobiol. Dis. 2003;13(3):273–282. doi: 10.1016/s0969-9961(03)00045-7. [http://dx.doi.org/10.1016/S0969-9961(03)00045-7]. [PMID: 12901842]. [DOI] [PubMed] [Google Scholar]
- 225.Van Kampen J.M., Kay D.G. Progranulin gene delivery reduces plaque burden and synaptic atrophy in a mouse model of Alzheimer’s disease. PLoS One. 2017;12(8):e0182896. doi: 10.1371/journal.pone.0182896. [http://dx.doi.org/10.1371/journal.pone.0182896]. [PMID: 28837568]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chen Y., Wang B., Liu D., Li J.J., Xue Y., Sakata K., Zhu L.Q., Heldt S.A., Xu H., Liao F.F. Hsp90 chaperone inhibitor 17-AAG attenuates Abeta-induced synaptic toxicity and memory impairment. J. Neurosci. 2014;34(7):2464–2470. doi: 10.1523/JNEUROSCI.0151-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wang R., Zhang Y., Li J., Zhang C. Resveratrol ameliorates spatial learning memory impairment induced by Aβ1-42 in rats. Neuroscience. 2017;344:39–47. doi: 10.1016/j.neuroscience.2016.08.051. [http://dx.doi.org/10.1016/j.neuroscience.2016.08.051]. [PMID: 27600946]. [DOI] [PubMed] [Google Scholar]
- 228.Kodali M., Parihar V.K., Hattiangady B., Mishra V., Shuai B., Shetty A.K. Resveratrol prevents age-related memory and mood dysfunction with increased hippocampal neurogenesis and microvasculature, and reduced glial activation. Sci. Rep. 2015;5:8075. doi: 10.1038/srep08075. [http://dx.doi.org/10.1038/srep08075]. [PMID: 25627672]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zhao H., Wang Q., Cheng X., Li X., Li N., Liu T., Li J., Yang Q., Dong R., Zhang Y., Zhang L. Inhibitive Effect of Resveratrol on the Inflammation in Cultured Astrocytes and Microglia Induced by Aβ1-42. Neuroscience. 2018;379:390–404. doi: 10.1016/j.neuroscience.2018.03.047. [http://dx.doi.org/10.1016/j.neuroscience.2018.03.047]. [PMID: 29627302]. [DOI] [PubMed] [Google Scholar]
- 230.Craft S., Baker L.D., Montine T.J., Minoshima S., Watson G.S., Claxton A., Arbuckle M., Callaghan M., Tsai E., Plymate S.R., Green P.S., Leverenz J., Cross D., Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 2012;69(1):29–38. doi: 10.1001/archneurol.2011.233. [http://dx.doi.org/10.1001/archneurol.2011.233]. [PMID: 21911655]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.De Felice F.G., Vieira M.N., Bomfim T.R., Decker H., Velasco P.T., Lambert M.P., Viola K.L., Zhao W.Q., Ferreira S.T., Klein W.L. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA. 2009;106(6):1971–1976. doi: 10.1073/pnas.0809158106. [http://dx.doi.org/10.1073/pnas.0809158106]. [PMID: 19188609]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Thompson A.J., Baranzini S.E., Geurts J., Hemmer B., Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622–1636. doi: 10.1016/S0140-6736(18)30481-1. [http://dx.doi.org/10.1016/S0140-6736(18)30481-1]. [PMID: 29576504]. [DOI] [PubMed] [Google Scholar]
- 233.Reich D.S., Lucchinetti C.F., Calabresi P.A. Multiple Sclerosis. N. Engl. J. Med. 2018;378(2):169–180. doi: 10.1056/NEJMra1401483. [http://dx.doi.org/10.1056/NEJMra1401483]. [PMID: 29320652]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Di Filippo M., de Iure A., Durante V., Gaetani L., Mancini A., Sarchielli P., Calabresi P. Synaptic plasticity and experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Brain Res. 2015;1621:205–213. doi: 10.1016/j.brainres.2014.12.004. [http://dx.doi.org/10.1016/j.brainres.2014.12.004]. [PMID: 25498984]. [DOI] [PubMed] [Google Scholar]
- 235.Mandolesi G., Gentile A., Musella A., Fresegna D., De Vito F., Bullitta S., Sepman H., Marfia G.A., Centonze D. Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nat. Rev. Neurol. 2015;11(12):711–724. doi: 10.1038/nrneurol.2015.222. [http://dx.doi.org/10.1038/nrneurol.2015.222]. [PMID: 26585978]. [DOI] [PubMed] [Google Scholar]
- 236.Ruano L., Portaccio E., Goretti B., Niccolai C., Severo M., Patti F., Cilia S., Gallo P., Grossi P., Ghezzi A., Roscio M., Mattioli F., Stampatori C., Trojano M., Viterbo R.G., Amato M.P. Age and disability drive cognitive impairment in multiple sclerosis across disease subtypes. Mult. Scler. 2017;23(9):1258–1267. doi: 10.1177/1352458516674367. [http://dx.doi.org/10.1177/1352458516674367]. [PMID: 27738090]. [DOI] [PubMed] [Google Scholar]
- 237.Achiron A., Polliack M., Rao S.M., Barak Y., Lavie M., Appelboim N., Harel Y. Cognitive patterns and progression in multiple sclerosis: construction and validation of percentile curves. J. Neurol. Neurosurg. Psychiatry. 2005;76(5):744–749. doi: 10.1136/jnnp.2004.045518. [http://dx.doi.org/10.1136/jnnp.2004.045518]. [PMID: 15834042]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Centonze D., Muzio L., Rossi S., Cavasinni F., De Chiara V., Bergami A., Musella A., D’Amelio M., Cavallucci V., Martorana A., Bergamaschi A., Cencioni M.T., Diamantini A., Butti E., Comi G., Bernardi G., Cecconi F., Battistini L., Furlan R., Martino G. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J. Neurosci. 2009;29(11):3442–3452. doi: 10.1523/JNEUROSCI.5804-08.2009. [http://dx.doi.org/10.1523/JNEUROSCI.5804-08.2009]. [PMID: 19295150]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Rossi S., Motta C., Studer V., Barbieri F., Buttari F., Bergami A., Sancesario G., Bernardini S., De Angelis G., Martino G., Furlan R., Centonze D. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult. Scler. 2014;20(3):304–312. doi: 10.1177/1352458513498128. [http://dx.doi.org/10.1177/1352458513498128]. [PMID: 23886826]. [DOI] [PubMed] [Google Scholar]
- 240.Kim D.Y., Hao J., Liu R., Turner G., Shi F.D., Rho J.M. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS One. 2012;7(5):e35476. doi: 10.1371/journal.pone.0035476. [http://dx.doi.org/10.1371/journal.pone.0035476]. [PMID: 22567104]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.DeLuca G.C., Yates R.L., Beale H., Morrow S.A. Cognitive impairment in multiple sclerosis: clinical, radiologic and pathologic insights. Brain Pathol. 2015;25(1):79–98. doi: 10.1111/bpa.12220. [http://dx.doi.org/10.1111/bpa.12220]. [PMID: 25521179]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Pitt D., Werner P., Raine C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000;6(1):67–70. doi: 10.1038/71555. [http://dx.doi.org/10.1038/71555]. [PMID: 10613826]. [DOI] [PubMed] [Google Scholar]
- 243.Calabrese M., Agosta F., Rinaldi F., Mattisi I., Grossi P., Favaretto A., Atzori M., Bernardi V., Barachino L., Rinaldi L., Perini P., Gallo P., Filippi M. Cortical lesions and atrophy associated with cognitive impairment in relapsing-remitting multiple sclerosis. Arch. Neurol. 2009;66(9):1144–1150. doi: 10.1001/archneurol.2009.174. [http://dx.doi.org/10.1001/archneurol.2009.174]. [PMID: 19752305]. [DOI] [PubMed] [Google Scholar]
- 244.Batista S., Zivadinov R., Hoogs M., Bergsland N., Heininen-Brown M., Dwyer M.G., Weinstock-Guttman B., Benedict R.H. Basal ganglia, thalamus and neocortical atrophy predicting slowed cognitive processing in multiple sclerosis. J. Neurol. 2012;259(1):139–146. doi: 10.1007/s00415-011-6147-1. [http://dx.doi.org/10.1007/s00415-011-6147-1]. [PMID: 21720932]. [DOI] [PubMed] [Google Scholar]
- 245.Bergsland N., Zivadinov R., Dwyer M.G., Weinstock-Guttman B., Benedict R.H. Localized atrophy of the thalamus and slowed cognitive processing speed in MS patients. Mult. Scler. 2016;22(10):1327–1336. doi: 10.1177/1352458515616204. [http://dx.doi.org/10.1177/1352458515616204]. [PMID: 26541795]. [DOI] [PubMed] [Google Scholar]
- 246.Steenwijk M.D., Geurts J.J., Daams M., Tijms B.M., Wink A.M., Balk L.J., Tewarie P.K., Uitdehaag B.M., Barkhof F., Vrenken H., Pouwels P.J. Cortical atrophy patterns in multiple sclerosis are non-random and clinically relevant. Brain. 2016;139(Pt 1):115–126. doi: 10.1093/brain/awv337. [http://dx.doi.org/10.1093/brain/awv337]. [PMID: 26637488]. [DOI] [PubMed] [Google Scholar]
- 247.Cocozza S., Petracca M., Mormina E., Buyukturkoglu K., Podranski K., Heinig M.M., Pontillo G., Russo C., Tedeschi E., Russo C.V., Costabile T., Lanzillo R., Harel A., Klineova S., Miller A., Brunetti A., Morra V.B., Lublin F., Inglese M. Cerebellar lobule atrophy and disability in progressive MS. J. Neurol. Neurosurg. Psychiatry. 2017;88(12):1065–1072. doi: 10.1136/jnnp-2017-316448. [http://dx.doi.org/10.1136/jnnp-2017-316448]. [PMID: 28844067]. [DOI] [PubMed] [Google Scholar]
- 248.Batista S., d’Almeida O.C., Afonso A., Freitas S., Macário C., Sousa L., Castelo-Branco M., Santana I., Cunha L. Impairment of social cognition in multiple sclerosis: Amygdala atrophy is the main predictor. Mult. Scler. 2017;23(10):1358–1366. doi: 10.1177/1352458516680750. [http://dx.doi.org/10.1177/1352458516680750]. [PMID: 28273767]. [DOI] [PubMed] [Google Scholar]
- 249.Sicotte N.L., Kern K.C., Giesser B.S., Arshanapalli A., Schultz A., Montag M., Wang H., Bookheimer S.Y. Regional hippocampal atrophy in multiple sclerosis. Brain. 2008;131(Pt 4):1134–1141. doi: 10.1093/brain/awn030. [http://dx.doi.org/10.1093/brain/awn030]. [PMID: 18375977]. [DOI] [PubMed] [Google Scholar]
- 250.Ziehn M.O., Avedisian A.A., Tiwari-Woodruff S., Voskuhl R.R. Hippocampal CA1 atrophy and synaptic loss during experimental autoimmune encephalomyelitis, EAE. Lab. Invest. 2010;90(5):774–786. doi: 10.1038/labinvest.2010.6. [http://dx.doi.org/10.1038/labinvest.2010.6]. [PMID: 20157291]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Wegner C., Esiri M.M., Chance S.A., Palace J., Matthews P.M. Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology. 2006;67(6):960–967. doi: 10.1212/01.wnl.0000237551.26858.39. [http://dx.doi.org/10.1212/01.wnl.0000237551.26858.39]. [PMID: 17000961]. [DOI] [PubMed] [Google Scholar]
- 252.Magliozzi R., Howell O.W., Reeves C., Roncaroli F., Nicholas R., Serafini B., Aloisi F., Reynolds R. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 2010;68(4):477–493. doi: 10.1002/ana.22230. [http://dx.doi.org/10.1002/ana.22230]. [PMID: 20976767]. [DOI] [PubMed] [Google Scholar]
- 253.Peterson J.W., Bö L., Mörk S., Chang A., Trapp B.D. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 2001;50(3):389–400. doi: 10.1002/ana.1123. [http://dx.doi.org/10.1002/ana.1123]. [PMID: 11558796]. [DOI] [PubMed] [Google Scholar]
- 254.Jurewicz A., Matysiak M., Tybor K., Kilianek L., Raine C.S., Selmaj K. Tumour necrosis factor-induced death of adult human oligodendrocytes is mediated by apoptosis inducing factor. Brain. 2005;128(Pt 11):2675–2688. doi: 10.1093/brain/awh627. [http://dx.doi.org/10.1093/brain/awh627]. [PMID: 16219674]. [DOI] [PubMed] [Google Scholar]
- 255.Vartanian T., Li Y., Zhao M., Stefansson K. Interferon-gamma-induced oligodendrocyte cell death: implications for the pathogenesis of multiple sclerosis. Mol. Med. 1995;1(7):732–743. [http://dx.doi.org/10.1007/BF03401888]. [PMID: 8612196]. [PMC free article] [PubMed] [Google Scholar]
- 256.Takeuchi H., Wang J., Kawanokuchi J., Mitsuma N., Mizuno T., Suzumura A. Interferon-gamma induces microglial-activation-induced cell death: a hypothetical mechanism of relapse and remission in multiple sclerosis. Neurobiol. Dis. 2006;22(1):33–39. doi: 10.1016/j.nbd.2005.09.014. [http://dx.doi.org/10.1016/j.nbd.2005.09.014]. [PMID: 16386911]. [DOI] [PubMed] [Google Scholar]
- 257.Rossi S., Motta C., Studer V., Macchiarulo G., Volpe E., Barbieri F., Ruocco G., Buttari F., Finardi A., Mancino R., Weiss S., Battistini L., Martino G., Furlan R., Drulovic J., Centonze D. Interleukin-1β causes excitotoxic neurodegeneration and multiple sclerosis disease progression by activating the apoptotic protein p53. Mol. Neurodegener. 2014;9:56. doi: 10.1186/1750-1326-9-56. [http://dx.doi.org/10.1186/1750-1326-9-56]. [PMID: 25495224]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Gilgun-Sherki Y., Melamed E., Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J. Neurol. 2004;251(3):261–268. doi: 10.1007/s00415-004-0348-9. [http://dx.doi.org/10.1007/s00415-004-0348-9]. [PMID: 15015004]. [DOI] [PubMed] [Google Scholar]
- 259.Nataf S., Carroll S.L., Wetsel R.A., Szalai A.J., Barnum S.R. Attenuation of experimental autoimmune demyelination in complement-deficient mice. J. Immunol. 2000;165(10):5867–5873. doi: 10.4049/jimmunol.165.10.5867. [http://dx.doi.org/10.4049/jimmunol.165.10.5867]. [PMID: 11067947]. [DOI] [PubMed] [Google Scholar]
- 260.Szalai A.J., Hu X., Adams J.E., Barnum S.R. Complement in experimental autoimmune encephalomyelitis revisited: C3 is required for development of maximal disease. Mol. Immunol. 2007;44(12):3132–3136. doi: 10.1016/j.molimm.2007.02.002. [http://dx.doi.org/10.1016/j.molimm.2007.02.002]. [PMID: 17353050]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Roostaei T., Sadaghiani S., Mashhadi R., Falahatian M., Mohamadi E., Javadian N., Nazeri A., Doosti R., Naser Moghadasi A., Owji M., Hashemi Taheri A.P., Shakouri Rad A., Azimi A., Voineskos A.N., Nazeri A., Sahraian M.A. Convergent effects of a functional C3 variant on brain atrophy, demyelination, and cognitive impairment in multiple sclerosis. Mult. Scler. 2018:1352458518760715. doi: 10.1177/1352458518760715. [PMID: 29485352]. [DOI] [PubMed] [Google Scholar]
- 262.Niculescu T., Weerth S., Niculescu F., Cudrici C., Rus V., Raine C.S., Shin M.L., Rus H. Effects of complement C5 on apoptosis in experimental autoimmune encephalomyelitis. J. Immunol. 2004;172(9):5702–5706. doi: 10.4049/jimmunol.172.9.5702. [http://dx.doi.org/10.4049/jimmunol.172.9.5702]. [PMID: 15100315]. [DOI] [PubMed] [Google Scholar]
- 263.Watkins L.M., Neal J.W., Loveless S., Michailidou I., Ramaglia V., Rees M.I., Reynolds R., Robertson N.P., Morgan B.P., Howell O.W. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J. Neuroinflammation. 2016;13(1):161. doi: 10.1186/s12974-016-0611-x. [http://dx.doi.org/10.1186/s12974-016-0611-x]. [PMID: 27333900]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Papadopoulos D., Dukes S., Patel R., Nicholas R., Vora A., Reynolds R. Substantial archaeocortical atrophy and neuronal loss in multiple sclerosis. Brain Pathol. 2009;19(2):238–253. doi: 10.1111/j.1750-3639.2008.00177.x. [http://dx.doi.org/10.1111/j.1750-3639.2008.00177.x]. [PMID: 18492094]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Jürgens T., Jafari M., Kreutzfeldt M., Bahn E., Brück W., Kerschensteiner M., Merkler D. Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain. 2016;139(Pt 1):39–46. doi: 10.1093/brain/awv353. [http://dx.doi.org/10.1093/brain/awv353]. [PMID: 26667278]. [DOI] [PubMed] [Google Scholar]
- 266.Dutta R., Chang A., Doud M.K., Kidd G.J., Ribaudo M.V., Young E.A., Fox R.J., Staugaitis S.M., Trapp B.D. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann. Neurol. 2011;69(3):445–454. doi: 10.1002/ana.22337. [http://dx.doi.org/10.1002/ana.22337]. [PMID: 21446020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Zhu B., Luo L., Moore G.R., Paty D.W., Cynader M.S. Dendritic and synaptic pathology in experimental autoimmune encephalomyelitis. Am. J. Pathol. 2003;162(5):1639–1650. doi: 10.1016/S0002-9440(10)64298-8. [http://dx.doi.org/10.1016/S0002-9440(10)64298-8]. [PMID: 12707048]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Michailidou I., Willems J.G., Kooi E.J., van Eden C., Gold S.M., Geurts J.J., Baas F., Huitinga I., Ramaglia V. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann. Neurol. 2015;77(6):1007–1026. doi: 10.1002/ana.24398. [http://dx.doi.org/10.1002/ana.24398]. [PMID: 25727254]. [DOI] [PubMed] [Google Scholar]
- 269.Koning N., Bö L., Hoek R.M., Huitinga I. Downregulation of macrophage inhibitory molecules in multiple sclerosis lesions. Ann. Neurol. 2007;62(5):504–514. doi: 10.1002/ana.21220. [http://dx.doi.org/10.1002/ana.21220]. [PMID: 17879969]. [DOI] [PubMed] [Google Scholar]
- 270.Han M.H., Lundgren D.H., Jaiswal S., Chao M., Graham K.L., Garris C.S., Axtell R.C., Ho P.P., Lock C.B., Woodard J.I., Brownell S.E., Zoudilova M., Hunt J.F., Baranzini S.E., Butcher E.C., Raine C.S., Sobel R.A., Han D.K., Weissman I., Steinman L. Janus-like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. J. Exp. Med. 2012;209(7):1325–1334. doi: 10.1084/jem.20101974. [http://dx.doi.org/10.1084/jem.20101974]. [PMID: 22734047]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Freria C.M., Zanon R.G., Santos L.M., Oliveira A.L. Major histocompatibility complex class I expression and glial reaction influence spinal motoneuron synaptic plasticity during the course of experimental autoimmune encephalomyelitis. J. Comp. Neurol. 2010;518(7):990–1007. doi: 10.1002/cne.22259. [http://dx.doi.org/10.1002/cne.22259]. [PMID: 20127802]. [DOI] [PubMed] [Google Scholar]
- 272.Vercellino M., Fenoglio C., Galimberti D., Mattioda A., Chiavazza C., Binello E., Pinessi L., Giobbe D., Scarpini E., Cavalla P. Progranulin genetic polymorphisms influence progression of disability and relapse recovery in multiple sclerosis. Mult. Scler. 2016;22(8):1007–1012. doi: 10.1177/1352458515610646. [http://dx.doi.org/10.1177/1352458515610646]. [PMID: 26447062]. [DOI] [PubMed] [Google Scholar]
- 273.Vercellino M., Grifoni S., Romagnolo A., Masera S., Mattioda A., Trebini C., Chiavazza C., Caligiana L., Capello E., Mancardi G.L., Giobbe D., Mutani R., Giordana M.T., Cavalla P. Progranulin expression in brain tissue and cerebrospinal fluid levels in multiple sclerosis. Mult. Scler. 2011;17(10):1194–1201. doi: 10.1177/1352458511406164. [http://dx.doi.org/10.1177/1352458511406164]. [PMID: 21613335]. [DOI] [PubMed] [Google Scholar]
- 274.De Riz M., Galimberti D., Fenoglio C., Piccio L.M., Scalabrini D., Venturelli E., Pietroboni A., Piola M., Naismith R.T., Parks B.J., Fumagalli G., Bresolin N., Cross A.H., Scarpini E. Cerebrospinal fluid progranulin levels in patients with different multiple sclerosis subtypes. Neurosci. Lett. 2010;469(2):234–236. doi: 10.1016/j.neulet.2009.12.002. [http://dx.doi.org/10.1016/j.neulet.2009.12.002]. [PMID: 19963041]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Kastenbauer S., Koedel U., Wick M., Kieseier B.C., Hartung H.P., Pfister H.W. CSF and serum levels of soluble fractalkine (CX3CL1) in inflammatory diseases of the nervous system. J. Neuroimmunol. 2003;137(1-2):210–217. doi: 10.1016/s0165-5728(03)00085-7. [http://dx.doi.org/10.1016/S0165-5728(03)00085-7]. [PMID: 12667665]. [DOI] [PubMed] [Google Scholar]
- 276.Mills J.H., Alabanza L.M., Mahamed D.A., Bynoe M.S. Extracellular adenosine signaling induces CX3CL1 expression in the brain to promote experimental autoimmune encephalomyelitis. J. Neuroinflammation. 2012;9:193. doi: 10.1186/1742-2094-9-193. [http://dx.doi.org/10.1186/1742-2094-9-193]. [PMID: 22883932]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Hulshof S., van Haastert E.S., Kuipers H.F., van den Elsen P.J., De Groot C.J., van der Valk P., Ravid R., Biber K. CX3CL1 and CX3CR1 expression in human brain tissue: noninflammatory control versus multiple sclerosis. J. Neuropathol. Exp. Neurol. 2003;62(9):899–907. doi: 10.1093/jnen/62.9.899. [http://dx.doi.org/10.1093/jnen/62.9.899]. [PMID: 14533779]. [DOI] [PubMed] [Google Scholar]
- 278.Huang D., Shi F.D., Jung S., Pien G.C., Wang J., Salazar-Mather T.P., He T.T., Weaver J.T., Ljunggren H.G., Biron C.A., Littman D.R., Ransohoff R.M. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006;20(7):896–905. doi: 10.1096/fj.05-5465com. [http://dx.doi.org/10.1096/fj.05-5465com]. [PMID: 16675847]. [DOI] [PubMed] [Google Scholar]
- 279.Kroksveen A.C., Guldbrandsen A., Vedeler C., Myhr K.M., Opsahl J.A., Berven F.S. Cerebrospinal fluid proteome comparison between multiple sclerosis patients and controls. Acta Neurol. Scand. Suppl. 2012;(195):90–96. doi: 10.1111/ane.12029. [http://dx.doi.org/10.1111/ane.12029]. [PMID: 23278663]. [DOI] [PubMed] [Google Scholar]
- 280.Blakely P.K., Hussain S., Carlin L.E., Irani D.N. Astrocyte matricellular proteins that control excitatory synaptogenesis are regulated by inflammatory cytokines and correlate with paralysis severity during experimental autoimmune encephalomyelitis. Front. Neurosci. 2015;9:344. doi: 10.3389/fnins.2015.00344. [http://dx.doi.org/10.3389/fnins.2015.00344]. [PMID: 26500475]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Vercellino M., Merola A., Piacentino C., Votta B., Capello E., Mancardi G.L., Mutani R., Giordana M.T., Cavalla P. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: Correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J. Neuropathol. Exp. Neurol. 2007;66(8):732–739. doi: 10.1097/nen.0b013e31812571b0. [http://dx.doi.org/10.1097/nen.0b013e31812571b0]. [PMID: 17882017]. [DOI] [PubMed] [Google Scholar]
- 282.Rossi S., Muzio L., De Chiara V., Grasselli G., Musella A., Musumeci G., Mandolesi G., De Ceglia R., Maida S., Biffi E., Pedrocchi A., Menegon A., Bernardi G., Furlan R., Martino G., Centonze D. Impaired striatal GABA transmission in experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2011;25(5):947–956. doi: 10.1016/j.bbi.2010.10.004. [http://dx.doi.org/10.1016/j.bbi.2010.10.004]. [PMID: 20940040]. [DOI] [PubMed] [Google Scholar]
- 283.Habbas S., Santello M., Becker D., Stubbe H., Zappia G., Liaudet N., Klaus F.R., Kollias G., Fontana A., Pryce C.R., Suter T., Volterra A. Neuroinflammatory TNFα Impairs Memory via Astrocyte Signaling. Cell. 2015;163(7):1730–1741. doi: 10.1016/j.cell.2015.11.023. [http://dx.doi.org/10.1016/j.cell.2015.11.023]. [PMID: 26686654]. [DOI] [PubMed] [Google Scholar]
- 284.Takeuchi H., Jin S., Wang J., Zhang G., Kawanokuchi J., Kuno R., Sonobe Y., Mizuno T., Suzumura A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006;281(30):21362–21368. doi: 10.1074/jbc.M600504200. [http://dx.doi.org/10.1074/jbc.M600504200]. [PMID: 16720574]. [DOI] [PubMed] [Google Scholar]
- 285.Yang G., Parkhurst C.N., Hayes S., Gan W.B. Peripheral elevation of TNF-α leads to early synaptic abnormalities in the mouse somatosensory cortex in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 2013;110(25):10306–10311. doi: 10.1073/pnas.1222895110. [http://dx.doi.org/10.1073/pnas.1222895110]. [PMID: 23733958]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.de Lago E., Moreno-Martet M., Cabranes A., Ramos J.A., Fernández-Ruiz J. Cannabinoids ameliorate disease progression in a model of multiple sclerosis in mice, acting preferentially through CB1 receptor-mediated anti-inflammatory effects. Neuropharmacology. 2012;62(7):2299–2308. doi: 10.1016/j.neuropharm.2012.01.030. [http://dx.doi.org/10.1016/j.neuropharm.2012.01.030]. [PMID: 22342378]. [DOI] [PubMed] [Google Scholar]
- 287.Rossi S., Furlan R., De Chiara V., Muzio L., Musella A., Motta C., Studer V., Cavasinni F., Bernardi G., Martino G., Cravatt B.F., Lutz B., Maccarrone M., Centonze D. Cannabinoid CB1 receptors regulate neuronal TNF-α effects in experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2011;25(6):1242–1248. doi: 10.1016/j.bbi.2011.03.017. [http://dx.doi.org/10.1016/j.bbi.2011.03.017]. [PMID: 21473912]. [DOI] [PubMed] [Google Scholar]
- 288.Shijie J., Takeuchi H., Yawata I., Harada Y., Sonobe Y., Doi Y., Liang J., Hua L., Yasuoka S., Zhou Y., Noda M., Kawanokuchi J., Mizuno T., Suzumura A. Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis in mice. Tohoku J. Exp. Med. 2009;217(2):87–92. doi: 10.1620/tjem.217.87. [http://dx.doi.org/10.1620/tjem.217.87]. [PMID: 19212100]. [DOI] [PubMed] [Google Scholar]
- 289.Stilo S.A., Murray R.M. The epidemiology of schizophrenia: replacing dogma with knowledge. Dialogues Clin. Neurosci. 2010;12(3):305–315. doi: 10.31887/DCNS.2010.12.3/sstilo. [PMID: 20954427]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Patel K.R., Cherian J., Gohil K., Atkinson D. Schizophrenia: overview and treatment options. P&T. 2014;39(9):638–345. [PMC free article] [PubMed] [Google Scholar]
- 291.Picchioni M.M., Murray R.M. Schizophrenia. BMJ. 2007;335(7610):91–95. doi: 10.1136/bmj.39227.616447.BE. [http://dx.doi.org/10.1136/bmj.39227.616447.BE]. [PMID: 17626963]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Johnstone E.C., Crow T.J., Frith C.D., Husband J., Kreel L. Cerebral ventricular size and cognitive impairment in chronic schizophrenia. Lancet. 1976;2(7992):924–926. doi: 10.1016/s0140-6736(76)90890-4. [http://dx.doi.org/10.1016/S0140-6736(76)90890-4]. [PMID: 62160]. [DOI] [PubMed] [Google Scholar]
- 293.Keilp J.G., Sweeney J.A., Jacobsen P., Solomon C., St Louis L., Deck M., Frances A., Mann J.J. Cognitive impairment in schizophrenia: specific relations to ventricular size and negative symptomatology. Biol. Psychiatry. 1988;24(1):47–55. doi: 10.1016/0006-3223(88)90120-5. [http://dx.doi.org/10.1016/0006-3223(88)90120-5]. [PMID: 3370277]. [DOI] [PubMed] [Google Scholar]
- 294.Vita A., De Peri L., Silenzi C., Dieci M. Brain morphology in first-episode schizophrenia: a meta-analysis of quantitative magnetic resonance imaging studies. Schizophr. Res. 2006;82(1):75–88. doi: 10.1016/j.schres.2005.11.004. [http://dx.doi.org/10.1016/j.schres.2005.11.004]. [PMID: 16377156]. [DOI] [PubMed] [Google Scholar]
- 295.Lawrie S.M., Abukmeil S.S. Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. Br. J. Psychiatry. 1998;172:110–120. doi: 10.1192/bjp.172.2.110. [http://dx.doi.org/10.1192/bjp.172.2.110]. [PMID: 9519062]. [DOI] [PubMed] [Google Scholar]
- 296.Goldman A.L., Pezawas L., Mattay V.S., Fischl B., Verchinski B.A., Chen Q., Weinberger D.R., Meyer-Lindenberg A. Widespread reductions of cortical thickness in schizophrenia and spectrum disorders and evidence of heritability. Arch. Gen. Psychiatry. 2009;66(5):467–477. doi: 10.1001/archgenpsychiatry.2009.24. [http://dx.doi.org/10.1001/archgenpsychiatry.2009.24]. [PMID: 19414706]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Thune J.J., Uylings H.B., Pakkenberg B. No deficit in total number of neurons in the prefrontal cortex in schizophrenics. J. Psychiatr. Res. 2001;35(1):15–21. doi: 10.1016/s0022-3956(00)00043-1. [http://dx.doi.org/10.1016/S0022-3956(00)00043-1]. [PMID: 11287052]. [DOI] [PubMed] [Google Scholar]
- 298.Selemon L.D., Rajkowska G., Goldman-Rakic P.S. Abnormally high neuronal density in the schizophrenic cortex. A morphometric analysis of prefrontal area 9 and occipital area 17. Arch. Gen. Psychiatry. 1995;52(10):805–818. doi: 10.1001/archpsyc.1995.03950220015005. [http://dx.doi.org/10.1001/archpsyc.1995.03950220015005]. [PMID: 7575100]. [DOI] [PubMed] [Google Scholar]
- 299.Selemon L.D., Rajkowska G., Goldman-Rakic P.S. Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of a three-dimensional, stereologic counting method. J. Comp. Neurol. 1998;392(3):402–412. [http://dx.doi.org/10.1002/(SICI)1096-9861(19980316)392:3<402:AID-CNE9>3.0.CO;2-5]. [PMID: 9511926]. [PubMed] [Google Scholar]
- 300.Roberts R.C., Conley R., Kung L., Peretti F.J., Chute D.J. Reduced striatal spine size in schizophrenia: a postmortem ultrastructural study. Neuroreport. 1996;7(6):1214–1218. doi: 10.1097/00001756-199604260-00024. [http://dx.doi.org/10.1097/00001756-199604260-00024]. [PMID: 8817535]. [DOI] [PubMed] [Google Scholar]
- 301.Rosoklija G., Toomayan G., Ellis S.P., Keilp J., Mann J.J., Latov N., Hays A.P., Dwork A.J. Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: preliminary findings. Arch. Gen. Psychiatry. 2000;57(4):349–356. doi: 10.1001/archpsyc.57.4.349. [http://dx.doi.org/10.1001/archpsyc.57.4.349]. [PMID: 10768696]. [DOI] [PubMed] [Google Scholar]
- 302.Garey L.J., Ong W.Y., Patel T.S., Kanani M., Davis A., Mortimer A.M., Barnes T.R., Hirsch S.R. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry. 1998;65(4):446–453. doi: 10.1136/jnnp.65.4.446. [http://dx.doi.org/10.1136/jnnp.65.4.446]. [PMID: 9771764]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Sweet R. A., Henteleff R. A., Zhang W., Sampson A. R., Lewis D. A. Reduced dendritic spine density in auditory cor-tex of subjects with schizophrenia. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2009;34(4):374–389. doi: 10.1038/npp.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Glantz L.A., Lewis D.A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry. 2000;57(1):65–73. doi: 10.1001/archpsyc.57.1.65. [http://dx.doi.org/10.1001/archpsyc.57.1.65]. [PMID: 10632234]. [DOI] [PubMed] [Google Scholar]
- 305.Konopaske G.T., Lange N., Coyle J.T., Benes F.M. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry. 2014;71(12):1323–1331. doi: 10.1001/jamapsychiatry.2014.1582. [http://dx.doi.org/10.1001/jamapsychiatry.2014.1582]. [PMID: 25271938]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 1982-1983;17(4):319–334. doi: 10.1016/0022-3956(82)90038-3. [http://dx.doi.org/10.1016/0022-3956(82) 90038-3]. [PMID: 7187776]. [DOI] [PubMed] [Google Scholar]
- 307.Davis J., Eyre H., Jacka F.N., Dodd S., Dean O., McEwen S., Debnath M., McGrath J., Maes M., Amminger P., McGorry P.D., Pantelis C., Berk M. A review of vulnerability and risks for schizophrenia: Beyond the two hit hypothesis. Neurosci. Biobehav. Rev. 2016;65:185–194. doi: 10.1016/j.neubiorev.2016.03.017. [http://dx.doi.org/10.1016/j.neubiorev. 2016.03.017]. [PMID: 27073049]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Sekar A., Bialas A.R., de Rivera H., Davis A., Hammond T.R., Kamitaki N., Tooley K., Presumey J., Baum M., Van Doren V., Genovese G., Rose S.A., Handsaker R.E., Daly M.J., Carroll M.C., Stevens B., McCarroll S.A. Schizophrenia Working Group of the Psychiatric Genomics Consortium. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530(7589):177–183. doi: 10.1038/nature16549. [http://dx.doi.org/10.1038/nature16549]. [PMID: 26814963]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Prasad K.M., Chowdari K.V., D’Aiuto L.A., Iyengar S., Stanley J.A., Nimgaonkar V.L. Neuropil contraction in relation to Complement C4 gene copy numbers in independent cohorts of adolescent-onset and young adult-onset schizophrenia patients-a pilot study. Transl. Psychiatry. 2018;8(1):134. doi: 10.1038/s41398-018-0181-z. [http://dx.doi.org/10.1038/s41398-018-0181-z]. [PMID: 30026462]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Laskaris L., Zalesky A., Weickert C.S., Di Biase M.A., Chana G., Baune B.T., Bousman C., Nelson B., McGorry P., Everall I., Pantelis C., Cropley V. Investigation of pe-ripheral complement factors across stages of psychosis. Schizophr. Res. 2018;204:30–37. doi: 10.1016/j.schres.2018.11.035. [PMID: 30527272]. [DOI] [PubMed] [Google Scholar]
- 311.Martins-de-Souza D., Gattaz W.F., Schmitt A., Rewerts C., Maccarrone G., Dias-Neto E., Turck C.W. Prefrontal cortex shotgun proteome analysis reveals altered calcium homeostasis and immune system imbalance in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2009;259(3):151–163. doi: 10.1007/s00406-008-0847-2. [http://dx.doi.org/10.1007/s00406-008-0847-2]. [PMID: 19165527]. [DOI] [PubMed] [Google Scholar]
- 312.Gupta D.S., McCullumsmith R.E., Beneyto M., Haroutunian V., Davis K.L., Meador-Woodruff J.H. Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse. 2005;57(3):123–131. doi: 10.1002/syn.20164. [http://dx.doi.org/10.1002/syn.20164]. [PMID: 15945063]. [DOI] [PubMed] [Google Scholar]
- 313.Ayoub M.A., Angelicheva D., Vile D., Chandler D., Morar B., Cavanaugh J.A., Visscher P.M., Jablensky A., Pfleger K.D., Kalaydjieva L. Deleterious GRM1 mutations in schizophrenia. PLoS One. 2012;7(3):e32849. doi: 10.1371/journal.pone.0032849. [http://dx.doi.org/10.1371/journal.pone.0032849]. [PMID: 22448230]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Kovács T., Kelemen O., Kéri S. Decreased fragile X mental retardation protein (FMRP) is associated with lower IQ and earlier illness onset in patients with schizophrenia. Psychiatry Res. 2013;210(3):690–693. doi: 10.1016/j.psychres.2012.12.022. [http://dx.doi.org/10.1016/j.psychres.2012.12.022]. [PMID: 23333116]. [DOI] [PubMed] [Google Scholar]
- 315.Pouget J.G., Goncalves V.F. Schizophrenia Working Group of the Psychiatric Genomics, C.; Spain, S. L.; Finucane, H. K.; Raychaudhuri, S.; Kennedy, J. L.; Knight, J., Genome-Wide Association Studies Suggest Limited Immune Gene Enrich-ment in Schizophrenia Compared to 5 Autoimmune Diseases. Schizophr. Bull. 2016;42(5):1176–1184. doi: 10.1093/schbul/sbw059. [http://dx.doi.org/10.1093/schbul/sbw059]. [PMID: 27242348]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Rajarajan P., Borrman T., Liao W., Schrode N., Flaherty E., Casiño C., Powell S., Yashaswini C., LaMarca E.A., Kassim B., Javidfar B., Espeso-Gil S., Li A., Won H., Geschwind D.H., Ho S.M., MacDonald M., Hoffman G.E., Roussos P., Zhang B., Hahn C.G., Weng Z., Brennand K.J., Akbarian S. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science. 2018;362(6420):eaat4311. doi: 10.1126/science.aat4311. [http://dx.doi.org/10.1126/science.aat4311]. [PMID: 30545851]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Schwarz E., Izmailov R., Liò P., Meyer-Lindenberg A. Protein Interaction Networks Link Schizophrenia Risk Loci to Synaptic Function. Schizophr. Bull. 2016;42(6):1334–1342. doi: 10.1093/schbul/sbw035. [http://dx.doi.org/10.1093/schbul/sbw035]. [PMID: 27056717]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Bergon A., Belzeaux R., Comte M., Pelletier F., Hervé M., Gardiner E.J., Beveridge N.J., Liu B., Carr V., Scott R.J., Kelly B., Cairns M.J., Kumarasinghe N., Schall U., Blin O., Boucraut J., Tooney P.A., Fakra E., Ibrahim E.C. CX3CR1 is dysregulated in blood and brain from schizophrenia patients. Schizophr. Res. 2015;168(1-2):434–443. doi: 10.1016/j.schres.2015.08.010. [http://dx.doi.org/10.1016/j.schres.2015.08.010]. [PMID: 26285829]. [DOI] [PubMed] [Google Scholar]
- 319.Ishizuka K., Fujita Y., Kawabata T., Kimura H., Iwayama Y., Inada T., Okahisa Y., Egawa J., Usami M., Kushima I., Uno Y., Okada T., Ikeda M., Aleksic B., Mori D., Someya T., Yoshikawa T., Iwata N., Nakamura H., Yamashita T., Ozaki N. Rare genetic variants in CX3CR1 and their contribution to the increased risk of schizophrenia and autism spectrum disorders. Transl. Psychiatry. 2017;7(8):e1184. doi: 10.1038/tp.2017.173. [http://dx.doi.org/10.1038/tp.2017.173]. [PMID: 28763059]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Stefansson H., Ophoff R.A., Steinberg S., Andreassen O.A., Cichon S., Rujescu D., Werge T., Pietilainen O.P., Mors O., Mortensen P.B., Sigurdsson E., Gustafsson O., Nyegaard M., Tuulio-Henriksson A., Ingason A., Hansen T., Suvisaari J., Lonnqvist J., Paunio T., Borglum A.D., Hartmann A., Fink-Jensen A., Nordentoft M., Hougaard D., Norgaard-Pedersen B., Bottcher Y., Olesen J., Breuer R., Moller H.J., Giegling I., Rasmussen H.B., Timm S., Mat-theisen M., Bitter I., Rethelyi J.M., Magnusdottir B.B., Sigmundsson T., Olason P., Masson G., Gulcher J.R., Har-aldsson M., Fossdal R., Thorgeirsson T.E., Thorsteinsdot-tir U., Ruggeri M., Tosato S., Franke B., Strengman E., Kiemeney L.A., Genetic R. Outcome in, P.; Melle, I.; Dju-rovic, S.; Abramova, L.; Kaleda, V.; Sanjuan, J.; de Frutos, R.; Bramon, E.; Vassos, E.; Fraser, G.; Ettinger, U.; Picchioni, M.; Walker, N.; Toulopoulou, T.; Need, A. C.; Ge, D.; Yoon, J. L.; Shianna, K. V.; Freimer, N. B.; Cantor, R. M.; Murray, R.; Kong, A.; Golimbet, V.; Carracedo, A.; Arango, C.; Costas, J.; Jonsson, E. G.; Terenius, L.; Agartz, I.; Petursson, H.; Nothen, M. M.; Rietschel, M.; Matthews, P. M.; Muglia, P.; Peltonen, L.; St Clair, D.; Goldstein, D. B.; Stefansson, K.; Collier, D. A. Common variants conferring risk of schizo-phrenia. Nature. 2009;460(7256):744–747. doi: 10.1038/nature08186. [http://dx.doi.org/10.1038/nature08186]. [PMID: 19571808]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O’Donovan M.C., Sullivan P.F., Sklar P. International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [http://dx.doi.org/10.1038/nature08185]. [PMID: 19571811]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Galimberti D., Dell’Osso B., Fenoglio C., Villa C., Cortini F., Serpente M., Kittel-Schneider S., Weigl J., Neuner M., Volkert J., Leonhard C., Olmes D.G., Kopf J., Cantoni C., Ridolfi E., Palazzo C., Ghezzi L., Bresolin N., Altamura A.C., Scarpini E., Reif A. Progranulin gene variability and plasma levels in bipolar disorder and schizophrenia. PLoS One. 2012;7(4):e32164. doi: 10.1371/journal.pone.0032164. [http://dx.doi.org/10.1371/journal.pone.0032164]. [PMID: 22505994]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Farhy-Tselnicker I., van Casteren A.C.M., Lee A., Chang V.T., Aricescu A.R., Allen N.J. Astrocyte-Secreted Glypican 4 Regulates Release of Neuronal Pentraxin 1 from Axons to Induce Functional Synapse Formation. Neuron. 2017;96(2):428–445. doi: 10.1016/j.neuron.2017.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Potkin S.G., Macciardi F., Guffanti G., Fallon J.H., Wang Q., Turner J.A., Lakatos A., Miles M.F., Lander A., Vawter M.P., Xie X. Identifying gene regulatory networks in schizophrenia. Neuroimage. 2010;53(3):839–847. doi: 10.1016/j.neuroimage.2010.06.036. [http://dx.doi.org/10.1016/j.neuroimage.2010.06.036]. [PMID: 20600988]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Laskaris L.E., Di Biase M.A., Everall I., Chana G., Christopoulos A., Skafidas E., Cropley V.L., Pantelis C. Microglial activation and progressive brain changes in schizophrenia. Br. J. Pharmacol. 2016;173(4):666–680. doi: 10.1111/bph.13364. [http://dx.doi.org/10.1111/bph.13364]. [PMID: 26455353]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Trépanier M.O., Hopperton K.E., Mizrahi R., Mechawar N., Bazinet R.P. Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol. Psychiatry. 2016;21(8):1009–1026. doi: 10.1038/mp.2016.90. [http://dx.doi.org/10.1038/mp.2016.90]. [PMID: 27271499]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Pasternak O., Kubicki M., Shenton M.E. In vivo imaging of neuroinflammation in schizophrenia. Schizophr. Res. 2016;173(3):200–212. doi: 10.1016/j.schres.2015.05.034. [http://dx.doi.org/10.1016/j.schres.2015.05.034]. [PMID: 26048294]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.van Kesteren C.F., Gremmels H., de Witte L.D., Hol E.M., Van Gool A.R., Falkai P.G., Kahn R.S., Sommer I.E. Immune involvement in the pathogenesis of schizophrenia: a meta-analysis on postmortem brain studies. Transl. Psychiatry. 2017;7(3):e1075. doi: 10.1038/tp.2017.4. [http://dx.doi.org/10.1038/tp.2017.4]. [PMID: 28350400]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Quidé Y., Bortolasci C.C., Spolding B., Kidnapillai S., Watkeys O.J., Cohen-Woods S., Berk M., Carr V.J., Walder K., Green M.J. Association between childhood trauma exposure and pro-inflammatory cytokines in schizophrenia and bipolar-I disorder. Psychol. Med. 2018:1–9. doi: 10.1017/S0033291718003690. [http://dx.doi.org/10.1017/S0033291718003690]. [PMID: 30560764]. [DOI] [PubMed] [Google Scholar]
- 330.Gallego J.A., Blanco E.A., Husain-Krautter S., Madeline Fagen E., Moreno-Merino P., Del Ojo-Jiménez J.A., Ahmed A., Rothstein T.L., Lencz T., Malhotra A.K. Cytokines in cerebrospinal fluid of patients with schizophrenia spectrum disorders: New data and an updated meta-analysis. Schizophr. Res. 2018;202:64–71. doi: 10.1016/j.schres.2018.07.019. [http://dx.doi.org/10.1016/j.schres.2018.07.019]. [PMID: 30025760]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Goldsmith D.R., Rapaport M.H., Miller B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry. 2016;21(12):1696–1709. doi: 10.1038/mp.2016.3. [http://dx.doi.org/10.1038/mp.2016.3]. [PMID: 26903267]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Miller B.J., Buckley P., Seabolt W., Mellor A., Kirkpatrick B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry. 2011;70(7):663–671. doi: 10.1016/j.biopsych.2011.04.013. [http://dx.doi.org/10.1016/j.biopsych.2011.04.013]. [PMID: 21641581]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Brown A.S. Prenatal infection as a risk factor for schizophrenia. Schizophr. Bull. 2006;32(2):200–202. doi: 10.1093/schbul/sbj052. [http://dx.doi.org/10.1093/schbul/sbj052]. [PMID: 16469941]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Mattei D., Ivanov A., Ferrai C., Jordan P., Guneykaya D., Buonfiglioli A., Schaafsma W., Przanowski P., Deuther-Conrad W., Brust P., Hesse S., Patt M., Sabri O., Ross T.L., Eggen B.J.L., Boddeke E.W.G.M., Kaminska B., Beule D., Pombo A., Kettenmann H., Wolf S.A. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl. Psychiatry. 2017;7(5):e1120. doi: 10.1038/tp.2017.80. [http://dx.doi.org/10.1038/tp.2017.80]. [PMID: 28485733]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Coiro P., Padmashri R., Suresh A., Spartz E., Pendyala G., Chou S., Jung Y., Meays B., Roy S., Gautam N., Alnouti Y., Li M., Dunaevsky A. Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain Behav. Immun. 2015;50:249–258. doi: 10.1016/j.bbi.2015.07.022. [http://dx.doi.org/10.1016/j.bbi.2015.07.022]. [PMID: 26218293]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Pendyala G., Chou S., Jung Y., Coiro P., Spartz E., Pad-mashri R., Li M., Dunaevsky A. Maternal immune Activation causes Behavioral impairments and altered cerebellar cytokine and synaptic protein expression. Neuropsychopharmacology. 2017;42(7):1435–1446. doi: 10.1038/npp.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Brennand K.J., Simone A., Jou J., Gelboin-Burkhart C., Tran N., Sangar S., Li Y., Mu Y., Chen G., Yu D., McCarthy S., Sebat J., Gage F.H. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473(7346):221–225. doi: 10.1038/nature09915. [http://dx.doi.org/10.1038/nature09915]. [PMID: 21490598]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Sellgren C.M., Gracias J., Watmuff B., Biag J.D., Thanos J.M., Whittredge P.B., Fu T., Worringer K., Brown H.E., Wang J., Kaykas A., Karmacharya R., Goold C.P., Sheridan S.D., Perlis R.H. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019;22(3):374–385. doi: 10.1038/s41593-018-0334-7. [http://dx.doi.org/10.1038/s41593-018-0334-7]. [PMID: 30718903]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Chen X., Xiong Z., Li Z., Yang Y., Zheng Z., Li Y., Xie Y., Li Z. Minocycline as adjunct therapy for a male patient with deficit schizophrenia. Neuropsychiatr. Dis. Treat. 2018;14:2697–2701. doi: 10.2147/NDT.S179658. [http://dx.doi.org/10.2147/NDT.S179658]. [PMID: 30349268]. [DOI] [PMC free article] [PubMed] [Google Scholar]