

Abstract
After more than a century from its discovery, valproic acid (VPA) still represents one of the most efficient antiepi-leptic drugs (AEDs). Pre and post-synaptic effects of VPA depend on a very broad spectrum of actions, including the regu-lation of ionic currents and the facilitation of GABAergic over glutamatergic transmission. As a result, VPA indirectly mod-ulates neurotransmitter release and strengthens the threshold for seizure activity. However, even though participating to the anticonvulsant action, such mechanisms seem to have minor impact on epileptogenesis. Nonetheless, VPA has been reported to exert anti-epileptogenic effects. Epigenetic mechanisms, including histone deacetylases (HDACs), BDNF and GDNF modulation are pivotal to orientate neurons toward a neuroprotective status and promote dendritic spines organization. From such broad spectrum of actions comes constantly enlarging indications for VPA. It represents a drug of choice in child and adult with epilepsy, with either general or focal seizures, and is a consistent and safe IV option in generalized convulsive sta-tus epilepticus. Moreover, since VPA modulates DNA transcription through HDACs, recent evidences point to its use as an anti-nociceptive in migraine prophylaxis, and, even more interestingly, as a positive modulator of chemotherapy in cancer treatment. Furthermore, VPA-induced neuroprotection is under investigation for benefit in stroke and traumatic brain injury. Hence, VPA has still got its place in epilepsy, and yet deserves attention for its use far beyond neurological diseases. In this review, we aim to highlight, with a translational intent, the molecular basis and the clinical indications of VPA.
Keywords: Valproic acid, epilepsy, epileptogenesis, neuroprotection, pharmacology, epigenetics
1. INTRODUCTION
Valproate was introduced in clinical practice about 50 years ago, and its efficacy and tolerability profiles have been well reported in preclinical and clinical settings. It is a mainstay of antiepileptic therapy because of a broad spectrum of effectiveness for a wide range of seizures and epileptic syndromes. Indeed, data from several clinical trials suggest that valproate has the broadest spectrum of anticonvulsant action compared to all currently available antiepileptic drugs (AEDs), both in adults and children suffering from epilepsy [1]. The aim of this review is to analyze both clinical and preclinical aspects regarding the use of valproic acid (N-dipropylacetic acid or VPA), moving from preclinical evidences of anticonvulsant activity and neuroprotection to clinical data on efficacy and tolerability profile both in the pediatric and adult population. We examined specific review articles, preclinical studies, systematic reviews, textbooks, meta-analysis, retrospective studies and evinced that because of a good compromise between multiple clinical uses and tolerability of side effects, beyond specific precautions in female of childbearing potential [2], VPA is a first choice drug to this day in the treatment of many epileptic and non-epileptic diseases, and still deserves attention for the implication of its epigenetic effect in several fields, from epileptogenesis to cancer treatment [3, 4] .
2. HISTORICAL BACKGROUND
VPA is a unique drug, and is to date one of the most prescribed AEDs worldwide. VPA is a branched short-chain fatty acid deriving from valeric acid, a low molecular weight carboxylic acid. In 1882, Beverley Burton, an American chemist, was the first to synthetize VPA, which was considered at first an organic solvent [5]. However, VPA antiepileptic property was discovered after more than 50 years, in 1962, when it was tested as a solvent for other molecules being checked for potential anticonvulsant activity [6]. After laboratory studies demonstrated its anticonvulsant activity, the first clinical trial using VPA in epilepsy was performed in 1964 [7]. The drug made its way to the European market under the brand Depakine in France in 1967, followed by the UK in 1973 and other European countries in the following decade. Being liquid, it is used as a coniugated sodium salt, constituting a water-soluble powder. After Food and Drug Administration (FDA) approval in 1978, it also became available in the US [8].
3. PHARMACOKINETICS AND PHARMACO- DYNAMICS
The bioavailability of VPA ranges from 96 to 100% for all commonly used formulations, which include normal and sustained-release tablets, film-coated tablets, capsules and oral or intravenous solutions. VPA bioavailability is maintained during chronic treatment when its absorption is even quicker compared to absorption after a single dose. Absorption is consistently delayed if the drug is taken 2 to 3 hours after a meal, which explains why it tends to be significantly slower in the afternoon, after lunch, than in the morning, after breakfast. VPA highly binds to proteins (87-95%), resulting in low clearance rates (6-20 ml/h/Kg) [8]. However, protein binding depends on VPA concentration, so that if the serum level of VPA is above the therapeutic range (>600 µmol/L, 80 µg/mL), protein binding may decrease up to 67%. Some conditions, usually linked with hypoproteinemia, are likely to reduce VPA protein bound fraction: renal disease, liver disease, old age, pregnancy and use of other protein-bound drugs, depending on competitor’s affinity to plasma proteins. In human beings, VPA is metabolized via three major pathways: glucuronidation, beta-oxidation and cytochrome P450 (CYP)-mediated oxidation. The first two pathways are predominant, since they made up for 50% and 40% of the VPA metabolized respectively, while CYP dependent oxidation represents a minor route, managing less than 10% of VPA [9]. Valproate glucuronide is the principal metabolite of VPA in urine (30-50%), and is not considered to be toxic for cells. On the contrary, some of the products of VPA metabolism produced by mitochondrial and non-mitochondrial pathways are known to be hepatotoxic. Indeed, a pivotal step in VPA metabolism is the CYP2C9, 2A6 and 2B6 mediated production of 4-ene-VPA, toxic to cells. VPA half-life ranges from 9 to 18 hours, with shorter timings, from 5 to 12 hours, only observed with concomitant treatment with enzyme-inducing drugs, including phenytoine (PHT), carbamazepine (CBZ) and barbiturates [10]. VPA enhances γ aminobutyric acid (GABA) synthesis and release, leading to the potentiation of GABA-ergic transmission in specific brain regions [11]. At the same time, it also reduces the release of the excitatory molecules, such as β-hydroxybutyric acid, and reduces neuronal excitation due to the activation of N-metyl-D-aspartate (NMDA) glutamate receptors [12]. In addition to these properties, the anticonvulsant effect of VPA also derives from the attenuation of the high frequency neuronal firing, obtained via the blockade of voltage gated ionic channels, including sodium, potassium and calcium channels [13]. Results from preclinical and clinical studies also suggest that VPA modulates dopaminergic (DA-ergic) and serotoninergic (5HT-ergic) transmission, paramount for its effectiveness in psychiatric disorders, as well as in neurological disorders beyond epilepsy [14]. Recently VPA has been shown to inhibit the histone deacetylases (HDAC), especially HDAC1, potentially regulating the expression of genes participating in apoptosis and antitumor action. Thus, VPA has been proposed as a possible anticancer drug, able to boost the effects of chemotherapeutics [15]. VPA is unique among all AEDs for the wide spectrum of effectiveness against all the types of seizure and epileptic syndromes, both in pediatric and adult patients. In particular, it has been tested in both generalized (tonic-clonic, absences and myoclonic) and focal seizures, and it has been found effective in Lennox-Gastaut, West and Dravet syndrome Table (1). The efficacy, safety and tolerability of intravenous (IV) VPA has also been evaluated in the management of generalized convulsive status epilepticus (GCSE) [16] Table (1). VPA is a multitarget drug with strong antinociceptive and anti-inflammatory action at low dosages, with such effects deriving from the inhibition of the TNF-α related pathways. Hence, it is currently used in migraine prophylaxis as well as for the treatment of several psychiatric diseases, including bipolar and mood disorders [3, 17]. In the last decades, the neuroprotective effects of VPA have been reported in a broad variety of acute CNS injuries models, including stroke and hypoxia, traumatic brain injury and spinal cord injury [18].
Table 1. VPA monotherapy in epilepsy from 1978 to 2018.
| Seizure Type | Adult/Infant | Patient Number | Evidences | Refs. |
|---|---|---|---|---|
| AS | I | 25 | ** | [165] |
| PGTC, FS | A | 181 | ** | [177] |
| GTC, FS | A | 88 | ** | [176] |
| AS | I | 7 | *** | [164] |
| SGTC, FIAS | A | 480 | **and* | [179] |
| GS, GTC, AS, LG | A | N/A (review) | ***and** | [165] |
| SGTC, FIAS | A | 143 | *** | [180] |
| VSE | A | N/A (review) | *** | [182] |
| GS | A | 716 (RCT) | *** | [178] |
| SE | A and I | N/A (review) | ** | [16] |
| AS | A | N/A (review) | *** | [166] |
Abbreviations: AS=absence seizure; GTC=generalised tonic clonic seizures; GS=generalized seizures, or myoclonic seizures, tonic clonic seizures, myoclonic astatic seizures; FIAS=focal impaired awareness seizures (previously known as complex partial seizures; LG=Lennox-Gastaut; PGTC=primary generalized tonic-clonic seizures; FS=focal seizures; SGTC=secondary generalised tonic-clonic seizures; SE=status epilepticus (convulsive and non convulsive); VSE=visual sensitive epilepsies: absences, myoclonic seizures, GTC seizures precipitated by photic stimulation in idiopathic generalized epilepsies and in Dravet syndrome. Evidences: *= inferior efficacy in seizure control compared to other antiepileptic drugs (AEDs); **=comparable efficacy in seizure control with that of other AEDs (not considering adverse reactions); ***=complete seizure control, or better efficacy than other drugs, or therapy of choice.
4. MECHANISM OF ACTION AND EFFECTS IN EXPERIMENTAL MODELS
4.1. Effect on GABA and Glutamate Systems
During the last half century, a broad spectrum of mechanisms have been discovered to participate in the anti-epileptic, mood-stabilizer and neuroprotective properties of VPA. One of the main mechanisms of VPA concerns a pre and post-synaptic modulation of GABA-ergic transmission Fig. (1). Specifically, VPA increases the inhibitory activity of gamma-aminobutyric acid (GABA) with both pre-synaptic and post-synaptic mechanisms, promoting the availability of synaptic GABA as well as facilitating GABA mediated responses [19]. Moreover, VPA directly acts on GABA receptors, enhancing the stimulus-induced responses of both GABA-A and GABA-B receptors [20]. In particular, through a direct interaction with the benzodiazepines regulatory regions of GABA receptors, VPA delays the extinction of the post-synaptic inhibitory potential due to the activation of GABA-A receptors, and also facilitates the tethering of baclofen to GABA-B receptors [11, 21, 14]. The selective enhancement of GABA-ergic mediated transmission explains the increase in brain GABA concentration under VPA treatment [22, 23]. Beyond influencing the inhibitory GABAergic system, VPA also modulates the transmission of excitatory aminoacids, such as glutamate. In rat models, VPA reduced the NMDA and kainate-induced excitatory responses within the medial prefrontal cortex [24]. Moreover, VPA increases the expression of NR2A and NR2B subunits of the NMDAR, promoting neural cortex neuroplasticity, though controlling excitotoxicity and apoptosis via the overexpression of associated calcium/calmodulin-dependent protein kinase II [25] Fig. (1). Moreover, NR2A/2B expression regulation allows an indirect regulation of Ras-ERK pathway and glutamate receptor trafficking [26]. In mice with Bassoon gene (Bsn) mutation, with irregular neuronal activity and spontaneous persistent cortical seizures also associated with altered NR2A/2B ratio in NMDAR, VPA has been reported to rebalance NR2A/2B ratio, rescue striatal synaptic plasticity, and reduce seizures and mortality [27]. Moreover, the chronic administration of VPA reduced the hippocampal neurons surface expression and synaptic localization of GluR1/2 [28] Fig. (1).
Fig. (1).
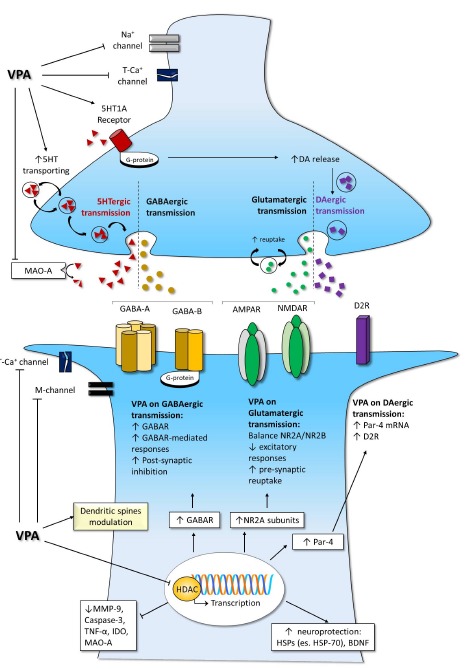
Valproate effects on synaptic cleft and intracellular pathways. The increase in GABAergic transmission pairs with a modulation other aminergic systems; the excitatory ionic channels, such as calcium and sodium channels, are negatively modulated, while potassium currents are enhanced. Moreover, epigenetic mechanisms influence receptor expression and neuroprotective pathways. Legend. AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid –AMPA- receptor; BDNF: brain derived neurotrophic factor; DA: dopamine; D2R: dopamine D2 receptor; GABA: gaba-amino-butirric acid; GABAR: GABA receptors; GABA-A: GABA receptor type A; GABA-B: GABA receptor type B; HSPs: heat shock proteins; HSP-70: heat shock protein 70; K+: potassium; IDO: indoleamine 2,3 dioxygenase; MAO-A: mono-amines oxidase type A; M-channel: low-threshold noninactivating voltage-gated potassium current; MMP-9: metalloproteinases-9; Na+ channel: sodium channel; NMDAR: N-metyl-D-aspartate -NMDA- receptor; NR2A: 2A subunit of NMDAR; NR2B: 2B subunit of NMDAR; Par-4: prostate apoptosis response-4; T-Ca+ channel: low-threshold T-calcium channel; TNF-α: tumor necrosis factor α; VPA: valproate; 5HT: serotonin.
4.2. Effect on Serotonin and Dopamine Systems
VPA has been shown to enhance the extracellular level of serotonin (5-HT) and dopamine (DA) in the hippocampus and striatum, even though this modulation seemed to be unrelated to the antiepileptic effect of VPA [29, 30]. On the contrary, VPA-induced modulation of the DA-ergic and serotoninergic systems was demonstrated relevant for the antipsychotic and neuropsychiatric actions of VPA. Moreover, VPA participates in DA signaling pathways via epigenetic mechanisms, increasing the transcription of the prostate apoptosis response-4 (Par-4) factor [31]. Since Par-4 positively modulates the intracellular DA D2 receptor (D2R) pathway [32], VPA indirectly enhances the activity of the D2R cascade [31]. Furthermore, VPA, activating the 5-HT1A receptors, increases DA-release in pre-frontal areas [1, 14, 30, 33] Fig. (1) and Table 2). Moreover, in animal models of depression, both up-regulation of 5-HT transporter and downregulation of monoamine oxidases type A (MAO-A) have been linked to the antidepressant action of VPA [34].
4.3. Effect on Ionic Channels
VPA action on neuronal firing is concentration and activity dependent. Indeed, VPA has been reported to limit high frequency repetitive firing in cultured neurons at concentrations much lower than those required to depress a normal neuronal cell activity [35]. This effect, critical for the anticonvulsant action of VPA, is linked to the modulation of sodium, calcium and potassium channels, especially with a use-dependent decrease in inward sodium currents [36]. Specifically, VPA blocks both persistent and fast sodium currents [37]. The limitation of persistent sodium channel-induced currents, being selective, confers VPA the property of protecting against seizures, at the same time having minimal interference with normal neuronal function [36]. In peripheral ganglion neurons, VPA has also been shown to block low threshold T calcium channels activity, acting on both pre and post-synaptic level [38]. Moreover, even though only achievable with high concentrations, VPA increases the amplitude of the late potassium outward currents, further increasing the threshold for epileptiform activity [39] Fig. (1). However, it should be noted that the ion channel regulation seems not to have a crucial role in VPA-mediated neuroprotection against ischemic damage in in vitro setting [40] or in experimental epileptogenesis models [41]. Further challenging the role of ionic channel regulation as primary to VPA action, a recent experimental study, addressing membrane permeability in an animal-derived hippocampal epilepsy model, reported that VPA effects were unrelated to the reduction in presynaptic sodium or calcium currents [42]. On the contrary, an anticonvulsant direct effect has been
postulated for the VPA-induced regulation of the M-channel, that generates a low-threshold voltage-gated potassium current restoring neuron rest potential. The preservation of M-channel induced currents has thus been proposed as a possible therapeutic target for antiepileptic drugs, especially VPA [43].
4.4. Epigenetics Effects
Beyond the direct modulation of excitatory and inhibitory synaptic pathways, VPA also has long-term effects. Since VPA effects may take long timing to manifest and persist despite discontinuation, it is unarguable that some of them derive from a central action, the key of which is gene expression modulation [44]. Epigenetic mechanism of VPA is determinant to its several action, such as the anticonvulsant activity, the neuroprotective effects and the ability to modulate neurogenesis [27]. The term “epigenetic” stands for an information that can be inherited during or after cell division, without being strictly related to the DNA pure sequence. Thus, “epigenetic” refers to a heritable DNA accessory data, such as its methylation or histone acetylation status [45, 46]. This epigenetic mechanism leads to a long-lasting over-generation modification in gene expression, crucial to regulatory processes in central nervous system [46]. VPA long-term action counts on epigenetic mechanisms, such as those related to the transcription and regulation of synaptic receptors and several signaling molecules, with high impact on excitotoxic and neuroprotective pathways [44, 47]. Main pathways of VPA epigenetic effects depend on the inhibition of the histone deacetylase (HDAC) and the modulation of brain-derived neurotrophic factor (BDNF) [48] Fig. (1). Indeed, VPA, via HDAC inhibition, modulates DNA demethylation and histone acetylation, resulting in a modification of nucleosome status and chromatin structure [9, 49, 50]. Chromatin decondensation is regulated by several associated proteins, such as Structural Maintenance of Chromatin proteins (SMC), SMC-associated proteins and DNA methyltransferases. With down-stream effects, VPA modulates all of these proteins, regulating the sensitivity of DNA to nucleases [44]. HDACs are divided into 4 classes, with VPA acting only on the first two [51]. DNA methylation, especially when it involves the “promoter” region, corresponds to the inhibition of gene transcription. The inhibition of HDACs leads to chromatin de-condensation and facilitation of DNA transcription. Thus, VPA, inhibiting HDACs, indirectly regulates gene expression [51]. Moreover, VPA can induce mono-, di- and tri-methylation on a lysine 9 region of histone3 (H3), with direct facilitation of transcriptional activity [52]. With different pathways, down-regulating SMCs, inhibiting HDACs and modulating H3 methylation, VPA consistently influences gene expression. VPA chronically enhances the interaction between the transcription factors, including c-Fos and c-Jun, and the activator protein 1 (AP-1), promoting the expression of AP-1 controlled genes [53, 54]. Moreover, AP-1 DNA binding is time and dose-dependently increased by VPA treatment [55]. Thus, VPA progressively furthers the transcription of AP-1 dependent genes, such as Bcl-2, serotonin-2A (5-HT2A) receptor and GAP-43 [55, 56]. Among all others transcription factors, VPA also modulates the CAMP response element-binding protein (CREB) and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB). VPA influence on NF-kB leads to a modulation of the immune response via the decrease of TNF-alpha and IL-6 [57]. More tangled is to date the relationship between VPA and CREB, a transcription factor implicated in phosphorylation in neuronal development, migration, memory and synaptic plasticity [44, 58]. Contrasting results emerged from studies on animal models, in which VPA was shown to decrease the activity of NF-kB in the frontal cortex [59], while other authors described an increase in the hippocampus and cortex mediated by the ERK-pathway, with a subsequent increase in brain derived neurotrophic factor (BDNF) [44]. BDNF is a brain-specific signalling molecule pivotal for cell survival, growth and migration [60]. The expression of BDNF highly varies according to developmental stage [61, 62]. VPA modulation of BDNF expression has been matter of several controversies, mainly related to the differences in study paradigms and to the deduction of BDNF expression from the levels of its codifying mRNA. In particular, rats receiving VPA in the prenatal period were first shown to express high BDNF mRNA levels in the neural tube [54], while in vitro cortical neurons have been shown to have low BDNF transcript when exposed to VPA [62-64]. BDNF protein levels have been only recently investigated by ELISA in mice models, suggesting that early post-natal exposure to VPA reduces BDNF expression by nearly 50%. Moreover, VPA has been shown to preserve a region-specific pattern, decreasing BDNF expression in the cerebellum while not impacting on hippocampal BDNF protein level [62] Fig. (1). As for BDNF, in vivo and in vitro researches showed VPA increases also the glial cell line-derived neurotrophic factor (GDNF), which takes part in the neuroprotective and cognitive-enhancer properties [44, 65]. The regulation of neuronal survival mediated by VPA also derives from the up-regulation of neuron-specific genes. In particular, VPA up-regulates NeuroD, a neuron-located transcription factor increasing the expression of genes promoting neuronal survival, such as Bad and Bcl-2, potentiating GABA-ergic transmission [44, 66]. What is more, VPA suppresses the transcription of both apoptotic (including Bax gene), and pro-inflammatory (such as IL6, Fas-L, and metalloproteinases) signals [67]. Epigenetic mechanisms also take part in the anticonvulsant action of VPA. Indeed, VPA has been reported to modulate the expression of SCN3A, a gene codifying for the α-isoform of the voltage-gated sodium channel III. This channel, that under normal circumstances presents only in the childhood [68], is overexpressed in epileptic patients and seizure animal models as well [9, 69]. SCN3A confers high neuronal excitability, thus it has been implicated in the development of epilepsy, and is considered as a target for interventions aimed at its down-regulation. Indeed, since methylation of SCN3A promoter critically regulates its transcription, it has been implicated in epileptogenesis too. VPA has been shown to downregulate SCN3A expression in a mice model, reducing the methylation of the gene promoter. Thus, VPA was suggested to achieve an anticonvulsant action also through epigenetic pathways based on the downregulation of SNCA3 expression [9]. Such properties are exclusive to VPA since no changes in SCN3A expression has been found with other anticonvulsants, such as CBZ or lamotrigine (LTG) [9]. VPA epigenetic pathways have been shown to participate in epileptogenesis. Antiepileptic drugs (AEDs) are very different one another both in terms of cellular mechanisms and in anticonvulsant activity and efficacy [42, 70]. Investigations both in seizure models and human beings have demonstrated that the anticonvulsant activity of AEDs could highly vary during prolonged treatment. For example, benzodiazepines efficacy decreases over time due to adaptive processes, mainly receptor modifications conferring “functional tolerance”. Older AEDs, such as phenobarbital (PB), can incur in metabolic tolerance, due to an adaptive remodeling of the enzymes deputed to drug elimination or metabolism [23, 70]. On the other side, some AEDs seem to increase their efficacy over time. If for some of them the mechanisms of such curious effect have been reported, such as for the tissue accumulation of PB in patients treated with primidone, for VPA no clear explanation has been proposed, even though epigenetic pathways and epileptogenesis modulation are crucial to it [23]. The term epileptogenesis refers to those artificial or spontaneous processes that lead to recurrent seizures. Stroke, traumatic brain injury, toxic insults and status epilepticus can all be the starting point of epileptogenesis [44, 71]. VPA has been shown to block seizure-induced neurogenesis, and such effect has been directly linked to the epigenetic mechanisms deriving from HDAC inhibition [48] (Table 2). What is more, BDNF expression control is crucial to seizure control and, therein, to epileptogenesis [48, 72-74]. Indeed, BDNF transcription increases in the foci of seizures both in animal models and patients [72, 75]. Moreover, both in animal models and patients, epileptiform activity has been reported to result in a consistent long-term up-regulation of tyrosin kinase tropomyosin-related kinase B (TrkB), the intra-cellular receptor binding BDNF. The consistent facilitation of BDNF binding leads to the promotion of several pathways controlling synaptic plasticity and neurite growth [48, 73, 74, 76, 77]. VPA role in influencing synaptic plasticity also derives from the regulation of the BDNF/TrkB pathway. This pathway has been shown to modulate striatal plasticity in the Bsn model, a mice model developing early-onset epilepsy. As soon as cortical seizures repeat, neuronal cells react with a specific changes in their plasticity patterns, resulting in the facilitation of the feedforward inhibition within the striatal circuitry [48]. TrkB/BDNF are crucial to the induction and preservation of the altered synaptic plasticity, which derives from a severe up-regulation of TrkB and an erroneous distribution of BDNF among fast-spiking interneurons and medium spiny neurons. VPA has been shown to restore the physiologic expression pattern of TrkB, and to promote the correct distribution of BDNF among striatal neurons. Thus, through seizure reduction and epigenetic mechanisms, VPA directly modulates synaptic plasticity [48] Fig. (1), Table 2).
Table 2. Effects of VPA in different animal models of epilepsy.
| VPA Effects in Animal Model | Maximal Electroshock Seizures (MES) |
Pentyl-enetetrazole
Seizure Test (PTZ) |
Amygdala-kindling Model | Kainate Model | Pilocarpine Model | Mice Lacking Bassoon Protein |
|---|---|---|---|---|---|---|
| Animal model characteristic |
acute tonic seizures | acute clonic seizures |
chronic focal seizures | chemically induced SE | chemically induced SE | genetic epilepsy |
| VPA Anti-epileptogenic effects | + | + | + | + | +/- | + |
| VPA Neuroprotective effects | NA | NA | + | + | +/- | + |
| VPA protection from seizure-induced damage | NA | NA | + | + | +/- | + |
| References | 1, 33, 90 | 1, 85, 87 | 92, 93, 94, 108 | 79, 95 | 79, 83, 84, 91 | 27, 28, 99 |
Legend: “NE”=not effective; “+”=VPA effective; “+/-“= inconclusive data on VPA effectiveness; “NA”=data not available.
5. THERAPEUTICAL ASPECTS: FROM EXPERIMENTAL TO CLINICAL EVIDENCES
The development of new AEDs, with improved effectiveness and better tolerability profiles, represents to date the most important goal of neuropharmacology in epilepsy, and relies on preclinical research on animal models. Despite the availability of countless animal models of epileptic seizures and epilepsy, pharmacological studies are rarely performed on the few of the chronic models of epilepsy at our disposal Table (1) [14, 23]. These animal models can be categorized, according to the timing in which they present seizures, in acute or chronic seizures models. However, both categories include paradigms characterized by the induction of seizures or epilepsy using chemical or electrical stimuli in previously healthy and “non-epileptic” animals, mostly rats [41, 71, 78, 79].
5.1. Antiepileptic and Antiepileptogenic Effects
Experimental studies performed in various models of generalized and focal seizures have widely confirmed the antiepileptic effect of VPA [23, 80] Table (2). Despite ineffective in preventing status epilepticus in a pilocarpine model [81, 82], VPA significantly increased the latency to seizure initiation [83, 84], although the observation period (72 hours) was too short to allow a thorough assessment of treatment effectiveness [84] Table (2). In the pentylenetetrazol (PTZ) model of epilepsy [81, 85, 86], VPA acute intraperitoneal administration significantly increased the seizure threshold, with anticonvulsant activity further potentiated by prolonged treatment [87-89] Table (2). The efficacy of VPA was confirmed also in the electroshock-induced seizures model (MES), where VPA dose-dependently reduced seizure frequency [90] Table (2). In an experimental design, based on a kindling model [91, 92], VPA dose-dependently increased the number of after-discharges (AD) required to induce epileptic seizures [93] Table (2). VPA decreased the AD duration and effectively prevented seizures regulating membrane permeability, blocking the voltage-dependent sodium channels and the T-type voltage-activated Ca2+ currents, and also potentiating GABA-mediated inhibition [94]. In the kindling model, VPA also counteracted the neuronal damage induced by prolonged seizures in the hippocampal formation, promoting neuroprotection and preventing the development of behavioral disturbances [80, 92]. Similar effects were reported in the kainate model of status epilepticus [95, 79], despite being not confirmed in the pilocarpine temporal lobe epilepsy (TLE) model [84] Table (2). Moreover, in a rat model of human temporal lobe epilepsy (TLE), VPA efficiently decreased mean seizure frequency and was able to bring to a seizure-free status [96]. In the mouse epilepsy model derived from the manipulation of the Bsn gene, characterized by frequent seizures but long survival [97, 98], VPA significantly reduced seizure frequency and mortality [99] Table (2). Nevertheless, despite suppressing seizures, the vast majority of AEDs is unable to limit the course of the underlying natural history of epilepsy. Thus, even though critical to seizure control, AEDs still lack a clear anti-epileptogenic potential [100]. Among all AEDs, though several have shown neuroprotective properties, only few have been reported to have anti-epileptogenic effects [101]. Indeed, data from the kindling pharmacoresistant epilepsy model, suggest that VPA prevents kindling development and blocks fully kindled seizures, thus acting both as a symptomatic as well as disease-modifying drug [93, 94] Table (2). The fine regulation of glutamatergic and GABAergic pathway and the neuroprotective properties of VPA are crucial to the modulation of epileptogenesis [101, 102]. In particular, epileptic seizures, especially status epilepticus (SE), can produce neuronal damage, and, later on, recurrent spontaneous seizure activity [101, 103]. Thus, the inhibition of neuronal damage, through neuroprotective mechanisms, represents a possible strategy to prevent epileptogenesis [104]. VPA has been widely investigated for its broad spectrum of effects peculiar to this indication. In the kainic acid model of SE, high doses of VPA for 40 days after SE induction, beyond protecting hippocampal neurons from seizure-induced damage, also inhibited following spontaneous activity [95]. Moreover, VPA exposure was associated with prevention of seizure induced cognitive impairment, highlighting the critical role of neuroprotection for both processes [95]. In the same setting, however, PB was ineffective in achieving both neuroprotection and antiepileptogenesis [95]. Even though neuroprotective effects were also provided with gabapentin, other AEDs, such as PB and levetiracetam (LEV), failed to reproduce the effects of VPA, with spontaneous seizure activity and behavioral deficit happening despite high dosages respectively in the kainic acid and amygdala electrical stimulation models [95, 105] Table (2). Further experimental evidences supporting VPA anti-epileptogenic effects derive from different SE models. In particular, in an SE model induced by a sustained electrical stimulation of the basolateral amygdala, VPA was reported to be as effective as a selective blocker of AMPA receptor subtype in preventing neuronal loss. Specific, VPA 24 hours continuous infusion after the end of electrical stimulation significantly provided neuronal hippocampal neuroprotection, comparable to an anti-excitotoxic and glutamate depleting agent such as NS1209 (8-methyl-5-(4-(N,N-dimethylsulfamoyl)phenyl)-6,7,8,9-tetrahydro-1H-pyrrolo [3,2-h]-iso-quinoline-2,3-dione-3-O-(4-hydroxybutyric acid-2-yl)oxime) [106]. Moreover, VPA efficacy has also been shown in a recently developed model of primary generalized seizure, obtained with repeated exposure to flurothyl, a GABA-A receptor antagonist. In this model, characterized by the generalized seizures that remit within 1 month from onset, VPA suppressed spontaneous seizures, blocking the evolution to more complex seizure types, further suggesting an inhibition of epileptogenetic processes [107]. Nevertheless, reports of VPA poor efficacy in preventing epileptogenesis render the issue still matter of debate. In 4 hours sustained SE rat model, achieved with electrical stimulation of amygdala, 4 weeks VPA treatment, even though preventing hippocampal neurodegeneration, did not significantly reduce spontaneous seizure activity [108] Table (2). VPA has also been shown ineffective in preventing further seizures in a pilocarpine-induced SE model [84]. However, poor homogeneity among experimental protocols seems a major issue to overcome to critically assess VPA effects. To date, an educated guess aroused as to whether the timing of VPA treatment might consistently influence epileptogenesis, with some authors suggesting a pivotal role of early VPA exposure to obtain significant neuroprotective effects [101]. Further studies with appropriately designed experimental protocols are needed to shed light on VPA effects and their time-dependence.
5.2. Neuroprotection from Neuronal Damage
“Neuroprotection” refers to the biochemically induced resources to prevent, resist or cope with a specific neuronal hazard. Pathways of neuroprotection can derive from clinical, structural, electrical and molecular cellular changes. Despite the huge research interest in the last decades, we still need to define the neuroprotective role of VPA with well-implemented clinical trials, translating the results of the experimental findings, often providing conflicting results, in the clinical field. The neuroprotective role of VPA in ischemic hazard has been reported to depend on the modulation of HDAC after hypoxic insults [40, 109-112]. Nonetheless, several pathways have been shown to take part in VPA-induced neuroprotection [113]. In rodent models of ischemic middle cerebral artery occlusion (MCAO), VPA produced an HDAC inhibition-mediated suppression of NF-kB and metalloproteinase-9 activation, leading to attenuated post-ischemic brain-blood barrier disruption and reduced brain edema [111, 114]. Furthermore, in the same MCAO model, VPA also reduced the infarct size and the clinical deficit via the up-regulation of heat-shock proteins, especially Hsp-70, and the suppression of caspase-3 expression and activation [114, 115]. The increase in Hsp-70 was also reported in the alternative model of animal brain ischemia, the four-vessel occlusion, together with a decrease in IL-1β and TNF-α, and the upregulation of gelsolin [114, 116, 117]. Evidences from in vitro study on ischemic insults also suggest VPA is highly neuroprotective. Specifically, in an in vitro model of oxygen and glucose deprivation, patch-clamp recording highlighted the protection from in vitro ischemia conferred by VPA treatment [40]. However, contrarily to CBZ and TPM, able to provide neuroprotection through the simultaneous limitation of fast sodium and high-voltage activated calcium channels, VPA reaches a significant neuroprotection at higher doses, and independently from ionic channels regulation, suggesting a prevalent role of intracellular mechanism, such as the regulation of caspases activity [40, 115]. A further pathway, involving microglia, has been implicated in VPA-induced neuroprotection [118]. Neuronal cells cultured in organotypic hippocampal slices (OHSC) have been demonstrated to be consistently affected by N-methyl-D-aspartate (NMDA) induced excitotoxicity [119, 120]. The depletion of microglia enhanced neuronal cell death, especially in dentate gyrus and CA3 region of hippocampal slices, thus proposing microglia to be determinant in neuroprotection [120]. In ischemic and toxic insults, as well as in epilepsy, neuronal death is due to the release of 5’-triphosphate (ATP) in the extracellular space from the heavily activated neurons [121]. VPA has been shown to upregulate the ATP-activated purine receptor P2X7, leading, in microglia, to the release of tumor necrosis factor α (TNF α), boosting the neuroprotective pathway [118]. Thus, even though it still remains to be clarified if such effects depend on HDACs inhibition or DNA demethylation, VPA has been shown to act also on microglia to deliver its neuroprotective effects [118].
Beyond epilepsy and toxic/ischemic insults, VPA-mediated neuroprotection has been widely reported in several animal models of neurodegenerative disorders. Indeed, VPA exerted neuroprotection in a Parkinson’s disease rat model obtained with ventral tegmental area and substantia nigra (SN) injury [122-124]. In this recently developed animal model of Parkinson’s disease, motor and cognitive features result from neuronal loss due to the effects of lactalysin, an irreversible ubiquitin proteasome system (UPS) inhibitor. Lactalysin causes intracytoplasmic accumulation of altered proteins, especially alpha-synuclein, leading to degeneration of both DA-ergic and non-DA-ergic neurons in the SN pars compacta (SNpc) [124]. Alpha-synuclein has been shown to prevent the acetylation of histone proteins, and an imbalance of HDACs and histone acetyltransferase (HATs), the other enzyme deputed to the regulation of histone protein acetylation, has been shown in neurodegenerative diseases [125]. Thus, HDACs have been implicated in Parkinson’s disease [124]. In particular, since HDACs are expressed in the SNpc, VPA has been investigated to assess if, inhibiting HDACs, it could confer neuroprotection. In the lactolysin rat model, the neuronal loss produced by the injection of the irreversible proteasome inhibitor was dose-dependently counteracted with VPA treatment, mainly via the upregulation of neurotrophic factors [123, 124]. Moreover, neuroprotection was not only exerted in the SNpc, but also in the ventral tegmental area [122-124]. Hence, VPA represents a prime line candidate for intra-nigral and extra-nigral neuroprotection [124]. VPA neuroprotective properties are not exclusive to Parkinson’s disease, but have been reported in several other neurodegenerative conditions and models, from Huntington’s disease to amyloid beta-peptide induced neurotoxicity and amyotrophic lateral sclerosis [126, 127]. Regarding the latter, in an ALS animal model, VPA was able to reduce the expression of Homer1b/c, modulating apoptosis and neuronal loss and promoting neuronal protection and survival [128]. Further studies are on the verge of assessing the clinical benefit of VPA use as a neuroprotective agent in humans.
5.3. Cognitive Impairment and Autism Spectrum Disorders
The association between epileptic activity and cognitive function has been highly debated in the last decades [129]. Epigenetic pathways have been implicated also in VPA antidepressant effect, as well as in the modifications in cognitive function. In particular, VPA has been reported to inhibit the expression of indoleamine 2,3 dioxygenase (IDO), modulating the kynurenine pathway of tryptophan metabolism [130, 34]. Nonetheless, the complex epigenetic cascade induced by VPA implies long-term beneficial as well as detrimental effects on neuronal homeostasis and plasticity [99, 131]. Bsn mutants mice have been often used as models, since the defective long-lasting synaptic plasticity in hippocampus, in particular in CA1 area, results in morphological alterations leading to hippocampal learning deficit. In this model, the treatment with VPA rescued physiologic LTP but was unable to restore either the morphological alterations of dendritic spines or the impairment of non-spatial hippocampal memory [99]. Sgobio and colleagues demonstrated that, in control animals, VPA treatment, when started early and longitudinally maintained, produced alterations in morphology of dendritic spines as well as impaired socially transmitted food preference. Such effects have been proposed as a possible explanation for cognitive dysfunction observed in children taking VPA [2, 99]. Both in vivo and in vitro models of epilepsy have confirmed dendritic morphology alterations, including spine loss, in the brain foci of seizures [132]. In epilepsy experimental models, alterations in dendritic morphology in the hippocampus (as well as in other brain areas) have been associated to memory deficits, which can be prevented controlling seizures [132-135]. Thus, hippocampal glutamate transport has been proposed as a possible future target for specifically designed drugs. Interestingly, chronic treatment with VPA leads to a dose-dependent increase in the hippocampal glutamate uptake, with an up-regulation of glutamate specific transporters [136]. Neuroprotection from glutamate induced excitotoxicity has been reported both in vivo and in vitro in several models of glutamatergic injury [126, 137]. In the Bsn mutant mice, the mutation of Bsn gene brings to the lack of a presynaptic structural protein, resulting in morphological alterations involving the pyramidal CA1 hippocampal neurons, including reduced dendrite length and branch nodes, as well as lower spine density on both apical and basal dendrites. VPA is unable to directly restore the original dendritic architecture but rescues the hippocampal synaptic plasticity, which participates in the epilepsy-induced memory deficits [99, 138]. This action, together with the facilitation of mitogen-activated protein kinases pathway, the modulation of neurogenesis and the blockade of cognitive decline induced by hippocampal seizure activity, have all been reported with VPA [139, 140]. In particular, VPA induced HDACs modulation results in inhibited neurogenesis in animal models of status epilepticus, preventing cognitive decline [140]. Positive cognitive effects were found in KA model, were treatment with VPA was superior to other AEDs in preserving spatial learning and memory function, which were evaluated through the modified Morris water-maze swim task [141]. However, in an animal study using the same Morris water-maze swim test paradigm, VPA failed to improve the cognitive performances of rats [108]. On the contrary, in an animal study using the novel object locating test to evaluate hippocampal driven spatial memory, a link between VPA and cognitive deficits has been postulated due to the suppression of hippocampal neurogenesis [142]. Beyond animal models, cognitive deficit was also addressed by studies on human beings, where prospective studies consistently documented that VPA is associated with an increased risk of cognitive impairment in young children, and of in utero malformations [2, 20, 143-147]. In particular, the Neurodevelopmental Effects of Antiepileptic Drugs (NEAD) study reported cognitive impact in children after fetal VPA exposure. Specific, in children of mothers treated with VPA monotherapy, a dose-dependent negative VPA impact was found for several cognitive tests, including IQ, verbal and non-verbal abilities, memory, and executive function [148]. Similar results were also confirmed by further studies, which pointed at VPA as the main factor influencing children IQ, even more than maternal IQ [149]. Following studies further provided evidences for VPA-induced cognitive impairment [133, 150-153]. Moreover, heading back to animal models, even though several risk factors might participate in the process, fetal exposure to VPA during the first trimester of pregnancy resulted in higher incidence of autism in the offspring [153, 154]. Epigenetic mechanisms have also been proposed as the basis of VPA-induced autism spectrum disorders (ASD). VPA exposed animals have become one of the most widely used models of ASD. In such model, ASD-like behavior, with impaired sociability and marble burying, persisted in the following generations when VPA treated animals were mated with naïve females to produce second and third generation [131]. Such effect derives from a transgenerational inheritance of epigenetic modifications. Indeed, VPA impacts on DNA transcription with epigenetic mechanisms, leading to an increased susceptibility of transmitting new traits to the subsequent generations [131]. Since ASD-like behavior has been linked to an imbalance between glutamatergic and GABAergic transmission, experimental models have been tested to investigate whether a modulation of these systems can provide clinical benefit. In particular, very recently, a single treatment of agmatine, an endogenous NMDA receptor antagonist, has been shown to resolve ASD-like behaviour in the VPA model. Thus, agmatine and NMDA receptor agonists have been proposed as targets to restore ERK1/2 activation and resolve ASD-like behaviour [155]. Considered all the provided evidences, the United States Food and Drug Administration and the European Medicines Agency recently revised the indication of VPA treatment, especially in female patients [156-158]. To date, VPA use has been consistently restricted according to the results of retrospective and prospective studies suggesting VPA as a risk factor for ASD and cognitive impairment [2, 146, 147]. Combining the data from these studies with further clinical evidences will help us to define the real weight of the effects of VPA on different stages of neurodevelopment.
5.4. Clinical Efficacy in Epilepsy
VPA is one of the most useful anticonvulsant drugs, and has one of the broadest profile of antiepileptic actions, as well as clinical indications. In fact, its efficacy in both focal and generalised seizures and epilepsy syndromes, especially in paediatric population, has been widely and accurately validated with randomised controlled trials and observational studies [159, 160]. Considering all available evidences, VPA stands out from other existing AEDs for its wide spectrum of activity against almost all seizure types. However, supporting evidences and clinical efficacy vary depending on different epilepsy syndromes. Since the late 1960s and early 1970s VPA has been used in many clinical trials proving its effectiveness in reducing the incidence of both generalized and focal seizures Table (2). Several clinical trials consistently validated the high efficacy of VPA in patients suffering from typical and atypical absence seizures both in childhood and adulthood, showing similar seizure control using VPA or other AEDs [161-164]. In a recent review, Glauser and colleagues [165] showed that, in children suffering from newly diagnosed or untreated absence seizures, VPA initial monotherapy led to satisfactory seizure control, or seizure freedom without intolerable side effects Table (1). Ethosuximide (ESM) and VPA have level A recommendation, while LTG has level C recommendation, for being used as initial monotherapy in newly diagnosed or untreated absence seizure in childhood. To date, the lack of data and the inconclusive ones deriving from the only ad-hoc phase III placebo-controlled trial do not allow us to recommend LEV for absence seizures, even though further studies might pave the way for the implementation of this drug in the near future [165, 166]. Recently, a Cochrane review tried to assess the evidence, deriving from placebo-controlled or drug-versus-drug controlled trials, supporting the effectiveness of ESM, VPA and LTG in treating absence seizures in children and adolescents [167]. Even though absence seizures are considered a relatively common seizure type among children, the authors identified only eight randomised controlled trials to be considered for analysis [165, 168-174], seven of them recruiting 20 to 48 participants. Only one study [165] included a much larger sample. This was a randomised, parallel double-blind controlled trial comparing ESM, LTG and VPA in children receiving a new diagnosis of absence epilepsy. A total of 453 previously untreated patients with typical absence seizure, between seven months and 12 years, were enrolled and followed up to 12 months. The review found some evidence, based on the eight small trials previously mentioned, suggesting that LTG treatment was more likely to result in seizure free status compared with placebo treatment. At the same time, these authors found clear evidence that patients taking ESM or VPA experienced significantly higher seizure free rates compared to patients taking LTG. However, considered the different adverse event profiles of the two drugs, ESM is preferred over VPA in children with absence epilepsy. Nevertheless, it must be underlined that if absence and generalised tonic-clonic seizures co-exist, VPA should be preferred [159]. In 1982 Turnbull and colleagues [175] found that treatment with PHT or VPA achieved a satisfactory control of both primary generalized tonic-clonic seizures (PGTC) and focal seizures, reporting however a lower effectiveness in the control of the latter ones Table (1). Such results were replicated in further studies with the achievement of a good seizure control through therapeutic levels of VPA Table (1) [176]. The “Standard and New Antiepileptic Drugs- SANAD study”, an unblinded randomized controlled trial performed in 2007 in the United Kingdom (UK) [177], estimated and confronted the efficacy and tolerability profile of VPA, LTG and topiramate (TPM) in generalized and unclassified epilepsy Table (1). In accordance with previous observations, these authors suggested that VPA represents the first line treatment for the majority of patients diagnosed with idiopathic generalized epilepsy or unclassifiable seizures, while LTG and TPM should be avoided because of lower effectiveness and tolerability profiles [177]. This study was criticized a few months later because the lack of blinding might have affected the time to treatment failure and because based on a non-prescriptive protocol allowing clinicians to decide drug dosages according to patient’s clinical conditions. Therefore we believe that the SANAD study was extremely useful to describe the drugs characteristics but it is necessary to analyze the longer term outcomes that are important in chronic diseases such as epilepsy. In a multicenter double-blind trial [178] that compared VPA versus CBZ in the treatment of adults with complex partial seizures (CPS) or secondarily generalized tonic-clonic seizures (SGTC), VPA and CBZ had similar efficacy profiles, even though a better control of CPS was achieved with CBZ Table (1). In conclusion, VPA should be considered a first-choice drug for the treatment of generalised onset seizures in adulthood, and its wide spectrum of efficacy recommends its use in several types of epilepsy, even in seizures difficult to classify. Further studies demonstrated VPA efficacy in refractory focal epilepsy, supporting its prescription as a very first-line AEDs to treat scarcely responsive patients [179] Table (1). A series of studies over the past years reported that convulsive and non-convulsive status epilepticus (SE) could be treated with intravenous (IV) VPA in young children [160]. This offered an advantage over more sedating agents, which could cause hypotension or respiratory depression. However, the limited number of available studies regarding the use of VPA in SE and their poor quality makes it difficult to obtain implications for clinical practice. More rigorous randomized controlled trials (RCTs) comparing VPA with other AEDs electively used in SE are needed to state its efficacy and tolerability in clinical practice. In addition VPA is efficient in rare syndromes including West syndrome, Lennox-Gastaut syndrome, myoclonic or myoclonic astatic seizures, absences, myoclonic idiopathic generalized epilepsies, generalized tonic-clonic seizures precipitated by photic stimulation, and in Dravet syndrome [180]. In some children with narrow photosensitivity ranges, preventive measures might avoid seizure onset, whereas in patients with severe photosensitivity, drug treatment is often mandatory. Covanis et al. suggested that VPA could be the first-line drug for photosensitivity associated epilepsies since 85% of patients reach seizure free status under VPA treatment [181] Table (1). Finally, it is important to underline that, in comparison to other AEDs (e.g. CBZ), VPA seems to possess little potential to worsen seizures. Indeed, only anecdotal reports highlighting seizure worsening with VPA are available in literature, with a reason for paradoxical effect of VPA remaining largely unclear [182]. In the last decades, thanks to the variety of mechanisms of action, VPA is gaining attention as a possible treatment for several conditions other than epilepsy. Recently, the U.S. Food and Drug Administration and the European Medicines Agency (FDA/EMA) approved VPA use for the treatment of bipolar disorders and for migraine prophylaxis, and it might also have potential implications in Alzheimer’s disease and in cancer therapy, especially for its epigenetic effects [47].
5.5. Clinical Efficacy in Cancer Treatment
Great interest has aroused in the last decades regarding the possible role of VPA as an adjunct treatment for solid tumours. Several evidences suggest VPA use as a treatment “booster” in several type of cancer. Preclinical data report anticancer effect of VPA over 20 solid tumours, from melanoma to colon cancer cells, and evidences seem to accumulate year after year [4]. In particular, in animal models, VPA, modulating the transcription of genes such as ABCA1, ABCA3 or ABCA7, increased the sensitivity to cisplatin among non-small cell lung cancer cells [183]. VPA epigenetic effects have been implied in the treatment of breast cancer, where, in the near future, its use as an HDAC inhibitors could be combined with chemotherapy to target oncogenes through different pathways [184]. The same aim recently aroused also for squamous cells neoplasms [185]. Moreover, VPA can increase the efficacy of radiotherapy in patients with glioblastoma, and, at the same time, to protect hippocampal neurons from radiotherapy induced sugranular zone apoptosis, preventing cognitive deficit associated with brain irradiation [186]. Quite the opposite, no benefit for glioblastoma treatment was achieved associating VPA with best medical therapy (chemotherapy) in a pooled cohort of patients diagnosed with glioblastoma, deriving from four randomized clinical trials [187]. Further clinical studies are needed in order to define VPA efficacy as an adjunct treatment for solid cancer.
6. MAIN ADVERSE EFFECTS
Although VPA is generally well-tolerated and has a favorable tolerability profile, its use might be limited by reduction in efficacy or by the onset of adverse drug reactions (ADR) [2, 10, 188]. Overall, compared to older and newer AEDs, central nervous system-related ADRs are usually rare; in fact, VPA is not generally associated with drowsiness and fatigability with loss of concentration, and also the detrimental effects on cognition are rare. Moreover, when compared to patients with migraine, epileptic patients seem to present ADRs of antiepileptic drugs, including VPA, far more rarely. In addition, when compared to LTG or TPM, VPA had significantly lower rates of ADRs among epileptic patients [189]. Many systematic reviews, clinical trials and post marketing literature reviews have evaluated the real extent of VPA side effects and their clinical implications including hepatic, gastrointestinal, neurological, hematological, cutaneous, teratogenic and metabolic disorders. The incidence of hepatotoxicity is <1% per 20.000 patients treated with VPA. However, hepatotoxicity is clearly age-dependent: its risk is indeed significantly higher (1:600- 1:800) in children below the age of 2 years, especially if suffering from severe seizure disorders or other neurological diseases, including brain damage and cognitive impairment [190]. Hepatotoxicity usually occurs within the first 6 months from VPA treatment initiation. Liver function tests can be carried out before the beginning of therapy, but often these evaluations are not useful because liver enzymes abnormalities do not always precede hepatotoxicity onset. Therefore, VPA is contraindicated in all conditions exposing to higher risk of hepatotoxicity, in particular in case of coexistence of metabolic disorders, including beta-oxidation or urea cycle impairment, and mitochondrial diseases, such as Leigh-syndrome, mitochondrial encephalopathy lactacidosis and stroke-like episodes, Alpers-Huttenlocher syndrome, ataxia-neuropathy spectrum, myoclonic epilepsy, myopathy and sensory ataxia and myoclonic epilepsy with ragged-red fibers [191, 192]. Clinical features of liver toxicity include apathy, somnolence or altered mental status, anorexia, vomiting, jaundice and possibly seizure worsening, especially during febrile status. Typical signs of VPA-induced hyperammonemic encephalopathy, that can rarely be fatal, are acute consciousness impairment, confusion, somnolence or lethargy, focal or bilateral neurological symptoms or signs as well as seizure worsening [193, 194]. Clinical picture can eventually progress towards severe ataxia, stupor and coma. Neurobehavioral alterations can be erroneously attributed to postictal effects, psychiatric disturbances or non-convulsive status epilepticus, leading to inappropriate increase in VPA dosages [195, 196]. Hyperammoniemic state can also be achieved with iv valproate administration [197]. In case the clinical picture raises the suspicion of urea cycle enzymatic defect, metabolic screening should be performed well in advance of starting VPA treatment [198]. In a recent randomized controlled clinical trial of newly diagnosed childhood absence epilepsy, VPA therapy was discontinued mostly because of its neurological, behavioral and psychiatric adverse effects [165]. Postural tremor is another adverse event of valproate, and often mimics essential tremor [10]. No definite relationship has been demonstrated between VPA dose and the onset of tremor, with observational data suggesting an increased prevalence of this adverse events among women [199]. Dizziness, memory problems, insomnia and nystagmus have been reported as common side effects that generally resolve with drug dose adjustments or discontinuation [200]. Headache, confusion, drowsiness, and tiredness have also been described with VPA [201] whereas delirium and dementia have been reported in a few case reports [202]. Gastrointestinal disturbances are relatively common in patients receiving VPA, with most frequently reported symptoms including nausea and vomiting, dysphagia and diarrhea [202, 203]. Abdominal pain and tenderness, independently from abdominal distension, vomiting, diarrhea, lethargy and apathy must be considered as alarm signs for possible pancreatitis, in particular in the pediatric population. Hence, screening with sensitive blood tests such as serum amylase is recommended, as well as dosage of serum lipases, more sensitive and lasting at elevated levels for prolonged time [204, 205]. This important and severe adverse effect develops generally within the first months of therapy, especially in young children, more often if treated with multiple therapies. Although no individual predisposition to VPA hematologic toxicity has been identified, a wide spectrum of hematologic abnormalities have been reported during this treatment ranging from transient immune thrombocytopenia to isolated neutropenia, red cell aplasia and bone marrow failure [206-210]. Hypersensitivity syndrome reactions (HSR) represent very rare idiosyncratic ADRs which can develop independently from dosage, timing, frequency, posology, but can depend on host peculiar conditions [200, 211]. Clinical manifestations of this rare and severe syndrome include fever, skin rash and multiple organ involvement, in particular liver impairment and nephritis. Even rarer adverse events are hair loss and hair texture alterations, which usually occur in the first month of treatment to find a spontaneous resolution in following months, gait disturbances [212], delirium [213], and parkinsonism [214].
6.1. Weight Changes and Endocrine Disturbances
Weight gain represents a possible adverse event of VPA and, being often marked, actually impacts on patient quality of life [215]. Despite being extremely heterogeneous among patients taking VPA, it is clear that in female patients, which are more prone to develop it especially during puberty [216], it can be frequent and disturbing, with significant detrimental effect on therapy compliance and quality of life. Moreover, sometimes the increase in weight can itself limit the prescription of VPA in clinical practice [217, 218]. Indeed, among adolescent girls, excessive weight gain can cause serious psychological disturbances as well as facilitate the development of important endocrinological abnormalities [219]. The mechanism through which VPA may induce an increase in body weight is still debated. However, among the several proposed hypotheses, the most supported are a dysregulation of the hypothalamic system, the effects on adipokine levels [220], hyperinsulinaemia and the increase in insulin resistance [196]. Many studies show a strict relationship between weight gain, hyperinsulinemia and insulin resistance during treatment with VPA both in children and adults [221-228]. Overall, several studies report an association between hyperinsulinaemia, dyslipidemia, obesity, especially visceral one, and insulin resistance. The onset of hyperinsulinaemia among obese patients taking VPA does not depend only on the development of insulin resistance after body weight increase and obesity. On the contrary, insulin resistance itself, once developed, might lead to weight gain, with visceral fat further facilitating insulin resistance closing a vicious circle. Indeed, VPA induced weight gain correlates with an increase in insulin, but with decreased glycemia able to foster appetite, and eventually, worsen weight gain itself [229]. Moreover, epigenetic mechanisms also participate in weight gain, with VPA inducing the transcription of genes coding for fat mass and obesity-associated (FTO) proteins, highly related to fat mass increase [9]. Nevertheless, the overexpression of FTO modulates the acetylation of SCN3A, reducing its expression; thus, FTO mediates weight gain as well as VPA anticonvulsant action [9]. Even though VPA probably is not able to provoke direct insulin secretion, it might still limit insulin hepatic metabolism, resulting in prolonged hyperinsulinaemia [196]. Thus, in VPA patients with obesity, hyperinsulinemia, insulin resistance and weight gain all participate maintaining one another {Lihn, 2005 #488} [230]. Moreover, a serious clinical condition related to weight gain, insulin resistance and hyperinsulinemia is metabolic syndrome, including atherogenic dyslipidemia and elevated blood pressure [196, 231]. Despite data are still lacking regarding the exact frequency of these adverse events, hyperandrogenism, menstrual irregularities, and polycystic ovary syndrome have all been related to the chronic use of VPA [232]. In particular, an increased prevalence of polycystic ovaries (PCO), hyperandrogenism and menstrual irregularities such adverse events are significantly more commonly reported among women receiving VPA compared to those receiving other AEDs [233]. In addition, anti-progestin action of VPA might also contribute to the higher frequency of anovulatory cycles [217]. Reproductive and sexual endocrine disorders can occur also in males treated with VPA. In male patients reproductive disorders have scarcely been analyzed. Altered sperm mobility, reduced testicular weight and infertility are possible reproductive disorders [234].
6.2. Pregnancy and Teratogenic Risk
VPA can cross the placenta and can cause a spectrum of congenital abnormalities. The vast majority of congenital malformations in children born from epileptic women receiving VPA during pregnancy seems to be due to the direct teratogenic effects of VPA rather than to the epileptic disorder affecting the mother [235]. Teratogenic effects are much higher when VPA is given as a co-medication with other anticonvulsants. The most frequent reported effects are neural tube or cardiovascular defects, orofacial cleft, hypospadia, atresia of the gastrointestinal tract, diaphragmatic hernia and craniosynostosis [200, 236]. For all these reasons, in 2014 the European Medicines Agency restricted the use of VPA in women and girls of childbearing potential, “unless other treatments are ineffective or not tolerated” [158]. In fact, the exposure to VPA in the prenatal period is associated with major congenital malformation. The large case-control study based on data collected from the “European Surveillance of Congenital Anomalies database” reported a consistent correlation between first trimester exposure to VPA monotherapy and augmented risk for spina bifida, craniosynostosis, cleft palate, hypospadia, atrial septal defect and polydactyly [237]. Similarly, a meta-analysis conducted in 2005 revealed that, compared to other commonly used AEDs, VPA prenatal exposure was associated with a 2 to 7 fold increase in the risk of major congenital malformations, including neural tube and cardiovascular defects, genitourinary or musculoskeletal abnormalities, and cleft lip or palate [238, 239]. A dose-dependent increase in the risk of teratogenic effects related to VPA exposure during pregnancy has been suggested according to data derived from both prospective cohort studies and national birth registers [146, 147, 240]. Results from the North American AED Pregnancy Registry, including pregnant women, enrolled between 1997
and 2011, pointed to a dose-dependent increase in risk of malformations with VPA and PB compared to lamotrigine and levetiracetam [241]. Recently, Tomson and colleagues have provided evidences from a large observational registry (EURAP) regarding teratogenic risk with eight different antiepileptic drugs [242]. Data from the largest prospective cohort study of pregnancies in women with epilepsy (EURAP registry) confirmed that the use of VPA is dose-dependently associated with an increased risk of congenital malformations at 1 year after birth. However, the study also allowed comparison between different antiepileptic drugs and dose regimens. From such comparison emerged that low dosage of valproate (≤650 mg/day) carried a risk of congenital malformations similar to high-dose carbamazepine (>700 mg/day) and lamotrigine (>325 mg/day) [242]. Overall, in women of childbearing potential, as well as in pregnancy, monotherapy with low doses of levetiracetam or lamotrigine seems the most appropriate choice to date.
Beyond the recommendations available in women of childbearing potential, specific attention should be paid for off-label use of VPA. Indeed, VPA is also used outside of the typical indications, with a potential risk of minor surveillance on the potential risk on both short and long-term [243].
CONCLUSION AND FUTURE PERSPECTIVE
From the marketing of VPA as an anticonvulsant in 1960s, after more than 50 years of experimental research and clinical use, VPA stands as one of the most efficient AEDs. VPA exerts its effect not only at the synaptic cleft, but also directly and indirectly inducing neuronal conformational adaptation. In particular, the balancing of glutamatergic and GABAergic transmission, the modulation of 5HT and DA-ergic pathways, together with the epigenetic mechanisms deriving from a direct modulation of HDAC, represent the basis of VPA functioning, promoting it as the antiepileptic drug with the widest spectrum of activity. The vast potential of VPA translates into clinical practice, where VPA is considered not only the AED of choice for epilepsy with generalized seizures, but also for other different types of seizure, from focal seizures to Lennox-Gastaut encephalopathy. Use of valproate in women of childbearing potential should be avoided, and, especially in pregnancy, low-dose lamotrigine or levetiracetam should be preferred over VPA.
Better understanding of the ways of VPA molecular functioning might shed light on different pathways to be targeted by new AEDs. Furthermore, beyond its use in bipolar disorder and migraine [47], VPA is earning its own space in cancer treatment, as an epigenetic modulator of gene expression. In particular, future research will look at VPA use as an HDAC modulator together with chemotherapeutics, in order to target different oncogenes and molecular pathways in a broad variety of solid tumors [183-185]. After 50 years of use, VPA still preserves its efficacy, and continues to show interesting opportunities, not only for the neurological field.
ACKNOWLEDGEMENTS
Declared none.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
No funding was received for this review by any of the involved Centers (University of Perugia, University of L’Aquila, Ospedale di Ancona and IRCCS S. Lucia, Rome).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Löscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs. 2002;16(10):669–694. doi: 10.2165/00023210-200216100-00003. [http://dx.doi.org/10.2165/00023210-200216100-00003]. [PMID: 12269861]. [DOI] [PubMed] [Google Scholar]
- 2.Tomson T., Battino D., Perucca E. Valproic acid after five decades of use in epilepsy: time to reconsider the indications of a time-honoured drug. Lancet Neurol. 2016;15(2):210–218. doi: 10.1016/S1474-4422(15)00314-2. [http://dx.doi.org/10.1016/S1474-4422(15)00314-2]. [PMID: 26655849]. [DOI] [PubMed] [Google Scholar]
- 3.Gobbi G., Debonnel G. What is a recommended treatment for aggression in a patient with schizophrenia? J. Psychiatry Neurosci. 2003;28(4):320. [PMID: 12921225]. [PMC free article] [PubMed] [Google Scholar]
- 4.Duenas-Gonzalez A., Candelaria M., Perez-Plascencia C., Perez-Cardenas E., de la Cruz-Hernandez E., Herrera L.A. Valproic acid as epigenetic cancer drug: Preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat. Rev. 2008;34(3):206–222. doi: 10.1016/j.ctrv.2007.11.003. [http://dx.doi.org/10.1016/j.ctrv.2007.11.003]. [PMID: 18226465]. [DOI] [PubMed] [Google Scholar]
- 5.Burton B.S. On the propyl derivatives and decomposition products of ethylacetoacetate. Am. Chem. J. 1882;3:385–395. [Google Scholar]
- 6.Henry T.R. The history of valproate in clinical neuroscience. Psychopharmacol. Bull. 2003;37(Suppl. 2):5–16. [PMID: 14624229]. [PubMed] [Google Scholar]
- 7.Lebreton S., Carraz G., Meunier H., Beriel H. Therapie. 1964;19:451–456. [Pharmacodynamic properties of 2,2-dipropylacetic acid. 2D report on its anti-epileptic properties]. [Pharmacodynamic Properties of 2,2-Dipropylacetic Acid. 2d Report on Its Anti-Epileptic Properties]. [PMID: 14138082]. [PubMed] [Google Scholar]
- 8.Peterson G.M., Naunton M. Valproate: a simple chemical with so much to offer. J. Clin. Pharm. Ther. 2005;30(5):417–421. doi: 10.1111/j.1365-2710.2005.00671.x. [http://dx.doi.org/10.1111/j.1365-2710.2005.00671.x]. [PMID: 16164485]. [DOI] [PubMed] [Google Scholar]
- 9.Tan N.N., Tang H.L., Lin G.W., Chen Y.H., Lu P., Li H.J., Gao M.M., Zhao Q.H., Yi Y.H., Liao W.P., Long Y.S. Epigenetic downregulation of Scn3a expression by valproate: a possible role in its anticonvulsant activity. Mol. Neurobiol. 2017;54(4):2831–2842. doi: 10.1007/s12035-016-9871-9. [SRC -.]. [http://dx.doi.org/10.1007/s12035-016-9871-9]. [PMID: 27013471]. [DOI] [PubMed] [Google Scholar]
- 10.Perucca E. Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs. 2002;16(10):695–714. doi: 10.2165/00023210-200216100-00004. [http://dx.doi.org/10.2165/00023210-200216100-00004]. [PMID: 12269862]. [DOI] [PubMed] [Google Scholar]
- 11.Löscher W. In vivo administration of valproate reduces the nerve terminal (synaptosomal) activity of GABA aminotransferase in discrete brain areas of rats. Neurosci. Lett. 1993;160(2):177–180. doi: 10.1016/0304-3940(93)90407-c. [http://dx.doi.org/10.1016/0304-3940(93)90407-C]. [PMID: 8247350]. [DOI] [PubMed] [Google Scholar]
- 12.Gean P.W., Huang C.C., Hung C.R., Tsai J.J. Valproic acid suppresses the synaptic response mediated by the NMDA receptors in rat amygdalar slices. Brain Res. Bull. 1994;33(3):333–336. doi: 10.1016/0361-9230(94)90202-x. [http://dx.doi.org/10.1016/0361-9230(94)90202-X]. [PMID: 7904890]. [DOI] [PubMed] [Google Scholar]
- 13.Johannessen C.U., Johannessen S.I. Valproate: past, present, and future. CNS Drug Rev. 2003;9(2):199–216. doi: 10.1111/j.1527-3458.2003.tb00249.x. [http://dx.doi.org/10.1111/j.1527-3458.2003.tb00249.x]. [PMID: 12847559]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Löscher W. Valproate: a reappraisal of its pharmacodynamic properties and mechanisms of action. Prog. Neurobiol. 1999;58(1):31–59. doi: 10.1016/s0301-0082(98)00075-6. [http://dx.doi.org/10.1016/S0301-0082(98)00075-6]. [PMID: 10321796]. [DOI] [PubMed] [Google Scholar]
- 15.Göttlicher M., Minucci S., Zhu P., Krämer O.H., Schimpf A., Giavara S., Sleeman J.P., Lo Coco F., Nervi C., Pelicci P.G., Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20(24):6969–6978. doi: 10.1093/emboj/20.24.6969. [http://dx.doi.org/10.1093/emboj/20.24.6969]. [PMID: 11742974]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brigo F., Storti M., Del Felice A., Fiaschi A., Bongiovanni L.G. IV IV Valproate in generalized convulsive status epilepticus: a systematic review. Eur. J. Neurol. 2012;19(9):1180–1191. doi: 10.1111/j.1468-1331.2011.03606.x. [http://dx.doi.org/10.1111/j.1468-1331.2011.03606.x]. [PMID: 22182304]. [DOI] [PubMed] [Google Scholar]
- 17.Ximenes J.C., de Oliveira G.D., Siqueira R.M., Neves K.R., Santos C.G., Correia A.O., Félix F.H., Leal L.K., de Castro Brito G.A., da Graça Naffah-Mazzacorati M., Viana G.S. Valproic acid: an anticonvulsant drug with potent antinociceptive and anti-inflammatory properties. Naunyn Schmiedebergs Arch. Pharmacol. 2013;386(7):575–587. doi: 10.1007/s00210-013-0853-4. [http://dx.doi.org/10.1007/s00210-013-0853-4]. [PMID: 23584602]. [DOI] [PubMed] [Google Scholar]
- 18.Chen S., Wu H., Klebe D., Hong Y., Zhang J. Valproic acid: a new candidate of therapeutic application for the acute central nervous system injuries. Neurochem. Res. 2014;39(9):1621–1633. doi: 10.1007/s11064-014-1241-2. [http://dx.doi.org/10.1007/s11064-014-1241-2]. [PMID: 24482021]. [DOI] [PubMed] [Google Scholar]
- 19.Rogawski M.A., Löscher W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004;5(7):553–564. doi: 10.1038/nrn1430. [http://dx.doi.org/10.1038/nrn1430]. [PMID: 15208697]. [DOI] [PubMed] [Google Scholar]
- 20.Cunningham M.O., Woodhall G.L., Jones R.S. Valproate modifies spontaneous excitation and inhibition at cortical synapses in vitro. Neuropharmacology. 2003;45(7):907–917. doi: 10.1016/s0028-3908(03)00270-3. [http://dx.doi.org/10.1016/S0028-3908(03)00270-3]. [PMID: 14573383]. [DOI] [PubMed] [Google Scholar]
- 21.Löscher W., Schmidt D. Increase of human plasma GABA by sodium valproate. Epilepsia. 1980;21(6):611–615. doi: 10.1111/j.1528-1157.1980.tb04314.x. [http://dx.doi.org/10.1111/j.1528-1157.1980.tb04314.x]. [PMID: 6777153]. [DOI] [PubMed] [Google Scholar]
- 22.Johannessen C.U. Mechanisms of action of valproate: a commentatory. Neurochem. Int. 2000;37(2-3):103–110. doi: 10.1016/s0197-0186(00)00013-9. [http://dx.doi.org/10.1016/S0197-0186(00)00013-9]. [PMID: 10812195]. [DOI] [PubMed] [Google Scholar]
- 23.Löscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure. 2011;20(5):359–368. doi: 10.1016/j.seizure.2011.01.003. [http://dx.doi.org/10.1016/j.seizure.2011.01.003]. [PMID: 21292505]. [DOI] [PubMed] [Google Scholar]
- 24.Gobbi G., Janiri L. Sodium- and magnesium-valproate in vivo modulate glutamatergic and GABAergic synapses in the medial prefrontal cortex. Psychopharmacology (Berl.) 2006;185(2):255–262. doi: 10.1007/s00213-006-0317-3. [http://dx.doi.org/10.1007/s00213-006-0317-3]. [PMID: 16496131]. [DOI] [PubMed] [Google Scholar]
- 25.Rinaldi T., Kulangara K., Antoniello K., Markram H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. USA. 2007;104(33):13501–13506. doi: 10.1073/pnas.0704391104. [http://dx.doi.org/10.1073/pnas.0704391104]. [PMID: 17675408]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim M.J., Dunah A.W., Wang Y.T., Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46(5):745–760. doi: 10.1016/j.neuron.2005.04.031. [http://dx.doi.org/10.1016/j.neuron.2005.04.031]. [PMID: 15924861]. [DOI] [PubMed] [Google Scholar]
- 27.Ghiglieri V., Picconi B., Sgobio C., Bagetta V., Barone I., Paillè V., Di Filippo M., Polli F., Gardoni F., Altrock W., Gundelfinger E.D., De Sarro G., Bernardi G., Ammassari-Teule M., Di Luca M., Calabresi P. Epilepsy-induced abnormal striatal plasticity in Bassoon mutant mice. Eur. J. Neurosci. 2009;29(10):1979–1993. doi: 10.1111/j.1460-9568.2009.06733.x. [http://dx.doi.org/10.1111/j.1460-9568.2009.06733.x]. [PMID: 19453636]. [DOI] [PubMed] [Google Scholar]
- 28.Du J., Creson T.K., Wu L.J., Ren M., Gray N.A., Falke C., Wei Y., Wang Y., Blumenthal R., Machado-Vieira R., Yuan P., Chen G., Zhuo M., Manji H.K. The role of hippocampal GluR1 and GluR2 receptors in manic-like behavior. J. Neurosci. 2008;28(1):68–79. doi: 10.1523/JNEUROSCI.3080-07.2008. [http://dx.doi.org/10.1523/JNEUROSCI.3080-07.2008]. [PMID: 18171924]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biggs C.S., Pearce B.R., Fowler L.J., Whitton P.S. Regional effects of sodium valproate on extracellular concentrations of 5-hydroxytryptamine, dopamine, and their metabolites in the rat brain: an in vivo microdialysis study. J. Neurochem. 1992;59(5):1702–1708. doi: 10.1111/j.1471-4159.1992.tb11001.x. [http://dx.doi.org/10.1111/j.1471-4159.1992.tb11001.x]. [PMID: 1402915]. [DOI] [PubMed] [Google Scholar]
- 30.Ichikawa J., Meltzer H.Y. Valproate and carbamazepine increase prefrontal dopamine release by 5-HT1A receptor activation. Eur. J. Pharmacol. 1999;380(1):R1–R3. doi: 10.1016/s0014-2999(99)00517-8. [http://dx.doi.org/10.1016/S0014-2999(99)00517-8]. [PMID: 10513560]. [DOI] [PubMed] [Google Scholar]
- 31.Lee S., Jeong J., Park Y.U., Kwak Y., Lee S.A., Lee H., Son H., Park S.K. Valproate alters dopamine signaling in association with induction of Par-4 protein expression. PLoS One. 2012;7(9):e45618. doi: 10.1371/journal.pone.0045618. [http://dx.doi.org/10.1371/journal.pone.0045618]. [PMID: 23029138]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park S.K., Nguyen M.D., Fischer A., Luke M.P., Affar B., Dieffenbach P.B., Tseng H.C., Shi Y., Tsai L.H. Par-4 links dopamine signaling and depression. Cell. 2005;122(2):275–287. doi: 10.1016/j.cell.2005.05.031. [http://dx.doi.org/10.1016/j.cell.2005.05.031]. [PMID: 16051151]. [DOI] [PubMed] [Google Scholar]
- 33.Löscher W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002;50(1-2):105–123. doi: 10.1016/s0920-1211(02)00073-6. [http://dx.doi.org/10.1016/S0920-1211(02)00073-6]. [PMID: 12151122]. [DOI] [PubMed] [Google Scholar]
- 34.Qiu H.M., Yang J.X., Jiang X.H., Fei H.Z., Liu D., Hu X.Y., Zhou Q.X. Upregulating serotonin transporter expression and downregulating monoamine oxidase-A and indoleamine 2, 3-dioxygenase expression involved in the antidepressant effect of sodium valproate in a rat model. Neuroreport. 2014;25(17):1338–1343. doi: 10.1097/WNR.0000000000000269. [http://dx.doi.org/10.1097/WNR.0000000000000269]. [PMID: 25304496]. [DOI] [PubMed] [Google Scholar]
- 35.McLean M.J., Macdonald R.L. Sodium valproate, but not ethosuximide, produces use- and voltage-dependent limitation of high frequency repetitive firing of action potentials of mouse central neurons in cell culture. J. Pharmacol. Exp. Ther. 1986;237(3):1001–1011. [PMID: 3086538]. [PubMed] [Google Scholar]
- 36.Van den Berg R.J., Kok P., Voskuyl R.A. Valproate and sodium currents in cultured hippocampal neurons. Exp. Brain Res. 1993;93(2):279–287. doi: 10.1007/BF00228395. [http://dx.doi.org/10.1007/BF00228395]. [PMID: 8387930]. [DOI] [PubMed] [Google Scholar]
- 37.Taverna S., Mantegazza M., Franceschetti S., Avanzini G. Valproate selectively reduces the persistent fraction of Na+ current in neocortical neurons. Epilepsy Res. 1998;32(1-2):304–308. doi: 10.1016/s0920-1211(98)00060-6. [http://dx.doi.org/10.1016/S0920-1211(98)00060-6]. [PMID: 9761329]. [DOI] [PubMed] [Google Scholar]
- 38.Kelly K.M., Gross R.A., Macdonald R.L. Valproic acid selectively reduces the low-threshold (T) calcium current in rat nodose neurons. Neurosci. Lett. 1990;116(1-2):233–238. doi: 10.1016/0304-3940(90)90416-7. [http://dx.doi.org/10.1016/0304-3940(90)90416-7]. [PMID: 2175404]. [DOI] [PubMed] [Google Scholar]
- 39.Walden J., Altrup U., Reith H., Speckmann E. J. Effects of valproate on early and late potassium currents of single neurons. European neuropsychopharmacology: the journal of the European College of Neuropsychopharmacology. 1993;3:137–141. doi: 10.1016/0924-977x(93)90265-n. [http://dx.doi.org/10.1016/0924-977X(93)90265-N] [DOI] [PubMed] [Google Scholar]
- 40.Costa C., Martella G., Picconi B., Prosperetti C., Pisani A., Di Filippo M., Pisani F., Bernardi G., Calabresi P. Multiple mechanisms underlying the neuroprotective effects of antiepileptic drugs against in vitro ischemia. Stroke. 2006;37(5):1319–1326. doi: 10.1161/01.STR.0000217303.22856.38. [http://dx.doi.org/10.1161/01.STR.0000217303.22856.38]. [PMID: 16574927]. [DOI] [PubMed] [Google Scholar]
- 41.Pitkänen A., Kharatishvili I., Karhunen H., Lukasiuk K., Immonen R., Nairismägi J., Gröhn O., Nissinen J. Epileptogenesis in experimental models. Epilepsia. 2007;48(Suppl. 2):13–20. doi: 10.1111/j.1528-1167.2007.01063.x. [http://dx.doi.org/10.1111/j.1528-1167.2007.01063.x]. [PMID: 17571349]. [DOI] [PubMed] [Google Scholar]
- 42.Sitges M., Chiu L.M., Reed R.C. Effects of levetiracetam, carbamazepine, phenytoin, valproate, lamotrigine, oxcarbazepine, topiramate, vinpocetine and sertraline on presynaptic hippocampal Na+ and Ca2+ channels permeability. Neurochem. Res. 2016;41(4):758–769. doi: 10.1007/s11064-015-1749-0. [http://dx.doi.org/10.1007/s11064-015-1749-0]. [PMID: 26542150]. [DOI] [PubMed] [Google Scholar]
- 43.Kay H.Y., Greene D.L., Kang S., Kosenko A., Hoshi N. M-current preservation contributes to anticonvulsant effects of valproic acid. J. Clin. Invest. 2015;125(10):3904–3914. doi: 10.1172/JCI79727. [http://dx.doi.org/10.1172/JCI79727]. [PMID: 26348896]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monti B., Polazzi E., Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr. Mol. Pharmacol. 2009;2(1):95–109. doi: 10.2174/1874467210902010095. [http://dx.doi.org/10.2174/1874467210902010095]. [PMID: 20021450]. [DOI] [PubMed] [Google Scholar]
- 45.Feinberg A.P. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–440. doi: 10.1038/nature05919. [http://dx.doi.org/10.1038/nature05919]. [PMID: 17522677]. [DOI] [PubMed] [Google Scholar]
- 46.Kobow K., Blümcke I. The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia. 2011;52(Suppl. 4):15–19. doi: 10.1111/j.1528-1167.2011.03145.x. [http://dx.doi.org/10.1111/j.1528-1167.2011.03145.x]. [PMID: 21732935]. [DOI] [PubMed] [Google Scholar]
- 47.Nalivaeva N.N., Belyaev N.D., Turner A.J. Sodium valproate: an old drug with new roles. Trends Pharmacol. Sci. 2009;30(10):509–514. doi: 10.1016/j.tips.2009.07.002. [http://dx.doi.org/10.1016/j.tips.2009.07.002]. [PMID: 19762089]. [DOI] [PubMed] [Google Scholar]
- 48.Ghiglieri V., Sgobio C., Patassini S., Bagetta V., Fejtova A., Giampà C., Marinucci S., Heyden A., Gundelfinger E.D., Fusco F.R., Calabresi P., Picconi B. TrkB/BDNF-dependent striatal plasticity and behavior in a genetic model of epilepsy: modulation by valproic acid. Neuropsychopharmacology. 2010;35(7):1531–1540. doi: 10.1038/npp.2010.23. [http://dx.doi.org/10.1038/npp.2010.23]. [PMID: 20200504]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cervoni N., Szyf M. Demethylase activity is directed by histone acetylation. J. Biol. Chem. 2001;276(44):40778–40787. doi: 10.1074/jbc.M103921200. [http://dx.doi.org/10.1074/jbc.M103921200]. [PMID: 11524416]. [DOI] [PubMed] [Google Scholar]
- 50.Cervoni N., Detich N., Seo S.B., Chakravarti D., Szyf M. The oncoprotein Set/TAF-1beta, an inhibitor of histone acetyltransferase, inhibits active demethylation of DNA, integrating DNA methylation and transcriptional silencing. J. Biol. Chem. 2002;277(28):25026–25031. doi: 10.1074/jbc.M202256200. [http://dx.doi.org/10.1074/jbc.M202256200]. [PMID: 11978794]. [DOI] [PubMed] [Google Scholar]
- 51.Chateauvieux S., Morceau F., Dicato M., Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. J. Biomed. Biotechnol. 2010;2010:479364. doi: 10.1155/2010/479364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milutinovic S., D’Alessio A.C., Detich N., Szyf M. Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis. 2007;28(3):560–571. doi: 10.1093/carcin/bgl167. [http://dx.doi.org/10.1093/carcin/bgl167]. [PMID: 17012225]. [DOI] [PubMed] [Google Scholar]
- 53.Chen G., Yuan P.X., Jiang Y.M., Huang L.D., Manji H.K. Valproate robustly enhances AP-1 mediated gene expression. Brain Res. Mol. Brain Res. 1999;64(1):52–58. doi: 10.1016/s0169-328x(98)00303-9. [http://dx.doi.org/10.1016/S0169-328X(98)00303-9]. [PMID: 9889318]. [DOI] [PubMed] [Google Scholar]
- 54.Fukuchi M., Nii T., Ishimaru N., Minamino A., Hara D., Takasaki I., Tabuchi A., Tsuda M. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci. Res. 2009;65(1):35–43. doi: 10.1016/j.neures.2009.05.002. [http://dx.doi.org/10.1016/j.neures.2009.05.002]. [PMID: 19463867]. [DOI] [PubMed] [Google Scholar]
- 55.Chen G., Yuan P., Hawver D.B., Potter W.Z., Manji H.K. Increase in AP-1 transcription factor DNA binding activity by valproic acid. Neuropsychopharmacology. 1997;16(3):238–245. doi: 10.1016/S0893-133X(96)00239-4. [http://dx.doi.org/10.1016/S0893-133X(96)00239-4]. [PMID: 9138440]. [DOI] [PubMed] [Google Scholar]
- 56.Sullivan N.R., Burke T., Siafaka-Kapadai A., Javors M., Hensler J.G. Effect of valproic acid on serotonin-2A receptor signaling in C6 glioma cells. J. Neurochem. 2004;90(5):1269–1275. doi: 10.1111/j.1471-4159.2004.02690.x. [http://dx.doi.org/10.1111/j.1471-4159.2004.02690.x]. [PMID: 15312182]. [DOI] [PubMed] [Google Scholar]
- 57.Ichiyama T., Okada K., Lipton J.M., Matsubara T., Hayashi T., Furukawa S. Sodium valproate inhibits production of TNF-alpha and IL-6 and activation of NF-kappaB. Brain Res. 2000;857(1-2):246–251. doi: 10.1016/s0006-8993(99)02439-7. [http://dx.doi.org/10.1016/S0006-8993(99)02439-7]. [PMID: 10700573]. [DOI] [PubMed] [Google Scholar]
- 58.Lonze B.E., Ginty D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35(4):605–623. doi: 10.1016/s0896-6273(02)00828-0. [http://dx.doi.org/10.1016/S0896-6273(02)00828-0]. [PMID: 12194863]. [DOI] [PubMed] [Google Scholar]
- 59.Chen B., Wang J.F., Hill B.C., Young L.T. Lithium and valproate differentially regulate brain regional expression of phosphorylated CREB and c-Fos. Brain Res. Mol. Brain Res. 1999;70(1):45–53. doi: 10.1016/s0169-328x(99)00125-4. [http://dx.doi.org/10.1016/S0169-328X(99)00125-4]. [PMID: 10381542]. [DOI] [PubMed] [Google Scholar]
- 60.Chao M.V. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003;4(4):299–309. doi: 10.1038/nrn1078. [http://dx.doi.org/10.1038/nrn1078]. [PMID: 12671646]. [DOI] [PubMed] [Google Scholar]
- 61.Baquet Z.C., Gorski J.A., Jones K.R. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J. Neurosci. 2004;24(17):4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [http://dx.doi.org/10.1523/JNEUROSCI.3920-03.2004]. [PMID: 15115821]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bath K. G., Pimentel T. Effect of early postnatal exposure to valproate on neurobehavioral development and regional BDNF expression in two strains of mice. Epilepsy Behav. 2017;70(Pt A):110–117. doi: 10.1016/j.yebeh.2017.02.026. [http://dx.doi.org/10.1016/j.yebeh.2017.02.026] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bittigau P., Sifringer M., Ikonomidou C. Antiepileptic drugs and apoptosis in the developing brain. Ann. N. Y. Acad. Sci. 2003;993:103–114. doi: 10.1111/j.1749-6632.2003.tb07517.x. [http://dx.doi.org/10.1111/j.1749-6632.2003.tb07517.x]. [PMID: 12853301]. [DOI] [PubMed] [Google Scholar]
- 64.Shi X.Y., Wang J.W., Cui H., Li B.M., Lei G.F., Sun R.P. Effects of antiepileptic drugs on mRNA levels of BDNF and NT-3 and cell neogenesis in the developing rat brain. Brain Dev. 2010;32(3):229–235. doi: 10.1016/j.braindev.2009.03.012. [http://dx.doi.org/10.1016/j.braindev.2009.03.012]. [PMID: 19394173]. [DOI] [PubMed] [Google Scholar]
- 65.Wu X., Chen P.S., Dallas S., Wilson B., Block M.L., Wang C.C., Kinyamu H., Lu N., Gao X., Leng Y., Chuang D.M., Zhang W., Lu R.B., Hong J.S. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int. J. Neuropsychopharmacol. 2008;11(8):1123–1134. doi: 10.1017/S1461145708009024. [http://dx.doi.org/10.1017/S1461145708009024]. [PMID: 18611290]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laeng P., Pitts R.L., Lemire A.L., Drabik C.E., Weiner A., Tang H., Thyagarajan R., Mallon B.S., Altar C.A. The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. J. Neurochem. 2004;91(1):238–251. doi: 10.1111/j.1471-4159.2004.02725.x. [http://dx.doi.org/10.1111/j.1471-4159.2004.02725.x]. [PMID: 15379904]. [DOI] [PubMed] [Google Scholar]
- 67.Sinn D.I., Kim S.J., Chu K., Jung K.H., Lee S.T., Song E.C., Kim J.M., Park D.K., Lee K. S.; Kim, M.; Roh, J.K. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol. Dis. 2007;26(2):464–472. doi: 10.1016/j.nbd.2007.02.006. [http://dx.doi.org/10.1016/j.nbd. 2007.02.006]. [PMID: 17398106]. [DOI] [PubMed] [Google Scholar]
- 68.Whitaker W.R., Faull R.L., Waldvogel H.J., Plumpton C.J., Emson P.C., Clare J.J. Comparative distribution of voltage-gated sodium channel proteins in human brain. Brain Res. Mol. Brain Res. 2001;88(1-2):37–53. doi: 10.1016/s0169-328x(00)00289-8. [http://dx.doi.org/10.1016/S0169-328X(00)00289-8]. [PMID: 11295230]. [DOI] [PubMed] [Google Scholar]
- 69.Guo F., Yu N., Cai J.Q., Quinn T., Zong Z.H., Zeng Y.J., Hao L.Y. Voltage-gated sodium channel Nav1.1, Nav1.3 and beta1 subunit were up-regulated in the hippocampus of spontaneously epileptic rat. Brain Res. Bull. 2008;75(1):179–187. doi: 10.1016/j.brainresbull.2007.10.005. [http://dx.doi.org/10.1016/j.brainresbull.2007.10.005]. [PMID: 18158113]. [DOI] [PubMed] [Google Scholar]
- 70.Löscher W., Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia. 2006;47(8):1253–1284. doi: 10.1111/j.1528-1167.2006.00607.x. [http://dx.doi.org/10.1111/j.1528-1167.2006.00607.x]. [PMID: 16922870]. [DOI] [PubMed] [Google Scholar]
- 71.Experimental models of status epilepticus; CRC Press: Boca Raton, 1998. T.E. (Eds.) ed. [Google Scholar]
- 72.Jankowsky J.L., Patterson P.H. The role of cytokines and growth factors in seizures and their sequelae. Prog. Neurobiol. 2001;63(2):125–149. doi: 10.1016/s0301-0082(00)00022-8. [http://dx.doi.org/10.1016/S0301-0082(00)00022-8]. [PMID: 11124444]. [DOI] [PubMed] [Google Scholar]
- 73.Wyneken U., Smalla K.H., Marengo J.J., Soto D., de la Cerda A., Tischmeyer W., Grimm R., Boeckers T.M., Wolf G., Orrego F., Gundelfinger E.D. Kainate-induced seizures alter protein composition and N-methyl-D-aspartate receptor function of rat forebrain postsynaptic densities. Neuroscience. 2001;102(1):65–74. doi: 10.1016/s0306-4522(00)00469-3. [http://dx.doi.org/10.1016/S0306-4522(00)00469-3]. [PMID: 11226670]. [DOI] [PubMed] [Google Scholar]
- 74.Wyneken U., Marengo J.J., Villanueva S., Soto D., Sandoval R., Gundelfinger E.D., Orrego F. Epilepsy-induced changes in signaling systems of human and rat postsynaptic densities. Epilepsia. 2003;44(2):243–246. doi: 10.1046/j.1528-1157.2003.17602.x. [http://dx.doi.org/10.1046/j.1528-1157.2003.17602.x]. [PMID: 12558581]. [DOI] [PubMed] [Google Scholar]
- 75.Takahashi M., Hayashi S., Kakita A., Wakabayashi K., Fukuda M., Kameyama S., Tanaka R., Takahashi H., Nawa H. Patients with temporal lobe epilepsy show an increase in brain-derived neurotrophic factor protein and its correlation with neuropeptide Y. Brain Res. 1999;818(2):579–582. doi: 10.1016/s0006-8993(98)01355-9. [http://dx.doi.org/10.1016/S0006-8993(98)01355-9]. [PMID: 10082852]. [DOI] [PubMed] [Google Scholar]
- 76.Binder D.K., Routbort M.J., McNamara J.O. Immunohistochemical evidence of seizure-induced activation of trk receptors in the mossy fiber pathway of adult rat hippocampus. J. Neurosci. 1999;19(11):4616–4626. doi: 10.1523/JNEUROSCI.19-11-04616.1999. [http://dx.doi.org/10.1523/JNEUROSCI.19-11-04616.1999]. [PMID: 10341259]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zuccato C., Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nature reviews Neurology, 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [http://dx.doi.org/10.1038/nrneurol.2009.54] [DOI] [PubMed] [Google Scholar]
- 78.Löscher W. Animal models of epilepsy and epileptic seizures. Berlin: Springer; 1999. [http://dx.doi.org/10.1007/978-3-642-60072-2_2] [Google Scholar]
- 79.Pitkänen A. Drug-mediated neuroprotection and antiepilepto-genesis: animal data. Neurology. 2002;59(9) Suppl. 5:S27–S33. doi: 10.1212/wnl.59.9_suppl_5.s27. [http://dx.doi.org/10.1212/WNL.59.9_suppl_5.S27]. [PMID: 12428029]. [DOI] [PubMed] [Google Scholar]
- 80.Löscher W., Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol. Rev. 2010;62(4):668–700. doi: 10.1124/pr.110.003046. [http://dx.doi.org/10.1124/pr.110.003046]. [PMID: 21079040]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kandratavicius L., Balista P.A., Lopes-Aguiar C., Ruggiero R.N., Umeoka E.H., Garcia-Cairasco N., Bueno-Junior L.S., Leite J.P. Animal models of epilepsy: use and limitations. Neuropsychiatr. Dis. Treat. 2014;10:1693–1705. doi: 10.2147/NDT.S50371. [http://dx.doi.org/ 10.2147/NDT.S50371]. [PMID: 25228809]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leite J.P., Garcia-Cairasco N., Cavalheiro E.A. New insights from the use of pilocarpine and kainate models. Epilepsy Res. 2002;50(1-2):93–103. doi: 10.1016/s0920-1211(02)00072-4. [http://dx.doi.org/10.1016/S0920-1211(02)00072-4]. [PMID: 12151121]. [DOI] [PubMed] [Google Scholar]
- 83.Curia G., Longo D., Biagini G., Jones R.S., Avoli M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods. 2008;172(2):143–157. doi: 10.1016/j.jneumeth.2008.04.019. [http://dx.doi.org/10.1016/j.jneumeth.2008.04.019]. [PMID: 18550176]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klitgaard H.V., Matagne A.C., Vanneste-Goemaere J., Margineanu D.G. Effects of prolonged administration of levetiracetam on pilocarpine-induced epileptogenesis in rats. Epilepsia. 2001;42:114–115. [Google Scholar]
- 85.Gasior M., Ungard J.T., Beekman M., Carter R.B., Witkin J.M. Acute and chronic effects of the synthetic neuroactive steroid, ganaxolone, against the convulsive and lethal effects of pentylenetetrazol in seizure-kindled mice: comparison with diazepam and valproate. Neuropharmacology. 2000;39(7):1184–1196. doi: 10.1016/s0028-3908(99)00190-2. [http://dx.doi.org/10.1016/S0028-3908(99)00190-2]. [PMID: 10760361]. [DOI] [PubMed] [Google Scholar]
- 86.White H.S. Preclinical development of antiepileptic drugs: past, present, and future directions. Epilepsia. 2003;44(Suppl. 7):2–8. doi: 10.1046/j.1528-1157.44.s7.10.x. [http://dx.doi.org/10.1046/j.1528-1157.44.s7.10.x]. [PMID: 12919332]. [DOI] [PubMed] [Google Scholar]
- 87.El-Azab M.F., Moustafa Y.M. Influence of calcium channel blockers on anticonvulsant and antinociceptive activities of valproic acid in pentylenetetrazole-kindled mice. Pharmacol. Rep. 2012;64(2):305–314. doi: 10.1016/s1734-1140(12)70769-7. [http://dx.doi.org/10.1016/S1734-1140(12)70769-7]. [PMID: 22661180]. [DOI] [PubMed] [Google Scholar]
- 88.Löscher W., Nau H., Marescaux C., Vergnes M. Comparative evaluation of anticonvulsant and toxic potencies of valproic acid and 2-en-valproic acid in different animal models of epilepsy. Eur. J. Pharmacol. 1984;99(2-3):211–218. doi: 10.1016/0014-2999(84)90243-7. [http://dx.doi.org/10.1016/0014-2999(84)90243-7]. [PMID: 6428923]. [DOI] [PubMed] [Google Scholar]
- 89.Löscher W., Hönack D. Comparison of anticonvulsant efficacy of valproate during prolonged treatment with one and three daily doses or continuous (“controlled release”) administration in a model of generalized seizures in rats. Epilepsia. 1995;36(9):929–937. doi: 10.1111/j.1528-1157.1995.tb01637.x. [http://dx.doi.org/10.1111/j.1528-1157.1995.tb01637.x]. [PMID: 7649133]. [DOI] [PubMed] [Google Scholar]
- 90.Luszczki J.J., Trojnar M.K., Ratnaraj N., Patsalos P.N., Czuczwar S.J. Interactions of stiripentol with clobazam and valproate in the mouse maximal electroshock-induced seizure model. Epilepsy Res. 2010;90(3):188–198. doi: 10.1016/j.eplepsyres.2010.04.006. [http://dx.doi.org/10.1016/j.eplepsyres.2010.04.006]. [PMID: 20493662]. [DOI] [PubMed] [Google Scholar]
- 91.Blanco M.M., dos Santos J.G., Jr, Perez-Mendes P., Kohek S.R., Cavarsan C.F., Hummel M., Albuquerque C., Mello L.E. Assessment of seizure susceptibility in pilocarpine epileptic and nonepileptic Wistar rats and of seizure reinduction with pentylenetetrazole and electroshock models. Epilepsia. 2009;50(4):824–831. doi: 10.1111/j.1528-1167.2008.01797.x. [http://dx.doi.org/10.1111/j.1528-1167.2008.01797.x]. [PMID: 19054404]. [DOI] [PubMed] [Google Scholar]
- 92.Bertram E. The relevance of kindling for human epilepsy. Epilepsia. 2007;48(Suppl. 2):65–74. doi: 10.1111/j.1528-1167.2007.01068.x. [http://dx.doi.org/10.1111/j.1528-1167.2007.01068.x]. [PMID: 17571354]. [DOI] [PubMed] [Google Scholar]
- 93.Silver J.M., Shin C., McNamara J.O. Antiepileptogenic effects of conventional anticonvulsants in the kindling model of epilespy. Ann. Neurol. 1991;29(4):356–363. doi: 10.1002/ana.410290404. [http://dx.doi.org/10.1002/ana.410290404]. [PMID: 1929206]. [DOI] [PubMed] [Google Scholar]
- 94.Srivastava A.K., White H.S. Carbamazepine, but not valproate, displays pharmacoresistance in lamotrigine-resistant amygdala kindled rats. Epilepsy Res. 2013;104(1-2):26–34. doi: 10.1016/j.eplepsyres.2012.10.003. [http://dx.doi.org/10.1016/j.eplepsyres.2012.10.003]. [PMID: 23158096]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bolanos A.R., Sarkisian M., Yang Y., Hori A., Helmers S.L., Mikati M., Tandon P., Stafstrom C.E., Holmes G.L. Comparison of valproate and phenobarbital treatment after status epilepticus in rats. Neurology. 1998;51(1):41–48. doi: 10.1212/wnl.51.1.41. [http://dx.doi.org/10.1212/WNL.51.1.41]. [PMID: 9674776]. [DOI] [PubMed] [Google Scholar]
- 96.Nissinen J., Pitkänen A. Effect of antiepileptic drugs on spontaneous seizures in epileptic rats. Epilepsy Res. 2007;73(2):181–191. doi: 10.1016/j.eplepsyres.2006.10.003. [http://dx.doi.org/10.1016/j.eplepsyres.2006.10.003]. [PMID: 17161937]. [DOI] [PubMed] [Google Scholar]
- 97.Angenstein F., Niessen H.G., Goldschmidt J., Lison H., Altrock W.D., Gundelfinger E.D., Scheich H. Manganese-enhanced MRI reveals structural and functional changes in the cortex of Bassoon mutant mice. Cereb. Cortex. 2007;17(1):28–36. doi: 10.1093/cercor/bhj121. [http://dx.doi.org/10.1093/cercor/bhj121]. [PMID: 16452644]. [DOI] [PubMed] [Google Scholar]
- 98.Ghiglieri V., Sgobio C., Costa C., Picconi B., Calabresi P. Striatum-hippocampus balance: from physiological behavior to interneuronal pathology. Prog. Neurobiol. 2011;94(2):102–114. doi: 10.1016/j.pneurobio.2011.04.005. [http://dx.doi.org/10.1016/j.pneurobio.2011.04.005]. [PMID: 21514357]. [DOI] [PubMed] [Google Scholar]
- 99.Sgobio C., Ghiglieri V., Costa C., Bagetta V., Siliquini S., Barone I., Di Filippo M., Gardoni F., Gundelfinger E.D., Di Luca M., Picconi B., Calabresi P. Hippocampal synaptic plasticity, memory, and epilepsy: effects of long-term valproic acid treatment. Biol. Psychiatry. 2010;67(6):567–574. doi: 10.1016/j.biopsych.2009.11.008. [http://dx.doi.org/10.1016/j.biopsych.2009.11.008]. [PMID: 20074705]. [DOI] [PubMed] [Google Scholar]
- 100.Shinnar S., Berg A.T. Does antiepileptic drug therapy prevent the development of “chronic” epilepsy? Epilepsia. 1996;37(8):701–708. doi: 10.1111/j.1528-1157.1996.tb00639.x. [http://dx.doi.org/10.1111/j.1528-1157.1996.tb00639.x]. [PMID: 8764806]. [DOI] [PubMed] [Google Scholar]
- 101.Radzik I., Miziak B., Dudka J. Chrościńska-Krawczyk, M.; Czuczwar, S.J. Prospects of epileptogenesis prevention. Pharmacol. Rep. 2015;67(3):663–668. doi: 10.1016/j.pharep.2015.01.016. [http://dx.doi.org/10.1016/j.pharep.2015.01.016]. [PMID: 25933984]. [DOI] [PubMed] [Google Scholar]
- 102.Ueda Y., Willmore L. J. Molecular regulation of glutamate and GABA transporter proteins by valproic acid in rat hippocampus during epileptogenesis. Exp Brain Res, 2000:334–339. doi: 10.1007/s002210000443. [http://dx.doi.org/10.1007/s002210000443] [DOI] [PubMed] [Google Scholar]
- 103.Cavalheiro E.A., Leite J.P., Bortolotto Z.A., Turski W.A., Ikonomidou C., Turski L. Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia. 1991;32(6):778–782. doi: 10.1111/j.1528-1157.1991.tb05533.x. [http://dx.doi.org/10.1111/j.1528-1157.1991.tb05533.x]. [PMID: 1743148]. [DOI] [PubMed] [Google Scholar]
- 104.K. Łukawski, M. Andres-Mach, M. Czuczwar, J.J. Łuszczki, K. Kruszyński, S.J. Czuczwar. Mechanisms of epileptogenesis and preclinical approach to antiepileptogenic therapies. Pharmacol. Rep. 2018;70(2):284–293. doi: 10.1016/j.pharep.2017.07.012. [http://dx.doi.org/10.1016/j.pharep.2017.07.012] [PMID: 29477036] [DOI] [PubMed] [Google Scholar]
- 105.Brandt C., Glien M., Gastens A.M., Fedrowitz M., Bethmann K., Volk H.A., Potschka H., Löscher W. Prophylactic treatment with levetiracetam after status epilepticus: lack of effect on epileptogenesis, neuronal damage, and behavioral alterations in rats. Neuropharmacology. 2007;53(2):207–221. doi: 10.1016/j.neuropharm.2007.05.001. [http://dx.doi.org/10.1016/j.neuropharm.2007.05.001]. [PMID: 17585956]. [DOI] [PubMed] [Google Scholar]
- 106.Langer M., Brandt C., Zellinger C., Löscher W. Therapeutic window of opportunity for the neuroprotective effect of valproate versus the competitive AMPA receptor antagonist NS1209 following status epilepticus in rats. Neuropharmacology. 2011;61(5-6):1033–1047. doi: 10.1016/j.neuropharm.2011.06.015. [http://dx.doi.org/10.1016/j.neuropharm.2011.06.015]. [PMID: 21736883]. [DOI] [PubMed] [Google Scholar]
- 107.Kadiyala S.B., Yannix J.Q., Nalwalk J.W., Papandrea D., Beyer B.S., Herron B.J., Ferland R.J. Eight flurothyl-induced generalized seizures lead to the rapid evolution of spontaneous seizures in mice: A model of epileptogenesis with seizure remission. J. Neurosci. 2016;36(28):7485–7496. doi: 10.1523/JNEUROSCI.3232-14.2016. [http://dx.doi.org/10.1523/JNEUROSCI.3232-14.2016]. [PMID: 27413158]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brandt C., Gastens A.M., Sun Mz., Hausknecht M., Löscher W. Treatment with valproate after status epilepticus: effect on neuronal damage, epileptogenesis, and behavioral alterations in rats. Neuropharmacology. 2006;51(4):789–804. doi: 10.1016/j.neuropharm.2006.05.021. [http://dx.doi.org/10.1016/j.neuropharm.2006.05.021]. [PMID: 16806297]. [DOI] [PubMed] [Google Scholar]
- 109.Mora A., González-Polo R.A., Fuentes J.M., Soler G., Centeno F. Different mechanisms of protection against apoptosis by valproate and Li+. Eur. J. Biochem. 1999;266(3):886–891. doi: 10.1046/j.1432-1327.1999.00919.x. [http://dx.doi.org/10.1046/j.1432-1327.1999.00919.x]. [PMID: 10583382]. [DOI] [PubMed] [Google Scholar]
- 110.Kim H.J., Rowe M., Ren M., Hong J.S., Chen P.S., Chuang D.M. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J. Pharmacol. Exp. Ther. 2007;321(3):892–901. doi: 10.1124/jpet.107.120188. [http://dx.doi.org/10.1124/jpet.107.120188]. [PMID: 17371805]. [DOI] [PubMed] [Google Scholar]
- 111.Wang Z., Leng Y., Tsai L.K., Leeds P., Chuang D.M. Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J. Cereb. Blood Flow Metab. 2011;31(1):52–57. doi: 10.1038/jcbfm.2010.195. [http://dx.doi.org/10.1038/jcbfm.2010.195]. [PMID: 20978517]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Caccamo D., Pisani L.R., Mazzocchetti P., Ientile R., Calabresi P., Pisani F., Costa C. Neuroprotection as a Potential Therapeutic Perspective in Neurodegenerative Diseases: Focus on Antiepileptic Drugs. Neurochem. Res. 2016;41(1-2):340–352. doi: 10.1007/s11064-015-1809-5. [http://dx.doi.org/10.1007/s11064-015-1809-5]. [PMID: 26721507]. [DOI] [PubMed] [Google Scholar]
- 113.Calabresi P., Cupini L.M., Centonze D., Pisani F., Bernardi G. Antiepileptic drugs as a possible neuroprotective strategy in brain ischemia. Ann. Neurol. 2003;53(6):693–702. doi: 10.1002/ana.10603. [http://dx.doi.org/10.1002/ana.10603]. [PMID: 12783414]. [DOI] [PubMed] [Google Scholar]
- 114.Picascia A., Grimaldi V., Iannone C., Soricelli A., Napoli C. Innate and adaptive immune response in stroke: Focus on epigenetic regulation. J. Neuroimmunol. 2015;289:111–120. doi: 10.1016/j.jneuroim.2015.10.013. [http://dx.doi.org/10.1016/j.jneuroim.2015.10.013]. [PMID: 26616880]. [DOI] [PubMed] [Google Scholar]
- 115.Ren M., Leng Y., Jeong M., Leeds P.R., Chuang D.M. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J. Neurochem. 2004;89(6):1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [http://dx.doi.org/10.1111/j.1471-4159.2004.02406.x]. [PMID: 15189338]. [DOI] [PubMed] [Google Scholar]
- 116.Yildirim F., Gertz K., Kronenberg G., Harms C., Fink K.B., Meisel A., Endres M. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp. Neurol. 2008;210(2):531–542. doi: 10.1016/j.expneurol.2007.11.031. [http://dx.doi.org/10.1016/j.expneurol.2007.11.031]. [PMID: 18234195]. [DOI] [PubMed] [Google Scholar]
- 117.Xuan A., Long D., Li J., Ji W., Hong L., Zhang M., Zhang W. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci. 2012;90(11-12):463–468. doi: 10.1016/j.lfs.2012.01.001. [http://dx.doi.org/10.1016/j.lfs.2012.01.001]. [PMID: 22285595]. [DOI] [PubMed] [Google Scholar]
- 118.Masuch A., Shieh C.H., van Rooijen N., van Calker D., Biber K. Mechanism of microglia neuroprotection: Involvement of P2X7, TNFα and valproic acid. Glia. 2016;64(1):76–89. doi: 10.1002/glia.22904. [http://dx.doi.org/10.1002/glia.22904]. [PMID: 26295445]. [DOI] [PubMed] [Google Scholar]
- 119.van Weering H.R., Boddeke H.W., Vinet J., Brouwer N., de Haas A.H., van Rooijen N., Thomsen A.R., Biber K.P. CXCL10/CXCR3 signaling in glia cells differentially affects NMDA-induced cell death in CA and DG neurons of the mouse hippocampus. Hippocampus. 2011;21(2):220–232. doi: 10.1002/hipo.20742. [http://dx.doi.org/10.1002/hipo.20742]. [PMID: 20082289]. [DOI] [PubMed] [Google Scholar]
- 120.Vinet J., Weering H.R., Heinrich A., Kälin R.E., Wegner A., Brouwer N., Heppner F.L., Rooijen Nv., Boddeke H.W., Biber K. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J. Neuroinflammation. 2012;9:27. doi: 10.1186/1742-2094-9-27. [http://dx.doi.org/10.1186/1742-2094-9-27]. [PMID: 22293457]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dale N., Frenguelli B.G. Release of adenosine and ATP during ischemia and epilepsy. Curr. Neuropharmacol. 2009;7(3):160–179. doi: 10.2174/157015909789152146. [http://dx.doi.org/10.2174/157015909789152146]. [PMID: 20190959]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harrison I.F., Anis H.K., Dexter D.T. Associated degeneration of ventral tegmental area dopaminergic neurons in the rat nigrostriatal lactacystin model of parkinsonism and their neuroprotection by valproate. Neurosci. Lett. 2016;614:16–23. doi: 10.1016/j.neulet.2015.12.052. [http://dx.doi.org/10.1016/j.neulet.2015.12.052]. [PMID: 26742637]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harrison I.F., Crum W.R., Vernon A.C., Dexter D.T. Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson’s is associated with histone acetylation and up-regulation of neurotrophic factors. Br. J. Pharmacol. 2015;172(16):4200–4215. doi: 10.1111/bph.13208. [http://dx.doi.org/10.1111/bph.13208]. [PMID: 26040297]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Harrison I.F., Dexter D.T. Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson’s disease? Pharmacol. Ther. 2013;140(1):34–52. doi: 10.1016/j.pharmthera.2013.05.010. [http://dx.doi.org/10.1016/j.pharmthera.2013.05.010]. [PMID: 23711791]. [DOI] [PubMed] [Google Scholar]
- 125.Rouaux C., Jokic N., Mbebi C., Boutillier S., Loeffler J.P., Boutillier A.L. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22(24):6537–6549. doi: 10.1093/emboj/cdg615. [http://dx.doi.org/10.1093/emboj/cdg615]. [PMID: 14657026]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mark R.J., Ashford J.W., Goodman Y., Mattson M.P. Anticonvulsants attenuate amyloid beta-peptide neurotoxicity, Ca2+ deregulation, and cytoskeletal pathology. Neurobiol. Aging. 1995;16(2):187–198. doi: 10.1016/0197-4580(94)00150-2. [http://dx.doi.org/10.1016/0197-4580(94)00150-2]. [PMID: 7777136]. [DOI] [PubMed] [Google Scholar]
- 127.Mishra J., Chaudhary T., Kumar A. Rosiglitazone synergizes the neuroprotective effects of valproic acid against quinolinic acid-induced neurotoxicity in rats: targeting PPARγ and HDAC pathways. Neurotox. Res. 2014;26(2):130–151. doi: 10.1007/s12640-014-9458-z. [http://dx.doi.org/10.1007/s12640-014-9458-z]. [PMID: 24566814]. [DOI] [PubMed] [Google Scholar]
- 128.Jiang H.Z., Wang S.Y., Yin X., Jiang H.Q., Wang X.D., Wang J., Wang T.H., Qi Y., Yang Y.Q., Wang Y., Zhang C.T., Feng H.L. Downregulation of Homer1b/c in SOD1 G93A Models of ALS: A Novel Mechanism of Neuroprotective Effect of Lithium and Valproic Acid. Int. J. Mol. Sci. 2016;17(12):E2129. doi: 10.3390/ijms17122129. [http://dx.doi.org/10.3390/ijms17122129]. [PMID: 27999308]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Avanzini G., Depaulis A., Tassinari A., de Curtis M. Do seizures and epileptic activity worsen epilepsy and deteriorate cognitive function? Epilepsia. 2013;54(Suppl. 8):14–21. doi: 10.1111/epi.12418. [http://dx.doi.org/10.1111/epi.12418]. [PMID: 24571112]. [DOI] [PubMed] [Google Scholar]
- 130.O’Connor J.C., André C., Wang Y., Lawson M.A., Szegedi S.S., Lestage J., Castanon N., Kelley K.W., Dantzer R. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J. Neurosci. 2009;29(13):4200–4209. doi: 10.1523/JNEUROSCI.5032-08.2009. [http://dx.doi.org/10.1523/JNEUROSCI.5032-08.2009]. [PMID: 19339614]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Choi C.S., Gonzales E.L., Kim K.C., Yang S.M., Kim J.W., Mabunga D.F., Cheong J.H., Han S.H., Bahn G.H., Shin C.Y. The transgenerational inheritance of autism-like phenotypes in mice exposed to valproic acid during pregnancy. Sci. Rep. 2016;6:36250. doi: 10.1038/srep36250. [http://dx.doi.org/10.1038/srep36250]. [PMID: 27819277]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fiala J.C., Spacek J., Harris K.M. Dendritic spine pathology: Cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 2002;39(1):29–54. doi: 10.1016/s0165-0173(02)00158-3. [http://dx.doi.org/10.1016/S0165-0173(02)00158-3]. [PMID: 12086707]. [DOI] [PubMed] [Google Scholar]
- 133.Meador K.J., Baker G.A., Browning N., Clayton-Smith J., Combs-Cantrell D.T., Cohen M., Kalayjian L.A., Kanner A., Liporace J.D., Pennell P.B., Privitera M., Loring D.W., Group N.S. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N. Engl. J. Med. 2009;360(16):1597–1605. doi: 10.1056/NEJMoa0803531. [http://dx.doi.org/10.1056/NEJMoa0803531]. [PMID: 19369666]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Carreño M., Donaire A., Sánchez-Carpintero R. Cognitive disorders associated with epilepsy: diagnosis and treatment. Neurologist. 2008;14(6) Suppl. 1:S26–S34. doi: 10.1097/01.nrl.0000340789.15295.8f. [http://dx.doi.org/10.1097/01.nrl.0000340789.15295.8f]. [PMID: 19225368]. [DOI] [PubMed] [Google Scholar]
- 135.Hermann B.P., Lin J.J., Jones J.E., Seidenberg M. The emerging architecture of neuropsychological impairment in epilepsy. Neurol. Clin. 2009;27(4):881–907. doi: 10.1016/j.ncl.2009.08.001. [http://dx.doi.org/10.1016/j.ncl.2009.08.001]. [PMID: 19853214]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hassel B., Iversen E.G., Gjerstad L., Taubøll E. Up-regulation of hippocampal glutamate transport during chronic treatment with sodium valproate. J. Neurochem. 2001;77(5):1285–1292. doi: 10.1046/j.1471-4159.2001.00349.x. [http://dx.doi.org/10.1046/j.1471-4159.2001.00349.x]. [PMID: 11389179]. [DOI] [PubMed] [Google Scholar]
- 137.Bruno V., Sortino M.A., Scapagnini U., Nicoletti F., Canonico P.L. Antidegenerative effects of Mg(2+)-valproate in cultured cerebellar neurons. Funct. Neurol. 1995;10(3):121–130. [PMID: 8557213]. [PubMed] [Google Scholar]
- 138.Lynch M., Sayin U., Bownds J., Janumpalli S., Sutula T. Long-term consequences of early postnatal seizures on hippocampal learning and plasticity. Eur. J. Neurosci. 2000;12(7):2252–2264. doi: 10.1046/j.1460-9568.2000.00117.x. [http://dx.doi.org/10.1046/j.1460-9568.2000.00117.x]. [PMID: 10947804]. [DOI] [PubMed] [Google Scholar]
- 139.Yuan P. X., Huang L. D., Jiang Y. M., Gutkind J. S., Manji H. K., Chen G. The mood stabilizer valproic acid activates mitogen activated protein kinases and promotes neurite growth. J. Biol. Chem, 2001;276:31674–31683. doi: 10.1074/jbc.M104309200. [DOI] [PubMed] [Google Scholar]
- 140.Jessberger S., Nakashima K., Clemenson G.D., Jr, Mejia E., Mathews E., Ure K., Ogawa S., Sinton C.M., Gage F.H., Hsieh J. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci. 2007;27(22):5967–5975. doi: 10.1523/JNEUROSCI.0110-07.2007. [http://dx.doi.org/10.1523/JNEUROSCI.0110-07.2007]. [PMID: 17537967]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Halbsgut L.R., Fahim E., Kapoor K., Hong H., Friedman L.K. Certain secondary antiepileptic drugs can rescue hippocampal injury following a critical growth period despite poor anticonvulsant activity and cognitive deficits. Epilepsy Behav. 2013;29(3):466–477. doi: 10.1016/j.yebeh.2013.08.019. [http://dx.doi.org/10.1016/j.yebeh.2013.08.019]. [PMID: 24103817]. [DOI] [PubMed] [Google Scholar]
- 142.Umka J., Mustafa S., ElBeltagy M., Thorpe A., Latif L., Bennett G., Wigmore P.M. Valproic acid reduces spatial working memory and cell proliferation in the hippocampus. Neuroscience. 2010;166(1):15–22. doi: 10.1016/j.neuroscience.2009.11.073. [http://dx.doi.org/10.1016/j.neuroscience.2009.11.073]. [PMID: 20006675]. [DOI] [PubMed] [Google Scholar]
- 143.Bromley R.L., Mawer G., Love J., Kelly J., Purdy L., McEwan L., Briggs M., Clayton-Smith J., Shi X., Baker G.A. Early cognitive development in children born to women with epilepsy: a prospective report. Epilepsia. 2010;51(10):2058–2065. doi: 10.1111/j.1528-1167.2010.02668.x. [http://dx.doi.org/10.1111/j.1528-1167.2010.02668.x]. [PMID: 20633039]. [DOI] [PubMed] [Google Scholar]
- 144.Thomas S. V., Sukumaran S., Lukose N., George A., Sarma P. S. Intellectual and language functions in children of mothers with epilepsy. Epilepsia. 2007;48 SRC -:2234–2240. doi: 10.1111/j.1528-1167.2007.01376.x. [DOI] [PubMed] [Google Scholar]
- 145.Thomas S.V., Ajaykumar B., Sindhu K., Nair M.K., George B., Sarma P.S. Motor and mental development of infants exposed to antiepileptic drugs in utero. Epilepsy Behav. 2008;13(1):229–236. doi: 10.1016/j.yebeh.2008.01.010. [http://dx.doi.org/10.1016/j.yebeh.2008.01.010]. [PMID: 18346940]. [DOI] [PubMed] [Google Scholar]
- 146.Tomson T., Battino D., Bonizzoni E., Craig J., Lindhout D., Perucca E., Sabers A., Thomas S.V., Vajda F., Group E.S. Dose-dependent teratogenicity of valproate in mono- and polytherapy: an observational study. Neurology. 2015;85(10):866–872. doi: 10.1212/WNL.0000000000001772. [http://dx.doi.org/10.1212/WNL.0000000000001772]. [PMID: 26085607]. [DOI] [PubMed] [Google Scholar]
- 147.Tomson T., Marson A., Boon P., Canevini M.P., Covanis A., Gaily E., Kälviäinen R., Trinka E. Valproate in the treatment of epilepsy in girls and women of childbearing potential. Epilepsia. 2015;56(7):1006–1019. doi: 10.1111/epi.13021. [http://dx.doi.org/10.1111/epi.13021]. [PMID: 25851171]. [DOI] [PubMed] [Google Scholar]
- 148.Meador K.J., Baker G.A., Browning N., Cohen M.J., Bromley R.L., Clayton-Smith J., Kalayjian L.A., Kanner A., Liporace J.D., Pennell P.B., Privitera M., Loring D.W., Group N.S. Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): a prospective observational study. Lancet Neurol. 2013;12(3):244–252. doi: 10.1016/S1474-4422(12)70323-X. [http://dx.doi.org/10.1016/S1474-4422(12)70323-X]. [PMID: 23352199]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Baker G.A., Bromley R.L., Briggs M., Cheyne C.P., Cohen M.J., García-Fiñana M., Gummery A., Kneen R., Loring D.W., Mawer G., Meador K.J., Shallcross R., Clayton-Smith J. IQ at 6 years after in utero exposure to antiepileptic drugs: a controlled cohort study. Neurology. 2015;84(4):382–390. doi: 10.1212/WNL.0000000000001182. [http://dx.doi.org/10.1212/WNL.0000000000001182]. [PMID: 25540307]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shallcross R., Bromley R.L., Irwin B., Bonnett L.J., Morrow J., Baker G.A., Liverpool Manchester Neurodevelopment G., Epilepsy U.K., Pregnancy R. Child development following in utero exposure: levetiracetam vs sodium valproate. Neurology. 2011;76(4):383–389. doi: 10.1212/WNL.0b013e3182088297. [http://dx.doi.org/10.1212/WNL.0b013e3182088297]. [PMID: 21263139]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Meador K.J., Baker G.A., Browning N., Cohen M.J., Clayton-Smith J., Kalayjian L.A., Kanner A., Liporace J.D., Pennell P.B., Privitera M., Loring D.W., Group N.S. Foetal antiepileptic drug exposure and verbal versus non-verbal abilities at three years of age. Brain. 2011;134(Pt 2):396–404. doi: 10.1093/brain/awq352. [http://dx.doi.org/10.1093/brain/awq352]. [PMID: 21224309]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nadebaum C., Anderson V., Vajda F., Reutens D., Barton S., Wood A. The Australian brain and cognition and antiepileptic drugs study: IQ in school-aged children exposed to sodium valproate and polytherapy. J. Int. Neuropsychol. Soc. 2011;17(1):133–142. doi: 10.1017/S1355617710001359. [http://dx.doi.org/10.1017/S1355617710001359]. [PMID: 21092354]. [DOI] [PubMed] [Google Scholar]
- 153.Roullet F.I., Lai J.K., Foster J.A. In utero exposure to valproic acid and autism--a current review of clinical and animal studies. Neurotoxicol. Teratol. 2013;36:47–56. doi: 10.1016/j.ntt.2013.01.004. [http://dx.doi.org/10.1016/j.ntt.2013.01.004]. [PMID: 23395807]. [DOI] [PubMed] [Google Scholar]
- 154.Schneider T. Przewłocki, R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology. 2005;30(1):80–89. doi: 10.1038/sj.npp.1300518. [http://dx.doi.org/10.1038/sj.npp.1300518]. [PMID: 15238991]. [DOI] [PubMed] [Google Scholar]
- 155.Kim J. W., Seung H., Kim K. C., Gonzales E. L. T., Oh H. A., Yang S. M., Ko M. J., Han S. H., Banerjee S., Shin C. Y. Agmatine rescues autistic behaviors in the valproic acid-induced animal model of autism. Neuropharmacology. 2017;113(Pt A):71–81. doi: 10.1016/j.neuropharm.2016.09.014. [http://dx.doi.org/10.1016/j.neuropharm.2016.09.014] [DOI] [PubMed] [Google Scholar]
- 156.administration. U. f. a. d. Information for healthcare professionals: risk of neural tube birth defects following prenatal exposure to valproate. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug SafetyInformationforPatientsandProviders/DrugSafetyInformation forHeathcareProfessionals/ucm192649.htm
- 157.Administration., U. F. a. D. Valproate products: drug safety communication — risk of impaired cognitive development in children exposed in utero (during pregnancy) http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm261610.htm
- 158.Agency., E. M. Assessment report. Procedure under Article 31 of Directive 2001/83/EC resulting from pharmacovigilance data. http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Valproate_and_related_substances_31/ Recommendation_provided_by_Pharmacovigilance_Risk_Assessment_Committee/WC500177352.pdf
- 159.Glauser T., Ben-Menachem E., Bourgeois B., Cnaan A., Guerreiro C., Kälviäinen R., Mattson R., French J.A., Perucca E., Tomson T., Guidelines I.S.A. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2013;54(3):551–563. doi: 10.1111/epi.12074. [http://dx.doi.org/10.1111/epi.12074]. [PMID: 23350722]. [DOI] [PubMed] [Google Scholar]
- 160.Guerrini R. Valproate as a mainstay of therapy for pediatric epilepsy. Paediatr. Drugs. 2006;8(2):113–129. doi: 10.2165/00148581-200608020-00004. [http://dx.doi.org/10.2165/00148581-200608020-00004]. [PMID: 16608372]. [DOI] [PubMed] [Google Scholar]
- 161.Maheshwari M.C., Jeavons P.M. Proceedings: The effect of sodium valproate (Epilim) on the EEG. Electroencephalogr. Clin. Neurophysiol. 1975;39(4):429. [PMID: 51737]. [PubMed] [Google Scholar]
- 162.Villarreal H.J., Wilder B.J., Willmore L.J., Bauman A.W., Hammond E.J., Bruni J. Effect of valproic acid on spike and wave discharges in patients with absence seizures. Neurology. 1978;28(9 Pt 1):886–891. doi: 10.1212/wnl.28.9.886. [http://dx.doi.org/10.1212/WNL.28.9.886]. [PMID: 99687]. [DOI] [PubMed] [Google Scholar]
- 163.Braathen G., Theorell K., Persson A., Rane A. Valproate in the treatment of absence epilepsy in children: a study of dose-response relationships. Epilepsia. 1988;29(5):548–552. doi: 10.1111/j.1528-1157.1988.tb03759.x. [http://dx.doi.org/10.1111/j.1528-1157.1988.tb03759.x]. [PMID: 3137019]. [DOI] [PubMed] [Google Scholar]
- 164.Davis R., Peters D.H., McTavish D. Valproic acid. A reappraisal of its pharmacological properties and clinical efficacy in epilepsy. Drugs. 1994;47(2):332–372. [http://dx.doi.org/10.2165/00003495-199447020-00008]. [PMID: 7512905]. [PubMed] [Google Scholar]
- 165.Glauser T.A., Cnaan A., Shinnar S., Hirtz D.G., Dlugos D., Masur D., Clark P.O., Adamson P.C. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy: initial monotherapy outcomes at 12 months. Epilepsia. 2013;54(1):141–155. doi: 10.1111/epi.12028. [http://dx.doi.org/10.1111/epi.12028]. [PMID: 23167925]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hwang H., Kim H., Kim S.H., Kim S.H., Lim B.C., Chae J.H., Choi J.E., Kim K.J., Hwang Y.S. Long-term effectiveness of ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. Brain Dev. 2012;34(5):344–348. doi: 10.1016/j.braindev.2011.08.007. [http://dx.doi.org/10.1016/j.braindev.2011.08.007]. [PMID: 21893390]. [DOI] [PubMed] [Google Scholar]
- 167.Brigo F., Igwe S.C. Ethosuximide, sodium valproate or lamotrigine for absence seizures in children and adolescents. Cochrane Database Syst. Rev. 2017;2:CD003032. doi: 10.1002/14651858.CD003032.pub3. [PMID: 28195639]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Basu S., Bhattacharyya K.B., Das K., Das D. Ethosuximide, sodium valproate or lamotrigine for absence seizures in children and adolescents. Cochrane Database Syst. Rev. 2017;2:CD003032. doi: 10.1002/14651858.CD003032.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Callaghan N., O’Hare J., O’Driscoll D., O’Neill B., Daly M. Comparative study of ethosuximide and sodium valproate in the treatment of typical absence seizures (petit mal). Dev. Med. Child Neurol. 1982;24(6):830–836. doi: 10.1111/j.1469-8749.1982.tb13703.x. [PMID: 6818076]. [DOI] [PubMed] [Google Scholar]
- 170.Frank L.M., Enlow T., Holmes G.L., Manasco P., Concannon S., Chen C., Womble G., Casale E.J. Lamictal (lamotrigine) monotherapy for typical absence seizures in children. Epilepsia. 1999;40(7):973–979. doi: 10.1111/j.1528-1157.1999.tb00805.x. [http://dx.doi.org/10.1111/j.1528-1157.1999.tb00805.x]. [PMID: 10403222]. [DOI] [PubMed] [Google Scholar]
- 171.Coppola G., Auricchio G., Federico R., Carotenuto M., Pascotto A. Lamotrigine versus valproic acid as first-line monotherapy in newly diagnosed typical absence seizures: An open-label, randomized, parallel-group study. Epilepsia. 2004;45(9):1049–1053. doi: 10.1111/j.0013-9580.2004.40903.x. [http://dx.doi.org/10.1111/j.0013-9580.2004.40903.x]. [PMID: 15329068]. [DOI] [PubMed] [Google Scholar]
- 172.Huang T.S., Zhu J.L., Li B., Hu Y., Chen L., Liao J.X. Zhongguo Dang Dai Er Ke Za Zhi. 2009;11(8):653–655. [Valproic acid versus lamotrigine as a monotherapy for absence epilepsy in children]. [PMID: 19695193]. [PubMed] [Google Scholar]
- 173.Martinovic Z. Comparison of ethosuximide with sodium valproate as monotherapies of absence seizures. New York: Raven Press; 1983. [Google Scholar]
- 174.Sato S., White B.G., Penry J.K., Dreifuss F.E., Sackellares J.C., Kupferberg H.J. Valproic acid versus ethosuximide in the treatment of absence seizures. Neurology. 1982;32(2):157–163. doi: 10.1212/wnl.32.2.157. [http://dx.doi.org/10.1212/WNL.32.2.157]. [PMID: 6798490]. [DOI] [PubMed] [Google Scholar]
- 175.Turnbull D. M., Rawlins M. D., Weightman D., Chadwicks D. W. A comparison of phenytoin and valproate in previously untreated adult epileptic patients Journal of Neurology Neurosurgery and Psychiatry. Journal of Neurology Neurosurgery and Psychiatry. 1982;45 SRC:55–59. doi: 10.1136/jnnp.45.1.55. [http://dx.doi.org/10.1136/jnnp.45.1.55] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Callaghan N., Kenny R.A., O’Neill B., Crowley M., Goggin T. A prospective study between carbamazepine, phenytoin and sodium valproate as monotherapy in previously untreated and recently diagnosed patients with epilepsy. J. Neurol. Neurosurg. Psychiatry. 1985;48(7):639–644. doi: 10.1136/jnnp.48.7.639. [http://dx.doi.org/10.1136/jnnp.48.7.639]. [PMID: 3928820]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Marson A.G., Al-Kharusi A.M., Alwaidh M., Appleton R., Baker G.A., Chadwick D.W., Cramp C., Cockerell O.C., Cooper P.N., Doughty J., Eaton B., Gamble C., Goulding P.J., Howell S.J., Hughes A., Jackson M., Jacoby A., Kellett M., Lawson G.R., Leach J.P., Nicolaides P., Roberts R., Shackley P., Shen J., Smith D.F., Smith P.E., Smith C.T., Vanoli A., Williamson P.R. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369(9566):1016–1026. doi: 10.1016/S0140-6736(07)60461-9. [http://dx.doi.org/10.1016/S0140-6736(07)60461-9]. [PMID: 17382828]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mattson R.H., Cramer J.A., Collins J.F. A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic-clonic seizures in adults. The Department of Veterans Affairs Epilepsy Cooperative Study No. 264 Group. N. Engl. J. Med. 1992;327(11):765–771. doi: 10.1056/NEJM199209103271104. [http://dx.doi.org/10.1056/NEJM199209103271104]. [PMID: 1298221]. [DOI] [PubMed] [Google Scholar]
- 179.Beydoun A., Sackellares J.C., Shu V. Safety and efficacy of divalproex sodium monotherapy in partial epilepsy: a double-blind, concentration-response design clinical trial. Neurology. 1997;48(1):182–188. doi: 10.1212/wnl.48.1.182. [http://dx.doi.org/10.1212/WNL.48.1.182]. [PMID: 9008516]. [DOI] [PubMed] [Google Scholar]
- 180.Guerrini R., Genton P. Epileptic syndromes and visually induced seizures. Epilepsia. 2004;45(Suppl. 1):14–18. doi: 10.1111/j.0013-9580.2004.451011.x. [http://dx.doi.org/10.1111/j.0013-9580.2004.451011.x]. [PMID: 14706039]. [DOI] [PubMed] [Google Scholar]
- 181.Covanis A., Stodieck S.R., Wilkins A.J. Treatment of photosensitivity. Epilepsia. 2004;45(Suppl. 1):40–45. doi: 10.1111/j.0013-9580.2004.451006.x. [http://dx.doi.org/10.1111/j.0013-9580.2004.451006.x]. [PMID: 14706045]. [DOI] [PubMed] [Google Scholar]
- 182.Belcastro V., Caraballo R.H., Romeo A., Striano P. Early-onset absence epilepsy aggravated by valproic acid: a video-EEG report. Epileptic Disord. 2013;15(4):440–443. doi: 10.1684/epd.2013.0616. [DOI] [PubMed] [Google Scholar]
- 183.Chen J.H., Zheng Y.L., Xu C.Q., Gu L.Z., Ding Z.L., Qin L., Wang Y., Fu R., Wan Y.F., Hu C.P. Valproic acid (VPA) enhances cisplatin sensitivity of non-small cell lung cancer cells via HDAC2 mediated down regulation of ABCA1. Biol. Chem. 2017;398(7):785–792. doi: 10.1515/hsz-2016-0307. [http://dx.doi.org/10.1515/hsz-2016-0307]. [PMID: 28002023]. [DOI] [PubMed] [Google Scholar]
- 184.Damaskos C., Garmpis N., Valsami S., Kontos M., Spartalis E., Kalampokas T., Kalampokas E., Athanasiou A., Moris D., Daskalopoulou A., Davakis S., Tsourouflis G., Kontzoglou K., Perrea D., Nikiteas N., Dimitroulis D. Histone deacetylase inhibitors: An attractive therapeutic strategy against breast cancer. Anticancer Res. 2017;37(1):35–46. doi: 10.21873/anticanres.11286. [http://dx.doi.org/10.21873/anticanres.11286]. [PMID: 28011471]. [DOI] [PubMed] [Google Scholar]
- 185.Caponigro F., Di Gennaro E., Ionna F., Longo F., Aversa C., Pavone E., Maglione M.G., Di Marzo M., Muto P., Cavalcanti E., Petrillo A., Sandomenico F., Maiolino P., D’Aniello R., Botti G., De Cecio R., Losito N.S., Scala S., Trotta A., Zotti A.I., Bruzzese F., Daponte A., Calogero E., Montano M., Pontone M., De Feo G., Perri F., Budillon A. Phase II clinical study of valproic acid plus cisplatin and cetuximab in recurrent and/or metastatic squamous cell carcinoma of Head and Neck-V-CHANCE trial. BMC Cancer. 2016;16(1):918. doi: 10.1186/s12885-016-2957-y. [http://dx.doi.org/10.1186/s12885-016-2957-y]. [PMID: 27884140]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Thotala D., Karvas R.M., Engelbach J.A., Garbow J.R., Hallahan A.N., DeWees T.A., Laszlo A., Hallahan D.E. Valproic acid enhances the efficacy of radiation therapy by protecting normal hippocampal neurons and sensitizing malignant glioblastoma cells. Oncotarget. 2015;6(33):35004–35022. doi: 10.18632/oncotarget.5253. [http://dx.doi.org/10.18632/oncotarget.5253]. [PMID: 26413814]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Happold C., Gorlia T., Chinot O., Gilbert M.R., Nabors L.B., Wick W., Pugh S.L., Hegi M., Cloughesy T., Roth P., Reardon D.A., Perry J.R., Mehta M.P., Stupp R., Weller M. Does valproic acid or levetiracetam improve survival in glioblastoma? A pooled analysis of prospective clinical trials in newly diagnosed glioblastoma. J. Clin. Oncol. 2016;34(7):731–739. doi: 10.1200/JCO.2015.63.6563. [http://dx.doi.org/10.1200/JCO.2015.63.6563]. [PMID: 26786929]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Schreur L., Middeljans-Tijssen C.W., Hengstman G.J., Olde Rikkert M.G. Tijdschr. Gerontol. Geriatr. 2009;40(1):29–33. doi: 10.1007/BF03088474. [Cognitive impairment and parkinsonism due to use of sodium valproate]. [http://dx.doi.org/10.1007/BF03088474]. [PMID: 19326700]. [DOI] [PubMed] [Google Scholar]
- 189.Romoli M., Costa C., Siliquini S., Corbelli I., Eusebi P., Bedetti C., Caproni S., Cupini L. M., Calabresi P., Sarchielli P. Antiepileptic drugs in migraine and epilepsy: Who is at increased risk of adverse events? Cephalalgia : an international journal of headache. 2018;38(2):274–282. doi: 10.1177/0333102416683925. [DOI] [PubMed] [Google Scholar]
- 190.Bryant A.E., III, Dreifuss F.E. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465–469. doi: 10.1212/wnl.46.2.465. [http://dx.doi.org/10.1212/WNL.46.2.465]. [PMID: 8614514]. [DOI] [PubMed] [Google Scholar]
- 191.Lin C.M., Thajeb P. Valproic acid aggravates epilepsy due to MELAS in a patient with an A3243G mutation of mitochondrial DNA. Metab. Brain Dis. 2007;22(1):105–109. doi: 10.1007/s11011-006-9039-9. [http://dx.doi.org/10.1007/s11011-006-9039-9]. [PMID: 17226098]. [DOI] [PubMed] [Google Scholar]
- 192.Wong H. Y., Chu T. S., Lai J. C., Fung K. P., Fok T. F., Fujii T. Sodium valproate inhibits glucose transport and exacerbates Glut-1 deficiency. vitro Biochem, 2005;96 SRC -:775–785. doi: 10.1002/jcb.20555. [DOI] [PubMed] [Google Scholar]
- 193.Hamer H.M., Knake S., Schomburg U., Rosenow F. Valproate-induced hyperammonemic encephalopathy in the presence of topiramate. Neurology. 2000;54(1):230–232. doi: 10.1212/wnl.54.1.230. [http://dx.doi.org/10.1212/WNL.54.1.230]. [PMID: 10636156]. [DOI] [PubMed] [Google Scholar]
- 194.LaBuzetta J.N., Yao J.Z., Bourque D.L., Zivin J. Adult nonhepatic hyperammonemia: a case report and differential diagnosis. Am. J. Med. 2010;123(10):885–891. doi: 10.1016/j.amjmed.2010.02.029. [http://dx.doi.org/10.1016/j.amjmed.2010.02.029]. [PMID: 20920686]. [DOI] [PubMed] [Google Scholar]
- 195.Verrotti A., Trotta D., Morgese G., Chiarelli F. Valproate-induced hyperammonemic encephalopathy. Metab. Brain Dis. 2002;17(4):367–373. doi: 10.1023/a:1021918104127. [http://dx.doi.org/10.1023/A:1021918104127]. [PMID: 12602513]. [DOI] [PubMed] [Google Scholar]
- 196.Verrotti A., Manco R., Agostinelli S., Coppola G., Chiarelli F. The metabolic syndrome in overweight epileptic patients treated with valproic acid. Epilepsia. 2010;51(2):268–273. doi: 10.1111/j.1528-1167.2009.02206.x. [http://dx.doi.org/10.1111/j.1528-1167.2009.02206.x]. [PMID: 19682024]. [DOI] [PubMed] [Google Scholar]
- 197.DeWolfe J.L., Knowlton R.C., Beasley M.T., Cofield S., Faught E., Limdi N.A. Hyperammonemia following intravenous valproate loading. Epilepsy Res. 2009;85(1):65–71. doi: 10.1016/j.eplepsyres.2009.02.012. [http://dx.doi.org/10.1016/j.eplepsyres.2009.02.012]. [PMID: 19299111]. [DOI] [PubMed] [Google Scholar]
- 198.Finsterer J., Zarrouk S. Epilepsy in mitochondrial disorders. Seizure. 2012;21(5):316–321. doi: 10.1016/j.seizure.2012.03.003. [http://dx.doi.org/10.1016/j.seizure.2012.03.003]. [PMID: 22459315]. [DOI] [PubMed] [Google Scholar]
- 199.Alonso-Juarez M., Torres-Russotto D., Crespo-Morfin P., Baizabal-Carvallo J.F. The clinical features and functional impact of valproate-induced tremor. Parkinsonism Relat. Disord. 2017;44:147–150. doi: 10.1016/j.parkreldis.2017.09.011. [http://dx.doi.org/10.1016/j.parkreldis.2017.09.011]. [PMID: 28941829]. [DOI] [PubMed] [Google Scholar]
- 200.Nanau R.M., Neuman M.G. Adverse drug reactions induced by valproic acid. Clin. Biochem. 2013;46(15):1323–1338. doi: 10.1016/j.clinbiochem.2013.06.012. [http://dx.doi.org/10.1016/j.clinbiochem.2013.06.012]. [PMID: 23792104]. [DOI] [PubMed] [Google Scholar]
- 201.Yu P.M., Zhu G.X., Wu X.Y., Li T., Xu L., Yue L., Wang B., Hong Z. A 6-month prospective study on efficacy safety and QOL profiles of extended-release formulation of valproate in patients with epilepsy. Seizure. 2011;20(1):23–26. doi: 10.1016/j.seizure.2010.09.014. [http://dx.doi.org/10.1016/j.seizure.2010.09.014]. [PMID: 20951067]. [DOI] [PubMed] [Google Scholar]
- 202.Penot J.P., Pradeau F. Iatrogenic dementia and extrapyramidal syndrome: rare adverse effect of valproic acid-aspirin combination. Presse Med. 2010;39(2):279–280. doi: 10.1016/j.lpm.2009.02.024. [http://dx.doi.org/10.1016/j.lpm.2009.02.024]. [PMID: 19419832]. [DOI] [PubMed] [Google Scholar]
- 203.Jahromi S.R., Togha M., Fesharaki S.H., Najafi M., Moghadam N.B., Kheradmand J.A., Kazemi H., Gorji A. Gastrointestinal adverse effects of antiepileptic drugs in intractable epileptic patients. Seizure. 2011;20(4):343–346. doi: 10.1016/j.seizure.2010.12.011. [http://dx.doi.org/10.1016/j.seizure.2010.12.011]. [PMID: 21236703]. [DOI] [PubMed] [Google Scholar]
- 204.Bale J.F., Jr, Gay P.E., Madsen J.A. Monitoring of serum amylase levels during valproic acid therapy. Ann. Neurol. 1982;11(2):217–218. doi: 10.1002/ana.410110226. [http://dx.doi.org/10.1002/ana.410110226]. [PMID: 6176180]. [DOI] [PubMed] [Google Scholar]
- 205.Grauso-Eby N.L., Goldfarb O., Feldman-Winter L.B., McAbee G.N. Acute pancreatitis in children from Valproic acid: case series and review. Pediatr. Neurol. 2003;28(2):145–148. doi: 10.1016/s0887-8994(02)00517-9. [http://dx.doi.org/10.1016/S0887-8994(02)00517-9]. [PMID: 12699868]. [DOI] [PubMed] [Google Scholar]
- 206.Acharya S., Bussel J.B. Hematologic toxicity of sodium valproate. J. Pediatr. Hematol. Oncol. 2000;22(1):62–65. doi: 10.1097/00043426-200001000-00012. [http://dx.doi.org/10.1097/00043426-200001000-00012]. [PMID: 10695824]. [DOI] [PubMed] [Google Scholar]
- 207.Chakraborty S., Chakraborty J., Mandal S., Ghosal M.K. A rare occurrence of isolated neutropenia with valproic acid: a case report. J. Indian Med. Assoc. 2011;109(5):345–346. [PMID: 22187773]. [PubMed] [Google Scholar]
- 208.Delgado M.R., Riela A.R., Mills J., Browne R., Roach E.S. Thrombocytopenia secondary to high valproate levels in children with epilepsy. J. Child Neurol. 1994;9(3):311–314. doi: 10.1177/088307389400900318. [http://dx.doi.org/10.1177/088307389400900318]. [PMID: 7930412]. [DOI] [PubMed] [Google Scholar]
- 209.Köse G., Arhan E., Unal B., Ozaydin E., Guven A., Sayli T.R. Valproate-associated coagulopathies in children during short-term treatment. J. Child Neurol. 2009;24(12):1493–1498. doi: 10.1177/0883073808331084. [http://dx.doi.org/10.1177/0883073808331084]. [PMID: 19482838]. [DOI] [PubMed] [Google Scholar]
- 210.Vasudev K., Keown P., Gibb I., McAllister-Williams R.H. Hematological effects of valproate in psychiatric patients: what are the risk factors? J. Clin. Psychopharmacol. 2010;30(3):282–285. doi: 10.1097/JCP.0b013e3181db2684. [http://dx.doi.org/10.1097/JCP.0b013e3181db2684]. [PMID: 20473063]. [DOI] [PubMed] [Google Scholar]
- 211.Knowles S.R., Shapiro L.E., Shear N.H. Anticonvulsant hypersensitivity syndrome: incidence, prevention and management. Drug Saf. 1999;21(6):489–501. doi: 10.2165/00002018-199921060-00005. [http://dx.doi.org/10.2165/00002018-199921060-00005]. [PMID: 10612272]. [DOI] [PubMed] [Google Scholar]
- 212.Evans M.D., Shinar R., Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509–511. doi: 10.1016/j.seizure.2011.02.009. [http://dx.doi.org/10.1016/j.seizure.2011.02.009]. [PMID: 21435910]. [DOI] [PubMed] [Google Scholar]
- 213.Ozen S., Bulbul I., Soyucok E. Valproate induced hypoactive delirium in a bipolar disorder patient with psychotic features. Turkish J. Psychiatry. 2010;21(1):79–84. [PubMed] [Google Scholar]
- 214.Ristić A.J.; Vojvodić N.; Janković S.; Sindelić A.; Sokić D. The frequency of reversible parkinsonism and cognitive decline associated with valproate treatment: a study of 364 patients with different types of epilepsy. Epilepsia. 2006;47(12):2183–2185. doi: 10.1111/j.1528-1167.2006.00711.x. [http://dx.doi.org/10.1111/j.1528-1167.2006.00711.x]. [PMID: 17201721]. [DOI] [PubMed] [Google Scholar]
- 215.Belcastro V., D’Egidio C., Striano P., Verrotti A. Metabolic and endocrine effects of valproic acid chronic treatment. Epilepsy Res. 2013;107(1-2):1–8. doi: 10.1016/j.eplepsyres.2013.08.016. [http://dx.doi.org/10.1016/j.eplepsyres.2013.08.016]. [PMID: 24076030]. [DOI] [PubMed] [Google Scholar]
- 216.El-Khatib F., Rauchenzauner M., Lechleitner M., Hoppichler F., Naser A., Waldmann M., Trinka E., Unterberger I., Bauer G., Luef G.J. Valproate, weight gain and carbohydrate craving: a gender study. Seizure. 2007;16(3):226–232. doi: 10.1016/j.seizure.2006.12.009. [http://dx.doi.org/10.1016/j.seizure.2006.12.009]. [PMID: 17210261]. [DOI] [PubMed] [Google Scholar]
- 217.Verrotti A., la Torre R., Trotta D., Mohn A., Chiarelli F. Valproate-induced insulin resistance and obesity in children. Horm. Res. 2009;71(3):125–131. doi: 10.1159/000197868. [http://dx.doi.org/10.1159/000197868]. [PMID: 19188736]. [DOI] [PubMed] [Google Scholar]
- 218.Verrotti A., D’Egidio C., Coppola G., Parisi P., Chiarelli F. Epilepsy, sex hormones and antiepileptic drugs in female patients. Expert Rev. Neurother. 2009;9(12):1803–1814. doi: 10.1586/ern.09.112. [http://dx.doi.org/10.1586/ern.09.112]. [PMID: 19951139]. [DOI] [PubMed] [Google Scholar]
- 219.Isojärvi J.I., Laatikainen T.J., Knip M., Pakarinen A.J., Juntunen K.T., Myllylä V.V. Obesity and endocrine disorders in women taking valproate for epilepsy. Ann. Neurol. 1996;39(5):579–584. doi: 10.1002/ana.410390506. [http://dx.doi.org/10.1002/ana.410390506]. [PMID: 8619542]. [DOI] [PubMed] [Google Scholar]
- 220.Greco R., Latini G., Chiarelli F., Iannetti P., Verrotti A. Leptin, ghrelin, and adiponectin in epileptic patients treated with valproic acid. Neurology. 2005;65(11):1808–1809. doi: 10.1212/01.wnl.0000187074.27586.d1. [http://dx.doi.org/10.1212/01.wnl.0000187074.27586.d1]. [PMID: 16344528]. [DOI] [PubMed] [Google Scholar]
- 221.Biton V., Mirza W., Montouris G., Vuong A., Hammer A.E., Barrett P.S. Weight change associated with valproate and lamotrigine monotherapy in patients with epilepsy. Neurology. 2001;56(2):172–177. doi: 10.1212/wnl.56.2.172. [http://dx.doi.org/10.1212/WNL.56.2.172]. [PMID: 11160951]. [DOI] [PubMed] [Google Scholar]
- 222.Jallon P., Picard F. Bodyweight gain and anticonvulsants: a comparative review. Drug Saf. 2001;24(13):969–978. doi: 10.2165/00002018-200124130-00004. [http://dx.doi.org/10.2165/00002018-200124130-00004]. [PMID: 11735653]. [DOI] [PubMed] [Google Scholar]
- 223.Kim J.Y., Lee H.W. Metabolic and hormonal disturbances in women with epilepsy on antiepileptic drug monotherapy. Epilepsia. 2007;48(7):1366–1370. doi: 10.1111/j.1528-1167.2007.01052.x. [http://dx.doi.org/10.1111/j.1528-1167.2007.01052.x]. [PMID: 17565596]. [DOI] [PubMed] [Google Scholar]
- 224.Luef G., Abraham I., Hoppichler F., Trinka E., Unterberger I., Bauer G., Lechleitner M. Increase in postprandial serum insulin levels in epileptic patients with valproic acid therapy. Metabolism. 2002;51(10):1274–1278. doi: 10.1053/meta.2002.34708. [http://dx.doi.org/10.1053/meta.2002.34708]. [PMID: 12370846]. [DOI] [PubMed] [Google Scholar]
- 225.Martin C.K., Han H., Anton S.D., Greenway F.L., Smith S.R. Effect of valproic acid on body weight, food intake, physical activity and hormones: results of a randomized controlled trial. J. Psychopharmacol. (Oxford) 2009;23(7):814–825. doi: 10.1177/0269881108091595. [http://dx.doi.org/10.1177/0269881108091595]. [PMID: 18583434]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Morrell M.J., Isojärvi J., Taylor A.E., Dam M., Ayala R., Gomez G., O’Neill F., Tennis P., Messenheimer J. Higher androgens and weight gain with valproate compared with lamotrigine for epilepsy. Epilepsy Res. 2003;54(2-3):189–199. doi: 10.1016/s0920-1211(03)00085-8. [http://dx.doi.org/10.1016/S0920-1211(03)00085-8]. [PMID: 12837570]. [DOI] [PubMed] [Google Scholar]
- 227.Sharpe C., Wolfson T., Trauner D.A. Weight gain in children treated with valproate. J. Child Neurol. 2009;24(3):338–341. doi: 10.1177/0883073808323023. [http://dx.doi.org/10.1177/0883073808323023]. [PMID: 19258293]. [DOI] [PubMed] [Google Scholar]
- 228.Wirrell E.C. Valproic acid-associated weight gain in older children and teens with epilepsy. Pediatr. Neurol. 2003;28(2):126–129. doi: 10.1016/s0887-8994(02)00505-2. [http://dx.doi.org/10.1016/S0887-8994(02)00505-2]. [PMID: 12699863]. [DOI] [PubMed] [Google Scholar]
- 229.Demir E., Aysun S. Weight gain associated with valproate in childhood. Pediatr. Neurol. 2000;22(5):361–364. doi: 10.1016/s0887-8994(00)00133-8. [http://dx.doi.org/10.1016/S0887-8994(00)00133-8]. [PMID: 10913727]. [DOI] [PubMed] [Google Scholar]
- 230.Lihn A.S., Pedersen S.B., Richelsen B. Adiponectin: action, regulation and association to insulin sensitivity. Obes. Rev. 2005;6(1):13–21. doi: 10.1111/j.1467-789X.2005.00159.x. [http://dx.doi.org/10.1111/j.1467-789X.2005.00159.x]. [PMID: 15655035]. [DOI] [PubMed] [Google Scholar]
- 231.Verrotti A., Greco R., Latini G., Chiarelli F. Endocrine and Metabolic Changes in Epileptic Patients Receiving Valproic Acid. J. Pediatric Endocrinol. Metabol. 2005:423–430. doi: 10.1515/jpem.2005.18.5.423. 18 SRC. [http://dx.doi.org/10.1515/JPEM.2005.18.5.423] [DOI] [PubMed] [Google Scholar]
- 232.Prabhakar S., Sahota P., Kharbanda P.S., Siali R., Jain V., Lal V., Khurana D. Sodium valproate, hyperandrogenism and altered ovarian function in Indian women with epilepsy: a prospective study. Epilepsia. 2007;48(7):1371–1377. doi: 10.1111/j.1528-1167.2007.01100.x. [http://dx.doi.org/10.1111/j.1528-1167.2007.01100.x]. [PMID: 17441994]. [DOI] [PubMed] [Google Scholar]
- 233.Bilo L., Meo R. Epilepsy and polycystic ovary syndrome: where is the link? Neurol. Sci. 2006;27(4):221–230. doi: 10.1007/s10072-006-0675-y. [http://dx.doi.org/10.1007/s10072-006-0675-y]. [PMID: 16998724]. [DOI] [PubMed] [Google Scholar]
- 234.Verrotti A., Loiacono G., Laus M., Coppola G., Chiarelli F., Tiboni G.M. Hormonal and reproductive disturbances in epileptic male patients: emerging issues. Reprod. Toxicol. 2011;31(4):519–527. doi: 10.1016/j.reprotox.2011.02.002. [http://dx.doi.org/10.1016/j.reprotox.2011.02.002]. [PMID: 21338669]. [DOI] [PubMed] [Google Scholar]
- 235.Genton P., Semah F., Trinka E. Valproic acid in epilepsy: pregnancy-related issues. Drug Saf. 2006;29(1):1–21. doi: 10.2165/00002018-200629010-00001. [http://dx.doi.org/10.2165/00002018-200629010-00001]. [PMID: 16454531]. [DOI] [PubMed] [Google Scholar]
- 236.Morrow J., Russell A., Guthrie E., Parsons L., Robertson I., Waddell R., Irwin B., McGivern R.C., Morrison P.J., Craig J. Malformation risks of antiepileptic drugs in pregnancy: a prospective study from the UK Epilepsy and Pregnancy Register. J. Neurol. Neurosurg. Psychiatry. 2006;77(2):193–198. doi: 10.1136/jnnp.2005.074203. [http://dx.doi.org/10.1136/jnnp.2005.074203]. [PMID: 16157661]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Jentink J., Loane M.A., Dolk H., Barisic I., Garne E., Morris J.K., de Jong-van den Berg L.T., Group E.A.S.W. Valproic acid monotherapy in pregnancy and major congenital malformations. N. Engl. J. Med. 2010;362(23):2185–2193. doi: 10.1056/NEJMoa0907328. [http://dx.doi.org/10.1056/NEJMoa0907328]. [PMID: 20558369]. [DOI] [PubMed] [Google Scholar]
- 238.Tanoshima M., Kobayashi T., Tanoshima R., Beyene J., Koren G., Ito S. Risks of congenital malformations in offspring exposed to valproic acid in utero: A systematic review and cumulative meta-analysis. Clin. Pharmacol. Ther. 2015;98(4):417–441. doi: 10.1002/cpt.158. [http://dx.doi.org/10.1002/cpt.158]. [PMID: 26044279]. [DOI] [PubMed] [Google Scholar]
- 239.Jackson A., Bromley R., Morrow J., Irwin B., Clayton-Smith J. In utero exposure to valproate increases the risk of isolated cleft palate. Arch. Dis. Child. Fetal Neonatal Ed. 2016;101(3):F207–F211. doi: 10.1136/archdischild-2015-308278. [http://dx.doi.org/10.1136/archdischild-2015-308278]. [PMID: 26408639]. [DOI] [PubMed] [Google Scholar]
- 240.Diav-Citrin O., Shechtman S., Bar-Oz B., Cantrell D., Arnon J., Ornoy A. Pregnancy outcome after in utero exposure to valproate: evidence of dose relationship in teratogenic effect. CNS Drugs. 2008;22(4):325–334. doi: 10.2165/00023210-200822040-00004. [http://dx.doi.org/10.2165/00023210-200822040-00004]. [PMID: 18336060]. [DOI] [PubMed] [Google Scholar]
- 241.Hernández-Díaz S., Smith C.R., Shen A., Mittendorf R., Hauser W.A., Yerby M., Holmes L.B. Comparative safety of antiepileptic drugs during pregnancy. Neurology. 2012;78(21):1692–1699. doi: 10.1212/WNL.0b013e3182574f39. [http://dx.doi.org/10.1212/WNL.0b013e3182574f39]. [PMID: 22551726]. [DOI] [PubMed] [Google Scholar]
- 242.Tomson T., Battino D., Bonizzoni E., Craig J., Lindhout D., Perucca E., Sabers A., Thomas S.V., Vajda F., Group E.S. Comparative risk of major congenital malformations with eight different antiepileptic drugs: a prospective cohort study of the EURAP registry. Lancet Neurol. 2018;17(6):530–538. doi: 10.1016/S1474-4422(18)30107-8. [http://dx.doi.org/10.1016/S1474-4422(18)30107-8]. [PMID: 29680205]. [DOI] [PubMed] [Google Scholar]
- 243.Virta L.J., Kälviäinen R., Villikka K., Keränen T. Declining trend in valproate use in Finland among females of childbearing age in 2012-2016 - a nationwide registry-based outpatient study. Eur. J. Neurol. 2018;25(6):869–874. doi: 10.1111/ene.13610. [http://dx.doi.org/10.1111/ene.13610]. [PMID: 29509301]. [DOI] [PubMed] [Google Scholar]