

Abstract
Migraine, an extremely disabling neurological disorder, has a strong genetic component. Since monogenic mi-graines (resulting from mutations or changes in a single gene) may help researchers discover migraine pathophysiology, transgenic mice models harboring gene mutations identified in Familial Hemiplegic Migraine (FHM) patients have been gen-erated. Studies in these FHM mutant mice models have shed light on the mechanisms of migraine and may aid in the identifi-cation of novel targets for treatment. More specifically, the studies shed light on how gene mutations, hormones, and other factors impact the pathophysiology of migraine. The models may also be of relevance to researchers outside the field of mi-graine as some of their aspects are relevant to pain in general. Additionally, because of the comorbidities associated with mi-graine, they share similarities with the mutant mouse models of epilepsy, stroke, and perhaps depression. Here, we review the experimental data obtained from these mutant mice and focus on how they can be used to investigate the pathophysiology of migraine, including synaptic plasticity, neuroinflammation, metabolite alterations, and molecular and behavioral mecha-nisms of pain.
Keywords: Familial hemiplegic migraine, mouse models, migraine with aura, cortical spreading depression, pathophysiology, neuroinflammation, treatment
1. INTRODUCTION
Migraine, the third-most common human disease in the world [1], is a neurobiological disorder with characteristics such as headache, generally combined with nausea and vomiting, and light and sound hypersensitivity [2]. The two types of migraines are migraine with aura (MA) and migraine without aura (MO) [2], of which the first occurs in more than one-third of the people. Aura may last for 5-60 minutes and has transient neurological symptoms, such as sensory, motor, visual, or speech difficulties that develop before the onset of headache. The comorbidity of migraine with other disorders including stroke [3, 4] and epilepsy [5-7] has been reported. The clinical symptoms in migraine are well known; however, its precise molecular pathophysiology is not yet well defined. A better understanding of migraine mechanisms has been possible with several animal studies which have helped in improving therapy [8, 9]. In migraineurs’ brains, there is a higher vulnerability to changes in the homeostasis of the ions [10-12]. As many clinical and experimental data suggest, cortical spreading depression (CSD) has a main role in triggering the headache mechanism [10-12] in migraine with aura. CSD is a wave of depolarization in neuronal and glial cells which spreads 3-6 mm/min in the cortex, and is well
known as the electrophysiological cause of aura in migraine patients [13]. After CSD, parenchymal neuroinflammatory cascades start, Pannexin1 channels in neurons open, and neuroinflammatory molecules such as high mobility group box-1 (HMGB1) and nuclear factor-κB (NF-κB) are released, leading to the activation of the trigeminovascular system [12]. CSD starts a range of events in the cortex, subcortex, [14] meninges, and brainstem, consistent with the development of headache in animal models [10, 12]. Migraine is still known as a complex disorder with few monogenic reasons identified [15]. Monogenic type of migraines are helpful models for the detailed study of the disease neurobiology because of the number of available transgenic mouse models and reported symptoms that overlap with common migraine. Here, by reviewing genetic animal models for migraine, we tried to provide a holistic image of the present knowledge on migraine pathophysiology, synaptic plasticity, the role of neuroinflammatory processes, metabolite alterations, and molecular and behavioral mechanisms of pain, besides the future studies. We have included studies on the first FHM mice models of monogenic migraine, and excluded clinical cases, patients, or in vitro studies.
1.1. Familial Hemiplegic Migraine Mutations as a Model for Disease
Genes and pathways for migraine vulnerability have been unraveled by studies involving mutations in monogenic migraine. Mutations leading to familial hemiplegic migraine (FHM) type 1, 2, and 3 provide the basis for the current transgenic mouse models of migraine. These studies show an increase in CSD susceptibility in these mouse models with multiple mutations and mechanisms leading to the phenomenon. This may be due to the enhanced cortical glutamatergic transmission observed in FHM1 mouse models [16]. Antiepileptic drugs suppressing neuronal excitability have highlighted the role of neuronal excitation in CSD susceptibility determination [17, 18]. Besides, studies in FHM1 [19], common migraine patients in the clinic [20, 21], and genome-wide association studies (GWAS) in subjects with common migraine [8] confirm the hyperexcitability of the cortex as an underlying cause for migraine.
FHM is one of the monogenic migraine types with aura, characterized by transient hemiparesis during aura events [2]. Since most of the patients with FHM present symptoms similar to common migraine along with typical hemiplegia [22], FHM can be included in migraine classification, and models that mimic this condition might be relevant for studying common migraine mechanisms. Three genes for FHM have been recognized: voltage-dependent calcium channel, alpha 1A subunit (CACNA1A) for FHM1; ATPase Na+/K+ pump, alpha 2 subunit (ATP1A2) for FHM2; and voltage-gated sodium channel, type 1 alpha subunit (SCN1A) for FHM3.
FHM1 gene, CACNA1A, located on chromosome 19p13, is responsible for coding a voltage-gated Cav2.1 Ca2+ channel subunit in neurons [22], the channels which regulate the release of neurotransmitters at central synapses in the brain [23]. FHM1 mutations show Cav2.1 channel gain-of-function in human cell systems [24] and in mutant mice neurons [25]. This effect increases Ca2+ influx following an action potential and increases the release of glutamate in the synaptic cleft [16, 26] causing enhancement in neuronal network activity, followed by CSD facilitation [27]. This may explain symptoms of migraine aura and headache in FHM1. In clinical perspective, the variability of FHM1 mutations changes from hemiplegia, as reported in patients with R192Q mutation [22] to severe deficits such as cerebellar ataxia, epilepsy, and fatal coma in S218L mutant patients [28].
The FHM2 gene, ATP1A2, located on chromosome 1q23, is responsible for encoding the α2 subunit of the Na+/K+ transporting ATPase [29] which forces Na+ ions outward of the glial cells and K+ ions inward, binds Na+, K+, and ATP, and utilizes ATP hydrolysis. Thus, the pump regulates glutamate and potassium re-uptake in synaptic cleft into glial cells. FHM2 mutations in glial cells cause loss of function in the Na+/K+ ATPase, leading to an increase in extracellular K+ and glutamate, and hyper-excitation in neuronal networks [30, 31]. In the clinic, FHM2 mutations show pure hemiplegia and FHM attacks [29, 32, 33], or the mixture of FHM symptoms with other brain diseases, including aphasia, cerebellar ataxia, behavioral changes, and coma [34-36].
The FHM3 gene, SCN1A, located on chromosome 2q24, encodes voltage-gated NaV1.1 sodium channel subunit [37] that has an essential role in the generation of an action potential and its propagation. Sometimes, FHM3 mutations are present with other clinical symptoms, including transient blindness and generalized tonic-clonic epilepsy [37-40], apart from migraine and hemiplegia. Reports from FHM3 patients with epilepsy suggest a common molecular mechanism between FHM and epilepsy indicated by seizures reported independently from attacks of hemiplegic migraine [38]. In FHM3, gain-of-function of NaV1.1 channels causes hyperexcitability of inhibitory interneurons. Reports of loss-of-function show the complex spectrum of NaV1.1 and the effects of its mutation FHM3.
Fig. (1) shows the summarized mechanisms of three FHM mutations.
Fig. (1).
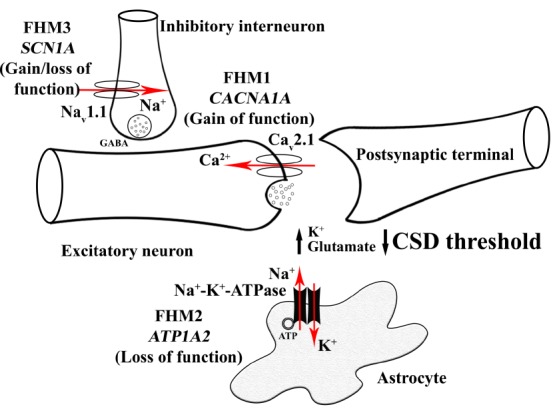
Familial Hemiplegic Migraine (FHM) mutations causing increased susceptibility of the brain to Cortical Spreading Depression (CSD). FHM1 mutations in CACNA1A gene encoding P/Q type neuronal Cav2.1 channels result in the gain of function of this calcium channel that increases synaptic glutamate release upon neuronal activity. Loss of function mutations in ATP1A2 gene encoding a2 subunit of Na+-K+-ATPase antiport carrier located on astrocyte membrane is responsible for FHM2. This mutation leads to an increased amount of K+ and glutamate in the synaptic cleft. FHM3 mutations in SCN1A gene encoding voltage-gated NaV1.1 sodium channel subunit result in hyperexcitability of GABAergic interneurons by increasing extracellular K+. All three types of FHM mutations cause increased K+ and glutamate in the synaptic cleft, hence facilitation of CSD, the neurophysiological correlate of migraine aura.
1.2. Mouse Models of FHM
So far, two mutant FHM1 knock-in (KI) mouse models with the R192Q or S218L gain-of-function mutations in the CACNA1A have been reported [25, 41]. Animals with the R192Q mutation present a milder form, [26] however, mice with the severe S218L mutation display an overt phenotype with cerebellar ataxia and seizures, [42] consistent with the phenotype of patients with FHM1 mutations. Studies with FHM1 KI mice revealed that besides enhancement in susceptibility to CSD [25, 41-44], as a triggering mechanism underlying migraine aura, the excitation/inhibition balance is changed [16, 45, 46]. Moreover, synaptic plasticity is altered [47] and pain signaling mediated by calcitonin gene-related peptide (CGRP) in trigeminal nuclei [48-50] is modified.
FHM2 mutant mice with W887R loss-of-function missense mutation in their Atp1a2 gene were generated. Homozygous FHM2 KI animals are lethal, same as homozygous Atp1a2 knock-out mice, hence only heterozygous animals could be used for research. Similarly, with FHM1 KI mice, heterozygous FHM2 mutants in vivo have enhanced CSD susceptibility [51]. In the α2 isoform KI mice, the α2 isoform protein is decreased in brain lysates in western blots to 60% in heterozygous α2+/W887R [51] and α2+/G301R mice [52] and to very low amounts in homozygous fetuses of α2G301R/G301R.
Recently, an abstract has been published in Cephalalgia reporting the first FHM3 KI mouse model which was generated using homologous recombination, with human mutation L1649Q which is located in the mouse orthologous Scn1a gene [53]. FHM3 mutant mice display an enhanced CSD susceptibility by an increased inhibitory activity of interneurons [53]. However, further data regarding these mice and the mechanisms of CSD susceptibility have not published yet so the validity of this model needs to be cleared.
2. FHM1
2.1. Cav2.1 Gain of Function and Increase in CSD Susceptibility in FHM1 Mice
Cerebral hyperexcitability seen in FHM1 patients can potentially occur due to FHM1 mutations leading to a Cav2.1 gain of function [19]. In brain slices of cortex and cultures of neurons, FHM1 R192Q [16, 45] and S218L [46] mutations have an increase in excitatory but not inhibitory neurotransmitter release. Moreover, the increase in synaptic transmission mediated by glutamatergic neurons in the cortex and increase in CSD susceptibility are associated, and are shown for R192Q mutant mice in vitro in brain slices [16]. Interestingly, by reducing excitatory transmission to wild-type (WT) levels in the cortex by partial inhibition of Ca2+ channels, the CSD susceptibility facilitation was turned back to normal level [16]. This finding may explain the link between the increased influx of Ca2+ into the cell, synaptic transmission mediated by glutamatergic neurons, and an increase in the susceptibility to CSD. Brain slices from FHM1 S218L mutant mice have increased neuronal Ca2+ levels at rest. Additionally, in vivo Ca2+ imaging in the somatosensory cortex of these mutants indicates a change in the synaptic morphology [54] which is compatible with the increase in their synaptic strength and a hyperexcitable phenotype. Vulnerability to CSD was also increased [41-44] in S218L compared to R192Q KI mice in vivo under anesthesia, and could be modulated by stress and sex hormones [43, 55]. The higher vulnerability to CSD in S218L/S218L compared to S218L/ WT mutant in vivo [41, 42] is due, in part, to cortical Cav2.1 channels in the synapses of S218L/S218L mutant, which are open at a membrane potential far below the threshold for action potential [46].
2.2. Neuronal Network-Dependent Synaptic Plasticity in FHM1 Mutant Mice
Cortical synapse proteomes of FHM1 R192Q KI mice and WT animals were analyzed by iTRAQ reagent-based Liquid Chromatography-Mass Spectrometry approach to identify the proteins and their differential expressions. Most of the differences in expression were subtle except for a few alterations in abundance levels of proteins involved in the presynaptic active zone or postsynaptic density [56]. This indicates that the R192Q mutation does not have a functional effect on synaptic molecular architecture, which is consistent with the earlier findings in the same mutant mice reporting no changes in the abundance level of different kinds of CaV channels [25] or in the releasable pool of neurotransmitters at cortical synapses [16]. However, 19 proteins involved in vesicle turnover, actin molecule dynamics, and neurite outgrowth, and finally, glutamate transporters were significantly different between genotypes. These data imply the possibility of compensatory mechanisms to counter-balance a dysregulated glutamatergic signaling taking part in the FHM pathophysiology at the level of synapses as an adaptation mechanism [56]. An increase in synaptic depression in the cortex of R192Q [16] and S218L mutant mice [46] has been shown by in vitro studies, indicating a decline in synaptic transmission efficacy after repetitive stimulation. Findings from both in vitro and in vivo studies of the synapse of the brainstem Calyx of Held in S218L/S218L mutant demonstrated a faster recovery after synaptic depression in comparison with WT [57]. This finding also was shown in the R192Q mutant in vitro [58]. In the cerebellum slices of R192Q and S218L KI mice, a reduction in short-term synaptic facilitation at the synapses of parallel fibers to Purkinje cells was observed. This may occur due to a facilitated state of KI Cav2.1 channels at presynaptic location [26], which damage the balance in the firing tuning of the Purkinje cell, causing cerebellar ataxia observed in FHM1. A recent in vivo study examining the function of the hippocampus in R192Q and S218L mutant showed an enhancement in long-term potentiation and no change in long-term depression associated with disrupted learning and memory as a result of changing the balance between depression and potentiation at the neuronal circuit scale [47]. How the mechanisms of the brain network modifications in the hippocampus, cerebellum, and brainstem cause FHM1-specific features of migraine-related neuronal networks, is not clear yet. It is interesting that a single mutation has different effects in the trigeminal ganglion (TG) [49] and brainstem compared to the cortex [59], leading to the idea that the effect of FHM1 mutations may depend on the type of neuron. For instance, a strong gain-of-function influence on FHM1 mutations is seen for excitatory pyramidal neurons in contrast to the absence of any influence of these mutations on fast-spiking interneurons of the cortex. This may be due to the presence of some CaV2.1 channels specific to interneurons with gating features not altered by the FHM1 mutation [45]. Differences in function between several types of neurons in healthy networks in the cortex [60] could be the reason for distinct effects of FHM1 mutations on various neuronal networks leading to dynamic perturbations in the excitatory/inhibitory balance in the brain. It has been reported recently that tissue anoxia could be the underlying mechanism for strong aura in FHM1 patients [61]. In this study, Ca2+ signals have been reduced during normal network activity in a FHM1 mouse model compared to WT mice, it has been proposed that this event could explain deficits in neurovascular responses in the FHM1 KI mice, leading to a brain weakness in patients with FHM1 mutations [61].
2.3. The Effect of FHM1 Mutations on Neuroinflammation
The first evidence of pro-inflammatory profile in the in vivo/in vitro studies using TG from R192Q mutant mice indicates neuroinflammation in this migraine transgenic mouse model. In TGs of KI or WT mice, most neurons co-express P2X3 and tumor necrosis factor alpha (TNFα) receptors, making them susceptible to inflammation mediated by TNFα and the pain signaling related to it [62, 63]. R192Q KI TGs have been reported to be enriched in activated macrophages, as indicated by both immunoreactivity to the CD11b (adhesion molecule marker for active microglia and macrophages), Iba1, and the macrophage antigen ED1, and also by macrophage morphology, compared to WT mice [63]. TGs of R192Q mutant mice expressed increased levels of mRNA of IL6, IL1b, and IL10 and the MCP-1 chemokine and TNFα cytokines. As TNFα is the main factor in TG sensitization, expression of TNFα and macrophage occurrence were significantly higher in the ganglia of R192Q KI compared to WT mice following an inflammatory reaction induced by LPS injection [63]. The complex molecular and cellular environment in KI TG reveals a novel phenotype of tissue consistent with a neuroinflammatory profile. This phenotype can aggregate the pathophysiology of trigeminal pain by a release of mediators such as TNFα and may establish crosstalk between resident glia and sensory neurons, which is the reason for the process of neuronal sensitization in FHM patients [63]. In a recent study, gene expression profile analysis from cortical tissue of R192Q KI mice revealed the molecular pathways affected by CSD [64]. CSD-induced up-regulation of a specific group of genes in R192Q mutant brains was the striking data of the study. These genes have a prominent functional enrichment for signaling genes of inflammatory pathways [64], including multiple genes of cluster 1 such as Cd53, Ccl2, Anxa2, Ms4a6d, C3ar1, Timp1, and Vim, which are the main initiators of inflammatory reactions [65]. Pathway analysis indicated that the exacerbated inflammation was related to interferon [64]. However, the way the interaction between R192Q mutation and CSD results in a definite inflammatory profile should be studied further. Investigations in epilepsy models may help to understand the close interplay between increased excitation and inflammation [66, 67], suggesting the elevated activity in glutamatergic pathways in R192Q KI mice [16] being more severe during CSD episodes, as a reason for enhanced inflammatory response. On the other hand, it can be interpreted that the CSD-induced elevation of the inflammatory response can be due to a pro-inflammatory state in the FHM1 R192Q brains which may already exist in naive animals [64]. Therefore, naive TG of R192Q KI mice could exhibit a pro-inflammatory phenotype with increased activated macrophages and microglia and, elevated levels of cytokine expression [50, 68-70]. As reported in wild-type mice, CSD initiated a signaling cascade among neurons under stress and trigeminal afferents via the opening of neuronal Pannexin1 channel after the activation of caspase-1 followed by the neuronal release of HMGB1 and NF-κB activation in astrocytes [12]. Therefore, additional studies are needed to investigate the CSD-induced parenchymal neuroinflammation pathway in FHM mutant mouse models. Recently, bilateral HMGB1 release and NF-κB activation have been shown in the cortical and subcortical regions of FHM1 R192Q KI mice compared to WT after CSD [71]. In the western blots from the cerebrospinal fluid of S218L KI mice, more HMGB1 release has been shown after 1-hour of multiple CSDs compared to sham-operated and WT mice [71]. Interestingly, a basal ongoing parenchymal neuroinflammatory process has been demonstrated in all areas of the brain in R192Q mice including primary motor cortex, primary somatosensory cortex, striatum, and thalamus, compared to WT [71]. The release in HMGB1 from neurons as an alarmin molecule has been confirmed to be only due to the basal parenchymal neuroinflammatory processes or CSD events in the brain of FHM1 mice and not due to the perfusion-hypoxic stress which causes neuronal swelling and chromatin margination leading to HMGB1 release [72]. Future studies are needed to reveal the mechanism of the bilateral parenchymal neuroinflammation in FHM1 mutant compared to WT, and to determine whether the basal parenchymal inflammatory state is due to hyperexcitability of these mice, or the reverse, whether the presence of ongoing neuroinflammation increases the hyperexcitability of the brain in the mutant.
2.4. Altered Brain and Plasma Metabolites after CSD in FHM1 Mice
CSD induces metabolic remodeling in R192Q mutant mice which is measurable in plasma [73]. These immediate changes after CSD leading to metabolic alterations can be better studied in R192Q KI mice and are much more difficult in patients. With different experiments including capillary electrophoresis, multivariate data analysis, and mass spectrometry, a considerable distinction between the profiles of R192Q mutant and WT mice after CSD has been established. Lysine and pipecolic acid were identified as two important metabolites that were changed. This change indicates an increase in GABAergic neurotransmission after excitation as a mechanism of compensation [73]. CSD waves can also change the repertoire of peptides and metabolites both in the cortex and the subcortical areas of the brain in R192Q mutant mice as indicated by large-scale matrix-assisted laser desorption/ionization mass spectrometry imaging [74]. The changes in the distribution of biomolecules have been observed only in R192Q KI mice after CSD. The biomolecules influenced by CSD remain to be identified [74].
2.5. Molecular Pain Mechanisms, Modified Trigeminovascular Processing, CGRP Release, and Purinergic Signaling in FHM1 Mice
In vitro glutamate and CGRP release from trigeminal neurons [75] and in vivo trigeminocervical complex (TCC) nociceptive transmission [76] are known to be facilitated by CaV2.1 channels. Investigation of the function of TG in FHM1 mice led to a molecular phenotype of pain mediated by modified CGRP-induced purinergic signaling. In TG of R192Q mice, the sensory neurons showed an increased purinergic activity of P2X3 receptor that is ATP-gated. This increase may be due to a change in the phosphorylation level of intracellular protein domains [68]. The hyperpolarization-activated conductance Ih, that is mediated by hyperpolarization activated cyclic nucleotide-gated channels (HCN), contributes to sub-threshold behavior and firing in WT and FHM1 KI neurons of TG [77]. Most WT and KI TG neurons expressed Ih current, which was blocked by the inhibitor ZD7288, however, Ih current was smaller in the neurons of KI mice although they showed similar activation and deactivation kinetics [77]. The low amplitude of Ih in FHM1 mutant TG neurons indicates that down-regulation of Ih current in sub-threshold behavior is a compensatory mechanism for limiting sensory hyperexcitability, which is observed after specific stressful stimuli [77]. Enhancement of Ca2+ influx in FHM1 mutant neurons may cause essential P2X3 receptor activity since the membrane expression of P2X3 was not changed. Algogenic mediator, bradykinin, augments both glial P2Y-mediated calcium responses and the percentage of responding cells. This indicates a particularly prominent modulation of purinergic signaling in satellite glial cells in mutant ganglia, which can be interpreted in the migraine pathophysiology [50]. Furthermore, neurons of R192Q KI mice TG showed more baseline release of different mediators such as CGRP, TNFα, and brain-derived neurotrophic factor that could contribute to P2X3 receptor potentiation and explain why an increased potentiation of P2X3 receptors
by exogenous TNFα or CGRP is not possible [78]. These observations suggest that KI cells have a basal neuroinflammatory profile with the possibility of facilitation of the release of endogenous mediators such as ATP, in order to activate P2X3 receptors that are constitutively hyper-functional and amplify nociceptive signaling by trigeminal sensory neurons [69]. The surge in extracellular TNFα may raise the activity of transient receptor potential vanilloid 1 (TRPV1) receptor, and therefore, it may amplify nociceptive transduction in the periphery, which is important in migraine pain [79]. Purinergic activity enhancement was related to abnormal profiles of chemokine/cytokine and an increase in active macrophages seen in FHM1 R192Q mice TG [63]. In R192Q KI mice TG, the release of CGRP increased while it was not changed in the dura; this is consistent with the lack of FHM1 mutation effect on CaV2.1 currents observed in TG neurons that are capsaicin-sensitive and consist the majority population of small dural afferents [49]. After co-culturing FHM1 TG neurons with satellite glial cells, the enhancement in the release of CGRP at baseline and after neuronal activity led to P2Y receptors potentiation in glial cells and subsequent activation of these cells [50]. However, decrease in the CGRP signal, as reported in another study, may explain the depletion of intracellular CGRP stores in the FHM1 KI mice due to an active trigeminovascular system (TVS) with an increase in the release of CGRP. Taken together, the specific neuroinflammatory condition in FHM1 KI mice TGs, with a constant neuronal and glial purinergic receptor activity, might facilitate pain signaling propagation, considering the important role of the mediators of inflammatory pain involved in meningeal nociceptors [80]. Examination of another FHM1 mutant mouse model like S218L KI, which also has a reduction in threshold for CSD induction [42], or studies in other mouse models for FHM2 and FHM3, can be helpful to understand if the above-mentioned differences are specified to the R192Q mutation or whether the molecular alterations occur as the consequence of the same pathophysiological mechanisms inherent to all FHM subtypes.
A recent report has shown that trigeminal neurons in WT mice have negative control over P2X3 receptors by brain natriuretic peptide pathway which is reflected in the suppression of P2X3 receptors-mediated excitability. In FHM1 KI cultures, the lack of this inhibition causes a hyperexcitability phenotype which could facilitate the trigeminal pain signaling in migraine [81]. The phenotype of TG neurons sensitization in FHM1 KI mice has been shown to be not due to an up-regulation of CGRP receptors, whereas it is likely caused by larger CGRP release. This data indicates that, in FHM1 TG, when the extracellular concentration of CGRP turns back to control level with targeted treatment the normal sensory neuron signaling can be restored [82]. In the trigeminocervical complex of WT mice, the number of Fos-expressed cells elevated significantly after dural stimulation compared to the sham group and declined after naratriptan treatment [83]. In R192Q KI animals, the stimulated and sham or naratriptan-treated mice were not significantly different. In FHM1 mutant mice, the number of Fos-positive cells in the stimulated group was significantly lower than the WT stimulated animals [83].
2.6. Modified Behavioral Pain Responses in FHM1 Mice
The mouse grimace scale, which measures the expression of facial pain in mice, indicates a baseline pain face in R192Q KI mice as compared to the wild-type mice. This baseline pain was reversed by rizatriptan (50 mg/kg) indicating the presence of endogenous pain in the KI mice [84]. When R192Q KI mice were subjected to novelty and/or restraint stress, several behavioral measures indicating a spontaneous head pain were found. These measures showed lateralization and increased frequency in KI mice but not in WT mice, and they were more frequent in females compared to males [85]. These spontaneous head pain behaviors were normalized by systemic administration of rizatriptan and morphine, different acute analgesics and anti-migraine drugs, in a dose-dependent manner. The behaviors included abnormal frequent head grooming with excess unilateral oculotemporal strokes and the elevated number of blink rates with one eye closed [85]. Some of these appeared more frequently and were more severe in the S218L than in the R192Q KI mice, consistent with the clinical symptoms observed in patients. The pain-expressing behavior normalized by administration of serotonergic anti-migraine drugs supports the involvement of trigeminal pain pathways [85]. The R192Q mice showed unchanged susceptibility to different noxious thermal, mechanical, and chemical stimuli applied exogenously with respect to WT ones. Additionally, the mutant mice had the tendency to avoid the bright but closed-safe arms in a modified version of the elevated plus maze compared to WT mice [85]. This indicates a photophobia-like behavior in both R192Q and S218L mutant mice. If the findings from CGRP alterations and TG function in vitro could be translated to the in vivo paradigm, FHM1 KI mice may serve as a precious animal model to study the effects of current and new anti-migraine drugs, which may be related to the CGRP modulation and neuroinflammatory mechanisms [86, 87]. Moreover, important insights regarding the pain behavior pathways could be obtained by the comparison of the behavioral alterations in FHM1 mutant to the photophobia-like behavior seen in a mutant mouse model sensitized to CGRP [88, 89].
FHM1 gain-of-function mutation enhances excitatory transmission and long-term potentiation (LTP) in the hippocampus; however, learning and memory are impaired in these mutant mice indicating a possible explanation for cognitive alterations in FHM [47]. R192Q mutant mice were studied for stimulus intensity-response curves determined for field potentials taken from hippocampal CA1 evoked from anterior commissure and evaluation for neuroplasticity by measuring LTP and long-term depression (LTD) under anesthesia. The hippocampal field potentials presented a significant enhancement in R192Q KI mice compared to the WT ones. There was a shift to the left in stimulus intensity–response curves and larger maxima in the mutants [47]. Besides, a two-fold augmentation in LTP was seen in R192Q KI mice, whereas there was no alteration in LTD compared with WT mice. Moreover, the mutant mice displayed significant deficits in their spatial memory as measured by Morris water maze and also contextual fear-conditioning experiments
with respect to WT controls. The test of novel object recognition was not disrupted in R192Q mice, but S218L mutants showed remarkable impairment in this test, implying a genotype-phenotype relationship [47]. These data suggest that the abnormal enhancement in plasticity can be as detrimental for efficient learning as decreased plasticity.
Another behavioral clue comes from sleep behavior changes in FHM1 mice. Knowing the G-protein-mediated effect on adenosine receptors and its effect on CaV2.1 channel function, R192Q FHM1 mice have been studied to understand if they are less sensitive to adenosinergic inhibition and if they show a sleep-specific phenotype [90]. These mutant mice have a faster adaptation to the light-dark cycle phase shifts in their sleep-wake rhythms and behaviors [90]. After applying a 6-hour sleep deprivation paradigm, and using caffeine as a general adenosine receptor antagonist, and cyclopentyladenosine as a specific A1 receptor agonist, R192Q mutant mice showed an enhanced waking pattern in the active dark period compared to WT. This may be due to decreased susceptibility to the adenosine A1 receptor inhibition as indicated by baseline recordings (EEG and electromyogram) [90]. These observations may shed light on the mechanism of the relationship between migraine and disrupted sleep in the clinic.
With respect to stress effect, 20 minutes and 3-hours restraint stress tests have no effect on CSD susceptibility in both R192Q mutant and WT mice, but there is an elevation in the levels of plasma corticosterone in KI ones [55]. However, subcutaneous injection of corticosterone (20 mg/kg) raises CSD frequency dramatically in the KI mice, while levels of corticosterone in plasma are similarly increased in KI and WT mice. Mifepristone as a glucocorticoid receptor (GR) antagonist normalized the effect of corticosterone on CSD frequency. This indicates that the GR activity induced by corticosteroid may increase CSD frequency in an individual who is genetically susceptible and may lead to a predisposition to migraine attacks [55]. Since FHM1 mutation is inherited dominantly, phenotype studies related to cellular and behavioral aspects of R192Q heterozygous KI mice are also worth studying.
2.7. Sex-Related Differences in FHM1
In the clinic, there is a strong sex effect in migraine patients, 18% of women suffer from migraine compared to 6% of men. Besides, women have more severe and more frequent migraine attacks than men. However, the limited number of investigations related to sex effect and difference of CSD consequences in female and male FHM1 mutant mice is a shortcoming of these models in migraine studies. The features of CSD phenotype, including velocity, amplitude, threshold, and frequency are more severe in female S218L and R192Q KI mice compared to male ones and disappear after ovariectomy [42]. Orchiectomy, on the other hand, elevates susceptibility to CSD in R192Q KI mice. Testosterone replacement chronically restores CSD susceptibility by an androgen receptor-based mechanism and leads to a new prophylactic target for migraine [43]. Reports also show that estradiol augments genetically-enhanced CSD susceptibility in both S218L and R192Q FHM1 mutant mice [42].
2.8. RNA Expression Changes in FHM1
There is a pronounced difference in the RNA expression in the cerebellum of S218L mutant mice compared to R192Q and WT mice [91]. In particular, tyrosine hydroxylase, which indicates the delayed cerebellar maturation, has been shown to be highly upregulated in S218L cerebella. However, a minimal difference in the expression level was reported in the caudal cortex of both FHM1 mutant strain [91].
2.9. Stroke Studies in FHM1
There is a high comorbidity between migraine and other diseases such as stroke [86-88], epilepsy [5, 6], and depression [89, 90], suggesting genetic or environmental-epigenetic-specific common pathogenic pathways. In this case, transgenic animal models of migraine could provide a significant opportunity to study in vivo pathophysiology of migraine. Epidemiological studies have suggested an enhanced risk of stroke or ischemia in migraine patients, especially in female MA [92-94]. Large- scale meta-analysis of genome-wide data recently demonstrated common genes for susceptibility between ischemic stroke and migraine [95]. However, to further increase our knowledge of migraine and stroke comorbidity and fill the gap in mechanisms related to translating these data to the molecular level, FHM1 transgenic mice as monogenic migraine models might represent a relevant preclinical tool [96]. The experimental induction of transient focal cerebral ischemia revealed larger infarcts in R192Q and S218L KI mice when compared to WT animals [97]. Interestingly, there was a perfect correlation between phenotype and genotype (in R192Q better than in S218L) and an allele-dosage influence (in heterozygotes better than in homozygotes). This can be explained by increased susceptibility to ischemic injury by the enhanced neuronal hyperexcitability observed in migraine pathogenesis. Experimentally induced cerebral ischemia triggers anoxic depolarization that is faster and increases the frequency of development of the peri-infarct depolarizations (PIDs), causing a more severe mismatch in metabolic supply-demand and faster growth of infarct [97]. MK-801 treatment as an antagonist to N-methyl-D-aspartate (NMDA) receptor blocked the infarct growth and reversed the effects on the WT mice, indicating CSD as a potential target for stroke treatment [97]. Migraine-prophylactic drugs, such as lamotrigine and topiramate, used chronically, decreased the number of ischemic depolarizations and caused an improvement in stroke consequences in FHM1 KI mice and WT [98], suggesting that the hyperexcitability induced by genetically increased glutamatergic neurotransmission could be a shared mechanism for migraine and ischemia. In FHM1 mice, enhanced oligemia and desaturation of hemoglobin after CSD [54], together with increased levels and velocity of (Ca2+) current surge in neurons during CSD, could also lead to the increased susceptibility to ischemia in migraine-vulnerable brains [97].
3. FHM2
3.1. FHM2 KI Mice Demonstrate Decreased Astrocytic α2 Na+, K+-ATPase Expression and Increased Susceptibility to CSD
FHM2 is a form of MA which is autosomal dominant and is produced by α2-subunit mutations of the Na+/K+ ATPase, which has a massive expression in the brain astrocytes of an adult animal. The first FHM2 KI mouse model with human W887R mutation in the Atp1a2 orthologous gene was created in 2011 [51]. Only heterozygous α2 KO and KI animals are viable, since the homozygous mutant mice are lethal immediately after birth [51, 99, 100]. In the α2 isoform KI mice, the α2 isoform protein is decreased in brain lysates in western blots to around 60% in heterozygous adult α2+/W887R [51] and α2+/G301R levels [52] and to very low levels in homozygous fetuses of α2G301R/G301R. The decreases could be related to the delayed and inefficient endoplasmic reticulum secretion and deficit in proteasomes degradation [51]. The α2Na+/K+ ATPase has a critical role in extra-synaptic K+ buffering on neuronal activation [101, 102]; however, additional direct evidence needs to be provided. Heterozygous FHM2 mice, similar to FHM1 mice, demonstrated a decreased threshold for CSD with increased CSD propagation velocity [51]. The increased CSD susceptibility in KI mice was shown to be due to reduced rates of K+ and glutamate clearance by cortical astrocytes during neuronal activation and decreased density of GLT1a glutamate transporters in cortical presynaptic astrocytic processes in FHM2 mutant mice [103].
3.2. Modified Behavioral Pain Responses in FHM2 Mice
In FHM2, the behavioral implications of introducing the W887R mutation in mouse were measured by a modified version of SHIRPA protocol [51]; however, the only significant change between α2+/W887R mice and WT was an increase in anxiety and fear behavior. Whereas, the behavior in the α2+/G301R KI mice was different. The sucrose preference test indicated that the α2+/G301R mice demonstrated stress-induced anhedonia and elevated acoustic startle response, indicating a high level of fear and anxiety. Moreover, the α2+/G301R mutants demonstrated depression-like behavior and increased levels of immobility in the tail suspension test when compared to WT animals [52]. Although these studies show that there are different pain behavior responses in FHM1 and FHM2 mutant mice, they also posit the behavioral changes seen in these mutant mice resemble those in humans [104, 105]. However, it should be cleared if the mutation in patients causes the psychiatric manifestations in a consistent manner, and if the mouse shows psychiatric manifestations similar to human.
3.3. Sex Effect on the Glutamate System and Behavioral-Emotional Expression in FHM2
In the FHM2 KI mice model, the female α2+/G301R mice show more abnormal behaviors than the male α2+/G301R mutant. The female α2+/G301R mice were hypoactive in the open field test when compared to WT females and male α2+/G301R mutant [52]. Moreover, in the open field test female α2+/G301R mutant showed excessive grooming behaviors, indicating compulsive behavior in these mice. Female α2+/G301R mice also demonstrated obsessive-compulsive disorder- (OCD)-like features on the marble-burying test, by burring a significantly higher number of marbles than both, WT and male α2+/G301R mutant animals. All these behaviors reversed to the normal state after a single progestin treatment [52]. This indicates a role for female hormone cycle in the changed behaviors of α2+/G301R females compared to WT females and male α2+/G301R animals. This may explain the effect of ovarian hormones on migraine and a higher prevalence of migraine in women compared to men. Moreover, NMDA receptor antagonists changed the female α2+/G301R OCD-like behaviors, increased startle response to acoustic stimuli, and hypoactivity, indicating that glutamatergic mechanisms are under the influence of mutated α2 isoform in these FHM2 mice. The elevation of glutamate levels in the main brain areas of α2+/G301R mice supports this idea when cultures of both astrocytes and neurons from α2G301R/G301R mutants showed a significant decrease in glutamate uptake with respect to similar cultures of WT animals [52]. This also suggests that the participation of the female hormone cycle affects the glutamate system and might relate it to the behavioral-emotional evidence in FHM2.
4. FHM3
4.1. FHM3 KI Mice Show Enhanced CSD Susceptibility by an Increased Inhibitory Interneuron Activity
The FHM3 mouse model study shows that both cortical and hippocampal fast-spiking interneurons demonstrate a significantly increased frequency of action potential firing in acute slices [53]. It indicates that these interneurons have gain-of-function because of point mutation L1649Q in the mouse orthologous Scn1a gene. Moreover, pyramidal neurons of the cortical layer V have significantly more inhibitory input [53]. In the in vivo experiments, FHM3 mutant mice show a significant increase in CSD frequency, like the other forms of FHM mutants [53]. Besides, these mutants have a significantly lower CSD induction threshold [53]. This newly described mouse model of FHM3 mutation reveals a potentially new mechanism of CSD susceptibility through the increase in inhibitory interneurons activity and could be further studied for drug targeting for this new pathway.
4.2. Need for Experiments on Behavioral Responses in FHM3 Mice
Data related to behavioral responses of L1649Q mutation is not available for FHM3. Results of behavioral tests in FHM3 mouse model would be important to compare the data with FHM1 and FHM2 mouse models, as FHM3 mutation causes increased activity of inhibitory interneurons [53] and enhances cortical inhibition as opposed to FHM1 and FHM2.
CONCLUSION
As described above, in order to decipher migraine mechanisms, animal models of monogenic forms of migraine have been beneficial to understand the excitatory/inhibitory balance alteration in different experimental paradigms and its consequences on neurophysiology and behavior in a migraine context. There are a few key shortcomings of FHM models for investigating the pathophysiology of migraine including 1- The lack of enough information about the mechanisms of the brain network modifications in the hippocampus, cerebellum and brainstem causing FHM1 specific features of migraine-related neuronal networks in this model;
2- The lack of enough study on the neuroinflammatory cascades involved in the dysregulated excitatory/inhibitory balance. 3- The lack of sufficient information about underlying mechanisms of increased CSD susceptibility in FHM2 and FHM3 mutant mice; 4- The lack of enough studies for the relevant data relating to the clinical manifestation of FHM2 and FHM3 mechanisms since the current mouse models reports have not shown significant results compared to FHM1 mouse models; and finally 5- S218L mutant mouse, with the more severe phenotype of FHM1, has been argued to be a sound model for studying migraine pathophysiology, since some researchers assume it as a model for epilepsy studies or a model for more complex neurological disorders. However, FHM mutant mice helped answer some clinically essential challenges including the link between aura and headache in a CSD model of migraine, and the relationship between gene-environment interplay in clinical phenotypes. The transgenic mice described in the present study represent a refined tool to develop pre-clinical studies relevant to the migraine field, due to the similarity of phenotypes between patients and the mutant mice. Additionally, they will aid in future studies to investigate pain behavior in FHM and common migraine. MA in FHM is a predominant symptom and a majority of preclinical research has linked CSD events as the underlying mechanism for migraine aura; however, just about one-third of migraine patients exhibit aura symptoms. Therefore, to understand if patients with MO also have CSD, more research is needed to investigate other trigeminovascular system stimulators in other models including CGRP models [88, 89] and new models of migraine in which aura is a less-prominent symptom [106, 107]. Additional investigations of various FHM mouse models, together with the clinical studies of different comparable FHM types of patients will contribute to deciphering the pathophysiology of migraine as a disorder with multiple altering symptoms, varying severity, and various functional consequences with different genetic backgrounds.
ACKNOWLEDGEMENTS
We would like to thank Prof. Dr. Arn M.J.M. van den Maagdenberg and Dr. Else A. Tolner for their precious advice and discussion to make the appropriate design and literature search.
AUTHOR CONTRIBUTIONS
AD contributed to the design, literature search, and writing of the review. HK contributed to the literature search and editing of the review.
Consent for Publication
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Vos T., Flaxman A.D., Naghavi M., Lozano R., Michaud C., Ezzati M., Shibuya K., Salomon J.A., Abdalla S., Aboyans V., Abraham J., Ackerman I., Aggarwal R., Ahn S.Y., Ali M.K., Alvarado M., Anderson H.R., Anderson L.M., Andrews K.G., Atkinson C., Baddour L.M., Bahalim A.N., Barker-Collo S., Barrero L.H., Bartels D.H., Basáñez M.G., Baxter A., Bell M.L., Benjamin E.J., Bennett D., Bernabé E., Bhalla K., Bhandari B., Bikbov B., Bin Abdulhak A., Birbeck G., Black J.A., Blencowe H., Blore J.D., Blyth F., Bolliger I., Bonaventure A., Boufous S., Bourne R., Boussinesq M., Braithwaite T., Brayne C., Bridgett L., Brooker S., Brooks P., Brugha T.S., Bryan-Hancock C., Bucello C., Buchbinder R., Buckle G., Budke C.M., Burch M., Burney P., Burstein R., Calabria B., Campbell B., Canter C.E., Carabin H., Carapetis J., Carmona L., Cella C., Charlson F., Chen H., Cheng A.T., Chou D., Chugh S.S., Coffeng L.E., Colan S.D., Colquhoun S., Colson K.E., Condon J., Connor M.D., Cooper L.T., Corriere M., Cortinovis M., de Vaccaro K.C., Couser W., Cowie B.C., Criqui M.H., Cross M., Dabhadkar K.C., Dahiya M., Dahodwala N., Damsere-Derry J., Danaei G., Davis A., De Leo D., Degenhardt L., Dellavalle R., Delossantos A., Denenberg J., Derrett S., Des Jarlais D.C., Dharmaratne S.D., Dherani M., Diaz-Torne C., Dolk H., Dorsey E.R., Driscoll T., Duber H., Ebel B., Edmond K., Elbaz A., Ali S.E., Erskine H., Erwin P.J., Espindola P., Ewoigbokhan S.E., Farzadfar F., Feigin V., Felson D.T., Ferrari A., Ferri C.P., Fèvre E.M., Finucane M.M., Flaxman S., Flood L., Foreman K., Forouzanfar M.H., Fowkes F.G., Franklin R., Fransen M., Freeman M.K., Gabbe B.J., Gabriel S.E., Gakidou E., Ganatra H.A., Garcia B., Gaspari F., Gillum R.F., Gmel G., Gosselin R., Grainger R., Groeger J., Guillemin F., Gunnell D., Gupta R., Haagsma J., Hagan H., Halasa Y.A., Hall W., Haring D., Haro J.M., Harrison J.E., Havmoeller R., Hay R.J., Higashi H., Hill C., Hoen B., Hoffman H., Hotez P.J., Hoy D., Huang J.J., Ibeanusi S.E., Jacobsen K.H., James S.L., Jarvis D., Jasrasaria R., Jayaraman S., Johns N., Jonas J.B., Karthikeyan G., Kassebaum N., Kawakami N., Keren A., Khoo J.P., King C.H., Knowlton L.M., Kobusingye O., Koranteng A., Krishnamurthi R., Lalloo R., Laslett L.L., Lathlean T., Leasher J.L., Lee Y.Y., Leigh J., Lim S.S., Limb E., Lin J.K., Lipnick M., Lipshultz S.E., Liu W., Loane M., Ohno S.L., Lyons R., Ma J., Mabweijano J., MacIntyre M.F., Malekzadeh R., Mallinger L., Manivannan S., Marcenes W., March L., Margolis D.J., Marks G.B., Marks R., Matsumori A., Matzopoulos R., Mayosi B.M., McAnulty J.H., McDermott M.M., McGill N., McGrath J., Medina-Mora M.E., Meltzer M., Mensah G.A., Merriman T.R., Meyer A.C., Miglioli V., Miller M., Miller T.R., Mitchell P.B., Mocumbi A.O., Moffitt T.E., Mokdad A.A., Monasta L., Montico M., Moradi-Lakeh M., Moran A., Morawska L., Mori R., Murdoch M.E., Mwaniki M.K., Naidoo K., Nair M.N., Naldi L., Narayan K.M., Nelson P.K., Nelson R.G., Nevitt M.C., Newton C.R., Nolte S., Norman P., Norman R., O’Donnell M., O’Hanlon S., Olives C., Omer S.B., Ortblad K., Osborne R., Ozgediz D., Page A., Pahari B., Pandian J.D., Rivero A.P., Patten S.B., Pearce N., Padilla R.P., Perez-Ruiz F., Perico N., Pesudovs K., Phillips D., Phillips M.R., Pierce K., Pion S., Polanczyk G.V., Polinder S., Pope C.A., III, Popova S., Porrini E., Pourmalek F., Prince M., Pullan R.L., Ramaiah K.D., Ranganathan D., Razavi H., Regan M., Rehm J.T., Rein D.B., Remuzzi G., Richardson K., Rivara F.P., Roberts T., Robinson C., De Leòn F.R., Ronfani L., Room R., Rosenfeld L.C., Rushton L., Sacco R.L., Saha S., Sampson U., Sanchez-Riera L., Sanman E., Schwebel D.C., Scott J.G., Segui-Gomez M., Shahraz S., Shepard D.S., Shin H., Shivakoti R., Singh D., Singh G.M., Singh J.A., Singleton J., Sleet D.A., Sliwa K., Smith E., Smith J.L., Stapelberg N.J., Steer A., Steiner T., Stolk W.A., Stovner L.J., Sudfeld C., Syed S., Tamburlini G., Tavakkoli M., Taylor H.R., Taylor J.A., Taylor W.J., Thomas B., Thomson W.M., Thurston G.D., Tleyjeh I.M., Tonelli M., Towbin J.A., Truelsen T., Tsilimbaris M.K., Ubeda C., Undurraga E.A., van der Werf M.J., van Os J., Vavilala M.S., Venketasubramanian N., Wang M., Wang W., Watt K., Weatherall D.J., Weinstock M.A., Weintraub R., Weisskopf M.G., Weissman M.M., White R.A., Whiteford H., Wiersma S.T., Wilkinson J.D., Williams H.C., Williams S.R., Witt E., Wolfe F., Woolf A.D., Wulf S., Yeh P.H., Zaidi A.K., Zheng Z.J., Zonies D., Lopez A.D., Murray C.J., AlMazroa M.A., Memish Z.A. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [http://dx.doi.org/10.1016/S0140-6736(12)61729-2]. [PMID: 23245607]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olesen J., Bes A., Kunkel R., Lance J.W., Nappi G., Pfaffenrath V., Rose F.C., Schoenberg B.S., Soyka D., Tfelt-Hansen P., Welch K.M.A. 2013. [Google Scholar]
- 3.Kurth T., Chabriat H., Bousser M.G. Migraine and stroke: a complex association with clinical implications. Lancet Neurol. 2012;11(1):92–100. doi: 10.1016/S1474-4422(11)70266-6. [http://dx.doi.org/10.1016/S1474-4422(11)70266-6]. [PMID: 22172624]. [DOI] [PubMed] [Google Scholar]
- 4.Spector J.T., Kahn S.R., Jones M.R., Jayakumar M., Dalal D., Nazarian S. Migraine headache and ischemic stroke risk: an updated meta-analysis. Am. J. Med. 2010;123(7):612–624. doi: 10.1016/j.amjmed.2009.12.021. [http://dx.doi.org/10.1016/j.amjmed.2009.12.021]. [PMID: 20493462]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keezer M.R., Bauer P.R., Ferrari M.D., Sander J.W. The comorbid relationship between migraine and epilepsy: a systematic review and meta-analysis. Eur. J. Neurol. 2015;22(7):1038–1047. doi: 10.1111/ene.12612. [http://dx.doi.org/10.1111/ene.12612]. [PMID: 25495495]. [DOI] [PubMed] [Google Scholar]
- 6.Bauer P.R., Carpay J.A., Terwindt G.M., Sander J.W., Thijs R.J., Haan J., Visser G.H. Headache and epilepsy. Curr. Pain Headache Rep. 2013;17(8):351. doi: 10.1007/s11916-013-0351-x. [http://dx.doi.org/10.1007/s11916-013-0351-x]. [PMID: 23801004]. [DOI] [PubMed] [Google Scholar]
- 7.McWilliams L.A., Goodwin R.D., Cox B.J. Depression and anxiety associated with three pain conditions: results from a nationally representative sample. Pain. 2004;111(1-2):77–83. doi: 10.1016/j.pain.2004.06.002. [http://dx.doi.org/10.1016/j.pain.2004.06.002]. [PMID: 15327811]. [DOI] [PubMed] [Google Scholar]
- 8.Ferrari M.D., Klever R.R., Terwindt G.M., Ayata C., van den Maagdenberg A.M. Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol. 2015;14(1):65–80. doi: 10.1016/S1474-4422(14)70220-0. [http://dx.doi.org/10.1016/S1474-4422(14)70220-0]. [PMID: 25496898]. [DOI] [PubMed] [Google Scholar]
- 9.Tolner E.A., Houben T., Terwindt G.M., de Vries B., Ferrari M.D., van den Maagdenberg A.M. From migraine genes to mechanisms. Pain. 2015;156(Suppl. 1):S64–S74. doi: 10.1097/01.j.pain.0000460346.00213.16. [http://dx.doi.org/10.1097/01.j.pain.0000460346.00213.16]. [PMID: 25789438]. [DOI] [PubMed] [Google Scholar]
- 10.Bolay H., Reuter U., Dunn A.K., Huang Z., Boas D.A., Moskowitz M.A. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat. Med. 2002;8(2):136–142. doi: 10.1038/nm0202-136. [http://dx.doi.org/10.1038/nm0202-136]. [PMID: 11821897]. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X., Levy D., Kainz V., Noseda R., Jakubowski M., Burstein R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann. Neurol. 2011;69(5):855–865. doi: 10.1002/ana.22329. [http://dx.doi.org/10.1002/ana.22329]. [PMID: 21416489]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karatas H., Erdener S.E., Gursoy-Ozdemir Y., Lule S., Eren-Koçak E., Sen Z.D., Dalkara T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science. 2013;339(6123):1092–1095. doi: 10.1126/science.1231897. [http://dx.doi.org/10.1126/science.1231897]. [PMID: 23449592]. [DOI] [PubMed] [Google Scholar]
- 13.Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117(Pt 1):199–210. doi: 10.1093/brain/117.1.199. [http://dx.doi.org/10.1093/brain/117.1.199]. [PMID: 7908596]. [DOI] [PubMed] [Google Scholar]
- 14.Eikermann-Haerter K., Yuzawa I., Qin T., Wang Y., Baek K., Kim Y.R., Hoffmann U., Dilekoz E., Waeber C., Ferrari M.D., van den Maagdenberg A.M., Moskowitz M.A., Ayata C. Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J. Neurosci. 2011;31(15):5755–5763. doi: 10.1523/JNEUROSCI.5346-10.2011. [http://dx.doi.org/10.1523/JNEUROSCI.5346-10.2011]. [PMID: 21490217]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wessman M., Terwindt G.M., Kaunisto M.A., Palotie A., Ophoff R.A. Migraine: a complex genetic disorder. Lancet Neurol. 2007;6(6):521–532. doi: 10.1016/S1474-4422(07)70126-6. [http://dx.doi.org/10.1016/S1474-4422(07)70126-6]. [PMID: 17509487]. [DOI] [PubMed] [Google Scholar]
- 16.Tottene A., Conti R., Fabbro A., Vecchia D., Shapovalova M., Santello M., van den Maagdenberg A.M., Ferrari M.D., Pietrobon D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron. 2009;61(5):762–773. doi: 10.1016/j.neuron.2009.01.027. [http://dx.doi.org/10.1016/j.neuron.2009.01.027]. [PMID: 19285472]. [DOI] [PubMed] [Google Scholar]
- 17.Mulleners W.M., Chronicle E.P., Vredeveld J.W., Koehler P.J. Visual cortex excitability in migraine before and after valproate prophylaxis: a pilot study using TMS. Eur. J. Neurol. 2002;9(1):35–40. doi: 10.1046/j.1468-1331.2002.00334.x. [http://dx.doi.org/10.1046/j.1468-1331.2002.00334.x]. [PMID: 11784374]. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X., Velumian A.A., Jones O.T., Carlen P.L. Modulation of high-voltage-activated calcium channels in dentate granule cells by topiramate. Epilepsia. 2000;41(Suppl. 1):S52–S60. doi: 10.1111/j.1528-1157.2000.tb02173.x. [http://dx.doi.org/10.1111/j.1528-1157.2000.tb02173.x]. [PMID: 10768302]. [DOI] [PubMed] [Google Scholar]
- 19.van der Kamp W. MaassenVanDenBrink, A.; Ferrari, M.D.; van Dijk, J.G. Interictal cortical excitability to magnetic stimulation in familial hemiplegic migraine. Neurology. 1997;48(5):1462–1464. doi: 10.1212/wnl.48.5.1462. [http://dx.doi.org/10.1212/WNL.48.5.1462]. [PMID: 9153495]. [DOI] [PubMed] [Google Scholar]
- 20.Brighina F., Bolognini N., Cosentino G., Maccora S., Paladino P., Baschi R., Vallar G., Fierro B. Visual cortex hyperexcitability in migraine in response to sound-induced flash illusions. Neurology. 2015;84(20):2057–2061. doi: 10.1212/WNL.0000000000001584. [http://dx.doi.org/10.1212/WNL.0000000000001584]. [PMID: 25888559]. [DOI] [PubMed] [Google Scholar]
- 21.Mulleners W.M., Chronicle E.P., Palmer J.E., Koehler P.J., Vredeveld J.W. Visual cortex excitability in migraine with and without aura. Headache. 2001;41(6):565–572. doi: 10.1046/j.1526-4610.2001.041006565.x. [http://dx.doi.org/10.1046/j.1526-4610.2001.041006565.x]. [PMID: 11437892]. [DOI] [PubMed] [Google Scholar]
- 22.Ophoff R.A., Terwindt G.M., Vergouwe M.N., van Eijk R., Oefner P.J., Hoffman S.M., Lamerdin J.E., Mohrenweiser H.W., Bulman D.E., Ferrari M., Haan J., Lindhout D., van Ommen G.J., Hofker M.H., Ferrari M.D., Frants R.R. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87(3):543–552. doi: 10.1016/s0092-8674(00)81373-2. [http://dx.doi.org/10.1016/S0092-8674(00)81373-2]. [PMID: 8898206]. [DOI] [PubMed] [Google Scholar]
- 23.Catterall W.A. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium. 1998;24(5-6):307–323. doi: 10.1016/s0143-4160(98)90055-0. [http://dx.doi.org/10.1016/S0143-4160(98)90055-0]. [PMID: 10091001]. [DOI] [PubMed] [Google Scholar]
- 24.Tottene A., Fellin T., Pagnutti S., Luvisetto S., Striessnig J., Fletcher C., Pietrobon D. Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc. Natl. Acad. Sci. USA. 2002;99(20):13284–13289. doi: 10.1073/pnas.192242399. [http://dx.doi.org/10.1073/pnas.192242399]. [PMID: 12235360]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Maagdenberg A.M., Pietrobon D., Pizzorusso T., Kaja S., Broos L.A., Cesetti T., van de Ven R.C., Tottene A., van der Kaa J., Plomp J.J., Frants R.R.A., Ferrari M.D.A. Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41(5):701–710. doi: 10.1016/s0896-6273(04)00085-6. [http://dx.doi.org/10.1016/S0896-6273(04)00085-6]. [PMID: 15003170]. [DOI] [PubMed] [Google Scholar]
- 26.Adams P.J., Rungta R.L., Garcia E., van den Maagdenberg A.M., MacVicar B.A., Snutch T.P. Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc. Natl. Acad. Sci. USA. 2010;107(43):18694–18699. doi: 10.1073/pnas.1009500107. [http://dx.doi.org/10.1073/pnas.1009500107]. [PMID: 20937883]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pietrobon D. Calcium channels and migraine. Biochimica et Biophysica Acta (BBA)-. Biomembranes. 2013;1828(7):1655–1665. doi: 10.1016/j.bbamem.2012.11.012. [http://dx.doi.org/10.1016/j.bbamem.2012.11.012]. [DOI] [PubMed] [Google Scholar]
- 28.Stam A.H., Luijckx G.J., Poll-Thé B.T., Ginjaar I.B., Frants R.R., Haan J., Ferrari M.D., Terwindt G.M., van den Maagdenberg A.M. Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J. Neurol. Neurosurg. Psychiatry. 2009;80(10):1125–1129. doi: 10.1136/jnnp.2009.177279. [http://dx.doi.org/10.1136/jnnp.2009.177279]. [PMID: 19520699]. [DOI] [PubMed] [Google Scholar]
- 29.De Fusco M., Marconi R., Silvestri L., Atorino L., Rampoldi L., Morgante L., Ballabio A., Aridon P., Casari G. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump α2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 2003;33(2):192–196. doi: 10.1038/ng1081. [http://dx.doi.org/10.1038/ng1081]. [PMID: 12539047]. [DOI] [PubMed] [Google Scholar]
- 30.Tavraz N.N., Friedrich T., Dürr K.L., Koenderink J.B., Bamberg E., Freilinger T., Dichgans M. Diverse functional consequences of mutations in the Na+/K+-ATPase α2-subunit causing familial hemiplegic migraine type 2. J. Biol. Chem. 2008;283(45):31097–31106. doi: 10.1074/jbc.M802771200. [http://dx.doi.org/10.1074/jbc.M802771200]. [PMID: 18728015]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pisano T., Spiller S., Mei D., Guerrini R., Cianchetti C., Friedrich T., Pruna D. Functional characterization of a novel C-terminal ATP1A2 mutation causing hemiplegic migraine and epilepsy. Cephalalgia. 2013;33(16):1302–1310. doi: 10.1177/0333102413495116. [http://dx.doi.org/10.1177/0333102413495116]. [PMID: 23838748]. [DOI] [PubMed] [Google Scholar]
- 32.Riant F., De Fusco M., Aridon P., Ducros A., Ploton C., Marchelli F., Maciazek J., Bousser M.G., Casari G., Tournier-Lasserve E. ATP1A2 mutations in 11 families with familial hemiplegic migraine. Hum. Mutat. 2005;26(3):281–281. doi: 10.1002/humu.9361. [http://dx.doi.org/10.1002/humu.9361]. [PMID: 16088919]. [DOI] [PubMed] [Google Scholar]
- 33.Vanmolkot K.R., Kors E.E., Turk U., Turkdogan D., Keyser A., Broos L.A., Kia S.K., van den Heuvel J.J., Black D.F., Haan J., Frants R.R., Barone V., Ferrari M.D., Casari G., Koenderink J.B., van den Maagdenberg A.M. Two de novo mutations in the Na,K-ATPase gene ATP1A2 associated with pure familial hemiplegic migraine. Eur. J. Hum. Genet. 2006;14(5):555–560. doi: 10.1038/sj.ejhg.5201607. [http://dx.doi.org/10.1038/sj.ejhg.5201607]. [PMID: 16538223]. [DOI] [PubMed] [Google Scholar]
- 34.Echenne B., Ducros A., Rivier F., Joutel A., Humbertclaude V., Roubertie A., Azaïs M., Bousser M.G., Tournier-Lasserve E. Recurrent episodes of coma: an unusual phenotype of familial hemiplegic migraine with linkage to chromosome 1. Neuropediatrics. 1999;30(4):214–217. doi: 10.1055/s-2007-973493. [http://dx.doi.org/10.1055/s-2007-973493]. [PMID: 10569214]. [DOI] [PubMed] [Google Scholar]
- 35.Spadaro M., Ursu S., Lehmann-Horn F., Veneziano L., Antonini G., Giunti P., Frontali M., Jurkat-Rott K.A. G301R Na+/K+ -ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics. 2004;5(3):177–185. doi: 10.1007/s10048-004-0183-2. [http://dx.doi.org/10.1007/s10048-004-0183-2]. [PMID: 15459825]. [DOI] [PubMed] [Google Scholar]
- 36.Jurkat-Rott K., Freilinger T., Dreier J.P., Herzog J., Göbel H., Petzold G.C., Montagna P., Gasser T., Lehmann-Horn F., Dichgans M. Variability of familial hemiplegic migraine with novel A1A2 Na+/K+-ATPase variants. Neurology. 2004;62(10):1857–1861. doi: 10.1212/01.wnl.0000127310.11526.fd. [http://dx.doi.org/10.1212/01.WNL.0000127310.11526.FD]. [PMID: 15159495]. [DOI] [PubMed] [Google Scholar]
- 37.Dichgans M., Freilinger T., Eckstein G., Babini E., Lorenz-Depiereux B., Biskup S., Ferrari M.D., Herzog J., van den Maagdenberg A.M., Pusch M., Strom T.M. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366(9483):371–377. doi: 10.1016/S0140-6736(05)66786-4. [http://dx.doi.org/10.1016/S0140-6736(05)66786-4]. [PMID: 16054936]. [DOI] [PubMed] [Google Scholar]
- 38.Castro M.J., Stam A.H., Lemos C., de Vries B., Vanmolkot K.R.J., Barros J., Terwindt G.M., Frants R.R., Sequeiros J., Ferrari M.D., Pereira-Monteiro J.M., van den Maagdenberg A.M. First mutation in the voltage-gated Nav1.1 subunit gene SCN1A with co-occurring familial hemiplegic migraine and epilepsy. Cephalalgia. 2009;29(3):308–313. doi: 10.1111/j.1468-2982.2008.01721.x. [http://dx.doi.org/10.1111/j.1468-2982.2008.01721.x]. [PMID: 19220312]. [DOI] [PubMed] [Google Scholar]
- 39.Vahedi K., Depienne C., Le Fort D., Riant F., Chaine P., Trouillard O., Gaudric A., Morris M.A., Leguern E., Tournier-Lasserve E., Bousser M.G. Elicited repetitive daily blindness: a new phenotype associated with hemiplegic migraine and SCN1A mutations. Neurology. 2009;72(13):1178–1183. doi: 10.1212/01.wnl.0000345393.53132.8c. [http://dx.doi.org/10.1212/01.wnl.0000345393.53132.8c]. [PMID: 19332696]. [DOI] [PubMed] [Google Scholar]
- 40.Parihar R., Ganesh S. The SCN1A gene variants and epileptic encephalopathies. J. Hum. Genet. 2013;58(9):573–580. doi: 10.1038/jhg.2013.77. [http://dx.doi.org/10.1038/jhg.2013.77]. [PMID: 23884151]. [DOI] [PubMed] [Google Scholar]
- 41.van den Maagdenberg A.M., Pizzorusso T., Kaja S., Terpolilli N., Shapovalova M., Hoebeek F.E., Barrett C.F., Gherardini L., van de Ven R.C., Todorov B., Broos L.A., Tottene A., Gao Z., Fodor M., De Zeeuw C.I., Frants R.R., Plesnila N., Plomp J.J., Pietrobon D., Ferrari M.D. High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann. Neurol. 2010;67(1):85–98. doi: 10.1002/ana.21815. [http://dx.doi.org/10.1002/ana.21815]. [PMID: 20186955]. [DOI] [PubMed] [Google Scholar]
- 42.Eikermann-Haerter K., Dileköz E., Kudo C., Savitz S.I., Waeber C., Baum M.J., Ferrari M.D., van den Maagdenberg A.M., Moskowitz M.A., Ayata C. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J. Clin. Invest. 2009;119(1):99–109. doi: 10.1172/JCI36059. [PMID: 19104150]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eikermann-Haerter K., Baum M.J., Ferrari M.D., van den Maagdenberg A.M., Moskowitz M.A., Ayata C. Androgenic suppression of spreading depression in familial hemiplegic migraine type 1 mutant mice. Ann. Neurol. 2009;66(4):564–568. doi: 10.1002/ana.21779. [http://dx.doi.org/10.1002/ana.21779]. [PMID: 19847904]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eikermann-Haerter K., Ayata C. Cortical spreading depression and migraine. Curr. Neurol. Neurosci. Rep. 2010;10(3):167–173. doi: 10.1007/s11910-010-0099-1. [http://dx.doi.org/10.1007/s11910-010-0099-1]. [PMID: 20425031]. [DOI] [PubMed] [Google Scholar]
- 45.Vecchia D., Tottene A., van den Maagdenberg A.M., Pietrobon D. Mechanism underlying unaltered cortical inhibitory synaptic transmission in contrast with enhanced excitatory transmission in CaV2.1 knockin migraine mice. Neurobiol. Dis. 2014;69:225–234. doi: 10.1016/j.nbd.2014.05.035. [http://dx.doi.org/10.1016/j.nbd.2014.05.035]. [PMID: 24907493]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vecchia D., Tottene A., van den Maagdenberg A.M., Pietrobon D. Abnormal cortical synaptic transmission in CaV2.1 knockin mice with the S218L missense mutation which causes a severe familial hemiplegic migraine syndrome in humans. Front. Cell. Neurosci. 2015;9:8. doi: 10.3389/fncel.2015.00008. [http://dx.doi.org/10.3389/fncel.2015.00008]. [PMID: 25741235]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dilekoz E., Houben T., Eikermann-Haerter K., Balkaya M., Lenselink A.M., Whalen M.J., Spijker S., Ferrari M.D., van den Maagdenberg A.M., Ayata C. Migraine mutations impair hippocampal learning despite enhanced long-term potentiation. J. Neurosci. 2015;35(8):3397–3402. doi: 10.1523/JNEUROSCI.2630-14.2015. [http://dx.doi.org/10.1523/JNEUROSCI.2630-14.2015]. [PMID: 25716839]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathew R., Andreou A.P., Chami L., Bergerot A., van den Maagdenberg A.M., Ferrari M.D., Goadsby P.J. Immunohistochemical characterization of calcitonin gene-related peptide in the trigeminal system of the familial hemiplegic migraine 1 knock-in mouse. Cephalalgia. 2011;31(13):1368–1380. doi: 10.1177/0333102411418847. [http://dx.doi.org/10.1177/0333102411418847]. [PMID: 21893556]. [DOI] [PubMed] [Google Scholar]
- 49.Fioretti B., Catacuzzeno L., Sforna L., Gerke-Duncan M.B., van den Maagdenberg A.M.J.M., Franciolini F., Connor M., Pietrobon D. Trigeminal ganglion neuron subtype-specific alterations of Ca(V)2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J. Physiol. 2011;589(Pt 23):5879–5895. doi: 10.1113/jphysiol.2011.220533. [http://dx.doi.org/10.1113/jphysiol.2011.220533]. [PMID: 22005682]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ceruti S., Villa G., Fumagalli M., Colombo L., Magni G., Zanardelli M., Fabbretti E., Verderio C., van den Maagdenberg A.M., Nistri A., Abbracchio M.P. Calcitonin gene-related peptide-mediated enhancement of purinergic neuron/glia communication by the algogenic factor bradykinin in mouse trigeminal ganglia from wild-type and R192Q Cav2.1 Knock-in mice: implications for basic mechanisms of migraine pain. J. Neurosci. 2011;31(10):3638–3649. doi: 10.1523/JNEUROSCI.6440-10.2011. [http://dx.doi.org/10.1523/JNEUROSCI.6440-10.2011]. [PMID: 21389219]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leo L., Gherardini L., Barone V., De Fusco M., Pietrobon D., Pizzorusso T., Casari G. Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet. 2011;7(6):e1002129. doi: 10.1371/journal.pgen.1002129. [http://dx.doi.org/10.1371/journal.pgen.1002129]. [PMID: 21731499]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bøttger P., Glerup S., Gesslein B., Illarionova N.B., Isaksen T.J., Heuck A., Clausen B.H., Füchtbauer E.M., Gramsbergen J.B., Gunnarson E., Aperia A., Lauritzen M., Lambertsen K.L., Nissen P., Lykke-Hartmann K. Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci. Rep. 2016;6:22047. doi: 10.1038/srep22047. [http://dx.doi.org/10.1038/srep22047]. [PMID: 26911348]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Auffenberg E., Hedrich U., Lerche H., Dichgans M., Plesnila N., Freilinger T. A novel mouse model for familial hemiplegic migraine type 3 reveals increased susceptibility for cortical spreading depression. Cephalalgia. 2017;•••:206–206. [Google Scholar]
- 54.Eikermann-Haerter K., Arbel-Ornath M., Yalcin N., Yu E.S., Kuchibhotla K.V., Yuzawa I., Hudry E., Willard C.R., Climov M., Keles F., Belcher A.M., Sengul B., Negro A., Rosen I.A., Arreguin A., Ferrari M.D., van den Maagdenberg A.M., Bacskai B.J., Ayata C. Abnormal synaptic Ca(2+) homeostasis and morphology in cortical neurons of familial hemiplegic migraine type 1 mutant mice. Ann. Neurol. 2015;78(2):193–210. doi: 10.1002/ana.24449. [http://dx.doi.org/10.1002/ana.24449]. [PMID: 26032020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shyti R., Eikermann-Haerter K., van Heiningen S.H., Meijer O.C., Ayata C., Joëls M., Ferrari M.D., van den Maagdenberg A.M., Tolner E.A. Stress hormone corticosterone enhances susceptibility to cortical spreading depression in familial hemiplegic migraine type 1 mutant mice. Exp. Neurol. 2015;263:214–220. doi: 10.1016/j.expneurol.2014.10.015. [http://dx.doi.org/10.1016/j.expneurol.2014.10.015]. [PMID: 25447936]. [DOI] [PubMed] [Google Scholar]
- 56.Klychnikov O.I., Li K.W., Sidorov I.A., Loos M., Spijker S., Broos L.A., Frants R.R., Ferrari M.D., Mayboroda O.A., Deelder A.M., Smit A.B., van den Maagdenberg A.M. Quantitative cortical synapse proteomics of a transgenic migraine mouse model with mutated Ca(V)2.1 calcium channels. Proteomics. 2010;10(13):2531–2535. doi: 10.1002/pmic.200900733. [http://dx.doi.org/10.1002/pmic.200900733]. [PMID: 20391530]. [DOI] [PubMed] [Google Scholar]
- 57.Di Guilmi M.N., Wang T., Inchauspe C.G., Forsythe I.D., Ferrari M.D., van den Maagdenberg A.M., Borst J.G.G., Uchitel O.D. Synaptic gain-of-function effects of mutant Cav2.1 channels in a mouse model of familial hemiplegic migraine are due to increased basal [Ca2+]i. J. Neurosci. 2014;34(21):7047–7058. doi: 10.1523/JNEUROSCI.2526-13.2014. [http://dx.doi.org/10.1523/JNEUROSCI.2526-13.2014]. [PMID: 24849341]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inchauspe C.G., Urbano F.J., Di Guilmi M.N., Ferrari M.D., van den Maagdenberg A.M., Forsythe I.D., Uchitel O.D. Presynaptic CaV2.1 calcium channels carrying familial hemiplegic migraine mutation R192Q allow faster recovery from synaptic depression in mouse calyx of Held. J. Neurophysiol. 2012;108(11):2967–2976. doi: 10.1152/jn.01183.2011. [http://dx.doi.org/10.1152/jn.01183.2011]. [PMID: 22956801]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Inchauspe C.G., Urbano F.J., Di Guilmi M.N., Forsythe I.D., Ferrari M.D., van den Maagdenberg A.M., Uchitel O.D. Gain of function in FHM-1 Cav2.1 knock-in mice is related to the shape of the action potential. J. Neurophysiol. 2010;104(1):291–299. doi: 10.1152/jn.00034.2010. [http://dx.doi.org/10.1152/jn.00034.2010]. [PMID: 20484531]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shu Y., Hasenstaub A., McCormick D.A. Turning on and off recurrent balanced cortical activity. Nature. 2003;423(6937):288–293. doi: 10.1038/nature01616. [http://dx.doi.org/10.1038/nature01616]. [PMID: 12748642]. [DOI] [PubMed] [Google Scholar]
- 61.Khennouf L., Gesslein B., Lind B.L., van den Maagdenberg A.M., Lauritzen M. Activity-dependent calcium, oxygen, and vascular responses in a mouse model of familial hemiplegic migraine type 1. Ann. Neurol. 2016;80(2):219–232. doi: 10.1002/ana.24707. [http://dx.doi.org/10.1002/ana.24707]. [PMID: 27314908]. [DOI] [PubMed] [Google Scholar]
- 62.Hullugundi S.K., Ansuini A., Ferrari M.D., van den Maagdenberg A.M.J.M., Nistri A. A hyperexcitability phenotype in mouse trigeminal sensory neurons expressing the R192Q Cacna1a missense mutation of familial hemiplegic migraine type-1. Neuroscience. 2014;266:244–254. doi: 10.1016/j.neuroscience.2014.02.020. [http://dx.doi.org/10.1016/j.neuroscience.2014.02.020]. [PMID: 24583041]. [DOI] [PubMed] [Google Scholar]
- 63.Franceschini A., Vilotti S., Ferrari M.D., van den Maagdenberg A.M., Nistri A., Fabbretti E. TNFα levels and macrophages expression reflect an inflammatory potential of trigeminal ganglia in a mouse model of familial hemiplegic migraine. PLoS One. 2013;8(1):e52394. doi: 10.1371/journal.pone.0052394. [http://dx.doi.org/10.1371/journal.pone.0052394]. [PMID: 23326332]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eising E., Shyti R., ’t Hoen P.A.C., Vijfhuizen L.S., Huisman S.M.H., Broos L.A.M., Mahfouz A., Reinders M.J.T., Ferrari M.D., Tolner E.A., de Vries B., van den Maagdenberg A.M.J.M.P., Vijfhuizen L.S., Huisman S.M., Broos L.A., Mahfouz A., Reinders M.J., Ferrari M.D., Tolner E.A., de Vries B. Cortical spreading depression causes unique dysregulation of inflammatory pathways in a transgenic mouse model of migraine. Mol. Neurobiol. 2017;54(4):2986–2996. doi: 10.1007/s12035-015-9681-5. [http://dx.doi.org/10.1007/s12035-015-9681-5]. [PMID: 27032388]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang I.M., Zhang B., Yang X., Zhu J., Stepaniants S., Zhang C., Meng Q., Peters M., He Y., Ni C., Slipetz D., Crackower M.A., Houshyar H., Tan C.M., Asante-Appiah E., O’Neill G., Luo M.J., Thieringer R., Yuan J., Chiu C.S., Lum P.Y., Lamb J., Boie Y., Wilkinson H.A., Schadt E.E., Dai H., Roberts C. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol. 2012;8(1):594. doi: 10.1038/msb.2012.24. [http://dx.doi.org/10.1038/msb.2012.24]. [PMID: 22806142]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vezzani A., French J., Bartfai T., Baram T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011;7(1):31–40. doi: 10.1038/nrneurol.2010.178. [http://dx.doi.org/10.1038/nrneurol.2010.178]. [PMID: 21135885]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vezzani A., Friedman A., Dingledine R.J. The role of inflammation in epileptogenesis. Neuropharmacology. 2013;69:16–24. doi: 10.1016/j.neuropharm.2012.04.004. [http://dx.doi.org/10.1016/j.neuropharm.2012.04.004]. [PMID: 22521336]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nair A., Simonetti M., Birsa N., Ferrari M.D., van den Maagdenberg A.M., Giniatullin R., Nistri A., Fabbretti E. Familial hemiplegic migraine Ca(v)2.1 channel mutation R192Q enhances ATP-gated P2X3 receptor activity of mouse sensory ganglion neurons mediating trigeminal pain. Mol. Pain. 2010;6(1):48. doi: 10.1186/1744-8069-6-48. [PMID: 20735819]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Franceschini A., Hullugundi S.K., van den Maagdenberg A.M., Nistri A., Fabbretti E. Effects of LPS on P2X3 receptors of trigeminal sensory neurons and macrophages from mice expressing the R192Q Cacna1a gene mutation of familial hemiplegic migraine-1. Purinergic Signal. 2013;9(1):7–13. doi: 10.1007/s11302-012-9328-1. [http://dx.doi.org/10.1007/s11302-012-9328-1]. [PMID: 22836594]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Franceschini A., Nair A., Bele T., van den Maagdenberg A.M., Nistri A., Fabbretti E. Functional crosstalk in culture between macrophages and trigeminal sensory neurons of a mouse genetic model of migraine. BMC Neurosci. 2012;13(1):143. doi: 10.1186/1471-2202-13-143. [http://dx.doi.org/10.1186/1471-2202-13-143]. [PMID: 23171280]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dehghani M.A. 2019. [Google Scholar]
- 72.Dehghani A., Karatas H., Can A., Erdemli E., Yemisci M., Eren-Kocak E., Dalkara T. Nuclear expansion and pore opening are instant signs of neuronal hypoxia and can identify poorly fixed brains. Sci. Rep. 2018;8(1):14770. doi: 10.1038/s41598-018-32878-1. [http://dx.doi.org/10.1038/s41598-018-32878-1]. [PMID: 30282977]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shyti R., Kohler I., Schoenmaker B., Derks R.J., Ferrari M.D., Tolner E.A., Mayboroda O.A., van den Maagdenberg A.M. Plasma metabolic profiling after cortical spreading depression in a transgenic mouse model of hemiplegic migraine by capillary electrophoresis--mass spectrometry. Mol. Biosyst. 2015;11(5):1462–1471. doi: 10.1039/c5mb00049a. [http://dx.doi.org/10.1039/C5MB00049A]. [PMID: 25856790]. [DOI] [PubMed] [Google Scholar]
- 74.Carreira R.J., Shyti R., Balluff B., Abdelmoula W.M., van Heiningen S.H., van Zeijl R.J., Dijkstra J., Ferrari M.D., Tolner E.A., McDonnell L.A., van den Maagdenberg A.M. Large-scale mass spectrometry imaging investigation of consequences of cortical spreading depression in a transgenic mouse model of migraine. J. Am. Soc. Mass Spectrom. 2015;26(6):853–861. doi: 10.1007/s13361-015-1136-8. [http://dx.doi.org/10.1007/s13361-015-1136-8]. [PMID: 25877011]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiao Y., Richter J.A., Hurley J.H. Release of glutamate and CGRP from trigeminal ganglion neurons: Role of calcium channels and 5-HT1 receptor signaling. Mol. Pain. 2008;4(1):12. doi: 10.1186/1744-8069-4-12. [http://dx.doi.org/10.1186/1744-8069-4-12]. [PMID: 18416824]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shields K.G., Storer R.J., Akerman S., Goadsby P.J. Calcium channels modulate nociceptive transmission in the trigeminal nucleus of the cat. Neuroscience. 2005;135(1):203–212. doi: 10.1016/j.neuroscience.2004.08.054. [http://dx.doi.org/10.1016/j.neuroscience.2004.08.054]. [PMID: 16084658]. [DOI] [PubMed] [Google Scholar]
- 77.Eroli F., Vilotti S., van den Maagdenberg A.M.J.M., Nistri A. Hyperpolarization-activated current Ih in mouse trigeminal sensory neurons in a transgenic mouse model of familial hemiplegic migraine type-1. Neuroscience. 2017;351:47–64. doi: 10.1016/j.neuroscience.2017.03.033. [http://dx.doi.org/10.1016/j.neuroscience.2017.03.033]. [PMID: 28363781]. [DOI] [PubMed] [Google Scholar]
- 78.Hullugundi S.K., Ferrari M.D., van den Maagdenberg A.M., Nistri A. The mechanism of functional up-regulation of P2X3 receptors of trigeminal sensory neurons in a genetic mouse model of familial hemiplegic migraine type 1 (FHM-1). PLoS One. 2013;8(4):e60677. doi: 10.1371/journal.pone.0060677. [http://dx.doi.org/10.1371/journal.pone.0060677]. [PMID: 23577145]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meents J.E., Neeb L., Reuter U. TRPV1 in migraine pathophysiology. Trends Mol. Med. 2010;16(4):153–159. doi: 10.1016/j.molmed.2010.02.004. [http://dx.doi.org/10.1016/j.molmed.2010.02.004]. [PMID: 20347391]. [DOI] [PubMed] [Google Scholar]
- 80.Zhang X.C., Kainz V., Burstein R., Levy D. Tumor necrosis factor-α induces sensitization of meningeal nociceptors mediated via local COX and p38 MAP kinase actions. Pain. 2011;152(1):140–149. doi: 10.1016/j.pain.2010.10.002. [http://dx.doi.org/10.1016/j.pain.2010.10.002]. [PMID: 21036476]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marchenkova A., van den Maagdenberg A.M., Nistri A. Loss of inhibition by brain natriuretic peptide over P2X3 receptors contributes to enhanced spike firing of trigeminal ganglion neurons in a mouse model of familial hemiplegic migraine type-1. Neuroscience. 2016;331:197–205. doi: 10.1016/j.neuroscience.2016.06.034. [http://dx.doi.org/10.1016/j.neuroscience.2016.06.034]. [PMID: 27346147]. [DOI] [PubMed] [Google Scholar]
- 82.Vilotti S., Vana N., Van den Maagdenberg A.M., Nistri A. Expression and function of calcitonin gene-related peptide (CGRP) receptors in trigeminal ganglia of R192Q Cacna1a knock-in mice. Neurosci. Lett. 2016;620:104–110. doi: 10.1016/j.neulet.2016.03.046. [http://dx.doi.org/10.1016/j.neulet.2016.03.046]. [PMID: 27021026]. [DOI] [PubMed] [Google Scholar]
- 83.Park J., Moon H., Akerman S., Holland P.R., Lasalandra M.P., Andreou A.P., Ferrari M.D., van den Maagdenberg A.M., Goadsby P.J. Differential trigeminovascular nociceptive responses in the thalamus in the familial hemiplegic migraine 1 knock-in mouse: a Fos protein study. Neurobiol. Dis. 2014;64:1–7. doi: 10.1016/j.nbd.2013.12.004. [http://dx.doi.org/10.1016/j.nbd.2013.12.004]. [PMID: 24355314]. [DOI] [PubMed] [Google Scholar]
- 84.Langford D.J., Bailey A.L., Chanda M.L., Clarke S.E., Drummond T.E., Echols S., Glick S., Ingrao J., Klassen-Ross T., Lacroix-Fralish M.L., Matsumiya L., Sorge R.E., Sotocinal S.G., Tabaka J.M., Wong D., van den Maagdenberg A.M., Ferrari M.D., Craig K.D., Mogil J.S. Coding of facial expressions of pain in the laboratory mouse. Nat. Methods. 2010;7(6):447–449. doi: 10.1038/nmeth.1455. [http://dx.doi.org/10.1038/nmeth.1455]. [PMID: 20453868]. [DOI] [PubMed] [Google Scholar]
- 85.Chanda M.L., Tuttle A.H., Baran I., Atlin C., Guindi D., Hathaway G., Israelian N., Levenstadt J., Low D., Macrae L., O’Shea L., Silver A., Zendegui E., Mariette Lenselink A., Spijker S., Ferrari M.D., van den Maagdenberg A.M., Mogil J.S. Behavioral evidence for photophobia and stress-related ipsilateral head pain in transgenic Cacna1a mutant mice. Pain. 2013;154(8):1254–1262. doi: 10.1016/j.pain.2013.03.038. [http://dx.doi.org/10.1016/j.pain.2013.03.038]. [PMID: 23673147]. [DOI] [PubMed] [Google Scholar]
- 86.Wrobel Goldberg S., Silberstein S.D. Targeting CGRP: A new era for migraine treatment. CNS Drugs. 2015;29(6):443–452. doi: 10.1007/s40263-015-0253-z. [http://dx.doi.org/10.1007/s40263-015-0253-z]. [PMID: 26138383]. [DOI] [PubMed] [Google Scholar]
- 87.Russo A.F. CGRP as a neuropeptide in migraine: lessons from mice. Br. J. Clin. Pharmacol. 2015;80(3):403–414. doi: 10.1111/bcp.12686. [http://dx.doi.org/10.1111/bcp.12686]. [PMID: 26032833]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Recober A., Kuburas A., Zhang Z., Wemmie J.A., Anderson M.G., Russo A.F. Role of calcitonin gene-related peptide in light-aversive behavior: implications for migraine. J. Neurosci. 2009;29(27):8798–8804. doi: 10.1523/JNEUROSCI.1727-09.2009. [http://dx.doi.org/10.1523/JNEUROSCI.1727-09.2009]. [PMID: 19587287]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Recober A., Kaiser E.A., Kuburas A., Russo A.F. Induction of multiple photophobic behaviors in a transgenic mouse sensitized to CGRP. Neuropharmacology. 2010;58(1):156–165. doi: 10.1016/j.neuropharm.2009.07.009. [http://dx.doi.org/10.1016/j.neuropharm.2009.07.009]. [PMID: 19607849]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deboer T., van Diepen H.C., Ferrari M.D., Van den Maagdenberg A.M., Meijer J.H. Reduced sleep and low adenosinergic sensitivity in cacna1a R192Q mutant mice. Sleep (Basel) 2013;36(1):127–136. doi: 10.5665/sleep.2316. [http://dx.doi.org/10.5665/sleep.2316]. [PMID: 23288979]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Vries B., Eising E., Broos L.A., Koelewijn S.C., Todorov B., Frants R.R., Boer J.M., Ferrari M.D., Hoen P.A., van den Maagdenberg A.M. RNA expression profiling in brains of familial hemiplegic migraine type 1 knock-in mice. Cephalalgia. 2014;34(3):174–182. doi: 10.1177/0333102413502736. [http://dx.doi.org/10.1177/0333102413502736]. [PMID: 23985897]. [DOI] [PubMed] [Google Scholar]
- 92.Kurth T., Chabriat H., Bousser M.G. Migraine and stroke: a complex association with clinical implications. Lancet Neurol. 2012;11(1):92–100. doi: 10.1016/S1474-4422(11)70266-6. [http://dx.doi.org/10.1016/S1474-4422(11)70266-6]. [PMID: 22172624]. [DOI] [PubMed] [Google Scholar]
- 93.Sacco S., Ornello R., Ripa P., Pistoia F., Carolei A. Migraine and hemorrhagic stroke: a meta-analysis. Stroke. 2013;44(11):3032–3038. doi: 10.1161/STROKEAHA.113.002465. [http://dx.doi.org/10.1161/STROKEAHA.113.002465]. [PMID: 24085027]. [DOI] [PubMed] [Google Scholar]
- 94.Spector J.T., Kahn S.R., Jones M.R., Jayakumar M., Dalal D., Nazarian S. Migraine headache and ischemic stroke risk: an updated meta-analysis. Am. J. Med. 2010;123(7):612–624. doi: 10.1016/j.amjmed.2009.12.021. [http://dx.doi.org/10.1016/j.amjmed.2009.12.021]. [PMID: 20493462]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Malik R., Freilinger T., Winsvold B.S., Anttila V., Vander Heiden J., Traylor M., de Vries B., Holliday E.G., Terwindt G.M., Sturm J., Bis J.C., Hopewell J.C., Ferrari M.D., Rannikmae K., Wessman M., Kallela M., Kubisch C., Fornage M., Meschia J.F., Lehtimäki T., Sudlow C., Clarke R., Chasman D.I., Mitchell B.D., Maguire J., Kaprio J., Farrall M., Raitakari O.T., Kurth T., Ikram M.A., Reiner A.P., Longstreth W.T., Jr, Rothwell P.M., Strachan D.P., Sharma P., Seshadri S., Quaye L., Cherkas L., Schürks M., Rosand J., Ligthart L., Boncoraglio G.B., Davey Smith G., van Duijn C.M., Stefansson K., Worrall B.B., Nyholt D.R., Markus H.S., van den Maagdenberg A.M., Cotsapas C., Zwart J.A., Palotie A., Dichgans M. International Headache Genetics Consortium. METASTROKE Collaboration of the International Stroke Genetics Consortium. Shared genetic basis for migraine and ischemic stroke: A genome-wide analysis of common variants. Neurology. 2015;84(21):2132–2145. doi: 10.1212/WNL.0000000000001606. [http://dx.doi.org/10.1212/WNL.0000000000001606]. [PMID: 25934857]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eikermann-Haerter K. Spreading depolarization may link migraine and stroke. Headache. 2014;54(7):1146–1157. doi: 10.1111/head.12386. [http://dx.doi.org/10.1111/head.12386]. [PMID: 24913618]. [DOI] [PubMed] [Google Scholar]
- 97.Eikermann-Haerter K., Lee J.H., Yuzawa I., Liu C.H., Zhou Z., Shin H.K., Zheng Y., Qin T., Kurth T., Waeber C., Ferrari M.D., van den Maagdenberg A.M., Moskowitz M.A., Ayata C. Migraine mutations increase stroke vulnerability by facilitating ischemic depolarizations. Circulation. 2012;125(2):335–345. doi: 10.1161/CIRCULATIONAHA.111.045096. [http://dx.doi.org/10.1161/CIRCULATIONAHA.111.045096]. [PMID: 22144569]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eikermann-Haerter K., Lee J.H., Yalcin N., Yu E.S., Daneshmand A., Wei Y., Zheng Y., Can A., Sengul B., Ferrari M.D., van den Maagdenberg A.M., Ayata C. Migraine prophylaxis, ischemic depolarizations, and stroke outcomes in mice. Stroke. 2015;46(1):229–236. doi: 10.1161/STROKEAHA.114.006982. [http://dx.doi.org/10.1161/STROKEAHA.114.006982]. [PMID: 25424478]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.James P.F., Grupp I.L., Grupp G., Woo A.L., Askew G.R., Croyle M.L., Walsh R.A., Lingrel J.B. Identification of a specific role for the Na, K-ATPase α2 isoform as a regulator of calcium in the heart. Mol. Cell, 1999,3(5), 555-563. Ikeda, K.; Onaka, T.; Yamakado, M.; Nakai, J.; Ishikawa, T.O.; Taketo, M.M. and Kawakami, K., Degeneration of the amygdala/piriform cortex and enhanced fear/anxiety behaviors in sodium pump α2 subunit (Atp1a2)-deficient mice. J. Neurosci. 2003;23(11):4667–4676. [PMID: 12805306]. [Google Scholar]
- 100.Ikeda K., Onaka T., Yamakado M., Nakai J., Ishikawa T.O., Taketo M.M., Kawakami K. Degeneration of the amygdala/piriform cortex and enhanced fear/anxiety behaviors in sodium pump α2 subunit (Atp1a2)-deficient mice. J. Neurosci. 2003;23(11):4667–4676. doi: 10.1523/JNEUROSCI.23-11-04667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.D’Ambrosio R., Gordon D.S., Winn H.R. Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K(+) in rat hippocampus. J. Neurophysiol. 2002;87(1):87–102. doi: 10.1152/jn.00240.2001. [http://dx.doi.org/10.1152/jn.00240.2001]. [PMID: 11784732]. [DOI] [PubMed] [Google Scholar]
- 102.Larsen B.R., MacAulay N. Kir4.1-mediated spatial buffering of K(+): experimental challenges in determination of its temporal and quantitative contribution to K(+) clearance in the brain. Channels (Austin) 2014;8(6):544–550. doi: 10.4161/19336950.2014.970448. [http://dx.doi.org/10.4161/19336950.2014.970448]. [PMID: 25483287]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Capuani C., Melone M., Tottene A., Bragina L., Crivellaro G., Santello M., Casari G., Conti F., Pietrobon D. Defective glutamate and K+ clearance by cortical astrocytes in familial hemiplegic migraine type 2. EMBO Mol. Med. 2016;8(8):967–986. doi: 10.15252/emmm.201505944. [http://dx.doi.org/10.15252/emmm.201505944]. [PMID: 27354390]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karner E., Delazer M., Benke T., Bösch S. Cognitive functions, emotional behavior, and quality of life in familial hemiplegic migraine. Cogn. Behav. Neurol. 2010;23(2):106–111. doi: 10.1097/WNN.0b013e3181c3a8a6. [http://dx.doi.org/10.1097/WNN.0b013e3181c3a8a6]. [PMID: 20535059]. [DOI] [PubMed] [Google Scholar]
- 105.Bøttger P., Doğanlı C., Lykke-Hartmann K. Migraine- and dystonia-related disease-mutations of Na+/K+-ATPases: relevance of behavioral studies in mice to disease symptoms and neurological manifestations in humans. Neurosci. Biobehav. Rev. 2012;36(2):855–871. doi: 10.1016/j.neubiorev.2011.10.005. [http://dx.doi.org/10.1016/j.neubiorev.2011.10.005]. [PMID: 22067897]. [DOI] [PubMed] [Google Scholar]
- 106.Eikermann-Haerter K., Yuzawa I., Dilekoz E., Joutel A., Moskowitz M.A., Ayata C. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy syndrome mutations increase susceptibility to spreading depression. Ann. Neurol. 2011;69(2):413–418. doi: 10.1002/ana.22281. [http://dx.doi.org/10.1002/ana.22281]. [PMID: 21387384]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hasan M., Fermaintt C.S., Gao N., Sakai T., Miyazaki T., Jiang S., Li Q.Z., Atkinson J.P., Morse H.C., III, Lehrman M.A., Yan N. Lehrman, M.A. and Yan, N. Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation. Immunity. 2015;43(3):463–474. doi: 10.1016/j.immuni.2015.07.022. [http://dx.doi.org/10.1016/j.immuni.2015.07.022]. [PMID: 26320659]. [DOI] [PMC free article] [PubMed] [Google Scholar]