

Abstract
The synchronized activity of neuronal networks under physiological conditions is mirrored by specific oscillatory patterns of the EEG that are associated with different behavioral states and cognitive functions. Excessive synchronization can, however, lead to focal epileptiform activity characterized by interictal and ictal discharges in epileptic patients and animal models. This review focusses on studies that have addressed epileptiform synchronization in temporal lobe regions by em-ploying in vitro and in vivo recording techniques. First, we consider the role of ionotropic and metabotropic excitatory glu-tamatergic transmission in seizure generation as well as the paradoxical role of GABAA signaling in initiating and perhaps maintaining focal seizure activity. Second, we address non-synaptic mechanisms (which include voltage-gated ionic currents and gap junctions) in the generation of epileptiform synchronization. For each mechanism, we discuss the actions of anti-epileptic drugs that are presumably modulating excitatory or inhibitory signaling and voltage-gated currents to prevent sei-zures in epileptic patients. These findings provide insights into the mechanisms of seizure initiation and maintenance, thus leading to the development of specific pharmacological treatments for focal epileptic disorders.
Keywords: Epileptiform synchronization, mesial temporal lobe epilepsy, interictal spikes, seizures, anti-epileptic drugs, excitatory transmission, inhibitory transmission, voltage-gated channels
1. INTRODUCTION
Neuronal synchronization represents the integrated activity occurring over time among neuronal networks that are located in one or more interconnected brain structures [1]. Under normal conditions, neuronal synchronization makes the brain generate specific EEG rhythms that are associated with different physiological states extending from cognitive functions (such as perception, formation and recall of memories) to specific sleep states; these EEG patterns include the theta rhythm, beta and gamma oscillations, sharp-wave ripples and sleep spindles [2, 3]. However, excessive and thus abnormal neuronal synchronization can cause the occurrence of both focal [4, 5] and generalized epileptic discharges [6-9].
Here, we will review the evolving concepts regarding the cellular and pharmacological mechanisms that cause the generation of focal epileptiform discharges; these studies encompass over fifty years of neuroscience research paralleling many of our advancements in understanding the processes
that regulate neuronal excitability and thus brain function. We will review data obtained from in vivo and in vitro animal models of focal epilepsy as well as from epileptic patients not responding to antiepileptic drugs and thus investigated with invasive electrophysiological recordings (including detection of single unit activity) before undergoing brain surgery. Most of these studies were performed in limbic brain structures that include the hippocampus, the rhinal cortices and the amygdala, which are known to play a role in mesial temporal lobe epilepsy (MTLE) [10-12].
An epileptic brain generates interictal discharges (i.e., short lasting electrographic events lasting less than one second that are not accompanied by any detectable clinical symptom) Fig. (1A) as well as ictal discharges (i.e., periods of abnormal, hypersynchronous activity lasting between a few tens of seconds and several minutes thus disrupting normal brain function) (Fig. 1B). Moreover, during the last few years, it has been shown that epileptiform synchronization is associated with the occurrence of high-frequency oscillations (HFOs) in the EEG (field) recordings (Fig. 1C). These HFOs - which are not visible in standard EEG recordings and can only be extracted by amplifying the appropriately filtered signal - have been categorised into two groups, based on their frequency content: (i) ripples, which include oscillatory events between 80 and 200 Hz [13] and (ii) fast ripples, namely oscillatory events between 250-500 Hz [14]. Both ripples and fast ripples have been observed in the epileptic tissue of animals and humans, in association with interictal spikes or seizures; moreover, fast ripples have been reported to occur more frequently in seizure onset zones [14-21]. Interictal and ictal discharges as well as HFOs may share some common synchronizing mechanisms [5].
Fig. (1).
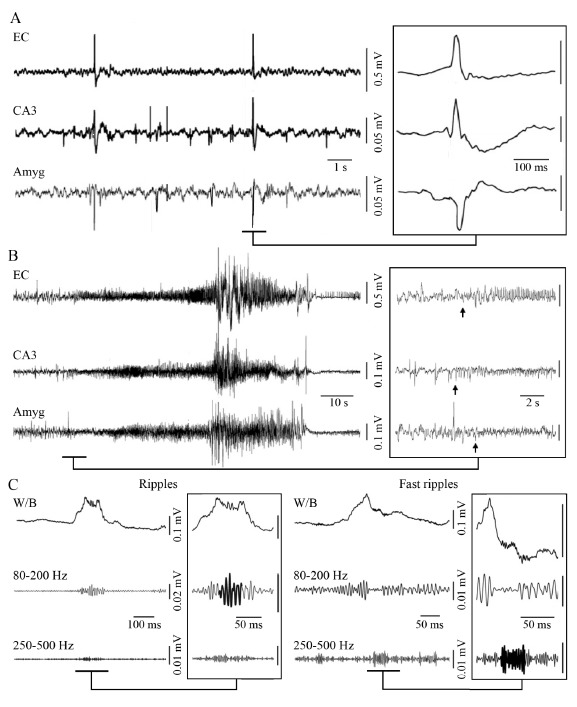
A: Interictal discharges recorded in the entorhinal cortex (EC), CA3 region of the hippocampus and amygdala (Amy) of a pilocarpine-treated epileptic rat. Note that some interictal spikes are only observed in some regions whereas other tend to propagate. B: Representative spontaneous seizure recorded in a pilocarpine-treated epileptic rat from the EC, CA3 region and amygdala. Low-voltage fast activity (arrows) marks the onset of the seizure, which is in this case first observed in the CA3 region. C: High-frequency activity (80-500 Hz) recorded in association with an interictal spike, from a pilocarpine-treated epileptic animal. The interictal spike is visible on the wideband signal (W/B) whereas high-frequency activity is only visible after the signal has been filtered between 80-200 Hz and between 250-500 Hz. Ripples include oscillatory events that are only visible in the 80-200 Hz frequency band (left panel) whereas fast ripples include events that are only visible in the 250-500 Hz frequency band (right panel).
This review will specifically focus on the role of (i) excitatory and (ii) inhibitory signaling as well as (iii) non-synaptic mechanisms (which include voltage-gated ionic currents and gap junctions) in epileptiform synchronization. In each of these sections, we will also take into consideration which of the current antiepileptic drugs are thought to target these mechanisms thus leading to reduction or prevention of seizures in patients (and animal models).
2. EXCITATORY SIGNALING
Interictal discharges represent the first epileptic phenomenon to be experimentally reproduced. Several studies performed in vivo in the 1950s and 1960s revealed that epileptic cortical foci, acutely induced by the topical application of convulsants (such as penicillin or strychnine) or local freezing, generate spikes in the EEG of animals that share features with those recorded from patients presenting with focal epileptic disorders (see for review Ayala et al., 1973 [22]). Intracellular recordings from neurons located in these foci demonstrated that interictal events were associated with paroxysmal depolarizing shifts (PDSs) of the neuron membrane potential causing bursts of action potential firing followed by a hyperpolarization [23-28].
These findings were later confirmed in experiments performed in brain slices maintained in vitro during bath application of drugs interfering with GABAA receptor signaling Fig. (2). These studies demonstrated that interictal spikes: (i) reflect the enhancement of synaptic excitation that results from the weakening of inhibition, and (ii) rest on recurrent excitation and regenerative Ca2+ currents that make cortical principal cells fire synchronously [29-31]. Shortly after, Daniel Johnston and his collaborators firmly demonstrated that the PDSs associated with interictal spikes represent network-driven giant synaptic potentials [32]; moreover, results obtained in further experiments performed in this laboratory showed that the relative contribution of excitatory (glutamatergic) and inhibitory (GABAergic) currents to the PDS (and thus to the interictal spike) generation depends on the specific pharmacological manipulations employed to induce them [33-35] (see also section 3).
Fig. (2).
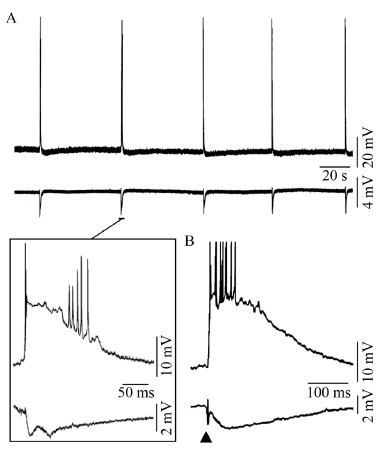
A: Spontaneous interictal-like events recorded in the perirhinal cortex in a rat brain slice maintained in vitro during application of picrotoxin. B: Interictal discharge recorded from the human neocortex in an in vitro brain slice preparation following a single-shock electrical stimulus (triangle) that was delivered through a stimulating electrode close to recording micropipettes. Upper and lower traces, in both A and B, correspond to intracellular and field potential recordings.
Ictal discharges, which are characterized by prolonged depolarizations of the neuronal membrane leading to the sustained firing of action potentials, were also reported to occur, but less frequently, in some of the original in vivo acute studies (cf., [25]). Moreover, ictal activity was recorded in vitro during the blockade of GABAA receptor signaling but only when immature brain slices (postnatal days 9 to 19) were employed [36]. As discussed below (section 3), ictal activity is often observed in adult brain tissue maintained in vitro during pharmacological procedures that do not solely interfere with GABAA receptor-mediated inhibition [5, 37]. In principle, this evidence suggests the contribution of ionotropic glutamatergic receptor-mediated inward (depolarizing) currents to both interictal and ictal discharge generation. Moreover, several studies have demonstrated the involvement of synaptic excitatory interactions in the generation of HFOs, and in particular those occurring at frequency higher than 200 Hz (the so called fast ripples) [38].
The availability of selective antagonists of ionotropic glutamate receptors in the 1980s led to the identification of the involvement of NMDA and non-NMDA - i.e., α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate - ionotropic glutamatergic receptor subtypes [39, 40] in both interictal and ictal discharges recorded from several cortical areas including the hippocampus, the subiculum, the rhinal, cingulate and insular cortices, the amygdala, and the neocortex [41-49] Fig. (3C)-(E). However, since ionotropic glutamate receptors are essential for normal brain activity and synaptic plasticity (i.e., in processes that underlie learning and memory) they cannot just be antagonized to achieve seizure control in epileptic patients. For instance, perampanel- which is the only approved antiepileptic drug with AMPA antagonism properties [50] - induces dizziness and motor impairment, perhaps due to inhibition of cerebellar AMPA receptors [51-53]. Therefore, recent attempts have been made to develop drugs that can block forebrain but not cerebellar AMPA receptors [54]. In addition, Olney et al. [55] have shown that NMDA receptor antagonism MK-801 could cause psychotomimetic and memory-impairing side effects, as well as neurotoxicity.
Fig. (3).
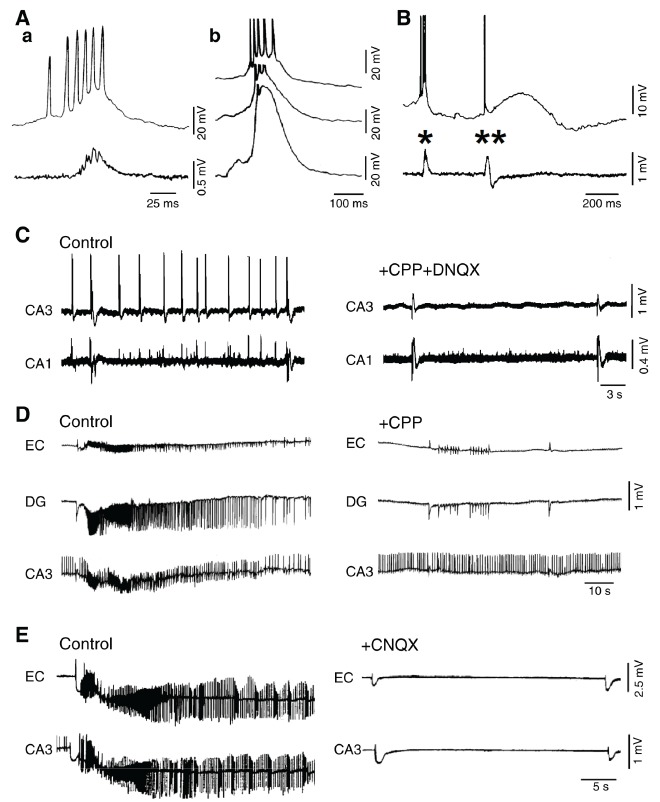
A: Spontaneous interictal-like epileptiform activity recorded in vitro from the CA3 area of a rat hippocampal slice during application of 4-aminopyridine. Simultaneous intracellular (upper trace) and field potential (lower trace) recordings are shown in a, while intracellular recordings performed at different membrane potential values (obtained by injecting steady intracellular current) are illustrated in b. B: Simultaneous intracellular (upper trace) and field potential (lower trace) recordings obtained from the CA3 area of a rat hippocampal slice during application of 4-aminopyridine; note that two types of interictal-like events occur: single asterisk identifies the ‘fast’ interictal spike (also depicted in panels Aa and Ab) while double asterisks point to the ‘slow’ interictal event that is characterized by a long-lasting depolarization. C: Effects induced by the ionotropic glutamate receptor antagonists (CPP and DNQX) on the spontaneous interictal-like epileptiform activity induced by 4-aminopyridine in a hippocampal brain slice during 4-aminopyridine application; note that blockade of glutamatergic ionotropic transmission abolishes only the “fast” interictal spikes. D and E: Effects induced by NMDA (CPP) and non-NMDA (CNQX) receptor antagonism on the ictal discharges recorded in an extended brain slice during application of 4-aminopyridine. Note that CPP abolishes the ictal activity without modifying the interictal discharges; in contrast, CNQX blocks both ictal and fast interictal discharges while the slow spikes continue to occur.
In spite of these limitations, several antiepileptic drugs can interfere with ionotropic glutamatergic functions. For instance, topiramate selectively inhibits kainate receptor-mediated excitatory postsynaptic responses in principal neurons of the rat basolateral amygdala [56], while protecting against seizures induced in vivo by 2-amino-3-(5-tert-butyl-3-hydroxy-4-isoxazolyl) propionic acid, which is a selective agonist of the GluR5 kainate receptor [57]. Braga et al. [58] reported that topiramate, by blocking presynaptic GluK1 kainate receptors, inhibits glutamate-mediated suppression of GABA release from interneurons, thus indirectly enhancing GABAA receptor-mediated inhibition. It has also been reported that the antiepileptic drug felbamate interacts with NMDA receptors in cultured rat hippocampal neurons [59]. Such action should reduce glutamate release by blocking
presynaptic NMDA receptors in the entorhinal cortex; this specific effect did not occur when phenytoin or gabapentin was used [60].
Recent studies have also focussed on the role of metabotropic glutamate receptors in seizure generation and epileptogenesis [61]. Activation of metabotropic group II and III receptors exerts anticonvulsant effects [62, 63]. In contrast, stimulation of metabotropic group 1 receptors in brain slices that were not generating spontaneous epileptiform activity under control conditions, elicits seizure-like discharges that continue to occur after washout of the agonist, and result from the activation of voltage-dependent cationic currents along with concomitant suppression of afterhyperpolarization [64-66]. These effects are similar to those reported to occur during activation of muscarinic receptors by the muscarinic agonist carbachol in subiculum and entorhinal cortex [67-69]. It has been indeed shown that the anti-epileptic drug topiramate can reduce these carbachol-induced excitatory effects [70]. To date, however, in spite of this evidence, little is known about the precise ability of antiepileptic drugs to target metabotropic glutamate receptors.
3. INHIBITORY SIGNALING
Inhibition in the brain results mainly from the activation of GABAA and GABAB receptors that are located pre-, post- and extra-synaptically on the membrane of principal cells and interneurons. GABAA receptors activate ionotropic anionic channels that are permeable to Cl- and HCO3-- while GABAB receptors act through second messengers leading to a K+ hyperpolarizing current [37, 71]. Early studies reported that interfering with GABA synthesis leads to convulsions [72] while treatment with exogenous GABA prevents seizures [73]. In addition, as mentioned in section 2, several convulsive drugs are GABAA receptor antagonists [29, 30], and inhibition is markedly reduced at the onset of electrographic seizures recorded in the hippocampus [74] or neocortex [75]. As originally reported by Sloviter [76], several studies have also suggested that decreased inhibition in an epileptic brain results from functional disconnection of interneurons from excitatory inputs (a.k.a., dormant interneuron hypothesis). In addition, decreased inhibition has been documented in mesial temporal lobe epilepsy and attributed to deficits in GABA transporter function [77] and/or alterations in GABAA receptor subunit composition [78-82]. Therefore, in the early 1990s, weakening of inhibition was considered by the majority of epilepsy researchers as the main mechanism leading to focal seizures and thus to epileptic disorders.
This view has been challenged by successive studies in which epileptiform activity was found to occur during pharmacological manipulations that do not interfere with GABAA receptor function and at times, paradoxically, enhance it. First, it was shown that epileptiform discharges induced by application of Mg2+ free-medium, were accompanied by preservation of inhibition [83, 84]. Second, it was found that cortical networks made hyperexcitable by blocking specific K+ currents generate two types of synchronous interictal spikes Fig. (3A), B) and that one of them continues to be generated even when AMPA and NMDA glutamatergic receptors were antagonized [85-87] Fig. (3C)-E). These interictal spikes were abolished by GABAA receptor antagonists and were associated with elevations in extracellular [K+] that depend on GABAA receptor activation [88-91]. GABAA receptor activation can indeed lead to accumulation of Cl- inside the postsynaptic neurons thus activating the co-transporter KCC2 that extrudes K+ and Cl- [92, 93]. In addition, GABAA receptor-mediated depolarizations in cortical neurons are contributed by HCO3−, an anion that goes through the activated GABAA receptor and has an equilibrium potential more positive than Cl− [94-96].
Around this time, several studies demonstrated that GABAA receptor activation can play a paradoxical role in initiating and maintaining focal seizure-like activity [88, 89, 97-105]. As illustrated in Fig. (4A)-C, pharmacological procedures that interfere with GABAA signaling, can abolish prolonged periods of epileptiform synchronization (which resemble ictal events) and replace them with a pattern of recurring, short-lasting interictal spikes. In addition, such ictal activity was potentiated by low doses of phenobarbital, a drug that enhances GABAA receptor function (Fig. 4D). The unexpected role played by GABAA receptors in initiating and maintaining focal seizures has been confirmed over the last two decades by several studies including those in which optogenetic activation of parvalbumin- or somatostatin-positive interneurons was reported to initiate ictal events similar to those occurring spontaneously [106, 107]. Interestingly, the role of GABAA signaling in promoting epileptiform synchronization is underscored by the ability of CA1 hippocampal networks to generate prolonged discharges following pharmacological blockade of both GABAB and ionotropic glutamatergic receptors [108]. Data obtained from models of mesial temporal lobe epilepsy also indicate that the onset of focal seizures is associated with increased activity of interneurons that release GABA thus silencing principal neurons [109-111]. In line with this evidence, the onset of seizures in epileptic patients undergoing presurgical electrophysiological investigations, is associated with marked reduction of unit firing [112, 113].
Fig. (4).
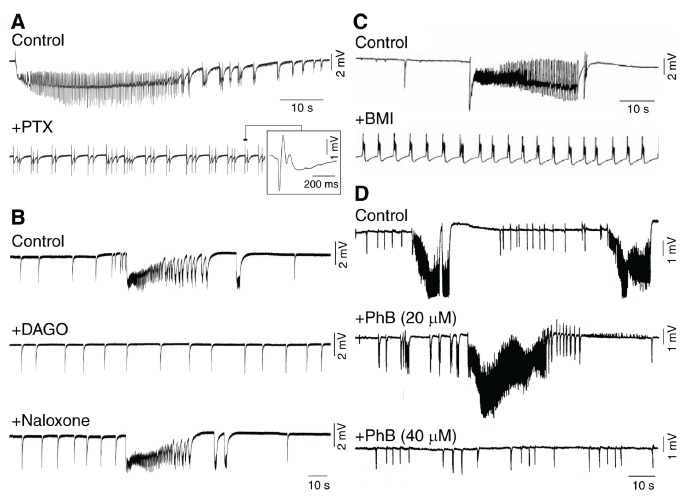
A and B: Effects induced by interfering with GABA receptor function on the epileptiform synchronization induced in vitro by 4-aminopyridine in the rodent piriform (A) or anterior cingulate cortex (B). Note that both antagonizing GABAA receptors with picrotoxin (PTX) or decreasing GABA release with the mu-opioid receptor agonist [d-Ala2,N-MePhe4-,Gly-ol5] enkephalin (DAGO) abolish ictal discharges; note also that the effects induced by DAGO are abolished by successive application of naloxone. C: Antagonizing GABAA receptors with bicuculline methiodide (BMI) blocks the occurrence of ictal discharges induced by 4-aminopyridine in slices of the human neocortex presenting with focal dysplasia. D: In these in vitro experiments, ictal discharges are increased in duration by low doses of phenobarbital (PhB, 20 µM), but abolished by higher concentrations of this barbiturate (PhB, 40 µM).
The evidence that increasing GABAA receptor function promotes epileptiform synchronization and thus focal seizure activity may explain the disappointingly limited efficacy of several antiepileptic drugs that were developed in the 1990s to potentiate GABAA signaling, to control seizures in epileptic patients. These compounds include γ-vinyl-GABA (which inhibits the breakdown of GABA by the enzyme GABA transaminase) [114], tiagabine (which increases GABA levels by inhibiting GABA reuptake) [115, 116] and progabide [117, 118]. Gabapentin, which is similar in structure to GABA but does not interact with GABA receptors [119], has also shown limited efficacy in controlling seizures. On the other hand, benzodiazepines can halt seizure activity and stop status epilepticus [120]. These compounds increase GABAA receptor function in the central nervous system by acting on an allosteric ‘benzodiazepine site’ located on most α subunit-containing GABAA receptors [121-123].
The roles played by metabotropic GABAB receptors remain to some extent unclear. Their activation, obtained by bath applying baclofen, promotes ictal-like discharges in hippocampal slices during exposure to 4-aminopyridine [124, 125]; but see also Mott et al [126]. Presumably, these effects depend on activity-dependent changes in hippocampal network excitability along with the weakening of GABAA receptor signaling [125, 126]. However, successive experiments demonstrated that blocking GABAB receptors, at least in the 4-aminopyridine model, did not consistently reduce epileptiform synchronization [127].
4. NON-SYNAPTIC MECHANISMS
Voltage-gated Na+ currents, which play a role in action potential firing and in generating persistent depolarizing currents, are by definition non-synaptic although they are triggered and sustained by synaptic depolarizations [128-130]. The repetitive firing of action potentials is a consistent feature of epileptiform discharges Fig. (5A), and therefore the activity of voltage-gated Na+ channel is relevant in the pathophysiology of both generalized and focal epileptic disorders [131, 132]. A significant increase in persistent Na+ currents occurs in models of mesial temporal lobe epilepsy [133, 134] and in neurons recorded from tissue resected from temporal lobe epileptic patients [135]. Findings obtained from transgenic mice have also indicated that an increase in persistent Na+ current is sufficient to cause chronic seizures [133, 135-137].
Fig. (5).
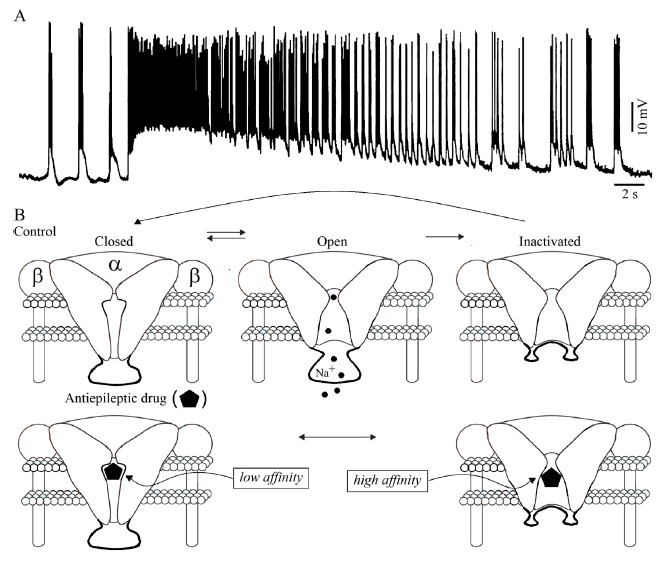
A: Intracellular activity generated by a presumptive principal cell in the rodent perirhinal cortex maintained in vitro during application of 4-minopyridine; note that the ictal activity is associated with sustained discharge of ‘fast’, Na+ dependent action potentials. B: Functional states of the voltage-gated Na+ channel under control conditions and in the presence of an antiepileptic drug such as phenytoin or lamotrigine. Na+ channels are in resting states at hyperpolarized membrane potentials. In response to membrane depolarization, they open and then inactivate. Antiepileptic Na+ channel blockers bind with low affinity to resting states but with higher affinity to inactivated states. This state-dependent action causes the voltage and activity-dependent inhibition that enables these drugs to be effective antiepileptic agents.
As illustrated in Fig. (5B), voltage-gated Na+ channels comprise a central α-subunit and two auxiliary β-subunits; these channels: (i) are in a closed state at hyperpolarized (around resting) membrane potentials; (ii) briefly open when the membrane is depolarized thus producing an inward Na+ current causing the ascending component of the action potential; and then (iii) convert to a non-conducting inactivated state [128]. Attenuation of voltage-gated Na+ current is induced, at therapeutic concentrations, by several antiepileptic drugs that are effective for treating epileptic patients presenting with focal and secondarily generalized seizures. Phenytoin and carbamazepine are the prototypic drugs exerting such an effect but several other antiepileptic compounds-which include valproate, lamotrigine, topiramate and lacosamide- may also act on this target (see for review [131, 138]).
As detailed in Fig. (5B), these antiepileptic drugs are weak blockers of voltage-gated Na+ channels when the neuron is at resting membrane potential but their action is greatly enhanced during sustained membrane depolarization or high-frequency channel activity, which are both occurring during seizure activity. These functional characteristics mirror the modulated receptor model that was developed to describe the mechanism of action of local anesthetics [128]. According to this model, the closed channels, which predominate at resting membrane potential, have a low affinity for these antiepileptic drugs while the inactivated channels, which are predominant at depolarized potentials, bind them with high affinity [114, 128, 139]. These characteristics explain why these antiepileptic drugs suppress seizures while having minimum effects on normal brain functions, i.e., when brain neuronal networks are normally functioning and thus not undergoing sustained prolonged states of depolarization.
Besides Na+, other voltage-gated channels can be the target of antiepileptic drugs. Accordingly, it has been reported that lamotrigine increases the hyperpolarization-activated cation current, also termed Ih, that is largely present in the dendrites of pyramidal cells of the hippocampus [140] (see for review Poolos et al., 2012 [141]). A similar effect has been identified with the antiepileptic drug gabapentin [142]. Lamotrigine has also been shown to inhibit N-type and P-type high-voltage-activated Ca2+ channels [143] that are instrumental in both excitatory and inhibitory synaptic transmission [144]. In keeping with the role of synaptic neurotransmitter release in the generation of seizure activity, a rather new and efficacious antiepileptic drug, levetiracetam, has been proposed to modulate presynaptic Ca2+ channel activity by acting on the synaptic vesicle glycoprotein SV2A, which interacts with synaptotagmin and results in neurotransmitter release modulation [145, 146]. Levitiracetam has anti-ictogenic effects in animal models of epilepsy such as in the amygdala kindling model [147], audiogenic kindling [148], spontaneously epileptic rats [149], the kainic acid model [150] and the pilocarpine model [151]. Since alterations in Ca2+ are correlated to epileptiform discharges, the antagonistic effects of levetiracetam and lamotrigine on Ca2+ signaling might represent the basis for their anticonvulsant efficacy. Beyond that, regulation of Ca2+ homeostasis could also preserve neuronal viability [152], and also lacosamide can provide protection against excitotoxicity without altering synaptic plasticity [153]. Lamotrigine as well as carbamazepine have also been reported to increase voltage-gated K+ outward (repolarizing) currents [154, 155].
Epileptiform synchronization also depends on non-synaptic interactions such as intercellular gap junctions, ephaptic interactions, and - as discussed above - elevations in extracellular [K+] (see for review Jefferys et al. [4]). Indeed, non-synaptic synchronizing mechanisms may play important roles in contributing to seizure activity, which is associated with decreases in extracellular [Ca2+] that are incompatible with fully efficient synaptic transmission [7, 156]. Gap junctions, which are constituted by connexins and pannexins [157, 158], may play significant roles in synchronizing neuronal networks under physiological and pathological conditions including epileptic disorders [159-162]. For instance, Draguhn et al. [163] have reported that spontaneous high-frequency activity at approximately 200 Hz (ripples) in the CA1 region of the hippocampus in vitro is generated by principal cell networks that are synchronized trough gap junctions. In vivo, in transgenic Connexin36 deficient mice, a reduction in the frequency of occurrence of ripples and epileptiform discharges induced with 4-aminopyridine was observed by the same group [164]. Oscillations at higher frequencies (> 250 Hz) are also reduced under carbenoxolone in hippocampal slices from epileptic patients [165] and in vivo in pilocarpine-treated mice [166]. Pharmacological procedures that block or enhance the function of gap junctions could therefore lead to a decrease or an increase of epileptiform synchronization, respectively (see for review Mylvaganam et al., 2014 [162]; Carlen et al., 2000 [167]). It has also been established that the astrocytic connexin 43 is elevated in human epileptic tissue [168, 169] while experiments performed in human neocortical tissue from patients with focal cortical dysplasia have shown that gap junction blockers reduce ictal-like events induced by 4-aminopyridine [170]. Although gap junction blockers depress synaptic and non-synaptic currents thus producing nonspecific effects (cf., [171, 172]), gap junctions remain an appropriate target for the development of antiepileptic drugs.
CONCLUDING REMARKS
We have presented here a short review of the studies addressing the fundamental cellular and pharmacological mechanisms that lead to the initiation, maintenance and spread of focal epileptiform discharges. We have highlighted the fact that while excitation and voltage-gated sodium are a main components of the synchronous firing that characterize an epileptiform event, GABAA receptor signaling emerges as a surprising, paradoxical mechanism that actively contributes to the initiation and maintenance of prolonged epileptiform phenomena (i.e., to ictogenesis). In addition, while voltage-gated sodium channels remain the best studied target for antiepileptic drugs, recent data obtained with levetiracetam suggest that its anti-ictogenic properties might be associated with the modulation of both excitatory and inhibitory transmission trough changes in calcium dynamics. Non-synaptic mechanisms, such as the activity of gap junctions, might also underlie the generation of epileptiform synchronization. Altogether, these findings-which have been obtained in vivo and in vitro over the last six decades - reveal a complex pattern of participating mechanisms to ictogenesis.
ACKNOWLEDGEMENTS
We acknowledge the participation of Drs. M. D’Antuono, P. de Guzman, G. Hwa, and P. Perreault, to the experiments from which the figures shown in this review originate; these experiments were performed at the MNI between 1987 and 2007. We also thank Dr. M. Mantegazza for providing inspiration for Fig. 5.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This review was also based on experiments supported by the Canadian Institutes of Health Research (CIHR grants 8109 and 74609), CURE and the Savoy Foundation.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Niedermeyer E., da Silva F.H.L. Electroencephalography: Basic Principles, Clinical Applications, and Related Fields. Lippincott Williams & Wilkins; 2005. [Google Scholar]
- 2.Steriade M., Gloor P., Llinás R.R., Lopes de Silva F.H., Mesulam M.M. Report of IFCN committee on basic Mechanisms. Basic mechanisms of cerebral rhythmic activities. Electroencephalogr. Clin. Neurophysiol. 1990;76(6):481–508. doi: 10.1016/0013-4694(90)90001-z. [http://dx.doi.org/10. 1016/0013-4694(90)90001-Z]. [PMID: 1701118]. [DOI] [PubMed] [Google Scholar]
- 3.Buzsáki G. Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus. 2015;25(10):1073–1188. doi: 10.1002/hipo.22488. [http://dx.doi.org/10.1002/hipo.22488]. [PMID: 26135716]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jefferys J.G.R., Jiruska P., de Curtis M., Avoli M. 2012. Limbic network Synchronization and temporal lobe epilepsy. [PubMed] [Google Scholar]
- 5.Avoli M., de Curtis M., Gnatkovsky V., Gotman J., Köhling R., Lévesque M., Manseau F., Shiri Z., Williams S. Specific imbalance of excitatory/inhibitory signaling establishes seizure onset pattern in temporal lobe epilepsy. J. Neurophysiol. 2016;115(6):3229–3237. doi: 10.1152/jn.01128.2015. [http://dx.doi.org/10.1152/jn.01128.2015]. [PMID: 27075542]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gloor P., Fariello R.G. Generalized epilepsy: some of its cellular mechanisms differ from those of focal epilepsy. Trends Neurosci. 1988;11(2):63–68. doi: 10.1016/0166-2236(88)90166-x. [http://dx.doi.org/10.1016/0166-2236(88)90166-X]. [PMID: 2465601]. [DOI] [PubMed] [Google Scholar]
- 7.Timofeev I., Steriade M. Neocortical seizures: initiation, development and cessation. Neuroscience. 2004;123(2):299–336. doi: 10.1016/j.neuroscience.2003.08.051. [http://dx.doi.org/10.1016/j.neuroscience.2003.08.051]. [PMID: 14698741]. [DOI] [PubMed] [Google Scholar]
- 8.Beenhakker M.P., Huguenard J.R. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62(5):612–632. doi: 10.1016/j.neuron.2009.05.015. [http://dx.doi.org/10.1016/ j.neuron.2009.05.015]. [PMID: 19524522]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crunelli V., Leresche N., Cope D.W. 2012. GABA-A Receptor function in typical absence seizures. [PubMed] [Google Scholar]
- 10.Engel J., Jr Introduction to temporal lobe epilepsy. Epilepsy Res. 1996;26(1):141–150. doi: 10.1016/s0920-1211(96)00043-5. [http://dx.doi.org/10.1016/S0920-1211(96) 00043-5]. [PMID: 8985696]. [DOI] [PubMed] [Google Scholar]
- 11.Gloor P. The Temporal Lobe and Limbic System. USA: Oxford University Press; 1997. [Google Scholar]
- 12.Engel J., Jr, McDermott M.P., Wiebe S., Langfitt J.T., Stern J.M., Dewar S., Sperling M.R., Gardiner I., Erba G., Fried I., Jacobs M., Vinters H.V., Mintzer S., Kieburtz K. Early surgical therapy for drug-resistant temporal lobe epilepsy: A randomized trial. JAMA. 2012;307(9):922–930. doi: 10.1001/jama.2012.220. [http://dx.doi.org/10.1001/ jama.2012.220]. [PMID: 22396514]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buzsáki G., Silva F.L. High frequency oscillations in the intact brain. Prog. Neurobiol. 2012;98(3):241–249. doi: 10.1016/j.pneurobio.2012.02.004. [http://dx.doi.org/ 10.1016/j.pneurobio.2012.02.004]. [PMID: 22449727]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bragin A., Engel J., Jr, Wilson C.L., Fried I., Buzsáki G. High-frequency oscillations in human brain. Hippocampus. 1999;9(2):137–142. doi: 10.1002/(SICI)1098-1063(1999)9:2<137::AID-HIPO5>3.0.CO;2-0. [http://dx.doi.org/10.1002/(SICI)1098-1063(1999)9:2 <137:AID-HIPO5>3.0.CO;2-0]. [DOI] [PubMed] [Google Scholar]
- 15.Staba R.J., Wilson C.L., Bragin A., Jhung D., Fried I., Engel J., Jr High-frequency oscillations recorded in human medial temporal lobe during sleep. Ann. Neurol. 2004;56(1):108–115. doi: 10.1002/ana.20164. [http://dx. doi.org/10.1002/ana.20164]. [PMID: 15236407]. [DOI] [PubMed] [Google Scholar]
- 16.Bragin A., Engel J., Jr, Wilson C.L., Fried I., Mathern G.W. Hippocampal and entorhinal cortex high-frequency oscillations (100--500 Hz) in human epileptic brain and in kainic acid--treated rats with chronic seizures. Epilepsia. 1999;40(2):127–137. doi: 10.1111/j.1528-1157.1999.tb02065.x. [PMID: 9952257]. [http://dx.doi.org/10.1111/j.1528-1157.1999.tb02065.x]. [DOI] [PubMed] [Google Scholar]
- 17.Lévesque M., Bortel A., Gotman J., Avoli M. High-frequency (80-500 Hz) oscillations and epileptogenesis in temporal lobe epilepsy. Neurobiol. Dis. 2011;42(3):231–241. doi: 10.1016/j.nbd.2011.01.007. [http://dx.doi.org/ 10.1016/j.nbd.2011.01.007]. [PMID: 21238589]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lévesque M., Salami P., Gotman J., Avoli M. Two seizure-onset types reveal specific patterns of high-frequency oscillations in a model of temporal lobe epilepsy. J. Neurosci. 2012;32(38):13264–13272. doi: 10.1523/JNEUROSCI.5086-11.2012. [http://dx.doi.org/10.1523/JNEUROSCI.5086-11.2012]. [PMID: 22993442]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jirsch J.D., Urrestarazu E., LeVan P., Olivier A., Dubeau F., Gotman J. High-frequency oscillations during human focal seizures. Brain. 2006;129(Pt 6):1593–1608. doi: 10.1093/brain/awl085. [http://dx.doi.org/10. 1093/brain/awl085]. [DOI] [PubMed] [Google Scholar]
- 20.Urrestarazu E., Jirsch J.D., LeVan P., Hall J., Avoli M., Dubeau F., Gotman J. High-frequency intracerebral EEG activity (100-500 Hz) following interictal spikes. Epilepsia. 2006;47(9):1465–1476. doi: 10.1111/j.1528-1167.2006.00618.x. [http://dx.doi.org/10.1111/j.1528-1167.2006.00618.x]. [PMID: 16981862]. [DOI] [PubMed] [Google Scholar]
- 21.Zijlmans M., Jacobs J., Kahn Y.U., Zelmann R., Dubeau F., Gotman J. Ictal and interictal high frequency oscillations in patients with focal epilepsy. Clin. Neurophysiol. 2011;122(4):664–671. doi: 10.1016/j.clinph.2010.09.021. [http://dx.doi.org/10.1016/j.clinph.2010.09.021]. [PMID: 21030302]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ayala G.F., Dichter M., Gumnit R.J., Matsumoto H., Spencer W.A. Genesis of epileptic interictal spikes. New knowledge of cortical feedback systems suggests a neurophysiological explanation of brief paroxysms. Brain Res. 1973;52:1–17. doi: 10.1016/0006-8993(73)90647-1. [http://dx. doi.org/10.1016/0006-8993(73)90647-1]. [PMID: 4573428]. [DOI] [PubMed] [Google Scholar]
- 23.Li C.L. Cortical intracellular potentials and their responses to strychnine. J. Neurophysiol. 1959;22(4):436–450. doi: 10.1152/jn.1959.22.4.436. [http://dx. doi.org/10.1152/jn.1959.22.4.436]. [PMID: 13673295]. [DOI] [PubMed] [Google Scholar]
- 24.Goldensohn E.S., Purpura D.P. Intracellular potentials of cortical neurons during focal epileptogenic discharges. Science. 1963;139(3557):840–842. doi: 10.1126/science.139.3557.840. [http://dx.doi.org/10.1126/science.139.3557. 840]. [PMID: 13948714]. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto H., Marsan C.A. Cortical cellular phenomena in experimental epilepsy: Interictal manifestations. Exp. Neurol. 1964;9:286–304. doi: 10.1016/0014-4886(64)90025-1. [http://dx.doi.org/10.1016/0014-4886(64)90025-1]. [PMID: 14145629]. [DOI] [PubMed] [Google Scholar]
- 26.Prince D.A. Inhibition in “epileptic” neurons. Exp. Neurol. 1968;21(3):307–321. doi: 10.1016/0014-4886(68)90043-5. [http://dx.doi.org/10.1016/0014-4886(68)90043-5]. [PMID: 5673646]. [DOI] [PubMed] [Google Scholar]
- 27.Dichter M., Spencer W.A. Penicillin-induced interictal discharges from the cat hippocampus. I. Characteristics and topographical features. J. Neurophysiol. 1969;32(5):649–662. doi: 10.1152/jn.1969.32.5.649. [http://dx.doi.org/ 10.1152/jn.1969.32.5.649]. [PMID: 4309021]. [DOI] [PubMed] [Google Scholar]
- 28.Dichter M., Spencer W.A. Penicillin-induced interictal discharges from the cat hippocampus. II. Mechanisms underlying origin and restriction. J. Neurophysiol. 1969;32(5):663–687. doi: 10.1152/jn.1969.32.5.663. [http://dx.doi. org/10.1152/jn.1969.32.5.663]. [PMID: 4309022]. [DOI] [PubMed] [Google Scholar]
- 29.Dingledine R., Gjerstad L. Reduced inhibition during epileptiform activity in the in vitro hippocampal slice. J. Physiol. 1980;305:297–313. doi: 10.1113/jphysiol.1980.sp013364. [http://dx.doi.org/10.1113/jphysiol.1980.sp013364]. [PMID: 7441555]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartzkroin P.A., Prince D.A. Changes in excitatory and inhibitory synaptic potentials leading to epileptogenic activity. Brain Res. 1980;183(1):61–76. doi: 10.1016/0006-8993(80)90119-5. [http://dx.doi.org/10.1016/0006-8993(80)90119-5]. [PMID: 6244050]. [DOI] [PubMed] [Google Scholar]
- 31.Traub R.D., Wong R.K. Cellular mechanism of neuronal synchronization in epilepsy. Science. 1982;216(4547):745–747. doi: 10.1126/science.7079735. [PMID: 7079735]. [http://dx.doi.org/10.1126/science.7079735]. [DOI] [PubMed] [Google Scholar]
- 32.Johnston D., Brown T.H. Giant synaptic potential hypothesis for epileptiform activity. Science. 1981;211(4479):294–297. doi: 10.1126/science.7444469. [http://dx.doi.org/10.1126/science.7444469]. [PMID: 7444469]. [DOI] [PubMed] [Google Scholar]
- 33.Rutecki P.A., Lebeda F.J., Johnston D. Epileptiform activity induced by changes in extracellular potassium in hippocampus. J. Neurophysiol. 1985;54(5):1363–1374. doi: 10.1152/jn.1985.54.5.1363. [http://dx.doi.org/10. 1152/jn.1985.54.5.1363]. [PMID: 2416891]. [DOI] [PubMed] [Google Scholar]
- 34.Rutecki P.A., Lebeda F.J., Johnston D. 4-Aminopyridine produces epileptiform activity in hippocampus and enhances synaptic excitation and inhibition. J. Neurophysiol. 1987;57(6):1911–1924. doi: 10.1152/jn.1987.57.6.1911. [http://dx.doi.org/10.1152/jn.1987.57.6.1911]. [PMID: 3037040]. [DOI] [PubMed] [Google Scholar]
- 35.Rutecki P.A., Lebeda F.J., Johnston D. Epileptiform activity in the hippocampus produced by tetra ethyl ammonium. J. Neurophysiol. 1990;64(4):1077–1088. doi: 10.1152/jn.1990.64.4.1077. [http://dx.doi.org/10.1152/jn. 1990.64.4.1077]. [PMID: 2258736]. [DOI] [PubMed] [Google Scholar]
- 36.Swann J.W., Brady R.J. Penicillin-induced epileptogenesis in immature rat CA3 hippocampal pyramidal cells. Brain Res. 1984;314(2):243–254. doi: 10.1016/0165-3806(84)90046-4. [http://dx.doi.org/10.1016/0165-3806(84)90046-4]. [PMID: 6704751]. [DOI] [PubMed] [Google Scholar]
- 37.Avoli M., de Curtis M. GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog. Neurobiol. 2011;95(2):104–132. doi: 10.1016/j.pneurobio.2011.07.003. [http://dx.doi.org/10.1016/j. pneurobio.2011.07.003]. [PMID: 21802488]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dzhala V.I., Staley K.J. Mechanisms of fast ripples in the hippocampus. J. Neurosci. 2004;24(40):8896–8906. doi: 10.1523/JNEUROSCI.3112-04.2004. [http://dx.doi.org/ 10.1523/JNEUROSCI.3112-04.2004]. [PMID: 15470156]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collingridge G. L., Lodge D., Mayer M., Turrigiano G., Frenguelli B. G. Ionotropic glutamate receptors: Still exciting after all these years. Neuropharmacology. 2017;112(Pt A):1–3. doi: 10.1016/j.neuropharm.2016.09.019. [DOI] [PubMed] [Google Scholar]
- 40.Zhu S., Gouaux E. Structure and symmetry inform gating principles of ionotropic glutamate receptors. Neuropharmacology. 2017;112(Pt A):11–15. doi: 10.1016/j.neuropharm.2016.08.034. [http://dx.doi.org/10.1016/j.neuropharm.2016.08. 034] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huberfeld G., Menendez de la Prida L., Pallud J., Cohen I., Le Van Q.M., Adam C., Clemenceau S., Baulac M., Miles R. Glutamatergic pre-ictal discharges emerge at the transition to seizure in human epilepsy. Nat. Neurosci. 2011;14(5):627–634. doi: 10.1038/nn.2790. [http://dx.doi.org/10.1038/nn.2790]. [PMID: 21460834]. [DOI] [PubMed] [Google Scholar]
- 42.Doherty J., Dingledine R. The roles of metabotropic glutamate receptors in seizures and epilepsy. Curr. Drug Targets CNS Neurol. Disord. 2002;1(3):251–260. doi: 10.2174/1568007023339355. [http://dx.doi.org/10.2174/ 1568007023339355]. [PMID: 12769618]. [DOI] [PubMed] [Google Scholar]
- 43.Wong R.K., Bianchi R., Taylor G.W., Merlin L.R. Role of metabotropic glutamate receptors in epilepsy. Adv. Neurol. 1999;79:685–698. [PMID: 10514855]. [PubMed] [Google Scholar]
- 44.Jones R.S., Heinemann U. Synaptic and intrinsic responses of medical entorhinal cortical cells in normal and magnesium-free medium in vitro. J. Neurophysiol. 1988;59(5):1476–1496. doi: 10.1152/jn.1988.59.5.1476. [http://dx.doi.org/10.1152/jn.1988.59.5.1476]. [PMID: 2898511]. [DOI] [PubMed] [Google Scholar]
- 45.Hwa G.G., Avoli M. The involvement of excitatory amino acids in neocortical epileptogenesis: NMDA and non-NMDA receptors. Exp. Brain Res. 1991;86(2):248–256. doi: 10.1007/BF00228949. [http://dx.doi.org/10.1007/ BF00228949]. [PMID: 1684548]. [DOI] [PubMed] [Google Scholar]
- 46.Benini R., D’Antuono M., Pralong E., Avoli M. Involvement of amygdala networks in epileptiform synchronization in vitro. Neuroscience. 2003;120(1):75–84. doi: 10.1016/s0306-4522(03)00262-8. [http://dx.doi.org/10.1016/S0306-4522(03)00262-8]. [PMID: 12849742]. [DOI] [PubMed] [Google Scholar]
- 47.Sudbury J.R., Avoli M. Epileptiform synchronization in the rat insular and perirhinal cortices in vitro. Eur. J. Neurosci. 2007;26(12):3571–3582. doi: 10.1111/j.1460-9568.2007.05962.x. [http://dx.doi.org/10.1111/j.1460-9568.2007. 05962.x]. [PMID: 18052975]. [DOI] [PubMed] [Google Scholar]
- 48.Panuccio G., Curia G., Colosimo A., Cruccu G., Avoli M. Epileptiform synchronization in the cingulate cortex. Epilepsia. 2009;50(3):521–536. doi: 10.1111/j.1528-1167.2008.01779.x. [http://dx.doi.org/10.1111/j.1528-1167.2008.01779. x]. [PMID: 19178556]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panuccio G., Sanchez G., Lévesque M., Salami P., de Curtis M., Avoli M. On the ictogenic properties of the piriform cortex in vitro. Epilepsia. 2012;53(3):459–468. doi: 10.1111/j.1528-1167.2012.03408.x. [http://dx.doi.org/10.1111/ j.1528-1167.2012.03408.x]. [PMID: 22372627]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Witkin J.M., Li J., Gilmour G., Mitchell S.N., Carter G., Gleason S.D., Seidel W.F., Eastwood B.J., McCarthy A., Porter W.J., Reel J., Gardinier K.M., Kato A.S., Wafford K.A. Electroencephalographic, cognitive, and neurochemical effects of LY3130481 (CERC-611), a selective antagonist of TARP-γ8-associated AMPA receptors. Neuropharmacology. 2017;126:257–270. doi: 10.1016/j.neuropharm.2017.07.028. [http://dx.doi.org/10.1016/j.neuropharm.2017.07.028]. [PMID: 28757050]. [DOI] [PubMed] [Google Scholar]
- 51.Zhuo C., Jiang R., Li G., Shao M., Chen C., Chen G., Tian H., Li J., Xue R., Jiang D. Efficacy and tolerability of second and third generation anti-epileptic drugs in refractory epilepsy: A network meta-analysis. Sci. Rep. 2017;7(1):2535. doi: 10.1038/s41598-017-02525-2. [http://dx. doi.org/10.1038/s41598-017-02525-2]. [PMID: 28566726]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanada T., Hashizume Y., Tokuhara N., Takenaka O., Kohmura N., Ogasawara A., Hatakeyama S., Ohgoh M., Ueno M., Nishizawa Y. Perampanel: a novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia. 2011;52(7):1331–1340. doi: 10.1111/j.1528-1167.2011.03109.x. [http://dx.doi.org/10.1111/j.1528-1167.2011.03109.x]. [PMID: 21635236]. [DOI] [PubMed] [Google Scholar]
- 53.Rogawski M.A., Hanada T. Preclinical pharmacology of perampanel, a selective non-competitive AMPA receptor antagonist. Acta Neurol. Scand. Suppl. 2013;(197):19–24. doi: 10.1111/ane.12100. [http://dx.doi.org/10. 1111/ane.12100]. [PMID: 23480152]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kato A.S., Burris K.D., Gardinier K.M., Gernert D.L., Porter W.J., Reel J., Ding C., Tu Y., Schober D.A., Lee M.R., Heinz B.A., Fitch T.E., Gleason S.D., Catlow J.T., Yu H., Fitzjohn S.M., Pasqui F., Wang H., Qian Y., Sher E., Zwart R., Wafford K.A., Rasmussen K., Ornstein P.L., Isaac J.T., Nisenbaum E.S., Bredt D.S., Witkin J.M. Forebrain-selective AMPA-receptor antagonism guided by TARP γ-8 as an antiepileptic mechanism. Nat. Med. 2016;22(12):1496–1501. doi: 10.1038/nm.4221. [http://dx.doi.org/10.1038/nm.4221]. [PMID: 27820603]. [DOI] [PubMed] [Google Scholar]
- 55.Olney J.W., Labruyere J., Wang G., Wozniak D.F., Price M.T., Sesma M.A. NMDA antagonist neurotoxicity: Mechanism and prevention. Science. 1991;254(5037):1515–1518. doi: 10.1126/science.1835799. [PMID: 1835799]. [http://dx.doi.org/10.1126/science.1835799]. [DOI] [PubMed] [Google Scholar]
- 56.Gryder D.S., Rogawski M.A. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J. Neurosci. 2003;23(18):7069–7074. doi: 10.1523/JNEUROSCI.23-18-07069.2003. [PMID: 12904467]. [http://dx.doi.org/10.1523/ JNEUROSCI.23-18-07069.2003]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaminski R.M., Banerjee M., Rogawski M.A. Topiramate selectively protects against seizures induced by ATPA, a GluR5 kainate receptor agonist. Neuropharmacology. 2004;46(8):1097–1104. doi: 10.1016/j.neuropharm.2004.02.010. [http://dx.doi.org/10.1016/j.neuropharm.2004.02.010]. [PMID: 15111016]. [DOI] [PubMed] [Google Scholar]
- 58.Braga M.F.M., Aroniadou-Anderjaska V., Li H., Rogawski M.A. Topiramate reduces excitability in the basolateral amygdala by selectively inhibiting GluK1 (GluR5) kainate receptors on interneurons and positively modulating GABAA receptors on principal neurons. J. Pharmacol. Exp. Ther. 2009;330(2):558–566. doi: 10.1124/jpet.109.153908. [http:// dx.doi.org/10.1124/jpet.109.153908]. [PMID: 19417176]. [DOI] [PubMed] [Google Scholar]
- 59.Subramaniam S., Rho J.M., Penix L., Donevan S.D., Fielding R.P., Rogawski M.A. Felbamate block of the N-methyl-D-aspartate receptor. J. Pharmacol. Exp. Ther. 1995;273(2):878–886. [PMID: 7752093]. [PubMed] [Google Scholar]
- 60.Yang J., Wetterstrand C., Jones R.S.G. Felbamate but not phenytoin or gabapentin reduces glutamate release by blocking presynaptic NMDA receptors in the entorhinal cortex. Epilepsy Res. 2007;77(2-3):157–164. doi: 10.1016/j.eplepsyres.2007.09.005. [http://dx.doi.org/10.1016/j.eplepsyres.2007.09. 005]. [PMID: 17980555]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bianchi R., Wong R.K.S., Merlin L.R. Glutamate receptors in epilepsy: Group I mGluR-mediated epileptogenesis. Jasper’s Basic mechanisms of the epilepsies. 2012 [PubMed] [Google Scholar]
- 62.Ghauri M., Chapman A.G., Meldrum B.S. Convulsant and anticonvulsant actions of agonists and antagonists of group III mGluRs. Neuroreport. 1996;7(9):1469–1474. doi: 10.1097/00001756-199606170-00005. [http://dx.doi.org/ 10.1097/00001756-199606170-00005]. [PMID: 8856700]. [DOI] [PubMed] [Google Scholar]
- 63.Kłodzińska A., Chojnacka-Wójcik E., Pilc A. Selective group II glutamate metabotropic receptor agonist LY354740 attenuates pentetrazole- and picrotoxin-induced seizures. Pol. J. Pharmacol. 1999;51(6):543–545. [PMID: 10817535]. [PubMed] [Google Scholar]
- 64.Wong R.K.S., Chuang S-C., Bianchi R. Metabotropic glutamate receptors and epileptogenesis. Epilepsy Curr. 2002;2(3):81–85. doi: 10.1046/j.1535-7597.2002.00031.x. [http://dx.doi.org/10.1046/j.1535-7597.2002.00031.x]. [PMID: 15309152]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park J-Y., Remy S., Varela J., Cooper D.C., Chung S., Kang H-W., Lee J-H., Spruston N. A post-burst after depolarization is mediated by group i metabotropic glutamate receptor-dependent upregulation of Ca(v)2.3 R-type calcium channels in CA1 pyramidal neurons. PLoS Biol. 2010;8(11):e1000534. doi: 10.1371/journal.pbio.1000534. [http://dx. doi.org/10.1371/journal.pbio.1000534]. [PMID: 21103408]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Congar P., Leinekugel X., Ben-Ari Y., Crépel V. A long-lasting calcium-activated nonselective cationic current is generated by synaptic stimulation or exogenous activation of group I metabotropic glutamate receptors in CA1 pyramidal neurons. J. Neurosci. 1997;17(14):5366–5379. doi: 10.1523/JNEUROSCI.17-14-05366.1997. [http://dx.doi.org/10.1523/ JNEUROSCI.17-14-05366.1997]. [PMID: 9204921]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kawasaki H., Palmieri C., Avoli M. Muscarinic receptor activation induces depolarizing plateau potentials in bursting neurons of the rat subiculum. J. Neurophysiol. 1999;82(5):2590–2601. doi: 10.1152/jn.1999.82.5.2590. [http://dx.doi.org/10.1152/jn.1999.82.5.2590]. [PMID: 10561429]. [DOI] [PubMed] [Google Scholar]
- 68.Klink R., Alonso A. Ionic mechanisms of muscarinic depolarization in entorhinal cortex layer II neurons. J. Neurophysiol. 1997;77(4):1829–1843. doi: 10.1152/jn.1997.77.4.1829. [http://dx.doi.org/10.1152/jn.1997.77.4.1829]. [PMID: 9114239]. [DOI] [PubMed] [Google Scholar]
- 69.Klink R., Alonso A. Muscarinic modulation of the oscillatory and repetitive firing properties of entorhinal cortex layer II neurons. J. Neurophysiol. 1997;77(4):1813–1828. doi: 10.1152/jn.1997.77.4.1813. [http://dx.doi.org/10.1152/ jn.1997.77.4.1813]. [PMID: 9114238]. [DOI] [PubMed] [Google Scholar]
- 70.D’Antuono M., Kawasaki H., Palmieri C., Curia G., Biagini G., Avoli M. Antiepileptic drugs and muscarinic receptor-dependent excitation in the rat subiculum. Neuropharmacology. 2007;52(5):1291–1302. doi: 10.1016/j.neuropharm.2007.01.008. [http://dx.doi.org/10.1016/j.neuropharm.2007.01.008]. [PMID: 17337018]. [DOI] [PubMed] [Google Scholar]
- 71.Avoli M., Krnjević K. The long and winding road to gamma-Amino-Butyric acid as neurotransmitter. Can. J. Neurol. Sci. 2016;43(2):219–226. doi: 10.1017/cjn.2015.333. [http://dx.doi.org/10.1017/cjn.2015.333]. [PMID: 26763167]. [DOI] [PubMed] [Google Scholar]
- 72.Coursin D.B. Convulsive seizures in infants with pyridoxine-deficient diet. J. Am. Med. Assoc. 1954;154(5):406–408. doi: 10.1001/jama.1954.02940390030009. [PMID: 13117629]. [http://dx.doi.org/10.1001/jama.1954.02940390030009]. [PMID: 13117629]. [DOI] [PubMed] [Google Scholar]
- 73.Hawkins J.E., Jr, Sarett L.H. On the efficacy of asparagine, glutamine, gamma-aminobutyric acid and 1-pyrroiidinone in preventing chemically induced seizures in mice. Clin. Chim. Acta. 1957;2(6):481–484. doi: 10.1016/0009-8981(57)90049-9. [http://dx.doi.org/10.1016/0009-8981(57)90049-9]. [PMID: 13500579]. [DOI] [PubMed] [Google Scholar]
- 74.Ben-Ari Y., Krnjević K., Reinhardt W. Hippocampal seizures and failure of inhibition. Can. J. Physiol. Pharmacol. 1979;57(12):1462–1466. [http://dx.doi.org/10.1139/y79-218]. [Google Scholar]
- 75.Kostopoulos G., Avoli M., Gloor P. Participation of cortical recurrent inhibition in the genesis of spike and wave discharges in feline generalized penicillin epilepsy. Brain Res. 1983;267(1):101–112. doi: 10.1016/0006-8993(83)91043-0. [http://dx.doi.org/10.1016/0006-8993(83)91043-0]. [PMID: 6860937]. [DOI] [PubMed] [Google Scholar]
- 76.Sloviter R.S. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235(4784):73–76. doi: 10.1126/science.2879352. [http://dx.doi.org/10.1126/science.2879352]. [PMID: 2879352]. [DOI] [PubMed] [Google Scholar]
- 77.Williamson A., Telfeian A.E., Spencer D.D. Prolonged GABA responses in dentate granule cells in slices isolated from patients with temporal lobe sclerosis. J. Neurophysiol. 1995;74(1):378–387. doi: 10.1152/jn.1995.74.1.378. [http://dx.doi.org/10.1152/jn.1995.74.1.378]. [PMID: 7472339]. [DOI] [PubMed] [Google Scholar]
- 78.McDonald J.W., Garofalo E.A., Hood T., Sackellares J.C., Gilman S., McKeever P.E., Troncoso J.C., Johnston M.V. Altered excitatory and inhibitory amino acid receptor binding in hippocampus of patients with temporal lobe epilepsy. Ann. Neurol. 1991;29(5):529–541. doi: 10.1002/ana.410290513. [http://dx.doi.org/10.1002/ana.410290513]. [PMID: 1650160]. [DOI] [PubMed] [Google Scholar]
- 79.Johnson E.W., de Lanerolle N.C., Kim J.H., Sundaresan S., Spencer D.D., Mattson R.H., Zoghbi S.S., Baldwin R.M., Hoffer P.B., Seibyl J.P. “Central” and “peripheral” benzodiazepine receptors: opposite changes in human epileptogenic tissue. Neurology. 1992;42(4):811–815. doi: 10.1212/wnl.42.4.811. [http://dx.doi.org/10.1212/WNL.42. 4.811]. [PMID: 1314342]. [DOI] [PubMed] [Google Scholar]
- 80.Olsen R.W., Bureau M., Houser C.R., Delgado-Escueta A.V., Richards J.G., Möhler H. GABA/benzodiazepine receptors in human focal epilepsy. Epilepsy Res. Suppl. 1992;8:383–391. doi: 10.1016/b978-0-444-89710-7.50053-7. [PMID: 1329826]. [PMID: 1329826]. [DOI] [PubMed] [Google Scholar]
- 81.Rice A., Rafiq A., Shapiro S.M., Jakoi E.R., Coulter D.A., DeLorenzo R.J. Long-lasting reduction of inhibitory function and gamma-aminobutyric acid type A receptor subunit mRNA expression in a model of temporal lobe epilepsy. Proc. Natl. Acad. Sci. USA. 1996;93(18):9665–9669. doi: 10.1073/pnas.93.18.9665. [http://dx.doi.org/10.1073/pnas. 93.18.9665]. [PMID: 8790388]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kamphuis W., De Rijk T.C., Lopes da Silva F.H. Expression of GABAA receptor subunit mRNAs in hippocampal pyramidal and granular neurons in the kindling model of epileptogenesis: An in situ hybridization study. Brain Res. Mol. Brain Res. 1995;31(1-2):33–47. doi: 10.1016/0169-328x(95)00022-k. [http://dx.doi.org/10.1016/0169-328X(95)00022-K]. [PMID: 7476032]. [DOI] [PubMed] [Google Scholar]
- 83.Mody I., Lambert J.D., Heinemann U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J. Neurophysiol. 1987;57(3):869–888. doi: 10.1152/jn.1987.57.3.869. [http://dx.doi.org/10.1152/jn.1987.57.3.869]. [PMID: 3031235]. [DOI] [PubMed] [Google Scholar]
- 84.Tancredi V., Hwa G.G., Zona C., Brancati A., Avoli M. Low magnesium epileptogenesis in the rat hippocampal slice: electrophysiological and pharmacological features. Brain Res. 1990;511(2):280–290. doi: 10.1016/0006-8993(90)90173-9. [http://dx.doi.org/10.1016/0006-8993(90)90173-9]. [PMID: 1970748]. [DOI] [PubMed] [Google Scholar]
- 85.Dickson C.T., Alonso A. Muscarinic induction of synchronous population activity in the entorhinal cortex. J. Neurosci. 1997;17(17):6729–6744. doi: 10.1523/JNEUROSCI.17-17-06729.1997. [http://dx.doi.org/10.1523/JNEUROSCI.17-17-06729.1997]. [PMID: 9254685]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Michelson H.B., Wong R.K. Synchronization of inhibitory neurones in the guinea-pig hippocampus in vitro. J. Physiol. 1994;477(Pt 1):35–45. doi: 10.1113/jphysiol.1994.sp020169. [http://dx.doi.org/10.1113/jphysiol.1994.sp020169]. [PMID: 8071887]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Perreault P., Avoli M. 4-aminopyridine-induced epileptiform activity and a GABA-mediated long-lasting depolarization in the rat hippocampus. J. Neurosci. 1992;12(1):104–115. doi: 10.1523/JNEUROSCI.12-01-00104.1992. [http:// dx.doi.org/10.1523/JNEUROSCI.12-01-00104.1992]. [PMID: 1309571]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Avoli M., Barbarosie M., Lücke A., Nagao T., Lopantsev V., Köhling R. Synchronous GABA-mediated potentials and epileptiform discharges in the rat limbic system in vitro. J. Neurosci. 1996;16(12):3912–3924. doi: 10.1523/JNEUROSCI.16-12-03912.1996. [http://dx.doi.org/10.1523/JNEUROSCI. 16-12-03912.1996]. [PMID: 8656285]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Avoli M., Louvel J., Kurcewicz I., Pumain R., Barbarosie M. Extracellular free potassium and calcium during synchronous activity induced by 4-aminopyridine in the juvenile rat hippocampus. J. Physiol. 1996;493(Pt 3):707–717. doi: 10.1113/jphysiol.1996.sp021416. [http://dx.doi.org/10.1113/ jphysiol.1996.sp021416]. [PMID: 8799893]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morris M.E., Obrocea G.V., Avoli M. Extracellular K+ accumulations and synchronous GABA-mediated potentials evoked by 4-aminopyridine in the adult rat hippocampus. Exp. Brain Res. 1996;109(1):71–82. doi: 10.1007/BF00228628. [http://dx.doi.org/10.1007/BF00228628]. [PMID: 8740210]. [DOI] [PubMed] [Google Scholar]
- 91.Barolet A.W., Morris M.E. Changes in extracellular K+ evoked by GABA, THIP and baclofen in the guinea-pig hippocampal slice. Exp. Brain Res. 1991;84(3):591–598. doi: 10.1007/BF00230971. [http://dx.doi.org/10.1007/ BF00230971]. [PMID: 1650707]. [DOI] [PubMed] [Google Scholar]
- 92.Di Cristo G., Awad P.N., Hamidi S., Avoli M. KCC2, epileptiform synchronization, and epileptic disorders. Prog. Neurobiol. 2018;162:1–16. doi: 10.1016/j.pneurobio.2017.11.002. [http://dx.doi.org/10.1016/j.pneurobio.2017.11. 002]. [PMID: 29197650]. [DOI] [PubMed] [Google Scholar]
- 93.Viitanen T., Ruusuvuori E., Kaila K., Voipio J. The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J. Physiol. 2010;588(Pt 9):1527–1540. doi: 10.1113/jphysiol.2009.181826. [http://dx.doi.org/10.1113/jphysiol.2009.181826]. [PMID: 20211979]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grover L.M., Lambert N.A., Schwartzkroin P.A., Teyler T.J. Role of HCO3- ions in depolarizing GABAA receptor-mediated responses in pyramidal cells of rat hippocampus. J. Neurophysiol. 1993;69(5):1541–1555. doi: 10.1152/jn.1993.69.5.1541. [http://dx.doi.org/10.1152/jn.1993.69.5.1541]. [PMID: 8389828]. [DOI] [PubMed] [Google Scholar]
- 95.Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog. Neurobiol. 1994;42(4):489–537. doi: 10.1016/0301-0082(94)90049-3. [http://dx.doi.org/10.1016/0301-0082(94)90049-3]. [PMID: 7522334]. [DOI] [PubMed] [Google Scholar]
- 96.Staley K.J., Soldo B.L., Proctor W.R. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269(5226):977–981. doi: 10.1126/science.7638623. [http://dx.doi.org/10.1126/science.7638623]. [PMID: 7638623]. [DOI] [PubMed] [Google Scholar]
- 97.Velazquez J.L., Carlen P.L. Synchronization of GABAergic interneuronal networks during seizure-like activity in the rat horizontal hippocampal slice. Eur. J. Neurosci. 1999;11(11):4110–4118. doi: 10.1046/j.1460-9568.1999.00837.x. [http://dx.doi.org/10.1046/j.1460-9568.1999.00837.x]. [PMID: 10583499]. [DOI] [PubMed] [Google Scholar]
- 98.Köhling R., Vreugdenhil M., Bracci E., Jefferys J.G. Ictal epileptiform activity is facilitated by hippocampal GABAA receptor-mediated oscillations. J. Neurosci. 2000;20(18):6820–6829. doi: 10.1523/JNEUROSCI.20-18-06820.2000. [PMID: 10995826]. [http://dx.doi.org/10.1523/JNEUROSCI.20-18-06820.2000]. [PMID: 10995826]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Timofeev I., Grenier F., Steriade M. The role of chloride-dependent inhibition and the activity of fast-spiking neurons during cortical spike-wave electrographic seizures. Neuroscience. 2002;114(4):1115–1132. doi: 10.1016/s0306-4522(02)00300-7. [PMID: 12379264]. [DOI] [PubMed] [Google Scholar]
- 100.D’Antuono M., Louvel J., Köhling R., Mattia D., Bernasconi A., Olivier A., Turak B., Devaux A., Pumain R., Avoli M. GABAA receptor-dependent synchronization leads to ictogenesis in the human dysplastic cortex. Brain. 2004;127(Pt 7):1626–1640. doi: 10.1093/brain/awh181. [http://dx.doi.org/10.1093/brain/awh181]. [PMID: 15175227]. [DOI] [PubMed] [Google Scholar]
- 101.Derchansky M., Jahromi S.S., Mamani M., Shin D.S., Sik A., Carlen P.L. Transition to seizures in the isolated immature mouse hippocampus: A switch from dominant phasic inhibition to dominant phasic excitation. J. Physiol. 2008;586(2):477–494. doi: 10.1113/jphysiol.2007.143065. [http:// dx.doi.org/10.1113/jphysiol.2007.143065]. [PMID: 17991696]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gnatkovsky V., Librizzi L., Trombin F., de Curtis M. Fast activity at seizure onset is mediated by inhibitory circuits in the entorhinal cortex in vitro. Ann. Neurol. 2008;64(6):674–686. doi: 10.1002/ana.21519. [http://dx.doi.org/10.1002/ana.21519]. [PMID: 19107991]. [DOI] [PubMed] [Google Scholar]
- 103.Fujiwara-Tsukamoto Y., Isomura Y., Imanishi M., Ninomiya T., Tsukada M., Yanagawa Y., Fukai T., Takada M. Prototypic seizure activity driven by mature hippocampal fast-spiking interneurons. J. Neurosci. 2010;30(41):13679–13689. doi: 10.1523/JNEUROSCI.1523-10.2010. [http://dx.doi.org/ 10.1523/JNEUROSCI.1523-10.2010]. [PMID: 20943908]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Uva L., Breschi G.L., Gnatkovsky V., Taverna S., de Curtis M. Synchronous inhibitory potentials precede seizure-like events in acute models of focal limbic seizures. J. Neurosci. 2015;35(7):3048–3055. doi: 10.1523/JNEUROSCI.3692-14.2015. [http://dx.doi.org/10.1523/JNEUROSCI.3692-14.2015]. [PMID: 25698742]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Librizzi L., Losi G., Marcon I., Sessolo M., Scalmani P., Carmignoto G., de Curtis M. Interneuronal network activity at the onset of seizure-like events in entorhinal cortex slices. J. Neurosci. 2017;37(43):10398–10407. doi: 10.1523/JNEUROSCI.3906-16.2017. [http://dx.doi.org/10.1523/JNEUROSCI. 3906-16.2017]. [PMID: 28947576]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shiri Z., Manseau F., Lévesque M., Williams S., Avoli M. Interneuron activity leads to initiation of low-voltage fast-onset seizures. Ann. Neurol. 2015;77(3):541–546. doi: 10.1002/ana.24342. [http://dx.doi.org/10. 1002/ana.24342]. [PMID: 25546300]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yekhlef L., Breschi G.L., Lagostena L., Russo G., Taverna S. Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. J. Neurophysiol. 2015;113(5):1616–1630. doi: 10.1152/jn.00841.2014. [http://dx.doi.org/10.1152/jn.00841.2014]. [PMID: 25505119]. [DOI] [PubMed] [Google Scholar]
- 108.Uusisaari M., Smirnov S., Voipio J., Kaila K. Spontaneous epileptiform activity mediated by GABA(A) receptors and gap junctions in the rat hippocampal slice following long-term exposure to GABA(B) antagonists. Neuropharmacology. 2002;43(4):563–572. doi: 10.1016/s0028-3908(02)00156-9. [PMID: 12367602]. [http://dx.doi.org/10.1016/S0028-3908(02) 00156-9]. [PMID: 12367602]. [DOI] [PubMed] [Google Scholar]
- 109.Grasse D.W., Karunakaran S., Moxon K.A. Neuronal synchrony and the transition to spontaneous seizures. Exp. Neurol. 2013;248:72–84. doi: 10.1016/j.expneurol.2013.05.004. [http://dx.doi.org/10.1016/j.expneurol.2013.05.004]. [PMID: 23707218]. [DOI] [PubMed] [Google Scholar]
- 110.Fujita S., Toyoda I., Thamattoor A.K., Buckmaster P.S. Preictal activity of subicular, CA1, and dentate gyrus principal neurons in the dorsal hippocampus before spontaneous seizures in a rat model of temporal lobe epilepsy. J. Neurosci. 2014;34(50):16671–16687. doi: 10.1523/JNEUROSCI.0584-14.2014. [http://dx.doi.org/10.1523/JNEUROSCI.0584-14.2014]. [PMID: 25505320]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Toyoda I., Fujita S., Thamattoor A.K., Buckmaster P.S. Unit Activity of hippocampal interneurons before spontaneous seizures in an animal model of temporal lobe epilepsy. J. Neurosci. 2015;35(16):6600–6618. doi: 10.1523/JNEUROSCI.4786-14.2015. [http://dx.doi.org/10.1523/JNEUROSCI. 4786-14.2015]. [PMID: 25904809]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Truccolo W., Donoghue J.A., Hochberg L.R., Eskandar E.N., Madsen J.R., Anderson W.S., Brown E.N., Halgren E., Cash S.S. Single-neuron dynamics in human focal epilepsy. Nat. Neurosci. 2011;14(5):635–641. doi: 10.1038/nn.2782. [http://dx.doi.org/10.1038/nn.2782]. [PMID: 21441925]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schevon C.A., Weiss S.A., McKhann G., Jr, Goodman R.R., Yuste R., Emerson R.G., Trevelyan A.J. Evidence of an inhibitory restraint of seizure activity in humans. Nat. Commun. 2012;3:1060. doi: 10.1038/ncomms2056. [http://dx.doi.org/10.1038/ncomms2056]. [PMID: 22968706]. [http://dx.doi.org/10.1038/ncomms2056]. [PMID: 22968706]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rogawski M.A., Löscher W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004;5(7):553–564. doi: 10.1038/nrn1430. [http://dx.doi.org/ 10.1038/nrn1430]. [PMID: 15208697]. [DOI] [PubMed] [Google Scholar]
- 115.Brodie M.J. Tiagabine pharmacology in profile. Epilepsia. 1995;36(Suppl. 6):S7–S9. doi: 10.1111/j.1528-1157.1995.tb06015.x. [http://dx.doi.org/10.1111/j.1528-1157. 1995.tb06015.x]. [PMID: 8595791]. [DOI] [PubMed] [Google Scholar]
- 116.Pollack M.H., Roy-Byrne P.P., Van Ameringen M., Snyder H., Brown C., Ondrasik J., Rickels K. The selective GABA reuptake inhibitor tiagabine for the treatment of generalized anxiety disorder: results of a placebo-controlled study. J. Clin. Psychiatry. 2005;66(11):1401–1408. doi: 10.4088/jcp.v66n1109. [http://dx.doi.org/10.4088/JCP.v66n1109]. [PMID: 16420077]. [DOI] [PubMed] [Google Scholar]
- 117.Lloyd K.G., Morselli P.L., Depoortere H., Fournier V., Zivkovic B., Scatton B., Broekkamp C., Worms P., Bartholini G. The potential use of GABA agonists in psychiatric disorders: evidence from studies with progabide in animal models and clinical trials. Pharmacol. Biochem. Behav. 1983;18(6):957–966. doi: 10.1016/s0091-3057(83)80021-5. [http://dx.doi.org/10.1016/S0091-3057(83)80021-5]. [PMID: 6351106]. [DOI] [PubMed] [Google Scholar]
- 118.Loiseau P., Bossi L., Guyot M., Orofiamma B., Morselli P.L. Double-blind crossover trial of progabide versus placebo in severe epilepsies. Epilepsia. 1983;24(6):703–715. doi: 10.1111/j.1528-1157.1983.tb04633.x. [http://dx.doi.org/10. 1111/j.1528-1157.1983.tb04633.x]. [PMID: 6357772]. [DOI] [PubMed] [Google Scholar]
- 119.Taylor C.P. Mechanisms of action of gabapentin. Rev. Neurol. (Paris) 1997;153(Suppl. 1):S39–S45. [PMID: 9686247]. [PubMed] [Google Scholar]
- 120.Pang T., Hirsch L.J. Treatment of convulsive and nonconvulsive status epilepticus. Curr. Treat. Options Neurol. 2005;7(4):247–259. doi: 10.1007/s11940-005-0035-x. [http://dx.doi.org/10.1007/s11940-005-0035-x]. [PMID: 15967088]. [DOI] [PubMed] [Google Scholar]
- 121.Costa E., Guidotti A., Mao C.C. Evidence for involvement of GABA in the action of benzodiazepines: studies on rat cerebellum. Adv. Biochem. Psychopharmacol. 1975;(14):113–130. [PMID: 242198]. [PMID: 242198]. [PubMed] [Google Scholar]
- 122.Choi D.W., Farb D.H., Fischbach G.D. Chlordiazepoxide selectively augments GABA action in spinal cord cell cultures. Nature. 1977;269(5626):342–344. doi: 10.1038/269342a0. [http://dx.doi.org/10.1038/269342a0]. [PMID: 561893]. [DOI] [PubMed] [Google Scholar]
- 123.Olsen R.W. Allosteric ligands and their binding sites define γ-aminobutyric acid (GABA) type A receptor subtypes. Adv. Pharmacol. 2015;73:167–202. doi: 10.1016/bs.apha.2014.11.005. [http://dx.doi.org/10.1016/bs.apha. 2014.11.005]. [PMID: 25637441]. [DOI] [PubMed] [Google Scholar]
- 124.Watts A.E., Jefferys J.G. Effects of carbamazepine and baclofen on 4-aminopyridine-induced epileptic activity in rat hippocampal slices. Br. J. Pharmacol. 1993;108(3):819–823. doi: 10.1111/j.1476-5381.1993.tb12884.x. [PMID: 8467367]. [http://dx.doi.org/10.1111/j.1476-5381.1993.tb12884.x]. [PMID: 8467367]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Motalli R., Louvel J., Tancredi V., Kurcewicz I., Wan-Chow-Wah D., Pumain R., Avoli M. GABA(B) receptor activation promotes seizure activity in the juvenile rat hippocampus. J. Neurophysiol. 1999;82(2):638–647. doi: 10.1152/jn.1999.82.2.638. [http://dx.doi.org/10.1152/jn. 1999.82.2.638]. [PMID: 10444662]. [DOI] [PubMed] [Google Scholar]
- 126.Mott D.D., Bragdon A.C., Lewis D.V., Wilson W.A. Baclofen has a proepileptic effect in the rat dentate gyrus. J. Pharmacol. Exp. Ther. 1989;249(3):721–725. [PMID: 2543809]. [PMID: 2543809]. [PubMed] [Google Scholar]
- 127.Motalli R., D’Antuono M., Louvel J., Kurcewicz I., D’Arcangelo G., Tancredi V., Manfredi M., Pumain R., Avoli M. Epileptiform synchronization and GABA(B) receptor antagonism in the juvenile rat hippocampus. J. Pharmacol. Exp. Ther. 2002;303(3):1102–1113. doi: 10.1124/jpet.102.040782. [http://dx.doi.org/10.1124/jpet.102.040782]. [PMID: 12438533]. [DOI] [PubMed] [Google Scholar]
- 128.Hille B. Ion Channels of excitable membranes: 9780878933211: Medicine & Health Science Books @ Amazon.com. https://www. amazon.com/Channels-Excitable-Membranes-Bertil-Hille/dp/
- 129.Crill W.E. Persistent sodium current in mammalian central neurons. Annu. Rev. Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [http://dx.doi. org/10.1146/annurev.ph.58.030196.002025]. [PMID: 8815799]. [DOI] [PubMed] [Google Scholar]
- 130.Magistretti J., Ragsdale D.S., Alonso A. High conductance sustained single-channel activity responsible for the low-threshold persistent Na(+) current in entorhinal cortex neurons. J. Neurosci. 1999;19(17):7334–7341. doi: 10.1523/JNEUROSCI.19-17-07334.1999. [PMID: 10460240]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mantegazza M., Curia G., Biagini G., Ragsdale D.S., Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010;9(4):413–424. doi: 10.1016/S1474-4422(10)70059-4. [http://dx.doi.org/10.1016/S1474-4422(10)70059-4]. [PMID: 20298965]. [DOI] [PubMed] [Google Scholar]
- 132.Kaplan D.I., Isom L.L., Petrou S. Role of sodium channels in epilepsy. Cold Spring Harb. Perspect. Med. 2016;6(6):a022814. doi: 10.1101/cshperspect.a022814. [http://dx.doi.org/10.1101/cshperspect.a022814]. [PMID: 27143702]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Agrawal N., Alonso A., Ragsdale D.S. Increased persistent sodium currents in rat entorhinal cortex layer V neurons in a post-status epilepticus model of temporal lobe epilepsy. Epilepsia. 2003;44(12):1601–1604. doi: 10.1111/j.0013-9580.2003.23103.x. [http://dx.doi.org/10.1111/j.0013-9580.2003. 23103.x]. [PMID: 14636336]. [DOI] [PubMed] [Google Scholar]
- 134.Blumenfeld H., Lampert A., Klein J.P., Mission J., Chen M.C., Rivera M., Dib-Hajj S., Brennan A.R., Hains B.C., Waxman S.G. Role of hippocampal sodium channel Nav1.6 in kindling epileptogenesis. Epilepsia. 2009;50(1):44–55. doi: 10.1111/j.1528-1167.2008.01710.x. [http://dx.doi.org/10. 1111/j.1528-1167.2008.01710.x]. [PMID: 18637833]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vreugdenhil M., Hoogland G., van Veelen C.W.M., Wadman W.J. Persistent sodium current in subicular neurons isolated from patients with temporal lobe epilepsy. Eur. J. Neurosci. 2004;19(10):2769–2778. doi: 10.1111/j.1460-9568.2004.03400.x. [http://dx.doi.org/10.1111/j.1460-9568.2004. 03400.x]. [PMID: 15147310]. [DOI] [PubMed] [Google Scholar]
- 136.Kearney J.A., Plummer N.W., Smith M.R., Kapur J., Cummins T.R., Waxman S.G., Goldin A.L., Meisler M.H. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102(2):307–317. doi: 10.1016/s0306-4522(00)00479-6. [http://dx.doi.org/10.1016/S0306-4522(00)00479-6]. [PMID: 11166117]. [DOI] [PubMed] [Google Scholar]
- 137.Stafstrom C.E. Persistent sodium current and its role in epilepsy. Epilepsy Curr. 2007;7(1):15–22. doi: 10.1111/j.1535-7511.2007.00156.x. [http://dx.doi.org/10.1111/j.1535-7511.2007.00156.x]. [PMID: 17304346]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Curia G., Longo D., Biagini G., Jones R.S.G., Avoli M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods. 2008;172(2):143–157. doi: 10.1016/j.jneumeth.2008.04.019. [http://dx.doi.org/10.1016/j.jneumeth. 2008.04.019]. [PMID: 18550176]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ragsdale D.S., Scheuer T., Catterall W.A. Frequency and voltage-dependent inhibition of type IIA Na+ channels, expressed in a mammalian cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs. Mol. Pharmacol. 1991;40(5):756–765. [PMID: 1658608]. [PubMed] [Google Scholar]
- 140.Poolos N.P., Migliore M., Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat. Neurosci. 2002;5(8):767–774. doi: 10.1038/nn891. [http://dx.doi.org/ 10.1038/nn891]. [PMID: 12118259]. [DOI] [PubMed] [Google Scholar]
- 141.Poolos N.P. 2012. [Google Scholar]
- 142.Surges R., Freiman T.M., Feuerstein T.J. Gabapentin increases the hyperpolarization-activated cation current Ih in rat CA1 pyramidal cells. Epilepsia. 2003;44(2):150–156. doi: 10.1046/j.1528-1157.2003.36802.x. [PMID: 12558567]. [http://dx.doi.org/10.1046/j.1528-1157.2003.36802.x]. [PMID: 12558567]. [DOI] [PubMed] [Google Scholar]
- 143.Stefani A., Spadoni F., Siniscalchi A., Bernardi G. Lamotrigine inhibits Ca2+ currents in cortical neurons: functional implications. Eur. J. Pharmacol. 1996;307(1):113–116. doi: 10.1016/0014-2999(96)00265-8. [PMID: 8831112]. [http://dx.doi.org/10.1016/0014-2999(96)00265-8]. [PMID: 8831112]. [DOI] [PubMed] [Google Scholar]
- 144.Catterall W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011;3(8):a003947. doi: 10.1101/cshperspect.a003947. [http://dx.doi.org/10.1101/ cshperspect.a003947]. [PMID: 21746798]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lynch B.A., Lambeng N., Nocka K., Kensel-Hammes P., Bajjalieh S.M., Matagne A., Fuks B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA. 2004;101(26):9861–9866. doi: 10.1073/pnas.0308208101. [http://dx. doi.org/10.1073/pnas.0308208101]. [PMID: 15210974]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vogl C., Mochida S., Wolff C., Whalley B.J., Stephens G.J. The synaptic vesicle glycoprotein 2A ligand levetiracetam inhibits presynaptic Ca2+ channels through an intracellular pathway. Mol. Pharmacol. 2012;82(2):199–208. doi: 10.1124/mol.111.076687. [http://dx.doi.org/10.1124/mol. 111.076687]. [PMID: 22554805]. [DOI] [PubMed] [Google Scholar]
- 147.Löscher W., Hönack D., Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J. Pharmacol. Exp. Ther. 1998;284(2):474–479. [PMID: 9454787]. [PubMed] [Google Scholar]
- 148.Vinogradova L.V., van Rijn C.M. Anticonvulsive and antiepileptogenic effects of levetiracetam in the audiogenic kindling model. Epilepsia. 2008;49(7):1160–1168. doi: 10.1111/j.1528-1167.2008.01594.x. [http://dx.doi.org/10.1111/ j.1528-1167.2008.01594.x]. [PMID: 18397292]. [DOI] [PubMed] [Google Scholar]
- 149.Yan H-D., Ji-qun C., Ishihara K., Nagayama T., Serikawa T., Sasa M. Separation of antiepileptogenic and antiseizure effects of levetiracetam in the spontaneously epileptic rat (SER). Epilepsia. 2005;46(8):1170–1177. doi: 10.1111/j.1528-1167.2005.35204.x. [http://dx.doi.org/10.1111/j.1528-1167. 2005.35204.x]. [PMID: 16060925]. [DOI] [PubMed] [Google Scholar]
- 150.Sugaya Y., Maru E., Kudo K., Shibasaki T., Kato N. Levetiracetam suppresses development of spontaneous EEG seizures and aberrant neurogenesis following kainate-induced status epilepticus. Brain Res. 2010;1352:187–199. doi: 10.1016/j.brainres.2010.06.061. [http://dx.doi.org/ 10.1016/j.brainres.2010.06.061]. [PMID: 20599805]. [DOI] [PubMed] [Google Scholar]
- 151.Lévesque M., Behr C., Avoli M. The anti-ictogenic effects of levetiracetam are mirrored by interictal spiking and high-frequency oscillation changes in a model of temporal lobe epilepsy. Seizure. 2015;25:18–25. doi: 10.1016/j.seizure.2014.11.008. [http://dx.doi.org/10.1016/j.seizure.2014.11.008]. [PMID: 25645630]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pisani A., Bonsi P., Martella G., De Persis C., Costa C., Pisani F., Bernardi G., Calabresi P. Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia. 2004;45(7):719–728. doi: 10.1111/j.0013-9580.2004.02204.x. [http://dx.doi.org/10.1111/j.0013-9580.2004.02204.x]. [PMID: 15230693]. [DOI] [PubMed] [Google Scholar]
- 153.Mazzocchetti P., Tantucci M., Bastioli G., Calabrese V., Di Filippo M., Tozzi A., Calabresi P., Costa C. Lacosamide protects striatal and hippocampal neurons from in vitro ischemia without altering physiological synaptic plasticity. Neuropharmacology. 2018;135:424–430. doi: 10.1016/j.neuropharm.2018.03.040. [http://dx.doi.org/10.1016/j.neuropharm.2018. 03.040]. [PMID: 29614316]. [DOI] [PubMed] [Google Scholar]
- 154.Zona C., Tancredi V., Palma E., Pirrone G.C., Avoli M. Potassium currents in rat cortical neurons in culture are enhanced by the antiepileptic drug carbamazepine. Can. J. Physiol. Pharmacol. 1990;68(4):545–547. doi: 10.1139/y90-079. [http://dx.doi.org/10.1139/y90-079]. [PMID: 2328457]. [DOI] [PubMed] [Google Scholar]
- 155.Zona C., Tancredi V., Longone P., D’Arcangelo G., D’Antuono M., Manfredi M., Avoli M. Neocortical potassium currents are enhanced by the antiepileptic drug lamotrigine. Epilepsia. 2002;43(7):685–690. doi: 10.1046/j.1528-1157.2002.51401.x. [http://dx.doi.org/10.1046/j.1528-1157.2002. 51401.x]. [PMID: 12102669]. [DOI] [PubMed] [Google Scholar]
- 156.Pumain R., Menini C., Heinemann U., Louvel J., Silva-Barrat C. Chemical synaptic transmission is not necessary for epileptic seizures to persist in the baboon Papio papio. Exp. Neurol. 1985;89(1):250–258. doi: 10.1016/0014-4886(85)90280-8. [http://dx.doi.org/10.1016/0014-4886(85)90280-8]. [PMID: 2988992]. [DOI] [PubMed] [Google Scholar]
- 157.Bruzzone R., Hormuzdi S.G., Barbe M.T., Herb A., Monyer H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA. 2003;100(23):13644–13649. doi: 10.1073/pnas.2233464100. [http://dx.doi.org/10.1073/pnas.2233464100]. [PMID: 14597722]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Söhl G., Maxeiner S., Willecke K. Expression and functions of neuronal gap junctions. Nat. Rev. Neurosci. 2005;6(3):191–200. doi: 10.1038/nrn1627. [http://dx.doi.org/10.1038/nrn1627]. [PMID: 15738956]. [DOI] [PubMed] [Google Scholar]
- 159.Dermietzel R., Spray D.C. Gap junctions in the brain: where, what type, how many and why? Trends Neurosci. 1993;16(5):186–192. doi: 10.1016/0166-2236(93)90151-b. [http://dx.doi.org/10.1016/0166-2236(93)90151-B]. [PMID: 7685944]. [DOI] [PubMed] [Google Scholar]
- 160.Bennett M.V.L., Zukin R.S. Electrical coupling and neuronal synchronization in the Mammalian brain. Neuron. 2004;41(4):495–511. doi: 10.1016/s0896-6273(04)00043-1. [http://dx.doi.org/10.1016/S0896-6273(04)00043-1]. [PMID: 14980200]. [DOI] [PubMed] [Google Scholar]
- 161.Nakase T., Naus C.C.G. Gap junctions and neurological disorders of the central nervous system. Biochim. Biophys. Acta. 2004;1662(1-2):149–158. doi: 10.1016/j.bbamem.2004.01.009. [http://dx.doi.org/10.1016/j.bbamem.2004.01. 009]. [http://dx.doi.org/10.1016/j.bbamem.2004.01.009]. [PMID: 15033585]. [DOI] [PubMed] [Google Scholar]
- 162.Mylvaganam S., Ramani M., Krawczyk M., Carlen P.L. Roles of gap junctions, connexins, and pannexins in epilepsy. Front. Physiol. 2014;5:172. doi: 10.3389/fphys.2014.00172. [http://dx.doi.org/10.3389/fphys.2014.00172]. [PMID: 24847276]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Draguhn A., Traub R.D., Schmitz D., Jefferys J.G.R. Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro. Nature. 1998;394(6689):189–192. doi: 10.1038/28184. [http://dx.doi.org/10. 1038/28184]. [PMID: 9671303]. [DOI] [PubMed] [Google Scholar]
- 164.Maier N., Güldenagel M., Söhl G., Siegmund H., Willecke K., Draguhn A. Reduction of high-frequency network oscillations (ripples) and pathological network discharges in hippocampal slices from connexin 36-deficient mice. J. Physiol. 2002;541(Pt 2):521–528. doi: 10.1113/jphysiol.2002.017624. [http://dx.doi.org/10.1113/jphysiol.2002.017624]. [PMID: 12042356]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Simon A., Traub R.D., Vladimirov N., Jenkins A., Nicholson C., Whittaker R.G., Schofield I., Clowry G.J., Cunningham M.O., Whittington M.A. Gap junction networks can generate both ripple-like and fast ripple-like oscillations. Eur. J. Neurosci. 2014;39(1):46–60. doi: 10.1111/ejn.12386. [http://dx.doi.org/10.1111/ejn.12386]. [PMID: 24118191]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ventura-Mejia C., Medina-Ceja L. Decreased fast ripples in the hippocampus of rats with spontaneous recurrent seizures treated with carbenoxolone and quinine. BioMed Res. Int. 2014;2014:282490. doi: 10.1155/2014/282490. [http://dx.doi.org/10.1155/2014/282490]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Carlen P.L., Skinner F., Zhang L., Naus C., Kushnir M., Perez Velazquez J.L. The role of gap junctions in seizures. Brain Res. Brain Res. Rev. 2000;32(1):235–241. doi: 10.1016/s0165-0173(99)00084-3. [PMID: 10751673]. [http://dx.doi.org/10.1016/S0165-0173(99)00084-3]. [PMID: 10751673]. [DOI] [PubMed] [Google Scholar]
- 168.Naus C.C., Bechberger J.F., Paul D.L. Gap junction gene expression in human seizure disorder. Exp. Neurol. 1991;111(2):198–203. doi: 10.1016/0014-4886(91)90007-y. [http://dx.doi.org/10.1016/0014-4886(91)90007-Y]. [PMID: 1846600]. [DOI] [PubMed] [Google Scholar]
- 169.Aronica E., Gorter J.A., Jansen G.H., Leenstra S., Yankaya B., Troost D. Expression of connexin 43 and connexin 32 gap-junction proteins in epilepsy-associated brain tumors and in the perilesional epileptic cortex. Acta Neuropathol. 2001;101(5):449–459. doi: 10.1007/s004010000305. [PMID: 11484816]. [DOI] [PubMed] [Google Scholar]
- 170.Gigout S., Louvel J., Kawasaki H., D’Antuono M., Armand V., Kurcewicz I., Olivier A., Laschet J., Turak B., Devaux B., Pumain R., Avoli M. Effects of gap junction blockers on human neocortical synchronization. Neurobiol. Dis. 2006;22(3):496–508. doi: 10.1016/j.nbd.2005.12.011. [http://dx.doi.org/10.1016/j.nbd.2005.12.011]. [PMID: 16478664]. [DOI] [PubMed] [Google Scholar]
- 171.Kurata Y., Marszalec W., Yeh J.Z., Narahashi T. Agonist and potentiation actions of n-octanol on gamma-aminobutyric acid type A receptors. Mol. Pharmacol. 1999;55(6):1011–1019. doi: 10.1124/mol.55.6.1011. [PMID: 10347242]. [DOI] [PubMed] [Google Scholar]
- 172.Vessey J.P., Lalonde M.R., Mizan H.A., Welch N.C., Kelly M.E.M., Barnes S. Carbenoxolone inhibition of voltage-gated Ca channels and synaptic transmission in the retina. J. Neurophysiol. 2004;92(2):1252–1256. doi: 10.1152/jn.00148.2004. [http://dx.doi.org/10.1152/jn.00148.2004]. [PMID: 15028741]. [DOI] [PubMed] [Google Scholar]