

Abstract
Three inhibitors of type-B monoamine oxidase (MAOB), selegiline, rasagiline, and safinamide, are used for the treatment of Parkinson’s disease (PD). All three drugs improve motor signs of PD, and are effective in reducing motor fluc-tuations in patients undergoing long-term L-DOPA treatment. The effect of MAOB inhibitors on non-motor symptoms is not uniform and may not be class-related. Selegiline and rasagiline are irreversible inhibitors forming a covalent bond within the active site of MAOB. In contrast, safinamide is a reversible MAOB inhibitor, and also inhibits voltage-sensitive sodium channels and glutamate release. Safinamide is the prototype of a new generation of multi-active MAOB inhibitors, which in-cludes the antiepileptic drug, zonisamide. Inhibition of MAOB-mediated dopamine metabolism largely accounts for the an-tiparkinsonian effect of the three drugs. Dopamine metabolism by MAOB generates reactive oxygen species, which contrib-ute to nigro-striatal degeneration. Among all antiparkinsonian agents, MAOB inhibitors are those with the greatest neuropro-tective potential because of inhibition of dopamine metabolism, induction of neurotrophic factors, and, in the case of safina-mide, inhibition of glutamate release. The recent development of new experimental animal models that more closely mimic the progressive neurodegeneration associated with PD will allow to test the hypothesis that MAOB inhibitors may slow the progression of PD.
Keywords: Selegiline, rasagiline, safinamide, MAOB, basal ganglia, Parkinson’s disease, glutamate release
1. INTRODUCTION
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by a progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) resulting into dopamine (DA) denervation in the caudate nucleus and putamen. Neurons in other pigmented nuclei of the brainstem, as well as autonomic neurons in peripheral organs (e.g., heart and gut) also degenerate in PD [1]. The complex pathophysiology of neuronal degeneration is what makes PD a heterogeneous disorder. Intracellular inclusions called Lewy bodies containing aggregates of the protein, α-synuclein are found in most cases of sporadic PD, and mutations of α-synuclein are associated with rare monogenic cases of PD with autosomal dominant transmission [2]. However, there are cases of early-onset PD, such as those caused by parkin and PINK-1 mutations, in which Lewy bodies are absent [3, 4]. Abnormalities in the retromer pathway, which controls the clearance of α-synuclein and other membrane proteins, and defects in the mitochondrial quality control are considered as major cytopathological determinants in PD [5-8].
Striatal DA denervation is the cause of the hallmark motor symptoms of PD, i.e., rigidity, bradykinesia, resting tremor, and postural instability, which occur at the time of clinical diagnosis. Nonmotor signs, such as constipation, hyposmia, REM sleep disorders, depression, and cognitive impairment, may precede the clinical onset of PD by several years. L-3,5-dihydroxyphenylalanine (L-DOPA) combined with peripheral inhibitors of L-aromatic aminoacid decarboxylase (LAAD) is the gold standard treatment of PD, and a good motor response to L-DOPA is pathognomonic of the disorder. In the long-term, however, L-DOPA treatment becomes suboptimal beause of the occurrence of motor fluctuations (e.g., wearing-off and on-off phenomena) and dyskinesias (L-DOPA-induced dyskinesias or LIDs). This reflects changes in the pharmacokinetic and pharmacodynamic profile of L-DOPA within the context of a progressive dopaminergic denervation [9, 10]. In addition, L-DOPA does not halt or attenuate PD progression, and, might even be detrimental to SNpc dopaminergic neurons. The long-term L-DOPA syndrome gave impetus to the design of other antiparkinsonian drugs, which help to delay the onset of L-DOPA treatment, or could be given in association with L-DOPA to optimize motor response and/or mitigate LIDs (for example, by allowing a reduction in the daily dose of L-DOPA). These classes of drugs include DA receptor agonists, anticholinergic drugs, catechol-oxy-methyltransferase (COMT) inhibitors, the N-methyl-D-aspartate (NMDA) receptor antagonist, amantadine, and type-B monoamine oxidase (MAOB) inhibitors [3,11]. Three MAOB inhibitors with a different pharmacological profile are currently used in the treatment of PD: selegiline, rasagiline, and safinamide. The aim of the present review is to comparatively discuss the preclinical and clinical pharmacology of the three drugs, focusing on their efficacy in relieving motor and non-motor symptoms of PD, and motor complications associated with L-DOPA treatment. In addition, we will briefly discuss the potential of the three MAOB inhibitors as disease-modifying drugs in PD.
2. METHODS
We searched for the following terms on Pubmed: selegiline, deprenyl, rasagiline, safinamide, MAOB, neuroprotection and Parkinson’s disease, basal ganglia motor circuit and glutamate, GDNF and Parkinson’s disease, glutamate and basal ganglia, NMDA receptor antagonists and Parkinson’s disease, mGlu5 receptor antagonists and Parkinson’s disease, Parkinson’s disease reviews (2015-2018).
3. COMPARATIVE PHARMACOLOGY OF DIFFERENT GENERATIONS OF MAOB INHIBITORS
3.1. MAOB and its Relevance to PD
MAOs are flavin-dependent enzymes bound to the outer mitochondrial membrane, with oxidase monoamines, like DA, noradrenaline, adrenaline, serotonin, and trace amines, reducing molecular oxygen to hydrogen peroxide. The two MAO isoforms, MAOA and MAOB, show a differential cellular localization in the CNS, with MAOA being mainly localized in neurons and MAOB in astrocytes. MAOA, which metabolizes catecholamines and serotonin, is an established target for antidepressant medication, and both irreversible MAOA/B inhibitors and reversible MAOA inhibitors (RIMA) have been developed for the treatment of unipolar depression. In contrast, MAOB inhibitors have been developed for the treatment of PD because of their ability to inhibit DA metabolism [12, 17].
DA released from nigro-striatal fibers is taken up by both the high-affinity DA transporter (DAT) present in axon terminals, and the reverse organic cation transporter (OCT3) present in astrocytes. The latter transport provides the main source for the DA metabolized by MAOB. DA oxidation by MAOB generates radical oxygen species (ROS). This by-product of MAOB activity might contribute to the pathophysiology of PD because of the high vulnerability of SNpc dopaminergic neurons to oxidative damage [3]. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which induces toxicological parkinsonism in mice and primates, is converted by MAOB into the active metabolite, 1-methyl-4-phenylpyridinium (MPP+). MPP+ kills SNpc neurons by inhibiting complex I of the mitochondrial respiratory chain [18]. Because a number of pesticides and other environmental toxins are structurally similar to MPTP, MAOB inhibition represents a potential strategy to limit the impact of the environment on the pathophysiology of PD [3].
3.2. Different Mode of MAOB Inhibition by Selegiline, Rasagiline, and Safinamide
Selegiline (L-deprenyl) and rasagiline are irreversible MAOB inhibitors. Crystallographic analysis showed that the active site of human MAOB is formed by two cavities, named substrate and entrance cavity, respectively [19]. Rasagiline, for example, forms a covalent adduct with the N5 atom of the flavin cofactor within the active site cavity [20], thereby irreversibly inhibiting MAOB. Selegiline also covalently binds to the flavin-protein complex within the active site of MAOB [21]. Hence, full recovery of MAOB activity requires several days or weeks after withdrawal of selegiline or rasagiline, a time necessary for a de novo synthesis of MAOB. For example, PET studies with 14C-L-deprenyl, which tracks brain MAOB levels, demonstrate that about 40 days are required for near-to-full recovery of MAOB in both normal subjects and patients affected by PD after selegiline withdrawal [22]. Similar recovery times were observed after rasagiline [23]. This long time of recovery should be taken into account when establishing the dose, duration of treatment, and pharmacodynamic or pharmacokinetic interactions with other drugs in PD patients treated with selegiline or rasagiline. In contrast, safinamide is a reversible MAOB inhibitor. Full recovery of MAOB activity is observed as early as 24 hours after a single i.p. injection of safinamide in the mouse brain, and within five days after a single oral administration of safinamide in human platelets [24, 25].
Potency and selectivity are two main issues when comparing different generations of MAOB inhibitors in the treatment of PD. Rasagiline displays a high potency as MAOB inhibitor, with an IC50 value of 4 and 14 nM in rat and human brain homogenates, respectively [26]. The IC50 value in the human brain is in the same range of the Cmax value found after a single administration of 2 mg rasagiline in PD patients, taking into account that about 60% of the drug is bound to plasma albumin [27]. Selegiline displays a similar potency as rasagiline as a MAOB inhibitor in rat and human brain homogenates, but is about 10-fold less potent than rasagiline on brain and liver MAOB activity when administered systemically to rats [26]. Safinamide shows an IC50 value of 98 and 79 nM on MAOB activity in extracts from rat and the human brain, respectively [24]. These values are in the same range of the Cmax values in human volunteers after a single oral administration of safinamide at 2.5 mg/kg, or to steady-state plasma concentrations after a 6-day treatment with 1.25 mg/kg safinamide (approximately 3 and 1.1 μM, respectively) [28], considering that 88-90% of safinamide binds to plasma albumin [28]. The same in vitro measurements carried out in extracts of rat and human brain show a high selectivity of safinamide towards MAOB with respect to MAOA (IC50 values: 0.098 vs. 485 μM, and 0.079 vs. 80 μM in rat and the human brain, respectively) [24]. Rasagiline has a lower selectivity towards MAOB vs. MAOA (IC50 values: 0.004 vs. 0.412 μM and 0.014 vs. 0.7 μM in rat and the human brain, respectively) [26]. This results into a brain MAOB/MAOA selectivity ratio of about 1,000 for safinamide and about 50 for rasagiline, although values of 0.7 μM largely exceed the peak and steady-state plasma concentrations of rasagiline with the usual therapeutic doses in PD patients.
The possibility that MAOA could be influenced by treatment with MAOB inhibitors was examined in a series of clinical trials in which PD patients received long-term treatment with MAOB inhibitors combined or not with L-DOPA plus LAAD inhibitors. The conclusion of a first study was that long-term treatment with irreversible MAOB inhibitors could reduce MAOA activity, as shown by measurements of enzymatic activity performed with the plasma collected from patients 4 hours after the last administration of either selegiline or rasagiline [30]. These findings were not replicated in another study in which peripheral MAOA and MAOB activities were measured in PD patients chronically treated with L-DOPA combined or not with 1 mg rasagiline or 50 or 100 mg safinamide [31]. No changes in MAOA activity were found in any of the experimental groups, suggesting that at least in this study, rasagiline behaved as a selective inhibitor of MAOB. The possibility that selectivity towards MAOB is lost after long-term treatment with high doses of rasagiline or selegiline cannot be excluded and warrants further investigation because inhibition of MAOA in PD patients might cause adverse effects (e.g., sympathomimetic effects induced by dietary tyramine).
3.3. Safinamide: A New-generation Multi-active Drug that Inhibits Ion Channels and Glutamate Release
Safinamide was originally developed as an antiepileptic drug because of its ability to inhibit voltage-sensitive sodium channels (VSSCs) and voltage-sensitive calcium channels (VSCCs). Safinamide was shown to interact with the batrachotoxin-sensitive site of VSSCs with greater affinity as compared to riluzole and the antiepileptic drugs, phenytoin, carbamazepine, and lamotrigine [32]. In cultured hippocampal and cortical neurons, inhibition of VSSCs by safinamide was voltage-, use-, and frequency-dependent [32, 33], a characteristic shared by many antiepileptic drugs. Hence, the VSSC blockade by safinamide becomes prominent during repetitive neuronal firing. Safinamide also inhibits high-threshold N- and L-type VSCCs [32], with a preferential action on N-type channels. L-type VSCCs are largely expressed in the heart and blood vessels, where calcium entry enhances dromotropism and inotropism and causes vasoconstriction, respectively. However, inhibition of L-type VSCCs occurs at relatively high concentrations of safinamide, and this explains the lack of significant effects of the drug on blood pressure and heart function in patients affected by PD. Interestingly, the pharmacodynamic profile of safinamide is shared by zonisamide, an antiepileptic drug that inhibits both VSSCs, T-type VSCCs, and MAOB [34-36]. Thus, safinamide and zonisamide can be considered as prototypes of a new generation of multi-active MAOB inhibitors [35].
As a result of VSSC and VSCC blockade, safinamide inhibits depolarization-evoked glutamate release in cultured neurons and synaptosomal preparation [24, 32]. In a recent article, Morari et al. (2018) examined the effect of safinamide on glutamate and GABA release assessed by in vivo microdialysis in freely moving rats. Safinamide inhibited depolarization-evoked glutamate and GABA release in the hippocampus. In the globus pallidus (GP), subthalamic nucleus (STN), and substantia nigra pars reticulata (SNpr) safinamide selectively inhibited depolarization-evoked glutamate release, whereas the drug had no effect in the dorsal striatum, at least at the selected dose of 15 mg/kg. Measurements of free brain concentrations of safinamide suggested that the drug restrained neurotransmitter release by inhibiting VSSCs rather than VSCCs [33]. These findings are extremely interesting and suggest that safinamide may reduce the excitatory overdrive associated with PD in some stations of the basal ganglia motor circuit.
3.4. Comparative Pharmacokinetic Profiles of Selegiline, Rasagiline, and Safinamide
Selegiline, rasagiline, and safinamide show remarkable differences in their pharmacokinetic profiles in terms of absorption, distribution volume (VD), metabolism, and elimination.
Selegiline is indicated for the treatment of PD at a daily dose of 10 mg, given either q.d. or b.i.d. At the oral dose of 10 mg, selegiline shows a bioavailability <10%, peak plasma concentrations of about 2.7 ng/ml, Tmax values of 0.5 hours, VD of 1854 liters, plasma albumin binding >90%, and elimination t1/2 of 1.5 hours [37, 38]. The low bioavailability after oral administration reflects an extensive first-pass hepatic metabolism mediated by CYP2B6 and, to a lesser extent, by CYP2A6 and CYP3A4 [39]. Selegiline metabolism generates L-methamphetamine and desmethylselegiline, the latter also behaving as an irreversible MAOB inhibitor [37]. Desmethylselegiline is in turn metabolized into L-amphetamine [37]. The generation of amphetamine metabolites confounds the pharmacodynamic profile of selegiline. CYP2B6 is highly polymorphic, with some genetic variants causing increases, and other reductions in enzymatic expression (CYP2B6*4/22, and CYP2B6*5/6/18, respectively). CYP2B6*6 shows a high frequency in Africans and Afro-Americans. Hence, it is more likely to find increased responses to selegiline in these ethnic groups. Drugs that could be associated with selegiline in the treatment of PD, such as the anticholinergic agent, orfenadrine, and the antidepressants, fluoxetine and paroxetine, are reversible inhibitors of CYP2B6. In addition, the thienopyridines, ticlopidine and clopidogrel, which are widely used to inhibit platelet aggregation, irreversibly inhibit CYP2B6 by forming a disulfide bond within the active core of the enzyme [40]. All these drugs may boost the activity of selegiline and increase the probability of dose-related adverse effects. Opposite effects might be produced by drugs that induce CYP2B6, such as phenobarbital and rifampicin.
At clinical oral doses of 1 or 2 mg (q.d.), rasagiline shows a bioavailability of about 35%, peak plasma concentrations of 2.5 and 4.9 ng/ml, respectively, Tmax values of 0.5 and 1 hour, respectively, VD of 87-243 liters, plasma albumin binding of 60%, and elimination t1/2 of 1.5-3.5 hours [41-43]. Rasagiline undergoes hepatic metabolism by CYP1A2, with the production of inactive metabolites (1R-aminoindane; 3-hydroxy-N-propargyl-1-aminoindane; and 3-hydroxyaminoindane). Although several genetic variants of CYP1A2 have been found in humans (www.CYP.ki.se https://www.pharmvar.org/htdocs/archive/cyp1a2.htm), these variants are not considered clinically relevant. CYP1A2 is induced by smoking and proton pump inhibitors (in particular, omeprazole, esomeprazole, and lansoprazole), and inhibited by fluoroquinolone antibiotics (e.g., ciprofloxacin), the selective serotonin reuptake inhibitor (SSRI), fluvoxamine, and oral contraceptives [44-46]. None of these drugs should be associated with rasagiline. This concern is particularly relevant for the antidepressant, fluvoxamine, in light of the comorbidity between PD and depression [47]. Both rasagline and selegiline should be avoided in patients with liver failure.
At clinically effective doses (50 or 100 mg, q.d.) oral safinamide has a high bioavailability (80-92%) [48], and a linear pharmacokinetic profile, with a Tmax value of 1.8-2.8 hours, a VD of 150 liters, plasma albumin binding of 92%, and elimination t1/2 of 21-24 hours [29]. Cmax values and steady-state plasma concentrations after treatment with 1.25 mg/kg safinamide are about 1,050 and 450 ng/ml, respectively [28]. Full inhibition of MAOB by safinamide is observed 2-3 hours after oral administration. Safinamide is not metabolized by cytochrome-P450, and this avoids major pharmacokinetic interactions with other drugs. The main metabolites of safinamide (acidic and N-dealkylated metabolites) are generated by amide hydrolases and MAOA, and are pharmacologically inert [48].
4. EFFICACY, SAFETY, AND TOLERABILITY OF MAOB INHIBITORS IN THE TREATMENT OF PD
4.1. Effects of MAOB Inhibitors on Motor Symptoms Associated with PD and their Impact on Motor Complications of L-DOPA Treatment
Motor symptoms associated with PD (rigidity, bradykinesia, resting tremor, postural abnormalities) reflect a defective network activity within the basal ganglia motor circuit, in which the neostriatum (caudate nucleus and putamen) represents the major input station. The neostriatum receives several inputs including glutamatergic fibers originating from the cerebral cortex, dopaminergic fibers originating from the SNpc, and serotonergic and noradrenergic fibers originating from brainstem nuclei. In humans, dopaminergic fibers are extremely long (axons can reach 4 meters of length) and make about two millions of synapses within the neostriatum. Medium spiny projecting neurons constitute the large majority (>90%) of striatal neurons. Large aspiny cholinergic interneurons and different population of GABAergic interneurons are also present in the neostriatum [49]. Striatal projection neurons use GABA as a neurotransmitter and give rise to the direct and indirect pathways of the basal ganglia motor circuit. Neurons of the direct pathway contain dynorphin and substance P and project to the internal globus pallidus (GPi) and SNpr, which constitute the output stations of the circuit and send inhibitory GABAergic projections to ventral motor thalamic nuclei (ventral anterior, ventromedial, and ventrolateral nuclei). Activation of the direct pathway by D1 dopamine receptors inhibits GPi and SNpr neurons, thereby stimulating ventral thalamic neurons projecting to the motor cortex. Striatal GABAergic projection neurons of the indirect pathway send axons to the external portion of the globus pallidus (GPe). GPe neurons send inhibitory GABAergic projections to the STN, which, in turn, send excitatory glutamatergic projections to the GPi and SNpr. Inhibition of striato-pallidal neurons of the indirect pathway mediated by D2 dopamine receptors restrains the activity of STN neurons, thereby reducing the excitatory drive to the GPi and SNpr and enhancing the activity of motor thalamic nuclei [49].
In PD, the progressive loss of striatal DA innervation results into a reduced activity of the direct pathway and an overactivity of the indirect pathway, which ultimately results into the hallmark motor signs of PD when striatal DA denervation exceeds 70-75% [3]. The glutamate released from cortico-striatal fibers stimulates both the direct and indirect pathways by activating ionotropic and metabotropic receptors postsynaptically localized on striatal projection neurons Fig. (1). An enhanced glutamate release at cortico-striatal and STN-GPi/SNpr synapses contributes to the pathophysiology of PD, and drugs that restrain the overactivity of glutamatergic fibers or block NMDA or mGlu5 receptors improve motor symptoms of PD [49].
Fig. (1).
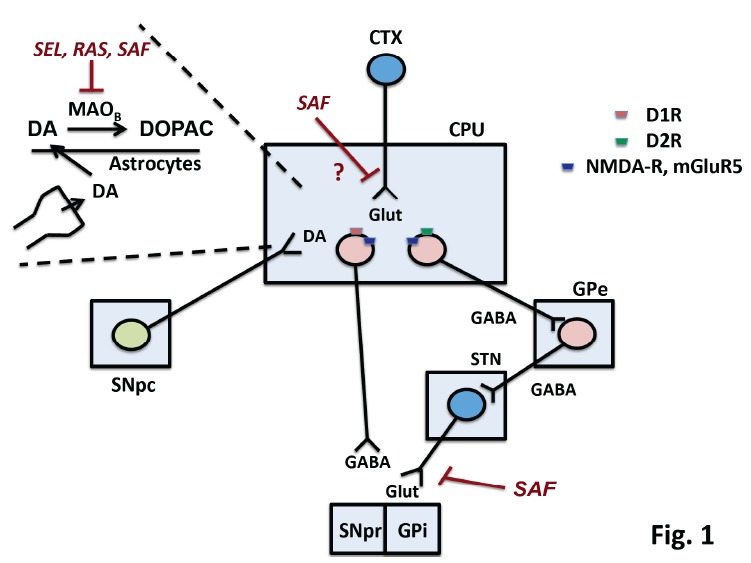
Mechanisms involved in the antiparkinsonian effect of selegiline (SEL), rasagiline (RAS), and safinamide (SAF). The Figure is a schematic drawing of the basal ganglia motor circuit, with dopaminergic neurons of the substantia nigra pars compacta (SNpc) projecting to the caudate nucleus and putamen (CPU). Striatal projection neurons of the direct and indirect pathway are stimulated and inhibited by D1 and D2 dopamine receptors (D1R, D2R), respectively. Both neurons express NMDA receptors (NMDA-R) and mGlu5 metabotroic glutamate receptors (mGluR5), which are activated by the glutamate released from cortico-striatal fibers (CTX = cerebral cortex). The external globus pallidus (GPe) and the subthatamic nucleus (STN) are the two stations of the indirect pathway. STN glutamatergic neurons send excitatory projections to the internal globus pallidus (GPi) and the substantia nigra pars reticulate (SNpr). Neurons of the direct patway send GABAergic projection to the GPi and SNpr. Dopamine (DA) released from nigro-striatal terminals is taken up by astrocytes, where it is transformed into DOPAC by MAOB. Inhibition of MAOB by SEL, RAS, and SAF enhances DA levels in the striatum. SAF can also inhibit glutamate release in the GP and SNpr. Inhibition of glutamate release in the striatum by SAF was not demonstrated by in vivo microdialysis experiments [33]. (The color version of the figure is available in the electronic copy of the article).
L-DOPA restores an efficient dopaminergic control of the basal ganglia motor circuit in the first years of treatment when plasma concentrations of the drug always fall inside the therapeutic window. Afterwards, L-DOPA treatment is complicated by the occurrence of motor fluctuations (e.g., early-morning off, wearing-off, delayed-on, no-on, and on-off phenomena) and LIDs. LIDs include tics, and choreiform, ballistic, and dystonic movements that more frequently occur at the time of peak concentrations of L-DOPA, but may be also present during the rising and declining phases of blood L-DOPA concentrations [50]. While changes in the pharmacokinetics of L-DOPA largely account for motor fluctuations, LIDs reflect mechanisms of maladaptive synaptic plasticity at the synapses between cortico-striatal fibers and striatal projection neurons of the direct pathway. Because activation of D1 receptors is critically involved in the induction and expression of LIDs, these adverse effects impose reductions in the daily dose and frequency of administration of L-DOPA, which may render the control of motor symptoms suboptimal [51].
By reducing brain DA metabolism, MAOB inhibitors have a mild-to-moderate effect on motor symptoms on their own and reduce the time spent in the off phase when combined with L-DOPA in patients showing motor fluctuations. In addition, inhibition of MAOB may allow a reduction of a daily dose of L-DOPA without compromising motor control in patients with LIDs. Inhibition of glutamate release by safinamide is an additional powerful mechanism that may highly contribute to improve motor symptoms (see below).
Selegiline was initially developed as a psychostimulant and antidepressant, and then used in the treatment of PD after the discovery that it behaves as an irreversible MAOB inhibitor [52]. Selegiline is used at daily doses of 5 or 10 mg both in monotherapy and in combination with L-DOPA (a dose of 10 mg/die is indicated in patients with motor fluctuations). The antiparkinsonian effect of an early treatment with selegiline in monotherapy was demonstrated in the DATATOP trial [53], which is discussed in further detail below. In the Sinemet-Deprenyl-Parlodel (SINDEPAR) study, selegiline treatment improved UPDRS score in PD patients when combined with either L-DOPA/carbidopa or with the DA receptor agonist, bromocriptine [54]. The UPDRS (Unified Parkinson’s Disease Rating Scale) is the most widely used scale in clinical studies of PD and is particularly helpful for the evaluation of the longitudinal course of the disease. In advanced PD, selegiline, added to pre-existing L-DOPA treatment, was able to improve tremor, rigidity, bradykinesia and dyskinesias, and was effective in reducing the frequency and severity of motor fluctuations [55].
Rasagiline has been approved as monotherapy for the early treatment of PD, and in association with L-DOPA in patients with motor fluctuations. The recommended daily dose is 1 mg. The therapeutic efficacy of rasagiline is largely discussed in two excellent reviews [15, 29]. Two clinical trials named TEMPO (TVP-1012 in Early Monotherapy for PD Outpatients) and ADAGIO (Attenuation of Disease Progression with Azilect Given Once-daily) showed the efficacy of rasagiline monotherapy in the early stages of PD as evaluated by UPDRS score and quality of life [56, 57]. A post-hoc analysis of the ADAGIO trial showed a good efficacy of rasagiline on tremor, bradykinesia, rigidity, and composite postural-instability-gait-difficulty, which was maintained for at least one year [58]. The efficacy of rasagiline in patients with advanced PD treated with L-DOPA and displaying motor fluctuations was examined in two clinical trials named LARGO (Lasting effect in Adjunct therapy with Rasagiline given Once-daily) and PRESTO (Parkinson’s Rasagiline: Efficacy and Safety in the Treatment of ‘Off’). In both trials, rasagiline was effective in reducing the time spent in the off phase [43, 59]. In the PRESTO trial, rasagiline was effective in reducing motor fluctuations regardless of the presence of other drugs combined with L-DOPA, such as DA receptor agonists or COMT inhibitors; however, rasagiline increased on-phase associated dyskinesias with respect to placebo [60]. A recent head-to-head 3-year retrospective case-control study evaluating the efficacy of selegiline and rasagiline in PD showed that treatment with either of the two MAOB inhibitors was associated with a lower daily dose of L-DOPA and a lower frequency of LIDs [61]. A reduced risk of LIDs during treatment with selegiline and rasagiline was confirmed in another retrospective cross-sectional cohort studies comparing patients treated with either of the two MAOB inhibitors for at least one year with patients who had never received MAOB inhibitors [62]. We wish to highlight that neither selegiline nor rasagiline improves LIDs through a direct mechanism, but they allow to reduce the daily dose od L-DOPA without a major impairment of motor control. Reversible Pisa syndrome, a postural deformity characterized by lateral trunk flexion, has been occasionally associated with rasagiline treatment in a patient affected by PD [63, 64]. Whether this rare complication is due to inhibition of DA metabolism or other mechanisms remains to be established.
Safinamide has been approved by FDA and EMA as an adjunctive therapy to L-DOPA and other antiparkinsonian drugs in patients affected by mid-advanced PD with motor fluctuations. A daily dose of 50 mg is required for reversible full inhibition of MAOB activity. At the dose of 100 mg safinamide also inhibits glutamate release, an effect that may contribute to the efficacy of the drug on motor and non-motor symptoms and fluctuations. In principle, this mechanism could restrain dyskinesias that might result from the inhibition of DA metabolism.
As outlined above, LIDs are the phenotypic expression of a maladaptive form of activity-dependent synaptic plasticity at excitatory synapses between cortico-striatal fibers and striatal projection neurons of the direct pathway. Paolo Calabresi and his Associates have shown that long-term potentiation (LTP) of excitatory synaptic transmission at cortico-striatal synapses requires the activation of D1, NMDA, mGlu1 and mGlu5 receptors [65]. In the pathophysiology of LIDs, striatal LTP likely underlies the priming effect, i.e., the re-occurrence of LIDs when L-DOPA is administered again after a period of withdrawal. In DA-denervated rats developing LIDs, striatal LTP becomes inflexible and cannot be depotentiated [66, 67]. In both animals and humans, LIDs are associated with an enhanced glutamate release in the neostriatum and SNpr [68]. Drugs that inhibit glutamate receptors, such as the NMDA receptor antagonist, amantandine, and a series of negative allosteric modulators of mGlu5 receptors (e.g., mavoglurant and dipraglurant) show efficacy in reducing LIDs in experimental animals and PD patients [51]. Safinamide is not a direct glutamate receptor antagonist, and cannot be considered as an antidyskinetic drug. However, by inhibiting glutamate release, safinamide may reduce the excitatory overdrive of the direct pathway underlying with LIDs, and, at the same time, may improve motor symptoms by reducing excitation of the indirect pathway. The latter effect may allow the reduction of the daily dose of L-DOPA, with a favourable effect on LIDs. Morari et al. [33] have found that safinamide reduces the evoked release of glutamate in the SNpr and GP, but not in the dorsal striatum, of freely moving rats (see above). However, it cannot be excluded that safinamide can also restrain glutamatergic transmission in the neostriatum under conditions of excitatory overdrive, as occurs in PD. Thus, the overall impact of safinamide on LIDs would result from the combination of the reduced DA metabolism (which is expected to enhance LIDs), the inhibition of glutamate release (which is expected to reduce LIDs), and the antiparkinsonian effect (which allows to reduce the daily dose of L-DOPA with beneficial effects on LIDs) Fig. (1). In “parkinsonian” monkeys safinamide showed great efficacy in reducing LIDs, and, as opposed to amantadine, could also prolong the antiparkinsonian effects of L-DOPA [69].
The efficacy of safinamide in patients with mid-advanced stage PD and motor fluctuations under treatment with L-DOPA or other dopaminergic agents has been examined in two phase-III clinical studies (Study 016 and SETTLE trials) [12, 13, 70, 71]. In both trials, a 24-week treatment with safinamide improved on- without troublesome dyskinesias and reduced off-times after 2 or 4 weeks of treatment. Safinamide also improved the overall clinical status of the patients in both studies. Remarkably, in the 18-months extension of Study 016 (“Study 018”) safinamide treatment significantly improved the DRS (dyskinesia rating scale) score in the subgroups of patients with moderate-to-severe dyskinesias [71]. This effect was seen with the daily dose of 100 mg, which should be effective in inhibiting glutamate release [72]. Interestingly, zonisamide, which shares with safinamide the ability to inhibit both VSSCs and MAOB, has shown efficacy in improving motor fluctuations in PD patients in a double-blind randomized study [73].
4.2. Effects of MAOB Inhibitors on Non-motor Symptoms Associated with PD
Non-motor symptoms are often considered as secondary symptoms in PD, but they critically affect the quality of life of patients, and are difficult to treat. PD is frequently associated with depression, fatigue, pain, and dysautonomic disorders such as orthostatic hypotension, constipation and genitourinary disorders. Several lines of evidence suggest that MAOB inhibitors can be useful in the management of all these symptoms.
Depression is observed in a large proportion of PD patients (up to 50%) and is clinically different with respect to depression not associated with PD. Anxiety and pessimism predominate in PD patients, whereas the feeling of guilt, self-reproach, and suicidal tendencies are rarely observed. Mood changes are often associated with motor fluctuation in L-DOPA-treated patients, with a reduction of mood being observed in the off phase [74, 75]. Selegiline has been shown to produce antidepressant-like effects in experimental animals. For example, selegiline treatment partially reverses the deficit in effort-related choice behavior induced by tetrabenazide, a drugs that inhibit the vesicular monoamine transporter, VMAT-2 [76]. Selegiline also improves depression-like behavior in mice subjected to maternal separation, a model of stress-related disorders endowed with face, construct, and pharmacological validity [77]. Interestingly, selegiline was also found to improve depression-like behavior in mice with genetic deletion of CD157/BST1, which is a risk factor for PD [75]. Transdermal delivery of selegiline is FDA-approved for the treatment of depression and might represent a treatment option for PD patients with depressive symptoms [78]. To what extent the metabolites L-methamphetamine and L-amphetamine contribute to the overall antidepressant effect of selegiline remains to be determined.
The effect of rasagiline on depressive symptoms in PD patients is less clear. The effect of a 12-week treatment with rasagiline on depressive and cognitive symptoms in non-demented PD patients was evaluated in a double-blind, placebo-controlled, clinical trial [74]. At week 12, rasagiline did not differ from placebo in improving depressive symptoms, as assessed by the Beck Depression Inventory scale; however, a significant difference in favour of rasagiline was found after 4 weeks of treatment [74]. A sub-analysis of the ADAGIO study showed a positive effect of rasagiline treatment on the fatigue [79], and rasagiline was also found to improve non-motor aspects of experiences of daily living [80].
The effect of safinamide (100 mg/die) on the emotional well-being domain of Parkinson’s Disease Questionnaire (PDQ-39) and on GRID-HAMD score (a standardization of the Hamilton Depression Rating Scale) was evaluated in PD patients with motor fluctuation. Safinamide displayed a beneficial effect on mood, and this has been ascribed to the improvement in wearing-off and to the effect of the drug on glutamatergic transmission [81]. Interestingly, safinamide improved painful cramps or spasms and hot- and cold-allodynia in PD patients (items 37 and 39 of PDQ-39), and allowed to reduce the use of concomitant pain treatments by >25% [82, 83]. Sleep disorders, such as RBD (REM sleep behavioural disorder), nocturnal apnoeas, and restless leg syndrome (RLS), are frequently associated with PD [3]. Three-month treatment with safinamide (100 mg/die) added to L-DOPA has been shown to markedly improve RLS in two patients with motor fluctuations [84].
4.3. Safety and Tolerability Profile of MAOB Inhibitors
Class-related adverse effects of the three MAOB inhibitors result from the enhancement of DAergic transmission in the CNS and in the periphery. In the case of selegiline, the formation of amphetamine-based metabolites might contribute to psychiatric, neurological, and cardiovascular effects.
Psychiatric adverse effects, such as delirium, hallucinations, and agitation, are more frequent with selegiline than with other antiparkinsonian drugs. Selegiline may also cause sedation or dyskinesias [85]. Cardiovascular adverse effects of selegiline include orthostatic hypotension, hypertension, atrial fibrillation, and other types of cardiac arrhythmias, as reported in the DATATOP study [86]. The risk of cardiovascular effects increases when selegiline is administered in combination with L-DOPA [87]. Amphetamine-based metabolites of selegiline may contribute to the psychiatric and cardiovascular adverse effects of the drug.
Data of TEMPO, LARGO and PRESTO trials (see below) indicate that rasagiline is well tolerated. In these trials, the adverse effects of rasagiline were classified as uncomfortable but not dangerous (e.g., asthenia, nausea, arthralgia, back pain, and headache). Two post hoc analyses of TEMPO and PRESTO studies showed an increased risk of age-related depression, postural hypotension and hallucinations in patients treated with rasagiline. Combination of rasagiline with other dopaminergic therapies is in general well tolerated and does not cause a decline in mental function [88]. The possibility that high doses of rasagiline (and selegiline) inhibit MAOA has been discussed above. However, tyramine-dietary restrictions are not recommended in patients treated with rasagiline; accordingly, tyramine challenge following a few weeks of rasagiline administration did not cause significant changes in blood pressure [88, 89]. In an open trial, a switch from selegiline (7.5 mg, daily) to rasagiline (1 mg, daily) improved motor behavior, motor complications, mood, and sleep in 30 patients affected by PD [90].
Safinamide treatment is well tolerated, and the reported adverse effects in clinical studies were mild-to-moderate and included gastro-intestinal effects, fever, hypertension, and back pain. The safety profile of the drug is excellent. Because of the potent and selective MAOB inhibition dyskinesias may occur as a result of the enhanced DA levels in the neostriatum. However, this risk is mitigated by the inhibition of glutamate release (see below). In the 018 extension study (see above), the incidence of dyskinesias and other DA-related adverse effects was similar between safinamide and placebo [2, 29, 70, 91-93].
Because of the comorbidity between PD and depression, MAOB inhibitors might be associated with SSRIs, serotonin and noradrenaline reuptake inhibitors (SNRIs), or other antidepressants that enhance serotonergic transmission. This association raises the potential concern of the serotonin syndrome, a rare but potentially fatal medical condition resulting from an excessive increase in central and peripheral serotonergic transmission. After reviewing the supporting evidence, Aboukarr and Giudice (2018) concluded that serotonin syndrome is infrequent and that combination of SSRIs and MAOB inhibitors is well tolerated providing that the SSRI dose is kept low [94]. In support to this statement, the serotonin syndrome was not observed when rasagiline was combined with SSRIs or other serotonergic antidepressants [88, 95-97].
Concerns related to pharmacokinetic interactions with other drugs have been discussed above. Again, safinamide differs from selegiline and rasagiline because it is not metabolized by, and does not influence the activity of, cytochrome-P450.
5. MAOB INHIBITORS AS POTENTIAL DISEASE-MODIFYING DRUGS IN PD
There are no treatments that halt the progression of PD perhaps because of the heterogeneity and redundance of the molecular events underlying the pathophysiology of neuronal death. However, among all classes of drugs that are currently used in the treatment of PD, MAOB inhibitors have consistently shown the greatest neuroprotective potential, suggesting that an early use of these drugs might have a beneficial effect on the disease course (for an elegant review, see Riederer and Müller, 2017) [98]. The DATATOP trial (Deprenyl and Tocopherol Antioxidant Therapy of Parkinsonism) was the first large multicentre clinical study examining the effect of early treatment with selegiline in patients affected by PD. Selegiline (10 mg/day), but not tocopherol, delayed the onset of L-DOPA treatment, suggesting that the drug was able to slow the progression of neurological disability in patients affected by PD [53, 99]. However, data of the DATATOP trial were confounded by the long-lasting symptomatic effect of selegiline (at that time not recognized), taking into account that the half-life of MAOB inhibition exceeded the duration of drug withdrawal in the study [100, 101]. Two subsequent clinical trials examined the protective and disease-modifying activity of rasagiline using the strategy of the “early vs. delayed start”. In the TEMPO trial rasagiline showed a greater therapeutic effect when administered for 12 months rather than in the last 6 months [56]. In the ADAGIO trial, 1176 patients were treated daily with rasagiline (1 or 2 mg) for 72 weeks or, alternatively, with placebo for the first 36 weeks followed by rasagiline in the remaining 36 weeks. The increase in UPDRS score at the end of the 72 weeks was smaller in the “early-rasagiline start” than in the “delayed-rasagiline start” group in patients treated with 1 mg of rasagiline [57]. In principle, these findings support the protective activity of rasagiline in PD. However, there are potential pitfalls that cannot be ignored. For example, the difference in the UPDRS score between the two groups treated with 1 mg rasagiline is very small (<2 points), the UPDRS scoring is not entirely objective (particularly when repeatedly applied to the same patient), and, more important, no changes between early and delayed starts were observed when rasagiline was administered at the dose of 2 mg/day [97]. It is the right time to perform comparative studies with selegiline, rasagiline, and safinamide on neuroprotection using more objective markers, such as longitudinal SPECT analysis of striatal dopaminergic terminals with 123I-ioflupane (DaTSCAN) or fluid biomarkers, such as light-chain neurofilaments in the blood or CSF, or α-synuclein in the CSF [102].
Most of preclinical studies exploring the neuroprotective potential of MAOB inhibitors were performed using the MPTP model in mice and monkeys. As highlighted above, the parkinsonian toxin, MPTP, crosses the blood-brain barrier and is then transformed into the active metabolite, MPP+. MPP+ is inwardly transported into dopaminergic nerve terminals, and then causes neurodegeneration by inhibiting the mitochondrial respiratory chain [18]. Of great relevance to the present review, conversion of MPTP into MPDP+, and then to MPP+, occurs in astrocytes and is catalyzed by MAOB [103, 104]. A large body of evidence indicates that all MAOB inhibitors, including zonisamide, protect SNpc neurons against damage caused by acute or subacute doses of MPTP in mice or monkeys [34, 105-116]. These models are extremely helpful for the screening of neuroprotective compounds although they do not mimic the slowly progressive degeneration of dopaminergic neurons occurring in PD. Interestingly, MAOB inhibitors were also able to rescue SNpc neurons when administered after MPTP, and displayed neuroprotective activity against damage caused by i.c.v. or intracerebral injection of MPP+ [114, 117-119]. This suggests that there are mechanisms other than inhibition of MPTP metabolism that contribute to neuroprotection by the three MAOB inhibitors. Accordingly, selegiline was found to protect against MPTP toxicity at low doses that are unable to inhibit MAOB in vivo [119]. Cell culture and in vivo studies demonstrate that selegiline and rasagiline enhance the production of neurotrophic factors, such as nerve growth factor (NGF), glial cell-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF) [119-125]. GDNF, in particular, supports the survival of SNpc dopaminergic neurons, and is considered as a potential target for disease-modifying drugs in PD [126-130]. Clinical studies with intraputaminal delivery of GDNF in PD have generated contrasting results [131], perhaps because exogenous GDNF has limited access to membrane receptors. By increasing endogenous production of GDNF MAOB inhibitors could overcome this limitation thereby boosting the pro-survival activity of GDNF.
CONCLUSION
Although safinamide is not indicated in monotherapy for the early treatment of PD, it has great neuroprotective potential owing to its ability to inhibit both MAOB activity and glutamate release. Again, PD is characterized by an increased excitatory drive in the basal ganglia motor circuit, and overactive excitatory fibers projecting from STN neurons to the SNpc might contribute to neuronal death through an excitotoxic mechanism. A role for glutamate in PD-associated neuronal death is suggested by the evidence that NMDA or mGlu5 receptor blockade is protective against neuronal damage caused by MPTP or its metabolite, MPP+ [132-137]. It will be interesting to examine the disease-modifying activity of safinamide in animal models that recapitulate the slowly progressive damage of SNpc neurons that is typical of PD, such as chronic MPTP models [138] or genetic mouse models harboring PD-associated mutations (e.g., LRRK2-G2019S) [139].
Acknowledgements
Declared none.
Consent for Publication
Not applicable.
Funding
The study was funded by Zambon, Italy.
Conflict of Interest
The authors declare no conflict of interest, financial or otherwise.
References
- 1.Titova N., Padmakumar C., Lewis S.J.G., Chaudhuri K.R. Parkinson’s: a syndrome rather than a disease? J. Neural Transm. (Vienna) 2017;124(8):907–914. doi: 10.1007/s00702-016-1667-6. [http://dx.doi.org/10.1007/s00702-016-1667-6]. [PMID: 28028643]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baltasar-Rodríguez L.M., Millán-Guerrero R.O., Aceves-Themsel R., Isais-Millán S., Delgado-Enciso I. Longitudinal study of three families with familial Parkinson’s disease. Gac. Med. Mex. 2006;142(5):387–391. [PMID: 17128818]. [PubMed] [Google Scholar]
- 3.Kalia L.V., Lang A.E. Parkinson’s disease. Lancet. 2015;386(9996):896–912. doi: 10.1016/S0140-6736(14)61393-3. [http://dx.doi.org/10.1016/S0140-6736(14) 61393-3]. [PMID: 25904081]. [DOI] [PubMed] [Google Scholar]
- 4.Schneider S.A., Alcalay R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017;32(11):1504–1523. doi: 10.1002/mds.27193. [http://dx.doi.org/10.1002/mds.27193]. [PMID: 29124790]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bose A., Beal M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016;139(Suppl. 1):216–231. doi: 10.1111/jnc.13731. [http://dx. doi.org/10.1111/jnc.13731]. [PMID: 27546335]. [DOI] [PubMed] [Google Scholar]
- 6.Gao G., Wang Z., Lu L., Duan C., Wang X., Yang H. Morphological analysis of mitochondria for evaluating the toxicity of α-synuclein in transgenic mice and isolated preparations by atomic force microscopy. Biomed. Pharmacother. 2017;96:1380–1388. doi: 10.1016/j.biopha.2017.11.057. [http://dx.doi.org/10.1016/j.biopha.2017.11.057]. [PMID: 29169728]. [DOI] [PubMed] [Google Scholar]
- 7.Plotegher N., Berti G., Ferrari E., Tessari I., Zanetti M., Lunelli L., Greggio E., Bisaglia M., Veronesi M., Girotto S., Dalla Serra M., Perego C., Casella L., Bubacco L. DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Sci. Rep. 2017;7:40699. doi: 10.1038/srep40699. [http://dx.doi.org/10.1038/ srep40699]. [PMID: 28084443]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J., Culp M.L., Craver J.G., Darley-Usmar V. Mitochondrial function and autophagy: integrating proteotoxic, redox, and metabolic stress in Parkinson’s disease. J. Neurochem. 2018;144(6):691–709. doi: 10.1111/jnc.14308. [http://dx.doi.org/10.1111/jnc.14308]. [PMID: 29341130]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinkle J.T., Perepezko K., Rosenthal L.S., Mills K.A., Pantelyat A., Mari Z., Tochen L., Bang J.Y., Gudavalli M., Yoritomo N., Butala A., Bakker C.C., Johnson V., Moukheiber E., Dawson T.M., Pontone G.M. Markers of impaired motor and cognitive volition in Parkinson’s disease: Correlates of dopamine dysregulation syndrome, impulse control disorder, and dyskinesias. Parkinsonism Relat. Disord. 2018;47:50–56. doi: 10.1016/j.parkreldis.2017.11.338. [http://dx.doi.org/10.1016/j.parkreldis. 2017.11.338]. [PMID: 29198499]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Picconi B., Hernández L.F., Obeso J.A., Calabresi P. Motor complications in Parkinson’s disease: Striatal molecular and electrophysiological mechanisms of dyskinesias. Mov. Disord. 2017;33(6):867–876. doi: 10.1002/mds.27261. [PMID: 29219207]. [DOI] [PubMed] [Google Scholar]
- 11.Morris J.G. The management of Parkinson’s disease. Aust. N. Z. J. Med. 1982;12(2):195–205. doi: 10.1111/j.1445-5994.1982.tb02460.x. [http://dx.doi.org/10.1111/j.1445-5994.1982.tb02460.x]. [PMID: 7046720]. [DOI] [PubMed] [Google Scholar]
- 12.Blair H.A., Dhillon S. Safinamide: A review in Parkinson’s disease. CNS Drugs. 2017;31(2):169–176. doi: 10.1007/s40263-017-0408-1. [http://dx.doi.org/10.1007/ s40263-017-0408-1]. [PMID: 28110399]. [DOI] [PubMed] [Google Scholar]
- 13.Bonuccelli U. Effects of safinamide on motor complications and pain in advancing Parkinson’s disease – Post Hoc analyses of pivotal trials. Eur. Neurol. Rev. 2015;10(2):1–7. [http://dx.doi. org/10.17925/ENR.2015.10.02.176]. [Google Scholar]
- 14.Fabbrini G., Abbruzzese G., Marconi S., Zappia M. Selegiline: a reappraisal of its role in Parkinson disease. Clin. Neuropharmacol. 2012;35(3):134–140. doi: 10.1097/WNF.0b013e318255838b. [http://dx.doi.org/10.1097/WNF.0b013e 318255838b]. [PMID: 22592509]. [DOI] [PubMed] [Google Scholar]
- 15.McCormack P.L. Rasagiline: A review of its use in the treatment of idiopathic Parkinson’s disease. CNS Drugs. 2014;28(11):1083–1097. doi: 10.1007/s40263-014-0206-y. [http://dx.doi.org/10.1007/s40263-014-0206-y]. [PMID: 25322951]. [DOI] [PubMed] [Google Scholar]
- 16.Miklya I. The significance of selegiline/(-)-deprenyl after 50 years in research and therapy (1965-2015). Mol. Psychiatry. 2016;21(11):1499–1503. doi: 10.1038/mp.2016.127. [http://dx.doi.org/10.1038/mp.2016.127]. [PMID: 27480491]. [DOI] [PubMed] [Google Scholar]
- 17.Stocchi F., Fossati C., Torti M. Rasagiline for the treatment of Parkinson’s disease: an update. Expert Opin. Pharmacother. 2015;16(14):2231–2241. doi: 10.1517/14656566.2015.1086748. [http://dx.doi.org/10.1517/14656566.2015. 1086748]. [PMID: 26364897]. [DOI] [PubMed] [Google Scholar]
- 18.Jenner P., Marsden C.D. The actions of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in animals as a model of Parkinson’s disease. J. Neural Transm. Suppl. 1986;20:11–39. [PMID: 3091760]. [PubMed] [Google Scholar]
- 19.Binda C., Li M., Hubalek F., Restelli N., Edmondson D.E., Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA. 2003;100(17):9750–9755. doi: 10.1073/pnas.1633804100. [http://dx.doi.org/10.1073/pnas.1633804100]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Binda C., Hubálek F., Li M., Herzig Y., Sterling J., Edmondson D.E., Mattevi A. Binding of rasagiline-related inhibitors to human monoamine oxidases: a kinetic and crystallographic analysis. J. Med. Chem. 2005;48(26):8148–8154. doi: 10.1021/jm0506266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagy J., Kalàsz H. Selegiline binding to monoamine oxidase enzyme in the rear view mirror. RJLBPCS. 2017;3(2):99–108. [Google Scholar]
- 22.Fowler J.S., Volkow N.D., Logan J., Wang G.J., MacGregor R.R., Schyler D., Wolf A.P., Pappas N., Alexoff D., Shea C. Slow recovery of human brain MAO B after L-deprenyl (Selegeline) withdrawal. Synapse. 1994;18(2):86–93. doi: 10.1002/syn.890180203. [http://dx.doi. org/10.1002/syn.890180203]. [PMID: 7839316]. [DOI] [PubMed] [Google Scholar]
- 23.Freedman N.M., Mishani E., Krausz Y., Weininger J., Lester H., Blaugrund E., Ehrlich D., Chisin R. In vivo measurement of brain monoamine oxidase B occupancy by rasagiline, using (11)C-l-deprenyl and PET. J. Nucl. Med. 2005;46(10):1618–1624. [PMID: 16204711]. [PubMed] [Google Scholar]
- 24.Caccia C., Maj R., Calabresi M., Maestroni S., Faravelli L., Curatolo L., Salvati P., Fariello R.G. Safinamide: from molecular targets to a new anti-Parkinson drug. Neurology. 2006;67(7) doi: 10.1212/wnl.67.7_suppl_2.s18. [DOI] [PubMed] [Google Scholar]
- 25.Leonetti F., Capaldi C., Pisani L., Nicolotti O., Muncipinto G., Stefanachi A., Cellamare S., Caccia C., Carotti A. Solid-phase synthesis and insights into structure-activity relationships of safinamide analogues as potent and selective inhibitors of type B monoamine oxidase. J. Med. Chem. 2007;50(20):4909–4916. doi: 10.1021/jm070725e. [http://dx.doi.org/10.1021/jm070725e]. [PMID: 17824599]. [DOI] [PubMed] [Google Scholar]
- 26.Youdim M.B., Gross A., Finberg J.P. Rasagiline N-propargyl-1R(+)-aminoindan, a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br. J. Pharmacol. 2001;132(2):500–506. doi: 10.1038/sj.bjp.0703826. [http://dx.doi.org/10.1038/sj.bjp.0703826]. [PMID: 11159700]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lecht S., Haroutiunian S., Hoffman A., Lazarovici P. Rasagiline - a novel MAO B inhibitor in Parkinson’s disease therapy. Ther. Clin. Risk Manag. 2007;3(3):467–474. [PMID: 18488080]. [PMC free article] [PubMed] [Google Scholar]
- 28.Marzo A., Dal Bo L., Monti N.C., Crivelli F., Ismaili S., Caccia C., Cattaneo C., Fariello R.G. Pharmacokinetics and pharmacodynamics of safinamide, a neuroprotectant with antiparkinsonian and anticonvulsant activity. Pharmacol. Res. 2004;50(1):77–85. doi: 10.1016/j.phrs.2003.12.004. [http://dx.doi.org/10.1016/j.phrs.2003.12.004]. [PMID: 15082032]. [DOI] [PubMed] [Google Scholar]
- 29.Stocchi F., Torti M. Adjuvant therapies for Parkinson’s disease: critical evaluation of safinamide. Drug Des. Devel. Ther. 2016;10:609–618. doi: 10.2147/DDDT.S77749. [http://dx.doi.org/10.2147/DDDT.S77749]. [PMID: 26917951]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartl J., Müller T., Grünblatt E., Gerlach M., Riederer P. Chronic monoamine oxidase-B inhibitor treatment blocks monoamine oxidase-A enzyme activity. J. Neural Transm. (Vienna) 2014;121(4):379–383. doi: 10.1007/s00702-013-1120-z. [http://dx.doi.org/10.1007/s00702-013-1120-z]. [PMID: 24272680]. [DOI] [PubMed] [Google Scholar]
- 31.Müller T., Riederer P., Grünblatt E. Determination of monoamine oxidase A and B activity in long-term treated patients with parkinson disease. Clin. Neuropharmacol. 2017;40(5):208–211. doi: 10.1097/WNF.0000000000000233. [http://dx.doi.org/10.1097/WNF.0000000000000233]. [PMID: 28682929]. [DOI] [PubMed] [Google Scholar]
- 32.Salvati P., Maj R., Caccia C., Cervini M.A., Fornaretto M.G., Lamberti E., Pevarello P., Skeen G.A., White H.S., Wolf H.H., Faravelli L., Mazzanti M., Mancinelli E., Varasi M., Fariello R.G. Biochemical and electrophysiological studies on the mechanism of action of PNU-151774E, a novel antiepileptic compound. J. Pharmacol. Exp. Ther. 1999;288(3):1151–1159. [PMID: 10027853]. [PubMed] [Google Scholar]
- 33.Morari M., Brugnoli A., Pisanò C.A., Novello S., Caccia C., Melloni E., Padoani G., Vailati S., Sardina M. Safinamide differentially modulates In Vivo glutamate and GABA release in the rat hippocampus and basal ganglia. J. Pharmacol. Exp. Ther. 2018;364(2):198–206. doi: 10.1124/jpet.117.245100. [http://dx.doi.org/10.1124/jpet.117.245100]. [PMID: 29167350]. [DOI] [PubMed] [Google Scholar]
- 34.Sonsalla P.K., Wong L.Y., Winnik B., Buckley B. The antiepileptic drug zonisamide inhibits MAO-B and attenuates MPTP toxicity in mice: clinical relevance. Exp. Neurol. 2010;221(2):329–334. doi: 10.1016/j.expneurol.2009.11.018. [http://dx.doi.org/10.1016/j.expneurol.2009.11.018]. [PMID: 19948168]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fox S.H., Katzenschlager R., Lim S.Y., Barton B., de Bie R.M.A., Seppi K., Coelho M., Sampaio C. International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson’s disease. Mov. Disord. 2018;33(8):1248–1266. doi: 10.1002/mds.27372. [http://dx.doi.org/10.1002/mds.27372]. [PMID: 29570866]. [DOI] [PubMed] [Google Scholar]
- 36.Uemura M.T., Asano T., Hikawa R., Yamakado H., Takahashi R. Zonisamide inhibits monoamine oxidase and enhances motor performance and social activity. Neurosci. Res. 2017;124:25–32. doi: 10.1016/j.neures.2017.05.008. [http://dx.doi.org/10.1016/j.neures.2017.05.008]. [PMID: 28624436]. [DOI] [PubMed] [Google Scholar]
- 37.Heinonen E.H., Anttila M.I., Karnani H.L., Nyman L.M., Vuorinen J.A., Pyykkö K.A., Lammintausta R.A. Desmethylselegiline, a metabolite of selegiline, is an irreversible inhibitor of monoamine oxidase type B in humans. J. Clin. Pharmacol. 1997;37(7):602–609. doi: 10.1002/j.1552-4604.1997.tb04342.x. [http://dx.doi.org/10.1002/j.1552-4604.1997.tb04342. x]. [PMID: 9243353]. [DOI] [PubMed] [Google Scholar]
- 38.Mahmood I. Clinical pharmacokinetics and pharmacodynamics of selegiline. An update. Clin. Pharmacokinet. 1997;33(2):91–102. doi: 10.2165/00003088-199733020-00002. [http://dx.doi.org/10.2165/00003088-199733020-00002]. [PMID: 9260033]. [DOI] [PubMed] [Google Scholar]
- 39.Benetton S.A., Fang C., Yang Y.O., Alok R., Year M., Lin C.C., Yeh L.T. P450 phenotyping of the metabolism of selegiline to desmethylselegiline and methamphetamine. Drug Metab. Pharmacokinet. 2007;22(2):78–87. doi: 10.2133/dmpk.22.78. [http://dx.doi.org/10.2133/dmpk.22.78]. [DOI] [PubMed] [Google Scholar]
- 40.Nishiya Y., Hagihara K., Ito T., Tajima M., Miura S., Kurihara A., Farid N.A., Ikeda T. Mechanism-based inhibition of human cytochrome P450 2B6 by ticlopidine, clopidogrel, and the thiolactone metabolite of prasugrel. Drug Metab. Dispos. 2009;37(3):589–593. doi: 10.1124/dmd.108.022988. [http://dx.doi.org/10.1124/dmd.108.022988]. [PMID: 19047469]. [DOI] [PubMed] [Google Scholar]
- 41.Chen J.J., Ly A.V. Rasagiline: A second-generation monoamine oxidase type-B inhibitor for the treatment of Parkinson’s disease. Am. J. Health Syst. Pharm. 2006;63(10):915–928. doi: 10.2146/ajhp050395. [http://dx. doi.org/10.2146/ajhp050395]. [PMID: 16675649]. [DOI] [PubMed] [Google Scholar]
- 42.New drug treatment for Parkinson’s disease. FDA Consum. 2006;40(4):7. [PMID: 17245831]. [PubMed] [Google Scholar]
- 43.Elmer L., Davis K., Fazzini E., Chin L., Scott B., Wilcox C., Colcher A., Reichwein S., Hauser R., Gauge L., Leehey M., Ondo W., Lewitt P., Kaminski P., Harrison M., Rost-Ruffner E., Racette B., Cooper P., Frei K., Luong N., Pahwa R., Dicarlo J., Lew M.F., Kawai C., Jennings D., Caplan K., Fink J.S., Novak P., Thomas C., Chouinard S., Beauvais C., Tabbal S., Uc E., Patterson J., Fernandez H., Bertoni J.M., Skrypnik L., Panisset M., Fortin M.J., Molho E., Nash J., Dalvi A., Schwieterman D., Goetz C., Janko K., Mendis T., Mahtat D., Gray P., Camicioli R., King P., Wojcieszek J., Kumar R., Judd D., Margolin D., Margolin J.J., Clem F., Gordon P., Marshall F., Berry D., Rottenberg D., Hansen J., Gordon M.F., Hassan M., Keltonic P., Hermanowicz N., Niswonger S., Jog M., Horn C., Shulman L., Cines M., Suchowersky O., Furtado S., Derwent L., Samanta J., Williamson K., Ramos C.S., Berrios L., Roque S., Sutton J., Young J., Waters C., Benabou R., Singer C., Koller W., Martin D., Sethi K., Carpenter J., Rao J., Cook M., Feigin A., Cox M., Riley D., Calabrese V., Standaert D., Tennis M., Manyam B., Whetteckey J., Wulbrecht B., Rajput A., Golbe L., Caputo D., Freeman A., McGinn L., Subramanian T., Ajax T., Aminoff M., Hevezi J., So J. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: The PRESTO study. Arch. Neurol. 2005;62(2):241–248. doi: 10.1001/archneur.62.2.241. [http://dx.doi.org/10.1001/archneur.62.2.241]. [PMID: 15710852]. [DOI] [PubMed] [Google Scholar]
- 44.Brouwers E.E., Söhne M., Kuipers S., van Gorp E.C., Schellens J.H., Koks C.H., Beijnen J.H., Huitema A.D. Ciprofloxacin strongly inhibits clozapine metabolism: two case reports. Clin. Drug Investig. 2009;29(1):59–63. doi: 10.2165/0044011-200929010-00006. [http://dx.doi.org/10.2165/0044011-200929010-00006]. [PMID: 19067475]. [DOI] [PubMed] [Google Scholar]
- 45.Spina E., Santoro V., D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin. Ther. 2008;30(7):1206–1227. doi: 10.1016/s0149-2918(08)80047-1. [http://dx.doi.org/ 10.1016/S0149-2918(08)80047-1]. [PMID: 18691982]. [DOI] [PubMed] [Google Scholar]
- 46.Zvyaga T., Chang S.Y., Chen C., Yang Z., Vuppugalla R., Hurley J., Thorndike D., Wagner A., Chimalakonda A., Rodrigues A.D. Evaluation of six proton pump inhibitors as inhibitors of various human cytochromes P450: focus on cytochrome P450 2C19. Drug Metab. Dispos. 2012;40(9):1698–1711. doi: 10.1124/dmd.112.045575. [http://dx. doi.org/10.1124/dmd.112.045575]. [PMID: 22648560]. [DOI] [PubMed] [Google Scholar]
- 47.Yapici Eser H., Bora H.A., Kuruoğlu A. Depression and Parkinson disease: Prevalence, temporal relationship, and determinants. Turk. J. Med. Sci. 2017;47(2):499–503. doi: 10.3906/sag-1603-101. [http://dx.doi.org/10. 3906/sag-1603-101]. [PMID: 28425238]. [DOI] [PubMed] [Google Scholar]
- 48.Onofrj M., Bonanni L., Thomas A. An expert opinion on safinamide in Parkinson’s disease. Expert Opin. Investig. Drugs. 2008;17(7):1115–1125. doi: 10.1517/13543784.17.7.1115. [http://dx.doi.org/10.1517/13543784.17.7.1115]. [PMID: 18549347]. [DOI] [PubMed] [Google Scholar]
- 49.Conn P.J., Battaglia G., Marino M.J., Nicoletti F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat. Rev. Neurosci. 2005;6(10):787–798. doi: 10.1038/nrn1763. [http://dx.doi.org/10.1038/nrn1763]. [PMID: 16276355]. [DOI] [PubMed] [Google Scholar]
- 50.Cenci M.A., Ohlin K.E., Odin P. Current options and future possibilities for the treatment of dyskinesia and motor fluctuations in Parkinson’s disease. CNS Neurol. Disord. Drug Targets. 2011;10(6):670–684. doi: 10.2174/187152711797247885. [http://dx.doi.org/10.2174/187152711797247885]. [PMID: 21838677]. [DOI] [PubMed] [Google Scholar]
- 51.Sebastianutto I., Cenci M.A. mGlu receptors in the treatment of Parkinson’s disease and L-DOPA-induced dyskinesia. Curr. Opin. Pharmacol. 2018;38:81–89. doi: 10.1016/j.coph.2018.03.003. [http://dx.doi.org/10.1016/j.coph. 2018.03.003]. [PMID: 29625424]. [DOI] [PubMed] [Google Scholar]
- 52.Magyar K. The pharmacology of selegiline. Int. Rev. Neurobiol. 2011;100:65–84. doi: 10.1016/B978-0-12-386467-3.00004-2. [http://dx.doi.org/10.1016/B978-0-12-386467-3.00004-2]. [PMID: 21971003]. [DOI] [PubMed] [Google Scholar]
- 53.Koller K., Olanow W., Rodnitzky R., Fink J.S., Growdon J.H., Paulson G., Kurlan R., Friedman J.H., Gancher S., Nutt J., Rajput A.H., Bennett J., Wooten J.F., LeWitt P., Goetz C., Tanner C., Shannon K., Klawans H., Suchowersky O., Brin M., Bressman S., Weiner W., Sanchez-Ramos J., Jankovic J., Penney J.B., Lang A., Hoehn M., Tetrud J., Grimes J.D., Pfeiffer R., Shults C., Thal L., Gauthier S., Golbe L.I., Perlmutter J.S., Moses H., III, Hurtig H.I., Stern M. Effect of deprenyl on the progression of disability in early Parkinson’s disease. N. Engl. J. Med. 1989;321(20):1364–1371. doi: 10.1056/NEJM198911163212004. [http://dx.doi.org/10.1056/ NEJM198911163212004]. [PMID: 2509910]. [DOI] [PubMed] [Google Scholar]
- 54.Olanow C.W., Hauser R.A., Gauger L., Malapira T., Koller W., Hubble J., Bushenbark K., Lilienfeld D., Esterlitz J. The effect of deprenyl and levodopa on the progression of Parkinson’s disease. Ann. Neurol. 1995;38(5):771–777. doi: 10.1002/ana.410380512. [http://dx.doi.org/10. 1002/ana.410380512]. [PMID: 7486869]. [DOI] [PubMed] [Google Scholar]
- 55.Myllylä V.V., Sotaniemi K., Mäki-Ikola O., Rinne U.K., Heinonen E.H. Role of selegiline in combination therapy of Parkinson’s disease. Neurology. 1996;47(6) Suppl. 3:S200–S209. doi: 10.1212/wnl.47.6_suppl_3.200s. [http:// dx.doi.org/10.1212/WNL.47.6_Suppl_3.200S]. [PMID: 8959989]. [DOI] [PubMed] [Google Scholar]
- 56.Shoulson F., Kieburtz S., Oakes S., Blindauer O., Goren L., Plumb E., Lew H., Lloyd H., Golbe W., Feigin O., Calabrese A., Marshall M.T., Mendis N., Mendis H., Pahwa M., Shulman R., Stacy B., Tuite K., Shannon T., Marek A., Dowling K., Sethi M. Adler, LeWitt.; Gordon, F. S.; and Salzman, M.D.; Shinaman, B.; Gauger, B.; Klimek, B.; Shannon, H.; Berry, G.; Del Rizzo, G.; Betcher, B.; Ewanishin, W.; Peterson, E.; Jaglin, K.; Stavris, DiMinno.; Richman, K.; McInnes, DeAngelis.; Winnick, T.; Evans, L.; Roberge, C.; Connolly, D.O.; and Josephson, and Mr. Chadwick, S., Shoulson, O., Fahn, K.; Blindauer, S.; Schwid, Lang.; White, N.; Cox, E.; Bausch, I.; Plumb, S.; Shoulson, O.; Kieburtz, F.; Blindauer, S.; Schwid, S.; Fahn, K.; Stern, O.; Siderowf, B.; Lew, H.; Loyd, H.; Golbe, W.; Feigin, O.; Calabrese, A.; Marshall, Miyasaki, T. M.N.; Mendis, H.; Pahwa, M.; Shulman, R.; Stacy, B.; Tuite, K. S.; Tanner, M.; Aminoff, D.; Kang, S.; Martin, A.; LeWitt, G.; Feldman, L.; White, S.; Cox, N.; Goren, O. S.; Levy, Mss, D.; Shinaman, B.; Gauger, B.; Klimek, B.; Shannon, H.; Berry, G.; Del Rizzo, G.; Betcher, B.; Ewanishin, W.; Peterson, E.; Jaglin, K.; Stavris, DiMinno, Richman, K.; McInnes, DeAngelis, Winnick, T.; Plumb, E.; Lind, R.; Conn, C.; Daigneault, O.; Josephson, I. E. B.; Mr Chadwick, O.; Cox, M. E.; Bausch, I.; Eyal, S. K.; Day, S.; Lew, H.; Loyd, H.; Golbe, W.; Feigin, O.; Calabrese, A.; Marshall, M.T.; Mendis, N.; Mendis, H.; Pahwa, M.; Shulman, R.; Stacy, B.; Tuite, K. S.; Tanner, M.; Aminoff, D.; Kang, S.; Martin, A.; LeWitt, G.; Feldman, L.; White, N.; Salzman, G. O.; Levy, B.; Gauger, B.; Klimek, B. Shannon, H.; Berry, G.; Del Rizzo, G.; Betcher, B.; Ewanishin, W.; Peterson, E.; Jaglin, K.; Stavris, DiMinno.; Richman, K.; McInnes, DeAngelis.; Winnick, T.; Evans, L.; Roberge, C.; Connolly, D.; Oliva, J.; Chadwick, S.; Siderowf, F.; Kieburtz, B.; Schwid, S.P. A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch. Neurol. 2004;61(4):561–566. doi: 10.1001/archneur.61.4.561. [http://dx.doi.org/10.1001/archneur.61.4.561]. [PMID: 15096406]. [DOI] [PubMed] [Google Scholar]
- 57.Olanow C.W., Rascol O., Hauser R., Feigin P.D., Jankovic J., Lang A., Langston W., Melamed E., Poewe W., Stocchi F., Tolosa E. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N. Engl. J. Med. 2009;361(13):1268–1278. doi: 10.1056/NEJMoa0809335. [http:// dx.doi.org/10.1056/NEJMoa0809335]. [PMID: 19776408]. [DOI] [PubMed] [Google Scholar]
- 58.Jankovic J., Berkovich E., Eyal E., Tolosa E. Symptomatic efficacy of rasagiline monotherapy in early Parkinson’s disease: post-hoc analyses from the ADAGIO trial. Parkinsonism Relat. Disord. 2014;20(6):640–643. doi: 10.1016/j.parkreldis.2014.02.024. [http://dx.doi.org/10.1016/j.parkreldis.2014. 02.024]. [PMID: 24637126]. [DOI] [PubMed] [Google Scholar]
- 59.Rascol O., Brooks D.J., Melamed E., Oertel W., Poewe W., Stocchi F., Tolosa E. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, lasting effect in adjunct therapy with Rasagiline given once daily, study): A randomised, double-blind, parallel-group trial. Lancet. 2005;365(9463):947–954. doi: 10.1016/S0140-6736(05)71083-7. [http://dx.doi.org/10.1016/S0140-6736(05)71083-7]. [PMID: 15766996]. [DOI] [PubMed] [Google Scholar]
- 60.Elmer L.W. Rasagiline adjunct therapy in patients with Parkinson’s disease: post hoc analyses of the PRESTO and LARGO trials. Parkinsonism Relat. Disord. 2013;19(11):930–936. doi: 10.1016/j.parkreldis.2013.06.001. [http://dx. doi.org/10.1016/j.parkreldis.2013.06.001]. [PMID: 23849501]. [DOI] [PubMed] [Google Scholar]
- 61.Cereda E., Cilia R., Canesi M., Tesei S., Mariani C.B., Zecchinelli A.L., Pezzoli G. Efficacy of rasagiline and selegiline in Parkinson’s disease: a head-to-head 3-year retrospective case-control study. J. Neurol. 2017;264(6):1254–1263. doi: 10.1007/s00415-017-8523-y. [http://dx.doi.org/10. 1007/s00415-017-8523-y]. [PMID: 28550482]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dashtipour K., Chen J.J., Kani C., Bahjri K., Ghamsary M. Clinical outcomes in patients with Parkinson’s disease treated with a monoamine oxidase Type-B inhibitor: A cross-sectional, cohort study. Pharmacotherapy. 2015;35(7):681–686. doi: 10.1002/phar.1611. [http://dx. doi.org/10.1002/phar.1611]. [PMID: 26139574]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fasano A., Di Matteo A., Vitale C., Squintani G., Ferigo L., Bombieri F., Santangelo G., Amboni M., Barone P., Tinazzi M. Reversible Pisa syndrome in patients with Parkinson’s disease on rasagiline therapy. Mov. Disord. 2011;26(14):2578–2580. doi: 10.1002/mds.23918. [http://dx.doi.org/10.1002/mds.23918]. [PMID: 22170277]. [DOI] [PubMed] [Google Scholar]
- 64.Valentino F., Cosentino G., Fierro B., Realmuto S., Mastrilli S., Savettieri G., D’Amelio M. Pisa syndrome after rasagiline therapy in a patient with Parkinson’s disease. Neurol. Sci. 2015;36(12):2305. doi: 10.1007/s10072-015-2374-z. [http://dx.doi.org/10.1007/s10072-015-2374-z]. [PMID: 26335016]. [DOI] [PubMed] [Google Scholar]
- 65.Gubellini P., Pisani A., Centonze D., Bernardi G., Calabresi P. Metabotropic glutamate receptors and striatal synaptic plasticity: implications for neurological diseases. Prog. Neurobiol. 2004;74(5):271–300. doi: 10.1016/j.pneurobio.2004.09.005. [http://dx.doi.org/10.1016/j.pneurobio.2004.09.005]. [PMID: 15582223]. [DOI] [PubMed] [Google Scholar]
- 66.Picconi B., Centonze D., Håkansson K., Bernardi G., Greengard P., Fisone G., Cenci M.A., Calabresi P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 2003;6(5):501–506. doi: 10.1038/nn1040. [http://dx.doi.org/10.1038/nn1040]. [PMID: 12665799]. [DOI] [PubMed] [Google Scholar]
- 67.Picconi B., Paillé V., Ghiglieri V., Bagetta V., Barone I., Lindgren H.S., Bernardi G., Angela C.M., Calabresi P. l-DOPA dosage is critically involved in dyskinesia via loss of synaptic depotentiation. Neurobiol. Dis. 2008;29(2):327–335. doi: 10.1016/j.nbd.2007.10.001. [http://dx. doi.org/10.1016/j.nbd.2007.10.001]. [PMID: 17997101]. [DOI] [PubMed] [Google Scholar]
- 68.Sgambato-Faure V., Cenci M.A. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog. Neurobiol. 2012;96(1):69–86. doi: 10.1016/j.pneurobio.2011.10.005. [http://dx.doi.org/10.1016/j. pneurobio.2011.10.005]. [PMID: 22075179]. [DOI] [PubMed] [Google Scholar]
- 69.Grégoire L., Jourdain V.A., Townsend M., Roach A., Di Paolo T. Safinamide reduces dyskinesias and prolongs L-DOPA antiparkinsonian effect in parkinsonian monkeys. Parkinsonism Relat. Disord. 2013;19(5):508–514. doi: 10.1016/j.parkreldis.2013.01.009. [http://dx.doi.org/10.1016/j.parkreldis. 2013.01.009]. [PMID: 23402994]. [DOI] [PubMed] [Google Scholar]
- 70.Borgohain R., Szasz J., Stanzione P., Meshram C., Bhatt M., Chirilineau D., Stocchi F., Lucini V., Giuliani R., Forrest E., Rice P., Anand R. Randomized trial of safinamide add-on to levodopa in Parkinson’s disease with motor fluctuations. Mov. Disord. 2014;29(2):229–237. doi: 10.1002/mds.25751. [http://dx.doi.org/10.1002/mds.25751]. [PMID: 24323641]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Borgohain R., Szasz J., Stanzione P., Meshram C., Bhatt M.H., Chirilineau D., Stocchi F., Lucini V., Giuliani R., Forrest E., Rice P., Anand R. Two-year, randomized, controlled study of safinamide as add-on to levodopa in mid to late Parkinson’s disease. Mov. Disord. 2014;29(10):1273–1280. doi: 10.1002/mds.25961. [http://dx.doi.org/10. 1002/mds.25961]. [PMID: 25044402]. [DOI] [PubMed] [Google Scholar]
- 72.Cattaneo C., Ferla R.L., Bonizzoni E., Sardina M. Long-Term Effects of safinamide on dyskinesia in mid- to late-stage Parkinson’s disease: A post-Hoc analysis. J. Parkinsons Dis. 2015;5(3):475–481. doi: 10.3233/JPD-150569. [http://dx.doi.org/10.3233/JPD-150569]. [PMID: 26406127]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Murata M., Hasegawa K., Kanazawa I., Fukasaka J., Kochi K., Shimazu R. Zonisamide improves wearing-off in Parkinson’s disease: A randomized, double-blind study. Mov. Disord. 2015;30(10):1343–1350. doi: 10.1002/mds.26286. [http://dx.doi.org/10.1002/mds.26286]. [PMID: 26094993]. [DOI] [PubMed] [Google Scholar]
- 74.Barone P., Santangelo G., Morgante L., Onofrj M., Meco G., Abbruzzese G., Bonuccelli U., Cossu G., Pezzoli G., Stanzione P., Lopiano L., Antonini A., Tinazzi M. A randomized clinical trial to evaluate the effects of rasagiline on depressive symptoms in non-demented Parkinson’s disease patients. Eur. J. Neurol. 2015;22(8):1184–1191. doi: 10.1111/ene.12724. [http://dx.doi.org/10.1111/ene.12724]. [PMID: 25962410]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kasai S., Yoshihara T., Lopatina O., Ishihara K., Higashida H. Selegiline ameliorates depression-like behavior in mice lacking the CD157/BST1 gene, a risk factor for Parkinson’s disease. Front. Behav. Neurosci. 2017;11:75. doi: 10.3389/fnbeh.2017.00075. [http://dx.doi.org/10.3389/ fnbeh.2017.00075]. [PMID: 28515684]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Contreras-Mora H., Rowland M.A., Yohn S.E., Correa M., Salamone J.D. Partial reversal of the effort-related motivational effects of tetrabenazine with the MAO-B inhibitor deprenyl (selegiline): Implications for treating motivational dysfunctions. Pharmacol. Biochem. Behav. 2018;166:13–20. doi: 10.1016/j.pbb.2018.01.001. [http://dx.doi.org/ 10.1016/j.pbb.2018.01.001]. [PMID: 29309800]. [DOI] [PubMed] [Google Scholar]
- 77.Amiri S., Amini-Khoei H., Mohammadi-Asl A., Alijanpour S., Haj-Mirzaian A., Rahimi-Balaei M., Razmi A., Olson C.O., Rastegar M., Mehdizadeh M., Zarrindast M.R. Involvement of D1 and D2 dopamine receptors in the antidepressant-like effects of selegiline in maternal separation model of mouse. Physiol. Behav. 2016;163:107–114. doi: 10.1016/j.physbeh.2016.04.052. [http://dx.doi.org/10.1016/j.physbeh.2016.04. 052]. [PMID: 27143252]. [DOI] [PubMed] [Google Scholar]
- 78.Cristancho M.A., Thase M.E. Critical appraisal of selegiline transdermal system for major depressive disorder. Expert Opin. Drug Deliv. 2016;13(5):659–665. doi: 10.1517/17425247.2016.1140145. [http://dx.doi.org/10.1517/ 17425247.2016.1140145]. [PMID: 26837935]. [DOI] [PubMed] [Google Scholar]
- 79.Stocchi F. Benefits of treatment with rasagiline for fatigue symptoms in patients with early Parkinson’s disease. Eur. J. Neurol. 2014;21(2):357–360. doi: 10.1111/ene.12205. [http://dx.doi.org/10.1111/ene.12205]. [PMID: 23790011]. [DOI] [PubMed] [Google Scholar]
- 80.Poewe W., Hauser R.A., Lang A. Effects of rasagiline on the progression of nonmotor scores of the MDS-UPDRS. Mov. Disord. 2015;30(4):589–592. doi: 10.1002/mds.26124. [http://dx.doi.org/10.1002/mds.26124]. [PMID: 25545629]. [DOI] [PubMed] [Google Scholar]
- 81.Cattaneo C., Müller T., Bonizzoni E., Lazzeri G., Kottakis I., Keywood C. Long-term effects of safinamide on mood fluctuations in Parkinson’s Disease. J. Parkinsons Dis. 2017;7(4):629–634. doi: 10.3233/JPD-171143. [http://dx.doi.org/10.3233/JPD-171143]. [PMID: 28777756]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cattaneo C., Kulisevsky J., Tubazio V., Castellani P. Long-term Efficacy of Safinamide on Parkinson’s Disease chronic pain. Adv. Ther. 2018;35(4):515–522. doi: 10.1007/s12325-018-0687-z. [http://dx.doi.org/10.1007/s12325-018-0687-z]. [PMID: 29542008]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cattaneo C., Barone P., Bonizzoni E., Sardina M. Effects of safinamide on pain in fluctuating Parkinson’s disease patients: A post-Hoc analysis. J. Parkinsons Dis. 2017;7(1):95–101. doi: 10.3233/JPD-160911. [http://dx.doi.org/10.3233/JPD-160911]. [PMID: 27802242]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liguori C., Mercuri N.B., Stefani A., Pierantozzi M. Effective treatment of restless legs syndrome by safinamide in Parkinson’s disease patients. Sleep Med. 2018;41:113–114. doi: 10.1016/j.sleep.2017.09.017. [http://dx.doi. org/10.1016/j.sleep.2017.09.017]. [PMID: 29268951]. [DOI] [PubMed] [Google Scholar]
- 85.Vermersch P., Petit H. Long-term selegiline tolerance in the treatment of Parkinson’s disease. Therapie. 1992;47(1):75–78. [PMID: 1523599]. [PubMed] [Google Scholar]
- 86.Mortality in DATATOP. A multicenter trial in early Parkinson’s disease. Ann. Neurol. 1998;43(3):318–325. doi: 10.1002/ana.410430309. [http://dx.doi.org/10. 1002/ana.410430309]. [PMID: 9506548]. [DOI] [PubMed] [Google Scholar]
- 87.Montastruc J.L., Chaumerliac C., Desboeuf K., Manika M., Bagheri H., Rascol O., Lapeyre-Mestre M. Adverse drug reactions to selegiline: a review of the French pharmacovigilance database. Clin. Neuropharmacol. 2000;23(5):271–275. doi: 10.1097/00002826-200009000-00006. [http://dx.doi. org/10.1097/00002826-200009000-00006]. [PMID: 11154095]. [DOI] [PubMed] [Google Scholar]
- 88.Chen J.J., Swope D.M., Dashtipour K. Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson’s disease. Clin. Ther. 2007;29(9):1825–1849. doi: 10.1016/j.clinthera.2007.09.021. [http://dx.doi.org/10.1016/j.clinthera.2007.09.021]. [PMID: 18035186]. [DOI] [PubMed] [Google Scholar]
- 89.deMarcaida J.A., Schwid S.R., White W.B., Blindauer K., Fahn S., Kieburtz K., Stern M., Shoulson I. Effects of tyramine administration in Parkinson’s disease patients treated with selective MAO-B inhibitor rasagiline. Mov. Disord. 2006;21(10):1716–1721. doi: 10.1002/mds.21048. [http://dx.doi.org/10.1002/mds.21048]. [PMID: 16856145]. [DOI] [PubMed] [Google Scholar]
- 90.Müller T., Hoffmann J.A., Dimpfel W., Oehlwein C. Switch from selegiline to rasagiline is beneficial in patients with Parkinson’s disease. J. Neural Transm. (Vienna) 2013;120(5):761–765. doi: 10.1007/s00702-012-0927-3. [http://dx.doi.org/10.1007/s00702-012-0927-3]. [PMID: 23196982]. [DOI] [PubMed] [Google Scholar]
- 91.Sharma T., Anand R., Stocchi F., Borgohain R., Rossetti R. 015 Study Group. Cognitive effects of Safinamide in early Parkinson’s disease patients.; International Congress of Parkinson’s Disease and Movement Disorders; June 3-7Istanbul, Turkey. 2007. [Google Scholar]
- 92.Stocchi F., Borgohain R., Onofrj M., Schapira A.H., Bhatt M., Lucini V., Giuliani R., Anand R. A randomized, double-blind, placebo-controlled trial of safinamide as add-on therapy in early Parkinson’s disease patients. Mov. Disord. 2012;27(1):106–112. doi: 10.1002/mds.23954. [http://dx.doi.org/10.1002/mds.23954]. [PMID: 21913224]. [DOI] [PubMed] [Google Scholar]
- 93.Schapira A.H., Stocchi F., Borgohain R., Onofrj M., Bhatt M., Lorenzana P., Lucini V., Giuliani R., Anand R. Long-term efficacy and safety of safinamide as add-on therapy in early Parkinson’s disease. Eur. J. Neurol. 2013;20(2):271–280. doi: 10.1111/j.1468-1331.2012.03840.x. [http://dx. doi.org/10.1111/j.1468-1331.2012.03840.x]. [PMID: 22967035]. [DOI] [PubMed] [Google Scholar]
- 94.Aboukarr A., Giudice M. Interaction between monoamine oxidase B inhibitors and selective serotonin reuptake inhibitors. Can. J. Hosp. Pharm. 2018;71(3):196–207. [http://dx.doi.org/10.4212/ cjhp.v71i3.2586]. [PMID: 29955193]. [PMC free article] [PubMed] [Google Scholar]
- 95.Chen J.J., Berchou R.C. Low incidence of cognitive and behavioral adverse events in rasagiline-treated patients with early to advanced Parkinson’s disease. Pkarmacotkerapy. 2005;25:1466. [Google Scholar]
- 96.Elmer L., Schwid S., Eberly S., Goetz C., Fahn S., Kieburtz K., Oakes D., Blindauer K., Salzman P., Oren S., Prisco U.L., Stern M., Shoulson I. Rasagiline-associated motor improvement in PD occurs without worsening of cognitive and behavioral symptoms. J. Neurol. Sci. 2006;248(1-2):78–83. doi: 10.1016/j.jns.2006.05.014. [http://dx.doi.org/ 10.1016/j.jns.2006.05.014]. [PMID: 16828804]. [DOI] [PubMed] [Google Scholar]
- 97.Goetz C.G., Schwid S.R., Eberly S.W., Oakes D., Shoulson I. Safety of rasagiline in elderly patients with Parkinson disease. Neurology. 2006;66(9):1427–1429. doi: 10.1212/01.wnl.0000210692.95595.1c. [http://dx.doi.org/10.1212/01.wnl. 0000210692.95595.1c]. [PMID: 16682679]. [DOI] [PubMed] [Google Scholar]
- 98.Riederer P., Müller T. Use of monoamine oxidase inhibitors in chronic neurodegeneration. Expert Opin. Drug Metab. Toxicol. 2017;13(2):233–240. doi: 10.1080/17425255.2017.1273901. [http://dx.doi.org/10.1080/17425255.2017. 1273901]. [PMID: 27998194]. [DOI] [PubMed] [Google Scholar]
- 99.Parkinson’s Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N. Engl. J. Med. 1993;328(3):176–183. doi: 10.1056/NEJM199301213280305. [http://dx.doi.org/10.1056/ NEJM199301213280305]. [PMID: 8417384]. [DOI] [PubMed] [Google Scholar]
- 100.Fowler J.S., Volkow N.D., Logan J., Wang G.J., MacGregor R.R., Schyler D., Wolf A.P., Pappas N., Alexoff D., Shea C. Slow recovery of human brain MAO B after L-deprenyl (Selegeline) withdrawal. Synapse. 1994;18(2):86–93. doi: 10.1002/syn.890180203. [http://dx.doi. org/10.1002/syn.890180203]. [PMID: 7839316]. [DOI] [PubMed] [Google Scholar]
- 101.Ahlskog J.E., Uitti R.J. Rasagiline, Parkinson neuroprotection, and delayed-start trials: still no satisfaction? Neurology. 2010;74(14):1143–1148. doi: 10.1212/WNL.0b013e3181d7d8e2. [http://dx.doi.org/10.1212/WNL.0b013e3181 d7d8e2]. [PMID: 20368634]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Majbour N.K., Vaikath N.N., Eusebi P., Chiasserini D., Ardah M., Varghese S., Haque M.E., Tokuda T., Auinger P., Calabresi P., Parnetti L., El-Agnaf O.M. Longitudinal changes in CSF alpha-synuclein species reflect Parkinson’s disease progression. Mov. Disord. 2016;31(10):1535–1542. doi: 10.1002/mds.26754. [http://dx.doi.org/10.1002/mds. 26754]. [PMID: 27548849]. [DOI] [PubMed] [Google Scholar]
- 103.Chiba K., Trevor A., Castagnoli N., Jr Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem. Biophys. Res. Commun. 1984;120(2):574–578. doi: 10.1016/0006-291x(84)91293-2. [http://dx. doi.org/10.1016/0006-291X(84)91293-2]. [PMID: 6428396]. [DOI] [PubMed] [Google Scholar]
- 104.Glover V., Gibb C., Sandler M. Monoamine oxidase B(MAO-B) is the major catalyst for 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) oxidation in human brain and other tissues. Neurosci. Lett. 1986;64(2):216–220. doi: 10.1016/0304-3940(86)90103-5. [http://dx.doi.org/10.1016/0304-3940 (86)90103-5]. [PMID: 3083305]. [DOI] [PubMed] [Google Scholar]
- 105.Akos K. MPTP, selegiline, and parkinsonism. Lancet. 1987;1(8523):38–39. doi: 10.1016/s0140-6736(87)90723-9. [http://dx.doi.org/10.1016/S0140-6736(87)90723-9]. [PMID: 2879111]. [DOI] [PubMed] [Google Scholar]
- 106.Cohen G., Pasik P., Cohen B., Leist A., Mytilineou C., Yahr M.D. Pargyline and deprenyl prevent the neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in monkeys. Eur. J. Pharmacol. 1984;106(1):209–210. doi: 10.1016/0014-2999(84)90700-3. [http://dx.doi.org/10. 1016/0014-2999(84)90700-3]. [PMID: 6442232]. [DOI] [PubMed] [Google Scholar]
- 107.Ebadi M., Sharma S., Shavali S., El Refaey H. Neuroprotective actions of selegiline. J. Neurosci. Res. 2002;67(3):285–289. doi: 10.1002/jnr.10148. [http://dx.doi.org/10.1002/jnr.10148]. [PMID: 11813232]. [DOI] [PubMed] [Google Scholar]
- 108.Fredriksson A., Palomo T., Archer T. Effects of MAO inhibitors upon MPTP mice chronically treated with suprathreshold doses of L-dopa. Behav. Pharmacol. 2000;11(7-8):571–581. doi: 10.1097/00008877-200011000-00004. [http://dx.doi. org/10.1097/00008877-200011000-00004]. [PMID: 11198128]. [DOI] [PubMed] [Google Scholar]
- 109.Fuller R.W., Hemrick-Luecke S.K., Perry K.W. Deprenyl antagonizes acute lethality of 1-methyl-4-phenyl-1,2,3,6-tetrahydro- pyridine in mice. J. Pharmacol. Exp. Ther. 1988;247(2):531–535. [PMID: 3141609]. [PubMed] [Google Scholar]
- 110.Gupta M., Wiener H.L. Effects of deprenyl on monoamine oxidase and neurotransmitters in the brains of MPTP-treated aging mice. Neurochem. Res. 1995;20(4):385–389. doi: 10.1007/BF00973091. [http://dx.doi.org/ 10.1007/BF00973091]. [PMID: 7544444]. [DOI] [PubMed] [Google Scholar]
- 111.Heikkila R.E., Hess A., Duvoisin R.C. Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) in the mouse: relationships between monoamine oxidase, MPTP metabolism and neurotoxicity. Life Sci. 1985;36(3):231–236. doi: 10.1016/0024-3205(85)90064-5. [http://dx.doi.org/10.1016/0024-3205(85)90064-5]. [PMID: 3917525]. [DOI] [PubMed] [Google Scholar]
- 112.Kupsch A., Sautter J., Götz M.E., Breithaupt W., Schwarz J., Youdim M.B., Riederer P., Gerlach M., Oertel W.H. Monoamine oxidase-inhibition and MPTP-induced neurotoxicity in the non-human primate: comparison of rasagiline (TVP 1012) with selegiline. J. Neural Transm. (Vienna) 2001;108(8-9):985–1009. doi: 10.1007/s007020170018. [http://dx.doi.org/10.1007/s007020170018]. [PMID: 11716151]. [DOI] [PubMed] [Google Scholar]
- 113.Muralikrishnan D., Samantaray S., Mohanakumar K.P. D-deprenyl protects nigrostriatal neurons against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurotoxicity. Synapse. 2003;50(1):7–13. doi: 10.1002/syn.10239. [http://dx.doi.org/10.1002/syn.10239]. [PMID: 12872288]. [DOI] [PubMed] [Google Scholar]
- 114.Sagi Y., Mandel S., Amit T., Youdim M.B. Activation of tyrosine kinase receptor signaling pathway by rasagiline facilitates neurorescue and restoration of nigrostriatal dopamine neurons in post-MPTP-induced parkinsonism. Neurobiol. Dis. 2007;25(1):35–44. doi: 10.1016/j.nbd.2006.07.020. [http://dx.doi.org/10.1016/j.nbd.2006.07.020]. [PMID: 17055733]. [DOI] [PubMed] [Google Scholar]
- 115.Tatton W.G., Greenwood C.E. Rescue of dying neurons: A new action for deprenyl in MPTP parkinsonism. J. Neurosci. Res. 1991;30(4):666–672. doi: 10.1002/jnr.490300410. [http://dx.doi.org/10.1002/jnr.490300410]. [PMID: 1686284]. [DOI] [PubMed] [Google Scholar]
- 116.Wu W.R., Zhu Z.T., Zhu X.Z. Differential effects of L-deprenyl on MPP+- and MPTP-induced dopamine overflow in microdialysates of striatum and nucleus accumbens. Life Sci. 2000;67(3):241–250. doi: 10.1016/s0024-3205(00)00628-7. [http://dx.doi.org/10.1016/S0024-3205(00)00628-7]. [PMID: 10983868]. [DOI] [PubMed] [Google Scholar]
- 117.Vizuete M.L., Steffen V., Ayala A., Cano J., Machado A. Protective effect of deprenyl against 1-methyl-4-phenylpyridinium neurotoxicity in rat striatum. Neurosci. Lett. 1993;152(1-2):113–116. doi: 10.1016/0304-3940(93)90496-8. [http://dx.doi.org/10.1016/0304-3940(93)90496-8]. [PMID: 8515861]. [DOI] [PubMed] [Google Scholar]
- 118.West B.D., Shughrue P.J., Vanko A.E., Ransom R.W., Kinney G.G. Amphetamine-induced locomotor activity is reduced in mice following MPTP treatment but not following selegiline/MPTP treatment. Pharmacol. Biochem. Behav. 2006;84(1):158–161. doi: 10.1016/j.pbb.2006.04.022. [http://dx.doi.org/10.1016/j.pbb.2006.04.022]. [PMID: 16757017]. [DOI] [PubMed] [Google Scholar]
- 119.Zhao Q., Cai D., Bai Y. Selegiline rescues gait deficits and the loss of dopaminergic neurons in a subacute MPTP mouse model of Parkinson’s disease. Int. J. Mol. Med. 2013;32(4):883–891. doi: 10.3892/ijmm.2013.1450. [http://dx.doi.org/10.3892/ijmm.2013.1450]. [PMID: 23877198]. [DOI] [PubMed] [Google Scholar]
- 120.Inaba-Hasegawa K., Shamoto-Nagai M., Maruyama W., Naoi M. Type B and A monoamine oxidase and their inhibitors regulate the gene expression of Bcl-2 and neurotrophic factors in human glioblastoma U118MG cells: different signal pathways for neuroprotection by selegiline and rasagiline. J. Neural Transm. (Vienna) 2017;124(9):1055–1066. doi: 10.1007/s00702-017-1740-9. [http://dx.doi.org/10.1007/s00702-017-1740-9]. [PMID: 28577058]. [DOI] [PubMed] [Google Scholar]
- 121.Maruyama W., Naoi M. “70th Birthday Professor Riederer” induction of glial cell line-derived and brain-derived neurotrophic factors by rasagiline and (-)deprenyl: a way to a disease-modifying therapy? J. Neural Transm. (Vienna) 2013;120(1):83–89. doi: 10.1007/s00702-012-0876-x. [http://dx.doi.org/10.1007/s00702-012-0876-x]. [PMID: 22892822]. [DOI] [PubMed] [Google Scholar]
- 122.Mandel S.A., Sagi Y., Amit T. Rasagiline promotes regeneration of substantia nigra dopaminergic neurons in post-MPTP-induced Parkinsonism via activation of tyrosine kinase receptor signaling pathway. Neurochem. Res. 2007;32(10):1694–1699. doi: 10.1007/s11064-007-9351-8. [http://dx. doi.org/10.1007/s11064-007-9351-8]. [PMID: 17701352]. [DOI] [PubMed] [Google Scholar]
- 123.Nagatsu T., Sawada M. Molecular mechanism of the relation of monoamine oxidase B and its inhibitors to Parkinson’s disease: possible implications of glial cells. J. Neural Transm. Suppl. 2006;(71):53–65. doi: 10.1007/978-3-211-33328-0_7. [PMID: 17447416]. [DOI] [PubMed] [Google Scholar]
- 124.Naoi M., Maruyama W. Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders. Curr. Pharm. Des. 2010;16(25):2799–2817. doi: 10.2174/138161210793176527. [http://dx.doi.org/10. 2174/138161210793176527]. [PMID: 20698822]. [DOI] [PubMed] [Google Scholar]
- 125.Naoi M., Maruyama W., Inaba-Hasegawa K. Revelation in the neuroprotective functions of rasagiline and selegiline: the induction of distinct genes by different mechanisms. Expert Rev. Neurother. 2013;13(6):671–684. doi: 10.1586/ern.13.60. [http://dx.doi.org/10.1586/ern.13.60]. [PMID: 23739004]. [DOI] [PubMed] [Google Scholar]
- 126.Kramer E.R., Liss B. GDNF-Ret signaling in midbrain dopaminergic neurons and its implication for Parkinson disease. FEBS Lett. 2015;589(24 Pt A):3760–3772. doi: 10.1016/j.febslet.2015.11.006. [http://dx.doi.org/10.1016/j. febslet.2015.11.006]. [PMID: 26555190]. [DOI] [PubMed] [Google Scholar]
- 127.Tenenbaum L., Humbert-Claude M. Glial cell line-derived neurotrophic factor gene delivery in Parkinson’s Disease: A delicate balance between neuroprotection, trophic effects, and unwanted Compensatory mechanisms. Front. Neuroanat. 2017;11:29. doi: 10.3389/fnana.2017.00029. [http://dx.doi.org/10.3389/fnana.2017.00029]. [PMID: 28442998]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gash D.M., Zhang Z., Gerhardt G. Neuroprotective and neurorestorative properties of GDNF. Ann. Neurol. 1998;44(3) Suppl. 1:S121–S125. doi: 10.1002/ana.410440718. [http://dx.doi.org/10.1002/ana.410440718]. [PMID: 9749583]. [DOI] [PubMed] [Google Scholar]
- 129.Kordower J.H. In vivo gene delivery of glial cell line--derived neurotrophic factor for Parkinson’s disease. Ann. Neurol. 2003;53(Suppl. 3):S120–S132. doi: 10.1002/ana.10485. [http://dx.doi.org/10.1002/ana.10485]. [PMID: 12666104]. [DOI] [PubMed] [Google Scholar]
- 130.Lin L.F., Doherty D.H., Lile J.D., Bektesh S., Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260(5111):1130–1132. doi: 10.1126/science.8493557. [http://dx. doi.org/10.1126/science.8493557]. [PMID: 8493557]. [DOI] [PubMed] [Google Scholar]
- 131.Gill S.S., Patel N.K., Hotton G.R., O’Sullivan K., McCarter R., Bunnage M., Brooks D.J., Svendsen C.N., Heywood P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 2003;9(5):589–595. doi: 10.1038/nm850. [http://dx.doi.org/ 10.1038/nm850]. [PMID: 12669033]. [DOI] [PubMed] [Google Scholar]
- 132.Brouillet E., Beal M.F. NMDA antagonists partially protect against MPTP induced neurotoxicity in mice. Neuroreport. 1993;4(4):387–390. doi: 10.1097/00001756-199304000-00011. [http://dx.doi.org/10.1097/00001756-199304000-00011]. [PMID: 8499594]. [DOI] [PubMed] [Google Scholar]
- 133.Lange K.W., Löschmann P.A., Sofic E., Burg M., Horowski R., Kalveram K.T., Wachtel H., Riederer P. The competitive NMDA antagonist CPP protects substantia nigra neurons from MPTP-induced degeneration in primates. Naunyn Schmiedebergs Arch. Pharmacol. 1993;348(6):586–592. doi: 10.1007/BF00167234. [http://dx.doi.org/10.1007/ BF00167234]. [PMID: 7907775]. [DOI] [PubMed] [Google Scholar]
- 134.Turski L., Bressler K., Rettig K.J., Löschmann P.A., Wachtel H. Protection of substantia nigra from MPP+ neurotoxicity by N-methyl-D-aspartate antagonists. Nature. 1991;349(6308):414–418. doi: 10.1038/349414a0. [http://dx.doi.org/10.1038/349414a0]. [PMID: 1846943]. [DOI] [PubMed] [Google Scholar]
- 135.Battaglia G., Fornai F., Busceti C.L., Aloisi G., Cerrito F., De Blasi A., Melchiorri D., Nicoletti F. Selective blockade of mGlu5 metabotropic glutamate receptors is protective against methamphetamine neurotoxicity. J. Neurosci. 2002;22(6):2135–2141. doi: 10.1523/JNEUROSCI.22-06-02135.2002. [http://dx.doi.org/10.1523/JNEUROSCI.22-06-02135.2002]. [PMID: 11896153]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Battaglia G., Busceti C.L., Molinaro G., Biagioni F., Storto M., Fornai F., Nicoletti F., Bruno V. Endogenous activation of mGlu5 metabotropic glutamate receptors contributes to the development of nigro-striatal damage induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. J. Neurosci. 2004;24(4):828–835. doi: 10.1523/JNEUROSCI.3831-03.2004. [http://dx.doi.org/10.1523/JNEUROSCI.3831-03.2004]. [PMID: 14749427]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Masilamoni G.J., Bogenpohl J.W., Alagille D., Delevich K., Tamagnan G., Votaw J.R., Wichmann T., Smith Y. Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain. 2011;134(Pt 7):2057–2073. doi: 10.1093/brain/awr137. [http://dx.doi.org/ 10.1093/brain/awr137]. [PMID: 21705423]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Muñoz-Manchado A.B., Villadiego J., Romo-Madero S., Suárez-Luna N., Bermejo-Navas A., Rodríguez-Gómez J.A., Garrido-Gil P., Labandeira-García J.L., Echevarría M., López-Barneo J., Toledo-Aral J.J. Chronic and progressive Parkinson’s disease MPTP model in adult and aged mice. J. Neurochem. 2016;136(2):373–387. doi: 10.1111/jnc.13409. [http://dx.doi.org/10.1111/jnc.13409]. [PMID: 26500044]. [DOI] [PubMed] [Google Scholar]
- 139.Tozzi A., Tantucci M., Marchi S., Mazzocchetti P., Morari M., Pinton P., Mancini A., Calabresi P. Dopamine D2 receptor-mediated neuroprotection in a G2019S Lrrk2 genetic model of Parkinson’s disease. Cell Death Dis. 2018;9(2):204. doi: 10.1038/s41419-017-0221-2. [http://dx. doi.org/10.1038/s41419-017-0221-2]. [PMID: 29434188]. [DOI] [PMC free article] [PubMed] [Google Scholar]