

Abstract
While the calpain system has now been discovered for over 50 years, there is still a paucity of information regard-ing the organization and functions of the signaling pathways regulated by these proteases, although calpains play critical roles in many cell functions. Moreover, calpain overactivation has been shown to be involved in numerous diseases. Among the 15 calpain isoforms identified, calpain-1 (aka µ-calpain) and calpain-2 (aka m-calpain) are ubiquitously distributed in most tissues and organs, including the brain. We have recently proposed that calpain-1 and calpain-2 play opposite functions in the brain, with calpain-1 activation being required for triggering synaptic plasticity and neuroprotection (Dr. Jekill), and calpain-2 limiting the extent of plasticity and being neurodegenerative (Mr. Hyde). Calpain-mediated cleavage has been ob-served in cytoskeleton proteins, membrane-associated proteins, receptors/channels, scaffolding/anchoring proteins, and pro-tein kinases and phosphatases. This review will focus on the signaling pathways related to local protein synthesis, cytoskele-ton regulation and neuronal survival/death regulated by calpain-1 and calpain-2, in an attempt to explain the origin of the op-posite functions of these 2 calpain isoforms. This will be followed by a discussion of the potential therapeutic applications of selective regulators of these 2 calpain isoforms.
Keywords: Calpain, signaling pathways, synaptic plasticity, neurodegeneration, neuroprotection
1. IMPORTANCE OF CALPAINS IN THE REGULATION OF CELL FUNCTIONS
The calpain system has now been discovered for over 50 years [1], and many studies have indicated that this family of calcium-dependent proteases plays important functions under physiological and pathological conditions in the brain and other organs [2, 3]. Nevertheless, there is still a paucity of information regarding the organization and functions of the signaling pathways regulated by these proteases. Among the 15 calpain isoforms identified, calpain-1 (aka µ-calpain) and calpain-2 (aka m-calpain) are ubiquitously distributed in most tissues and organs, including the brain. These 2 isoforms exhibit a high degree of homology Fig. (1), and the major difference consists in their calcium requirement for activation, with calpain-1 requiring micromolar calcium concentrations and calpain-2 millimolar calcium concentrations. Another significant difference between these 2 isoforms is the existence of different PDZ binding sequences in their C-terminal domains, which impart differing binding partners for these 2 isoforms [4]. Calpain-2 exhibits a typical class I PDZ binding domain, while calpain-1 exhibits a class II PDZ-binding domain. PDZ proteins have been found to be central to separating signal transduction pathways and eliminating cross-talk between signal transduction pathways [5]. We recently identified the tyrosine phosphatase, Protein Tyrosine Phosphatase, Non-receptor Type 13 (PTPN13), aka FAP-1, as the PDZ binding partner of calpain-2, and we showed that activation of calpain-2 but not calpain-1 cleaves PTPN13, and inactivates its phosphatase activity, resulting in c-Abl activation and tau tyrosine phosphorylation [6]. While the PDZ-binding partner of calpain-1 has not been identified, we previously found that calpain-1 but not calpain-2 can be immunoprecipitated with NR2A subunits of NMDA receptors following their activation [7]. This suggests that calpain-1 but not calpain-2 is associated with the large cluster of proteins linked to synaptic NMDA receptors. On the other hand, calpain-2 appears to be activated following stimulation of extrasynaptic NMDA receptors, and as mentioned above, is associated with PTPN13. While PTPN13 has been shown to play a role in cancer, little is known regarding its functions in the brain.
Fig. (1).
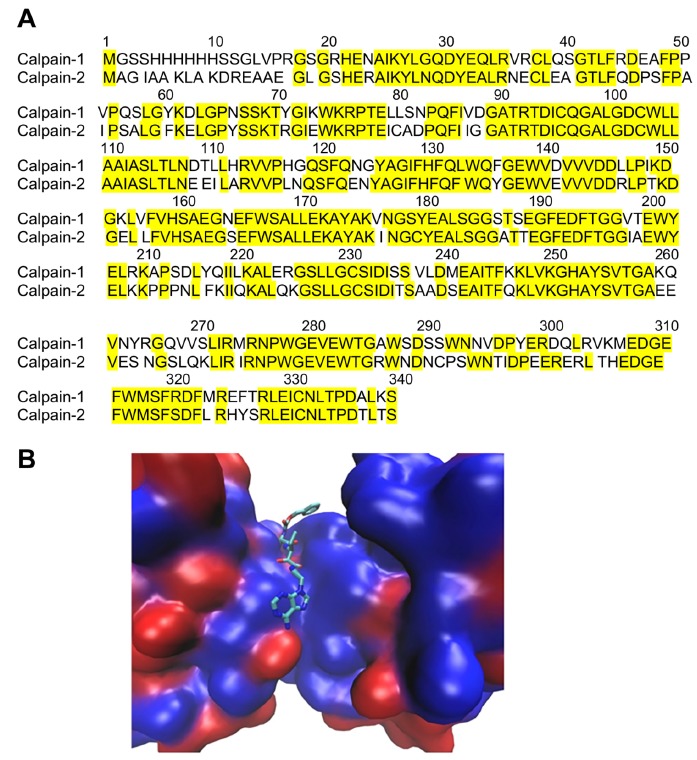
Comparison of the catalytic sites of calpain-1 and calpain-2. A. Amino acid sequences of the catalytic sites of calpain-1 and calpain-2. Calpain-1: PDB: 2ARY_B. Calpain-2: GenBank: EAW93251.1. Identical amino acids are highlighted and amount for more than 75% of the sequences. B. Protein backbones shown in surface mode with blue color indicating identical residues and red color non-identical residues. A ketoamide ligand is shown bound to the catalytic site. From [56]. (The color version of the figure is available in the electronic copy of the article).
We have recently proposed that calpain-1 and calpain-2 play opposite functions in the brain, with calpain-1 activation being required for triggering synaptic plasticity and neuroprotection, and calpain-2 limiting the extent of plasticity and being neurodegenerative [4]. We have also proposed that these opposite functions of calpain-1 and calpain-2 are related to their association with different signaling pathways, as a result of their binding to different PDZ-binding partners. However, the precise nature of the signaling pathways regulated by calpain-1 and calpain-2 remains largely unknown, thus preventing a clear understanding of their functions under both physiological and pathological conditions.
2. TARGETS OF CALPAIN-1 AND CALPAIN-2
Calpain-mediated cleavage has been observed in cytoskeleton proteins, membrane-associated proteins, receptors/channels, scaffolding/anchoring proteins, and protein kinases and phosphatases [8-10]. However, it is important to stress the difference between in vitro substrates and in vivo substrates. While many proteins can certainly be cleaved by calpain in cell-free systems, the real calpain substrates are the proteins that are actually cleaved under various conditions following calpain activation inside cells. To limit the scope of the review, we will focus on calpain substrates belonging to three regulatory pathways, i) local protein synthesis, ii) actin cytoskeleton regulation, and iii) neuronal survival/cell death.
2.1. Local Protein Synthesis
Among all the intracellular signaling pathways involved in local translational control, activation of the mammalian target of rapamycin (mTOR) plays a central role in activity-dependent synaptic plasticity at CA3-CA1 synapses [11, 12]. Binding of brain-derived neurotrophic factor (BDNF) to tyrosine receptor kinase B (TrkB) receptor initiates a signal transduction cascade, which induces the stimulation of phosphoinositide-3-kinase (PI3K)- and Akt-mediated mTOR phosphorylation leading to the stimulation of several translation factors, including the ribosomal protein S6 and the eukaryotic initiation factor 4E (eIF4) [13]. We recently identified a novel mechanism for BDNF-induced stimulation of mTOR-dependent protein synthesis in dendrites, which involves calpain-2-mediated degradation of phosphatase and tensin homolog (PTEN) [11], a negative regulator of PI3K/Akt signaling. Noteworthy, PTEN represents the first isoform-specific calpain substrate ever identified in vitro and in vivo, which has been used to selectively monitor calpain-2 activity under different physiological or pathological conditions [11, 14-16]. Furthermore, we showed that calpains cleave tuberous sclerosis proteins 1 and 2 (TSC1 and TSC2), also known as hamartin and tuberin, respectively, and this effect was found to contribute to BDNF-mediated mTOR activation [11], as both TSC1 and TSC2 function are suppressors of mTOR activity. Calpains have also been reported to cleave the regulatory and catalytic subunits of PI3K, thereby inhibiting Akt/mTOR signaling, although these effects were observed under non physiological conditions (i.e., serum starvation) and in cell lines [17]. Whether a similar phenomenon occurs in neurons remains to be explored.
2.2. Actin Cytoskeleton Regulation
Spectrin isoform αII, also known as brain spectrin or α-fodrin, was the first-identified substrate of calpains and is the principle component of the neuronal sub-membrane cytoskeleton [18]. αII-spectrin is anchored to the plasma membrane and binds to actin, calmodulin, and microtubules. Cleavage of spectrin by calpain alters the dynamic organization of membrane domains and membrane trafficking events [19] and thus changes synaptic integrity and stability. Other preferred cytoskeletal substrates of calpains include: microtubule-associated proteins (MAPs), neurofilament proteins [20], actin [21-24], cortactin [25], drebrin [26], RhoA [27, 28], integrin [29, 30], talin [31] and myristoylated alanine-rich C-kinase substrate (MARCKS) [32]. At this point, it is not known whether these proteins are differentially cleaved by calpain-1 and calpain-2.
2.3. Neuronal Death
While there is an abundant literature supporting the role of calpain in neurodegeneration, there is a paucity of information regarding the respective roles of calpain-1 and calpain-2 in this process, as well as the nature of the calpain targets that participate in neurodegeneration. Furthermore, while overactivation of calpain has been implicated in a wide range of pathological states, including stroke, epilepsy, traumatic nerve injury, neurodegenerative disorders, and aging [33-35], a number of studies have reported opposite findings, indicating that calpain activation could also provide neuroprotection under certain conditions [8, 36, 37]. As mentioned above, we reported that calpain-1 is neuroprotective due to calpain-1-mediated truncation of PHLPP1 and the resulting activation of the Akt pathway [4]. We showed that calpain-2 is associated with a multi-protein complex, which includes extrasynaptic NMDARs, and that activation of extrasynaptic NMDARs results in calpain-2 activation [7]. NR2B subunits are enriched in extrasynaptic NMDARs [38], and their activation is critical for excitotoxicity [39, 40]. Furthermore, NR2B directly binds RasGRF1, which provides a link between NMDAR activation and ERK activation [41]. As mentioned above, ERK activation directly phosphorylates and activates calpain-2 [42]; thus, this pathway is likely responsible for the prolonged activation of calpain-2 following stimulation of extrasynaptic NMDA receptors. Numerous studies have shown that calpain cleaves striatal-enriched tyrosine phosphatase (STEP), generating inactive fragments, resulting in activation of p38 and downstream cell death signaling pathways [43, 44].
3. POTENTIAL THERAPEUTIC APPLICATIONS OF SELECTIVE REGULATORS OF CALPAIN-1 AND CALPAIN-2
Based on the findings presented above, it follows that a selective calpain-1 activator could enhance synaptic plasticity and be neuroprotective. Calpain-1 would act as the good Dr. Jekill. On the other hand, calpain-2 would seem to act as the evil Mr. Hyde and a selective calpain-2 inhibitor could enhance synaptic plasticity and be neuroprotective Fig. (2).
Fig. (2).
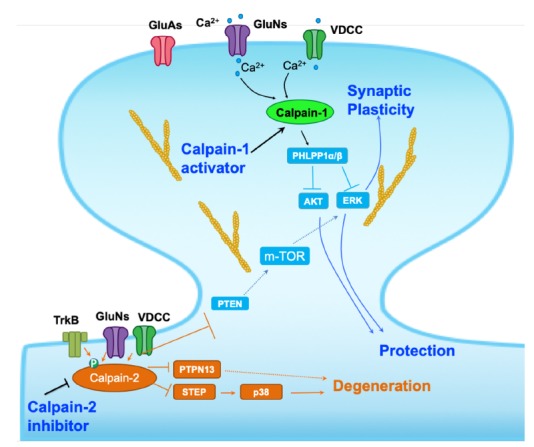
Schematic representation of some of the signaling pathways regulated by calpain-1 and calpain-2. Calpain-1 is downstream of the synaptic NMDA receptors and its activation following NMDA receptor stimulation results in the cleavage of PH domain and leucine-rich repeat protein phosphatase 1 (PHLPP1) and the activation of the Akt and Extracellular signal Regulated Kinase (ERK) pathways, which are responsible for linking calpain-1 to synaptic plasticity and to neuroprotection. On the other hand, calpain-2 is downstream of the extrasynaptic NMDA receptors, and its activation leads to the cleavage of STEP and PTPN13, thus linking calpain-2 to neurodegeneration. In addition, calpain-2 cleaves PTEN, resulting in mTOR activation and placing a break on synaptic plasticity (modified from [2]). As discussed in the text, a selective calpain-1 activator or a selective calpain-2 inhibitor would enhance synaptic plasticity and stimulate neuroprotective mechanisms.
3.1. Potential Therapeutic Applications of a Selective Calpain-1 Activator
Calpain-1 is downstream of the synaptic NMDA receptors and is therefore briefly activated following stimulation of synaptic NMDA receptors [7]. As mentioned above, calpain-1 activation is necessary for certain forms of synaptic plasticity and is neuroprotective in several models of acute neurodegeneration [15, 16, 45]. While calpain-1 is normally activated following an increase in calcium concentration, there are potentially several possibilities to enhance calpain-1 activation. First, calpain-1 is inhibited by the endogenous calpain inhibitor, calpastatin [46]. While the mechanism regulating calpastatin binding to calpain is not clearly understood, it might be possible to design small molecules inhibiting this binding, which would therefore function as calpain activators. However, because calpastatin inhibits both calpain-1 and calpain-2 such approach might be difficult to implement as the small molecules would need to selectively inhibit the binding of calpastatin to calpain-1. We reported that calpain-1 and calpain-2 exhibit different PDZ biding domains, with calpain-2 presenting a typical PDZ binding domain, and calpain-1 an atypical PDZ binding domain [4]. This finding suggested that calpain-1 and calpain-2 bind to different clusters of signaling proteins, which could account for their differential functions. It is thus conceivable that, by targeting these different PDZ binding domains, it could be possible to selectively manipulate calpain-1 and calpain-2. We have indeed found that selectively targeting the PDZ binding domain of calpain-1 leads to LTP enhancement (unpublished results). Whether this also results in increased neuroprotection following acute injury remains to be determined. In any event, such an approach could be beneficial to treat a variety of disorders associated with learning impairment, including various forms of learning disabilities.
3.2. Potential Therapeutic Applications of a Selective Calpain-2 Inhibitor
If calpain-2 activation limits the magnitude of TBS-induced LTP and is neurodegenerative, a selective calpain-2 inhibitor would be expected to enhance the magnitude of LTP and possibly facilitate learning and be neuroprotective [47]. We have verified these 2 predictions and found that such an inhibitor enhanced learning in two models of learning, novel object recognition and fear conditioning [48]. It is thus conceivable that a selective calpain-2 inhibitor could have therapeutic applications in a variety of diseases associated with learning impairment.
We have also reported that injection of a selective calpain-2 inhibitor (C2I) following increased intraocular pressure or traumatic brain injury provides neuroprotection [45]. In the case of acute glaucoma, we showed that systemic or intraocular administration of C2I 2 h after the increase in intraocular pressure prevented the death of retinal ganglion cells and maintained vision. These results suggest that a selective calpain-2 inhibitor could be developed for the treatment of acute glaucoma.
Traumatic brain injury (TBI) is the leading cause of death and disability in the age group 1-44. While more than 1 million people experience TBI each year in the USA, about 235,000 of them require hospitalization and 50,000 die [49, 50]. Those who survive often experience long-term disabilities, which impose a heavy financial and emotional cost to the entire society. While many mechanisms have been showed to participate in the long-term consequences of TBI, there is currently no consensus regarding the critical events that lead to neuronal damage and neuronal death, which take place during the hours/days following the injury [51-53]. While the calcium-dependent protease, calpain, has been repeatedly proposed to play a critical role in neuronal degeneration associated with a variety of insults, including TBI, stroke, and other neurodegenerative diseases, there have been conflicting reports regarding the effectiveness of calpain inhibitors in providing protection in stroke or TBI. Part of the difficulty in understanding the role of calpain in neurodegeneration and in TBI was due to lack of understanding of the functions of calpain-1 and calpain-2 in the brain. Results obtained in our laboratory over the last 5 years have provided a much better picture of the selective roles of calpain-1 and calpain-2 in various brain mechanisms, including in TBI. Thus, calpain-1 is rapidly but transiently activated following TBI, while calpain-2 activation is delayed and prolonged. Our results also showed that calpain-2 activation is closely associated with cell death following TBI and that a selective calpain-2 inhibitor, NA101, provides a significant degree of protection when administered systemically after TBI. Finally, our results not only account for the previous conflicting results regarding the involvement of calpain in neuronal death and in particular in TBI, but also strengthen the notion that calpain-2 is a good target to develop new neuroprotective molecules [47]. Our results also showed that calpain-2 activation around the lesion site was not significant until 8 h after TBI, suggesting the existence of an 8 h window post-TBI during which to deliver calpain-2 inhibitors. Systemic injection of NA101 at 1 or 4 h after TBI significantly reduced calpain-2 activation around the lesion area 24 h after TBI, indicating delivery efficacy and a relatively long half-life of NA101 in mice. We also showed that daily injection for 7 days after TBI resulted in a large decrease in brain lesion and in improvement in cognitive and motor function assessed 1 month after TBI [15]. We conclude that these results make a strong case to develop a selective calpain-2 inhibitor for the treatment of military and civilian population experiencing TBI.
Sports-related concussion (SRC) is widely recognized as a major form of mild traumatic brain injury, and as such could well represent another indication where a selective calpain-2 inhibitor could be beneficial. Interestingly, a calpain-generated fragment of brain spectrin, SNTF, has been reported to represent a blood biomarker for mild traumatic brain injury [54]. Furthermore, we also found that calpain-mediated truncation of PTPN13, the calpain-2 PDZ binding partner, represents a new link between calpain-2 activation and tau phosphorylation, which is a hallmark of chronic traumatic encephalopathy [55]. This finding also supports the notion that a selective calpain-2 inhibitor could be beneficial for the prevention of brain damage resulting from repeated mild traumatic brain injury.
Finally, as there is evidence that the same mechanism is activated under several conditions associated with acute neuronal injury, this treatment could be expanded to many other indications outside of TBI/repeated concussions.
CONCLUSION
It is now apparent that calpain-1 and calpain-2 play opposite roles in the regulation of a variety of functions in the Central Nervous System (CNS). As discussed above, this is the result of the association of these 2 calpain isoforms to different signaling pathways through their binding to different PDZ domain-binding partners. In particular, we showed that calpain-1 is downstream of synaptic NMDA receptors and its activation is critical for certain forms of synaptic plasticity and for its neuroprotective effects. In contrast, calpain-2 is associated with extrasynaptic NMDA receptors and its activation limits the extent of synaptic plasticity and is neurodegenerative. These features of calpain-1 and calpain-2 support the idea that a selective calpain-2 inhibitor could be very useful to provide neuroprotection under a variety of conditions associated with acute neuronal injury, such as traumatic brain injury and repeated concussions. It could also be valuable to reverse learning and memory impairments found in several neurological and neuropsychiatric diseases.
ACKNOWLEDGEMENTS
The author wants to acknowledge the contributions of all the collaborators who have worked on the roles of calpain in the brain in the author’s laboratory over the last 25 years.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by grant P01NS045260-01 from NINDS (PI: Dr. C.M. Gall) and grant R01NS057128 from NINDS to MB.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Guroff G. A neutral, calcium-activated proteinase from the soluble fraction of rat brain. J. Biol. Chem. 1964;239:149–155. [PMID: 14114836]. [PubMed] [Google Scholar]
- 2.Mehendale H.M., Limaye P.B. Calpain: A death protein that mediates progression of liver injury. Trends Pharmacol. Sci. 2005;26(5):232–236. doi: 10.1016/j.tips.2005.03.008. [http://dx.doi.org/10.1016/j.tips.2005.03.008]. [PMID: 15860369]. [DOI] [PubMed] [Google Scholar]
- 3.Potz B.A., Abid M.R., Sellke F.W. Role of calpain in pathogenesis of human disease processes. J. Nat. Sci. 2016;2(9):e218. [PMID: 27747292]. [PMC free article] [PubMed] [Google Scholar]
- 4.Baudry M., Bi X. Calpain-1 and calpain-2: The Yin and Yang of synaptic plasticity and neurodegeneration. Trends Neurosci. 2016;39(4):235–245. doi: 10.1016/j.tins.2016.01.007. [http://dx.doi.org/10.1016/j.tins.2016.01.007]. [PMID: 26874794]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loane D.J., Byrnes K.R. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7(4):366–377. doi: 10.1016/j.nurt.2010.07.002. [http://dx.doi.org/10.1016/j.nurt.2010.07.002]. [PMID: 20880501]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y., Hall R.A., Lee M., Kamgar-Parsi A., Bi X., Baudry M. The tyrosine phosphatase PTPN13/FAP-1 links calpain-2, TBI and tau tyrosine phosphorylation. Sci. Rep. 2017;7(1):11771. doi: 10.1038/s41598-017-12236-3. [http://dx.doi.org/10.1038/s41598-017-12236-3]. [PMID: 28924170]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y., Briz V., Chishti A., Bi X., Baudry M. Distinct roles for μ-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration. J. Neurosci. 2013;33(48):18880–18892. doi: 10.1523/JNEUROSCI.3293-13.2013. [http://dx.doi.org/10.1523/JNEUROSCI.3293-13.2013]. [PMID: 24285894]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H.Y., Lynch D.R. Calpain and synaptic function. Mol. Neurobiol. 2006;33(3):215–236. doi: 10.1385/MN:33:3:215. [http://dx.doi.org/10.1385/MN:33:3:215]. [PMID: 16954597]. [DOI] [PubMed] [Google Scholar]
- 9.Croall D.E., Ersfeld K. The calpains: modular designs and functional diversity. Genome Biol. 2007;8(6):218. doi: 10.1186/gb-2007-8-6-218. [http://dx.doi.org/10.1186/gb-2007-8-6-218]. [PMID: 17608959]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ono Y., Sorimachi H. Calpains: An elaborate proteolytic system. Biochim. Biophys. Acta. 2012;1824(1):224–236. doi: 10.1016/j.bbapap.2011.08.005. [http://dx.doi.org/10.1016/j.bbapap.2011.08.005]. [PMID: 21864727]. [DOI] [PubMed] [Google Scholar]
- 11.Briz V., Hsu Y.T., Li Y., Lee E., Bi X., Baudry M. Calpain-2-mediated PTEN degradation contributes to BDNF-induced stimulation of dendritic protein synthesis. J. Neurosci. 2013;33(10):4317–4328. doi: 10.1523/JNEUROSCI.4907-12.2013. [http://dx.doi.org/10.1523/JNEUROSCI.4907-12.2013]. [PMID: 23467348]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panja D., Bramham C.R. BDNF mechanisms in late LTP formation: A synthesis and breakdown. Neuropharmacology. 2014;76(Pt C):664–676. doi: 10.1016/j.neuropharm.2013.06.024. [http://dx.doi.org/10.1016/j.neuropharm.2013.06.024]. [PMID: 23831365]. [DOI] [PubMed] [Google Scholar]
- 13.Takei N., Inamura N., Kawamura M., Namba H., Hara K., Yonezawa K., Nawa H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci. 2004;24(44):9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004. [http://dx.doi.org/10.1523/JNEUROSCI.1427-04.2004]. [PMID: 15525761]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briz V., Baudry M. Estrogen regulates protein synthesis and actin polymerization in hippocampal neurons through different molecular mechanisms. Front. Endocrinol. (Lausanne) 2014;5:22. doi: 10.3389/fendo.2014.00022. [http://dx.doi.org/10.3389/fendo.2014.00022]. [PMID: 24611062]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y., Liu Y., Lopez D., Lee M., Dayal S., Hurtado A., Bi X., Baudry M. Protection against TBI-induced neuronal death with post-treatment with a selective calpain-2 inhibitor in mice. J. Neurotrauma. 2018;35(1):105–117. doi: 10.1089/neu.2017.5024. [http://dx.doi.org/10.1089/neu.2017.5024]. [PMID: 28594313]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y., Zhu G., Briz V., Hsu Y.T., Bi X., Baudry M. A molecular brake controls the magnitude of long-term potentiation. Nat. Commun. 2014;5:3051. doi: 10.1038/ncomms4051. [http://dx.doi.org/10.1038/ncomms4051]. [PMID: 24394804]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beltran L., Chaussade C., Vanhaesebroeck B., Cutillas P.R. Calpain interacts with class IA phosphoinositide 3-kinases regulating their stability and signaling activity. Proc. Natl. Acad. Sci. USA. 2011;108(39):16217–16222. doi: 10.1073/pnas.1107692108. [http://dx.doi.org/10.1073/pnas.1107692108]. [PMID: 21930956]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlin R.K., Bartelt D.C., Siekevitz P. Identification of fodrin as a major calmodulin-binding protein in postsynaptic density preparations. J. Cell Biol. 1983;96(2):443–448. doi: 10.1083/jcb.96.2.443. [http://dx.doi.org/10.1083/jcb.96.2.443]. [PMID: 6833363]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett V. Spectrin-based membrane skeleton: a multipotential adaptor between plasma membrane and cytoplasm. Physiol. Rev. 1990;70(4):1029–1065. doi: 10.1152/physrev.1990.70.4.1029. [http://dx.doi.org/10.1152/physrev.1990.70.4.1029]. [PMID: 2271059]. [DOI] [PubMed] [Google Scholar]
- 20.Ma M. Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon. Neurobiol. Dis. 2013;60:61–79. doi: 10.1016/j.nbd.2013.08.010. [http://dx.doi.org/10.1016/j.nbd.2013.08.010]. [PMID: 23969238]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banik N.L., Matzelle D., Terry E., Hogan E.L. A new mechanism of methylprednisolone and other corticosteroids action demonstrated in vitro: inhibition of a proteinase (calpain) prevents myelin and cytoskeletal protein degradation. Brain Res. 1997;748(1-2):205–210. doi: 10.1016/s0006-8993(96)01302-9. [http://dx.doi.org/10.1016/S0006-8993(96)01302-9]. [PMID: 9067463]. [DOI] [PubMed] [Google Scholar]
- 22.Fischer I., Romano-Clarke G., Grynspan F. Calpain-mediated proteolysis of microtubule associated proteins MAP1B and MAP2 in developing brain. Neurochem. Res. 1991;16(8):891–898. doi: 10.1007/BF00965538. [http://dx.doi.org/10.1007/BF00965538]. [PMID: 1787878]. [DOI] [PubMed] [Google Scholar]
- 23.Johnson G.V., Litersky J.M., Jope R.S. Degradation of microtubule-associated protein 2 and brain spectrin by calpain: A comparative study. J. Neurochem. 1991;56(5):1630–1638. doi: 10.1111/j.1471-4159.1991.tb02061.x. [http://dx.doi.org/10.1111/j.1471-4159.1991.tb02061.x]. [PMID: 2013758]. [DOI] [PubMed] [Google Scholar]
- 24.Potter D.A., Tirnauer J.S., Janssen R., Croall D.E., Hughes C.N., Fiacco K.A., Mier J.W., Maki M., Herman I.M. Calpain regulates actin remodeling during cell spreading. J. Cell Biol. 1998;141(3):647–662. doi: 10.1083/jcb.141.3.647. [http://dx.doi.org/10.1083/jcb.141.3.647]. [PMID: 9566966]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perrin B.J., Amann K.J., Huttenlocher A. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol. Biol. Cell. 2006;17(1):239–250. doi: 10.1091/mbc.E05-06-0488. [http://dx.doi.org/10.1091/mbc.e05-06-0488]. [PMID: 16280362]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chimura T., Launey T., Yoshida N. Calpain-mediated degradation of drebrin by excitotoxicity In vitro and In vivo. PLoS One. 2015;10(4):e0125119. doi: 10.1371/journal.pone.0125119. [http://dx.doi.org/10.1371/journal.pone.0125119]. [PMID: 25905636]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulkarni S., Goll D.E., Fox J.E. Calpain cleaves RhoA generating a dominant-negative form that inhibits integrin-induced actin filament assembly and cell spreading. J. Biol. Chem. 2002;277(27):24435–24441. doi: 10.1074/jbc.M203457200. [http://dx.doi.org/10.1074/jbc.M203457200]. [PMID: 11964413]. [DOI] [PubMed] [Google Scholar]
- 28.Briz V., Zhu G., Wang Y., Liu Y., Avetisyan M., Bi X., Baudry M. Activity-dependent rapid local RhoA synthesis is required for hippocampal synaptic plasticity. J. Neurosci. 2015;35(5):2269–2282. doi: 10.1523/JNEUROSCI.2302-14.2015. [http://dx.doi.org/10.1523/JNEUROSCI.2302-14.2015]. [PMID: 25653381]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du X., Saido T.C., Tsubuki S., Indig F.E., Williams M.J., Ginsberg M.H. Calpain cleavage of the cytoplasmic domain of the integrin beta 3 subunit. J. Biol. Chem. 1995;270(44):26146–26151. doi: 10.1074/jbc.270.44.26146. [http://dx.doi.org/10.1074/jbc.270.44.26146]. [PMID: 7592818]. [DOI] [PubMed] [Google Scholar]
- 30.Pfaff M., Du X., Ginsberg M.H. Calpain cleavage of integrin beta cytoplasmic domains. FEBS Lett. 1999;460(1):17–22. doi: 10.1016/s0014-5793(99)01250-8. [http://dx.doi.org/10.1016/S0014-5793(99)01250-8]. [PMID: 10571053]. [DOI] [PubMed] [Google Scholar]
- 31.Kerstein P.C., Jacques-Fricke B.T., Rengifo J., Mogen B.J., Williams J.C., Gottlieb P.A., Sachs F., Gomez T.M. Mechanosensitive TRPC1 channels promote calpain proteolysis of talin to regulate spinal axon outgrowth. J. Neurosci. 2013;33(1):273–285. doi: 10.1523/JNEUROSCI.2142-12.2013. [http://dx.doi.org/10.1523/JNEUROSCI.2142-12.2013]. [PMID: 23283340]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dulong S., Goudenege S., Vuillier-Devillers K., Manenti S., Poussard S., Cottin P. Myristoylated alanine-rich C kinase substrate (MARCKS) is involved in myoblast fusion through its regulation by protein kinase Calpha and calpain proteolytic cleavage. Biochem. J. 2004;382(Pt 3):1015–1023. doi: 10.1042/BJ20040347. [http://dx.doi.org/10.1042/BJ20040347]. [PMID: 15239673]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klimaviciusa L., Safiulina D., Kaasik A., Klusa V., Zharkovsky A. The effects of glutamate receptor antagonists on cerebellar granule cell survival and development. Neurotoxicology. 2008;29(1):101–108. doi: 10.1016/j.neuro.2007.09.006. [http://dx.doi.org/10.1016/j.neuro.2007.09.006]. [PMID: 17981335]. [DOI] [PubMed] [Google Scholar]
- 34.Coronado V.G., Xu L., Basavaraju S.V. 2011. [Google Scholar]
- 35.Vosler P.S., Brennan C.S., Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 2008;38(1):78–100. doi: 10.1007/s12035-008-8036-x. [http://dx.doi.org/10.1007/s12035-008-8036-x]. [PMID: 18686046]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jourdi H., Hamo L., Oka T., Seegan A., Baudry M. BDNF mediates the neuroprotective effects of positive AMPA receptor modulators against MPP+-induced toxicity in cultured hippocampal and mesencephalic slices. Neuropharmacology. 2009;56(5):876–885. doi: 10.1016/j.neuropharm.2009.01.015. [http://dx.doi.org/10.1016/j.neuropharm.2009.01.015]. [PMID: 19371576]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pannaccione A., Secondo A., Molinaro P., D’Avanzo C., Cantile M., Esposito A., Boscia F., Scorziello A., Sirabella R., Sokolow S., Herchuelz A., Di Renzo G., Annunziato L. A new concept: Aβ1-42 generates a hyperfunctional proteolytic NCX3 fragment that delays caspase-12 activation and neuronal death. J. Neurosci. 2012;32(31):10609–10617. doi: 10.1523/JNEUROSCI.6429-11.2012. [http://dx.doi.org/10.1523/JNEUROSCI.6429-11.2012]. [PMID: 22855810]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papouin T., Oliet S.H. Organization, control and function of extrasynaptic NMDA receptors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014;369(1654):20130601. doi: 10.1098/rstb.2013.0601. [http://dx.doi.org/10.1098/rstb.2013.0601]. [PMID: 25225095]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chazot P.L. The NMDA receptor NR2B subunit: A valid therapeutic target for multiple CNS pathologies. Curr. Med. Chem. 2004;11(3):389–396. doi: 10.2174/0929867043456061. [http://dx.doi.org/10.2174/0929867043456061]. [PMID: 14965239]. [DOI] [PubMed] [Google Scholar]
- 40.Hardingham G.E., Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010;11(10):682–696. doi: 10.1038/nrn2911. [http://dx.doi.org/10.1038/nrn2911]. [PMID: 20842175]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krapivinsky G., Krapivinsky L., Manasian Y., Ivanov A., Tyzio R., Pellegrino C., Ben-Ari Y., Clapham D.E., Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40(4):775–784. doi: 10.1016/s0896-6273(03)00645-7. [http://dx.doi.org/10.1016/S0896-6273(03)00645-7]. [PMID: 14622581]. [DOI] [PubMed] [Google Scholar]
- 42.Zadran S., Jourdi H., Rostamiani K., Qin Q., Bi X., Baudry M. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J. Neurosci. 2010;30(3):1086–1095. doi: 10.1523/JNEUROSCI.5120-09.2010. [http://dx.doi.org/10.1523/JNEUROSCI.5120-09.2010]. [PMID: 20089917]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu J., Kurup P., Zhang Y., Goebel-Goody S.M., Wu P.H., Hawasli A.H., Baum M.L., Bibb J.A., Lombroso P.J. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neurosci. 2009;29(29):9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [http://dx.doi.org/10.1523/JNEUROSCI.2212-09.2009]. [PMID: 19625523]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gladding C.M., Sepers M.D., Xu J., Zhang L.Y.J., Milnerwood A.J., Lombroso P.J., Raymond L.A. Calpain and STriatal-Enriched protein tyrosine phosphatase (STEP) activation contribute to extrasynaptic NMDA receptor localization in a Huntington’s disease mouse model. Hum. Mol. Genet. 2012;21(17):3739–3752. doi: 10.1093/hmg/dds154. [http://dx.doi.org/10.1093/hmg/dds154]. [PMID: 22523092]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y., Lopez D., Davey P.G., Cameron D.J., Nguyen K., Tran J., Marquez E., Liu Y., Bi X., Baudry M. Calpain-1 and calpain-2 play opposite roles in retinal ganglion cell degeneration induced by retinal ischemia/reperfusion injury. Neurobiol. Dis. 2016;93:121–128. doi: 10.1016/j.nbd.2016.05.007. [http://dx.doi.org/10.1016/j.nbd.2016.05.007]. [PMID: 27185592]. [DOI] [PubMed] [Google Scholar]
- 46.Hanna R.A., Campbell R.L., Davies P.L. Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature. 2008;456(7220):409–412. doi: 10.1038/nature07451. [http://dx.doi.org/10.1038/nature07451]. [PMID: 19020623]. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y., Bi X., Baudry M. Calpain-2 as a therapeutic target for acute neuronal injury. Expert Opin. Ther. Targets. 2018;22(1):19–29. doi: 10.1080/14728222.2018.1409723. [http://dx.doi.org/10.1080/14728222.2018.1409723]. [PMID: 29168923]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y., Wang Y., Zhu G., Sun J., Bi X., Baudry M. A calpain-2 selective inhibitor enhances learning & memory by prolonging ERK activation. Neuropharmacology. 2016;105:471–477. doi: 10.1016/j.neuropharm.2016.02.022. [http://dx.doi.org/10.1016/j.neuropharm.2016.02.022]. [PMID: 26907807]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Binder S., Corrigan J.D., Langlois J.A. The public health approach to traumatic brain injury: an overview of CDC’s research and programs. J. Head Trauma Rehabil. 2005;20(3):189–195. doi: 10.1097/00001199-200505000-00002. [http://dx.doi.org/10.1097/00001199-200505000-00002]. [PMID: 15908819]. [DOI] [PubMed] [Google Scholar]
- 50.Dewan M.C., Rattani A., Gupta S., Baticulon R.E., Hung Y.C., Punchak M. Agrawal.A., Adeley, A.O., Shime, M.G., Rubiano, A.M., Rosenfeld, J.V. and Park, K.B. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018:1–18. doi: 10.3171/2017.10.JNS17352. [DOI] [PubMed] [Google Scholar]
- 51.Mustafa A.G., Alshboul O.A. Pathophysiology of traumatic brain injury. Neurosciences (Riyadh) 2013;18(3):222–234. [PMID: 23887212]. [PubMed] [Google Scholar]
- 52.Bauer D., Tung M.L., Tsao J.W. Mechanisms of traumatic brain injury. Semin. Neurol. 2015;35(1):e14–e22. doi: 10.1055/s-0035-1549095. [http://dx.doi.org/10.1055/s-0035-1549095]. [PMID: 25816125]. [DOI] [PubMed] [Google Scholar]
- 53.Quillinan N., Herson P.S., Traystman R.J. Neuropathophysiology of brain injury. Anesthesiol. Clin. 2016;34(3):453–464. doi: 10.1016/j.anclin.2016.04.011. [http://dx.doi.org/10.1016/j.anclin.2016.04.011]. [PMID: 27521191]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siman R., Giovannone N., Hanten G., Wilde E.A., McCauley S.R., Hunter J.V., Li X., Levin H.S., Smith D.H. Evidence that the blood biomarker SNTF predicts brain imaging changes and persistent cognitive dysfunction in mild TBI patients. Front. Neurol. 2013;4:190. doi: 10.3389/fneur.2013.00190. [http://dx.doi.org/10.3389/fneur.2013.00190]. [PMID: 24302918]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vile A.R., Atkinson L. Chronic traumatic encephalopathy: The cellular sequela to repetitive brain injury. J. Clin. Neurosci. 2017;41:24–29. doi: 10.1016/j.jocn.2017.03.035. [http://dx.doi.org/10.1016/j.jocn.2017.03.035]. [PMID: 28347679]. [DOI] [PubMed] [Google Scholar]
- 56.Chatterjee P., Botello-Smith W.M., Zhang H., Qian L., Alsamarah A., Kent D., Lacroix J.J., Baudry M., Luo Y. Can relative binding free energy predict selectivity of reversible covalent inhibitors? J. Am. Chem. Soc. 2017;139(49):17945–17952. doi: 10.1021/jacs.7b08938. [http://dx.doi.org/10.1021/jacs.7b08938]. [PMID: 29124934]. [DOI] [PMC free article] [PubMed] [Google Scholar]