

Abstract
Misfolding and aggregation of prion protein are related to several neurodegenerative diseases in humans such as Creutzfeldt-Jakob disease, fatal familial insomnia, and Gerstmann-Straussler-Scheinker disease. A growing number of applications in the prion field including assays for detection of PrPSc and methods for production of PrPSc de novo require recombinant prion protein (PrP) of high purity and quality. Here, we report an experimental procedure for expression and purification of full-length mammalian prion protein. This protocol has been proved to yield PrP of extremely high purity that lacks PrP adducts, oxidative modifications, or truncation, which is typically generated as a result of spontaneous oxidation or degradation. We also describe methods for preparation of amyloid fibrils from recombinant PrP in vitro. Recombinant PrP fibrils can be used as a noninfectious synthetic surrogate of PrPSc for development of prion diagnostics including generation of PrPSc-specific antibody.
Keywords: Recombinant prion protein, Inclusion body, Protein purification, Amyloid fibrils, Conformational transition, Prion diseases, IMAC, HPLC
1. Introduction
Recombinant prion protein (PrP) expressed in E. coli. Has been used extensively in prion research for various applications. These applications include modeling of prion conversion in vitro, utilization of PrP as immunogen for generating anti-PrP antibody, development of anti-prion therapeutic strategies that involve active immunization using PrP refolded in β-sheet-rich conformations, screening of anti-prion drugs using in vitro conversion assays, and others. These applications require PrP of very high purity with minimal amounts of chemical modifications or degradation. While several methods for purification and refolding of recombinant PrP have been previously described by different groups [1–6], some of the previously developed protocols required a fusion of PrP to histidine tags or produced PrP of insufficient purity or partially degraded. Inconsistent results in converting of PrP into β-sheet-rich conformations described in the past are attributed, at least in part, to differences in experimental protocols for expression and purification of PrP employed by different laboratories. Here, we describe a reliable experimental protocol for expression of tag-free full-length recombinant PrP of high purity and with minimal amount of chemical modifications or degradation. This protocol yields approximately 10 mg of mouse PrP or 6–8 mg of hamster PrP per liter of bacterial culture.
The current chapter also describes experimental protocols for converting full-length recombinant PrP into amyloid fibrils developed by our group in the past [7–10]. While recombinant PrP fibrils were able to induce transmissible prion diseases in wild-type animals, their infectivity was found to be very low [11]. Nevertheless, the immunoconformational assay that utilized conformational, PrPSc-specific antibodies and a broad panel of non-conformational antibodies revealed that the PrP fibrils produced in vitro acquired a surface structure similar to that of PrPSc [12]. In this regard, PrP fibrils appear to be a suitable synthetic surrogate of PrPSc and can be utilized for the development of prion diagnostics, high-throughput screening of anti-prion drugs, development of anti-prion decontamination procedures or an antigen for generating PrPSc-specific antibody, and other important applications in the field.
2. Materials
All solutions are prepared with deionized water purified using Synergy 185 UV Ultrapure Water System (Millipore, Bedford, MA). Water and solutions for desalting and HPLC are de-gassed under vacuum. HPLC buffers are purged with helium. Shaking procedures at 37 °C were performed in an Innova 4300 incubator (New Brunswick Scientific) set at 200 rpm.
2.1. Protein Expression
Plasmid DNA encoding mouse PrP 23–230, Syrian hamster PrP 23–231, or human PrP 23–231 (129M or 129V variants) (see Note 1) in pET101/D-T0P0 (Invitrogen).
Competent BL21 Star (DE3) One Shot E. coli cells and their SOC medium (Invitrogen).
Luria-Bertani (LB) Broth.
100 mg/ml carbenicillin disodium salt in water and stored in aliquots at −20 °C.
Two 2800 ml baffled PYREX flasks.
Terrific Broth (TB) medium composition for 1200 ml (see Note 2): 14.4 g Bacto tryptone (BD Biosciences, Sparks, MD), 28.8 g Bacto yeast extract (BD Biosciences), 4.8 ml glycerol, water to adjust 1080 ml. TB medium needs to be autoclaved and then supplemented with 120 ml filter-sterilized solution of 0.17 M KH2PO4 and 0.72 M K2HPO4 and 100 μg/ml carbenicillin.
1 M isopropyl-beta-D-thiogalactopyranoside (IPTG) in water and stored in aliquots at −20 °C.
2.2. Isolation of Inclusion Bodies
Cell lysis buffer: 50 mM Tris-HCl, 1 mM ethylenediaminete- traacetic acid (EDTA), 100 mM NaCl, pH 8.0.
9 mg/ml phenylmethylsulfonyl fluoride (PMSF) in acetonitrile and stored at −20 °C.
Lysozyme solution: prepare at 10 mg/ml in lysis buffer. Store in aliquots at −20 °C.
Deoxycholic acid.
2 mg/ml deoxyribonuclease I (DNase I, type II) in water and stored in aliquots at −20 °C.
2.3. Immobilized Metal Ion Affinity Chromatography (IMAC) and Oxidative Refolding
9 M urea. After urea is dissolved in deionized water, 10 g/l, add mixed bed Amberlite (MB-150), and stir further the solution for at least 1 h. Before use, 005Fthe solution is filtered using disposable filter units with polyethersulfone membrane. 9 M urea can be stored at −20 °C.
Chelating Sepharose Fast Flow (GE Healthcare).
0.2 M nickel sulfate (NiSO4).
Acidic buffer for elution of loosely bound ions from Sepharose: 0.02 M Na acetate, 0.5 M NaCl, pH 3.0.
IMAC buffer A: 8 M urea, 0.1 M Na2HPO4,10 mM Tris-HCl, 10 mM reduced glutathione, pH 8.0.
IMAC buffer B: 8 M urea, 0.1 M Na2HPO4,10 mM Tris-HCl, 10 mM reduced glutathione, pH 4.5.
0.5 M ethylene glycol bis(2-aminoethyl ether)-N,N,N’N’-tet-raacetic acid (EGTA), pH 8.0.
Desalting buffer: 6 M urea, 0.1 M Tris-HCl, pH 7.5.
50 mM oxidized glutathione and stored in aliquots at −20 °C.
Solutions for Sepharose regeneration and preservation: 2 M NaCl; 1 M NaOH; molecular grade ethanol.
XK chromatography column (GE Healthcare).
HiPrep 26/10 desalting column (GE Healthcare).
FPLC system (ÄKTA prime, GE Healthcare).
13 × 100 mm tubes.
2.4. High- Performance Liquid Chromatography (HPLC)
HPLC buffer A: 0.1% trifluoroacetic acid in water.
HPLC buffer B: 0.1% trifluoroacetic acid in acetonitrile.
Protein C4 HPLC column, particle size 10 μm, inner diameter 22 mm, length 250 mm; column guard, particle size 12 μm, cartridge 10 mm (Vydac).
Polyethersulfone (PES) membrane disposable filter units.
Shimadzu HPLC system (Columbia, MD) operated with EZStart 7.3 SP1 software.
2.5. Amyloid Fibril Formation (Manual Setup)
0.5 M 2-(N-Morpholino)ethanesulfonic acid (MES) buffer, pH 6.0.
10 mM sodium acetate, pH 5.0.
0.5 M thiourea pH adjusted to 6.0.
6 M guanidine hydrochloride, pH adjusted to 6.0.
Dialysis tubing (Spectra/Por, molecular weight cutoff [MWCO] 2000) and clips.
1 mM thioflavin T stock in water, stored in the dark at +4 °C.
Delfia plate shaker (Perkin Elmer, Wellmix, or similar) with a microcentrifuge tube rack attached, Clay Adams Nutator Mixer (model 1105, Becton Dickinson & Co.)
Bath sonicator (Branson 2510, Bransonic, Danbury, CT).
Plastic tubes, 1.5 ml.
Recombinant full-length prion protein (PrP) (see below).
Dialysis tubing (MWCO 2000).
2.6. Amyloid Fibril Formation (Semiautomated Setup).
In addition to the reagents described above (see Subheading 2.5):
Teflon spheres (3/32 in diameter, McMaster-Carr).
96-well flat bottom nontreated polystyrene assay plates.
Transparent plate sealers.
Microplate fluorescence reader equipped with 444 nm excitation and 485 nm emission filters.
2.7. Epifluorescence Microscopy
Inverted fluorescent microscope (Nikon Eclipse TE2000-U) equipped with the illumination system, sets of objectives, filters (excitation filter 485DF22, beam splitter 505DRLP02, and emission filter 510LP (Omega Optical, Inc., Brattleboro, VT) and a charge-coupled device (CCD) camera.
Immersion oil type FF.
Glass Coplin staining jar.
Microscope cover glass no. 1.
Isopropanol.
Acetone.
Sulfuric acid.
Hydrogen peroxide.
3. Methods
Researchers might face the following technical challenges during expression and purification of PrP:
Difficulties in achieving complete solubilization of PrP inclusion bodies.
Precipitation and irreversible binding of PrP to the IMAC matrix.
Copper-dependent self-cleavage of PrP.
Incomplete oxidative refolding of PrP.
Spontaneous formation of oxidative adducts.
Spontaneous methionine oxidation.
To minimize these problems and to achieve successful purification of PrP of high purity, the protocol described below needs to be closely followed.
3.1. PrP Production
3.1.1. Transformation of Bacterial Cells
Chemical transformation of BL21 Star (DE3) One Shot E. coli is based on the protocol described in the pET Directional TOPO Expression Kit Instruction Manual (Invitrogen).
Thaw on ice one vial of cells.
Add 1 μl (20 ng) plasmid DNA into the vial of cells, and mix by stirring gently with the pipette tip.
Incubate on ice for 30 min.
Heat-shock the cells for 30 s at 42 °C.
Immediately transfer the tube on ice.
Add 250 μl of room temperature SOC medium.
Tape the tube on its side to the bottom of incubator, and shake at 37 °C at 200 rpm for 30 min.
Add the entire transformation reaction into 50 ml centrifuge tube containing 10 ml of LB supplemented with 100 μg/ml carbenicillin.
Shake at 37 °C at 200 rpm for 3–5 h.
Add the entire volume into the 500 ml flask containing 90 ml of LB supplemented with 100 μg/ml carbenicillin.
Shake overnight at 37 °C at200 rpm.
3.1.2. Induction of PrP Expression
Supplement 1080 ml autoclaved TB media with 120 ml filter sterilized solution of phosphates (0.17 M KH2PO4 and 0.72 M K2HPO4) and 100 μg/ml carbenicillin. Mix and save 1 ml of resulting mixture as an absorbance reference for cell growth monitoring.
Add 60 ml overnight cell culture, mix, and divide equally between two baffled flasks (use sterile 1 L cylinder).
Incubate flasks with cell culture shaking at 37 °C at 200 rpm until the absorbance at 600 nm reached 0.Dilute with fresh TB, if overgrown.
Induce expression by adding 1 mM IPTG.
Continue incubation for 4–5 h.
3.1.3. Cell Harvesting
To be able to determine cell pellet mass, weigh empty centrifuge bottles.
Divide bacterial culture between four 500 ml centrifuge bottles, and centrifuge at 2975 × g maximum RCF in Beckman rotor JLA-10.500 for 10 min at 4 °C.
Discard supernatant, and calculate cell pellet mass.
At this point, cells can be stored overnight at −20 °C.
3.1.4. Cell Lysis and Isolation of Inclusion Bodies
Thoroughly resuspend the pellet in lysis buffer (8.7 ml of buffer per each g of bacterial pellet) by vortex and pipetting up and down with 25 ml pipette.
Freeze at −80 °C, and thaw the cells at least one time to ensure cell lysis. At −80 °C, cells freeze in about 10 min. Room temperature water bath is used to thaw the pellet quickly.
Pour cell lysate into a beaker, add 2 μl PMSF and 20 μl lysozyme per 1 ml lysis buffer, and stir at room temperature for 20–40 min (seeNote 3).
Add deoxycholic acid, 1 mg/ml, and stir for 20–30 min until the liquid becomes viscous.
Add DNase I to 5 μg/ml, and stir for additional 30–45 min.
Divide the lysate between four 50 ml centrifuge tubes, centrifuge at 12,000 × g for 30 min at 4 °C, and decant the supernatant.
Thoroughly resuspend the pellet in 15 ml lysis buffer by vortex and pipetting up and down.
Repeat DNase I treatment: DNase I to 5 μg/ml to each centrifuge tube, incubate on rotating platform for 20 min.
Centrifuge at 12,000 × g for 30 min at 4 °C, and decant the supernatant.
Dilute lysis buffer with water 1:9. Thoroughly resuspend the pellet in 20 ml of diluted buffer. This step removes the excess of EDTA from the inclusion bodies to allow proper binding to Ni2+-charged chromatography column.
Centrifuge at 12,000 ×g for 20 min at 4 °C, and decant the supernatant. Resulting pellet contains recombinant PrP precipitated in the form of inclusion bodies, which can be stored frozen at − 80 °C for at least 1 month.
3.2. PrP Purification
3.2.1. Immobilized Meta lon Affinity Chromatography (IMAC)
IMAC purification is performed using a XK chromatography column (GE Healthcare), packed with 20 ml of Chelating Sepharose Fast Flow. Loading the Sepharose with Ni ions and protein binding are performed in solution. The same Sepharose can be reused several times for purification of the same PrP variant (see Note 4). Desalting column is stored at 4 °C; however, it should be equilibrated to room temperature before use.
Preparing Ni-Charged Sepharose
Take the required aliquot of the Sepharose into two 50 ml centrifuge tubes.
Let the Sepharose settle down by gravity, and remove preservative solution.
To wash the Sepharose, add water to the top of the tube, cover the tube, and gently resuspend the Sepharose by inverting the tube several times. Let the Sepharose settle down (it takes about 20 min), and remove water.
With the same procedure, wash with water again.
Remove water, and charge the Sepharose by adding 2 ml 0.2 M NiSO4.
Wash the excess of ions with water twice.
Elute the loosely bound ions, washing with acidic buffer, pH 3.0.
Wash two times with water.
Equilibrate the Sepharose by washing twice with IMAC buffer A. Keep the Sepharose under buffer until the protein is solubilized and ready for binding.
Protein Solubilization and Binding
Add 10 ml of IMAC buffer A to each tube of inclusion bodies (see Notes 2 and 5).
Thoroughly resuspend the pellet.
Incubate on rotating platform 1–1.5 h at room temperature.
Centrifuge at 12,000 × g for 15 min at 4 °C.
Remove equilibration buffer from the Ni-charged Sepharose.
Add the supernatant containing solubilized recombinant PrP to the Sepharose.
Gently rotate the mixture of the Sepharose and the protein at room temperature allowing 30–40 min for binding of the protein.
IMAC and Desalting
Secure empty chromatography column on a holder nearby chromatographer.
Close column outlet, and load the mixture of Sepharose and protein solution.
Open the outlet, and drain the excess of liquid from the column, collecting it as IMAC flow-through for the analysis of binding efficiency (Figs. 1 and 2). Make sure not to drain the slurry completely: insert the adaptor, and lock it above the Sepharose as soon as the liquid front reaches the surface of the Sepharose.
Connect the column to the FPLC system (AKTA prime, GE Healthcare). Set flow rate to 2 ml/min and fraction size to 5 ml. Wash unbound proteins with IMAC buffer A until the UV readings from the chromatographer reach low plato (Fig. 1; see Notes 4 and 5).
Switch to the IMAC buffer B, pH 4.5, to start elution of PrP. Fractions with protein are collected into borosilicate glass (13 × 100 mm tubes) containing EGTA; the final concentration of EGTA in each tube should be 5 mM after fraction is collected. Typical profile of IMAC purification is shown on Fig. 1.
Combine fractions containing PrP (see Fig. 1) in a 50 ml centrifuge tube. Typically, we collect 30–35 ml of protein solution and proceed with desalting immediately.
To remove the Sepharose from the chromatography column, add water to the column, gently resuspend the slurry with the 25 ml pipette, and transfer the Sepharose with water to a new 50 ml tube for regeneration.
Attach HiPrep 26/10 desalting column to the FPLC system, wash out storage solution, and equilibrate with desalting buffer: 6 M urea, 0.1 M Tris-HCl, pH 7.5 (see Note 6).
Desalting step is used to separate the protein from glutathione. Protein solution is loaded through the FPLC super loop. Because the total volume of the protein collected after IMAC exceeds column capacity (14 ml for HiPrep 26/10 desalting column), desalting is performed in two runs. First, 14 ml of the protein solution is loaded and desalted. Then, after the salt is washed out and the column is re-equilibrated once more, the rest of the protein (~11 ml) is run through the column (Fig. 3).
Wash desalting column immediately after the last run. Wash with water until the conductivity is at the baseline level. Disconnect the column from the FPLC system and reconnect it upside down. Wash with 0.2 M NaOH until the conductivity is on high plato. Wash with water again. Finally, fill out the column with 20% ethanol, disconnect, close, and store at 4 °C.
Combine the fractions containing recombinant PrP in a new 50 ml tube, and mix. To estimate protein concentration (C), prepare 1:5 dilution of protein solution, measure absorbance at 280 nm, and calculate the concentration using the following equation: C(mg/ml) = A280 × 5 × 0.37(formousePrP23 −230; 0.37 mg/ml = 1 o . e . at 280 nm). To minimize formation of dimers during oxidative refolding of PrP, dilute PrP solution with the desalting buffer (6 M urea, 0.1 M Tris-HCl, pH 7.5) to such extent that the concentration of PrP does not exceed 0.3 mg/ml.
Supplement the PrP solution with 5 mM EGTA and 0.2 mM oxidized glutathione, and gently rotate at room temperature overnight (see Note 3).
Fig. 1.
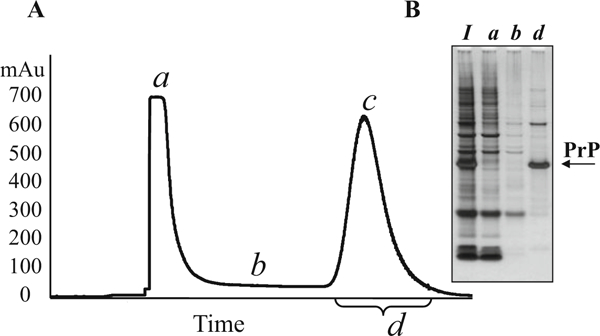
Typical IMAC profile of mouse recombinant PrP purification. (a) IMAC profile. The peak of unbound proteins (stage a) typically reaches about 700 mAu. After this peak drops to the baseline (below 50 mAu, stage b), the IMAC buffer A (pH 8.0) is changed to the IMAC buffer B (pH 4.5). The PrP peak typically reaches approximately 600 mAu (stage c). Fractions with UV values above 50 mAu (stage d) are combined for subsequent purification. (b) Analysis of IMAC fractions in SDS-PAGE (10% bis-tris) following by silver staining. I, solubilized inclusion bodies; a, b, d, fractions collected at the stages a, b, and d, respectively
Fig. 2.
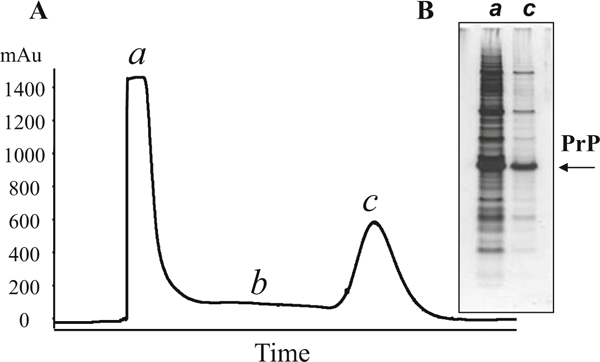
Leakage of proteins from the IMAC column. (a) IMAC profile. Overloading of IMAC column results in higher than normal UV values of unbound proteins (>1400 mAu, stage a) and subsequent baseline higher than 100 mAu (stage b). The height of the PrP peak remains at typical level of 600 mAu (stage c). (a) Analysis of IMAC fractions in SDS-PAGE (10% bis-tris) confirms the presence of high amount of PrP in the flow through (lane a)
Fig. 3.
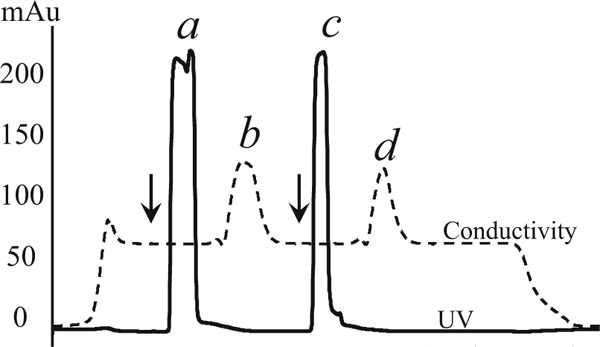
Desalting profile. PrP collected after IMAC were divided into two parts and desalted using gel filtration chromatography. The arrows mark the points of loading of PrP onto the desalting column. During desalting, the protein (peaks a and c) is separated from glutathione (peaks b and d)
Chelating Sepharose Regeneration
After the Sepharose is transferred from the chromatographer column to a 50 ml centrifuge tube, add water to the top of the tube, cover the tube, and gently resuspend the Sepharose by inverting the tube several times. Let the Sepharose settle down, and remove water.
Wash with 2 M NaCl using the same procedure.
Wash with water three times.
Wash with 1 M NaOH.
Wash with water two times. When removing water after the last wash, leave 2 ml above the Sepharose, and add 2 ml of pure ethanol for preservation. Keep at 4 °C.
High-Pressure Liquid Chromatography (HPLC)
To perform C4 HPLC, we use Shimadzu HPLC system operated with EZStart 7.3 SP1 software.
Prepare HPLC buffers A and B (1 l each).
Degas buffers A and B for 15–20 min by stirring under vacuum, and then keep under constant purging of helium.
Before connecting the column, wash the tubing and pumps of HPLC system with running buffers A and B consecutively at 5 ml/min for 10 min.
Connect the C4 column, and equilibrate with buffer A.
Visible precipitation of PrP occurs after overnight oxidation. To remove these precipitates, centrifuge the protein solution at 12,000 × g for 30 min, and then filter supernatant using disposable filter units with polyethersulfone (PES) membrane.
To reduce urea concentration, dilute the protein solution with HPLC buffer A (1:2 v/v), and load onto C4 column (see Note 7).
Wash unbound proteins from the C4 column with HPLC buffer A at the flow rate of 5 ml/min; monitor UV absorbance at 220 and 280 nm. When the baseline is reached, start the gradient (see Note 8).
Using HPLC profile as guidance, manually collect PrP fractions into borosilicate glass tubes. An a-PrP monomer is eluted in major peak between 52.5 and 54.5 min (Fig. 4a). Slow gradient separates correctly folded α-PrP monomer from PrP adducts.
At the end of the run, wash the column with HPLC buffer A until the baseline of UV absorbance is reached (see Note 9).
The quality of the purified protein is checked by SDS-PAGE (Fig. 4b) and by mass spectroscopy (see Note 10).
Freeze collected fractions at −80 °C. Lyophilize (we use Free Zone 2.5 Plus freeze dry system from Labconco (Kansas City, MO)). The protein is stored as lyophilized powder at −20 °C. The expression/purification yield depends on a species ofrPrP. When mouse, Syrian hamster, or human rPrP is expressed, purification typically resulted in 12–15 mg, 7–8 mg, or just 1 mg of protein, respectively, per 1200 ml bacterial culture.
In our experience, the longevity of HPLC C4 column is limited to ~25–30 preparations. A change in PrP retention time and significant tailing indicate that the column has reached the end of its lifetime and needs to be replaced. The column longevity depends on a species of PrP it is used to purify. The tendency for aggregation is much higher for human recombinant PrP than that for mouse or hamster PrP. As a result, the longevity of a column used for human PrP is shorter than that used for mouse or hamster PrP.
Fig. 4.
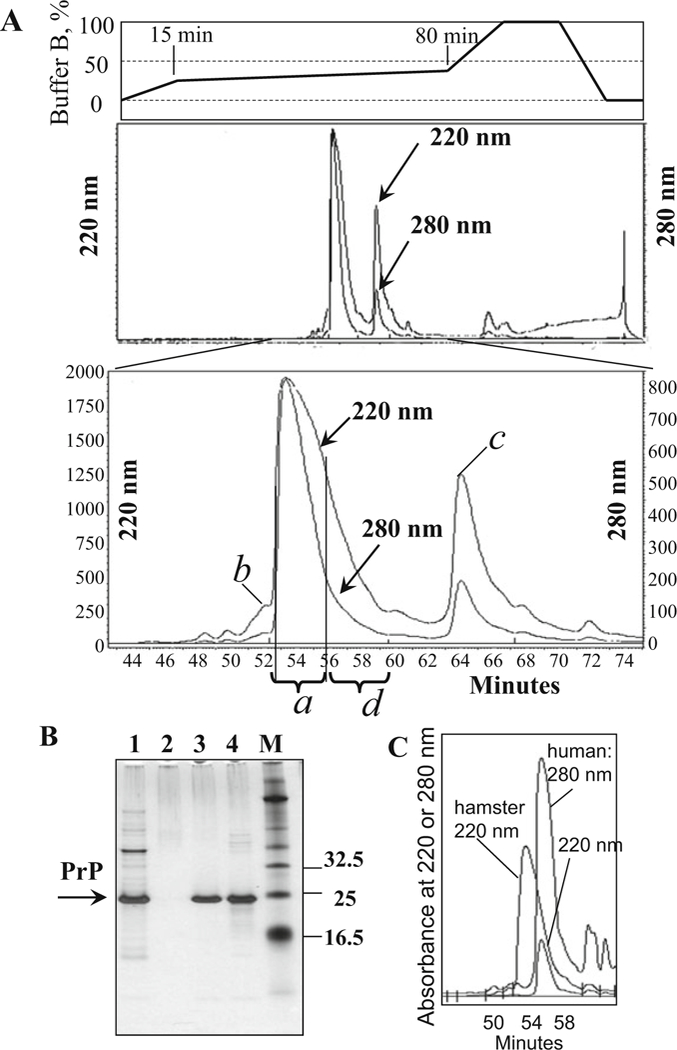
Purification of PrP on C4 column. (a) Typical HPLC profile of elution of mouse recombinant PrP. Major peak contains pure PrP (peak a); it is separated from the sub-peak containing PrP with oxidized methionines (peak b), from the peaks containing PrP with double glutathione adducts (peak c), and from other impurities. The right shoulder of the major HPLC peak (fractions d eluted at 56–60 min) is not collected; this shoulder may contain products of PrP degradation (see lane 4 in panel b). (b) Analysis of HPLC fractions in SDS-PAGE followed by silver staining: lane 1, PrP collected after IMAC and loaded onto C4 column; lane 2, HPLC flowthrough; lane 3, pure PrP collected from the major HPLC peak a; lane 4, right shoulder of the major peak (referred to as fraction d); and lane M, molecular marker. Lane 4 shows minor amounts of PrP degradation products that occur due to self-cleavage (seen as a smear with Mw < 23 kDa). The extent of PrP degradation may vary from preparation to preparation (see Note 3). (c) Comparative HPLC profile of elution of hamster and human recombinant PrP. The purification yield of human recombinant PrP (detection at 220 and 280 nm) is substantially lower than that of hamster PrP (detection at 220 nm)
3.3. Conversion of Full-Length PrP into Amyloid Fibrils in Manual Setup
Full-length PrP is capable of forming a variety of aggregated forms in vitro [7, 9, 13, 4]. Formation of amyloid fibrils is highly dependent on the reaction conditions. Ideal reaction conditions for fibril formation combine neutral or slightly acidic pH (between 5.0 and 7.5) and moderate concentrations of denaturants such as guanidine hydrochloride (up to 2 M) or urea (up to 4 M). Possible complications during fibril formation include inhibition of fibrillization by Cu2+ [13], copper-mediated and/or spontaneous N-terminal truncation of the protein [15–17], and side-chain oxidation [4]. In order to minimize these problems, copper ions are removed from the protein during purification, and 10 mM thiourea is added during fibril formation. Additional problem may arise if recombinant PrP used for conversion is of low pu1rity. The conversion conditions described here and referred to as standard are 2 M GdnHCl, pH However, fibrillations can be also performed in the absence of GdnHCl or at concentrations lower than M GdnHCl as previously described [18, 19].
Prepare stock solution of recombinant PrP (130 μM, 3 mg/ml) in 6 M GdnHCl, pH 6.0. This solution can be stored at −20 °C for up to 1 week (see Note 11). Alternatively, the protein can be dissolved directly in the MES buffer (50 mM, pH 6) at lower concentration (0.5–1 mg/ml). This solution, however, cannot be stored and must be used for fibril formation immediately (within several hours).
To prepare 500 μl reaction (see Note 12), mix the following reagents in the conical plastic tube: water (273.3 μl), GdnHCl (6 M, 83.4 μl), MES buffer (0.5 M, pH 6.0, 50 μl), and thiourea (0.5 M, 10 μl). Then add stock solution of PrP in 6M GdnHCl (3 mg/ml, 83.3 μl). Mix reagents gently, and avoid introducing air bubbles.
If you are using previously formed fibrils as seeds, sonicate them for at least 10 s in the bath sonicator, and add to the reaction mixture before adding the PrP stock. Seeding capacity of fibrils decreases upon prolonged storage. Small amounts of seeds (as little as 0.1% of the amount of protein) are sufficient to significantly decrease the lag phase of conversion.
Incubate the tube with continuous shaking at 600 rpm using a plate shaker or rotation at 24 rpm using Nutator Mixer at 37 °C (Fig. 5; see Note 13).
Monitor the kinetics of fibril formation using a thioflavin T binding assay. For this assay, withdraw the aliquots (4 μl) during the incubation. Before taking aliquots, pipette the reaction mixture gently each time (strong pipetting might perturb the kinetics of fibril formation). Dilute each aliquot into 10 mM Na acetate buffer (pH 5.0) to a final concentration of PrP of 0.3 μM, and then add thioflavin T to a final concentration of 10 μM. Record three emission spectra (from 460 to 520 nm) for each time point in 0.4 cm rectangular cuvettes with excitation at 445 nm on a microplate fluorescence reader (we use FluoroMax-3 fluorimeter, Jobin Yvon, Edison, NJ) keeping excitation and emission slits at 4 nm. Average the spectra, and determine the fluorescence intensity at emission maximum (usually around 482 nm). Emission will remain low for a few hours or days, then will start rising, and eventually will rise 10- to 40-fold of the original baseline level (see Note 14).
After the fibril formation has reached a plateau, the fibrils should be dialyzed for prolonged storage. Place the suspension of fibrils in the bag prepared from the dialysis tubing (MWCO 2000), and dialyze against a large volume of 10 mM Na acetate buffer (pH 5.0) with several buffer changes. Fibrils should be stored at +4 °C. Prolonged storage of fibrils at room temperature or at higher pH may lead to their aggregation and coppermediated protein self-cleavage. Freezing and thawing may cause fragmentation of fibrils into short pieces.
Fig. 5.

Plate shaker with a microcentrifuge tube rack used for the conversion reaction. Place plastic tubes next to each other to prevent their rotation inside the rack during shaking
3.4. Conversion of Full-Length PrP into Amyloid Fibrils in Semiautomated Setup
Perform the conversion of PrP into amyloid fibrils in semiautomated setup at least in triplicate to ensure reproducibility. Add three Teflon spheres (3/32 in. diameter) per well of 96- well assay plate. Mix the following reagents in the conical plastic tube: water (268.3 μl), GdnHCl (6 M, 163.7 μl), MES buffer (0.5 M, pH 6.0, 50 μl), thioflavin T (1 mM, 5 μl), and thiourea (0.5 M, 10 μl). Add stock solution of PrP in 6 M GdnHCl (3 mg/ml, 5 μl). After thorough mixing, divide the reaction mixture between three wells of the 96-well plate (160 μl per well), and cover the plate with the plate sealer. If previously formed fibrils are used as seeds, they should be sonicated for at least 10 s in the bath sonicator and added to the reaction mixture before addition of the PrP stock.
Insert the 96-well plate into the microplate reader. Set up incubation at 37 °C with shaking at 900 rpm (shaking diameter 1 mm) and fluorescence measurements every 5 or 10 min with excitation at 444 nm and emission at 485 nm (Fig. 6).
After the completion of the experiment, transfer the data to a graphing and data analysis program (we use Origin (OriginLab)), and fit to the following equation:
Fig. 6.
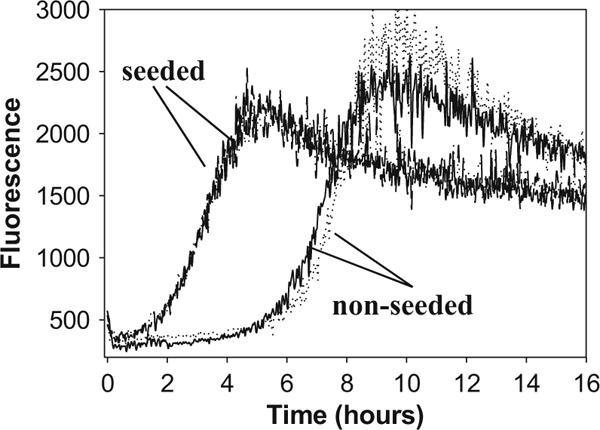
Kinetics of fibril formation from PrP (1 μM) carried out in semiautomated format under standard solvent conditions. Two repeats for non-seeded and seeded conversion reactions are shown. After reaching a plato, the ThT fluorescence often shows a decline that appears to be due to sorption of fibrils to plate walls and partial aggregation of fibrils into clumps
where A is the initial level of thioflavin T fluorescence, B is the increase in thioflavin T fluorescence during conversion, k is the rate constant of amyloid accumulation (h−1), tm is the midpoint of conversion of PrP to amyloid, and c is an empirical parameter describing changes in fluorescence after fibril formation. The lag time (tl) can be calculated as
3.5. Epifluorescence Microscopy
While ThT fluorescence assay is convenient for measuring the kinetics of fibril formation, this assay, however, is not sufficient for providing a definite poof as to whether amyloid fibrils were formed in the reaction mixture. Relatively small (two- to threefold) increase in ThT fluorescence may indicate formation of non-fibrillar PrP isoforms such as soluble β-oligomers, which also bind ThT. Several techniques including electron microscopy and atomic force microscopy have been used for confirming the formation of fibrils; however, their application is laborious and requires costly equipment. In our experience, epifluorescence microscopy in the presence of ThT serves as a rapid and reliable test for the presence of the amyloid fibrils in the sample, for assessing their quality, size, aggregation status, and even growth rate [7, 8, 10, 13, 20]. Here, we describe the experimental procedure for imaging amyloid fibrils using inverted fluorescence microscopy in the presence of ThT.
Deposit several cover glasses into a staining jar, and clean them by sonicating it in isopropanol (1 min), then in acetone (1 min), and again in isopropanol (1 min). Wash the cover glass with water, and incubate it in the mixture of sulfuric acid (70%) and hydrogen peroxide (30%) for at least 1 h. Rinse with water several times. Store in water, and dry with compressed air before use.
Add 0.5 μl of suspension of PrP fibrils (0.5 mg/ml) in 10 mM acetate buffer (pH 5.0) to the same buffer (99.5 μl) containing 10 μM thioflavin T. Incubate the solution in the plastic tube at 25 °C for 5 min.
Place 10–20 μl of the solution on the cover glass. Allow fibrils to sediment on the glass surface for 1–2 min. Examine the sample with an inverted fluorescence microscope (Nikon Eclipse TE2000-U) using 60× or 100× objective. The emission can be isolated from Rayleigh and Raman-shifted light by a combination of filters: an excitation filter 485DF22, a beam splitter 505DRLP02, and an emission filter 510LP (Omega Optical, Inc.). Digital images can be acquired using a CCD camera.
Shape and size of PrP fibrils vary depending on the conditions of their formation including shaking modes and purity of the protein (see Note 14). Under the standard conditions, PrP yields long (>1 μm) and straight fibrils, if highly pure (Fig. 7a). Fibrils formed at higher pH (pH > 7.0) tend to aggregate into large clusters. Exposure of fibrils formed at pH < 6 to pH > 6.5 or addition of salt induced aggregation into clumps of various size (Fig. 7b). Presence of impurities in the protein (substantial methionine oxidation or N-terminal truncation) can result in formation of shorter fibrils (<1 μm) that often appear bent or very short, dot-like fibrils (Fig. 7c).
Fig. 7.
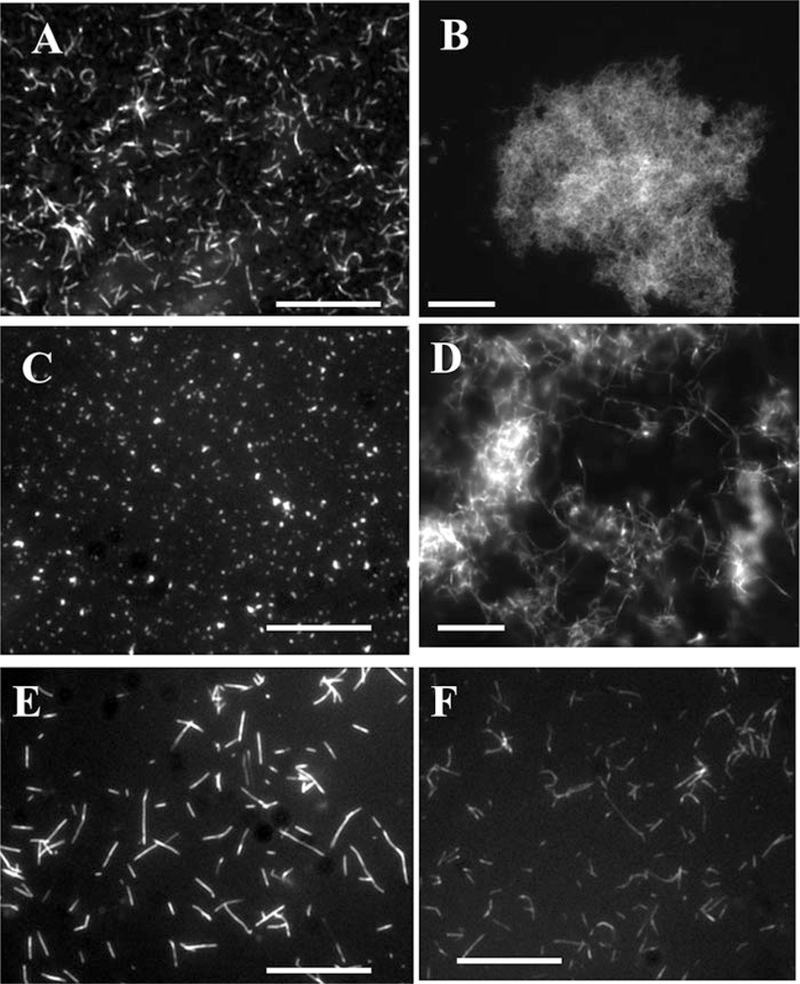
Epifluorescence microscopy of PrP fibrils stained with thioflavin T. (a) Fibrils of mouse PrP prepared under standard conditions (shaking, pH 6). (b) Fibrils of mouse PrP prepared under standard conditions and incubated for 24 h in PBS, pH 7.(c) PrP (mouse) of insufficient purity or with substantial methionine oxidation converts into very short or dot-like fibrils under standard conditions. (d) Fibrils of mouse PrP prepared under standard conditions and digested with PK for 1 h at 37 °C. (e) Fibrils of mouse PrP prepared under standard solvent conditions upon rotation. (f) Fibrils of Syrian hamster PrP prepared under standard solvent conditions upon rotation. When stained with ThT, mouse fibrils show substantially brighter fluorescence than hamster fibrils. Scale bars = 10 μm
4. Notes
Plasmids for expression of mouse PrP 23–230, Syrian hamster PrP 23–231, or human PrP 23–231 (129M or 129V) were cloned from mouse, Syrian hamster, or human cDNA, respectively, as described [7, 14].
We have found it convenient to grow bacteria in two flasks each containing 600 ml TB media. This typically results in total of 12 g bacterial pellet used for isolation of inclusion bodies. To prevent overloading of HPLC column, only one half of inclusion bodies should be used for IMAC. Another half is stored at −80 °C.
Recombinant PrP is prone to copper-dependent self-cleavage [15,16]. Performing cell lysis steps at lower temperature could slow down degradation; however, this would require prolonged incubation time. Therefore, the cell lysis steps are carried out at room temperature. EDTA and EGTA are added to the lysis buffer and during oxidative refolding, respectively, to minimize protein degradation.
Lack of Ni ions on the Sepharose may cause the leakage of protein during wash step. For better performance, recharge the column with Ni ions each time before reuse.
Loading the column with excessive amounts of protein (Fig. 2) or incomplete solubilization of inclusion bodies (Fig. 8) results in high UV readings during the wash with IMAC buffer A. To achieve proper solubilization of the protein, it is very important to prepare IMAC buffers A and B fresh, on the same day the IMAC purification is performed. Instead of keeping reduced glutathione in concentrated stock solution, we dissolved it directly in the IMAC buffers to the final concentration of 10 mM.
Make sure to follow recommendations on pressure limit and buffer compatibility for the specific column. Our desalting column cannot tolerate urea at the concentrations above 6 M; therefore, desalting buffer contains less urea than IMAC buffers. Depending on solution viscosity, flow rate needs to be adjusted during the run to keep pressure under the limit.
Because after overnight oxidation PrP solution contains urea, loading the protein onto C4 column requires pressure above Shimadzu HPLC system limits. To avoid high pressure, we dilute PrP solution threefold with buffer A and use external HPLC pump 2010 from Varian (Walnut Creek, CA) to load the protein. We wash the pump with water and with 20 ml of buffer A before loading PrP. We load protein solution at flow rate 5 ml/min and finish by loading 20 ml of HPLC buffer A, to ensure that no protein is left in the pump. After loading is completed, we disconnect and thoroughly wash the loading pump with water.
In our gradient method, the percentage of HPLC buffer B (0.1% trifluoroacetic acid in acetonitrile) grows from 0% to 25% during the first 15 min (at flow rate 5 ml/min). Then, gentle gradient is applied (from 25% to 35% of buffer B in the next 65 min) to ensure efficient separation of PrP adducts (Fig. 4a). Correctly folded, oxidized, α-helical monomeric form of PrP is eluted between 52.5 and 54.5 min. Then, the percentage of buffer B grows from 35% to 100% in 15 min; the column is washed with 100% of buffer B for 15 min, and the gradient drops down to 0% within 10 min.
If the same C4 column is used for purification of different PrP variants, running washing program with extended time of 100% acetonitrile will ensure the absence of crosscontamination. In our washing program, buffer B concentration grows from 0% to 100% within 20 min, remains constant 100% for 25 min, and then goes down to 0% during the last 20 min. The run is finished by equilibration of the C4 column with buffer A for 25 min, which prepares the column for loading of new protein.
To perform mass spectroscopy, a small amount of recombinant PrP (as a lyophilized powder) is dissolved in 20 μl of 1:1 water/ methanol mixture containing 1% acetic acid. The solution is then analyzed on a single quadrupole mass spectrometer operated in positive ion mode.
After PrP is purified, it is preferable to store the protein as a lyophilized powder at −20 °C. Storage in 6 M GdnHCl at −20°C is possible for up to 1 week. However, prolonged storage at 6 M GdnHCl leads to formation of the β-oligomeric nonfibrillar form. Prolonged storage (longer than 24 h) in solution at neutral pH and in the absence of denaturants is not advisable due to self-cleavage and nonspecific aggregation.
Fibril formation can proceed sluggishly if the volume of the solution in the tube is too low (<300 μl).
Make sure that the plastic tube fits tightly within the rack attached to the platform and the tube does not rotate inside the rack during orbital shaking (Fig. 5). End-over-end rotation on Nutator Mixer or similar device can also be used as an alternative way for fibrillation reactions. However, structurally different types of fibrils are formed under different shaking/ rotation modes even when identical solvent conditions are employed for fibrillation reactions [10, 21].
Exact value of thioflavin T fluorescence depends on several factors including purity of the protein and conditions of fibril formation. Differences in intensity of ThT fluorescence could be due to several factors including differences in ThT binding affinity for fibrils prepared at different solvent conditions, different yields of fibril formation, or different propensities of fibrils to aggregate into large clumps. The yield of ThT fluorescence of fibrils prepared under nonstandard conditions is often lower than that for standard fibrils. ThT fluorescence of fibrils prepared under different shaking modes or from different PrP variants (e.g., mouse vs. hamster PrP) can also vary [10] (Fig. 7e, f). Values of ThT fluorescence threefold or more above background usually indicate the presence of amyloid fibrils. In addition to ThT fluorescence assay, fluorescence microscopy helps to analyze the yield and quality of fibrils (Fig. 7).
Fig. 8.
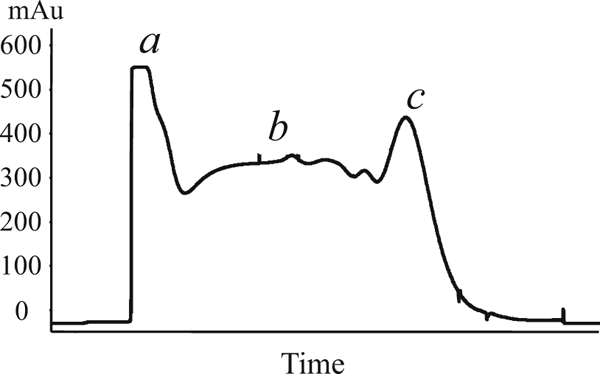
IMAC profile of incompletely solubilized protein. Reduced glutathione degraded during preparation of stock solution (see Note 5). As a result, recombinant PrP was incompletely solubilized and leaked from the Sepharose during the wash step. The peak of unbound proteins (stage a) was below 600 mAu; the UV values during the wash steps were above 300 mAu (stage b), and the yield of eluted PrP was reduced (stage c)
Acknowledgments
This work was supported by the National Institutes of Health grants R01 NS045585 and NS074998.
References
- 1.Zahn R, von Schroetter C, Wuthrich K (1997) Human prion protein expression in Escherichia coli and purified by high-affinity column refolding. FEBS Lett 417:400–404 [DOI] [PubMed] [Google Scholar]
- 2.Mehlhorn I, Groth D, StÖckel J et al. (1996) High-level expression and characterization of a purified 142-residue polypeptide of the prion protein. Biochemistry 35:5528–5537 [DOI] [PubMed] [Google Scholar]
- 3.Rezaei H, Marc D, Choiset Yet al (2000) High yield purification and physico-chemical properties of full-length recombinant allelic variants of sheep prion protein linked to scrapie susceptibility. Eur J Biochem 267:2833–2839 [DOI] [PubMed] [Google Scholar]
- 4.Hornemann S, Korth C, Oesch B et al. (1997) Recombinant full-length murine prion protein, mPrP(23–231): purification and spectroscopic characterization. FEBS Lett 413:277–281 [DOI] [PubMed] [Google Scholar]
- 5.Jackson GS, Hil AF, Joseph C et al. (1999) Multiple folding pathways for heterologously expressed human prion protein. Biochim Biophys Acta 1431(1):1–13 [DOI] [PubMed] [Google Scholar]
- 6.Yin SM, Zheng Y, Tien P (2003) On-column purification and refolding of recombinant bovine prion protein: using its octarepeat sequences as a natural affinity tag. Protein Expr Purif 32:104–109 [DOI] [PubMed] [Google Scholar]
- 7.Bocharova OV, Breydo L, Parfenov AS et al. (2005) In vitro conversion of full length mammalian prion protein produces amyloid form with physical property of PrPSc. J Mol Biol 346:645–659 [DOI] [PubMed] [Google Scholar]
- 8.Baskakov IV, Bocharova OV (2005) In vitro conversion of mammalian prion protein into amyloid fibrils displays unusual features. Biochemistry 44:2339–2348 [DOI] [PubMed] [Google Scholar]
- 9.Bocharova OV, Makarava N, Breydo L et al. (2005) Annealing PrP amyloid fibrils at high temperature results in extension of a proteinase K resistant core. J Biol Chem 281:2373–2379 [DOI] [PubMed] [Google Scholar]
- 10.Makarava N, Baskakov IV (2008) The same primary structure of the prion protein yields two distinct self-propagating states. J Biol Chem 283:15988–15996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makarava N, Kovacs GG, Bocharova OV et al. (2010) Recombinant prion protein induces a new transmissible prion disease in wild type animals. Acta Neuropathol 119:177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novitskaya V, Makarava N, Bellon A et al. (2006) Probing the conformation of the prion protein within a single amyloid fibril using a novel immunoconformational assay. J Biol Chem 281:15536–15545 [DOI] [PubMed] [Google Scholar]
- 13.Bocharova OV, Breydo L, Salnikov VV, Baskakov IV (2005) Cu(II) inhibits in vitro conversion of prion protein into amyloid fibrils. Biochemistry 44:6776–6787 [DOI] [PubMed] [Google Scholar]
- 14.Breydo L, Bocharova OV, Makarava N et al. (2005) Methionine oxidation interferes with conversion of the prion protein into the fibrillar proteinase K-resistant conformation. Biochemistry 44:15534–15543 [DOI] [PubMed] [Google Scholar]
- 15.McMahon HEM, Mange A, Nishida N et al. (2001) Cleavage of the amino terminus of the prion protein by reactive oxygen species. J Biol Chem 276:2286–2291 [DOI] [PubMed] [Google Scholar]
- 16.Mange A, Beranger F, Peoc’h K et al. (2004) Alpha- and beta-cleavages of the aminoterminus of the cellular prion protein. Biol Cell 96:125–132 [DOI] [PubMed] [Google Scholar]
- 17.Ostapchenko VG, Makarava N, Savtchenko R, Baskakov IV (2009) The polybasic N-terminal region of the prion protein controls the physical properties of both the cellular and fibrillar forms of PrP. J Mol Biol 383:1210–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y, Makarava N, Lee CI et al. (2008) Conformational stability of PrP amyloid firbils controls their smallest possible fragment size. J Mol Biol 376:1155–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almstedt K, Nystrom S, Nilsson KPR, Ham- marstrom P (2009) Amyloid fibrils of human prion protein are spun and woven from morphologically disordered aggregates. Prion 3:224–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bocharova OV, Breydo L, Salnikov VV et al. (2005) Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob disease. Protein Sci 14:1222–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ostapchenko VG, Sawaya MR, Makarava N et al. (2010) Two amyloid states of the prion protein display significantly different folding patterns. J Mol Biol 400:908–921 [DOI] [PMC free article] [PubMed] [Google Scholar]