

Abstract
Chronic neuroinflammation is recognized as a major neuropathological hallmark in a broad spectrum of neurodegenerative diseases including Alzheimer’s, Parkinson’s, Frontal Temporal Dementia, Amyotrophic Lateral Sclerosis, and prion diseases. Both microglia and astrocytes exhibit region-specific homeostatic transcriptional identities, which under chronic neurodegeneration, transform into reactive phenotypes in a region- and disease-specific manner. Little is known about region-specific identity of glia in prion diseases. The current study was designed to determine whether the region-specific homeostatic signature of glia changes with the progression of prion diseases, and whether these changes occur in a region-dependent or universal manner. Also of interest was whether different prion strains give rise to different reactive phenotypes. To answer these questions, we analyzed gene expression in the thalamus, cortex, hypothalamus and hippocampus of mice infected with 22L and ME7 prion strains using a Nanostring Neuroinflammation panel at the subclinical, early clinical and advanced stages of the disease. We found that at the preclinical stage of the disease, the region-specific homeostatic identities were preserved. However, with the appearance of clinical signs, the region-specific signatures were partially lost and replaced with a neuroinflammation signature. While the same sets of genes were activated by both prion strains, the timing of neuroinflammation and the degree of activation in different brain regions was strain-specific. Changes in astrocyte function scored at the top of the activated pathways. Moreover, clustering analysis suggested that the astrocyte function pathway responded to prion infection prior to the Activated Microglia or Neuron and Neurotransmission pathways. The current work established neuroinflammation gene expression signature associated with prion diseases. Our results illustrate that with the disease progression, the region-specific homeostatic transcriptome signatures are replaced by the region-independent neuroinflammation signature, which is common for prion strains with different cell tropism. The prion-associated neuroinflammation signature identified in the current study overlapped only partially with the microglia degenerative phenotype and the disease-associated microglia phenotype reported for animal models of other neurodegenerative diseases.
Keywords: Neurodegenerative diseases, Prion diseases, Chronic neuroinflammation, Reactive microglia, Reactive astrocytes, Thalamus, Prion strains, Gene expression
1. Introduction
Chronic neuroinflammation is recognized as one of the major neuropathological hallmarks of neurodegenerative diseases including Alzheimer’s, Parkinson’s, Frontal Temporal Dementia, Amyotrophic Lateral Sclerosis, and prion diseases (Stephenson, Nutma, van der Valk, & Amor, 2018). Chronic neuroinflammation manifests itself as a sustained activation of glial cells and the transformation of their homeostatic phenotype into reactive phenotypes (Li & Barres, 2017; Liddelow & Barres, 2017). Transcriptome analysis and single-cell RNA-sequencing revealed a considerable region-specific homeostatic heterogeneity in microglia and astrocyte phenotypes under normal conditions as well as dynamic phenotypic transformations in aging and neurodegenerative diseases (Boisvert, Erikson, Shokhirev, & Allen, 2018; Clarke et al., 2018; Grabert et al., 2016; Mathys et al., 2017; Soreq et al., 2017; Zeisel et al., 2018). While incredible progress has been made in characterizing diversity of glial phenotypes using mouse models of neurodegenerative diseases, concerns whether mouse models faithfully recapitulate key aspects of disease in humans have been raised on numerous occasions (Dawson, Golde, & Lagier-Tourenne, 2018; Friedman et al., 2018; Morrissette, Parachikova, Green, & LaFerla, 2009).
For elucidating mechanisms behind chronic neuroinflammation and neurodegeneration, prion disease offers several advantages over other neurodegenerative disorders. The most obvious reason behind choosing prion disease is that animals inoculated with prions develop actual bona fide prion disease, not a disease model (Watts & Prusiner, 2014). Inbred mice infected with prions recapitulate neuropathological and biochemical features associated with naturally occurring prion diseases including prion diseases in humans. Prion diseases can be efficiently transmitted between wild-type animals or inbred laboratory mice; the process does not rely on the expression or over-expression of modified human genes.
Prions, or PrPSc, are proteinaceous infectious agents that consist of misfolded, self-replicating states of a sialoglycoprotein called the prion protein or PrPC (Legname et al., 2004; Prusiner, 1982). Prion diseases display a diverse range of disease phenotypes, a feature attributed to the ability of PrPC to acquire multiple, conformationally distinct, self-replicating PrPSc states referred to as prion strains or subtypes (Caughey, Raymond, & Bessen, 1998; Collinge & Clarke, 2007; Thomzig, Spassov, Friedrich, Naumann, & Beekes, 2004). In addition to differences in structure, prion or PrPSc strains exhibit different patterns of terminal carbohydrate groups and a variable density of sialic acid residues on their surfaces (Baskakov & Katorcha, 2016; Baskakov, Katorcha, & Makarava, 2018; Katorcha, Makarava, Savtchenko, & Baskakov, 2015; Makarava, Savtchenko, & Baskakov, 2015). The differences in surface-exposed carbohydrate epitopes are believed to be due to a selective strain-specific recruitment of PrPC molecules among a large pool of more than 400 PrPC sialoglycoforms expressed by a cell (Baskakov, 2017; Baskakov & Katorcha, 2016; Katorcha et al., 2015). Considering the structural diversity of PrPSc and the diversity of carbohydrate epitopes on the surfaces of PrPSc particles, it is not surprising that prion strains exhibit selective strain-specific tropism with respect to brain region and cell type (Ayers et al., 2011; Carroll et al., 2016; Karapetyan et al., 2009). All prion strains used (22L and ME7) or discussed in the current work (RML, 301V and 263K) are of natural origin, i.e. isolated from animals that succumbed to transmissible spongiform encephalopathy and then serially passaged in mice (strains 22L, ME7, RML and 301V) or hamsters (strain 263K) (Bruce & Dickinson, 1987; Kimberlin, Cole, & Walker, 1987; Kimberlin, Walker, & Fraser, 1989).
Previous studies analyzed transcriptome changes using laboratory inbred mice infected with prions (Hwang et al., 2009; A. Majer et al., 2012; Anna Majer et al., 2019; Skinner et al., 2006; Sorensen et al., 2008; Vincenti et al., 2016; Xiang et al., 2004). The whole-transcriptome analysis, in combination with the analyses of selective gene sets, identified activation of microglia with strong proinflammatory characteristics as a common signature of chronic neuroinflammation associated with prion diseases (Baker & Manuelidis, 2003; Carroll et al., 2016; Lu, Baker, & Manuelidis, 2004; Anna Majer et al., 2019; Sorensen et al., 2008; Tribouillard-Tanvier, Striebel, Peterson, & Chesebro, 2009; Vincenti et al., 2016). While these studies revealed a variety of differentially activated genes and pathways, our knowledge about reactive phenotypes of microglia and astrocytes in prion diseases is very limited in comparison to other neurodegenerative diseases. In the majority of previous studies, whole brain tissues were used for transcriptome analysis leaving region specific identities concealed. In the current study, we asked whether prion strains give rise to different reactive phenotypes in glia and whether these phenotypes are influenced by region-specific homeostatic signatures. To address these questions, we analyzed gene expression in four brain regions of mice infected with two prion strains, 22L and ME7, that have different cell- and region-specific tropism (Carroll et al., 2016), using a Nanostring Neuroinflammation panel. The 22L strain is mainly associated with astrocytes, whereas the ME7 strain is predominantly found in association with neurons (Carroll et al., 2016). At the preclinical stage of the disease, the region-specific homeostatic identity of glia was well preserved. However, with the disease progression, the region-specific homeostatic signatures were partially lost and replaced with a neuroinflammation signature. The same genes were activated by both prion strains, however, the timing of neuroinflammation and the degree of activation in different brain regions were strain-specific. Global significance scoring of the differentially expressed genes identified the Astrocyte Function pathway at the top of the list followed by the Inflammatory Signaling, Matrix Remodeling, and Activated Microglia. Moreover, clustering analysis of the gene expression patterns suggested that the Astrocyte Function pathway responded to prion infection prior to the Activated Microglia or the Neuron and Neurotransmission pathways. The current work established Neuroinflammation gene expression signature associated with prion diseases and demonstrated that it was independent of the brain region or prion cell tropism.
2. Methods and Materials
2.1. Animal experiments and brain tissue collection
Using isoflurane anesthesia, six-week-old C57BL/6J female mice were inoculated intraperitoneally with 200 μl of 1% 22L or ME7 brain homogenate in PBS, pH 7.4. A control group was inoculated with PBS only. Animals were regularly scored for signs of neurological impairment and disease progression: progressive difficulty walking on a beam, hind limb clasping, and weight loss. Pre-symptomatic 22L and ME7 samples were collected at 153 – 154 days post-inoculation (dpi) from animals showing no clinical signs and no weight loss. The early clinical 22L samples (the 2nd time point) were collected upon consistent observation of mild motor impairment signs for two weeks, which were the first clinical signs observed. No significant weight loss was observed for these animals. For ME7, the 2nd time point samples were collected at 224 dpi from mice displaying no signs of neurological impairment or weight loss. ME7 mice started to develop the first clinical signs of the disease at 280 – 343 dpi. Within 15 – 31 days after the first clinical signs, 22L and ME7 mice became unable to walk on a beam, developed abnormal gait and became lethargic. Mice were considered terminally ill when they were unable to rear and/or lost 20% of their weight. At this time, the advanced clinical (3rd) point samples were collected. Mice were euthanized by CO2 asphyxiation and decapitation.
After euthanasia, brains were immediately extracted and kept ice-cold during dissection. Brains were sliced using rodent brain slicer matrix (Zivic Instruments, Pittsburg, PA). 2 mm central coronal sections of each brain were used to collect individual regions. Allen Brain Atlas digital portal (http://mouse.brain-map.org/static/atlas) was used as a reference. The hypothalami (HTh), as well as left and right thalami (Th), hippocampi (Hp) and cortexes (Ctx) were collected into RNase-free sterile tubes, frozen in liquid nitrogen, and stored at −80°C until the RNA isolation procedure. The anterior part of the brain and left half of the posterior part of the brain were saved in 10% buffered formalin for immunohistochemistry. Dissection remnants were frozen in separate tubes for Western blot with anti-PrP antibody ab3531 (Abcam, Cambridge, MA)
2.2. RNA isolation
Brain tissue samples were homogenized within RNase-free 1.5 ml tubes in 200 μl of Trizol (Thermo Fisher Scientific, Waltham, MA, USA), using RNase-free disposable pestles (Fisher scientific, Hampton, NH). After homogenization, an additional 600 μl of Trizol was added to each homogenate, and the samples were centrifuged at 11,400 x g for 5 min at 4°C. The supernatant was collected, incubated for 5 min at room temperature, then supplemented with 160 μl of cold chloroform and vigorously shaken for 30 sec by hand. After an additional 5 min incubation at room temperature, the samples were centrifuged at 11,400 x g for 15 min at 4°C. The top layer was transferred to new RNase-free tubes and mixed with an equal amount of 70% ethanol. Subsequent steps were performed using an Aurum Total RNA Mini Kit (Bio-Rad, Hercules, CA, USA) following the manufacturer’s instructions. Isolated total RNA was subjected to DNase I digestion. RNA purity and concentrations were estimated using a NanoDrop One Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
2.3. NanoString
Samples were processed by the Institute for Genome Center at the University of Maryland School of Medicine using the nCounter Mouse Neuroinflammation Panel. Only samples with an RNA integrity number RIN > 7.2 were used for Nanostring. All data passed QC, with no imaging, binding, positive control, or CodeSet content normalization flags. The analysis of data was performed using nSolver Analysis Software 4.0, including nCounter Advanced Analysis (version 2.0.115). For agglomerative clusters and heat maps, genes with less than 10% of samples above 20 counts were excluded. Z-score transformation was performed for genes. Clustering was done using Euclidian distance, linkage method was Average.
2.4. Histopathological study
Formalin-fixed brain sections were submerged for 1 hour in 95% formic acid to deactivate prion infectivity before being embedded in paraffin. Subsequent 4 μm sections produced using a Leica RM2235 microtome were mounted on slides and processed for immunohistochemistry. To expose epitopes, slides were subjected to 20 min of hydrated autoclaving at 121°C in trisodium citrate buffer, pH 6.0, with 0.05% Tween 20. Rabbit anti-Iba1 (Wako, Richmond, VA) was used to stain for microglia. Chicken polyclonal anti-GFAP (Sigma-Aldrich, St. Louis, MO) was used to stain for astrocytes. For detection of disease-associated PrP, a 5 min treatment with 88% formic acid was used following autoclaving. PrP was stained with anti-prion antibody SAF-84 (Cayman Chemical, Ann Arbor, MI). Detection was performed by using DAB Quanto chromogen and substrate (VWR, Radnor, PA).
3. Results
3.1. Experimental design
C57Black/6J mice were intraperitoneally (IP) inoculated with 22L or ME7 mouse-adapted prion strains (200 μl, 1% brain homogenate) at 5 weeks old and euthanized at three time points post-inoculation (Fig. S1A, Table S1). Animals infected with 22L prions were euthanized at the preclinical (1st time-point, 153 days post-inoculation (dpi)), early clinical (2nd time-point, 186 - 197 dpi) and advanced clinical stages of the disease (3rd time-point, 168 - 225 dpi) (Table S1). ME7-infected animals were euthanized at the early preclinical (1st time-point, 154 dpi), late preclinical (2nd time-point, 224 dpi) and advanced clinical stages of the disease (3rd time-point, 295 - 363 dpi) (Table S1). For identifying early stages, advanced clinical stages and monitoring progression of the disease, the disease scoring protocol was employed as described in Methods. IP inoculation allowed us to avoid effects related to brain trauma, yet this route of infection had some drawbacks such as differences in the onset of the disease and relatively poor cooperativity in disease progression within an animal group. In animals inoculated with 22L prions, the timing of the early and advanced clinical stages overlapped between groups due to variations of the disease onset and the rate of the disease progression (Fig. S1A, Table S1). As control groups, C57Black/6J mice were inoculated IP with PBS and euthanized at 151 dpi (controls for the 1st time-point for 22L and ME7), 197 - 223 dpi (controls for the 2nd time-point for 22L and ME7, and for the 3rd time-point for 22L) and 295 - 363 dpi (controls for the 3rd time-point for ME7) (Fig. S1A, Table S1).
For assessing the region-specific neuroinflammation status, four brain regions - thalamus (Th), hypothalamus (HTh), cortex (Ctx) and hippocampus (Hp) (n=3 individual animals per group) were selected based on previous studies (Carroll et al., 2016; Karapetyan et al., 2009; Sandberg et al., 2014; Srivastava et al., 2018) (Fig. S1B). Analysis of the expression of genes associated with neuroinflammation was conducted using the nCounter Nanostring Neuroinflammation panel (Table S2) that analyzes expression of 757 genes (including 13 housekeeping genes), which assess 23 pathways including Activated Microglia, Innate Immune Response, Adaptive Immune Response, Growth Factor Signaling, Inflammatory Signaling, Apoptosis, Autophagy, Astrocyte Function and others (the full list is in Fig. S2).
3.2. At the preclinical stage, the neuroinflammation gene expression profile displays a region-specific identity
Agglomerative hierarchical clustering of all data collected at the first, preclinical time point reveled that four brain regions displayed region-specific gene expression profiles, illustrating homeostatic signatures of individual regions (Fig. 1). Four distinct clusters corresponding to the Th, HTh, Ctx and Hp were observed (Fig. 1). With the exception of the Th cluster, 22L and ME7 datasets did not segregate into separate sub-clusters in remaining regions, but were mixed with normal controls. This result suggests that the thalamus might be the first region affected by neuroinflammation. A small subset of genes covered by the panel showed minor, yet statistically significant up- or down-regulation at preclinical stages (Table S3). However, these changes were not sufficient to override region-specific homeostatic identity. In summary, at the preclinical stage, the region-specific homeostatic identities were well-preserved in all brain regions (Fig. 1). The largest proportion of genes covered by the Neuroinflammation panel reports on microglia phenotype and their activation state (Fig. S2). As such, the region-specific homeostatic signatures are indicative of differences in microglia phenotypes in the four brain regions (Fig. 1).
Figure 1. Agglomerative hierarchical cluster analysis of all data collected for the first, preclinical time point.
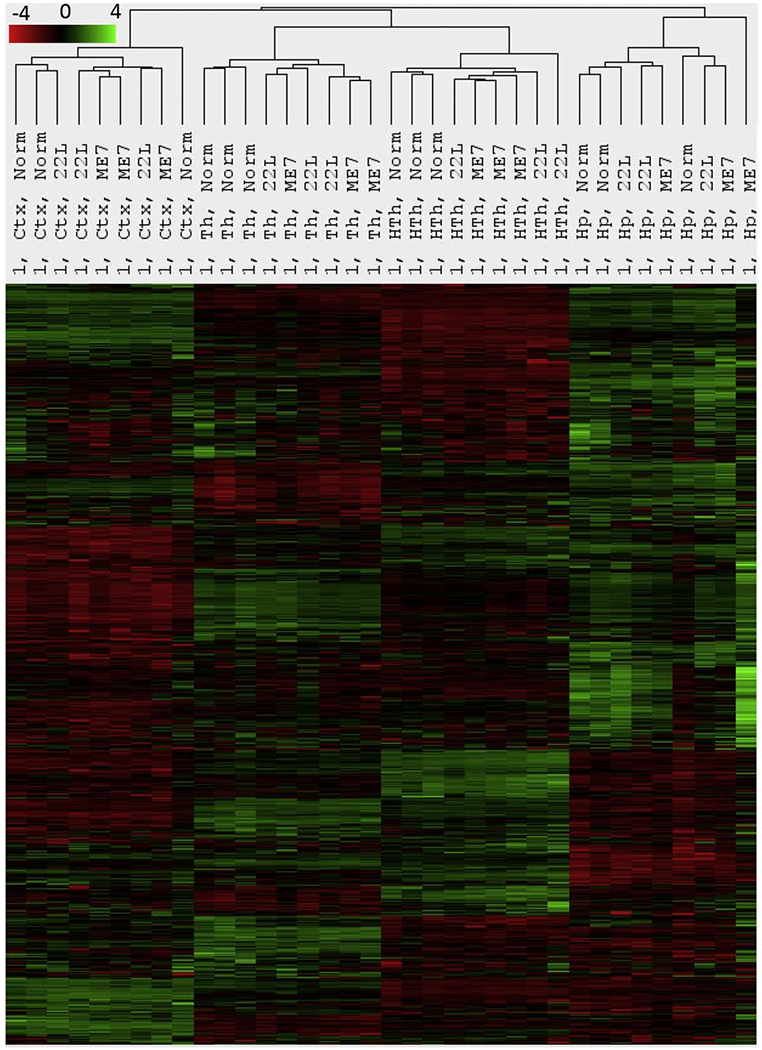
Four well-defined clusters formed strictly according to the brain region presenting region-specific homeostatic signatures. Within each region-specific cluster, 22L- and ME7-infected animals did not cluster away from the age-matched control (Norm) animals with the exception of thalamus.
3.3. Gradual loss of the region-specific homeostatic signatures with the disease progression
Agglomerative hierarchical clustering of all data collected at the second time point revealed a group of upregulated genes (Fig. S3, orange frame). Th and HTh from one 22L-infected animal (animal #4) clustered separately from all other samples, forming a well-separated branch (Fig. S3, red shading). Within its group, the animal #4 showed the highest amounts of PrPSc on Western blot (Fig. S1C) and the most pronounced inflammation of microglia as assessed by immunostaining of brain sections using microglia-specific marker Iba1 (Fig. S4). The Ctx of this animal also showed upregulation of the same set of genes, although to a lower degree (Fig. S3). Upregulation of the same genes were also visible in the thalamus of 22L-infected animal #5, although this sample remains in the cluster with the other sampled thalami. Notably, the ranking order between individual animals of the 22L group was the same (the most affected #4>#5>#6) regardless of whether it was assessed by differential gene expression, the amounts of PrPSc by Western blot, or microglia activation by immunostaining with Iba1 (Fig. S1B, S3, S4). In ME7-infected animals, which were all asymptomatic at the second time point, the thalami also showed upregulation of the same set of genes as 22L-affected thalami , with one ME7-affected Th (animal #13) clustering with 22L-infected Th (animal #5) (brown shading in Fig. S3). To summarize, at the time when signs of the disease began to appear, expression of a subset of genes started to dominate over the region-specific homeostatic signature in animals that were most advanced in the disease progression. The same set of genes appears to be upregulated regardless of brain region or prion strain.
The trend observed at the second time point was strengthened further at the third time point. At the advanced stage, 22L Th and HTh from all three animals of the group, now joined by the three ME7 Th of its group, clustered away from all other samples (Fig. 2, red shading). Notably, the upregulation of the gene set (defined by a red frame) was so profound that it overrode the region-specific transcriptome signatures still recognizable in the 22L Th+HTh and ME7 Th sample sets (Fig. 2). The set of genes that drove separation of 22L Th+HTh and ME7 Th into a distant cluster will be referred to as a neuroinflammation signature associated with the prion disease (marked by red frame in Fig. 2). The Ctx of all 22L- and ME7-infected animals also showed upregulation of the genes within the neuroinflammatory signature block, yet to a lower degree, and formed a sub-cluster within the Ctx cluster (Fig. 2). These results suggest that at the advanced stage of the disease, in the brain regions that are most strongly affected by prions, glia partially lose their region-specific homeostatic signature and merge into a highly reactive phenotype. This reactive phenotype is characterized by upregulation of the same set of genes as defined by the neuroinflammatory signature block. The full list of differentially expressed genes is presented in Table S4. The genes within the neuroinflammation signature block were common for all regions, although the extent of upregulation varied in a region-specific manner as discussed below.
Figure 2. Agglomerative hierarchical cluster analysis of all data collected for the third time point.
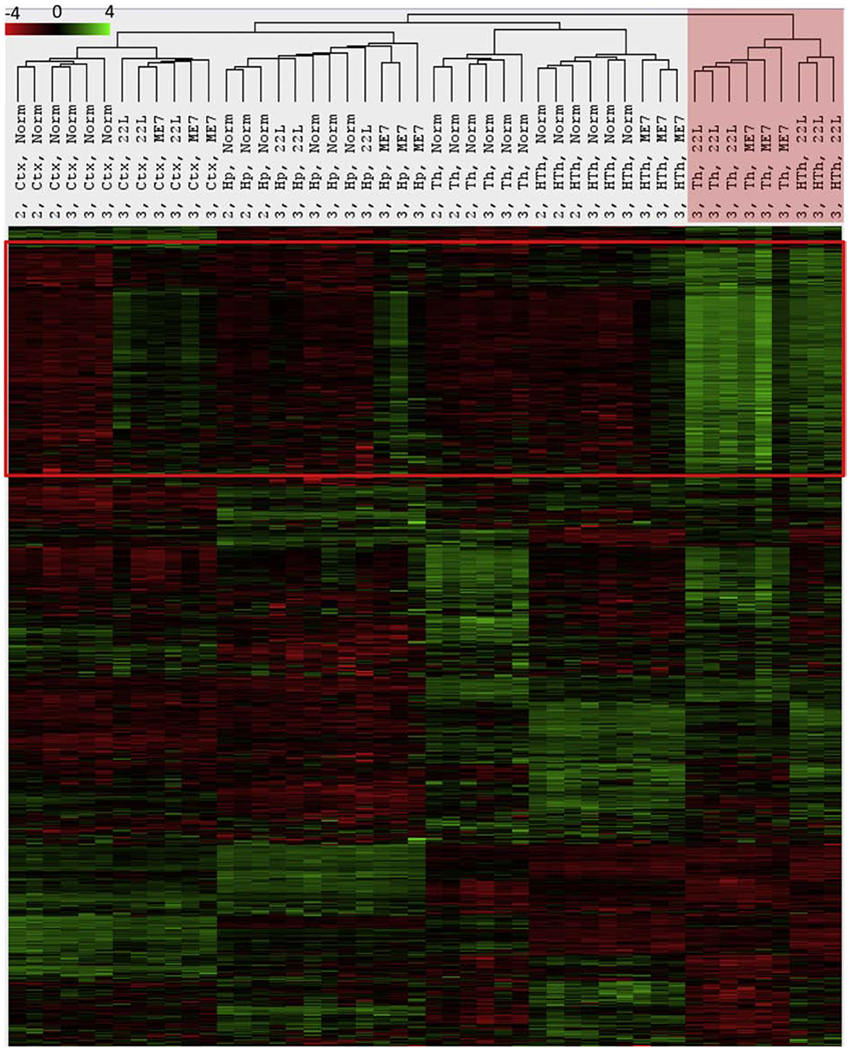
The red frame define genes forming the neuroinflammatory signature. 22L Th, 22L HTh and ME7 Th clustered away from all other samples as indicated by red shading. This separation is driven by a strong upregulation of genes within the neuroinflammatory signature block. Notably, 22L Ctx and ME7 Ctx, Hp and HTh also show upregulation of the same genes, which separates these samples from normal controls within the corresponding regional clusters.
3.4. Region- and strain-specific dynamics of neuroinflammation
Agglomerative clustering of grouped samples (n=3 per group) collected for four brain regions in both strains at three time points showed the same dynamics as the clustering of individual samples (Fig. 3). Again, clear separation of 22L Th, 22L HTh and ME7 Th into a highly distinctive cluster was evident in samples from the advanced stage of the disease (Fig. 3). Notably, the Ctx, Hp and HTh showed upregulation of the same sets of genes as those found in the Th, although to a lesser degree than in the Th, and to a different extent in 22L compared to ME7 (Fig. 3).
Figure 3. Agglomerative clustering analysis of grouped samples collected for all three time points.
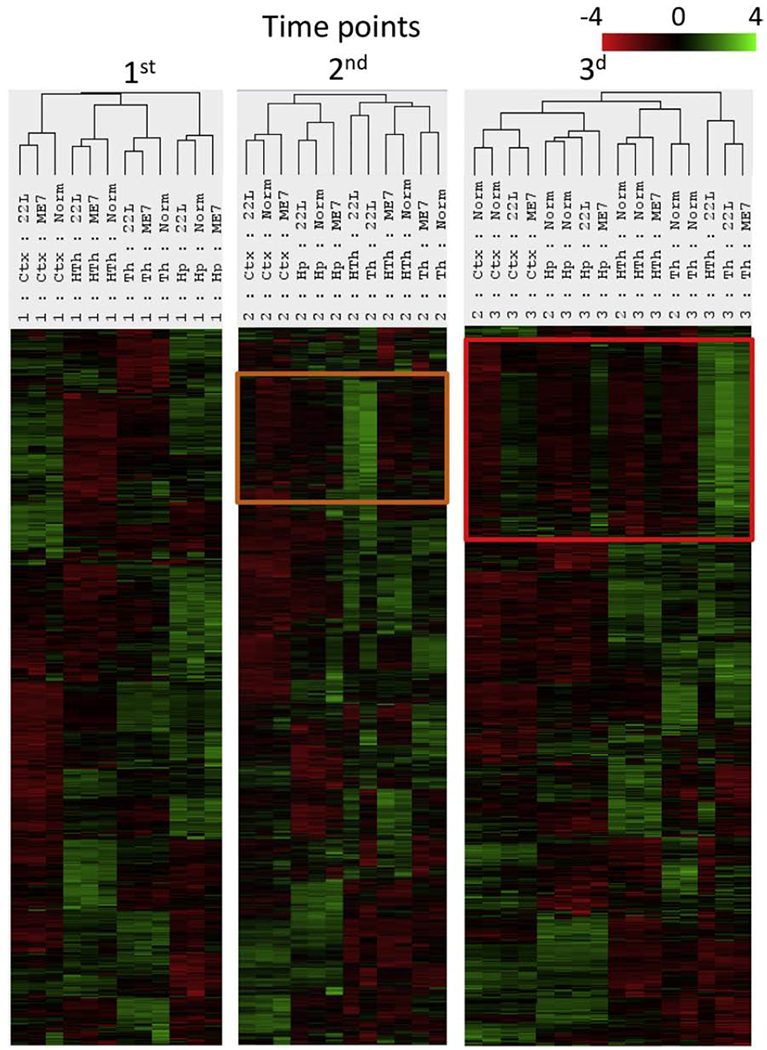
To group samples, the geometric mean of expression levels for all samples from each group (n=3) were calculated. The set of genes upregulated at the second time point (orange frame) expands further at third time point (red frame).
Analysis of genes within the neuroinflammation signature block revealed that the majority of genes belonged to the Astrocyte Function, Microglia Activation, Inflammatory Signaling and Autophagy pathways (Fig. 4). The genes within the neuroinflammation signature block responded to prion infection in a coherent manner and showed the same dynamics, as assessed across four brain regions, three time points and two prion strains (Fig. 4).
Figure 4. Gene composition within the neuroinflammation signature block.
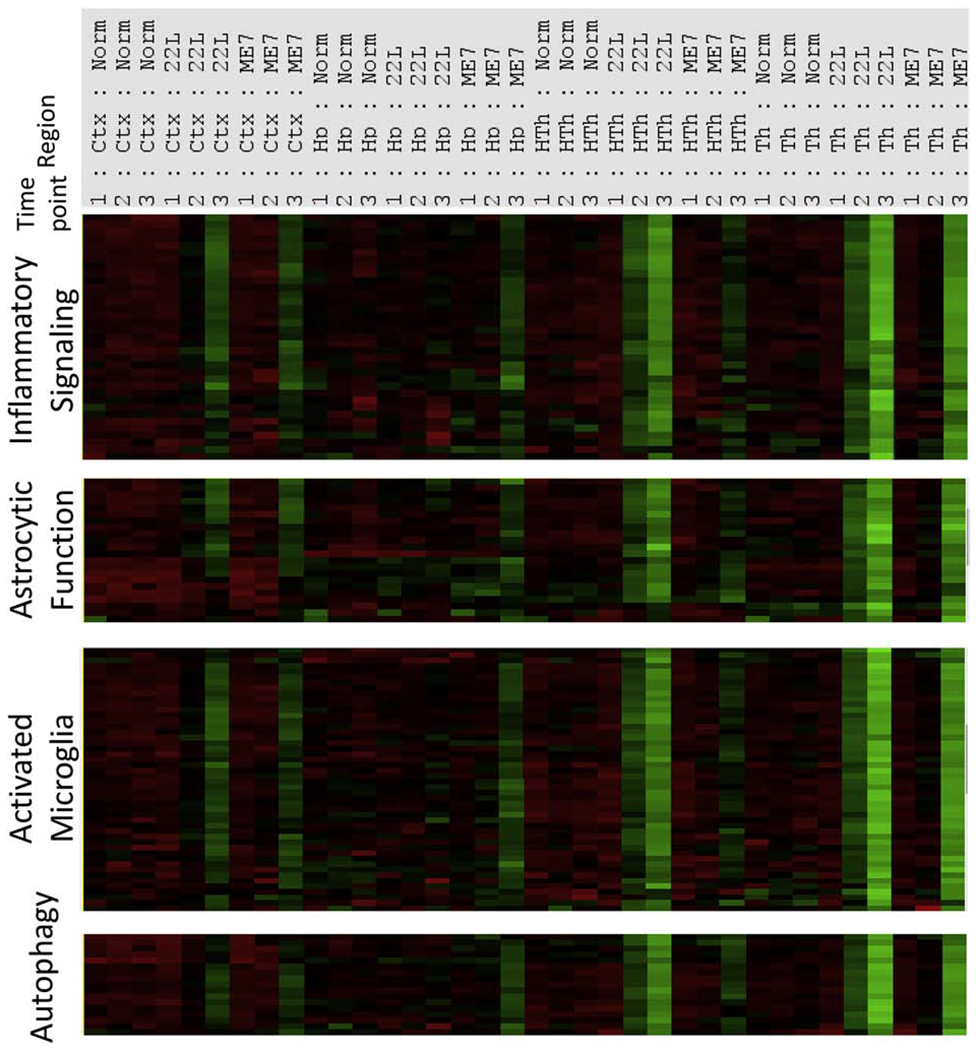
Distribution of upregulated genes within the neuroinflammation signature block according to pathways. Grouped samples corresponding to four brain regions in animals infected with 22L or ME7 strains or non-infected controls (Norm) and collected at three time points are presented.
Close comparison of 22L and ME7 sets revealed that the same genes were activated by both strains. However, the timing of neuroinflammation and the degree of activation in different brain regions were strain-specific. To establish a strain-specific ranking order with respect to (i) the temporal spread of neuroinflammation across the brain and (ii) the extent to which brain regions were affected at the advanced stage, we first assessed the intensity of changes within the genes of the neuroinflammation signature block. For 22L, the ranking order was Th>HTh>Ctx>>Hp (where Th was the earliest and most affected region), whereas for ME7, the ranking order was Th>Ctx>Hp=HTh (Fig 5A). The ranking order established by the gene expression correlated well with the region-specific deposition of PrPSc, reactive microgliosis and astroglyosis, as assessed by staining for Iba1 and GFAP, respectively (Fig. S5). Counting a number of differentially expressed genes at the advanced stage of disease (P< 0.05, fold change > +/−1.2) showed the same ranking order as assessed by the intensity of differential gene expression (Fig 5B). This ranking order was also confirmed upon examination of region-specific expression of individual genes including Cxcl10, Serpina3n, Cd68 and Clec7a (Fig. S6).
Figure 5. Region- and strain-specific dynamics of neuroinflammation.
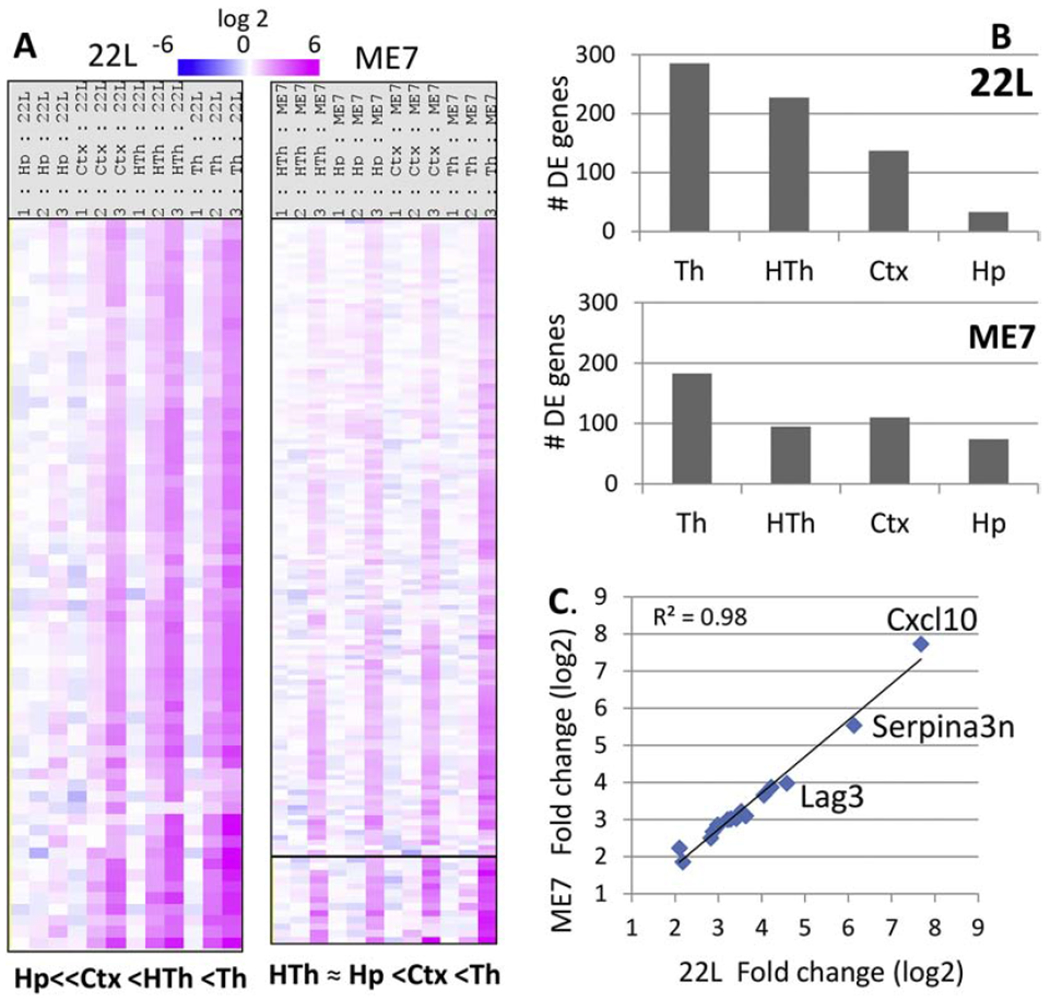
(A) Strain-specific ranking order with respect to the temporal spread of neuroinflammation across four brain areas and the degree to which the brain regions were affected at the advanced stage of the disease, as judged by the sum intensity of changes. The data presented as heatmap of grouped samples (n=3) showing log2 fold change in 22L- or ME7-infected mice over normal controls. (B) Number of differentially expressed (DE) genes in four brain regions in 22L- and ME7-infected mice at the advanced stage of the disease. (C) Correlation between log2 fold change of gene expression between ME7- and 22L-infected animals for the top 20 differentially expressed genes at the advance stage of the disease.
Analysis of top activated genes in 22L Th and ME7 Th revealed excellent correlation between the two strains with R2 = 0.98 (Fig. 5C, Table 1). Moreover, 22L and ME7 did not separate into different sub-clusters illustrating a lack of strain-specificity in the overall pattern of gene activation (Fig. 2). For instance, the 22L Ctx and ME7 Ctx displayed similar pattern of gene activation and together formed a sub-cluster within the Ctx cluster (Fig. 2). To summarize, these data strongly indicate a lack of strain-specificity in neuroinflammatory response. The strain-specificity consisted of the differences in tropism to different brain regions and the degree of gene activation rather than the activation of different subsets of genes.
Table 1.
Top differentially expressed genes in 22L- and ME7-infected animals at the advanced stage of the disease.
| mRNA | 22L 3rd point Th | ME7 3rd point Th | Pathways | ||||
|---|---|---|---|---|---|---|---|
| Log2 fold change | std error (log2) | P-value | Log2 fold change | std error (log2) | P-value | ||
| Cxcl10 | 7.68 | 0.589 | 3.77E-07 | 7.73 | 0.589 | 3.55E-07 | Astrocyte Function, Cytokine Signaling, Inflammatory Signaling, Innate Immune Response, Microglia Function |
| Serpina3n | 6.13 | 0.359 | 3.60E-08 | 5.54 | 0.359 | 8.81E-08 | Astrocyte Function |
| Lag3 | 4.58 | 0.284 | 6.10E-08 | 3.98 | 0.285 | 2.09E-07 | |
| Fcgr2b | 4.22 | 0.307 | 2.38E-07 | 3.87 | 0.307 | 4.99E-07 | Adaptive Immune Response, Autophagy |
| C4a | 4.05 | 0.243 | 4.56E-08 | 3.66 | 0.243 | 1.11E-07 | Astrocyte Function |
| Slamf9 | 3.63 | 0.264 | 2.40E-07 | 3.1 | 0.266 | 9.87E-07 | Microglia Function |
| C1qa | 3.53 | 0.248 | 1.78E-07 | 3.22 | 0.248 | 3.95E-07 | Innate Immune Response |
| Trem2 | 3.41 | 0.258 | 3.40E-07 | 3.03 | 0.259 | 9.41E-07 | Adaptive Immune Response, Inflammatory Signaling, Microglia Function |
| C1qb | 3.29 | 0.23 | 1.74E-07 | 3.02 | 0.23 | 3.57E-07 | Innate Immune Response |
| Tyrobp | 3.27 | 0.268 | 6.62E-07 | 3.01 | 0.268 | 1.34E-06 | Adaptive Immune Response, Innate Immune Response |
| C1qc | 3.21 | 0.237 | 2.73E-07 | 3 | 0.237 | 4.94E-07 | Innate Immune Response |
| Fcrls | 3 | 0.237 | 4.94E-07 | 2.84 | 0.237 | 7.81E-07 | Microglia Function |
| Mpeg1 | 2.98 | 0.251 | 8.32E-07 | 2.86 | 0.251 | 1.21E-06 | Inflammatory Signaling |
| Psmb8 | 2.87 | 0.232 | 5.99E-07 | 2.67 | 0.233 | 1.14E-06 | Adaptive Immune Response, Angiogenesis, Apoptosis, Astrocyte Function, Cell Cycle, Cytokine Signaling, Growth Factor Signaling, Inflammatory Signaling, Insulin Signaling, Microglia Function, NF-kB, Wnt |
| Ifi30 | 2.82 | 0.218 | 4.01E-07 | 2.5 | 0.22 | 1.21E-06 | Adaptive Immune Response, Inflammatory Signaling |
| Stat1 | 2.18 | 0.147 | 1.22E-07 | 1.86 | 0.148 | 5.18E-07 | Cytokine Signaling, Growth Factor Signaling, Inflammatory Signaling, Microglia Function |
| Olfml3 | 2.1 | 0.169 | 5.84E-07 | 2.23 | 0.169 | 3.32E-07 | Matrix Remodeling |
3.5. Change in astrocyte function scores at the top of the pathways analyzed.
Among the pathways covered by the Neuroinflammation panel, we wanted to know what pathways were affected the most. To answer this question, we focused on the thalamus, which showed the strongest activation among the four brain regions analyzed. The heatmap of pathway scores revealed that three pathways (Neuron and Neurotransmission, Epigenetic regulation, and Oligodendrocyte function) were downregulated, whereas the remaining pathways were strongly upregulated in both 22L and ME7 relative to the controls (Fig. 6). Undirected global significance scores of differences between prion-infected and control animals identified the Astrocyte Function pathway at the top of the list followed by the Inflammatory Signaling, Matrix Remodeling, Adaptive Immune Response and Microglia Function pathways for both prion strains (Table 2). Innate Immune Response, NF-kB and Autophagy pathways also scored highly. The undirected global significance scores of the genes related to the Inflammatory Signaling, Astrocyte Functions and Activated Microglia pathways were much higher than the scores for the Neurons and Neurotransmission pathway that was close to the bottom of the list (Table 2).
Figure 6. Heatmap of the pathway scores.
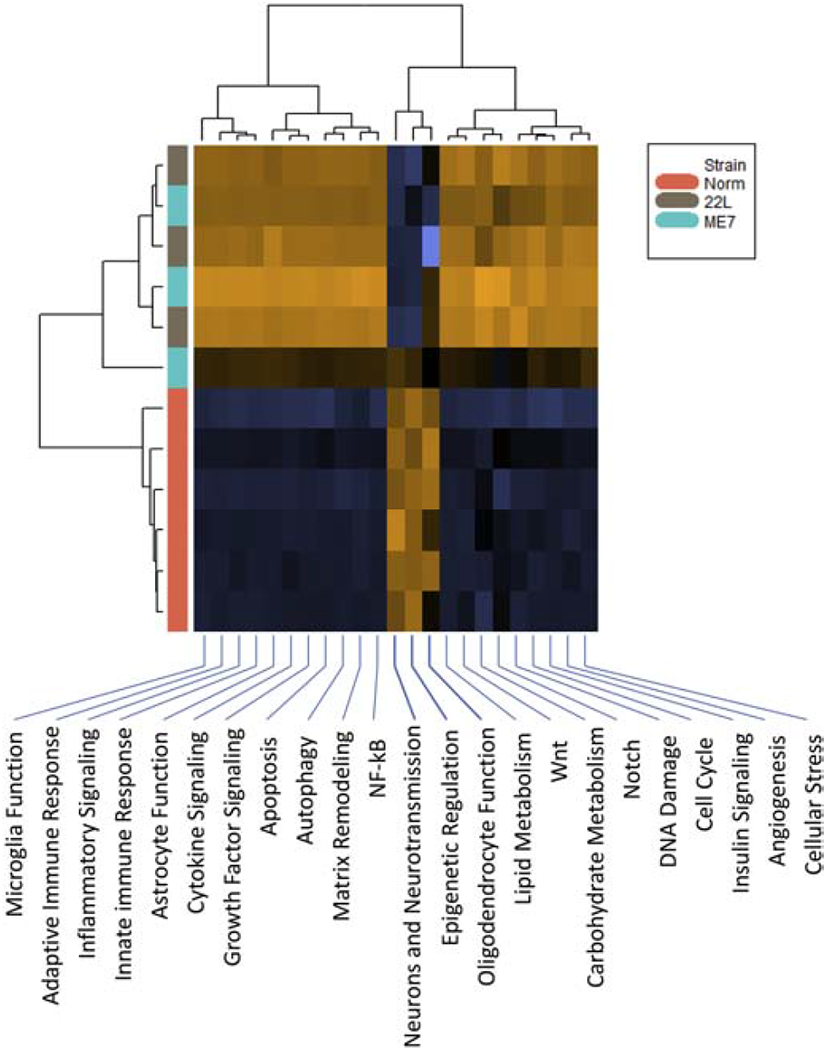
The pathways were scored based on the datasets generated for the thalamus at the third time point. Three pathways (Neurons and Neurotransmission, Epigenetic Regulation, and Oligodendrocyte Function) were downregulated, whereas remaining pathways were upregulated in 22L- and ME7-infected animals. Upregulated pathways are shaded brown, downregulated pathways are shaded blue.
Table 2.
Undirected and directed Global Significance Scores of 22 pathways analyzed by the Neuroinflammation panel
| Gene sets | Undirected GSS * | Directed GSS * | ||
|---|---|---|---|---|
| 22L vs. Norm | ME7 vs. Norm | 22L vs. Norm | ME7 vs. Norm | |
| Astrocyte Function | 6.42 | 5.626 | 5.875 | 5.558 |
| Inflammatory Signaling | 6.003 | 5.455 | 5.891 | 5.429 |
| Matrix Remodeling | 5.689 | 5.091 | 5.241 | 4.942 |
| Adaptive Immune Response | 5.512 | 4.976 | 4.861 | 4.666 |
| Autophagy | 5.06 | 4.422 | 4.388 | 4.04 |
| Microglia Function | 5.055 | 4.465 | 4.729 | 4.234 |
| Innate Immune Response | 5.035 | 4.491 | 4.582 | 4.314 |
| Cytokine Signaling | 4.778 | 4.274 | 4.241 | 4.026 |
| NF-kB | 4.684 | 4.275 | 4.529 | 4.236 |
| Insulin Signaling | 4.557 | 3.863 | 3.301 | 3.272 |
| Angiogenesis | 4.556 | 3.851 | 3.305 | 3.168 |
| Wnt | 4.363 | 3.256 | 1.396 | 2.421 |
| Growth Factor Signaling | 4.327 | 3.733 | 3.426 | 3.289 |
| Lipid Metabolism | 3.916 | 3.146 | 2.803 | 2.625 |
| Apoptosis | 3.469 | 2.879 | 2.074 | 2.327 |
| Cellular Stress | 3.22 | 2.678 | 1.889 | 1.818 |
| Neurons and Neurotransmission | 3.002 | 2.503 | -1.052 | 1.241 |
| Cell Cycle | 2.997 | 2.368 | 2.019 | 2.002 |
| Notch | 2.73 | 2.053 | 1.786 | 1.764 |
| Carbohydrate Metabolism | 2.655 | 2.402 | 1.597 | 2.259 |
| DNA Damage | 2.162 | 1.639 | 1.304 | 1.325 |
| Epigenetic Regulation | 2.116 | 1.469 | -0.968 | 0.834 |
In prion diseases, chronic inflammation is accompanied by proliferation of microglia (Gomez-Nicola & Perry, 2015). To test whether the global significance scores of the top pathways reflect possible changes in cell type composition in addition to differential gene expression per se, next we compared cell population scores of microglia, astrocytes and neurons (Fig. 7). In 22L- and ME7-infected animals, microglial cell-specific markers scored significantly higher relative to the controls. This result is consistent with significant proliferation and/or infiltration of microglia. As a result of proliferation/infiltration, the specific gene set scores for the Activated Microglia pathway and other pathways dominated by microglia-specific genes may have been inflated (Fig. 7). Despite a substantial increase in the specific gene set score for the Astrocyte Function pathway, an increase in the cell population score for astrocytes in infected relative to control animals was considerably less profound in comparison to those of microglia (Fig. 7). Unlike microglia, astrocytes do not proliferate in prion diseases (Asuni et al., 2014). A modest increase in scoring of the astrocyte markers is likely to be attributed to astrocyte hypertrophy. A global score of neuron-specific markers did not show a significant drop, yet a notable downregulation of the Neurons and Neurotransmission pathway was detected in prion-infected animals (Fig. 7). However, close examination of the fold change in expression of individual genes associated with the Neurons and Neurotransmission pathway was mostly small and of low statistical significance (Fig. S7). Few genes in the Neurons and Neurotransmission pathway showed strong upregulation. These genes also belong to other pathways and their activation was most probably related to the upregulation of these other pathways (C3ar1 and P2rx7 – activated microglia, S1pr2 – astrocyte function, etc., Fig.S7).
Figure 7. Cell population scores and pathway scores.
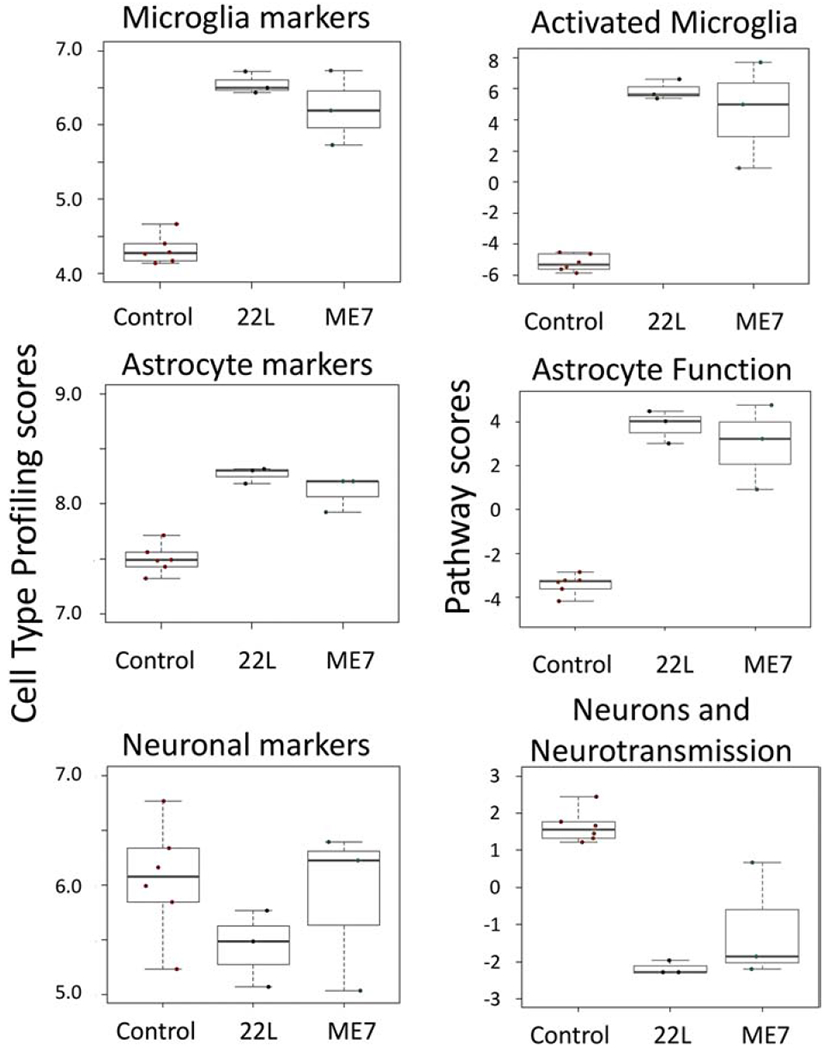
Cell population scores for microglia, astrocytes and neurons, as well as scores for the Activated Microglia, Astrocyte Function and Neuron and Neurotransmission pathways were generated based on the datasets taken for thalami at the third time point.
To find what cell types (microglia, astrocytes or neurons) respond to prion infection at the preclinical stage of disease, an nCounter Advanced Analysis was employed to cluster samples based on gene expression pattern in the Astrocyte Function, Microglia Function and Neurons and Neurotransmission pathways (Fig. 8). Clustering based on the genes in the Astrocyte Function pathways revealed that the thalami from normal and disease-affected animals separated into two clusters already at the first time point and continued to cluster away from each other for the second and third time points (Fig. 8). In contrast, when genes associated with the Microglia Function or Neurons and Neurotransmission were assessed, heatmaps demonstrated that normal and prion-affected thalami clustered away from each other only at the third, advanced stage of the disease (Fig. 8).
Figure 8. Hierarchical cluster analysis of the pathways.
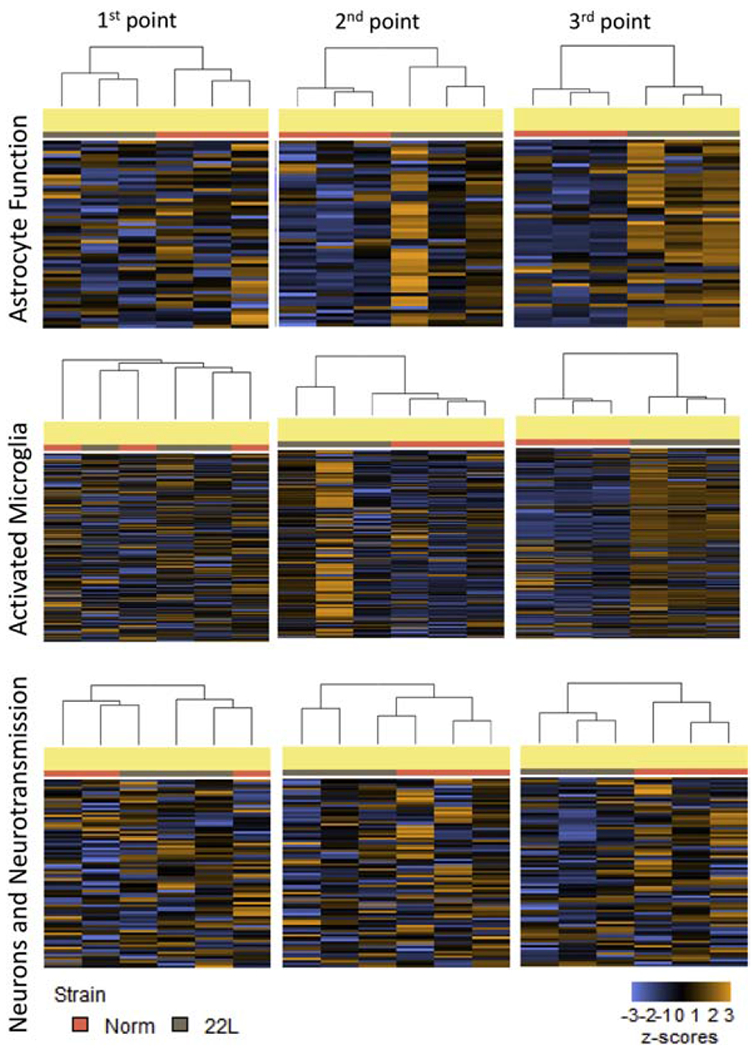
Hierarchical cluster analysis of the Astrocyte Function, Activated Microglia and Neuron and Neurotransmission pathways was performed based on the datasets generated for thalami of 22L-infected animals and normal controls for three time points.
In contrast to the Astrocyte Function or Microglia Activation pathways (Fig. 4), the Neuron and Neurotransmission pathway showed a very subtle response to prion infection (Fig. S7). Such subtle response could be, in part, due to a limited number of genes related to neurons and neurotransmission in the Neuroinflammation panel (80 genes), which may not capture neuronal dysfunction to the full extent. Nevertheless, only a minor down- or up-regulation of individual neuronal function-related genes, of which many lack statistical significance, argue against a substantial loss of neuronal population even at the terminal stage of disease.
To summarize, the Astrocyte Function pathway scored at the top of the list for both prion strains among the pathways analyzed by the Neuroinflammation panel. Moreover, changes in the genes associated with the Astrocyte Function pathways were already detectable at the preclinical stage of the disease. While the Microglia Activation pathway scored very highly too, its score is likely to be inflated due to microglia proliferation.
3.6. A1-, A2- and PAN-reactive markers are upregulated
Previous studies established that, depending on activation stimuli, astrocytes can acquire two opposite reactive phenotypes: proinflammatory, neurotoxic A1 state and neuroprotective A2 state (Liddelow et al., 2017; Zamanian et al., 2012). The concept of A1/A2 phenotypes has been employed for characterizing astrocyte activation states under pathological conditions or normal aging (Boisvert et al., 2018; Clarke et al., 2018). We found that among the A1-, A2- and PAN-specific markers included in the Neuroinflammation panel, the majority of PAN-reactive markers (Osmr, Vim, Serpina3n, Cxcl10, Timp1, S1pr3, Lcn2, Hspb1, Cp) as well as several A1-specific (Psmb8, Gbp2, H2-T23, Serping1) and A2-specific (Tgm1, Cd14, S100a10, Ptx3, Cd109) markers were upregulated at the advanced stage of prion disease (Fig. 9). Both prion strains showed upregulation of the same markers suggesting that the reactive astrocyte phenotype lacked prion strain specificity (Fig. 9). However, the extent to which markers were upregulated in the four brain regions mirrored the strain-specific dynamics of neuroinflammation: Th>HTh>Ctx>Hp for 22L, and Th> Ctx>Hp>HTh for ME7.
Figure 9. Heatmap of the A1-, A2- and PAN-specific markers.
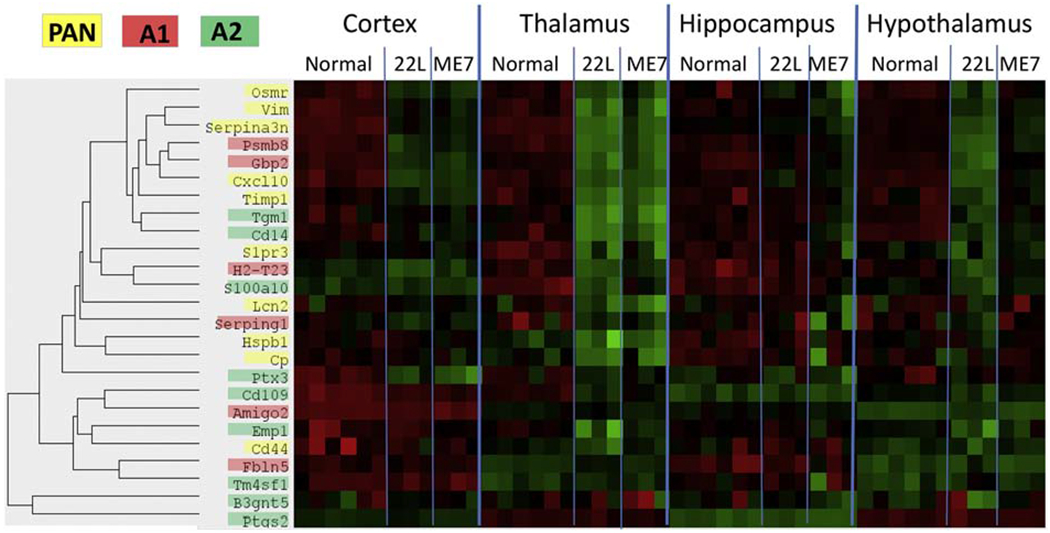
Heatmap analysis of A1-, A2- and PAN-specific markers in four brain regions of 22L-, ME7-infected animals and normal controls assessed for the third time point and shown for individual animals.
4. Discussion
Recent advances in transcriptome analysis, single-cell RNA-sequencing and single-cell cytometry revealed considerable heterogeneity of glial phenotypes under normal conditions as well as dynamic changes in aging and neurodegenerative diseases. Single-cell transcriptional profiling of 1/2 million cells identified seven molecularly distinct and regionally restricted astrocyte types, in which regional specialization was found to be defined developmentally (Zeisel et al., 2018). Single cell cytometry mapped distinct subsets of microglia populations providing insight into phenotypic heterogeneity among CNS-resident myeloid cells (Ajami et al., 2018; Korin et al., 2017; Mrdjen et al., 2018). Moreover, the genome-wide transcriptome analysis demonstrated that, like astrocytes, microglia too have distinct region-dependent homeostatic transcriptional identities (Grabert et al., 2016). Transcriptome profiling of human brains documented that, in astrocytes, the region-specific expression patterns undergo a major shift with normal aging (Soreq et al., 2017). Remarkably, the region-specific differences in the expression of astrocyte-specific genes were found to largely disappear with old age (Soreq et al., 2017). Likewise, microglia isolated from mouse brains also showed diminishing of the region-specific homeostatic signatures with normal aging (Grabert et al., 2016). Moreover, upregulation of microglia-specific genes and, in particular, those involved in immune and inflammatory functions (C1q, Trem) were found with age in humans (Soreq et al., 2017). Notably, a global shift in the expression pattern of glial-specific genes predicted age with greater precision than the expression of neuron-specific genes, underscoring the role of glia in normal aging (Soreq et al., 2017). Both microglia and astrocytes age in a regionally dependent manner showing variable aging rates in different regions (Boisvert et al., 2018; Grabert et al., 2016).
In the current study, we asked whether the region-specific homeostatic identity of glial cells changes with the progression of prion diseases, and if so, whether these changes occur in a region-dependent or uniform manner. Four brain regions of mice infected with the 22L strain, which is mainly associated with astrocytes, or the ME7 strain, which is found in association with neurons (Carroll et al., 2016), were examined at three time points. For both strains, all data sets collected for the first, preclinical time point separated into four distinct clusters in strict accordance to brain region, illustrating region-specific homeostatic signatures. However, the appearance of the first clinical signs was accompanied by an overexpression of a subset of genes forming a neuroinflammation signature, which started to dominate over the region-specific homeostatic signatures. A departure from a homeostatic region-specific identity strengthened further at the advanced stage of disease. While manifested to a different extent, the same neuroinflammation signature was observed in all four regions examined. Moreover, both the astrocyte-associated 22L and neuron-associated ME7 strains showed the same neuroinflammation signature, suggesting that it is independent of strain-specific cell tropism. Nevertheless, while the neuroinflammation signature was region- and strain-independent, the neuroinflammation spread across these four brain regions in a strain-specific manner. In fact, at the advanced stage of the disease, the four brain regions were affected to a different extent in 22L- and ME7-infected animals showing a strain-specific ranking order with respect to the severity of neuroinflammation. This study illustrates that, in a manner resembling normal aging, glia lose their region-specific identity with the progression of prion diseases, although, this process occurs at a much faster rate in animals infected with prions.
Comparison of the top differentially expressed genes from the current work (Cxcl10, Serpina3n, Lag3, Fcgr2b, C1qa, C1qb, C1qc, C4a, Stat1, Trem2, see Table 1) with those in previous studies showed excellent agreement (Carroll et al., 2016; Hwang et al., 2009; A. Majer et al., 2012; Anna Majer et al., 2019; Vincenti et al., 2016; Xiang et al., 2004). At the top of the list was Cxcl10, a proinflammatory chemokine that can contribute to neurotoxicity and apoptosis. Cxcl10 was identified in previous studies that employed global gene expression approaches or targeted approaches (Carroll & Chesebro, 2019; Carroll et al., 2016; Xiang et al., 2004). Serpina3n, a member of a large family of serine protease inhibitors, is a part of the astrocytic PAN-reactive gene panel and is upregulated in normal aging (Boisvert et al., 2018; Clarke et al., 2018). Mouse Serpina3n was activated in ME7-, RML- and 301V-infected mice (Hwang et al., 2009; Xiang et al., 2004). Its human homolog Serpina3 was strongly upregulated at the mRNA and protein levels in human prion diseases including variant CJD, sporadic CJD, iatrogenic CJD, familial CJD, Fatal Familial Insomnia and Gerstmann-Straussler-Scheinker syndrome, as well as in BSE-infected macaques (Barbisin et al., 2014; Vanni et al., 2017). Expression of Lag3, a lymphocyte activation protein 3 also known as CD223, was also found to increase in prion-infected brains, yet its knockout failed to modify disease progression (Liu, Sorce, Nuvolone, Domange, & Aguzzi, 2018). Fcgr2b is involved in natural killer cell-mediated neurotoxicity, and was previously shown to be upregulated in ME7- and RML-infected mice (Hwang et al., 2009; Xiang et al., 2004). C1qa, C1qb and C1qc, the subcomponents of the complement cascade factor C1q, and C4a component of the complement cascade were found to be upregulated in 22L-, RML-, ME7- and 301V-infected mice and 263K-infected hamsters (Carroll, Race, Williams, & Chesebro, 2018; Hwang et al., 2009; Lv et al., 2015; Xiang et al., 2004). In periphery, PrPSc interaction with C1q is required for the sequestration of prions by spleen and infection of follicular dendritic cells (Klein et al., 2001). Deficiency of C1q delays the onset of the disease upon peripheral infection (Mabbott, Bruce, Botto, Walport, & Pepys, 2001). Upregulation of Stat1, a pro-inflammatory transcription factor involved in the JAK-STAT pathway that mediates cellular response to cytokines and interleukins (including those produced in prion diseases), was found in 22L- or ME7-infected mice (Carroll, Striebel, Race, Phillips, & Chesebro, 2015; Na et al., 2007). Trem2, a triggering receptor expressed on myeloid cells-2, is a major genetic risk factor and a main player in Alzheimer’s disease (reviewed in (Gratuze, Leyns, & Holtzman, 2018)). Upregulation of Trem2 was found in RML-inoculated mice (Zhu et al., 2015). Yet, its depletion, while attenuating markers of activated microglia, did not affect the incubation time or survival of prion-infected mice (Zhu et al., 2015).
This study did not aim to identify new differentially expressed genes. However, comparison of the 22L thalamus at the third time point, the region with the most severe neuroinflammation, with the previous results on the global gene expression in whole brain tissues (Hwang et al., 2009; Vincenti et al., 2016) identified 37 new upregulated (P<0.05, fold change >1.5) and 11 new downregulated genes (P<0.05, fold change <0.66 fold) (Table S4). Improved sensitivity of detection could be due to a few factors. First, analysis of a brain region versus the whole brain might improve detection of genes that are up- or down-regulated in a region-specific manner or genes that display significantly different levels of basal expression between different regions. Secondly, the genes that respond regardless of a brain region but display statistically significant differential expression only in the most affected region might have a better chance of detection. Finally, Nanostring might provide a more sensitive way for detecting genes expressed at low levels than other approaches, as it directly counts the number of mRNA copies.
The prion-associated Neuroinflammation signature identified in the current work consists of genes that largely belong to the Inflammatory Signaling, Activated Microglia, Astrocyte Function and Autophagy pathways, but not the Neuron and Neurodegeneration pathway, which showed a very modest response. Among the neuron-specific genes, it is worth mentioning the upregulation of Cidea (Cell Death Inducing DFFA Like Effector A), which activates apoptosis, and downregulation of Ngf (Nerve Growth Factor) that helps neurons grow and survive. A modest scoring of the Neurons and Neurotransmission pathway was consistent with previous results on the transcriptome analysis that revealed relatively few degenerating neurons even at the advanced stages of the disease (A. Majer et al., 2012). The group of activated genes in the Inflammatory Signaling, Astrocyte Function, Activated Microglia and Autophagy pathways showed very similar expression dynamics in response to prion infection, as assessed in four brain regions monitored at three time points for two prion strains (Fig. 4). Such coherent dynamics is remarkable, as it suggests that the same mechanism is involved in responding to prion infection regardless of a brain region or a cell tropism of the prion strain.
A number of previous studies that employed animal models, post-mortem human brains or cells cultured in vitro aimed at defining the role of microglia in prion diseases. Consistent with the current studies, activation and proliferation of microglia were found to mirror PrPSc accumulation with respect to the affected brain regions and timing of PrPSc accumulation (Bate, Boshuizen, Langeveld, & Williams, 2002; Everbroeck et al., 2004; Giese et al., 1998; Greenlee et al., 2016; Kercher, Favara, Striebel, LaCasse, & Chesebro, 2007; Puoti et al., 2005; Sandberg et al., 2014; Vincenti et al., 2016; Williams, Lucassen, Ritchie, & Bruce, 1997). Using purified, brain-derived PrPSc, we previously showed that PrPSc can directly trigger a proinflammatory response in primary microglia, and that the chemical nature of the carbohydrate groups on the N-linked glycans of PrPSc is important for microglial activation (Srivastava et al., 2018). Nevertheless, the precise role of glia in chronic neurodegeneration associated with prion disease has been under extensive debate and remains controversial (Brown & Neher, 2014; Gomez-Nicola & Perry, 2015; Lampron, Elali, & Rivest, 2013; Zhu et al., 2015). Microglia activation was shown to occur at much earlier stages than synaptic loss (Baker & Manuelidis, 2003; Carroll et al., 2016; Gomez-Nicola, Fransen, Suzzi, & Perry, 2013; Lu et al., 2004; Sandberg et al., 2014), which is considered to be an early neuron-specific pathological sign (Hilton, Cunningham, Reynolds, & Perry, 2013; Jeffrey et al., 2000). Solid evidence in support of both neuroprotective phenotypes and inflammatory or neurotoxic phenotypes have been presented over the years (Carroll, Race, Williams, Striebel, & Chesebro, 2018; Everbroeck et al., 2004; Giese et al., 1998; Gomez-Nicola et al., 2013; Grizenkova, Akhtar, Brandner, Collinge, & Lloyd, 2014; Hughes, Field, Perry, Murray, & Cunningham, 2010; Kercher et al., 2007; Kranich et al., 2010; Muth et al., 2016; Riemer et al., 2008; Sakai et al., 2013; Sorce et al., 2014; Zhu et al., 2016). It is likely that multiple reactive phenotypes co-exist and undergo changes with disease progression. Several microglial transcripts upregulated in the prion-infected mice were shared with those in aged mice (Irf7, Stat1, Ifitm family) (Grabert et al., 2016), or shared with aged mice together with the APP/PS1 model of Aβ amyloidosis and the AAV-TauP301L model of tauopathy (Apoe, C3, Ccl3, Clec7a, Itgax, Lilrb4, Spp1) (Kang et al., 2018). Moreover, the neuroinflammation signature identified in prion-infected animals in the current study partially overlapped with the microglia degenerative phenotype (MGnD) and the disease-associated microglia phenotype (DAM) reported previously in mouse models of other neurodegenerative diseases (Butovsky & Weiner, 2018; Keren-Shaul et al., 2017; Krasemann et al., 2017). All three microglia phenotypes showed upregulation of the following genes: Apoe, Axl, Clec7a, Csf1, Fabp5, Grn, Lilrb4, Spp1, Trem2, Tyrobp and downregulation of Egr1. Yet, significant differences were also found. Prion-infected animals upregulated the following genes that were downregulated in DAM and MGnD: P2ry12, Ccr5, Csf1r, Cx3crl, Gpr34, Tgfbl, Tgfbrl, Mertk, Tmemll9, Sall1, Mafb. Vice versa, the gene Vegfa was downregulated in prion-infected animals while upregulated in DAM and MGnD (Butovsky & Weiner, 2018; Keren-Shaul et al., 2017; Krasemann et al., 2017). Together, these results suggest that prion disease is characterized by a prion-specific neuroinflammation signature, which only partially overlaps with those associated with other chronic neurodegenerative conditions.
Growing evidence suggests that autophagy plays a role in prion clearance on one hand, and regulates the release of prions via exosomes on the other hand (Abdelaziz, Abdulrahman, Glitch, & Schatzl, 2019; Abdulrahman et al., 2017; Abdulrahman, Abdelaziz, & Schatzl, 2018). Upregulation of the Autophagy pathway genes was found to occur in parallel with the genes of the Activated Microglia and Astrocyte function pathways, suggesting that microglia and/or astrocytes are responsible for autophagosomal clearance. However, ontological analysis of genes in microglia isolated from prion-infected mice in a previous study did not identify autophagy among pathways activated in microglia (Vincenti et al., 2016). Moreover, unlike astrocytes and neurons, microglial cells do not replicate prions and were found to exhibit very limited efficiency in the phagocytosis of PrPSc leading to question whether they are involved in PrPSc clearance (Hughes et al., 2010). Microglia serve as the main guard for protecting the CNS against pathogens. To discriminate between mammalian host cells and invading pathogens, microglia employ the same strategy as macrophages that involves sensing sialylated and non-sialylated glycans on a cell surface (Linnartz-Gerlach, Schuy, Shahraz, Tenner, & Neumann, 2016; Linnartz, Kopatz, Tenner, & Neumann, 2012; Varki, 2011). The majority of pathogens can synthetize only non-sialylated glycans and, instead of sialic acid, expose galactose at the terminal positions of glycans, which triggers a phagocytic “eat me” signal in microglia (Varki, 2008). Like mammalian cells, the PrPSc surface is heavily sialylated due to sialic acid residues at the terminal positions of the PrP N-linked glycans (Baskakov & Katorcha, 2016; Baskakov et al., 2018; Katorcha, Makarava, Savtchenko, D’Azzo, & Baskakov, 2014). Previously, we showed that the sialylation status of PrPSc is essential in determining the fate of prion infection in an organism (Katorcha et al., 2016; Katorcha et al., 2014; Srivastava et al., 2017; Srivastava et al., 2015). The reduction of PrPSc sialylation was found to fully abolish its infectivity upon administration to animals (Katorcha et al., 2016; Katorcha et al., 2014; Srivastava et al., 2017). It is not known, whether microglia can phagocytize PrPSc with a normal sialylation status. A human astrocyte cell line was found to be capable of taking up and degrading PrPSc (Choi, Head, Ironside, & Priola, 2014). However, it is not known whether astrocytes sense the same cues as the cells of myeloid origin for activating their phagocytic machinery. It remains to be determined whether astrocytes play a major role in the clearance of PrPSc in vivo and whether PrPSc clearance involves phagocytosis and autophagy.
In the current study, several components of a complement cascade including C1qa, C1qb, C1qc, C4a and C3ar1 (receptor for C3a) were found to be strongly upregulated in prion-infected mice. The components of a complement system including C1q, C3 and C4 play a critical role in synapse pruning during normal brain development (Schafer et al., 2012; Stevens et al., 2007). The developmental mechanisms of synaptic pruning involve the tagging of synapses by C1q, their opsonization by C3, followed by their engulfment and phagocytosis via an interaction with the C3 receptor expressed by microglia. Growing evidence suggests that similar mechanisms of synapse elimination are activated in reactive microglia in neurodegenerative diseases including Alzhemer’s disease, frontotemporal dementia, as well as normal aging (Hansen, Hanson, & Sheng, 2018; Hong et al., 2016; Lui et al., 2016; Stephan et al., 2013). Another gene that might be involved in neurotoxic activity of microglia is Axl, a TAM tyrosine kinase receptor. Under normal conditions, Axl drives microglial phagocytosis of apoptotic cells during adult neurogenesis (Fourgeaud et al., 2016). However, expression of Axl was found to be upregulated in mouse models of Parkinson’s disease (Fourgeaud et al., 2016) as well as in prion-infected mice in the current study. Upregulation of the components of a complement cascade observed in the current work suggest that non-cell autonomous mechanisms might be responsible for synaptic loss and dysfunction in prion diseases.
Reactive astrogliosis, routinely observed as an increase in GFAP signal, is one of the central features of prion diseases, yet information about the role of astrocytes in prion disease is scarce. Astrocytes can replicate and accumulate PrPSc independently of neurons (Aguzzi & Liu, 2017; Cronier, Laude, & Peyrin, 2004; Krejciova et al., 2017; Raeber, Race, Priola, & Sailer, 1997). Furthermore, the expression of PrPc on astrocytes was found to be sufficient for prion-induced chronic neurodegeneration (Jeffrey, Goodsir, Race, & Chesebro, 2004; Kercher, Favara, Chan, Race, & Chesebro, 2004). However, it remained unclear whether the normal physiological functions of astrocytes are altered as a result of reactive astrogliosis. Unexpectedly, the current study found that the Astrocyte Function pathway scored at the top of the tested pathways. While 22L does replicate in astrocytes, ME7 is predominantly neurotropic. Therefore, change in the expression of genes associated with astrocyte function cannot be attributed directly to the replication of PrPSc by astrocytes. In their homeostatic state, astrocytes are responsible for a variety of physiological functions including modulation of neurotransmission, formation and elimination of synapses, regulation of blood flow, the supply of energy and metabolic support to neurons, maintenance of the blood-brain-barrier, synthesis of cholesterol and much more (Dallérac, Zapata, & Rouach, 2018; Santello, Toni, & Volterra, 2019). Because the representation of genes that report on astrocytic function in the Neuroinflammation panel is limited, the current work cannot identify specific functions that are disrupted or upregulated in reactive astrocytes. We also do not know how changes in astrocyte function affects neurons.
Clustering analysis of the global scores at the first, preclinical point suggests that astrocytes might respond to prion infection ahead of microglia. While unexpected, this result is consistent with the previous findings that in mice infected with 22L, ME7 or RML, GFAP was upregulated prior of Gpr84, the marker of activated microglia (Carroll et al., 2016). While these findings need to be confirmed using a more detailed analysis of the preclinical stage, this result suggests that the relationship between reactive microglia and astrocytes appears to be more complex than one could envision based on the Barres’s hypothesis. According to this hypothesis, microglial cells in the proinflammatory reactive phenotype release astrocyte-activating signals (Il-1α, TNG-α and C1q) that induce pro-inflammatory A1 neurotoxic states in astrocytes (Liddelow & Barres, 2017; Liddelow et al., 2017). In support of this hypothesis, blocking of reactive microglia stimuli that induce the A1 astrocytic phenotype was found to be neuroprotective in mouse models of Parkinson’s disease (Yun et al., 2018). Moreover, consistent with this hypothesis, previous studies demonstrated attenuation of astrocytic gliosis and a delay of clinical onset of prion diseases in mice deficient of the interleukin-1 receptor, which is activated by proinflammatory cytokines produced by microglia (Schultz et al., 2004). However, contrary to expectation, the progression of prion disease was significantly accelerated in the triple (Il1α−/−, TNFα−/− and C1q−/−) knockout mice infected with prions (Hartmann et al., 2019). This finding contradicts the Barres’s hypothesis and raises the question of whether astrocytes are followers or drivers of the glial proinflammatory phenotype. The findings in the triple-knockout mice are unexpected, yet it could be reconciled with the Barres’s hypothesis if one assumes that the type of reactive phenotype acquired by astrocytes in prion diseases upregulate phagocytosis, which might be important for prion clearance. Indeed, in brains subjected to ischemia, reactive astrocytes were found to upregulate the genes responsible for phagocytosis and acquire a phagocytic phenotype (Morizawa et al., 2017).
In a manner similar to M1 and M2 macrophage nomenclature, reactive phenotypes of astrocytes were formally classified as A1 (pro-inflammatory or toxic) or A2 (neurotrophic or neuroprotective) (Ferrer, 2017; Liddelow & Barres, 2017). In the current study, the majority of PAN-specific markers, as well as several A1- and A2-specific markers, were upregulated in both 22L- and ME7-infected groups. The extent of activation in four brain regions correlated well with the strain-specific degree of neuroinflammation in these regions. Analysis of A1-, A2- or PAN-specific markers in crude brain tissues should be considered with great caution, as most of the markers are not exclusively astrocyte-specific, but are also expressed by other cell types. As such, these markers might be useful for reporting changes in region-specific microenvironments that govern astrocytic response under specific disease conditions. Nevertheless, the result of the current work is consistent with the recent study that also reported mixed astrocyte phenotype in prion disease (Hartmann et al., 2019). It is not clear whether the appearance of mixed phenotype, if such exist, could arise due to co-existing mixtures of pure A1 and A2 states, multiple distinct activation states in addition to the classical A1 and A2 states, co-expression of markers of different phenotypes within individual cells, or all of the above. The observation of mixed astrocyte phenotype in prion disease is not entirely unexpected considering that the reactive phenotype of microglia only partially overlaps with those described in mouse models of other neurodegenerative diseases.
In future studies, it would be interesting to examine the extent to which the neuroinflammation signature observed in prion infected animals resembles that in normal aging. Another area of considerable interest involves assessing changes in the functional states of astrocytes and determining whether these changes are protective against neurodegeneration or contribute to neurodegeneration. The Neuroinflammation panel is dominated by the genes that report on microglial activation, so further analyses that focus on astrocyte function are warranted. Designing a panel with an emphasis on astrocyte-specific genes and/or analysis of acutely isolated astrocytes could shed light on the role of astrocytes in chronic neurodegeneration. Defining causative relationship between the reactive states of microglia and astrocytes is also of considerable interest.
5. Conclusions
A growing number of studies demonstrated phenotypic heterogeneity in microglia and astrocytes under normal conditions as well as dynamic region- and subregion-specific changes in glial phenotypes under aging or neurodegenerative conditions. While region- and subregion-specific changes in glial phenotype have been documented using a number of animal models of neurodegenerative diseases, the extent to which animal models recapitulate neurodegenerative disease in humans has been under intense debate. Among neurodegenerative diseases, prion disease is the only one that can be faithfully reproduced in wild type animals. Indeed, non-transgenic, inbred mice infected with prions develop actual bona fide prion disease, and not a disease model. Yet, a significant gap in our understanding of glial phenotype in prion diseases exists. Previous transcriptome studies of prion-infected animals employed whole brain tissues for differential gene expression analysis, leaving region specific identities concealed. To fill the gap, the current study is the first to analyze temporal changes in neuroinflammation transcriptome in prion diseases in a region-specific manner. The current work revealed that (i) region-specific homeostatic identities of glia were preserved at the preclinical stages of prion disease. (ii) With the progression of clinical signs, the region-specific signatures were lost and replaced with a uniform neuroinflammation signature. (iii) The neuroinflammation transcriptome signature was not only region-independent but also uniform for prion strains with different cell tropism. (iv) Changes in astrocyte function scored at the top of activated pathways analyzed. Moreover, the astrocyte function pathway responded to prion infection prior to activated microglia. (v) The prion-associated neuroinflammation signature identified in the current study overlapped only partially with the microglia degenerative phenotype and the disease-associated microglia phenotype reported for animal models of other neurodegenerative diseases.
Supplementary Material
Figure S1. Experimental design. (A) Schematic representation of three animal groups inoculated IP with PBS, 22L or ME7 strains (200 μl, 1% brain homogenate) and euthanized at three time points post inoculation as indicated. (B) Coronal section of the brain showing four brain regions (Ctx - cortex, Hp - hippocampus, Th - thalamus, HTh - hypothalamus) collected for gene expression analysis. (C) Analysis of PrPSc amounts in animals used for the gene expression analysis. Western blots were stained with ab3531 anti-PrP antibody.
Figure S2. List of pathways with corresponding gene numbers analyzed by the Neuroinflammation panel.
Figure S3. Agglomerative hierarchical cluster analysis of all data collected for the second time point. Animals from the second time point showed high variation in the degree of profile changes, which is particularly visible for a set of genes in the orange frame. Th and HTh from the most affected 22L-infected animal (#4) clustered away from other animals and brain regions (red shading), whereas Th from the least affected 22L-infected animal remained within the sub-cluster with normal Th. Th from 22L-infected, animal #5, and the most affected among ME7 group, animal #13, formed a separate sub-cluster within a Th cluster (brown shading).
Figure S4. Histopathological analysis of 22L-infected animals from the second time point. Immunostaining of individual 22L-infected animals using antibody to microglia-specific marker, Ibal. Scale bars = 200 μm.
Figure S5. Histopathological analysis of 22L-infected animals from the third time point. Deposition of PrPSc probed by SAF-84 anti-PrP antibody, staining of microglia with anti-Iba1 or astrocytes with anti-GFAP in hippocampus, cortex and thalamus. Scale bars = 200 μm.
Figure S6. Region- and strain-specific pattern of expression of top differentially expressed genes. Normalized gene expression of Cxcl10, Serpina3n, Cd68, Clec7a in four brain regions in 22L- and ME7-infected animals and normal controls. Gene expression in individual animals is shown.
Figure S7. Differentially expressed genes in Neurons and Neurotransmission pathway. (A) Agglomerative clustering of the differentially expressed genes in the Neuron and Neurotransmission pathway. Downregulated (top) and upregulated (bottom) clusters for thalami at three time points are shown. (B) List of differentially expressed genes in the Neuron and Neurotransmission pathway with P<0.05 for thalami at the third time point.
Table S1. List of animals used for gene expression analysis.
Table S2. nCounter® Mouse Neuroinflammation Panel (in separate file)
Table S3. Top differentially expressed genes detected for the first time point (in separate file). Differentially expressed genes with P<0.05 are listed according to the prion strain and brain region.
Table S4. List of all differentially expressed genes in 22L- and ME7-infected animals in advanced stage of the disease (in separate file). Differentially expressed genes with P<0.05 are listed according to prion strain and brain region. Newly identified differentially expressed genes (P<0.05, fold change >1.5 or <0.66) are highlighted in yellow boxes.
Highlights.
In the preclinical stages of prion disease, glia preserve their regional homeostatic identities
With prion disease, a uniform signature of neuroinflammation replaces the regional identities
The same signature of inflammatory activation occurs with strains with different cellular tropism
Changes in pathways related to astrocytic function were the most frequent transcriptional alterations
Acknowledgments
We thank Institute for Genome Sciences at the University of Maryland School of Medicine for processing samples using the nCounter Nanostring platform.
Funding
This work was supported by the National Institute of Health grants R01 NS045585 (IVB) and R01 AI128925 (IVB).
Abbreviations
- 22L
mouse adapted prion strain 22L
- 263K
hamster adapted prion strain 263K
- 301V
mouse adapted prion strain 301V
- Ctx
cortex
- DE
differentially expressed
- GFAP
Glial fibrillary acidic protein
- Hp
hippocampus
- HTh
hypothalamus
- ME7
mouse adapted prion strain ME7
- PrPc
normal, cellular form of the prion protein
- PrPSc
transmissible, disease-associated form of the prion protein
- PBS
phosphate-buffered saline
- RML
mouse adapted prion strain RML
- Th
thalamus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics approval and consent to participate
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal protocol was approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore (Assurance Number A32000-01; Permit Number: 0215002).
Consent for publication
Not applicable
Availability of data and materials
All data generated or analyzed in this study are included in this published article [and its supplementary information files].
Competing interests
The authors declare that they have no competing interests.
References
- Abdelaziz DH, Abdulrahman BA, Glitch S, & Schatzl HM (2019). Autophagy pathways in the treatment of prion diseases. Curr Opin Pharmacol, 44, 46–52. doi: doi: 10.1016/j.coph.2019.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulrahman BA, Abdelaziz D, Thapa S, Lu L, Jain S, Gilch S, … Schatzl HM (2017). The celecoxib derivatives AR-12 and AR-14 induce autophagy and clear prion-infected cells from prions. Scientific Reports, 7(1), 17565. doi: 10.1038/s41598-017-17770-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulrahman BA, Abdelaziz DH, & Schatzl HM (2018). Autophagy regulates exosomal release of prions in neuronal cells. Journal of Biological Chemistry, 293(23), 8956–8968. doi: 10.1074/jbc.RA117.000713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A, & Liu Y (2017). A role for astroglia in prion diseases. J Exp Med, 214(12), 3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami B, Samusik N, Wieghofer P, Ho PP, Crotti A, Bjornson Z, … Steinman L (2018). Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nature Neuroscience, 21(4), 541–551. doi: 10.1038/s41593-018-0100-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuni AA, Gray B, Bailey J, Skipp P, Perry VH, & O’Connor V (2014). Analysis of the hippocampal proteome in ME7 prion disease reveals a predominant astrocytic signature and highlights the brain-restricted production of clusterin in chronic neurodegeneration. J Biol Chem, 289(7), 4532–4545. doi: 10.1074/jbc.M113.502690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers JL, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, & Bartz JC (2011). The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLOS Pathog., 7(3), e1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CA, & Manuelidis L (2003). Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA, 100(2), 675–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbisin M, Vanni S, Schmadicke A-C, Montag J, Motzkus D, Opitz L, … Legname G (2014). Gene expression profiling of brains from bovine spongiform encephalopathy (BSE)-infected cynomolgus macaques. BMC Genomics, 15. doi: doi: 10.1186/1471-2164-15-434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskakov IV (2017). Limited understanding of the functional diversity of N-linked glycans as a major gap of prion biology. Prion, 11(2), 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskakov IV, & Katorcha E (2016). Multifaceted role of sialylation in prion diseases. Front. Neurosci, 10, 358. doi: 10.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskakov IV, Katorcha E, & Makarava N (2018). Prion Strain-Specific Structure and Pathology: A View from the Perspective of Glycobiology. Viruses, 10(12), 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate C, Boshuizen RS, Langeveld JPM, & Williams A (2002). Temporal and spatial relationship between the death of PrP-damaged neurons and microglial activation. Neuroreport, 13(13), 1695–1700. [DOI] [PubMed] [Google Scholar]
- Boisvert MM, Erikson GA, Shokhirev MN, & Allen NJ (2018). The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep, 22, 269–285. doi: 10.1016/j.celrep.2017.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, & Neher JJ (2014). Microglial phagocytosis of live neurons. Nat Rev Neuroscience, 15, 209–216. [DOI] [PubMed] [Google Scholar]
- Bruce ME, & Dickinson AG (1987). Biological evidence that the scrapie agent has an independent genome. J.Gen.Virol, 68, 79–89. [DOI] [PubMed] [Google Scholar]
- Butovsky O, & Weiner HL (2018). Microglial signatures and their role in health and disease. Nature Reviews Neuroscience, 19(10), 622–635. doi: 10.1038/s41583-018-0057-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, & Chesebro B (2019). Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses, 11(1). doi: 10.3390/v11010065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, Race B, Williams K, & Chesebro B (2018). Toll-like receptor 2 confers partial neuroprotection during prion disease. PlOS One, 13, e0208559. doi: 10.1371/journal.pone.0208559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, Race B, Williams K, Striebel J, & Chesebro B (2018). Microglia Are Critical in Host Defense against Prion Disease. Journal of virology, 92(15), e00549–00518. doi: 10.1128/JVI.00549-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, Striebel JF, Race B, Phillips K, & Chesebro B (2015). Prion Infection of Mouse Brain Reveals Multiple New Upregulated Genes Involved in Neuroinflammation or Signal Transduction. 89(4), 2388–2404. doi: 10.1128/JVI.02952-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, Striebel JF, Rangel A, Woods T, Phillips K, Peterson KE, … Chesebro B (2016). Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog, 12(4), e1005551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Raymond GJ, & Bessen RA (1998). Strain-dependent differences in b-sheet conformations of abnormal prion protein. J.Biol.Chem, 273(48), 32230–32235. [DOI] [PubMed] [Google Scholar]
- Choi YP, Head MW, Ironside JW, & Priola SA (2014). Uptake and Degradation of Protease-Sensitive and -Resistant Forms of Abnormal Human Prion Protein Aggregates by Human Astrocytes. Am J Pathol, 184 (12), 3299–3307. doi: 10.1016/j.ajpath.2014.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Munich AE, Heiman M, & Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA, 115(8), E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, & Clarke AR (2007). A General Model of Prion Strains and Their Pathogenicity. Science, 318, 930–936. [DOI] [PubMed] [Google Scholar]
- Cronier S, Laude H, & Peyrin JM (2004). Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death. Proc Natl Acad Sci USA, 101(33), 12271–12276. doi: 10.1073/pnas.0402725101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallérac G, Zapata J, & Rouach N (2018). Versatile control of synaptic circuits by astrocytes: where, when and how? Nature Reviews Neuroscience, 19(12), 729–743. doi: 10.1038/s41583-018-0080-6 [DOI] [PubMed] [Google Scholar]
- Dawson TM, Golde TE, & Lagier-Tourenne C (2018). Animal models of neurodegenerative diseases. Nature Neuroscience, 21(10), 1370–1379. doi: 10.1038/s41593-018-0236-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everbroeck BV, Dobbeleir I, Waele MD, Leenheir ED, Lubke U, Martin J-J, & Cras P (2004). Extracellular protein deposition correlates with glial activation and oxidative stress in Creutzfeldt-Jakob and Alzheimer’s disease. Acta Neuropath, 108, 194–200. [DOI] [PubMed] [Google Scholar]
- Ferrer I (2017). Diversity of astroglial responses across human neurodegenerative disorders and brain aging. Brain Pathology, 27(5), 645–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L, Través PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, … Lemke G (2016). TAM receptors regulate multiple features of microglial physiology. Nature, 532, 240. doi: 10.1038/nature17630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin FL, Huntley MA, … Hansen DV (2018). Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep, 22(3), 832–847. [DOI] [PubMed] [Google Scholar]
- Giese A, Brown DR, Groschup MH, Feldmann C, Haist F, & Kretzschmar HA (1998). Role of microglia in neuronal cell death in prion disease. Brain Pathol, 8, 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Nicola D, Fransen NL, Suzzi S, & Perry VH (2013). Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci, 33(6), 2481–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Nicola D, & Perry VH (2015). Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. The Neuroscientist, 21(2), 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabert K, Michoel T, Karavolos ΜH, Clohisey S, Baillie JK, Stevens MP, … McColl BW (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nature Neuroscience, 19, 504. doi: 10.1038/nn.4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratuze M, Leyns CEG, & Holtzman DM (2018). New insights into the role of TREM2 in Alzheimer’s disease. Molecular Neurodegeneration, 13(1), 66. doi: 10.1186/sl3024-018-0298-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlee ΜHW, Lind M, Kokemuller R, Mammadova N, Kondru N, Manne S, … Greenlee J (2016). Temporal resolution of misfolded prion protein transport, accumulation, glial activation , and neuronal death in the retinas of mice inoculated with scrapi q. Am J Pathol, 186(9), 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grizenkova J, Akhtar S, Brandner S, Collinge J, & Lloyd SE (2014). Microglial Cx3crl knockout reduces prion disease incubation time in mice. BMC Neurosci, 75(44), 15–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Hanson JE, & Sheng M (2018). Microglia in Alzheimer’s disease. The Journal of Cell Biology, 217(2), 459–472. doi: 10.1083/jcb.201709069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann K, Sepulveda-Falla D, Rose IVL, Madore C, Muth C, Matschke J, …Krasemann S (2019). Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia, [journal article], Acta Neuropathologica Communications, 7(1), 83. doi: 10.1186/s40478-019-0735-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton KJ, Cunningham C, Reynolds RA, & Perry VH (2013). Early Hippocampal Synaptic Loss Precedes Neuronal Loss and Associates with Early Behavioural Deficits in Three Distinct Strains of Prion Disease. PLoS One, 8(6), e68062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, … Stevens B (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352(6286), 712–716. doi: 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ΜM, Field RH, Perry VH, Murray CL, & Cunningham C (2010). Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia, 55(16), 2017–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D, Lee TY, Yoo H, Gehlenborg N, Cho JH, Petritis B, … Hood LE (2009). A systems approach to prion disease. J Molecular System Biology, 5(1), 252. doi: 10.1038/msb.2009.10 %J Molecular Systems Biology [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey M, Goodsir CM, Race RE, & Chesebro B (2004). Scrapie-specific neuronal lesions are independent of neuronal PrP expression. Ann Neurol, 55(6), 781–792. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Halliday WG, Bell J, Johnston AR, MacLeod NK, Ingham C, … Fraser JR (2000). Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Nenropath.Appl.Nenrobiol, 26 41–54. [DOI] [PubMed] [Google Scholar]
- Kang SS, Ebbert MTW, Baker KE, Cook C, Wang X, Sens JP, … Fryer JD (2018). Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. The Journal of Experimental Medicine, 215(9), 2235–2245. doi: 10.1084/jem.20180653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karapetyan YE, Saa P, Mahal SP, Sferrazza GF, Sherman A, Sales N, … Lasmezas CI (2009). Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One, 4(5), e5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katorcha E, Daus ML, Gonzalez-Montalban N, Makarava N, Lasch P, Beekes M, & Baskakov IV (2016). Reversible off and on switching of prion infectivity via removing and reinstalling prion sialylation. Sci Rep, 6, 33119. doi: 10.1038/srep33119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katorcha E, Makarava N, Savtchenko R, & Baskakov IV (2015). Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci Rep, 5, 16912. doi: 10.1038/srepl6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katorcha E, Makarava N, Savtchenko R, DAzzo A, & Baskakov IV (2014). Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLOS Pathog., 10(9), el004366. doi: doi: 10.1371/joumal.ppat.1004366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kercher L, Favara C, Chan CC, Race R, & Chesebro B (2004). Differences in scrapie- induced pathology of the retina and brain in transgenic mice that express hamster prion protein in neurons, astrocytes, or multiple cell types. Am J Pathol, 165(6), 2055–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kercher L, Favara C, Striebel JF, LaCasse R, & Chesebro B (2007). Prion protein expression differences in microglia and astroglia influence scrapie-induced neurodegeneration in the retina and brain of transgenic mice. J Virol, 81(19), 10340–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, … Amit I (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell, 169(1), 1276–1290.el217. doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Kimberlin RH, Cole S, & Walker CA (1987). Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J.Gen.Virol, 68, 1875–1881. [DOI] [PubMed] [Google Scholar]
- Kimberlin RH, Walker CA, & Fraser H (1989). The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J.Gen.Virol, 70, 2017–2025. [DOI] [PubMed] [Google Scholar]
- Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, Zinkernagel RM, … Aguzzi A (2001). Complement facilitates early prion pathogenesis. NatMed, 7, 488–492. [DOI] [PubMed] [Google Scholar]
- Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, … Rolls A (2017). High-dimensional, single-cell characterization of the brain's immune compartment. Nature Neuroscience, 20, 1300–1309. doi: 10.1038/nn.4610 [DOI] [PubMed] [Google Scholar]
- Kranich J, Krautler NJ, Falsig J, Ballmer B, Li S, Hutter G, … Aguzzi A (2010). Engulfment of cerebral apoptotic bodies controls the course of prion disease in a mouse strain-dependent manner. J Exp Med, 207(10), 2271–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, … Butovsky O (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 77(3), 566–581.e569. doi: 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejciova Z, Alibhai J, Zhao C, Krencik R, Rzechorzek NM, Lillian EM, … Chandran S (2017). Human stem cell-derived astrocytes replicate human prions in a PRNP genotype-dependent manner. J Exp Med, 277(12), 3481–3495. doi: 10.1084/jem.20161547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampron A, Elali A, & Rivest S (2013). Innate Immunity in the CNS: Redefining the Relationship between the CNS and Its Environment. Neuron, 78, 214–232. [DOI] [PubMed] [Google Scholar]
- Legname G, Baskakov IV, Nguyen ΗΟB, Riesner D, Cohen FE, DeArmond SJ, & Prusiner SB (2004). Synthetic mammalian prions. Science, 305, 673–676. [DOI] [PubMed] [Google Scholar]
- Li Q, & Barres BA (2017). Microglia and macrophages in brain homeostasis and disease. Nat RevImmun, 18, 225–242. doi: 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, & Barres BA (2017). Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity, 16(6), 957–967. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, … Barres BA (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnartz-Gerlach B, Schuy C, Shahraz A, Tenner AJ, & Neumann H (2016). Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage- neuron Co-Culture System. Glia, 67(1), 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnartz B, Kopatz J, Tenner AJ, & Neumann H (2012). Sialic acid on the neuronal glycocalyx prevents complement Cl binding and complement receptor-3-mediated removal by microglia. J Neurosci, 32(3), 946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sorce S, Nuvolone M, Domange J, & Aguzzi A (2018). Lymphocyte activation gene 3 (Lag3) expression is increased in prion infections but does not modify disease progression. Scientific Reports, 5(1), 14600. doi: 10.1038/s41598-018-32712-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZY, Baker CA, & Manuelidis L (2004). New Molecular Markers of Early and Progressive CJD Brain Infection. J. Cell Biochem, 93, 644–652. [DOI] [PubMed] [Google Scholar]
- Lui H, Zhang J, Makinson Stefanie R., Cahill Michelle K., Kelley Kevin W., Huang H-Y, … Huang Eric J. (2016). Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell, 765(4), 921–935. doi: 10.1016/j.cell.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Y, Chen C, Zhang BY, Xiao K, Wang J, Chen LN, … Dong XP (2015). Remarkable Activation of the Complement System and Aberrant Neuronal Localization of the Membrane Attack Complex in the Brain Tissues of Scrapie-Infected Rodents. Mol Neurobiol, 52(3), 1165–1179. [DOI] [PubMed] [Google Scholar]
- Mabbott NA, Bruce ME, Botto M, Walport MJ, & Pepys MB (2001). Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat.Med, 7, 485–487. [DOI] [PubMed] [Google Scholar]
- Majer A, Medina SJ, Niu Y, Abrenica B, Manguiat KJ, Frost KL, … Booth SA (2012). Early Mechanisms of Pathobiology are Revealed by Transcriptional Temporal Dynamics in Hippocampal CA1 Neurons of Prion Infected Mice. PLOS Path, 5(11), el003002. doi: 10.1371/journal.ppat.1003002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majer A, Medina SJ, Sorensen D, Martin MJ, Frost KL, Phillipson C, … Booth SA (2019). The cell type resolved mouse transcriptome in neuron-enriched brain tissues from the hippocampus and cerebellum during prion disease. Scientific Reports, 9(1), 1099. doi: 10.1038/s41598-018-37715-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarava N, Savtchenko R, & Baskakov IV (2015). Two alternative pathways for generating transmissible prion disease de novo. Acta Neuropathologica Communications, 3, 69. doi: 10.1186/s40478-015-0248-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, … Tsai L-H (2017). Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Reports, 21(2), 366–380. doi: 10.1016/j.celrep.2017.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizawa YM, Hirayama Y, Ohno N, Shibata S, Shigetomi E, Sui Y, … Koizumi S (2017). Reactive astrocytes function as phagocytes after brain ischemia via ABCA1- mediated pathway. Nature Communications, 5(1), 28. doi: 10.1038/s41467-017-00037-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissette DA, Parachikova A, Green KN, & LaFerla FM (2009). Relevance of Transgenic Mouse Models to Human Alzheimer Disease. Journal of Biological Chemistry, 254(10), 6033–6037. doi: 10.1074/jbc.R800030200 [DOI] [PubMed] [Google Scholar]
- Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, … Becher B (2018). High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity, 45(3), 599. doi: 10.1016/j.immuni.2018.02.014 [DOI] [PubMed] [Google Scholar]
- Muth C, Schrock K, Madore C, Hartmann K, Fanek Z, Butkovsky O, … Krasemann S (2016). Activation of microglia by retroviral infection correlates with transient clearance of prions from the brain but does not change incubation time. Brain Pathol, Epub ahead of print. doi: 10.1111/bpa.12441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na Y-J, Jin J-K, Kim J-I, Choi E-K, Carp RI, & Kim Y-S (2007). JAK-STAT signaling pathway mediates astrogliosis in brains of scrapie-infected mice. Journal of Neurochemistry, 103(2), 637–649. doi: 10.1111/j.1471-4159.2007.04769.x [DOI] [PubMed] [Google Scholar]
- Prusiner SB (1982). Novel proteinaceous infectious particles cause scrapie. Science, 216, 136–144. [DOI] [PubMed] [Google Scholar]
- Puoti G, Giaccone G, Mangieri M, Limido L, Fociani P, Zerbi P, … Tagliavini F (2005). Sporadic Creutzfeldt-Jakob Disease: The Extent of Microglia Activation Is Dependent on the Biochemical Type of PrPSc. J Neuropathol Exp Neurol, 64(10), 902–909. [DOI] [PubMed] [Google Scholar]
- Raeber AJ, Race RE, Priola S, & Sailer SA (1997). Astrocyte-specific expressoin of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J, 16(20), 6057–6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer C, Schultz J, Burwinkel M, Schwarz A, Mok SWF, Gultner S, … Baier M (2008). Accelerated prion replication in, but prolonged survival times of, prion-infected CXCR3−/− mice. J Virol, 52(24), 12464–12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Hasebe R, Takahashi Y, Song C-H, Suzuki A, Yamasaki T, & Horiuchi M (2013). Absence of CD14 Delays Progression of Prion Diseases Accompanied by Increased Microglial Activation. J Virol, 57(24), 13433–13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg MK, Al-Doujaily H, Sharps B, De Oliveira MW, Schmidt C, Richard-Londt A, … Collinge J (2014). Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat Commun, 5, e4347. doi: 10.1038/ncomms5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Toni N, & Volterra A (2019). Astrocyte function from information processing to cognition and cognitive impairment. Nature Neuroscience, 22(2), 154–166. doi: 10.1038/s41593-018-0325-8 [DOI] [PubMed] [Google Scholar]
- Schafer Dorothy P., Lehrman Emily K., Kautzman Amanda G., Koyama R, Mardinly Alan R., Yamasaki R, … Stevens B (2012). Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron, 74(4), 691–705. doi: 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Schwarz A, Neidhold S, Burwinkel M, Riemer C, Simon D, … Baier M (2004). Role of interleukin-1 in prion disease-associated astrocyte activation. Am. J. Pathol, 165, 671–678. doi: 10.1016/S0002-9440(10)63331-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner PJ, Abbassi H, Chesebro B, Race RE, Reilly C, & Haase AT (2006). Gene expression alterations in brains of mice infected with three strains of scrapie. [journal article]. BMC Genomics, 7(1), 114. doi: 10.1186/1471-2164-7-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorce S, Nuvolone M, Keller A, Falsig J, Varol A, Schwarz P, … Aguzzi A (2014). The role of the NADH oxidase NOX2 in prion pathogenesis. PLoS Pathog, 10(12), e1004531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen G, Medina S, Parchaliuk D, Phillipson C, Robertson C, & Booth SA (2008). Comprehensive transcriptional profiling of prion infection in mouse models reveals networks of responsive genes. [journal article]. BMC Genomics, 9(1), 114. doi: 10.1186/1471-2164-9-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreq L, Consortium UBE, Consortium NABE, Rose J, Soreq E, Hardy J, … Ule J (2017). Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep, 18(2), 557–570. doi: 10.1016/j.celrep.2016.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Katorcha E, Daus ML, Lasch P, Beekes M, & Baskakov IV (2017). Sialylation controls prion fate in vivo. J Biol Chem, 292(6), 2359–2368. doi: 10.1074/jbc.M116.768010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Katorcha E, Makarava N, Barrett JP, Loane DJ, & Baskakov IV (2018). Inflammatory response of microglia to prions is controlled by sialylation of PrPSc. Sci Rep, 8(1), e11326. doi: 10.1038/s41598-018-29720-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Makarava N, Katorcha E, Savtchenko R, Brossmer R, & Baskakov IV (2015). Post-conversion sialylation of prions in lymphoid tissues. Proc Acad Natl Sci U S A, 112(48), E6654–6662. doi: 10.1073/pnas.1517993112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, … Barres BA (2013). A Dramatic Increase of C1q Protein in the CNS during Normal Aging. The Journal of Neuroscience, 22(33), 13460–13474. doi: 10.1523/jneurosci.1333-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson J, Nutma E, van der Valk P, & Amor S (2018). Inflammation in CNS neurodegenerative diseases. Immunology, 154(2), 204–219. doi: 10.1111/imm.12922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Zazquez LE, Howell GR, Christopherson KS, Nouri N, … Barres BA (2007). The classical complement cascade mediates CNS synapse elimination. Cell, 121(6), 1164–1178. [DOI] [PubMed] [Google Scholar]
- Thomzig A, Spassov S, Friedrich M, Naumann D, & Beekes M (2004). Discriminating Scrapie and Bovine Spongiform Encephalopathy Isolates by Infrared Spectroscopy of Pathological Prion Protein. J.Biol.Chem, 279 (33847), 33854. [DOI] [PubMed] [Google Scholar]
- Tribouillard-Tanvier D, Striebel JF, Peterson KE, & Chesebro B (2009). Analysis of Protein Levels of 24 Cytokines in Scrapie Agent-Infected Brain and Glial Cell Cultures from Mice Differing in Prion Protein Expression Levels. Journal of Virology, 55(21), 11244–11253. doi: 10.1128/jvi.01413-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanni S, Moda F, Zattoni M, Bistaffa E, De Cecco E, Rossi M, … Legname G (2017). Differential overexpression of SERPINA3 in human prion diseases. Scientific Reports, 7(1), 15637. doi: 10.1038/s41598-017-15778-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A (2008). Sialic acids in human health and disease. Trends in Molecular Medicine, 14(8), 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A (2011). Since there are PAMPs and DAMPs, there must be SAMPs? Glycan “self-associated molecular patterns” dampen innate immunity, but pathogens can mimic them. Glycobiology, 27(9), 1121–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenti JE, Murphy L, Grabert K, McColl BW, Cancellotti E, Freeman TC, & Manson JC (2016). Defining the Microglia Response during the Time Course of Chronic Neurodegeneration. J Virol, 90(6), 3003–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, & Prusiner SB (2014). Mouse Models for Studying the Formation and Propagation of Prions. Journal of Biological Chemistry, 289(29), 19841–19849. doi: 10.1074/jbc.R114.550707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Lucassen PJ, Ritchie D, & Bruce M (1997). PrP Deposition, Microglial Activation, and Neuronal Apoptosis in Murine Scrapie. Exp Neurology, 144, 433–438. [DOI] [PubMed] [Google Scholar]
- Xiang W, Windl O, Wiinsch G, Dugas M, Kohlmann A, Dierkes N, … Kretzschmar HA (2004). Identification of Differentially Expressed Genes in Scrapie-Infected Mouse Brains by Using Global Gene Expression Technology. 75(20), 11051–11060. doi: 10.1128/JVI.78.20.11051-11060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SP, Kam T-T, Panicker N, Kim S, Oh Y, Park J-S, … Ko HS (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nature Medicine, 24(1), 931–938. doi: 10.1038/s41591-018-0051-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, & Barres BA (2012). Genomic Analysis of Reactive Astrogliosis. The Journal of Neuroscience, 32(18), 6391–6410. doi: 10.1523/jneurosci.6221-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van derZwan J, … Linnarsson S (2018). Molecular Architecture of the Mouse Nervous System. Cell, 174(4), 999–1014. doi: 10.1016/j.cell.2018.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Herrmann US, Falsig J, Abakumova I, Nuvolone M, Schwarz P, … Aguzzi A (2016). A neuroprotective role for microglia in prion diseases. J Exp Med, 213(6), 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Herrmann US, Li B, Abakumova I, Moos R, Schwarz P, … Aguzzi A (2015). Triggering receptor expressed on myeloid cells-2 is involved in prion-induced microglial activation but does not contribute to prion pathogenesis in mouse brains. Neurobiol Aging, 36(5), 1994–2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Experimental design. (A) Schematic representation of three animal groups inoculated IP with PBS, 22L or ME7 strains (200 μl, 1% brain homogenate) and euthanized at three time points post inoculation as indicated. (B) Coronal section of the brain showing four brain regions (Ctx - cortex, Hp - hippocampus, Th - thalamus, HTh - hypothalamus) collected for gene expression analysis. (C) Analysis of PrPSc amounts in animals used for the gene expression analysis. Western blots were stained with ab3531 anti-PrP antibody.
Figure S2. List of pathways with corresponding gene numbers analyzed by the Neuroinflammation panel.
Figure S3. Agglomerative hierarchical cluster analysis of all data collected for the second time point. Animals from the second time point showed high variation in the degree of profile changes, which is particularly visible for a set of genes in the orange frame. Th and HTh from the most affected 22L-infected animal (#4) clustered away from other animals and brain regions (red shading), whereas Th from the least affected 22L-infected animal remained within the sub-cluster with normal Th. Th from 22L-infected, animal #5, and the most affected among ME7 group, animal #13, formed a separate sub-cluster within a Th cluster (brown shading).
Figure S4. Histopathological analysis of 22L-infected animals from the second time point. Immunostaining of individual 22L-infected animals using antibody to microglia-specific marker, Ibal. Scale bars = 200 μm.
Figure S5. Histopathological analysis of 22L-infected animals from the third time point. Deposition of PrPSc probed by SAF-84 anti-PrP antibody, staining of microglia with anti-Iba1 or astrocytes with anti-GFAP in hippocampus, cortex and thalamus. Scale bars = 200 μm.
Figure S6. Region- and strain-specific pattern of expression of top differentially expressed genes. Normalized gene expression of Cxcl10, Serpina3n, Cd68, Clec7a in four brain regions in 22L- and ME7-infected animals and normal controls. Gene expression in individual animals is shown.
Figure S7. Differentially expressed genes in Neurons and Neurotransmission pathway. (A) Agglomerative clustering of the differentially expressed genes in the Neuron and Neurotransmission pathway. Downregulated (top) and upregulated (bottom) clusters for thalami at three time points are shown. (B) List of differentially expressed genes in the Neuron and Neurotransmission pathway with P<0.05 for thalami at the third time point.
Table S1. List of animals used for gene expression analysis.
Table S2. nCounter® Mouse Neuroinflammation Panel (in separate file)
Table S3. Top differentially expressed genes detected for the first time point (in separate file). Differentially expressed genes with P<0.05 are listed according to the prion strain and brain region.
Table S4. List of all differentially expressed genes in 22L- and ME7-infected animals in advanced stage of the disease (in separate file). Differentially expressed genes with P<0.05 are listed according to prion strain and brain region. Newly identified differentially expressed genes (P<0.05, fold change >1.5 or <0.66) are highlighted in yellow boxes.