

Abstract
Aging is characterized by a progressive decline in the normal physiological functions of an organism, ultimately leading to mortality. Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor that plays a critical role in mitochondrial energy production as well as many enzymatic redox reactions. Age-associated decline in NAD+ is implicated as a driving factor in several categories of age-associated disease, including metabolic and neurodegenerative disease, as well as deficiency in the mechanisms of cellular defense against oxidative stress. The kynurenine metabolic pathway is the sole de novo NAD+ biosynthetic pathway, generating NAD+ from ingested tryptophan. Altered kynurenine pathway activity is associated with both aging and a variety of age-associated diseases. Kynurenine pathway interventions can extend lifespan in both fruit flies and nematodes, and altered NAD+ metabolism represents one potential mediating mechanism. Recent studies demonstrate that supplementation with NAD+ or NAD+-precursors increase longevity and promote healthy aging in fruit flies, nematodes, and mice. NAD+ levels and the intrinsic relationship to mitochondrial function have been widely studied in the context of aging. Mitochondrial function and dynamics have both been are implicated in longevity determination in a range of organisms from yeast to humans, at least in part due to their intimate link to regulating an organism’s cellular energy economy and capacity to resist oxidative stress. Recent findings support the idea that complex communication between the mitochondria and the nucleus orchestrates a series of events and stress responses involving mitophagy, mitochondrial number, mitochondrial unfolded protein response (UPRmt), and mitochondria fission and fusion events. In this review, we discuss how mitochondrial morphological changes and dynamics operate during aging, and how altered metabolism of tryptophan to NAD+ through the kynurenine pathway interacts with these processes.
Keywords: tryptophan, kynurenine pathway, NAD, mitochondria, oxidative stress
Graphical Abstract
1. Introduction
Aging is tightly coupled to metabolism, and research into specific metabolic processes has proven a productive strategy to develop novel treatments and preventative measures for age-associated disease. Accumulating evidence in recent years implicates altered tryptophan catabolism through the kynurenine pathway as a potential causative factor in numerous forms of age-associated disease (Kim et al., 2019; Sorgdrager et al., 2019). Complementary work has identified several intervention targets in the kynurenine pathway that extend lifespan in invertebrates (Oxenkrug, 2010; Oxenkrug et al., 2011) and improve outcomes in models of neurodegeneration (Chang et al., 2018; Lim et al., 2017; Rejdak et al., 2011; Sorgdrager et al., 2019), cardiovascular disease (Song et al., 2017), and acute inflammatory or autoimmune disease (Baumgartner et al., 2019; Lytton et al., 2019; Prendergast et al., 2014). The metabolic processing of tryptophan through the kynurenine pathway produces a range of biologically active intermediate metabolites. One branch of the pathway ultimately leads to de novo synthesis of nicotinamide adenine dinucleotide (NAD+). NAD+ is an essential cofactor that plays a critical role in many enzymatic redox reactions and in mitochondrial energy production. NAD levels decrease with age in a variety of tissues. This decline has been implicated as a driving factor in the pathophysiology of several categories of age-associated disease. This review explores the complex interplay between kynurenine metabolism, NAD+ production, and mitochondrial function in the context of aging and age-associated disease.
1.1. The kynurenine pathway in aging and disease
Kynurenine metabolism is the major catabolic route for ingested tryptophan and is highly conserved throughout the Eukaryotic lineage from yeast to humans. The pathway has two major branches, terminating in the production of the neuroactive metabolite kynurenic acid (KA) or NAD+, respectively (Figure 1). Each branch is active is different tissues and cell types. The dual roles of NAD+ as an enzymatic cofactor and as an energy carrier have made it a major focus of aging research recent years and has been the topic of many recent reviews (Johnson and Imai, 2018; Rajman et al., 2018; Yaku et al., 2018). The neuroactive properties of KA make it a target of interest in neurodegenerative disease as well as other neurological disorders not associated with aging (Schwarcz et al., 2012). Beyond the metabolic endpoints of the kynurenine pathway, many of the intermediate metabolites in the major branches, as well as several metabolites produced in alternative branches (e.g. xanthurenic acid, XA, and cinnabarinic acid, CA), are biologically active and represent additional potential intervention targets in the context of aging and age-associated disease. Of particular interest for this review are intermediate metabolites with redox properties that may influence mitochondrial function or the consequences of impaired mitochondrial function, namely 3-hydroxykynurenine (3HK), 3-hyrdoxyanthranilic acid (3HAA), and quinolinic acid (QA).
Figure 1. The kynurenine pathway represents one of three routes for NAD+ production or recycling.
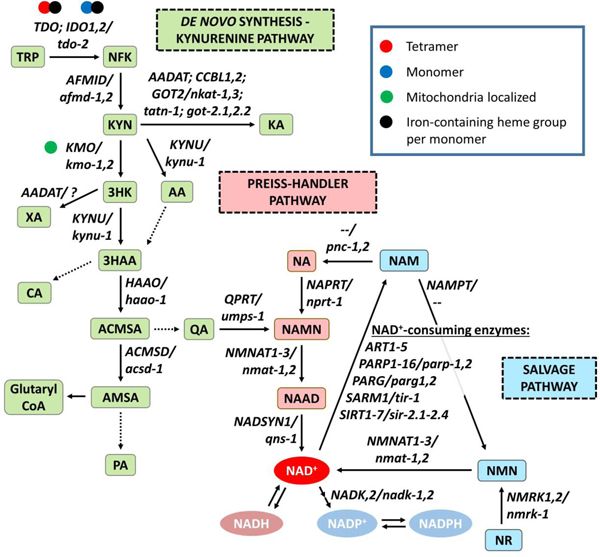
The enzymatic degradation of the essential amino acid tryptophan (TRP) through the series of reactions catalyzed by rate-limiting enzymes culminates in de novo synthesis of NAD+ constitutes one of the two major branches of the kynurenine pathway. The other major branch converts kynurenine (KYN) to the neuroactive metabolite kynurenic acid (KA). Cells can also generate NAD+ from nicotinic acid (NA) through the Preiss-Handler pathway, or from nicotinamide riboside (NR) through the salvage pathway. Note that mammalian genomes contain the enzyme nicotinamide phosphoribosyltransferase (NAMPT), which converts nicotinamide (NAM) to nicotinamide mononucleotide (NMN), but not the enzyme nicotinamides (NAMase), which converts NAM to NA, while the invertebrate C. elegans and D. melanogaster genomes contain NAMase but not NAMPT. Thus mammals recycle NAM through the salvage pathway, while invertebrates recycle NAM through the Preiss-Handler pathway. Each catalytic step is annotated with the human or C. elegans gene encoding the corresponding enzyme. Enzymes (enzyme symbol: human gene/worm gene): indoleamine 2,3-dioxygenase (IDO: IDO1,2/--); tryptophan 2,3-dioxygenase (TDO: TDO2/tdo-2); arylformamidase (AFMID: AFMID/afmd-1,2); kynurenine aminotransferase (KYAT: AADAT, CCBL1,2/nkat-1,3, tatn-1); glutamic-oxaloacetic transaminase (GOT: GOT2/got-2.1,2.2); kynurenine 3-monooxygenase (KMO: KMO/kmo-1,2); kynureninase (KYNU: KYNU/kynu-1); 3-hydroxyanthranilate 3,4-dioxygenase (HAAO: HAAO/haao-1); and aminocarboxymuconate-semialdehyde decarboxylase (ACMSD: ACMSD/acsd-1), quinolinate phosphoribosyl transferase (QPRT: QPRT/umps-1), nicotinate phosphoribosyltransferase (NAPRT: NAPRT/nprt-1), nicotinamide nucleotide adenylyltransferase (NMNAT: NMNAT1,2,3/nmat-1,2), NAD synthetase (NADSYN: NADSYN1/qns-1), NAD kinase (NADK: NADK,2/nadk-1,2), nicotinamide riboside kinase (NMRK: NMRK1,2/nmrk-1), nicotinamide phosphoribosyltransferase (NAMPT: NAMPT/-), ADP-ribosyltransferase (ART: ART1–5/--), poly(ADP-ribose polymerase 1–16 (PARP: PARP1–16/parp-1,2), poly(ADP-ribose) glycohydrolase (PARG: PARG/parg-1,2), sterile alpha and TIR motif containing (SARM: SARM1/tir-1), sirtuin NAD-dependent protein deacetylase (SIRT: SIRT1-7/sir-2.1,2.2,2.3,2.4). Metabolites: tryptophan (TRP); Nformylkynurenine (NFK); kynurenine (KYN); kynurenic acid (KA); 3-hydroxykynurenine (3HK); 3-hydroxyanthranilic acid (3HAA); anthranilic acid (AA); xanthurenic acid (XA); 2-amino-3-carboxymuconic semialdehyde (ACMSA); 2-aminomuconic semialdehyde (AMSA); and quinolinic acid (QA); glutaryl-coenzyme A (Glutaryl CoA); picolinic acid (PA); nicotinic acid (NA); nicotinic acid mononucleotide (NAMN); nicotinic acid adenine dinucleotide (NAAD); nicotinamide adenine dinucleotide (NAD+/NADH); nicotinamide adenine dinucleotide phosphate (NADP+/NADPH); nicotinamide (NAM); nicotinamide mononucleotide (NMN); nicotinamide riboside (NR).
Entry into the kynurenine pathway begins with the conversion of tryptophan (TRP) into N-formylkynurenine (NFK) by one of three enzymes: indoleamine 2,3dioxygenase 1, 2 (IDO1,2) and tryptophan 2,3-dioxygenase (TDO2) (Figure 1). TDO2 usually functions as a tetramer while IDO functions as a monomer, and both functional enzymes contain a non-covalently bound iron-containing heme group per monomer. The iron-atom present in the heme group catalyzes the redox reaction of TRP with molecular oxygen (O2) to open the 5-member ring in TRP to form NFK (Nelp et al., 2018). The enzyme arylformamidase (AFMID) next removes the formyl group, converting NFK to the pathway’s namesake metabolite, kynurenine (KYN). KYN represents the major branch point in the pathway, and can be converted to kynurenic acid (KA) by one of the three isoforms of kynurenine aminotransferase (KYAT1–3) or glutamic-oxaloacetic transaminase 2 (GOT2), to anthranilic acid (AA) by kynureninase (KYNU), or to 3HK by the mitochondrial-associated enzyme kynurenine 3-monooxygenase (KMO). 3HK is converted either to 3HAA by KYNU or to XA by the KYAT enzymes (forming a minor side branch of the pathway). AA is also converted to 3HAA through a non-enzymatic reaction. 3HAA is then converted to 2-amino-3-carboxymuconic semialdehyde (ACMSA) by 3HAA dioxygenase (HAAO), a cytosolic monomeric enzyme containing non-heme ferrous iron. 3HAA can also form cinnabarinic acid (CA) by oxidation, forming a second side branch). ACMSA spontaneously converts to QA, which is processed by QA phosphoribosyl transferase (QPRT) to the NAD+ precursor nicotinic acid mononucleotide (NAMN). In third side branch, ACMSA can alternatively be converted to 2-aminomuconic semialdehyde (AMSA) by the enzyme aminocarboxymuconate semialdehyde decarboxylase (ACMSD), which in turn can be processed to picolinic acid (PA), or to glutaryl coenzyme A and feed into glycolysis (Schwarcz et al., 2012).
Evidence in invertebrate models points to a direct role for the kynurenine pathway in aging. The earliest reports of lifespan extension directly related to components of the kynurenine pathway came from Gregory Oxenkrug at Tufts University, who showed that genetic (Oxenkrug, 2010) or pharmacological (Oxenkrug, 2013) inhibition of either TDO (encoded by the vermillion gene) or TRP transport into cells extended lifespan in the fruit fly Drosophila melanogaster. In subsequent studies, we and others have reported that knockdown of tdo-2 (encoding TDO) (Sutphin et al., 2017; van der Goot et al., 2012), kynu-1 (encoding KYNU) (Sutphin et al., 2017), or acsd-1 (encoding ACMSD) (Katsyuba et al., 2018), or supplementation with TRP (Edwards et al., 2015), similarly extend lifespan in the nematode Caenorhabditis elegans. van der Goot et al. (2012) present evidence that at least some of the beneficial effects of tdo-2 knockdown are mediated by increased TRP, while data presented by Katsyuba et al. (2018) suggest that the benefits of acsd-1 knockdown are mediated by increased NAD+ production. The mechanism mediating lifespan extension from kynu-1 is currently unclear; however, kynu-1 knockdown does not increase TRP levels in worms (Sutphin et al., 2017) and should prevent de novo NAD+ synthesis (Figure 1), suggesting a distinct mechanism of action.
While kynurenine pathway interventions have yet to be directly tested in the context of mammalian lifespan, benefits have been reported for a number of specific disease models. The types of disease targeted are closely tied to the tissue-specific expression patterns of kynurenine pathway enzymes—the pathway is most active in brain, liver, kidney, pancreas, and the immune system—and relevant properties of intermediate kynurenine pathway metabolites.
Kynurenine metabolism in the immune system.
Immune function is a major focus of disease-specific kynurenine pathway work and the topic of many detailed reviews (e.g. (Kim and Jeon, 2018; M. Liu et al., 2018; Mbongue et al., 2015; Routy et al., 2015, 2016)). Kynurenine pathways enzymes are widely expressed in immune cells, including microglial cells, macrophages (Guillemin et al., 2003), antigen presenting cells (APCs) such as dendritic cells (DCs) (Heng et al., 2016), B cells (Shinde et al., 2015), and natural killer (NK) cells (Routy et al., 2016).
Entry of tryptophan into the kynurenine pathway in immune cells is largely regulated by expression of IDO1 in response to pro-inflammatory signaling. Multiple immune signaling pathways—interferon gamma (IFNγ), interferon beta (IFNβ), tumor necrosis factor (TNF), Toll-like, transforming growth factor beta (TGFβ), and aryl hydrocarbon receptor (AhR)—either activate or maintain IDO1 expression (reviewed by Mbongue et al. (2015)). Elevated IDO1 activity influences surrounding cells and tissues in two ways: by limiting local TRP availability and producing kynurenine metabolites.
Activation of IDO1 depletes TRP from the local cellular microenvironment. This depletion has several immune suppressive effects. TRP depletion results in the accumulation of uncharged TRP-tRNA, which binds and activates the nutrient-responsive kinase GCN2, which in turn inhibits the eukaryotic initiation factor 2α kinase by phosphorylation. The resulting decrease in transcription and translation pushes T effector cells lineages in particular toward cell cycle arrest and apoptosis (Munn et al., 2005; Ravishankar et al., 2015). TRP depletion further suppresses the mechanistic target of rapamycin (mTOR), promoting T cell anergy by increasing autophagy (Metz et al., 2012; Xie et al., 2012). In combination, GCN2 activation and mTOR suppression promote Treg differentiation while suppressing Th1, Th2, and Th17 effector differentiation (Eleftheriadis et al., 2016), with the caveat that some aspects of the role of mTOR are an area of active debate (Pollizzi and Powell, 2015).
Downstream of TRP, elevated KYN directly binds and activates AhR (Opitz et al., 2011). Activated AhR both provides positive feedback by upregulating IDO1 expression (Vogel et al., 2008) and, when bound to KYN, contributes to immune suppression by promoting Treg differentiation (Grohmann and Puccetti, 2015). Activation of IDO1 by inflammatory signaling is of interest to the immune-oncology community due to immunosuppressive effects that result from depleting the local cellular environment of TRP (Hornyák et al., 2018; Labadie et al., 2019; Lee et al., 2010; M. Liu et al., 2018; Sforzini et al., 2019). Clinical trials of IDO or TDO inhibitors individually or in combination with inhibitors of immune checkpoint proteins—for example PD-1 (Nivolumab) (Bristol-Myers Squibb, 2011, 2012a) or CTLA-4 (Ipilimumab) (Bristol-Myers Squibb, 2012b)—demonstrate the efficacy of these drugs against metastatic clear-cell renal carcinoma and unresectable or advanced melanoma (Li et al., 2019; Stein et al., 2019). Overexpression of IDO1 is associated with poor patient survival in cancer patients and co-treatment with the IDO1 inhibitor Epacadostat show promising results in phase III clinical trials (Komiya and Huang, 2018). Another ongoing clinical trial is evaluating the potential of IDO inhibition as a first line therapy for patients with liver cancer by blocking tumor growth and metastasis (Edward Kim, 2018). Vaccines against IDO peptides are well-tolerated in patients with metastatic melanoma (Inge Marie Svane, 2012) and non-small cell lung carcinomas (Inge Marie Svane, 2010), and significantly improve median survival in metastatic lung cancer patients (Iversen et al., 2014). IDO/TDO inhibition is also being pursued as adjuvant therapy. NewLink Genetics Corporation is conducting two clinical trials, one for metastatic breast cancer patients treated with docetaxel or paclitaxel in combination with the IDO inhibitor 1-methyl-D-tryptophan (1MT) (NewLink Genetics Corporation, 2013) and a second for pediatric progressive primary malignant tumors using temozolomide in combination with 1MT (NewLink Genetics Corporation, 2015); however, the results are not yet posted. Elevated de novo NAD+ production resulting from increased kynurenine activity may further promote tumor chemoresistance through increased activity of the NAD+-dependent poly(ADP-ribose) polymerase-1 (PARP1), which facilitates repair of DNA oxidative damage (Heng et al., 2016; Sahm et al., 2013).
Downstream of KYN, there is accumulating evidence that 3HAA has anti-inflammatory properties, perhaps acting as a feedback mechanism within the kynurenine pathway following activation of IDO1 by pro-inflammatory cytokines (Krause et al., 2011; Lee et al., 2016; Zhang et al., 2012). Multiple pre-clinical studies show promise for 3HAA as a treatment target in diseases with a primary inflammatory or autoimmune character: (1) Yan et al. (2010) found that 3HAA intraperitoneal (IP) injections reduced clinical severity in mice with autoimmune encephalomyelitis, a common model of multiple sclerosis (MS), by limiting cytokine production, including IL-6 and INFγ, and promoting a shift toward regulatory T cell fate determination; (2) Hayashi et al. (2007) found that intratracheal treatment with 3HAA reduced allergic airway hyper-responsiveness and inhibited both eosinophil infiltration and cytokine production (IL-5 and IL-13) in the bronchial alveolar lavage fluid of mice with experimentally-induced asthma; (3) recently, Parrott et al. (2016) demonstrated that Haao knockout mice are protected against behavioral depression and working memory impairment induced by an acute inflammatory response with lipopolysaccharide (LPS); and finally, (4) Zhang et al. (2012) found that 3HAA IP injections reduced atherosclerotic lesion size and markers of local and systemic inflammation in Ldlr−/− mice fed a high-fat diet.
Kynurenine metabolism in the brain.
The two major branches of the kynurenine pathway are segregated by cell type in the brain, with the KA branch active primarily in astrocytes and the NAD+ branch active primarily in microglia (Schwarcz et al., 2012). This localized expression pattern to the resident innate immune cells—including expression of IDO1—means that kynurenine pathway activity is correlated with the elevated neuroinflammation in many diseases of the central nervous system (CNS) (Sühs et al., 2019). Anti-inflammatory benefits of kynurenine pathway inhibition may be offset by reduced NAD+ production in the central nervous system due to the high energy demands. Indeed, Braidy et al. (2011a) found that inhibition of either IDO or QPRT decreased NAD+ levels in cultured primary astrocytes and neurons, lending some credibility to this concern.
The two terminal metabolites in the major branches of the kynurenine pathway have opposing neuroactive properties that have made them a focal point for research into neurological consequences of altered kynurenine metabolism. KA is an antagonist for both α7 nicotinic acetylcholine (α7nACh) and N-methyl-D-aspartate (NMDA) receptors, while QA is and NMDA receptor agonist. 3HK levels are selectively increased in striatum, cortex and cerebellum of Hungtington’s disease (HD) mouse models, potentially linked to neuronal loss and reactive oxygen species (ROS) formation (Guidetti et al., 2006). Thus, the ratio between KA and 3HK+QA has been of interest in brain research, and impairing this balance is associated with dysfunctional or vulnerable neurons. Schwarcz et al. (2012) propose a model in which reducing the KA/3HK+QA ratio may benefit cognitive diseases like schizophrenia, while increasing the KA/3HK+QA may improve outcomes in age-associated neurodegeneration.
Kynurenine metabolism in other tissues.
Outside of the immune system, expression of kynurenine pathway genes is highest in liver, kidney, and pancreas, particularly the enzymes HAAO, KYNU, QPRT and ACMSD (Lim et al., 2013; Zheng et al., 2019). Entry of TRP into the kynurenine pathway in these tissues is controlled primarily by the non-immune responsive TDO, rather than IDO1 (Labadie et al., 2019). Liver is the major site of de novo NAD+ synthesis from TRP through the kynurenine pathway, and the likely primary target for kynurenine pathway interventions designed to alter systemic NAD+ production (Katsyuba et al., 2018; Okabe et al., 2019).
Tissue-specific processes related to kynurenine pathway activity depend not just on the levels of kynurenine metabolites or enzymes in that tissue, but also on the relationship of these components to other pathways. For instance, IDO1 expression is induced by pro-inflammatory cytokines, while TDO expression is induced by corticosteroids and glucagon (Lestage et al., 2002) (though there is evidence that TDO expression can be indirectly induced by inflammation through activation of the glucocorticoid receptor (Walker et al., 2013)). The kynurenine metabolite XA can inhibit insulin/IGF-1 signaling in pancreatic islets, while suppression of endogenously synthesized XA by long-term administration of pyridoxine results in minimal glycaemia and a less pronounced decrease in insulin (Meyramov et al., 2015; Oxenkrug, 2015). In an alternative metabolic route to producing NAD+, ACMSA can be enzymatically converted to AMSA, which is subsequently converted to either picolinic acid, which has not been extensively studied, or glutaryl-CoA, which regulates glycolysis among other processes in the cell (Davis et al., 2018; Palzer et al., 2018) (Figure 1).
1.2. NAD+ metabolism in aging and disease
Cells produce NAD+ through one of three metabolic pathways (Figure 1). The kynurenine pathway is the sole route for de novo NAD+ synthesis (from ingested TRP). Alternatively, cells can produce NAD+ from nicotinic acid (NA) via the Preiss-Handler pathway, or from nicotinamide riboside (NR) through the salvage pathway. The NAD+ branch of the kynurenine pathway concludes with the production of quinolinic acid (QA), which is converted into nicotinic acid mononucleotide (NAMN) by the enzyme quinolate phosphoribosyltransferase (QPRT). NAMN is converted to nicotinic acid adenine dinucleotide (NAAD) through a reaction catalyzed by NAMN adenylyltransferases (NMNATs). The metabolite NAAD is converted to NAD+ by the glutamine-dependent NAD+ synthetase (NADSYN) (Katsyuba et al., 2018). Cells can also generate NAD+ from nicotinic acid (NA) through the Preiss-Handler pathway, or from nicotinamide riboside (NR) through the salvage pathway. In the Preiss-Handler pathway, NA is converted by the enzyme nicotinate phosphoribosyltransferase (NAPRT) to NAMN, where it converges with de novo synthesis. In the salvage pathway, NR is converted to nicotinamide mononucleotide (NMN) by nicotinamide riboside kinases (NMRKs), and then to NAD+ by NMNATs.
The Preiss-Handler and salvage pathways generate NAD+ by recycling the nicotinamide (NAM) produced when NAD+ is consumed by one of a variety of NAD+-dependent enzymes (e.g. ART1, CD38, PARP1, PARG1, SARM1, SIRT1–7). Which pathway recycles NAM to NAD+ differs by genetic lineages. Mammalian genomes contain the enzyme nicotinamide phosphoribosyltransferase (NAMPT), which converts nicotinamide (NAM) to nicotinamide mononucleotide (NMN), but not the enzyme nicotinamides (NAMase), which converts NAM to NA. The invertebrate C. elegans and D. melanogaster genomes contain NAMase but not NAMPT. Thus mammals recycle NAM through the salvage pathway, while invertebrates recycle NAM through the Preiss-Handler pathway.
One proposed set of models implicates NAD+ degradation as a primary driver of NAD+ decline with age. The two main products of the hydrolysis-mediated degradation of NAD+ are ADP-ribose and nicotinamide (NAM). ADP-ribose is consumed during post-translational modification of proteins by PARPs, producing the concatenated poly(ADP-ribose). NAD+ hydrolysis can be caused in vitro by thermal degradation at higher temperatures or very-low pH (Hachisuka et al., 2017; Oppenheimer, 1994) or enzymatically mediated by any of the NAD+-consuming enzymes previously mentioned above. Among these, both the ADPase CD38 and PARP1 have been implicated in the age-associated depletion of NAD+. Increased PARP1 activity following age-associated accumulation of DNA damage has been proposed as a primary driver of NAD+ decline (Imai and Guarente, 2014). In support of this idea, deletion of Parp1 in mice or pharmacological inhibition of PARP1 in cells both lead to elevated NAD+ levels (Bai et al., 2011). However, both increased and decreased PARP1 activity have been observed during normal aging or in progeroid syndromes (Bakondi et al., 2011; Braidy et al., 2011b; Noren Hooten et al., 2012; Scheibye-Knudsen et al., 2014; Zhang et al., 2014), suggesting that the role of elevated PARP1 activity in age-associated NAD+ decline may be context-dependent. The role of PARP1 in aging and longevity has yet to be fully explored, and potential benefits in NAD+ availability and metabolic function resulting from PARP1 inhibition may be offset by reduced DNA-repair capacity. CD38 is likely the major NADase in mammalian tissue (Aksoy et al., 2006; Young et al., 2006) and deletion (Barbosa et al., 2007; Chiang et al., 2015) or pharmacological inhibition (Escande et al., 2013; Haffner et al., 2015) of CD38 dramatically increases NAD+ levels in normal and obese mice, respectively. One recent study reports that CD38 expression and activity both increase with age in mice, and that CD38 was required for the age-dependent decline in NAD+ (Camacho-Pereira et al., 2016). They further report an increase in CD38 expression in human adipose tissue (Camacho-Pereira et al., 2016). Like PARP1, potential benefits of CD38 inhibition in the context of NAD+ availability may be counteracted by negative impacts on other processes, including neurological function related to social behavior (Lopatina et al., 2012) and immune function (Partida-Sánchez et al., 2001), and more research is needed to fully clarify the role of CD38 in aging and longevity. The relationship between aging, NAD+ degradation, PARP1, and CD38 is discussed in greater detail elsewhere(Aman et al., 2018; Chini et al., 2017; Rajman et al., 2018).
NAD+ is both an energy carrier and an enzyme cofactor that plays a critical role in regulating cellular metabolism in eukaryotic cells and is therefore involved in many fundamental biological processes in cell signaling, regulation of gene expression, DNA repair pathways, and protein homeostasis. Many individual reactions in the Krebs cycle (aka the tricarboxylic acid cycle) and glycolysis are tightly regulated by the bioavailability of NAD+. Glycolysis requires NAD+ for the activity of the enzymes glyceraldehyde-3phosphate dehydrogenase (G3PDH), lactate dehydrogenase (LDH), and the pyruvate dehydrogenase (PDH) complex. The TCA cycle requires NAD+ malate dehydrogenase (MDH), a-ketoglutarate dehydrogenase (α-KGDH), and isocitrate dehydrogenase (IDH), and in regulating complex I (Yang and Sauve, 2016).
NAD+ deficiency during aging and age-associated disease.
Deficiency in NAD+ has been implicated in human disease ranging from congenital defects (Shi et al., 2017) to a range of age-associated pathologies—diabetes, cerebral and myocardial ischemia, neurodegeneration (Johnson and Imai, 2018; Zhang and Ying, 2019). Consistent with these links to disease, NAD+ levels decline with age and this decline has been implicated in many of the “hallmarks of aging” (as defined by López-Otín et al. (2013)), including epigenetic alterations, DNA damage/genomic instability, and mitochondrial dysfunction (Aman et al., 2018). One apparent cause for this decline in mammals is an age-associated decline in salvage pathway activity resulting from decreasing NAMPT expression at both the mRNA and protein level across multiple tissues (Stein and Imai, 2014; Yoshino et al., 2011). Exacerbating the decline in NAD+ production, levels of NAD+-consuming enzymes, such as CD38 and PARP1 (discussed above), increase with age in multiple tissues as well in the context specific diseases (Yang and Sauve, 2016). NAD+ homeostasis during aging is thus challenged by both decreasing production and increasing consumption.
Like kynurenine metabolism, NAD+ decline and the severity of the downstream consequences will be different in different tissues. For example, tissues with more active mitochondrial metabolism, such as brain or liver, may be more sensitive to declining NAD+. Specific ablation of the NAD+ biosynthetic enzyme NAMPT recapitulates the same decline in hippocampal NAD+ levels and NAMPT enzyme that occurs naturally during age (Stein and Imai, 2014). Neural stem/progenitor cell (NSPC) proliferation and self-renewal is impaired in adult NSPC-specific tamoxifen-inducible Nampt-knockout (Stein and Imai, 2014). Hepatic NAD+ levels also decreased with age in mice and humans by compromised dysfunction of NAMPT-mediated NAD+ salvage pathway. Deficiency in liver NAD+ pools impairs lipid homeostasis and induces moderate inflammation and fibrosis in a diet-induced non-alcoholic fatty liver disease (NAFLD) mouse model (Zhou et al., 2016).
Strategies to combat age-associated NAD+ decline with age.
Developing strategies to combat this decline in NAD+ availability with age is an ongoing goal of aging research directed at NAD+ metabolism. Recent studies demonstrate that supplementation with NAD+ or NAD+-precursors is sufficient improve health and promote longevity. Several studies have now reported lifespan extension or other health benefits from these supplements in both invertebrate and mammalian models.
In C. elegans, supplementation with NAD+ (Hashimoto et al., 2010), NR (Fang et al., 2016; Mouchiroud et al., 2013), NA (Schmeisser et al., 2013), or NAM (Mouchiroud et al., 2013; Schmeisser et al., 2013) significantly boosts NAD+ levels and extends lifespan. Mouchiroud et al. (2013) further show that elevating NAD+ by inhibiting the NAD+-consuming enzyme PARP with AZD2281 or ABT-888 also extends lifespan in C. elegans. The longevity benefits of NAD (Hashimoto et al., 2010), NR (Mouchiroud et al., 2013), and AZD2281 (Mouchiroud et al., 2013), but not NAM (Schmeisser et al., 2013), all required functional sir-2.1. NAD+ (Hashimoto et al., 2010), NA (Schmeisser et al., 2013), and AZD2281 (Mouchiroud et al., 2013), but not NR (Mouchiroud et al., 2013), required the FOXO-family transcription factor, DAF-16, for lifespan extension. Both AZD2281 and NR promoted DAF-16 activation (as measured by DAF-16 nuclear localization and expression of the transcriptional target, SOD-3), suggesting a role for insulin/IGF-1-like signaling in these benefits (Mouchiroud et al., 2013). All supplements provided protection against oxidative stress. These findings suggest that increased activity of NAD+-dependent enzymes and inhibition of insulin/IGF-1-like signaling each play a positive role in these benefits, but that there are likely subtle differences in the method for boosting NAD+ that are not yet well understood. In Drosophila, nicotinamidase (D-NAAM) overexpression extends lifespan (requiring the sirtuin ortholog Sir2) (Balan et al., 2008). Schmeisser et al. (2013) similarly report that overexpression of anmt-1, encoding NMNAT, is sufficient to extend lifespan. These results suggest that elevating NAD+ recycling from NAM through the Preiss-Handler pathway promotes longevity in invertebrates.
In mice, a single report to dates describes lifespan extension from a NAD+ precursor, NR, starting in 2 year old C57BL/6 mice (Zhang et al., 2016). The NR-supplemented mice also enjoyed improved muscle function and muscle stem cell retention. NR supplementation is now being evaluated for lifespan extension by the National Institute on Aging Interventions Testing Program (NIA ITP) in genetically heterogeneous UM-HET3 mice. Beyond lifespan extension, NR supplementation has been shown to improve or delay pathology in mouse models of numerous specific pathologies, including mitochondrial myopathy (Khan et al., 2014), HFD-induced obesity (Cantó et al., 2012), muscular dystrophy (Zhang et al., 2016), ataxia telangiectasia (Fang et al., 2016), and dilated cardiomyopathy (Diguet et al., 2018).
A second study found that NAM extended healthspan (as indicated by measures of glucose metabolism and oxidative stress in the liver, as well as motor control/behavior) of male mice fed a high-fat diet (HFD), but did not alter lifespan in male mice fed either a standard diet (SD) or HFD (Mitchell et al., 2018). Yoshida et al. (2019) employed a different approach to elevating NAD+, supplementing mice with extracellular NAMPT (eNAMPT), the enzyme regulating the first step in NAD+ salvage (Figure 1), to mice in extracellular vesicles. eNAMPT is contained exclusively in these vesicles in both mice and humans and levels decline with age. Mice supplemented with eNAMPT in this manner starting at 26 months of age had increased physical activity and increased lifespan relative to vehicle controls.
Mouse lifespan studies for NMN have yet to be reported; however, one study examined the impact of long-term NMN supplementation and found improvements in many areas of health, including protection against age-associated weight gain, improved insulin sensitivity and lipid profiles, improved eye function, increased bone mineral density, and increased immune function (Mills et al., 2016). NMN supplementation in mice has further been observed to protect against weight gain and changes in multiple metabolic measures in mice fed HFD (Uddin et al., 2017, 2016; Yoshino et al., 2011) and age-associated vascular dysfunction and oxidative stress (de Picciotto et al., 2016), cerebrovascular endothelial dysfunction (Tarantini et al., 2019), and susceptibility to acute kidney injury (Guan et al., 2017).
1.3. Mitochondrial function in aging and disease
Mitochondria are the major site for energy production in cells, but also serve as a hub for signaling, ROS production, and maintenance of cellular homeostasis. Like NAD+ metabolism, the mitochondria is a major focus of aging research and the topic of numerous detailed reviews (Giorgi et al., 2018; Kauppila et al., 2017; Srivastava, 2017; Sun et al., 2016; Theurey and Pizzo, 2018). Mitochondrial dysfunction—characterized by progressive changes in function, abundance, mitochondria DNA (mtDNA) mutation load, structure, and production of both energy and ROS—increases with age and has earned its own category in the “hallmarks of aging” (López-Otín et al., 2013). Mitochondrial dysfunction has further been causally implicated across a wide range of age-associated diseases, including neurodegeneration (Grimm and Eckert, 2017), cardiovascular disease (Kiyuna et al., 2018; Siasos et al., 2018), diabetes (Montgomery, 2019), and cancer (Porporato et al., 2018). Here we focus on aspects of mitochondrial dysfunction with links to kynurenine and NAD+ metabolism, specifically ROS production, turnover, and dynamics (Figure 2).
Figure 2. NAD+ synthesis and mitochondrial fitness.
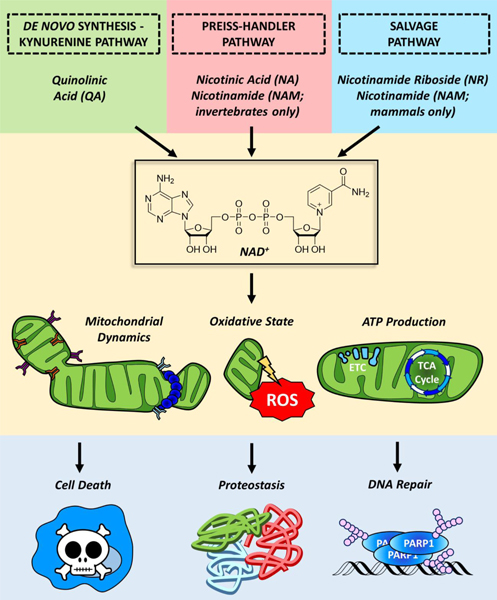
NAD+ is synthesized in the cell through the kynurenine/de novo biosynthetic pathway using quinolinic acid as a primary precursor. Cells also possess additional systems for producing NAD+ from alternative precursors. The Priess-Handler pathway generates NAD+ from nicotinic acid (NA) while the salvage pathway generates NAD+ from nicotinamide riboside (NR). Invertebrates recycle NAM generated from consuming NAD+ through the Priess-Handler pathway, while mammals recycle NAM through the salvage pathway. NAD+ regulates a variety of cellular process that modulates mitochondrial morphology, fitness, and function, which in turn impacts downstream processes including as cell death, proteostasis and DNA repair.
Mitochondria-derived reactive oxygen species.
The mitochondria, as the organelle that produces the vast majority of cellular ROS, plays a critical role in the regulating the generation and response to oxidative stress, as well as ROS-mediated communication with other organelles and the nucleus. Compared to nuclear DNA, mtDNA is particularly susceptible to oxidative damage because of both its proximity to the high levels of mitochondrial ROS production and its relatively poor defense against damage. Healthy mitochondria contribute to oxidative stress resistance by increasing respiratory capacity, increasing levels of NAD+, and producing ATP (Khan et al., 2017), which activates an ROS-dependent oxidative stress response mediated by the PI3K/Akt/ERK axis (Cruz et al., 2007). The antioxidant response is mediated, in part, by the forkhead box transcription factor, FOXO3A, which induces expression of the mitochondrial manganese superoxide dismutase, SOD2 (Wang et al., 2019), and further regulates several other aspects of mitochondrial function including mitochondrial abundance (measured by mtDNA copy number), expression of nuclear encoded mitochondrial proteins, and the expression and activity of respiratory complexes (Ferber et al., 2012). While early theories placed ROS squarely at the mechanistic center of aging, recent evidence suggests that this model is overly simplistic. While high-levels of ROS production likely drive age-associated decline though damage to macromolecules, lowlevels of ROS can produce a net benefit by activating systemic oxidative stress pathways in a process termed “mitohormesis” (Bárcena et al., 2018; Gonzalez-Freire et al., 2015; Ristow and Schmeisser, 2014). Both kynurenine- and NAD+-targeted interventions impact mitochondrial ROS production and related signaling pathways, as we discuss further in Section 2.2.
Mitochondrial integrity and turnover.
Due to its bacterial origin, mitochondria contain their own genome in the form of a extranuclear double-stranded circular DNA (mtDNA) ~16,500 bp in size and encoding 37 genes, 22 tRNAs, 13 proteins, and 2 rRNAs (Krishnan et al., 2007). Only about 1% of the ~1,200 proteins required for the normal function of mitochondria are encoded by the mtDNA. As previously noted, unlike the nuclear genome, mtDNA lacks of protective histones and enjoys less efficient DNA repair mechanisms (López-Otín et al., 2013), resulting in a relatively high mutation rate in mtDNA. Point and deletion mutations in mtDNA are associated with human longevity (De Benedictis et al., 1999) and accumulate in different tissues with age, and mtDNA mutation is thought to be one driver of mitochondrial dysfunction and downstream pathology with age.
One mechanism employed by cells to maintain mitochondrial fitness is mitochondrial recycling. Recycling is mediated by mitochondrial biogenesis and mitophagy; the latter removes damaged mitochondria while the former replicates functional mitochondria. The result is a cellular complement of mitochondria with improved efficiency and reduced ROS production. NAD+-levels promote mitochondrial biogenesis via SIRT1-mediated activation of PGC-1α (Cantó et al., 2009), while accumulation of mtDNA mutations may deplete mitochondrial NAD+ pools through high PARP activity (Clark-Matott et al., 2015).
Mitochondria morphology and dynamics.
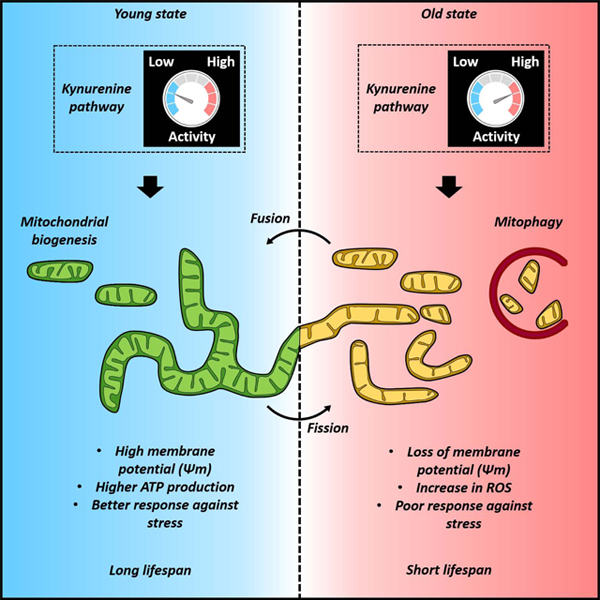
The function and turnover of mitochondria are tightly regulated by changes in its morphology and 3D structure. Morphological regulation occurs as a normal process in the cell, for example during progression through the cell cycle. Before the cell divides, it must segregate the mitochondria into small segments by a process called fission. When the two daughter cells are formed, the mitochondria tend to reassemble into the previous network-like morphology by a process called fusion. Outside the context of division, cells alter mitochondrial morphology in response to numerous molecular queues from the environment (Wai and Langer, 2016). Fission is promoted by excess nutrients, severe cellular stress and dysfunction (e.g. during cancer and obesity), and impaired oxidative phosphorylation. The resulting fragmented mitochondria are generally associated with metabolic dysfunction and disease, and are more susceptible to mitophagy (Weir et al., 2017). Fusion is promoted by nutrient withdrawal, mild stress, and increased oxidative phosphorylation. Hyperfused mitochondria are protected from mitophagy and thought to preserve cellular integrity in response to metabolic stress and other insults.
Mitochondrial fusion and fission events are tightly regulated by a small number of proteins that bind to the mitochondrial membrane and regulate physical changes to the membranes that govern the interconnectivity of the mitochondrial network. Dynamin 1 Like (DNM1L), also referred as dynamin-related protein 1 (DRP1), is a GTPase member of the dynamin protein superfamily. DRP1 promotes mitochondrial fragmentation by binding to and constricting the outer mitochondrial membrane (OMM) in a process similar to cytokinesis. As a consequence of this restriction, the mitochondria is segregated into two smaller segments. A second protein, Fission Mitochondrial 1 (FIS1), is anchored to the OMM and recruits DRP1. On the fusion side, the GTPases OPA1—localized to the inner mitochondrial membrane (IMM)—and mitofusin 1 and 2 (MFN1/2)—localized to the OMM—act in concert to bind and fuse the membranes of two mitochondrial segments (Byrne et al., 2019; Weir et al., 2017).
Mitochondrial dynamics have been directly implicated in aging. In yeast, promoting mitochondrial fusion by deleting of the DNM1L ortholog DNM1 extends replicative lifespan in a manner dependent on the presence of OPA1 ortholog MGM1 (Bernhardt et al., 2015). In Drosophila, promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila (Rana et al., 2017). In C. elegans, knocking out drp-1 extends lifespan, but only in a context where insulin signaling is impaired (Yang et al., 2011). Dysfunctional mitochondrial dynamics have been associated with age-associated disease and healthspan in mammals (reviewed by Sebastián et al. (2017)), though the ability to promote longevity by targeting mitochondrial fission or fusion has yet to be demonstrated. As discussed in Section 2.2, interventions in kynurenine or NAD+ metabolism that increase lifespan have been observed to alter mitochondrial dynamics.
2. Linking kynurenine metabolism to NAD+ and mitochondrial function
Mechanistic work on the role of the kynurenine pathway in disease has focused on the role of TRP and other TRP-related processes (Cervenka et al., 2017), the immune-responsive and immunomodulatory role of TRP depletion or intermediate pathway metabolites (Wang et al., 2015), the interplay between distinct neuroactive kynurenine metabolites (Schwarcz et al., 2012), or the pro- or anti-oxidant properties of intermediate kynurenine pathway metabolites (González Esquivel et al., 2017). With the recent resurgence of NAD+ and related processes as major targets in aging and age-associated disease (Imai and Guarente, 2014; Johnson and Imai, 2018), the kynurenine pathway’s alter ego as the de novo NAD+ synthesis pathway has increased in prominence as a mechanistic mediator of kynurenine-based interventions (Figure 2). Kynurenine metabolism influences NAD+-related processes directly by modifying NAD+ production. NAD+, in turn, regulates the TCA cycle and mitochondrial function, the epigenetic landscape (through modulation of sirtuin activity), DNA repair (through regulation of PARPs), and the hypoxic response (by tuning the energetic state of the cell). Intermediate kynurenine pathway activity can also influence NAD+ metabolism and mitochondrial function by modulating levels of ROS. Here we discuss potential models linking kynurenine and NAD+ in the context of aging and age-associated disease, and published evidence in support of these models.
2.1. The impact of altered kynurenine pathway activity on NAD+ production
The observations that supplementing NAD+ or NAD+-precursors can extend lifespan in worms (Fang et al., 2016; Hashimoto et al., 2010; Mouchiroud et al., 2013; Schmeisser et al., 2013) and mice (Zhang et al., 2016) suggests that increasing de novo NAD+ production by increasing metabolic flux through the kynurenine pathway should also increase lifespan. Consistent with this model, supplementing C. elegans with TRP has reported to elevate NAD+ levels (Katsyuba et al., 2018) while both increasing lifespan in wild type C. elegans (Edwards et al., 2015; Katsyuba et al., 2018) and delay pathology in C. elegans models of α-synuclein toxicity (van der Goot et al., 2012). Even more compelling, knockdown of the gene acsd-1, encoding ACMSD, shifts metabolism of ACMSA toward QA, resulting in elevated NAD+ production and increased lifespan (Katsyuba et al., 2018). Complicating the picture, knocking down either tdo-2 (encoding TDO) or kynu-1 (encoding KYNU)—and thus blocking de novo metabolism of NAD+ from TRP (Figure 1)—also increases lifespan to a similar or greater degree than TRP supplementation or acsd-1 knockdown (Sutphin et al., 2017; van der Goot et al., 2012). One solution to this apparent paradox would be a compensatory upregulation of Preiss-Handler or salvage pathway activity in response. A second possibility is that TRP, which accumulates when tdo-2 is knocked down (van der Goot et al., 2012), and KYN or 3HK, which accumulate when kynu-1 is knocked down (Sutphin et al., 2017), have prolongevity properties independent of NAD+ function. These possibilities are not mutually exclusive, which may suggest that combining TDO or KYNU inhibition with an NAD+ precursor may produce synergistic benefits (discussed further below). These models have yet to be tested.
The impact of interventions targeting one or more components of kynurenine and NAD+ metabolism is likely to be tissue-dependent. While a subset of the enzymes in these pathways are widely expressed, the major sites of kynurenine activity are liver, kidney, and the immune system. In the central nervous system, the KA- and NAD+-producing branches of the pathway are largely segregated to astrocytes and microglia, respectively, and only a subset of kynurenine pathway metabolites (TRP, KYN, 3HK) readily cross the blood brain barrier (Schwarcz et al., 2012). The entry of TRP into the kynurenine pathway is mediated by distinct tissue-expression patterns of IDO1 (primarily immune system), IDO2 (wide, low-level expression), and TDO2 (primarily liver) (Cervenka et al., 2017). The primary NAD+ precursor in the kynurenine pathway, QA, is largely not retained in liver, even after TRP loading, suggesting rapid processing to NAD+ in this tissue (L. Liu et al., 2018). QA does accumulate in activated immune cells and may act as reservoir for local NAD+ production, providing substrate for PARP activity needed to combat DNA damage from increased oxidative damage during immune activity (Moffett and Namboodiri, 2003). Alternatively, immune cells may excrete QA and utilize its pro-oxidant properties to attack invading pathogens (Heyes et al., 1995). This inherent complexity is a double-edged sword, providing numerous potential intervention targets for disease in different tissues while elevating the risk for unintended side-effects.
2.2. NAD+ synthesis and reactive oxygen species link kynurenine metabolism to modulation of mitochondrial function and morphology
The role of kynurenine metabolism in de novo NAD+ production provides one avenue for kynurenine pathway interventions to influence mitochondria function. A second potential link exists in the oxidant properties of intermediate kynurenine pathway metabolites (e.g. 3HK and 3HAA).
Cellular NAD+ levels affect key aspects of mitochondrial function, including ATP production, mitochondrial dynamics, and the production of ROS (Figure 2). As an important co-factor of the ETC and the TCA cycle, NAD+-levels affect ATP production by providing a necessary substrate for critical reactions in these processes. In C. elegans, elevating NAD+ by supplementing an NAD+ precursor (NR or NAM) (Mouchiroud et al., 2013), pharmacologically inhibiting PARP (Mouchiroud et al., 2013), or knocking down acsd-1 (Katsyuba et al., 2018) both extends lifespan and improves mitochondrial function, as measured by increased oxygen consumption, mtDNA content, electron transport chain gene expression, and ATP content. NAD+ precursor supplementation and PARP1 inhibition further produced temporal changes in mitochondrial dynamics and related processes, increasing mitochondrial fission and the mitochondrial unfolded protein response (UPRmt) in the short-term, and shifting toward hyper-fused mitochondria with increased oxidative stress resistance while maintaining elevated UPRmt in the long-term (Mouchiroud et al., 2013). These changes in mitochondrial dynamics were driven by changes in expression of fusion proteins OPA1 and MFN1/2, encoded by opa-1 (aka eat-3) and fzo-1, respectively, rather than expression of fission protein DRP1 (encoded by drp-1) (Mouchiroud et al., 2013). These effects of NAD+ on mitochondria were largely recapitulated in mammalian cells (Katsyuba et al., 2018; Mouchiroud et al., 2013). Supporting these observations, boosting NAD+ levels by NR supplementation in a mouse model of mitochondrial myopathy displaying a pseudo-starvation response, even when mice were well-fed, and delayed disease progression in by elevating mitochondrial biogenesis, reducing mitochondrial structural abnormalities, preventing mtDNA deletions, and stimulating the mitochondrial unfolded protein response (Khan et al., 2014).
NR and NMN supplementation have both been shown to reverse multiple aspects of mitochondrial dysfunction in a mouse model of ataxia telangiectasia (Fang et al., 2016), with NR enhancing survival. The observed shift in mitochondrial structure in the short- vs. long-term response to elevated NAD+ hints at potentially critical aspects of temporal mitochondrial dynamics that have yet to be explored in detail. Uddin et al. (2016) showed that NMN supplementation increased NAD+ levels in muscle and liver, ameliorated HFD-induced reduction of citrate synthase activity, and improved glucose tolerance in 5 month old mice, potentially by regulation of mitochondrial biogenesis and mtDNA copy number. A later study by the same group demonstrated that NMN can reverse HFD-induced gain in fat mass, improve glucose tolerance, and increase mitochondrial activity and fat catabolism (Uddin et al., 2017).
While these studies indicate that NAD+ can influence cellular stress response and longevity via changes in mitochondria structure and function, the mechanism by which NAD+ influences mitochondrial processes, and the implications for kynurenine-based interventions, remain to be fully explored. In the context of mitochondrial recycling, NAD+ can indirectly impact mitochondrial biogenesis through SIRT1. In mouse primary muscle cells, AMPK stimulates SIRT1 activity by elevating cellular NAD+ levels, promoting SIRT1 activity. SIRT1 deacetylates PGC-1α, which stimulates mitochondria biogenesis (Cantó et al., 2009). As a second possible mechanism, NAD+ may influence the fission and fusion processes by modulating ATP content, and downstream generation of other energy-related molecules such as GTP. OPA1 and DRP1 are mitochondrial GTPases required to maintain the mitochondrial cycle between fused and fragmented mitochondria (Long et al., 2017). Interventions that inhibit de novo NAD+ synthesis (e.g. inhibition of TDO or KYNU) may produce a state of “energy stress” by limiting available NAD+, thus promoting mitochondrial fission as a compensatory mechanism to promote resistance against oxidative stress. While this remains speculation, this resistance may mediate, at least in part, the prolonged lifespan observed in C. elegans (Sutphin et al., 2017; van der Goot et al., 2012) and Drosophila (Oxenkrug, 2010; Oxenkrug et al., 2011) subjected to genetic or pharmacological inhibition of the NAD+ branch of the kynurenine pathway, as well as the improved pathology observed in response to KMO or KYNU inhibition in mouse models of neurodegeneration (Zwilling et al., 2011).
Another potential benefit from kynurenine pathway interventions, besides regulating NAD+ synthesis, is the generation of antioxidant metabolites such as 3HAA and the reduction of pro-oxidant metabolites as QA. Elevated 3HAA may act as an ROS scavenger and work in conjunction with altered NAD+ levels to promote a healthy mitochondria. 3HK, like 3HAA, has been shown to have antioxidant properties in silico, in vitro, and in vivo, reducing lipid peroxidation in rat cerebral cortex and C6 glioma cells (Christen et al., 1990; Leipnitz et al., 2007; Zhuravlev et al., 2016). In contrast, QA has potent pro-oxidant properties, generating ROS via the Fenton reaction catalyzed by complex formation with iron ([Fe(III)]) (Kubicova et al., 2013).
2.3. Multiple mechanistic links to aging open the possibility for synergy from combined interventions
The observation that inhibition of kynurenine pathway activity—thus reducing de novo NAD+ production from TRP—and NAD+ precursor supplementation both increase lifespan suggests that optimal benefit may be derived by combining one or more therapies (Figure 3). The benefits of increasing NAD+ production and on mitochondrial function and activation of NAD+-dependent enzymes may produce synergistic benefits with the NAD+-independent biological activity of intermediate kynurenine metabolites. KA and QA are neuroactive, modulating activity of α7nACh and NMDA receptors (Schwarcz et al., 2012) and GPR35 (Cervenka et al., 2017). These properties are of interest in treating various neurological disorders, including neurodegeneration (Schwarcz et al., 2012). Elevating local TRP levels by inhibiting IDO1, IDO2, or TDO has potential benefits in both promoting immune-surveillance of cancer cells (Cervenka et al., 2017) and combatting neurodegeneration (van der Goot et al., 2012; van der Goot and Nollen, 2013). The antioxidant properties of 3HK and 3HAA (Chobot et al., 2015; Christen et al., 1990; Leipnitz et al., 2007; Thomas et al., 1996; Zhuravlev et al., 2016)—which can convert to pro-oxidant depending on the concentration of metal ions and environmental pH (Goldstein et al., 2000; Pérez-González et al., 2017)–and the immunomodulatory activity of 3HK, 3HAA, and QA (Krause et al., 2011) have potential benefits in a wide range of age-associated disease.
Figure 3. Cellular and molecular mechanisms regulated by the kynurenine -NAD+-mitochondria axis.
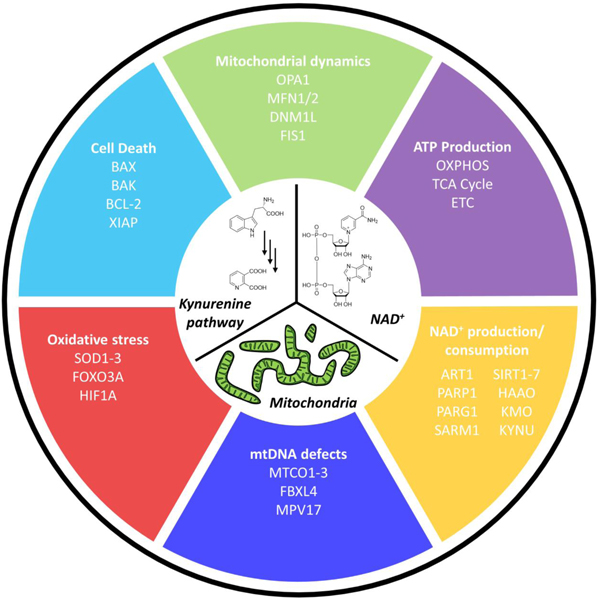
The kynurenine pathway and its interaction with NAD+ metabolism and mitochondrial fitness affect many cellular processes. Highlighted are processes and associated genes and systems with a known function in aging and age-associated disease. Oxidative phosphorylation (OXPHOS), TCA (tricarboxylic acid) cycle, electron transport chain (ETC), optic atrophy 1 (OPA1), mitofusin 1/2 (MFN1/2), dynamin-1 like (DNM1L), mitochondrial fission 1 (FIS1), kynurenine 3-monooxygenase (KMO), kynureninase (KYNU), 3-hydroxyanthranilate 3,4-dioxygenase (HAAO), superoxide dismutase 1–3 (SOD1–3), forkhead box O3 (FOXO3A), cytochrome c oxidase subunit 1–3 (MTCO1–3), F-box and leucine rich repeat 4 (FBXL4), mitochondrial inner membrane protein MPV17 (MPV17), ADP-ribosyltransferase 1 (ART1), Poly [ADP-ribose] polymerase 1 (PARP-1), Poly(ADP-ribose) glycohydrolase (PARG), Sterile Alpha and TIR Motif Containing 1 (SARM1), sirtuin 1–7 (SIRT1–7).
A straight-forward starting point would be to combine IDO inhibition—to prevent local TRP depletion and immune suppression—with an NAD+ precursor supplement to maintain high levels of NAD+ production in the absence of de novo synthesis through the kynurenine pathway and garner the benefits of increased NAD+ on mitochondrial function and activation of NAD+ dependent enzymes. Depending on the specific disease or tissue of interest, more complex therapies might include combinations of two or more of the following: targeted inhibition of one or more kynurenine pathway enzymes (in particular, TDO/IDO, KYNU, HAAO, or ACMSD), TRP supplementation, supplementation with one or more intermediate kynurenine pathway metabolite (in particular, 3HK, 3HAA, or KA), NAD+ precursor supplementation, drugs directly targeting one or more aspects of mitochondrial function (e.g. mitochondrial biogenesis).
The idea behind combining kynurenine inhibition with NAD supplementation has some support in the literature. Shi et al. (Shi et al., 2017) demonstrated that mice lacking either Kynu or Haao produce only inviable embryos when fed a diet lacking in niacin, suggesting that kynurenine metabolism is critical for normal development when a dietary NAD+ precursor is not present. Supplementing mice with NA rescued this phenotype. Feedback between different aspects of NAD+ production will also impact the optimal combination of interventions. For instance, Mitchell et al. (2018) observed that NAM supplementation in mice did not extend lifespan but did result in a rebalancing of hepatic NAD+ metabolism, suppressing salvage pathway expression while elevating de novo pathway enzymes.
3. Clinical evidence for targeting kynurenine metabolism in aging
As discussed in Section 1.1, most efforts to target kynurenine metabolism in a clinical setting use IDO/TDO inhibitors to re-sensitize cancer cells to immune-surveillance. Beyond this specific application in cancer, clinical interventions targeting kynurenine metabolism are lacking for treatment of age-associated pathology; however, kynurenine pathway components are getting some clinical attention in non-aging contexts. For example, metabolite levels of serotonin (5-HT), TRP, and KYN, as well as the enzyme activity of monoamine oxidases (MAO) and IDO, have been examined in the context of both septic shock (Versailles Hospital, 2004) and stroke (Versailles Hospital, 2012). A recent clinical trial is evaluating the ability of N-acetylcysteine to inhibit KYAT in patients with Schizophrenia, preventing the conversion of ingested TRP to KA and limiting the deleterious consequences of elevated KA on glutamate and dopamine signaling in this disease (University of Maryland, 2019). On the NAD+ front, NAD+, NA, NR, NAM, and NMN are all being tested in clinical trials for a variety of conditions, including a range of age-associated diseases, as are non-NAD+ compounds targeting various aspects of mitochondrial dynamics and biogenesis. To date, none of these studies is examining potential cross-over effects between kynurenine metabolism, NAD+ production, and mitochondrial function. This interplay remains ripe for both pre-clinical and clinical evaluation.
4. Conclusions and future directions
The role of NAD+ metabolism and mitochondrial function remain major areas of focus in aging research. Kynurenine metabolism is a more recent entrant to this stage, and mechanisms linking altered kynurenine pathway activity to longevity, healthy aging, and the onset and progression of age-associated disease are just beginning to emerge. The interplay between kynurenine metabolism, NAD+ production, and mitochondrial function in the context of aging has been examined by only a handful of studies to date, and we anticipate the coming years will see a more detailed examination across the spectrum of invertebrate and mammalian models. As evidence linking kynurenine metabolism to aging continues to grow, we anticipate expanded clinical interest in targeting pathway enzymes and metabolites for age-associated disease. Particularly promising is the prospect of combining interventions that target kynurenine, NAD+, and mitochondrial metabolism to achieve synergy and optimally increase healthy lifespan. For example, knockdown of intermediate kynurenine pathway enzymes, such as KYNU, HAAO and KMO, may beneficially increase specific metabolites in the pathway that exert antioxidant activity or initiate pro-health signaling pathways, with a concurrent detrimental decreased in NAD+ production. Combining this inhibition with NAD+ precursors may achieve the benefits of upregulating kynurenine pathway metabolites without the consequences of reducing NAD+ availability. Another approach to achieving additive or synergistic benefits may be to combine IDO/TDO inhibitors—maintaining local tryptophan levels and preventing T cell apoptosis—with direct supplementation of beneficial intermediate kynurenine metabolites or NAD+ precursors.
Highlights.
The kynurenine pathway has recently been identified as a promising target to increase healthy longevity.
Targeted inhibition of kynurenine pathway activity may alleviate several pathological conditions and promote healthspan.
Changes to the production and recycling of NAD+ is a likely mediator of the beneficial effects of kynurenine pathway interventions.
Mitochondrial function and dynamics represent NAD+-dependent processes downstream of kynurenine metabolism that may mediate benefits during aging.
Acknowledgments
This work was supported by a grant from the National Institute of General Medical Sciences of the National Institutes of Health (award number R35GM133588 to G. L. S.), a Glenn Foundation for Medical Research and American Federation for Aging Research (AFAR) Grant for Junior Faculty to G. L. S., a Pilot Award to G. L. S. from the University of Washington Nathan Shock Center for Excellence in the Basic Biology of Aging, and startup funding to G. L. S. through the Technology and Research Initiative Fund, which is administered by the Arizona Board of Regents.
Footnotes
Competing interests:
The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, Chini EN, 2006. Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem. Biophys. Res. Commun. 349, 353–359. 10.1016/j.bbrc.2006.08.066 [DOI] [PubMed] [Google Scholar]
- Aman Y, Qiu Y, Tao J, Fang EF, 2018. Therapeutic potential of boosting NAD+ in aging and age-related diseases. Transl. Med. Aging 2, 30–37. 10.1016/j.tma.2018.08.003 [DOI] [Google Scholar]
- Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier-de Murcia J, Auwerx J, 2011. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468. 10.1016/j.cmet.2011.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakondi E, Catalgol B, Bak I, Jung T, Bozaykut P, Bayramicli M, Ozer NK, Grune T, 2011. Age-related loss of stress-induced nuclear proteasome activation is due to low PARP-1 activity. Free Radic. Biol. Med. 50, 86–92. 10.1016/j.freeradbiomed.2010.10.700 [DOI] [PubMed] [Google Scholar]
- Balan V, Miller GS, Kaplun L, Balan K, Chong Z-Z, Li F, Kaplun A, VanBerkum MFA, Arking R, Freeman DC, Maiese K, Tzivion G, 2008. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J. Biol. Chem. 283, 27810–27819. 10.1074/jbc.M804681200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa MTP, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN, 2007. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 21, 3629–3639. 10.1096/fj.07-8290com [DOI] [PubMed] [Google Scholar]
- Bárcena C, Mayoral P, Quirós PM, 2018. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 340, 35–77. 10.1016/bs.ircmb.2018.05.002 [DOI] [PubMed] [Google Scholar]
- Baumgartner R, Forteza MJ, Ketelhuth DFJ, 2019. The interplay between cytokines and the Kynurenine pathway in inflammation and atherosclerosis. Cytokine 122, 154148. 10.1016/j.cyto.2017.09.004 [DOI] [PubMed] [Google Scholar]
- Bernhardt D, Müller M, Reichert AS, Osiewacz HD, 2015. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci. Rep. 5, 7885 10.1038/srep07885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Grant R, 2011a. Effects of Kynurenine Pathway Inhibition on NAD+ Metabolism and Cell Viability in Human Primary Astrocytes and Neurons. Int. J. Tryptophan Res. IJTR 4, 29–37. 10.4137/IJTR.S7052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R, 2011b. Age Related Changes in NAD+ Metabolism Oxidative Stress and Sirt1 Activity in Wistar Rats. PLoS ONE 6, e19194. 10.1371/journal.pone.0019194 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Squibb Bristol-Myers, 2012a. Study of Nivolumab (BMS-936558) vs. Everolimus in Pre-Treated Advanced or Metastatic Clear-cell Renal Cell Carcinoma (CheckMate 025) [WWW Document]. Clin. Internet Identifier NCT01668784. URL Available from: https://clinicaltrials.gov/ct2/show/NCT01668784?term=NCT01668784&draw=2&rank=1 [Google Scholar]
- Squibb Bristol-Myers, 2012b. PH 1 Biomarker Study of Nivolumab and Ipilimumab and Nivolumab in Combination With Ipilimumab in Advanced Melanoma (PD-1) [WWW Document]. Clin. Internet Identifier NCT01621490. URL https://clinicaltrials.gov/ct2/show/study/NCT01621490?term=NCT01621490&draw=2&r ank=1 [Google Scholar]
- Squibb Bristol-Myers, 2011. Phase I Biomarker Study (BMS-936558) [WWW Document]. Clin. Internet Identifier NCT01358721. URL Available from: https://clinicaltrials.gov/ct2/show/NCT01358721?term=NCT01358721&draw=2&rank=1 [Google Scholar]
- Byrne JJ, Soh MS, Chandhok G, Vijayaraghavan T, Teoh J-S, Crawford S, Cobham AE, Yapa NMB, Mirth CK, Neumann B, 2019. Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell. Mol. Life Sci. 76, 1967–1985. 10.1007/s00018-019-03024-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EN, 2016. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 23, 1127–1139. 10.1016/j.cmet.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J, 2009. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060. 10.1038/nature07813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, Auwerx J, 2012. The NAD+ Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity. Cell Metab. 15, 838–847. 10.1016/j.cmet.2012.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervenka I, Agudelo LZ, Ruas JL, 2017. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 357, eaaf9794. 10.1126/science.aaf9794 [DOI] [PubMed] [Google Scholar]
- Chang K-H, Cheng M-L, Tang H-Y, Huang C-Y, Wu Y-R, Chen C-M, 2018. Alternations of Metabolic Profile and Kynurenine Metabolism in the Plasma of Parkinson’s Disease. Mol. Neurobiol. 55, 6319–6328. 10.1007/s12035-017-0845-3 [DOI] [PubMed] [Google Scholar]
- Chiang S-H, Harrington WW, Luo G, Milliken NO, Ulrich JC, Chen J, Rajpal DK, Qian Y, Carpenter T, Murray R, Geske RS, Stimpson SA, Kramer HF, Haffner CD, Becherer JD, Preugschat F, Billin AN, 2015. Genetic Ablation of CD38 Protects against Western Diet-Induced Exercise Intolerance and Metabolic Inflexibility. PloS One 10, e0134927. 10.1371/journal.pone.0134927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini CCS, Tarragó MG, Chini EN, 2017. NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 455, 62–74. 10.1016/j.mce.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chobot V, Hadacek F, Weckwerth W, Kubicova L, 2015. Iron chelation and redox chemistry of anthranilic acid and 3-hydroxyanthranilic acid: A comparison of two structurally related kynurenine pathway metabolites to obtain improved insights into their potential role in neurological disease development. J. Organomet. Chem. 782, 103–110. 10.1016/j.jorganchem.2015.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen S, Peterhans E, Stocker R, 1990. Antioxidant activities of some tryptophan metabolites: possible implication for inflammatory diseases. Proc. Natl. Acad. Sci. 87, 2506–2510. 10.1073/pnas.87.7.2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark-Matott J, Saleem A, Dai Y, Shurubor Y, Ma X, Safdar A, Beal MF, Tarnopolsky M, Simon DK, 2015. Metabolomic analysis of exercise effects in the POLG mitochondrial DNA mutator mouse brain. Neurobiol. Aging 36, 2972–2983. 10.1016/j.neurobiolaging.2015.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz CM, Rinna A, Forman HJ, Ventura ALM, Persechini PM, Ojcius DM, 2007. ATP Activates a Reactive Oxygen Species-dependent Oxidative Stress Response and Secretion of Proinflammatory Cytokines in Macrophages. J. Biol. Chem. 282, 2871–2879. 10.1074/jbc.M608083200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis I, Yang Y, Wherritt D, Liu A, 2018. Reassignment of the human aldehyde dehydrogenase ALDH8A1 (ALDH12) to the kynurenine pathway in tryptophan catabolism. J. Biol. Chem. 293, 9594–9603. 10.1074/jbc.RA118.003320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Benedictis G, Rose G, Carrieri G, De Luca M, Falcone E, Passarino G, Bonafé M, Monti D, Baggio G, Bertolini S, Mari D, Mattace R, Franceschi C, 1999. Mitochondrial DNA inherited variants are associated with successful aging and longevity in humans. FASEB J. 13, 1532–1536. 10.1096/fasebj.13.12.1532 [DOI] [PubMed] [Google Scholar]
- de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, Imai SI, Seals DR, 2016. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15, 522–530. 10.1111/acel.12461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, Gouge A, Gressette M, Manoury B, Blanc J, Breton M, Decaux J-F, Lavery GG, Baczkó I, Zoll J, Garnier A, Li Z, Brenner C, Mericskay M, 2018. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 137, 2256–2273. 10.1161/CIRCULATIONAHA.116.026099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Edward, 2018. BMS-986205 and Nivolumab as First Line Therapy in Treating Patients With Liver Cancer [WWW Document]. Clin. Internet Identifier NCT03695250. URL https://clinicaltrials.gov/ct2/show/NCT03695250?term=NCT03695250&draw=2&rank=1 [Google Scholar]
- Edwards C, Canfield J, Copes N, Brito A, Rehan M, Lipps D, Brunquell J, Westerheide SD, Bradshaw PC, 2015. Mechanisms of amino acid-mediated lifespan extension in Caenorhabditis elegans. BMC Genet. 16, 8 10.1186/s12863-015-0167-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eleftheriadis T, Pissas G, Antoniadi G, Liakopoulos V, Tsogka K, Sounidaki M, Stefanidis I, 2016. Differential effects of the two amino acid sensing systems, the GCN2 kinase and the mTOR complex 1, on primary human alloreactive CD4+ T-cells. Int. J. Mol. Med. 37, 1412–1420. 10.3892/ijmm.2016.2547 [DOI] [PubMed] [Google Scholar]
- Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O’Neil L, White TA, Sinclair DA, Chini EN, 2013. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 62, 1084–1093. 10.2337/db12-1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S, Bollineni RC, Wilson MA, Iser WB, Wollman BN, Morevati M, Li J, Kerr JS, Lu Q, Waltz TB, Tian J, Sinclair DA, Mattson MP, Nilsen H, Bohr VA, 2016. NAD + Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 24, 566–581. 10.1016/j.cmet.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferber EC, Peck B, Delpuech O, Bell GP, East P, Schulze A, 2012. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 19, 968–979. 10.1038/cdd.2011.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, Marchi S, Simoes ICM, Ren Z, Morciano G, Perrone M, Patalas-Krawczyk P, Borchard S, Jędrak P, Pierzynowska K, Szymański J, Wang DQ, Portincasa P, Węgrzyn G, Zischka H, Dobrzyn P, Bonora M, Duszynski J, Rimessi A, Karkucinska-Wieckowska A, Dobrzyn A, Szabadkai G, Zavan B, Oliveira PJ, Sardao VA, Pinton P, Wieckowski MR, 2018. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 340, 209–344. 10.1016/bs.ircmb.2018.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LE, Leopold MC, Huang X, Atwood CS, Saunders AJ, Hartshorn M, Lim JT, Faget KY, Muffat JA, Scarpa RC, Chylack LT, Bowden EF, Tanzi RE, Bush AI, 2000. 3-Hydroxykynurenine and 3-hydroxyanthranilic acid generate hydrogen peroxide and promote alpha-crystallin cross-linking by metal ion reduction. Biochemistry 39, 7266–7275. [DOI] [PubMed] [Google Scholar]
- González Esquivel D, Ramírez-Ortega D, Pineda B, Castro N, Ríos C, Pérez de la Cruz V, 2017. Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 112, 331–345. 10.1016/j.neuropharm.2016.03.013 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Freire M, de Cabo R, Bernier M, Sollott SJ, Fabbri E, Navas P, Ferrucci L, 2015. Reconsidering the Role of Mitochondria in Aging. J. Gerontol. A. Biol. Sci. Med. Sci. 70, 1334–1342. 10.1093/gerona/glv070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm A, Eckert A, 2017. Brain aging and neurodegeneration: from a mitochondrial point of view. J. Neurochem. 143, 418–431. 10.1111/jnc.14037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann U, Puccetti P, 2015. The Coevolution of IDO1 and AhR in the Emergence of Regulatory T-Cells in Mammals. Front. Immunol. 6, 58 10.3389/fimmu.2015.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Wang S-R, Huang X-Z, Xie Q-H, Xu Y-Y, Shang D, Hao C-M, 2017. Nicotinamide Mononucleotide, an NAD+ Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. JASN 28, 2337–2352. 10.1681/ASN.2016040385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, Slow EJ, Wheeler VC, Woodman B, Schwarcz R, 2006. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol. Dis. 23, 190–197. 10.1016/j.nbd.2006.02.011 [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Smith DG, Smythe GA, Armati PJ, Brew GJ, 2003. Expression of The Kynurenine Pathway Enzymes in Human Microglia and Macrophages, in: Allegri G, Costa CVL, Ragazzi E, Steinhart H, Varesio L (Eds.), Developments in Tryptophan and Serotonin Metabolism Springer US, Boston, MA, pp. 105–112. 10.1007/978-1-4615-0135-0_12 [DOI] [PubMed] [Google Scholar]
- Hachisuka S, Sato T, Atomi H, 2017. Metabolism Dealing with Thermal Degradation of NAD+ in the Hyperthermophilic Archaeon Thermococcus kodakarensis. J. Bacteriol. 199 10.1128/JB.00162-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, Deaton DN, Guo Y, Harrington W, Henke BR, Jeune MR, Kaldor I, Milliken N, Petrov KG, Preugschat F, Schulte C, Shearer BG, Shearer T, Smalley TL, Stewart EL, Stuart JD, Ulrich JC, 2015. Discovery, Synthesis, and Biological Evaluation of Thiazoloquin(az)olin(on)es as Potent CD38 Inhibitors. J. Med. Chem. 58, 3548–3571. 10.1021/jm502009h [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Horikawa M, Nomura T, Sakamoto K, 2010. Nicotinamide adenine dinucleotide extends the lifespan of Caenorhabditis elegans mediated by sir-2.1 and daf-16. Biogerontology 11, 31–43. 10.1007/s10522-009-9225-3 [DOI] [PubMed] [Google Scholar]
- Hayashi T, Mo J-H, Gong X, Rossetto C, Jang A, Beck L, Elliott GI, Kufareva I, Abagyan R, Broide DH, Lee J, Raz E, 2007. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc. Natl. Acad. Sci. 104, 18619–18624. 10.1073/pnas.0709261104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng B, Lim CK, Lovejoy DB, Bessede A, Gluch L, Guillemin GJ, 2016. Understanding the role of the kynurenine pathway in human breast cancer immunobiology. Oncotarget 7 10.18632/oncotarget.6467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes MP, Saito K, Milstien S, Schiff SJ, 1995. Quinolinic acid in tumors, hemorrhage and bacterial infections of the central nervous system in children. J. Neurol. Sci. 133, 112–118. 10.1016/0022-510X(95)00164-W [DOI] [PubMed] [Google Scholar]
- Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, Halmos G, Székvölgyi L, 2018. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 9, 151 10.3389/fimmu.2018.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L, 2014. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471. 10.1016/j.tcb.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inge Marie Svane, 2012. Peptide Vaccine and Temozolomide for Metastatic Melanoma Patients [WWW Document]. Clin. Internet Identifier NCT01543464. URL https://clinicaltrials.gov/ct2/show/study/NCT01543464?term=NCT01543464&draw=2&r ank=1 [Google Scholar]
- Inge Marie Svane, 2010. IDO Peptid Vaccination for Stage III-IV Non Small-cell Lung Cancer Patients. (IDOvaccine) [WWW Document]. Clin. Internet Identifier NCT01219348. URL https://clinicaltrials.gov/ct2/show/study/NCT01219348?term=NCT01219348&draw=2&r ank=1 [Google Scholar]
- Iversen TZ, Engell-Noerregaard L, Ellebaek E, Andersen R, Larsen SK, Bjoern J, Zeyher C, Gouttefangeas C, Thomsen BM, Holm B, thor Straten P, Mellemgaard A, Andersen MH, Svane IM, 2014. Long-lasting Disease Stabilization in the Absence of Toxicity in Metastatic Lung Cancer Patients Vaccinated with an Epitope Derived from Indoleamine 2,3 Dioxygenase. Clin. Cancer Res. 20, 221–232. 10.1158/1078-0432.CCR-13-1560 [DOI] [PubMed] [Google Scholar]
- Johnson S, Imai S, 2018. NAD + biosynthesis, aging, and disease. F1000Research 7 10.12688/f1000research.12120.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsyuba E, Mottis A, Zietak M, De Franco F, van der Velpen V, Gariani K, Ryu D, Cialabrini L, Matilainen O, Liscio P, Giacchè N, Stokar-Regenscheit N, Legouis D, de Seigneux S, Ivanisevic J, Raffaelli N, Schoonjans K, Pellicciari R, Auwerx J, 2018. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 563, 354–359. 10.1038/s41586-018-0645-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppila TES, Kauppila JHK, Larsson N-G, 2017. Mammalian Mitochondria and Aging: An Update. Cell Metab. 25, 57–71. 10.1016/j.cmet.2016.09.017 [DOI] [PubMed] [Google Scholar]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsström S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, Suomalainen A, 2014. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 6, 721–731. 10.1002/emmm.201403943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, Kivelä R, Pessia A, Velagapudi V, Suomalainen A, 2017. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 26, 419–428.e5. 10.1016/j.cmet.2017.07.007 [DOI] [PubMed] [Google Scholar]
- Kim B-J, Hamrick MW, Yoo HJ, Lee SH, Kim SJ, Koh J-M, Isales CM, 2019. The Detrimental Effects of Kynurenine, a Tryptophan Metabolite, on Human Bone Metabolism. J. Clin. Endocrinol. Metab. 104, 2334–2342. 10.1210/jc.2018-02481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-K, Jeon SW, 2018. Neuroinflammation and the Immune-Kynurenine Pathway in Anxiety Disorders. Curr. Neuropharmacol. 16, 574–582. 10.2174/1570159X15666170913110426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyuna LA, Albuquerque R.P. e, Chen C-H, Mochly-Rosen D, Ferreira JCB, 2018. Targeting mitochondrial dysfunction and oxidative stress in heart failure: challenges and opportunities. Free Radic. Biol. Med. 129, 155–168. 10.1016/j.freeradbiomed.2018.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya T, Huang CH, 2018. Updates in the Clinical Development of Epacadostat and Other Indoleamine 2,3-Dioxygenase 1 Inhibitors (IDO1) for Human Cancers. Front. Oncol 8, 423 10.3389/fonc.2018.00423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause D, Suh H-S, Tarassishin L, Cui QL, Durafourt BA, Choi N, Bauman A, Cosenza-Nashat M, Antel JP, Zhao M-L, Lee SC, 2011. The Tryptophan Metabolite 3-Hydroxyanthranilic Acid Plays Anti-Inflammatory and Neuroprotective Roles During Inflammation. Am. J. Pathol. 179, 1360–1372. 10.1016/j.ajpath.2011.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KJ, Greaves LC, Reeve AK, Turnbull DM, 2007. Mitochondrial DNA Mutations and Aging. Ann. N. Y. Acad. Sci. 1100, 227–240. 10.1196/annals.1395.024 [DOI] [PubMed] [Google Scholar]
- Kubicova L, Hadacek F, Chobot V, 2013. Quinolinic Acid: Neurotoxin or Oxidative Stress Modulator? Int. J. Mol. Sci. 14, 21328–21338. 10.3390/ijms141121328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labadie BW, Bao R, Luke JJ, 2019. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan–Kynurenine–Aryl Hydrocarbon Axis. Clin. Cancer Res. 25, 1462–1471. 10.1158/1078-0432.CCR-18-2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Kwak J-H, Pyo S, 2016. Inhibition of LPS-induced inflammatory mediators by 3hydroxyanthranilic acid in macrophages through suppression of PI3K/NF-κB signaling pathways. Food Funct. 7, 3073–3082. 10.1039/C6FO00187D [DOI] [PubMed] [Google Scholar]
- Lee S-M, Lee Y-S, Choi J-H, Park S-G, Choi I-W, Joo Y-D, Lee W-S, Lee J-N, Choi I, Seo S-K, 2010. Tryptophan metabolite 3-hydroxyanthranilic acid selectively induces activated T cell death via intracellular GSH depletion. Immunol. Lett. 132, 53–60. 10.1016/j.imlet.2010.05.008 [DOI] [PubMed] [Google Scholar]
- Leipnitz G, Schumacher C, Dalcin KB, Scussiato K, Solano A, Funchal C, Dutra-Filho CS, Wyse ATS, Wannmacher CMD, Latini A, Wajner M, 2007. In vitro evidence for an antioxidant role of 3-hydroxykynurenine and 3-hydroxyanthranilic acid in the brain. Neurochem. Int. 50, 83–94. 10.1016/j.neuint.2006.04.017 [DOI] [PubMed] [Google Scholar]
- Lestage J, Verrier D, Palin K, Dantzer R, 2002. The enzyme indoleamine 2,3-dioxygenase is induced in the mouse brain in response to peripheral administration of lipopolysaccharide and superantigen. Brain. Behav. Immun. 16, 596–601. [DOI] [PubMed] [Google Scholar]
- Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bossé D, Lalani A-KA, Gopal S, Jin C, Horak C, Wind-Rotolo M, Signoretti S, McDermott DF, Freeman GJ, Van Allen EM, Schreiber SL, Stephen Hodi F, Sellers WR, Garraway LA, Clish CB, Choueiri TK, Giannakis M, 2019. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat. Commun. 10, 4346 10.1038/s41467-019-12361-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CK, Fernández-Gomez FJ, Braidy N, Estrada C, Costa C, Costa S, Bessede A, Fernandez-Villalba E, Zinger A, Herrero MT, Guillemin GJ, 2017. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 155, 76–95. 10.1016/j.pneurobio.2015.12.009 [DOI] [PubMed] [Google Scholar]
- Lim CK, Yap MMC, Kent SJ, Gras G, Samah B, Batten JC, De Rose R, Heng B, Brew BJ, Guillemin GJ, 2013. Characterization of the Kynurenine Pathway and Quinolinic Acid Production in Macaque Macrophages. Int. J. Tryptophan Res. 6, IJTR.S11789. 10.4137/IJTR.S11789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Su X, Quinn WJ, Hui S, Krukenberg K, Frederick DW, Redpath P, Zhan L, Chellappa K, White E, Migaud M, Mitchison TJ, Baur JA, Rabinowitz JD, 2018. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 27, 1067–1080.e5. 10.1016/j.cmet.2018.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, Li Y, 2018. Targeting the IDO1 pathway in cancer: from bench to bedside. J. Hematol. Oncol.J Hematol Oncol 11 10.1186/s13045-018-0644-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A, Klimova N, Kristian T, 2017. Mitochondrial NUDIX hydrolases: A metabolic link between NAD catabolism, GTP and mitochondrial dynamics. Neurochem. Int. 109, 193–201. 10.1016/j.neuint.2017.03.009 [DOI] [PubMed] [Google Scholar]
- Lopatina O, Inzhutova A, Salmina AB, Higashida H, 2012. The roles of oxytocin and CD38 in social or parental behaviors. Front. Neurosci. 6, 182 10.3389/fnins.2012.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G, 2013. The Hallmarks of Aging. Cell 153, 1194–1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton SD, Osiecki M, Woźniak Małgorzata, Cukrowska B, Wierzbicka A, Goliszek M, Socha P, Janczyk W, Dayanakli D, Abendroth D, Kramp S, Fechner K, Scheper T, Mahler M, Bentow C, Bogdanos D, Fuchs D, Woynarowski M, 2019. Tryptophan-kynurenine profile in pediatric autoimmune hepatitis. Immunol. Res. 67, 39–47. 10.1007/s12026-019-9068-1 [DOI] [PubMed] [Google Scholar]
- Mbongue JC, Nicholas DA, Torrez TW, Kim N-S, Firek AF, Langridge WHR, 2015. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines 3, 703–729. 10.3390/vaccines3030703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, Link CJ, Prendergast GC, 2012. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 1, 1460–1468. 10.4161/onci.21716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyramov GG, Kohnert К-D, Kikimbaeva АА, Аitkulov АМ, Кystaubaeva ZT, Тykeshanova GM, Dupont O-N, Laryushina ЕМ, Меyramova АG, Zhuzbaeva GO, Коvalenko ОL, Shaybek AS, 2015. Histological Changes in Pancreatic Islets of Animals with Experimental Diabetes Caused by Xanthurenic Acid under Condition of Suppression of Its Endogenous Synthesis. Bull. Exp. Biol. Med. 159, 680–684. 10.1007/s10517-015-3046-y [DOI] [PubMed] [Google Scholar]
- Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J, Imai S-I, 2016. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 24, 795–806. 10.1016/j.cmet.2016.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Bernier M, Aon MA, Cortassa S, Kim EY, Fang EF, Palacios HH, Ali A, Navas-Enamorado I, Di Francesco A, Kaiser TA, Waltz TB, Zhang N, Ellis JL, Elliott PJ, Frederick DW, Bohr VA, Schmidt MS, Brenner C, Sinclair DA, Sauve AA, Baur JA, de Cabo R, 2018. Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab. 27, 667–676.e4. 10.1016/j.cmet.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett JR, Namboodiri MA, 2003. Tryptophan and the immune response. Immunol. Cell Biol. 81, 247–265. 10.1046/j.1440-1711.2003.t01-1-01177.x [DOI] [PubMed] [Google Scholar]
- Montgomery MK, 2019. Mitochondrial Dysfunction and Diabetes: Is Mitochondrial Transfer a Friend or Foe? Biology 8 10.3390/biology8020033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo Y-S, Viswanathan M, Schoonjans K, Guarente L, Auwerx J, 2013. The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154, 430–441. 10.1016/j.cell.2013.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL, 2005. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22, 633–642. 10.1016/j.immuni.2005.03.013 [DOI] [PubMed] [Google Scholar]
- Nelp MT, Kates PA, Hunt JT, Newitt JA, Balog A, Maley D, Zhu X, Abell L, Allentoff A, Borzilleri R, Lewis HA, Lin Z, Seitz SP, Yan C, Groves JT, 2018. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. 115, 3249–3254. 10.1073/pnas.1719190115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NewLink Genetics Corporation, 2015. Study of the IDO Pathway Inhibitor, Indoximod, and Temozolomide for Pediatric Patients With Progressive Primary Malignant Brain Tumors [WWW Document]. Clin. Internet Identifier NCT02502708. URL https://clinicaltrials.gov/ct2/show/NCT02502708?term=NCT02502708&draw=2&rank=1 [Google Scholar]
- NewLink Genetics Corporation, 2013. Study of Chemotherapy in Combination With IDO Inhibitor in Metastatic Breast Cancer [WWW Document]. Clin. Internet Identifier NCT01792050. URL https://clinicaltrials.gov/ct2/show/NCT01792050?term=NCT01792050&draw=2&rank=1 [Google Scholar]
- Noren Hooten N, Fitzpatrick M, Kompaniez K, Jacob KD, Moore BR, Nagle J, Barnes J, Lohani A, Evans MK, 2012. Coordination of DNA repair by NEIL1 and PARP-1: a possible link to aging. Aging 4, 674–685. 10.18632/aging.100492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe K, Yaku K, Tobe K, Nakagawa T, 2019. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 26, 34 10.1186/s12929-019-0527-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M, 2011. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203. 10.1038/nature10491 [DOI] [PubMed] [Google Scholar]
- Oppenheimer NJ, 1994. NAD hydrolysis: Chemical and enzymatic mechanisms. Mol. Cell. Biochem. 138, 245–251. 10.1007/BF00928468 [DOI] [PubMed] [Google Scholar]
- Oxenkrug G, 2013. Insulin Resistance and Dysregulation of Tryptophan–Kynurenine and Kynurenine–Nicotinamide Adenine Dinucleotide Metabolic Pathways. Mol. Neurobiol. 48, 294–301. 10.1007/s12035-013-8497-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenkrug GF, 2015. Increased Plasma Levels of Xanthurenic and Kynurenic Acids in Type 2 Diabetes. Mol. Neurobiol. 52, 805–810. 10.1007/s12035-015-9232-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenkrug GF, 2010. The extended life span of Drosophila melanogaster eye-color (white and vermilion) mutants with impaired formation of kynurenine. J. Neural Transm. 117, 23–26. 10.1007/s00702-009-0341-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenkrug GF, Navrotskaya V, Voroboyva L, Summergrad P, 2011. Extension of life span of Drosophila melanogaster by the inhibitors of tryptophan-kynurenine metabolism. Fly (Austin) 5, 307–309. 10.4161/fly.5.4.18414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palzer L, Bader JJ, Angel F, Witzel M, Blaser S, McNeil A, Wandersee MK, Leu NA, Lengner CJ, Cho CE, Welch KD, Kirkland JB, Meyer RG, Meyer-Ficca ML, 2018. Alpha-Amino-Beta-Carboxy-Muconate-Semialdehyde Decarboxylase Controls Dietary Niacin Requirements for NAD+ Synthesis. Cell Rep. 25, 1359–1370.e4. 10.1016/j.celrep.2018.09.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott JM, Redus L, Santana-Coelho D, Morales J, Gao X, O’Connor JC, 2016. Neurotoxic kynurenine metabolism is increased in the dorsal hippocampus and drives distinct depressive behaviors during inflammation. Transl. Psychiatry 6, e918–e918. 10.1038/tp.2016.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, Randall TD, Lund FE, 2001. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 7, 1209–1216. 10.1038/nm1101-1209 [DOI] [PubMed] [Google Scholar]
- Pérez-González A, Alvarez-Idaboy JR, Galano A, 2017. Dual antioxidant/pro-oxidant behavior of the tryptophan metabolite 3-hydroxyanthranilic acid: a theoretical investigation of reaction mechanisms and kinetics. New J. Chem. 41, 3829–3845. 10.1039/C6NJ03980D [DOI] [Google Scholar]
- Pollizzi KN, Powell JD, 2015. Regulation of T cells by mTOR: the known knowns and the known unknowns. Trends Immunol. 36, 13–20. 10.1016/j.it.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato PE, Filigheddu N, Pedro JMB-S, Kroemer G, Galluzzi L, 2018. Mitochondrial metabolism and cancer. Cell Res. 28, 265–280. 10.1038/cr.2017.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast GC, Metz R, Muller AJ, Merlo LMF, Mandik-Nayak L, 2014. IDO2 in Immunomodulation and Autoimmune Disease. Front. Immunol. 5 10.3389/fimmu.2014.00585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajman L, Chwalek K, Sinclair DA, 2018. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 27, 529–547. 10.1016/j.cmet.2018.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Oliveira MP, Khamoui AV, Aparicio R, Rera M, Rossiter HB, Walker DW, 2017. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 8, 448 10.1038/s41467-017-00525-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravishankar B, Liu H, Shinde R, Chaudhary K, Xiao W, Bradley J, Koritzinsky M, Madaio MP, McGaha TL, 2015. The amino acid sensor GCN2 inhibits inflammatory responses to apoptotic cells promoting tolerance and suppressing systemic autoimmunity. Proc. Natl. Acad. Sci. U. S. A. 112, 10774–10779. 10.1073/pnas.1504276112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rejdak R, Junemann A, Grieb P, Thaler S, Schuettauf F, Chorągiewicz T, Żarnowski T, Turski WA, Zrenner E, 2011. Kynurenic acid and kynurenine aminotransferases in retinal aging and neurodegeneration. Pharmacol. Rep. 63, 1324–1334. 10.1016/S1734-1140(11)70697-1 [DOI] [PubMed] [Google Scholar]
- Ristow M, Schmeisser K, 2014. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose-Response 12, 288–341. 10.2203/dose-response.13-035.Ristow [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routy J-P, Mehraj V, Vyboh K, Cao W, Kema I, Jenabian M-A, 2015. Clinical Relevance of Kynurenine Pathway in HIV/AIDS: An Immune Checkpoint at the Crossroads of Metabolism and Inflammation. AIDS Rev. 17, 96–106. [PubMed] [Google Scholar]
- Routy J-P, Routy B, Graziani GM, Mehraj V, 2016. The Kynurenine Pathway is a Double-Edged Sword in Immune-Privileged Sites and in Cancer: Implications for Immunotherapy. Int. J. Tryptophan Res. 9, IJTR.S38355. 10.4137/IJTR.S38355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahm F, Oezen I, Opitz CA, Radlwimmer B, von Deimling A, Ahrendt T, Adams S, Bode HB, Guillemin GJ, Wick W, Platten M, 2013. The endogenous tryptophan metabolite and NAD+ precursor quinolinic acid confers resistance of gliomas to oxidative stress. Cancer Res. 73, 3225–3234. 10.1158/0008-5472.CAN-12-3831 [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan-Olive MM, Mangerich A, Wilson MA, Mattson MP, Bergersen LH, Cogger VC, Warren A, Le Couteur DG, Moaddel R, Wilson DM, Croteau DL, de Cabo R, Bohr VA, 2014. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855. 10.1016/j.cmet.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser K, Mansfeld J, Kuhlow D, Weimer S, Priebe S, Heiland I, Birringer M, Groth M, Segref A, Kanfi Y, Price NL, Schmeisser S, Schuster S, Pfeiffer AFH, Guthke R, Platzer M, Hoppe T, Cohen HY, Zarse K, Sinclair DA, Ristow M, 2013. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 9, 693–700. 10.1038/nchembio.1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Bruno JP, Muchowski PJ, Wu H-Q, 2012. Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477. 10.1038/nrn3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastián D, Palacín M, Zorzano A, 2017. Mitochondrial Dynamics: Coupling Mitochondrial Fitness with Healthy Aging. Trends Mol. Med. 23, 201–215. 10.1016/j.molmed.2017.01.003 [DOI] [PubMed] [Google Scholar]
- Sforzini L, Nettis MA, Mondelli V, Pariante CM, 2019. Inflammation in cancer and depression: a starring role for the kynurenine pathway. Psychopharmacology (Berl.). 10.1007/s00213-019-05200-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Enriquez A, Rapadas M, Martin EMMA, Wang R, Moreau J, Lim CK, Szot JO, Ip E, Hughes JN, Sugimoto K, Humphreys DT, McInerney-Leo AM, Leo PJ, Maghzal GJ, Halliday J, Smith J, Colley A, Mark PR, Collins F, Sillence DO, Winlaw DS, Ho JWK, Guillemin GJ, Brown MA, Kikuchi K, Thomas PQ, Stocker R, Giannoulatou E, Chapman G, Duncan EL, Sparrow DB, Dunwoodie SL, 2017. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N. Engl. J. Med. 377, 544–552. 10.1056/NEJMoa1616361 [DOI] [PubMed] [Google Scholar]
- Shinde R, Shimoda M, Chaudhary K, Liu H, Mohamed E, Bradley J, Kandala S, Li X, Liu K, McGaha TL, 2015. B Cell–Intrinsic IDO1 Regulates Humoral Immunity to T Cell–Independent Antigens. J. Immunol. 195, 2374–2382. 10.4049/jimmunol.1402854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siasos G, Tsigkou V, Kosmopoulos M, Theodosiadis D, Simantiris S, Tagkou NM, Tsimpiktsioglou A, Stampouloglou PK, Oikonomou E, Mourouzis K, Philippou A, Vavuranakis M, Stefanadis C, Tousoulis D, Papavassiliou AG, 2018. Mitochondria and cardiovascular diseases—from pathophysiology to treatment. Ann. Transl. Med. 6 10.21037/atm.2018.06.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Ramprasath T, Wang H, Zou M-H, 2017. Abnormal kynurenine pathway of tryptophan catabolism in cardiovascular diseases. Cell. Mol. Life Sci. 74, 2899–2916. 10.1007/s00018-017-2504-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgdrager FJH, Vermeiren Y, Van Faassen M, van der Ley C, Nollen EAA, Kema IP, De Deyn PP, 2019. Age- and disease-specific changes of the kynurenine pathway in Parkinson’s and Alzheimer’s disease. J. Neurochem. 10.1111/jnc.14843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, 2017. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 8 10.3390/genes8120398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JE, Soni A, Danilova L, Cottrell TR, Gajewski TF, Hodi FS, Bhatia S, Urba WJ, Sharfman WH, Wind-Rotolo M, Edwards R, Lipson EJ, Taube JM, 2019. Major pathologic response on biopsy (MPRbx) in patients with advanced melanoma treated with anti-PD-1: evidence for an early, on-therapy biomarker of response. Ann. Oncol. 30, 589–596. 10.1093/annonc/mdz019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Imai S, 2014. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 33, 1321–1340. 10.1002/embj.201386917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sühs K-W, Novoselova N, Kuhn M, Seegers L, Kaever V, Müller-Vahl K, Trebst C, Skripuletz T, Stangel M, Pessler F, 2019. Kynurenine Is a Cerebrospinal Fluid Biomarker for Bacterial and Viral Central Nervous System Infections. J. Infect. Dis. 220, 127–138. 10.1093/infdis/jiz048 [DOI] [PubMed] [Google Scholar]
- Sun N, Youle RJ, Finkel T, 2016. The Mitochondrial Basis of Aging. Mol. Cell 61, 654–666. 10.1016/j.molcel.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutphin GL, Backer G, Sheehan S, Bean S, Corban C, Liu T, Peters MJ, van Meurs JBJ, Murabito JM, Johnson AD, Korstanje R, the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium Gene Expression Working Group, 2017. Caenorhabditis elegans orthologs of human genes differentially expressed with age are enriched for determinants of longevity. Aging Cell 16, 672–682. 10.1111/acel.12595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantini S, Valcarcel-Ares MN, Toth P, Yabluchanskiy A, Tucsek Z, Kiss T, Hertelendy P, Kinter M, Ballabh P, Süle Z, Farkas E, Baur JA, Sinclair DA, Csiszar A, Ungvari Z, 2019. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 24, 101192. 10.1016/j.redox.2019.101192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurey P, Pizzo P, 2018. The Aging Mitochondria. Genes 9 10.3390/genes9010022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SR, Witting PK, Stocker R, 1996. 3-Hydroxyanthranilic Acid Is an Efficient, Cellderived Co-antioxidant for α-Tocopherol, Inhibiting Human Low Density Lipoprotein and Plasma Lipid Peroxidation. J. Biol. Chem. 271, 32714–32721. 10.1074/jbc.271.51.32714 [DOI] [PubMed] [Google Scholar]
- Uddin GM, Youngson NA, Doyle BM, Sinclair DA, Morris MJ, 2017. Nicotinamide mononucleotide (NMN) supplementation ameliorates the impact of maternal obesity in mice: comparison with exercise. Sci. Rep. 7, 15063 10.1038/s41598-017-14866-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin GM, Youngson NA, Sinclair DA, Morris MJ, 2016. Head to Head Comparison of Short-Term Treatment with the NAD+ Precursor Nicotinamide Mononucleotide (NMN) and 6 Weeks of Exercise in Obese Female Mice. Front. Pharmacol. 7 10.3389/fphar.2016.00258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- University of Maryland, 2019. The Effects of Kynurenine Aminotransferase Inhibition in People With Schizophrenia (TrypNAC-II) [WWW Document]. Clin. Internet Identifier NCT04013555. URL https://clinicaltrials.gov/ct2/show/NCT04013555?term=kynurenine&draw=2&rank=6 [Google Scholar]
- van der Goot AT, Nollen EAA, 2013. Tryptophan metabolism: entering the field of aging and age-related pathologies. Trends Mol. Med. 19, 336–344. 10.1016/j.molmed.2013.02.007 [DOI] [PubMed] [Google Scholar]
- van der Goot AT, Zhu W, Vazquez-Manrique RP, Seinstra RI, Dettmer K, Michels H, Farina F, Krijnen J, Melki R, Buijsman RC, Ruiz Silva M, Thijssen KL, Kema IP, Neri C, Oefner PJ, Nollen EAA, 2012. Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc. Natl. Acad. Sci. 109, 14912–14917. 10.1073/pnas.1203083109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versailles Hospital, 2012. TSK (Tryptophan - Serotonin - Kynurenine) Biomarkers Assessment in Stroke [WWW Document]. Clin. Internet Identifier NCT02963545. URL https://clinicaltrials.gov/ct2/show/NCT02963545?term=kynurenine&draw=2&rank=5 [Google Scholar]
- Versailles Hospital, 2004. Tryptophan, Serotonin and Kynurenine in Septic Shock (TSK) [WWW Document]. Clin. Internet Identifier NCT00684736. URL https://clinicaltrials.gov/ct2/show/NCT00684736?term=kynurenine&draw=2&rank=4 [Google Scholar]
- Vogel CFA, Goth SR, Dong B, Pessah IN, Matsumura F, 2008. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 375, 331–335. 10.1016/j.bbrc.2008.07.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Langer T, 2016. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. TEM 27, 105–117. 10.1016/j.tem.2015.12.001 [DOI] [PubMed] [Google Scholar]
- Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, Kelley KW, Dantzer R, 2013. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 38, 1609–1616. 10.1038/npp.2013.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Webster P, Chen L, Fisher AL, 2019. Cell-autonomous and non-autonomous roles of daf-16 in muscle function and mitochondrial capacity in aging C. elegans. Aging. 10.18632/aging.101914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liu D, Song P, Zou M-H, 2015. Tryptophan-kynurenine pathway is dysregulated in inflammation, and immune activation. Front. Biosci. Landmark Ed. 20, 1116–1143. 10.2741/4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, Laboy R, Hirschey MD, Mair WB, 2017. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab. 26, 884–896.e5. 10.1016/j.cmet.2017.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie D-L, Wu J, Lou Y-L, Zhong X-P, 2012. Tumor suppressor TSC1 is critical for T-cell anergy. Proc. Natl. Acad. Sci. U. S. A. 109, 14152–14157. 10.1073/pnas.1119744109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaku K, Okabe K, Nakagawa T, 2018. NAD metabolism: Implications in aging and longevity. Ageing Res. Rev. 47, 1–17. 10.1016/j.arr.2018.05.006 [DOI] [PubMed] [Google Scholar]
- Yan Y, Zhang G-X, Gran B, Fallarino F, Yu S, Li H, Cullimore ML, Rostami A, Xu H, 2010. IDO Upregulates Regulatory T Cells via Tryptophan Catabolite and Suppresses Encephalitogenic T Cell Responses in Experimental Autoimmune Encephalomyelitis. J. Immunol. 185, 5953–5961. 10.4049/jimmunol.1001628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CC, Chen D, Lee SS, Walter L, 2011. The dynamin-related protein DRP-1 and the insulin signaling pathway cooperate to modulate Caenorhabditis elegans longevity: DRP1 and insulin signaling cooperate to modulate aging. Aging Cell 10, 724–728. 10.1111/j.1474-9726.2011.00711.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Sauve AA, 2016. NAD + metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta BBA - Proteins Proteomics 1864, 1787–1800. 10.1016/j.bbapap.2016.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Satoh A, Lin JB, Mills KF, Sasaki Y, Rensing N, Wong M, Apte RS, Imai S-I, 2019. Extracellular Vesicle-Contained eNAMPT Delays Aging and Extends Lifespan in Mice. Cell Metab. 30, 329–342.e5. 10.1016/j.cmet.2019.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S, 2011. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14, 528–536. 10.1016/j.cmet.2011.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GS, Choleris E, Lund FE, Kirkland JB, 2006. Decreased cADPR and increased NAD+ in the Cd38−/− mouse. Biochem. Biophys. Res. Commun. 346, 188–192. 10.1016/j.bbrc.2006.05.100 [DOI] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, DAmico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ, Auwerx J, 2016. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. 10.1126/science.aaf2693 [DOI] [PubMed] [Google Scholar]
- Zhang H, Xiong Z-M, Cao K, 2014. Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc. Natl. Acad. Sci. U. S. A. 111, E2261–2270. 10.1073/pnas.1320843111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ovchinnikova O, Jönsson A, Lundberg AM, Berg M, Hansson GK, Ketelhuth DFJ, 2012. The tryptophan metabolite 3-hydroxyanthranilic acid lowers plasma lipids and decreases atherosclerosis in hypercholesterolaemic mice. Eur. Heart J. 33, 2025–2034. 10.1093/eurheartj/ehs175 [DOI] [PubMed] [Google Scholar]
- Zhang M, Ying W, 2019. NAD+ Deficiency Is a Common Central Pathological Factor of a Number of Diseases and Aging: Mechanisms and Therapeutic Implications. Antioxid. Redox Signal. 30, 890–905. 10.1089/ars.2017.7445 [DOI] [PubMed] [Google Scholar]
- Zheng X, Zhang A, Binnie M, McGuire K, Webster SP, Hughes J, Howie SEM, Mole DJ, 2019. Kynurenine 3-monooxygenase is a critical regulator of renal ischemia–reperfusion injury. Exp. Mol. Med. 51, 15 10.1038/s12276-019-0210-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C-C, Yang X, Hua X, Liu J, Fan M-B, Li G-Q, Song J, Xu T-Y, Li Z-Y, Guan Y-F, Wang P, Miao C-Y, 2016. Hepatic NAD+ deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br. J. Pharmacol. 173, 2352–2368. 10.1111/bph.13513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuravlev AV, Zakharov GA, Shchegolev BF, Savvateeva-Popova EV, 2016. Antioxidant Properties of Kynurenines: Density Functional Theory Calculations. PLOS Comput. Biol. 12, e1005213. 10.1371/journal.pcbi.1005213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwilling D, Huang S-Y, Sathyasaikumar KV, Notarangelo FM, Guidetti P, Wu H-Q, Lee J, Truong J, Andrews-Zwilling Y, Hsieh EW, Louie JY, Wu T, Scearce-Levie K, Patrick C, Adame A, Giorgini F, Moussaoui S, Laue G, Rassoulpour A, Flik G, Huang Y, Muchowski JM, Masliah E, Schwarcz R, Muchowski PJ, 2011. Kynurenine 3-Monooxygenase Inhibition in Blood Ameliorates Neurodegeneration. Cell 145, 863–874. 10.1016/j.cell.2011.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]