

Abstract
High protein diets are commonly utilized for weight loss, yet have been reported to raise cardiovascular risk. The mechanisms underlying this risk are unknown. Here, we show that dietary protein drives atherosclerosis and lesion complexity. Protein ingestion acutely elevates amino acid levels in blood and atherosclerotic plaques, stimulating macrophage mTOR signaling. This is causal in plaque progression as the effects of dietary protein are abrogated in macrophage-specific Raptor-null mice. Mechanistically, we find amino acids exacerbate macrophage apoptosis induced by atherogenic lipids, a process that involves mTORC1-dependent inhibition of mitophagy, accumulation of dysfunctional mitochondria, and mitochondrial apoptosis. Using macrophage-specific mTORC1- and autophagy-deficient mice we confirm this amino acid-mTORC1-autophagy signaling axis in vivo. Our data provide the first insights into the deleterious impact of excessive protein ingestion on macrophages and atherosclerotic progression. Incorporation of these concepts in clinical studies will be important to define the vascular effects of protein-based weight loss regimens.
INTRODUCTION
The immense risk lipids pose to atherosclerosis and cardiovascular events is now dogma in medicine. Modern atherogenic diets superimposed on a substrate of genetic modifications affecting lipid metabolism drive the hyperlipidemia that contributes to plaque progression. The undeniable efficacy of statins and now the new PCSK9 inhibitors in lowering LDL, and in turn cardiovascular events has established lipid therapy as a first-line therapeutic intervention. Mechanisms by which lipids promote atherogenesis are varied and include the conversion of plaque macrophages to foam cells and the modification of lipids to cytotoxic and proinflammatory species such as oxidized LDL1. Years of atherosclerosis research has focused on dissecting this complex interaction between lipid homeostasis and downstream sequelae in the expanding atherosclerotic plaque.
In contrast to lipids, the effect of dietary protein on cardiovascular disease is poorly defined and conflicting. High protein diets have been advocated for decades as a means of weight loss and the prevention of obesity and its metabolic sequelae2. The weight loss benefits of high protein diets came into vogue in the 1960s and have remained popular in to the present time. Beyond weight loss, much research has been devoted to the concomitant metabolic benefits these diets provide, spanning from enhanced insulin sensitivity to reduced fatty liver disease. Although the long-term risks of such diets on cardiovascular risk are largely unknown, it has been assumed that their effectiveness in preventing obesity and its metabolic sequelae would also extend to cardiovascular benefits. This was buttressed in several retrospective studies in the last decade suggesting a lack of association between high protein diets and coronary heart disease3,4. But more recently, when this question is assessed prospectively, a different conclusion is reached. For example, the prospective Swedish women’s study following patients for 15.7 years, found an increased CVD risk in women with diets enriched in protein5.
When animal studies conducted over the past few decades are taken in aggregate, diets high in protein actually favor increased atherogenesis6. Such results also hold in genetically tractable mouse models7. Surprisingly, aside from a cursory evaluation of plaque size these studies largely neglect mechanistic evaluation of the observed atherosclerosis phenotypes. Thus, in contrast to the field of lipid metabolism, links between dietary protein and cardiovascular disease remain associative and non-mechanistic.
In this work, we take advantage of commonly used pro-atherogenic mouse models and primary macrophages to dissect the association between dietary protein and atherosclerosis. We find that elevation of dietary protein to the 40 kCal%-range, a level often used in weight loss regimens, is sufficient to exacerbate atherosclerosis. Dietary protein especially increases lesion apoptosis and necrotic core formation, surrogates of the complex/unstable plaque. Mechanistically, the mTORC1 complex is classically known to integrate information about cellular nutrient status, including amino acid levels, to alter cellular signaling through a broad range of downstream targets8. We show that ingestion of protein can sufficiently raise blood and tissue amino acid levels to stimulate mTORC1 activation, particularly in macrophages of the atherosclerotic plaque. Mice with macrophage deletion of the critical mTORC1 component, Raptor, have reduced atherosclerosis and are no longer susceptible to high protein diet-induced atherosclerosis. The predominant effect of mTORC1 activation in macrophages is to suppress mitophagy, leading to accumulation of dysfunctional mitochondria, triggering macrophage apoptosis, and contributing to plaque complexity. We buttress these findings in vivo using mice with genetic perturbations in macrophage autophagy in conjunction with mTORC1.
RESULTS
High protein diets increase atherosclerotic plaque formation and plaque complexity despite having salutary effects on body weight and glucose tolerance.
The protein content of various high-protein diets commonly used for weight loss range between 30–64% (kcal%)9,10. In order to study atherosclerosis in mouse models, a diet with high fat content is used in conjunction with hyperlipidemia-prone ApoE-null or LDLR-null mice. A popular version of this so-called Western Diet is composed of 42% fat and 15% protein11,12. Thus, we chose to compare the metabolic phenotype and atherogenic potential of this “standard” Western diet (Std. WD) to one with similar fat content but three times more protein (43% fat, 46% protein) which we call “high protein” Western diet (HP WD) in a cohort of ApoE-null mice (Figure 1a,b). Over a 2-month period, we recapitulated the predicted satiating effects and weight loss benefits of high protein diets noting reduced food intake, resistance to diet-induced obesity, reduction in whole body fat content, and concomitant improvement in glucose disposal as gauged by glucose tolerance tests (Figure 1c, Supplemental Figure 1a–c). However, despite the salutary metabolic effects, high-protein fed mice developed increased atherosclerotic plaques at the level of the aortic root (Figure 1d). Further evaluation of these lesions showed a disproportionate increase in plaque macrophages and features of plaque complexity including significantly elevated markers of apoptosis and increased necrotic core burden (Figure 1e–g). Importantly, this phenotype was observed despite similar increases in serum cholesterol levels and other common serum metabolites (Supplemental Figure 1d). Prolonged feeding of this high protein Western diet (16 weeks) continued to result in significantly increased lesion area and plaque complexity (Supplemental Figure 1e–i).
Figure 1. High Protein diets increase atherosclerotic plaque formation and plaque complexity.
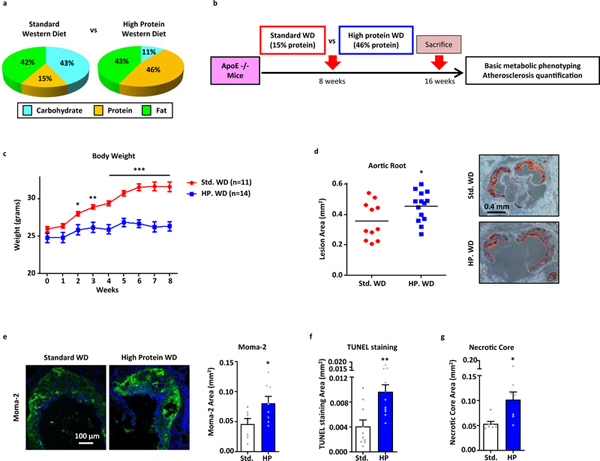
(a,b) Comparison between the carbohydrate, protein, and fat content in standard and high protein Western diets (kcal%) (a) and summary of experimental protocol for in vivo assessment of atherosclerosis (b). (c) Total body weight of ApoE-null mice fed standard or high protein Western diets for 8 weeks (Std. WD: n=11; HP WD: n=14). (d) Quantification of atherosclerotic plaque burden using Oil Red O-stained aortic root sections from mice fed standard or high protein Western diets for 8 weeks; representative roots shown on right. (e-g) Plaque composition quantified by immunofluorescence staining of aortic root sections for (e) macrophage (MOMA-2+) (Std. WD: n=7; HP WD: n=8), (f) apoptosis (TUNEL+) (Std. WD: n=10; HP WD: n=12), (g) and necrotic core (acellular) (Std. WD: n=7; HP WD: n=7). Data are presented as mean ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed unpaired t-test.
High protein intake elevates levels of select amino acids in vivo and acutely activates plaque macrophage mTORC1 signaling
The atherosclerotic plaque phenotyping above suggested a link between dietary protein and perturbations of plaque macrophage function. Thus we next assessed the downstream consequences of high protein ingestion quantifying the rise in amino acid levels and its effects on macrophage intracellular signaling. First, we found that although protein-rich diets lead to marginally increased total serum amino acid levels, certain amino acids such as leucine are significantly elevated whereas others such as glutamine maintain relatively high baseline levels that are resistant to fluctuation (Figure 2a; Supplemental Figure 2a shows levels of 8 amino acids selected from the various amino acid classes). In order to determine the kinetics and downstream effects of both circulating and tissue/cellular levels of amino acids after high protein feeding, we conducted timed oral gavage experiments composed of 3.2g/kg protein (Figure 2b). In this acute setting, as would be seen with the ingestion of a high protein drink or meal, we observed a rise in total serum amino acids peaking at 1 hour with leucine being one of the most robustly upregulated (Figure 2c,d, Supplemental Figure 2b).
Figure 2. High Protein diets elevate amino acids levels in vivo and activate mTOR signaling in plaque macrophages.
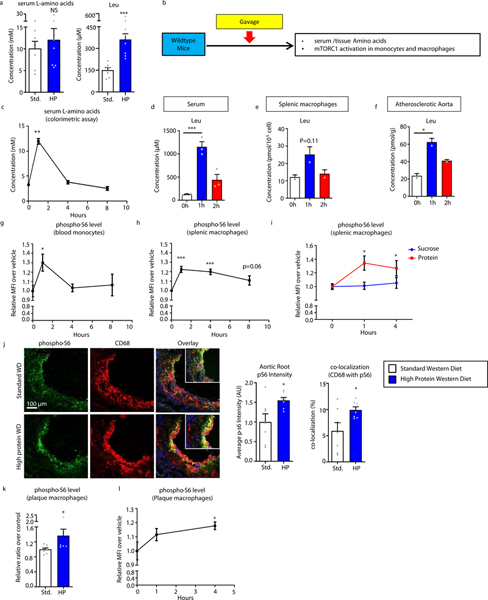
(a) Serum levels of total L-amino acids (colorimetric assay) and leucine (mass spectrometry) from mice fed standard (n=7) or high protein (n=8) Western diets for 8 weeks. (b) Summary of gavage/time course experimental protocol for determining acute effects of high protein intake. (c) Serum levels of total L-amino acids (colorimetric assay) after high protein gavage for indicated time (n=3). (d-f) Leucine levels in serum (n=3) (d), splenic macrophages (n=2) (e), and atherosclerotic aortas (n=2) (f) by mass spectrometry after high protein gavage for indicated times. (g,h) FACS analysis of pS6 levels in blood monocytes (n=3) (g) and splenic macrophages (n=5) (h) after high protein gavage for indicated times. (i) Comparison of pS6 levels in splenic macrophages after gavage with water or equal calories of sucrose (n=4) or protein (n=4) at 0, 1, and 4 hours. (j) Immunofluorescence analysis of pS6 levels and co-localization of pS6 with the macrophage marker CD68 in atherosclerotic plaques from mice fed standard (n=7) or high protein (n=7) Western diets for 8 weeks. Representative images are shown on left and quantification on right. (k,l) FACS analysis of pS6 levels in atherosclerotic plaque macrophages from (k) mice fed standard (n=6) or high protein (n=6) Western diet for 8 weeks or (l) after high protein gavage for indicated times (n=3). For (Figures 2g-i, 2l), graphs represent relative MFI over vehicle control. Data are presented as mean ±SEM. *P < 0.05, ***P < 0.001, two-tailed unpaired t-test for a, j, k, one-way ANOVA with Dunnett’s test for c-i, l.
Paralleling a rise in the serum, we observed similar increases of intracellular leucine and other amino acids in splenic macrophages and atherosclerotic aortic tissue with levels peaking at 1 hour and remaining detectable up to 2 hours (Figure 2e,f, Supplemental Figure 2c,d). This translated to an increased mTORC1 activation in blood monocytes and splenic macrophages as assessed by phosphorylation of the downstream target S6, with maximal activation at 1 hour and continued activation up to 8 hours (Figure 2g,h). Activation of mTORC1 was clearly dependent on protein intake as a high sucrose gavage of equal caloric content had no such effects (Figure 2i). We also evaluated the activation of mTORC1 in atherosclerotic plaques and macrophages by conducting immunofluorescence staining of aortic roots from high protein Western diet-fed ApoE-null mice showing increased plaque phospho-S6 intensity that highly co-localized with the macrophage marker CD68 (Figure 2j). This was corroborated by demonstration of increased phospho-S6 in macrophages isolated by FACS from atherosclerotic aortas of mice fed a high protein diet (Figure 2k) or gavaged with protein (Figure 2l).
The lysosome has recently been identified as a nexus for mTORC1 activation, where amino acids are thought to concentrate in lysosomes resulting in recruitment and activation of a Raptor-dependent mTOR protein complex at the lysosomal surface13,14. Although certain amino acids have been determined to be classical activators of mTORC1 through this mechanism, this has not been fully evaluated in macrophages. Using phosphorylation of S6K and S6 as well as co-localization of mTOR with the lysosomal marker Lamp2, we first verified that amino acid-sufficient media is indeed able to activate mTORC1 in primary macrophages (thioglycollate-elicited peritoneal macrophages – PMACs and bone marrow-derived macrophages - BMDMs) (Supplemental Figures 3a–c). We next incubated PMACs with a panel of 20 amino acids to determine the predominant mTORC1 activators in macrophages. Clear differences were seen between the amino acids, ranging from robust inducers (particularly leucine) to poor inducers (e.g. glutamine) (Supplemental Figures 3d,e). The top three inducers and non-inducers were further confirmed by co-localization of mTOR with Lamp2 (Supplemental Figures 3f,g ). For the rest of our studies, we focused on leucine as the quintessential mTORC1 inducer in macrophages.
High protein diet-induced atherosclerosis is dependent on macrophage mTORC1
In order to study the specific effects of amino acids and dietary protein on macrophage mTORC1 signaling and atherosclerosis, we developed macrophage-specific Raptor-null mice (called mϕ-Raptor-KO)15. Macrophages from these mice indeed showed leucine’s activation of mTORC1 to be Raptor-dependent as gauged by blunted activation of classic downstream targets as well as abrogated lysosomal mTOR recruitment (Supplemental Figures 4a,b).
We used mϕ-Raptor-KO mice (on a pro-atherogenic ApoE-null background) to study lesion formation after 8 weeks of standard Western diet feeding (42% fat, 15% protein) (Figure 3a). The absence of macrophage Raptor led to significantly decreased atherosclerotic lesion formation with concomitant reductions in plaque macrophage content, apoptotic cells, and necrotic core area (Figure 3b–e) without affecting body weight or serum lipid parameters (Supplemental Figure 4c,d). This data is in line with previously described bone marrow transplant studies of Raptor-deficient myeloid cells in LDLR-null mice16 and suggests that even at lower dietary protein levels, macrophage mTORC1 signaling plays an atherogenic role.
Figure 3. High protein diets accelerate atherogenesis through macrophage mTORC1 signaling.
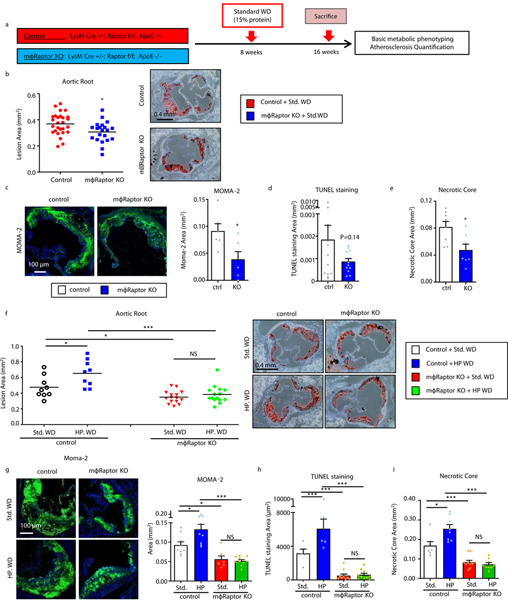
(a) Summary of experimental protocol for in vivo assessment of atherosclerosis in control and macrophage-specific Raptor-null (mϕ-Raptor-KO) mice (all on ApoE-KO background). (b) Quantification of atherosclerotic plaque burden using Oil Red O-stained aortic root sections from mice fed standard or high protein Western diets for 8 weeks; representative roots shown on right (Control: n=26; mϕRaptor-KO: n=21). (c-e) Plaque composition quantified by immunofluorescence staining of aortic root sections for (c) macrophage (MOMA-2+) (Control: n=6; mϕRaptor-KO: n=6), (d) apoptosis (TUNEL+) (Control: n=13; mϕRaptor-KO: n=13), (e) and necrotic core (acellular) (Control: n=8; mϕRaptor-KO: n=7). (f) Quantification of atherosclerotic plaque burden using Oil Red O-stained aortic root sections from Control and macrophage-specific Raptor-null (mϕRaptor-KO, on ApoE-KO background) mice fed standard or high protein Western diets for 8 weeks; representative roots shown on right (Control Std. WD: n=9, HP WD: n=9; mϕRaptor-KO Std. WD: n=15, HP WD: n=15). (g-i) Plaque composition quantified by immunofluorescence staining of aortic root sections for (g) macrophage (MOMA-2+) (Control Std. WD: n=9, HP WD: n=9; mϕ-Raptor-KO Std. WD: n=7, HP WD: n=10), (h) apoptosis (TUNEL+) (Control Std. WD: n=5, HP WD: n=5; mϕRaptor-KO Std. WD: n=10, HP WD: n=10), and (i) necrotic core (acellular) (Control Std. WD: n=7, HP WD: n=8; mϕRaptor-KO Std. WD: n=10; HP WD: n=9). Data are presented as mean ±SEM. *P < 0.05, **P < 0.01, two-tailed unpaired t-test for b-e, two-way ANOVA for f-i.
We next evaluated the link between dietary protein, macrophage mTORC1 activation, and atherosclerosis by challenging a cohort of Control and mϕ-Raptor-KO mice with either standard or high protein Western diets used previously. Although the absence of macrophage Raptor had no impact on common serum metabolic parameters (Supplemental Figure 4e), it completely abrogated the ability of dietary protein to increase atherosclerosis including its effects on plaque macrophage content, apoptosis, and necrotic core formation (Figures 3f–i). Taken together, these results establish macrophage mTORC1 signaling as the predominant effector of high protein diet-induced atherosclerosis and atherosclerotic plaque complexity.
Amino acids synergize with atherogenic stimuli to instigate mitochondrial dysfunction and apoptosis in macrophages
A characteristic feature of high protein diet-induced plaques appears to be increased apoptosis leading to lesion complexity. Thus, we evaluated the effect of amino acids on macrophage apoptosis under atherogenic conditions. Although leucine was unable to significantly induce macrophage apoptosis in isolation, it synergistically increased cell death when co-incubated with pro-apoptotic atherogenic lipids 7-KC or cholesterol crystals (Figures 4a,b, Supplemental Figures 5a,b). This effect was dependent on mTORC1 as Raptor-KO macrophages negated the pro-apoptotic response (Figure 4c). Importantly, leucine-enhanced apoptosis appeared to be mediated by the mitochondrial-dependent intrinsic pathway as caspase-9 activity was elevated with no changes in caspase-8 activity (Figures 4d). Given the pleiotropic effects of atherogenic lipids such as 7-KC, we conducted similar assays using the pro-apoptotic mitochondrial uncoupling agent FCCP. Leucine again synergistically activated macrophage apoptosis in the presence of FCCP, a process that was mTORC1-dependent (Figures 4e). In keeping with a mitochondrial-specific effect, caspase-9 activity and cleavage (but not caspase-8) were again increased in the presence of leucine (Figures 4f,g).
Figure 4. Leucine-mediated activation of mTORC1 synergizes with atherogenic lipids to induce mitochondrial-dependent apoptosis of macrophages.
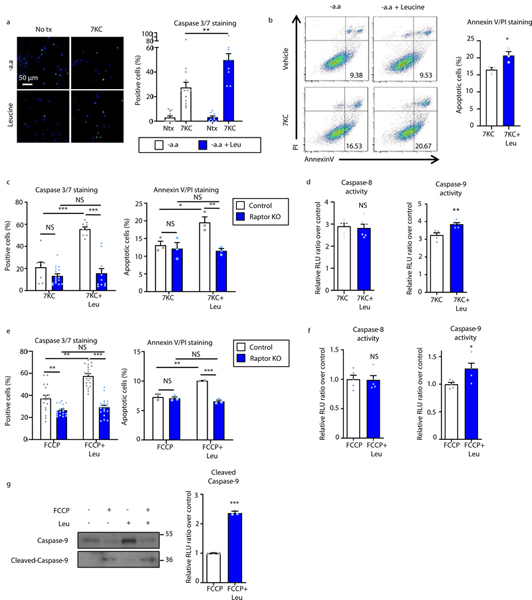
(a,b) Macrophages were treated with vehicle, 50μM 7-ketocholesterol (7KC) with or without leucine (1.91 mM) and apoptosis assessed by (a) Caspase-3/7 immunofluorescence staining (-aa: n=11; -aa+7KC: n=13; Leu: n=10; Leu+7KC: n=10) and (b) flow cytometry analysis of Annexin V/propidium iodide (PI) staining (7KC: n=3; Leu+7KC: n=3). Quantification is shown on the right of representative images and plots. (c) Control and Raptor KO macrophages subjected to similar Caspase-3/7 (Control 7KC: n=8, Leu+7KC: n=9; Raptor KO 7KC: n=16, Leu+7KC: n=11) and Annexin V/PI (n=3/group) assays to (a,b). (d) Activity of caspase-8 (n=6/group) and caspase-9 (n=5/group) in macrophages incubated with 7KC +/− leucine using a luminescence assay. (e-g) Measurement of the synergistic effect of leucine in FCCP-induced apoptosis. (e) Control and Raptor KO were treated with FCCP +/− leucine and apoptosis assessed by Caspase-3/7 immunofluorescence (Control FCCP: n=18, Leu+FCCP: n=20; Raptor KO FCCP: n=18, Leu+FCCP: n=19) and flow cytometry of AnnexinV/PI staining (Control FCCP: n=2, Leu+FCCP: n=2; Raptor KO FCCP: n=3, Leu+FCCP: n=3). (f) Activity of caspase-8 and caspase-9 in macrophages incubated with FCCP +/− leucine using a luminescence assay (n=5/group). (g) Immunoblot analysis and quantification of caspase-9 and cleaved caspase-9 in macrophages incubated with FCCP +/− leucine (normalized to Ponceau S staining as a loading control) (n=3/group). Data are presented as mean ±SEM. *P < 0.05, ***P < 0.001, NS=not significant, one-way ANOVA with Tukey’s test for a, two-tailed unpaired t-test for b,d,f,g, two-way ANOVA for c,e.
Since mitochondrial dysfunction is a prominent trigger for the intrinsic pathway of apoptosis, we also evaluated leucine’s effect on macrophages treated with the mitochondrial respiratory chain inhibitor rotenone17. Leucine synergistically enhanced mitochondrial uncoupling and superoxide generation when incubated with rotenone, a process that was Raptor-dependent (Figures 5a–f). Increased mitochondrial dysfunction was also observed in macrophages isolated from high protein diet-induced atherosclerotic plaques suggesting the occurrence of a similar process in vivo (Figures 5g,h).
Figure 5. Leucine-mediated activation of mTORC1 leads to accumulation of dysfunctional mitochondria and ROS production in macrophages.
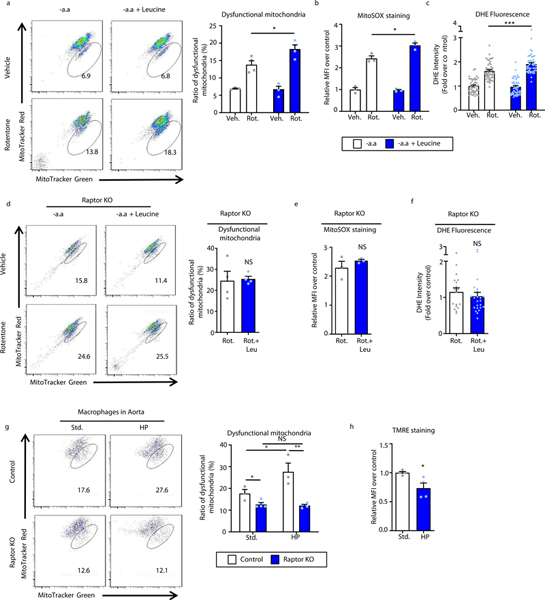
(a) Macrophages were treated with vehicle or 40μM rotenone with or without leucine (1.91 mM) for 6 hours and mitochondrial dysfunction evaluated as a ratio of reduced mitochondrial membrane potential (Δψm) (MitoTracker Red) vs mitochondrial mass (MitoTracker Green) by FACS analysis. Quantification is shown on the right of representative plots (-aa: n=3; -aa+Rot: n=4; Leu: n=4; Leu+Rot: n=4). (b) FACS analysis of mitochondria ROS (as assessed by MitoSOX labeling) in macrophages treated with rotenone +/− leucine for 24h (n=3/group). (c) Quantification of intracellular ROS levels using fluorescence microscopy of DHE-stained macrophages treated with rotenone +/− leucine for 12 hours (-aa: n=45; -aa+Rot: n=49; Leu: n=49; Leu+Rot: n=49 cells). (d-f) Raptor KO macrophages subjected to similar mitochondrial assays to (a-c) including (d) membrane potential Δψm (n=4/group), (e) mitoSOX (n=3/group), and (f) DHE-staining (Rot: n=18; Leu+Rot: n=23 cells). (g) Plaque macrophages were isolated from atherosclerotic aortas of Control and macrophage-specific Raptor-KO mice (mϕ-Raptor-KO, on ApoE-KO background) fed 2 months of standard or high protein Western diets and the degree of mitochondrial dysfunction determined by labeling with MitoTracker Red (Δψm) and MitoTracker Green (mitochondrial mass) followed by FACS analysis (Control: n=3/group; mϕRaptor-KO: n=7/group). (h) Similar experiments as in (g) conducted on a cohort of ApoE-KO mice and mitochondrial dysfunction determined by labeling for TMRE followed by FACS analysis (n=4/group). For all graphs, data are presented as mean ±SEM. Data are presented as mean ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001, NS=not significant, one-way ANOVA with Tukey’s test for a-c, two-tailed unpaired t-test for d,e,f,h, two-way ANOVA for g.
Inhibition of mitophagy is a critical mechanism by which macrophage mTORC1 signaling drives mitochondrial dysfunction and apoptosis
Extensive prior work by us and others has revealed a progressive dysfunction of autophagy in macrophages of the atherosclerotic plaque resulting in aberrant handling of cytotoxic protein aggregates, inflammatory signals, and lipids11,12,18–21. Mice with genetic deletion of macrophage autophagy (mϕ-ATG5-KO) have significantly elevated atherosclerosis with highly complex lesions characteristically rich in apoptotic macrophages and necrotic cores11,19. Since autophagy is a major target of inhibition by mTORC1 particularly in relation to nutritional regulation and mitochondrial autophagy (mitophagy) is a prominent mechanism by which cells clear pro-apoptotic/dysfunctional mitochondria22, we focused on the amino acid-mTORC1-autophagy link in macrophages in the context of atherosclerosis.
In cultured macrophages, we found that leucine has a potent ability to regulate autophagy as demonstrated by increased phosphorylation of the mTORC1 target and autophagy regulator Ulk1 and concomitant suppression of autophagosome formation (gauged by blunted LC3-I to LC3-II conversion on immunoblots and reductions in LC3 puncta by immunofluorescence microscopy) (Supplemental Figures 6a–c). This amino acid-mediated inhibition of macrophage autophagy was entirely mTORC1-dependent as Raptor-deficient macrophages had blunted phospho-Ulk1 levels and increased autophagy flux (i.e. enhanced LC3-II conversion upon concomitant treatment with the lysosomal inhibitor Bafilomycin) by immunoblot as well as abrogated suppression of LC3 puncta on immunofluorescence imaging (Supplemental Figures 6d–f ).
Due to the importance of mitophagy in clearing dysfunctional mitochondria and mitigating mitochondrial apoptosis, we evaluated the role of amino acids and mTORC1 in this selective form of autophagy. First, using the co-localization of the mitochondrial marker COX IV and the autophagic marker LC3, we confirmed leucine’s negative regulatory effects on mitophagy and showed its dependence on mTORC1 (Figures 6a). The pH-sensitive mitochondrial-specific dye mt-Keima allows tracking of mitochondria into acidic lysosomes and is regarded one of the most robust methods of measuring mitophagy23. Leucine also blunted the acidification of mt-Keima in control but not Raptor-null macrophages, supporting the inhibitory role of amino acid-induced mTORC1 on mitophagy (Figure 6b,c). Finally, we assessed the intensity of LC3 and its co-localization with COX IV in aortic root atherosclerotic plaques from standard versus high protein Western diet-fed animals. High protein feeding suppressed both the autophagy marker LC3 as well as its co-localization with COX IV in plaque macrophages corroborating our in vitro observations of a autophagy/mitophagy defect (Figure 6d,e).
Figure 6. Leucine-mediated activation of mTORC1 inhibits mitophagy in macrophages.
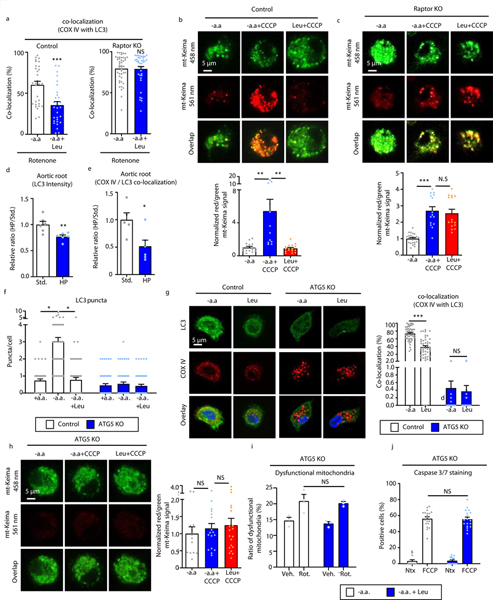
(a) The degree of LC3 co-localization with the mitochondrial marker COXIV determined by immunofluorescence microscopy of control and Raptor KO macrophages treated with rotenone for 3 hours (Control Rot.: n=30, Leu+ Rot.: n=30; Raptor KO Rot.: n=44, Leu+ Rot.: n=39 cells). (b,c) Live fluorescence imaging of Control (b) and Raptor KO (c) macrophages transduced with mt-Keima lentiviral vector followed by incubation with amino acid-free medium or the addition of CCCP +/− leucine. Representative images shown on left and quantification shown on right (Control -aa: n=15, -aa+CCCP: n=12, Leu+CCCP: n=16; Raptor KO -aa: n=24; -aa+CCCP: n=15, Leu+CCCP: n=15). (d,e) Aortic root sections from ApoE−/− mice fed a standard or high protein Western diet for 2 months were immunostained for LC3 and COXIV. The degree of LC3 intensity (d) and LC3 / COXIV co-localization (e) in aortic macrophages determined by immunofluorescence microscopy (n=6 mice/group). (f) Quantification number of LC3 puncta by immunofluorescence microscopy in control and ATG5-KO macrophages incubated with regular medium or amino acid-free medium with and without leucine for 30 minutes (Control +aa: n=52, -aa: n=48, Leu: n=52; ATG5 KO +aa: n=51, -aa: n=52, Leu: n=50). (g) The degree of LC3 co-localization with the mitochondrial marker COXIV was determined by immunofluorescence microscopy of control and ATG5-KO macrophages treated with rotenone for 3 hours (Control Rot.: n=52, Leu+Rot.: n=46; ATG5 KO Rot.: n=30, Leu+Rot.: n=30). (h) Live fluorescence imaging of ATG5-KO macrophages transduced with mt-Keima lentiviral vector followed by incubation with amino acid-free medium or the addition of CCCP +/− leucine. Representative images shown on left and quantification shown on right (aa: n=13; -aa+CCCP: n=23; Leu+CCCP: n=34). (i) ATG5-KO macrophages were treated with vehicle or 40μM rotenone with or without leucine for 6 hours and mitochondrial dysfunction quantified as a ratio of reduced mitochondrial membrane potential (Δψm) (MitoTracker Red) vs mitochondrial mass (MitroTracker Green) by FACS analysis (n=3/group). (j) ATG5-KO macrophages were co-incubated with vehicle or FCCP +/− leucine and percent of caspase 3/7-positive cells were quantified in three independent experiments (-aa: n=16; -aa+CC: n=19; Leu: n=15; Leu+CC: n=18). Data are presented as mean ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed unpaired t-test for a,d,e, one-way ANOVA with Tukey’s test for b,c,f-j.
As a confirmation of the autophagy-dependence of the deleterious effects of amino acids, we also conducted similar assays using autophagy-deficient (ATG5-KO) macrophages. As expected, in the absence of ATG5, leucine was unable to alter the development of LC3 puncta, co-localization of COX IV and LC3, or acidification of mt-Keima (Figures 6f–h, Supplemental Figure 6g). Furthermore, leucine’s synergistic effects with either rotenone or atherogenic lipids to induce mitochondrial dysfunction and apoptosis were abrogated in the autophagy-deficient setting (Figure 6i,j, Supplemental Figure 6h).
Inhibition of autophagy is a critical mechanism by which macrophage mTORC1 signaling and high protein diets drive atherosclerosis
In order to test the links between macrophage mTORC1 and autophagy on atherosclerosis, we compared lesion formation in Control, mϕ-Raptor-KO, mϕ-ATG5-KO, and dual mϕ-Raptor/ATG5-KO (DKO) mice (on a pro-atherogenic ApoE-null background) after 8 weeks of standard Western diet feeding (42% fat, 15% protein) (Supplemental Figure 7a,b). The atheroprotective phenotype of mϕ-Raptor-KO was entirely abrogated in the absence of autophagy (Figure 7a). The critical role for macrophage autophagy in mTORC1 signaling was also confirmed when the reductions in lesion macrophage content, degree of apoptosis, and necrotic core formation noted in mϕ-Raptor-KO were negated in the absence of autophagy (Figures 7b–d).
Figure 7. Autophagy deficiency reverses the atheroprotective effect of Raptor silencing in macrophages.
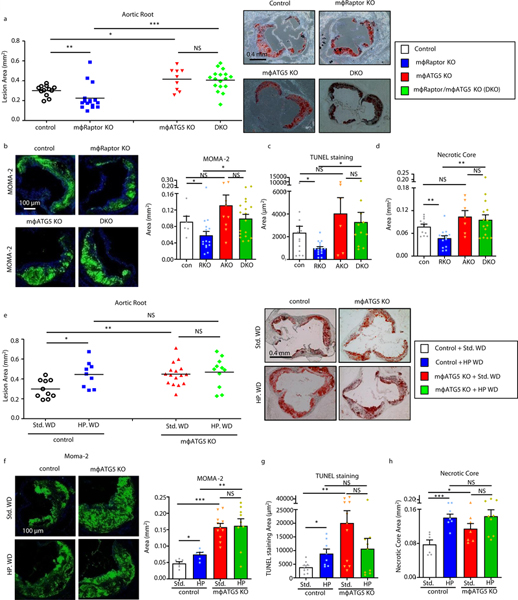
(a) Quantification of atherosclerotic plaque burden in Control, mϕRaptor-KO, mϕATG5-KO, and dual mϕRaptor/ mϕATG5-KO (DKO) mice (all ApoE-null background) mice fed a standard Western diet for 8 weeks using Oil Red O-stained aortic root sections with representative roots shown on right (Control: n=11; mϕRaptor-KO: n=16; mϕATG5-KO: n=9; DKO: n=17). (b-d) Plaque composition quantified by immunofluorescence staining of aortic root sections for (b) macrophage (MOMA-2+) (Control: n=6; mϕRaptor-KO: n=16; mϕATG5-KO: n=9; DKO: n=18),, (c) apoptosis (TUNEL+) (Control: n=14; mϕRaptor-KO: n=16; mϕATG5-KO: n=6; DKO: n=11),, (d) and necrotic core (acellular) (Control: n=11; mϕRaptor-KO: n=14; mϕATG5-KO: n=7; DKO: n=15). (e) Quantification of atherosclerotic plaque burden using Oil Red O-stained aortic root sections from Control and mϕATG5-KO mice (all on ApoE-null background) fed standard or high protein Western diets for 8 weeks; representative roots shown on right (Control Std. WD: n=10, HP WD: n=9; mϕ-ATG5-KO Std. WD: n=16, HP WD: n=11). (f-h) Plaque composition quantified by immunofluorescence staining of aortic root sections for (f) macrophage (MOMA-2+) (Control Std. WD: n=6, HP WD: n=6; mϕ-ATG5-KO Std. WD: n=11, HP WD: n=9), (g) apoptosis (TUNEL+) (Control Std. WD: n=9, HP WD: n=8; mϕ-ATG5-KO Std. WD: n=11, HP WD: n=19), (h) and necrotic core (acellular) (Control WD: n=6, HP WD: n=7; mϕ-ATG5-KO Std. WD: n=7, HP WD: n=9). Data presented as mean ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001, NS=not significant, two-way ANOVA for a-h.
As confirmation of the importance of macrophage autophagy in ameliorating high protein diet-induced atherogenesis, we next challenged a cohort of Control and mϕ-ATG5-KO mice with either standard or high protein Western diets. The absence of macrophage autophagy prevented the ability of dietary protein to further increase atherosclerosis (Figure 7e). In particular, the deleterious effects of dietary protein on plaque macrophage content, apoptosis, and necrotic core formation were uniformly abrogated (Figures 7f–h). Together with the dual mϕ-Raptor/ATG5-KO experiments above, these results confirm that macrophage autophagy is the critical downstream pathway that is perturbed by high protein diet-induced atherosclerosis and atherosclerotic plaque complexity.
Taken together, our data support the notion that excessive ingestion of dietary protein raises circulating amino acids to levels that impact plaque macrophage mTORC1-autophagy signaling resulting in apoptosis. We desired to ascertain the physiological relevance of a rise in circulating amino acids to perturbations of macrophage signaling and function. We had determined the concentration of circulating leucine in mice fed standard and high protein Western diets to be in the range of 50–100 μM and 400–1000 μM respectively (Figure 2a,d). Using these concentrations as a guide, the lowest dose at which leucine can activate mTORC1, suppress autophagy, elevate mitochondrial dysfunction, and exacerbate macrophage apoptosis requires a concentration of at least 400 μM (Figure 8). This is indeed on par with circulating levels of leucine after ingestion of a high protein meal and supports the pathophysiologic link between dietary protein excess and hyperactivation of deleterious macrophage signaling.
Figure 8. Leucine activates mTORC1 signaling and regulates downstream targets in a dosage dependent manner.
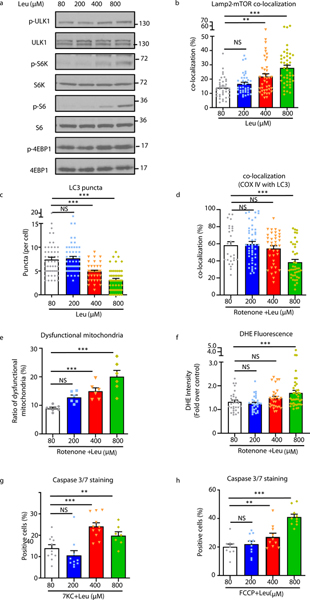
(a-h) Macrophage were incubated with amino acid-free media supplemented with increasing concentrations of leucine (80, 200, 400, and 800 uM) and various assays conducted as follows: (a) Western blot analysis of macrophage mTORC1 activity after 30min of incubation with leucine, (b) Quantification of the co-localization between mTOR and Lamp2 by immunofluorescence imaging of macrophages after 15min incubation with leucine (cells per group: 80μM: n=38; 200μM: n=39; 400μM: n=40; 800 μM: n=43), (c,d) Quantification of LC3 puncta (c) (cells per group: 80μM: n=51; 200μM: n=51; 400μM: n=43; 800 μM: n=51) and co-localization of the mitochondrial marker COX IV with LC3 (d) (cells per group: 80μM: n=28; 200μM: n=49; 400μM: n=37; 800 μM: n=40). (e) Quantification of mitochondrial dysfunction evaluated as a ratio of reduced mitochondrial membrane potential (Δψm) (MitoTracker Red) vs mitochondrial mass (MitoTracker Green) by FACS analysis in macrophage treated with 40μM rotenone with or without leucine (n=6/group). (f) Quantification of intracellular ROS levels using fluorescence microscopy of DHE-stained macrophages treated with rotenone +/− leucine for 12 hours (cells per group: 80μM: n=31; 200μM: n=31; 400μM: n=29; 800 μM: n=39). (g.h) Apoptosis assessed by Caspase-3/7 immunofluorescence staining of macrophages treated with (g) 50μM 7-ketocholesterol (7KC) +/− leucine (quantified images fields for 80μM: n=11; 200μM: n=10; 400μM: n=11; 800 μM: n=7) and (h) FCCP +/− leucine (quantified image fields for 80μM: n=10; 200μM: n=11; 400μM: n=10; 800 μM: n=10). Data are presented as mean ±SEM. **P < 0.01, ***P < 0.001, NS=not significant, one-way ANOVA with Tukey’s test.
DISCUSSION
In this study, we provide the first detailed evaluation of the atherogenic potential of high protein diets, implicating perturbation of the macrophage mTORC1-autophagy signaling axis as a critical mechanism. Using mouse models we show that diets with a protein composition comparable to commonly used weight loss regimens exacerbate atherosclerosis especially with regard to apoptosis and lesion complexity, indicators of plaque instability. The intake of a protein meal acutely raises levels of amino acids in the blood stream, circulating monocytes, and tissue macrophages including those residing in the atherosclerotic plaque, in turn leading to acute elevation of mTORC1 signaling. We find only select amino acids (leucine in particular) are potent inducers of mTORC1 in macrophages. When mTORC1 signaling is abolished, mice have reduced atherosclerosis and lesion complexity and are no longer susceptible to the atherogenic effects of high protein intake. Counter to the assumption that mTORC1 activation would favor cell survival, leucine acts synergistically with atherogenic lipids to increase macrophage mitochondrial dysfunction and apoptosis. These effects are due to mTORC1-dependent inhibition of autophagy/mitophagy, preventing the removal of dysfunctional mitochondria. Finally, this amino acid-mTORC1-autophagy axis is mechanistically relevant in vivo as mice with loss of macrophage autophagy abrogate the atheroprotective effect of mTORC1-deficiency and develop complex lesions that are not exacerbated further by elevations in dietary protein. Our dissection of this pathway spanning from ingestion of a protein meal to effects on plaque macrophage function are summarized in Figure 9. Several issues pertaining to this model are worthy of discussion.
Figure 9. Graphical summary of the progression of events from ingestion of a protein meal to deleterious effects on atherosclerotic plaque complexity.
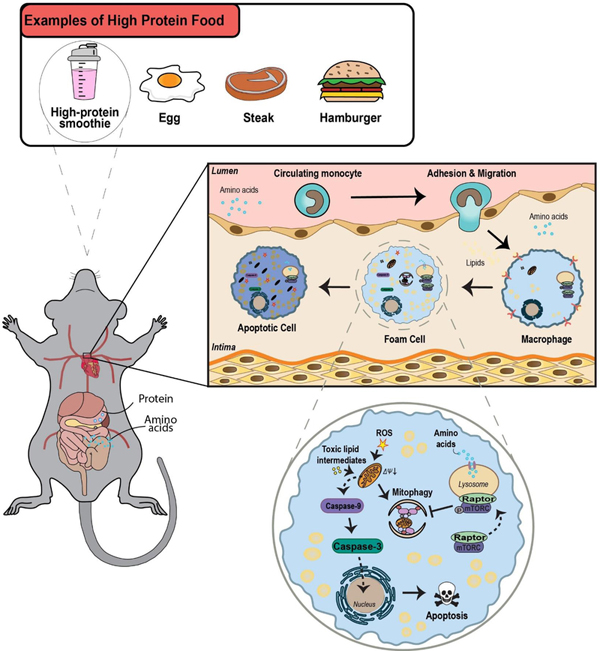
Ingestion and digestion of dietary protein first leads to an acute rise in blood amino acid levels and in turn tissue amino acid levels (including the atherosclerotic plaque). Upon exposure to rising amino acid levels, mTORC1 is activated in plaque macrophages. A critical downstream effect of activated mTORC1 is inhibition of mitochondrial autophagy (mitophagy). The resultant buildup of dysfunctional mitochondria triggers intrinsic apoptosis pathway. Enhanced apoptosis of plaque macrophages contributes to necrotic core formation and a rise in plaque complexity (a surrogate of the vulnerable plaque).
First, the activation of mTORC1 by amino acids initiates the phosphorylation of numerous downstream targets involved in not only limiting autophagy but promoting cellular biosynthetic pathways, proliferation, and growth24. Thus, although our data supports the inhibition of macrophage autophagy/mitophagy as a prominent underlying etiology of high protein diet-induced atherosclerosis, this phenotype is likely also contributed to by other mTORC1-dependent pathways. For example, we clearly see a 2-fold increase in the degree of apoptosis and necrotic core progression in the high protein setting whereas lesion size and macrophage content are only elevated by 30%. Macrophage apoptosis and its contribution to plaque complexity is the predominant feature exacerbated by autophagy inhibition. However, increases in lesion macrophage content and size might not solely be dependent on blunted autophagy and alternate mTORC1-dependent processes such as cellular proliferation should also be considered.
We found only certain amino acids (with leucine leading the list) to be potent activators of mTORC1 in macrophages. This raises the question of whether dietary proteins proportionally enriched in leucine could drive macrophage activation and atherogenesis. Interestingly, leucine content is highest in meats and lowest in vegetable and grain sources25. Since meats are considered more atherogenic than vegetable/grain sources, it is possible the extra leucine load provided by chronic meat intake is an additional mTORC1 stimulus and enhances risk for plaque progression. This can be addressed by conducting future studies similar to those we performed with modification of proteins based on the content of leucine and other amino acids.
Although our data demonstrates that the amino acid load provided by high protein feeding is a critical stimulus for mTORC1 signaling in macrophages and a driver of atherosclerosis, we cannot exclude the interactive roles of other dietary nutrients in plaque progression. Most recently, cholesterol loading derived from lipoprotein hydrolysis in lysosomes was found to be an independent activator of the mTORC1 complex in a mechanism not unlike lysosomal amino acid sensing14,26. Indeed, we observed leucine was not alone sufficient to induce macrophage apoptosis, but could synergistically enhance apoptosis with 7-ketocholesterol or cholesterol crystals. Since the presence of dietary lipids including cholesterol is essential for atherogenesis in both animal models and humans, it is intriguing to consider whether the high protein-induced atherosclerosis can be synergistically exacerbated in the setting of increased dietary cholesterol. This has relevance clinically since there is common consensus that “red meat” protein sources (which have high contents of both proteins and lipids) pose the most significant cardiovascular risk27. Moreover, high dietary protein might also need to be evaluated in the context of low dietary carbohydrates. Elevated dietary ratio of protein to carbohydrates (P:C) is an important exacerbator of cardiometabolic indices such as blood pressure, glucose intolerance, and circulating lipid levels which would all contribute to cardiovascular risk 28. It would also be interesting to consider the relevance of the P:C ratio on macrophage mTORC1 signaling and plaque progression.
Many popular dietary regimens designed for weight loss or body building rely on elevated protein content. Although effective at achieving weight loss, the cardiovascular risk of such diets has been proposed but remains controversial. We now provide a detailed mechanistic basis underlying this cardiovascular risk and pinpoint the mTORC1-autophagy signaling axis in macrophages as a critical mediator of this risk. The modulation of mTORC1 signaling in disease states is an area of therapeutic interest in many fields including cardiovascular disease. Rapamycin is a well-known mTORC1 inhibitor with atheroprotective properties29. Although systemic side-effects preclude its practical therapeutic use for chronic disease such as atherosclerosis, the critical role of amino acids on macrophage mTORC1 signaling suggests that therapies aimed at reducing macrophage amino acid load, lysosomal amino acid sensing, mTORC1 recruitment, or selective mTORC1 enzymatic activity should all be focus areas for discovery of novel therapeutics.
MATERIALS AND METHODS
Animals
Animal protocols were approved by the Washington University Animal Studies Committee. All mice used in this study were on C57BL/6J background (>N7). Mice with tissue-specific Raptor-deficiency (Raptor-KO) and ATG5-deficiency (ATG5-KO) have been extensively characterized15,30. These mice were crossed with Cre-recombinase transgenic mice under the control of the Lysozyme-M promoter (LysM-Cre) to generate macrophage-specific Raptor-KO (mϕRaptor-KO) and ATG5-KO (mϕATG5-KO) mice11,12,18,21. Crosses between mϕRaptor-KO, mϕATG5-KO, and ApoE-KO mice generated mice with mϕRaptor-KO, mϕATG5-KO, mϕRaptor-KO/ATG5-KO, or littermate controls on an ApoE-KO background. Raptor, ATG5, Cre, and ApoE genotyping was performed using standard PCR techniques12,15,30,31. Mice housed in a specific pathogen–free barrier facility were weaned at 3 weeks of age to a standard mouse chow providing 6% calories as fat. For in vivo experiments, male mice of the stated genotype or littermate Controls (flox/flox without LysM-Cre) were started at ~8 weeks of age with Standard (TD88137: 0.15% cholesterol, fat / protein / carbohydrate calories (kcal%) at 42% / 15% / 43% respectively) or High Protein (TD04524: similar cholesterol/fat content but with fat / protein / carbohydrate calories (kcal%) at 43% / 46% / 11%) Western-type diets (all from Harlan). For in vitro experiments, macrophages derived from mice aged between 2 to 6 months were used and within each experiment only mice with the same sex and similar age were compared.
Amino acid starvation and stimulation of cells
Amino acid-rich RPMI 1640 (Sigma, R8758) and amino acid-free RPMI 1640 (USBiological Life Science, R8999–04A) media containing 10% dialyzed FBS (Sigma, F0392) were used for macrophage cell culture experiments. When testing the specific effect of individual amino acids on mTORC1 signaling and autophagy, amino acid-free RPMI 1640 was supplemented with an amino acid concentration 2x (0.76 mM) of what is present in RPMI 1640. Only for alanine (which is not normally present in RPMI 1640 media), the 2x concentration was based on levels present in an alternate RPMI medium (Sigma, R2405)32. When testing the specific effect of individual amino acids on apoptotic cell death, mitochondrial dysfunction, and mitophagy, amino acid-free RPMI 1640 medium was supplemented with an amino acid concentration 5x (1.91 mM) of what is present in RPMI 164032.
Macrophage Culture and Treatment
Standard techniques were used to isolate thioglycollate-elicited peritoneal macrophages and bone marrow-derived macrophages12,21,33. Briefly, mice were injected with 4% sterile thioglycollate media (Sigma, T9032) intraperitoneally, and 4 days later, lavaged peritoneal macrophages were counted and plated (DMEM with 10% fetal bovine serum). Where indicated, the following treatments were performed on macrophages: 7-ketocholesterol (50μM; Sigma, C2394), FCCP (20 μM; Sigma, C2920), rotenone (40 μM; Sigma, R8875), bafilomycin (100 nM; Sigma, B1793), and cholesterol crystals (500 μg/ml)18. Cholesterol crystals were generated by ethanol precipitation of cholesterol powder. Cholesterol powder (Sigma C8667) was dissolved in 100% ethanol at a concentration of 10 mg/ml by heating to 60°C. Crystals were allowed to form at −20°C, sedimented by centrifugation, resuspended in PBS at 50 mg/ml, and sonicated extensively. Treated cells were harvested at various times for protein isolation using standard techniques, stained with antibodies for FACS analysis, or fixed with 4% paraformaldehyde for immunofluorescence microscopy.
Isolation of Splenic Macrophages
Dissected spleens were minced and single cell suspensions prepared using a 70-μm cell strainer. Cells were washed with 1% BSA in PBS and re-suspended in MACS buffer (0.5%BSA, 2mM EDTA in PBS) followed by incubation with CD11b microbeads (MiltenyiBiotec 130–049-601) for 15 min at 4 °C. Cells were washed, centrifuged at 500 × g for 10 min at 4 °C, re-suspended in MACS buffer, and transferred to an equilibrated MS separation column (MiltenyiBiotec 130–041-301) located in a MACS magnetic separator (MiltenyiBiotec 130–092-168). The column was washed with MACS buffer, bound CD11b+ fraction collected by a plunger, and after centrifugation (500 × g for 5 min at 4 °C), the cell pellet was used for RNA isolation.
Fluorescence Activated Cell Sorting (FACS)
FACS analysis for mitochondria dysfunction and ROS levels was performed on cultured macrophages and macrophages derived from atherosclerotic mouse aortas. Cultured macrophages were plated (Greiner 665102), treated with the indicated reagents, and incubated with MitoTracker Red CMXRos (250 nM; Life Technologies, M7512), MitoTracker Green FM (200 nM; Life Technologies, M7514) at 37°C for 30min, or MitoSOX Red mitochondrial superoxide indicator (5 μM; Life Technologies, M36008) at 37°C for 10min. Cells were collected and resuspended in FACS buffer (2% FBS in PBS) for subsequent flow cytometry. For aortic macrophages, dissected/cleaned aortas (extending from the aortic root to the abdominal aorta at the level of the renal arteries) were incubated at 37ºC for 60min in a digestion buffer consisting of RPMI, 2.5 μg/mL Liberase (Roche 05401127001), 125 μg/mL DNAse 1 (Sigma D4527), and 0.8 mg/mL hyaluronidase (Sigma H3506). Single cell suspensions were prepared using a 70-μm cell strainer and incubated with MitoTrackerR Red CMXRos and MitoTrackerR Green FM at 37°C for 30min, or MitoStatus TMRE (Tetramethylrhodamine ethyl ester) (200 nM; BD Pharmingen™, 564696) at 37°C for 20min. Cells were then labeled with Pacific Blue-conjugated CD45 (BioLengend 103126, 1:200), FITC-conjugated F4/80 (BioLengend 123108, 1:200), and PerCP-Cy5.5-conjugated CD11b (BioLengend 101228, 1:200) antibodies on ice for 30min, washed, and resuspended in FACS buffer for analysis.
FACS analysis for phosphorylated S6 ribosomal protein (phospho-S6) was performed on mouse spleens and aortas. For splenic macrophages, spleens were minced, filtered to single cell suspensions using a 70μm cell strainer, subjected to red blood cell lysis, and stained with fluorochrome-conjugated macrophage markers CD45, F4/80, and CD11b (as above). Atherosclerotic macrophages were isolated and stained as above. Cells were then fixed with 4% paraformaldehyde, permeabilized with 0.3% saponin, and incubated with p-S6 (Cell Signaling Technology 4856S, 1:200) followed by Alexa Fluor® 488 secondary antibody (Invitrogen A11008; 1:500).
FACS analysis for apoptosis was performed on cultured macrophages. Macrophages were plated, treated and harvested as described above, washed twice with annexin-binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4) and stained with Alexa Fluor® 488 Annexin V (Life Technologies, A133201; 1:50) and propidium iodide (500ng/ml; Sigma, P4170) at room temperature for 15min.
All samples were analyzed using the BD Biosciences Canto II or LSR II flow cytometer and quantified using FlowJo software. Gating strategy for splenic and aortic macrophages used sequential gating on SSC-A/FSC-A, FSC-H/FSC-W, SSC-H/SSC-W, CD45/FSC-A, F4–80/CD11b followed by gating for the specific marker of interest (i.e. pS6, Mitotracker, and TMRE). Gating strategy for cultured PMACs and BMDMs used SSC-A/FSC-A followed by gating for the specific marker of interest (i.e. Annexin V/PI, Mitotracker, MitoSox, and TMRE).
Immunofluorescence Microscopy
Immunofluorescence (IF) imaging of macrophages and frozen-tissue sections was performed follows34. Cells or tissues were fixed with 4% paraformaldehyde, blocked and permeabilized (1% BSA, 0.2 % milk powder, 0.3% Triton X-100 in TBS; pH 7.4), and incubated with the antibodies sequentially. Specificity of staining was tested in control experiments either by omitting primary antibodies or using samples from knock-out mice where available. The following primary antibodies were used in 1:250 dilutions: p62 (Progen Biotechnik, GP62-C), MOMA2 (AbDSerotec, MCA519C), LC3 (MBL, PM036), p-S6 (Cell Signaling Technology 4856), mTOR (Cell Signaling Technology, 2983), Lamp2 (Abcam, ab13524), COX IV (Abcam, ab14744) and CD68 (Biorad, MCA1957). Species-specific fluorescent secondary antibodies were obtained from Invitrogen/Life Technologies (1:250). CellEvent Caspase-3/7 Green Detection Reagent (Life Technologies, C10423), DeadEndFluorometric TUNEL System (Promega, G3250), Dihydroethidium (DHE) (Life Technologies, D11347) were used according to the manufacturer’s protocol. A Zeiss LSM-700 confocal microscope was used for image acquisition and images quantified using ZEN microscope software (Carl Zeiss AG).
Assessment of Mitophagy using mt-Keima
For live imaging analysis of mitophagy, macrophages were plated on glass-bottom culture dishes (Mattek Corporation, P35G-1.5–10-C) and were transduced with mt-Keima lentiviral vector35,36. Macrophages were then treated with indicated reagents and live fluorescence imaging conducted using a Nikon A1Rsi Confocal Microscope with Tokai-hit stage-top incubator at 37°C and 5% CO2. Cells were imaged with 458nm (shown in green) or 561nm (shown in red) light excitation. Image analysis and quantification of 561/458nm fluorescence ratio was performed using ImageJ software, regions of interest with thresholds were determined, and signal over threshold was quantified.
Western Blotting
Cells were lysed in a standard RIPA lysis buffer (150 mM NaCl; 10 mM Tris-HCl, pH 7.2; 0.1% Triton X-100; 1% sodium deoxycholate; 5 mM EDTA) containing protease inhibitor cocktail (Sigma-Aldrich, 4693132001) and phosphatase inhibitors on ice. Lysed samples were centrifuged at 10,000 g for 10 min. Standard techniques was used for protein quantification, separation, transfer, and blotting34. The following primary antibodies were used: LC3 (Novus Biologicals, NB100–2220, 1:1500), ULK1 (Sigma, A7481, 1:2000), β-actin (Sigma, A2066, 1:2000). p-ULK1 (14202, 1:2000), p-S6K (9234, 1:1000 ), S6K (2708, 1:2000), p-S6 (2215, 1:1000), S6 (2217, 1:2000), cleaved Caspase-9 (9509, 1:1000), and Caspase-9 (9504, 1:2000) were all from Cell Signaling Technology.
Quantification of Amino Acids Levels by Mass spectrometry
GC-MS (Gas chromatography-mass spectrometry) was used to quantify L-amino acid levels from mouse serum, isolated splenic macrophages, and aorta. The corresponding isotopic-labeled amino acids were used as internal standards during sample preparation. Samples were extracted into isopropanol:acetonitrile:water (3:3:2), centrifuged at 18,000g for 15min, and dried under N2 gas. N-Methyl-N-(Trimethysilyl) trifluoroacetamide (MSTFA) with 10% pyridine in CH3CN was then used to derivatize samples for analysis by GC-MS using an Agilent 7890A gas chromatograph interfaced to Agilent 5975C mass spectrometer and HP-5ms gas chromatography column (30 m/0.25-mm internal diameter/0.25-μm film coating). A temperature gradient (initially 80°C for 2 min) was linearly increasing by 10°C/min up to 300°C and held for 2 min. Samples were subjected to electron ionization (EI) mode using source temperature (200°C), electron energy (70 eV), emission current (300 μA), and injector/transfer line temperatures (250°C).
Caspase-8/9 Activity Assay
Caspase-8/9 activity was measured using Caspase-Glo® 8 and Caspase-Glo® 9 Assay Systems (Promega, G8200 and G8210). Generally, 2.5× 104 macrophages were plated inside 96 well plates and treated as described inside the figure legend. 100μl Reagent was added to each well including blanks and mixed with contents of wells using a plate shaker at 300–500rpm for 0.5–2 min. Incubate at room temperature for 30 min. Luminescence was recorded via a microplate reader (Tecan Group Ltd, infinite M200 PRO).
Analytical Procedures and Lesion Quantification
Serum metabolites (cholesterol, triglycerides, glucose, free fatty acids) were assayed from blood obtained from mice after 6-hour fast per manufacturer’s protocols (Thermo Scientific TR13421, TR22421, TR15408, and Wako Diagnostics HR Series NEFA-HR(2)).
For glucose tolerance test, mice were fasted for 6 hours, injected with 10% D-glucose (1 g/kg), and tail vein blood obtained at different time points was analyzed using a glucometer (Contour, Bayer Healthcare, Mishawaka, IN).
Serum L-amino acid were measured from mice fed standard or high protein Western diets for 2 month or from fasted mice given an oral protein gavage for the indicated time points using a colorimetric L-amino acid assay kit (Abcam, ab65347) following the manufacturer’s protocol.
Quantification of atherosclerosis at the aortic root was as follows12,21. PBS-perfused hearts were placed in a cryostat mold containing tissue freezing medium. 10 μm thick sections were taken from the samples beginning just caudal to the aortic sinus and extending into the proximal aorta. Slides were fixed with 4% paraformaldehyde and stained with Oil Red O. Images were taken by EVOS XL Core Cell Imaging system and Oil Red O positive regions were quantified using ZEN microscope software (Carl Zeiss AG).
Statistical Analyses
Statistical significance of differences was calculated using the Student unpaired t test or ANOVA (for multiple groups) followed by either Dunnett’s test (when multiple groups are compared with a single control) or Tukey’s multiple comparison test. Graphs containing error bars show the mean +/− standard error of the mean (SEM). Statistical significance is represented as follows: *P < 0.05, **P < 0.01, ***P < 0.001, NS=not significant. The raw data the support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH R01 HL125838, VA MERIT I01 BX003415, American Diabetes Association #1–18-IBS-029, Washington University Diabetic Cardiovascular Disease Center and Diabetes Research Center Grant (P30 DK020579), Washington University Mass Spectrometry core grants (P41GM103422 and P30DK056341), and the Foundation for Barnes-Jewish Hospital.
Footnotes
COMPETING FINANCIAL INTEREST
The authors declare no competing financial interest.
Data Availability
The data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1.Moore KJ & Tabas I Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355, doi: 10.1016/j.cell.2011.04.005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardner CD et al. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA : the journal of the American Medical Association 297, 969–977, doi: 10.1001/jama.297.9.969 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Halton TL et al. Low-carbohydrate-diet score and the risk of coronary heart disease in women. The New England journal of medicine 355, 1991–2002, doi: 10.1056/NEJMoa055317 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Hu FB et al. Dietary protein and risk of ischemic heart disease in women. Am J Clin Nutr 70, 221–227 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Lagiou P et al. Low carbohydrate-high protein diet and incidence of cardiovascular diseases in Swedish women: prospective cohort study. BMJ 344, e4026, doi: 10.1136/bmj.e4026 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Debry G Dietary proteins and atherosclerosis. (CRC Press, 2004). [Google Scholar]
- 7.Foo SY et al. Vascular effects of a low-carbohydrate high-protein diet. Proceedings of the National Academy of Sciences of the United States of America 106, 15418–15423, doi: 10.1073/pnas.0907995106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolfson RL & Sabatini DM The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab 26, 301–309, doi: 10.1016/j.cmet.2017.07.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Y et al. A dietary quality comparison of popular weight-loss plans. Journal of the American Dietetic Association 107, 1786–1791, doi: 10.1016/j.jada.2007.07.013 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson JW, Konz EC & Jenkins DJ Health advantages and disadvantages of weight-reducing diets: a computer analysis and critical review. Journal of the American College of Nutrition 19, 578–590 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Razani B et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 15, 534–544, doi: 10.1016/j.cmet.2012.02.011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sergin I et al. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Science signaling 9, ra2, doi: 10.1126/scisignal.aad5614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sancak Y et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303, doi: 10.1016/j.cell.2010.02.024 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zoncu R et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683, doi: 10.1126/science.1207056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sengupta S, Peterson TR, Laplante M, Oh S & Sabatini DM mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468, 1100–1104, doi: 10.1038/nature09584 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Ai D et al. Disruption of mammalian target of rapamycin complex 1 in macrophages decreases chemokine gene expression and atherosclerosis. Circulation research 114, 1576–1584, doi: 10.1161/CIRCRESAHA.114.302313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li N et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. The Journal of biological chemistry 278, 8516–8525, doi: 10.1074/jbc.M210432200 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Emanuel R et al. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arteriosclerosis, thrombosis, and vascular biology 34, 1942–1952, doi: 10.1161/ATVBAHA.114.303342 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao X et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 15, 545–553, doi: 10.1016/j.cmet.2012.01.022 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouimet M et al. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 13, 655–667, doi: 10.1016/j.cmet.2011.03.023 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sergin I et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nature communications 8, 15750, doi: 10.1038/ncomms15750 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans TD, Sergin I, Zhang X & Razani B Target acquired: Selective autophagy in cardiometabolic disease. Science signaling 10, doi: 10.1126/scisignal.aag2298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun N et al. Measuring In Vivo Mitophagy. Molecular cell 60, 685–696, doi: 10.1016/j.molcel.2015.10.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laplante M & Sabatini DM mTOR signaling in growth control and disease. Cell 149, 274–293, doi: 10.1016/j.cell.2012.03.017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.[FAO: nutritional studies: amino-acid content of foods and biological data on proteins]. FAO nutritional studies, 1–285 (1970). [PubMed] [Google Scholar]
- 26.Castellano BM et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355, 1306–1311, doi: 10.1126/science.aag1417 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernstein AM et al. Major dietary protein sources and risk of coronary heart disease in women. Circulation 122, 876–883, doi: 10.1161/CIRCULATIONAHA.109.915165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solon-Biet SM et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab 19, 418–430, doi: 10.1016/j.cmet.2014.02.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurdi A, De Meyer GR & Martinet W Potential therapeutic effects of mTOR inhibition in atherosclerosis. British journal of clinical pharmacology 82, 1267–1279, doi: 10.1111/bcp.12820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hara T et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889, doi:nature04724 [pii] 10.1038/nature04724 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Razani B et al. Fatty acid synthase modulates homeostatic responses to myocardial stress. The Journal of biological chemistry 286, 30949–30961, doi: 10.1074/jbc.M111.230508 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jewell JL et al. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198, doi: 10.1126/science.1259472 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Febbraio M et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 105, 1049–1056, doi: 10.1172/JCI9259 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Razani B et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276, 38121–38138, doi: 10.1074/jbc.M105408200 [pii] (2001). [DOI] [PubMed] [Google Scholar]
- 35.Katayama H, Kogure T, Mizushima N, Yoshimori T & Miyawaki A A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chemistry & biology 18, 1042–1052, doi: 10.1016/j.chembiol.2011.05.013 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Sun N et al. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nature protocols 12, 1576–1587, doi: 10.1038/nprot.2017.060 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.