

SUMMARY
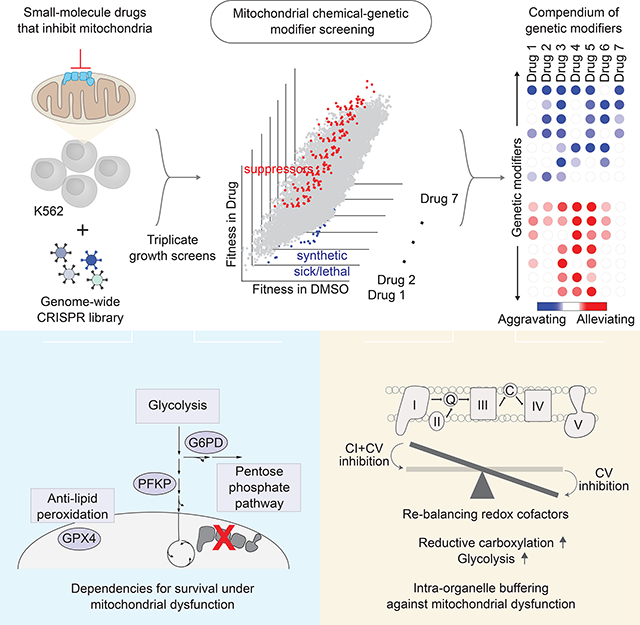
Mitochondrial dysfunction is associated with a spectrum of human conditions, ranging from rare, inborn errors of metabolism to the aging process. To identify pathways that modify mitochondrial dysfunction, we performed genome-wide CRISPR screens in the presence of small-molecule mitochondrial inhibitors. We report a compendium of chemical-genetic interactions involving 191 distinct genetic modifiers, including 38 that are synthetic sick/lethal and 63 that are suppressors. Genes involved in glycolysis (PFKP), pentose phosphate pathway (G6PD), and defense against lipid peroxidation (GPX4) scored high as synthetic sick/lethal. A surprisingly large fraction of suppressors are pathway-intrinsic and encode mitochondrial proteins. A striking example of such an “intra-organelle” buffering is the alleviation of a chemical defect in complex V by simultaneous inhibition of complex I, which benefits cells by re-balancing redox cofactors, increasing reductive carboxylation, and promoting glycolysis. Perhaps paradoxically, certain forms of mitochondrial dysfunction may best be buffered with “second site” inhibitors to the organelle.
Graphical Abstract
INTRODUCTION
Defects in mitochondrial function accompany a spectrum of conditions, ranging from rare inborn errors of metabolism to the aging process itself. To date, nearly 300 rare, monogenic forms of mitochondrial disease have been identified (Frazier et al., 2017) – each presenting typically in a devastating manner with tissue-specific pathology – making these disorders a very common collection of inborn errors of metabolism. Mitochondrial disorders typically present in a devastating manner during childhood with tissue-specific pathology, and there are no proven effective therapies available. Many common diseases, including Parkinson’s disease and type 2 diabetes, are also characterized by mitochondrial dysfunction, though an important question is whether organelle dysfunction is a cause or consequence of end pathology (Vafai and Mootha, 2012). A quantitative decline in mitochondrial abundance and activity has emerged as a signature of the aging process across different organisms (Trounce et al., 1989; Zahn et al., 2007).
How mitochondrial defects lead to such pleiotropic and heterogeneous pathologies is a major unsolved problem, and addressing this problem is challenging for several reasons. First, the organelle is highly complex, housing numerous coupled metabolic pathways that tend to be densely interconnected. Lesions at different points within the same mitochondrial pathway can often have highly non-uniform biochemical consequences. For example, defects in complex I and III both cause a decline in ATP production by oxidative phosphorylation (OXPHOS), but have opposite effects on de novo pyrimidine biosynthesis (Shaham et al., 2010). Second, the organelle’s machinery is coupled, often with redundancy, to metabolic and regulatory pathways in other cellular compartments. For example, in cultured cells glycolysis is capable of compensating for ATP production from OXPHOS as long as glucose is provided in the media. Third, mitochondrial dysfunction can activate stress responses (Bao et al., 2016), and in some instances buffering against toxicity, while in other instances serving as effectors of end pathology. Fourth, non-linear feedback loops help to ensure energetic and redox homeostasis acutely in response to stressors and over long-term, adaptive timescales (Balaban et al., 1986; Chance and Williams, 1955; Cogliati et al., 2013). It is clear that genetic networks support mitochondrial function and can buffer against or contribute to pathology. However, these networks have not been systematically mapped.
Genetic modifier screening on a sensitized chemical background presents a powerful means to decipher cellular networks (Hillenmeyer et al., 2008). Inspired by the concept of genetic interactions, the goal of such “chemical-genetics” approaches is to compare the combined effects of a genetic perturbation and a chemical insult. Such approaches can help to reveal genetic circuitry centered on the targets of well-studied drugs. While genome-wide chemical-genetic approaches have been applied extensively in yeast, the technology has only recently become available to extend these approaches to mammalian cells (Hart et al., 2015; Shalem et al., 2014; Wang et al., 2014). Recently, several studies have showcased the power of targeted CRISPR screening (Birsoy et al., 2015) and even genome-scale CRISPR screening (Arroyo et al., 2016) in mammalian cells to uncover new aspects of mitochondrial biology. The genome-scale CRISPR approach has even helped to nominate hypoxia as a novel and seemingly counterintuitive therapeutic approach for mitochondrial dysfunction (Jain et al., 2016). As these studies demonstrated, genome-scale approaches afford the opportunity to identify cellular pathways operating inside or outside of mitochondria that control the organelle, without prior assumptions of the nature of interactors.
Here we report the creation of a genome-scale compendium of chemical-genetic interactions focused on mitochondrial biology. We use a collection of mitochondrial inhibitors that allows us to model distinct, canonical modes of “mitochondrial dysfunction,” including defects in replication and translation of the mitochondrial genome (mtDNA), defects in individual complexes of the electron transport chain, impaired ATP formation by OXPHOS via the F1Fo-ATPase, and deficits in membrane potential polarization. We perform genome-wide CRISPR screens in triplicate to identify chemical-genetic interactions that are either synthetic sick/lethal or alleviating. We report a compendium of chemical-genetic interactions involving 191 distinct nuclear genes. These interactions fall into different categories, including genetic modifiers that suppress mitochondrial dysfunction, as well as those that aggravate it. About 65% of these modifiers themselves encode mitochondrial-localized proteins underscoring dense connectivity within the organelle. We illustrate how our resource can be useful by reporting four key results, including the surprising finding that the loss of complex I activity (genetically or with drugs such as metformin or piericidin) is capable of suppressing cellular defects stemming from chemical inhibition of complex V.
RESULTS
Genome-wide CRISPR Modifier Screens in the Presence of Mitochondrial Inhibitors
To systematically uncover genetic pathways that aggravate or suppress mitochondrial dysfunction, we combined genome-wide CRISPR growth screening on sensitized backgrounds in which mitochondrial physiology is inhibited at distinct nodes (Figure 1A). We modeled mitochondrial dysfunction using small molecule inhibitors that target distinct aspects of mitochondrial physiology: respiratory chain complex I (piericidin), complex III (antimycin), complex V (oligomycin), mitochondrial membrane potential (antimycin+oligomycin), mitochondrial DNA replication (ethidium bromide), and mitochondrial protein translation (chloramphenicol). We also included metformin, whose proposed targets includes complex I (Owen et al., 2000; Wheaton et al., 2014), but may include other targets as well (Madiraju et al., 2014). We chose the minimal drug concentrations that were sufficient to blunt OXPHOS activity in K562 cells (see the legend of Figure 1B for drug doses) while allowing cell proliferation. We confirmed complete OXPHOS inhibition by the observation of massive cell death in galactose but not glucose (Arroyo et al., 2016) in K562 cells (Figure S1A). We observed a high dosage requirement for metformin to inhibit respiration (Figure S1B), consistent with what has been seen in prior studies (Wheaton et al., 2014). In addition, we confirmed the efficacy of ethidium bromide in depleting mtDNA (Figure S1C), chloramphenicol in inhibition of mitochondrial translation (Figure S1D), and antimycin+oligomycin in collapsing membrane potential (Figure S1E).
Figure 1. Genome-Wide CRISPR Screens to Identify Modifiers of Mitochondrial Dysfunction.

A. Mitochondrial chemical inhibitors used in this study: piericidin (complex I), antimycin (complex III), oligomycin (complex V), antimycin+oligomycin (ΔΨm), ethidium bromide (mitochondrial DNA replication), chloramphenicol (mitochondrial translation), and metformin (complex I is among the proposed targets).
B. Schematic overview of the genome-wide CRISPR screens in K562 cells. The following drug dosages were used: 0.1% DMSO, 10 mM metformin (Met), 10 nM piericidin (Pier), 100 nM antimycin (Anti), 10 nM oligomycin (Oligo), 10 nM antimycin + 10 nM oligomycin (AO), 100 ng/mL ethidium bromide (EtBr), and 10 μg/mL chloramphenicol (CAP).
C. Growth curves for cumulative differences in growth under drug treatments. Growth curves for individual replicates over 15 days are shown. Gray arrows denote time points at which samples were harvested.
D. Categories of genetic interactions and the number of genetic modifiers associated with each category. Scatter plots of Z-scores showing knockouts that are enriched or depleted in each drug vs. DMSO. Knockouts are scored by ΔZDrug = (ZDrug−ZDMSO)/√2 (see Methods). Knockout enrichment (shown in red or magenta, ΔZDrug > 4.8) and depletion (shown in blue, ΔZDrug < −2.4) are used to define buffering and synthetic sick/lethal interactions, respectively. Among buffering interactions, suppressors (ZDrug > 2.4) are shown in red. Known genetic modifiers (vHL and ATPIF1 as genetic suppressors of OXPHOS inhibitors, and GOT1 loss as aggravator) are highlighted. The gray dotted lines represent the cutoffs for interactions.
We performed genome-wide CRISPR modifier screens in biological triplicate in K562 cells using an optimized sgRNA library (Doench et al., 2016) (Figure 1B). Seven days after infection, cells were split and treated with DMSO or one of 7 different drug treatments, and cultured for an additional 15 days in media containing high glucose (25 mM), pyruvate and uridine. To proliferate, cells lacking a functional electron transport chain are dependent on glucose to fuel high glycolytic activity for ATP generation, exogenous pyruvate for recycling NADH into NAD+ through lactate dehydrogenase, and exogenous uridine for pyrimidine salvage (King and Attardi, 1989; Morais et al., 1980). The sgRNAs were PCR amplified from the genomic DNA of harvested cells. Next-generation sequencing was performed and the abundance of each sgRNA in the drug treatment on day 15 was compared to the day 0 baseline. The growth curves for cumulative differences show moderate growth defects – i.e. slower but steady proliferation – in cells under metformin, piericidin, antimycin, and chloramphenicol (Figure 1C).
On the other hand, oligomycin, antimycin+oligomycin and ethidium bromide treatments appear to impact proliferation more significantly (Figure 1C). The growth curves demonstrate consistent effects of each drug across the biological triplicates. Of note, two of our “day 15 samples”, one for DMSO and one for metformin, were lost during the PCR step, yielding a total of 22 (out of 24) successful 15-day screens (Table S2).
Of the 19,000 genes targeted by our CRISPR library, 11,102 genes are expressed in K562 cells and could be scored for their chemical-genetic interactions (Figure 1D). By comparing the fitness of a gene’s loss in the presence of a drug (Zdrug) to the fitness of that same gene’s loss in the absence of a drug (ZDMSO), we can define the relative fitness of a knockout in drug vs. DMSO simply as ΔZDrug = (ZDrug − ZDMSO)/√2 (see Table S1 and Methods). We classify 38 hits as “synthetic sick/lethal” with ΔZDrug < −2.4 and 154 genes as “buffering” with ΔZDrug > 4.8 (we use a higher threshold on this side of the screen, since in a growth screen, knockouts are much more likely to be depleted by chance alone than enriched because of drop outs). Amongst the 154 genes involved in buffering interactions, we find a subset of 63 genes are additionally enriched on day 15 as compared with baseline on day 0 (i.e., ZDrug > 2.4). These 63 genetic modifiers with proliferative advantages are termed “suppressors,” whereas the other 91 are characterized as “epistatic buffering” (Figure 1D), a term that is used to describe an interaction between two deleterious effects (Jasnos and Korona, 2007). Of note, only loss of CYC1 (complex III component) shows opposing effects in two treatments – it aggravates antimycin+oligomycin, but epistatically buffers (without alleviating) oligomycin. To buttress the above analysis pipeline, we independently scored the genes using the model-based MAGeCK algorithm (Li et al., 2014) (Table S1 and Methods). The results from MAGeCK are highly consistent with the ΔZDrug scores. The fact that two different data analysis methods arrive largely at the same “hits” indicates that our results are not sensitive to the analysis type.
Of the 38 distinct synthetic sick/lethal hits that aggravate mitochondrial dysfunction, 23 (61%) interact with only one drug, and 15 (39%) have interactions shared by multiple drugs (Figure 2A). Of the 63 distinct suppressors, 31 (49%) interact with only one drug, and 32 (51%) have interactions shared by multiple drugs (Figure 2B). Importantly, the synthetic sick/lethal hits and suppressors are considered candidate genetic modifiers until proven in targeted experiments outside the context of a pooled screen.
Figure 2. Genetic Modifiers of Distinct Modes of Mitochondrial Dysfunction.

A. The 38 synthetic sick/lethal hits that score in ≥ 1 drug (ΔZDrug < −2.4).
B. The 63 suppressors that score in ≥ 1 drug (ΔZDrug > 4.8, and ZDrug > 2.4)
For (A) and (B), genes are further divided into two panels. The left panel contains genes that have interactions with multiple drugs, and is ordered by the number of drugs under which interactions occur. The right panel contains genes with only drug-specific interactions, and genes are listed by the specific drug and ordered by the strength of interaction. Gene-drug pairs that do not score are colored white (−2.4 ≤ DZDrug ≤ 4.8).
C. Counts of modifiers that are implicated in intra-mitochondrial vs. extra-mitochondrial interactions. The expected fraction of “intra-mitochondrial” in each category is 6%, as only 6% of all nuclear genes encode mitochondrial proteins.
Four observations suggest the robustness of the screening results. First, the guide counts between biological replicates are well-correlated as visualized by principal component analysis (Figure S1F). Second, the negative-controls guides (1,000 non-targeting sgRNAs, and sgRNAs for 3,726 non-expressed genes in K562) remain tightly distributed over the course of the screens among all treatments, whereas the guides for the core essential genes (Hart et al., 2015) are as expected strongly depleted at the endpoint (day 15) of the screen (Figure S1G, Table S3). Third, we recover the three published human mitochondrial chemical-genetic interactions, including those that involve vHL (Jain et al., 2016), ATPIF1 (Chen et al., 2014), and GOT1 (Birsoy et al., 2015) (Figure 1D). Fourth, we validated newly identified aggravating and buffering interactions in targeted experiments (Figures 3–6).
Figure 3. Losses of Genes Involved in Glycolysis or Pentose Phosphate Pathway are Synthetic Sick/Lethal with OXPHOS Dysfunction.

A. Top categories of KEGG pathways among depleted knockouts by Gene Set Enrichment Analysis (GSEA). The 11,102 genes was ranked by ΔZDrug in descending order and GSEA was run in the negative mode. Pathways are ordered by the maximum of −log10 FDR across 7 drugs.
B. Growth phenotypes of K562, A375, or HT-29 cells with sgRNAs directed against the genes indicated under piericidin or antimycin treatment. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). *p<0.05, **p<0.01 or ****p<0.0001 indicates two-tailed Student’s t-test p-value.
Figure 6. Suppression of Oligomycin’s Toxicity by Loss of Complex I Activity.

A. Scatter plot of the fitness of a gene knockout in oligomycin (ZOligo) vs. its fitness in DMSO (ZDMSO). Gray dots denote all 11,102 genes in the analysis and the red dots denote 50 nuclear genes that encode structural subunits and assembly factors of complex I.
B. Validation of NDUFA9 loss, piericidin, and metformin as suppressors of oligomycin in HAP1, 293T, HT-29, HeLa, and K562 cells. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3).
C. Validation of piericidin as suppressors of oligomycin in K562 in the presence (+Uridine) or absence (-Uridine) of 50 mg/mL uridine supplementation. Dialyzed serum was used for -Uridine growth. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). In (B) and (C), *p<0.05 or **p<0.01 indicates two-tailed Student’s t-test p-value.
D. Oxygen consumption as assessed by the Seahorse Extracellular Flux (XFe96) Analyzer in K562 cells after 3 days of drug treatment (average +/− SEM, n = 12).
E. Corrected extracellular acidification rate as assessed by the Seahorse Extracellular Flux (XFe96) Analyzer in K562 after 3 days of drug treatment (average +/− SEM, n = 12).
F. Media lactate levels in K562 using targeted mass spectrometry after 3 days of drug treatment (average +/− SEM, n = 3). *p<0.05 indicates two-tailed Student’s t-test p-value.
G. Volcano plot comparing piericidin+oligomycin to oligomycin from full-scan metabolomics in K562 using mass spectrometry after 3 days of drug treatment (average of n = 3). Highlighting in red indicates *p<0.05 two-tailed Student’s t-test p-value.
H. Schematics for stable isotope labeling with [U-13C]glutamine to monitor 13C incorporation in key metabolites.
I. Total metabolite pools and 13C labeling pattern of citrate, 2-hydroxyglutarate, and proline over the course of 8 hours upon tracer analysis with [U-13C]glutamine in K562 cells. Cells were pretreated in drugs in uridine-free medium 48 hours prior to [U-13C]glutamine supplementation (average +/− SEM, n = 3). *p<0.05 or **p<0.01 indicates two-tailed Student’s t-test p-value.
J. Growth phenotypes of K562 cells with sgRNAs directed against G6PD under oligomycin or piericidin+oligomycin treatment. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). **p<0.01 indicates two-tailed Student’s t-test p-value.
K. Growth phenotypes of cells stably expressing a vector vehicle or cytosolic TPNOX under the specified treatment in K562. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). ns p>0.05 or **p<0.01 indicates two-tailed Student’s t-test p-value. Top: immunoblots for FLAG and the TOMM20 loading control for cells expressing either the vector vehicle or cytosolic TPNOX.
Among the treatments that lead to a more severe growth defect, reduced depletion of sgRNAs targeting essential genes might represent an artifact. That is, sgRNAs may have depleted more in DMSO than in the drug-samples simply due to a large difference in growth rates, which would be misidentified as epistatic buffering interactions under the drug. However, it is unlikely the case as (i) drugs that lead to severe growth defect (oligomycin, antimycin+oligomycin, and ethidium bromide) have a large number of unique genetic interactions (Figure S2A), and (ii) generally essential genes, such as those encoding the cytosolic translation machinery, are not buffered under severe growth defect (Figure S4D). This is in stark contrast with genes that encode the mitochondrial translation machinery, which are significantly buffered under oligomycin and ethidium bromide treatment (Figure S4D).
Of the drugs we tested, metformin is of notable interest since it is one of the most widely used drugs in the world to treat type 2 diabetes, and is now in clinical trials for aging (Barzilai et al., 2016), yet its direct target(s) of action is still debated. Classical studies have clearly demonstrated that complex I is a target (Owen et al., 2000), but there may be additional sites of action (Madiraju et al., 2014). Piericidin and metformin are the most similar with respect to their genome-wide profiles across the battery of drugs we tested (Figures S1F, S2A), consistent with complex I being a target of metformin.
Intra-mitochondrial Chemical-Genetic Interactions are Pervasive
All of the chemicals used in this screen have targets localized within mitochondria (Figure 1A), and hence we can classify chemical-genetic interactions as “intra-mitochondrial” if the identified genetic modifier encodes a mitochondrial protein, or “extra-mitochondrial” if the identified genetic modifier encodes a protein localized outside of mitochondria. Of the 38 synthetic sick/lethal hits (Figure 2A), 20 (53%) are involved in intra-mitochondrial interactions. Of the 63 suppressors (Figure 2B), 46 (73%) are involved in intra-mitochondrial interactions. Of the 91 epistatic buffering hits (Figure S2C), 64 (70%) are involved in intra-mitochondrial interactions (Figure 2C). As genes encoding mitochondrial proteins only account for 6% of the human proteome (Calvo et al., 2016), intra-mitochondrial interactions are dramatically overrepresented among both synthetic sick/lethal hits (Fisher p value = 3.5×10−15) and suppressors (Fisher p value = 8.2×10−43). The overrepresentation of genes encoding mitochondrial proteins among epistatic buffering interactions could be a consequence of the fact that gene losses are redundant with chemical inhibition of the same target. The rationale for overrepresentation of intra-mitochondrial interactions among synthetic sick/lethal hits and suppressors is not obvious, and such interactions may point to dense functional connectivity that occurs inside the organelle, consistent with global yeast genetic interaction screens, where functionally related genes tend to show genetic interactions (Costanzo et al., 2016; Hoppins et al., 2011).
Genes whose Loss Aggravates the Toxic Effects of Mitochondrial Inhibitors
We identify 38 unique genes whose loss aggravates growth defects for ≥ 1 mitochondrial inhibitor (Figure 2A), i.e., they are synthetic sick or lethal with mitochondrial dysfunction. As expected, we recovered GOT1, which has previously been identified to be essential for growth under phenformin treatment (Birsoy et al., 2015). Here, we report the chemical-context specificity of this interaction, as GOT1 loss is aggravated by piericidin and antimycin, but not by the other drugs. Pathway analysis (Figure 3A, Table S4) identifies glycolysis and the pentose phosphate pathway as being enriched. The top scoring multi-drug synthetic sick/lethal hits are G6PD (involved in pentose phosphate) and PFKP (glycolysis). A priori, genes associated with glycolysis are expected to be essential under OXPHOS inhibition, and we recovered multiple genes involved in this pathway (GPI, ALDOA, HK2 and PFKP) or in its regulation (PGP) among the aggravators. Two genes in the pentose phosphate pathway (G6PD and RPE) scored, suggesting an increased demand for NADPH or ribose-5-phosphate in the setting of OXPHOS inhibition.
We validated three of the newly identified synthetic lethal interactions, including GPX4, which we focus on next, G6PD and PFKP. Loss of G6PD or PFKP increases toxicity of multiple mitochondrial inhibitors, including piericidin and antimycin, which we validated in multiple cell lines (Figure 3B). G6PD deficiency represents one of the most common enzymopathies (Cappellini and Fiorelli, 2008) and is polymorphic in hundreds of millions of individuals worldwide. To our knowledge, the synthetic sick/lethal interaction between G6PD loss and mitochondrial dysfunction has not been previously reported. PFKP encodes one of the three isozymes of the Phosphofructokinase 1 (PFK-1) expressed in human cells. Of note, the loss of the other two isozymes (PFKM, PFKL) is tolerated under OXPHOS inhibitions, even when all three isozymes are highly expressed in K562 (Table S1).
GPX4, whose Loss is Synthetic Lethal, is Induced by Mitochondrial Dysfunction to Guard Against Ferroptosis
Glutathione peroxidase 4 (GPX4) scored as one of the strongest hits among our synthetic sick/lethal interactions: loss of this enzyme enhances the toxicity of antimycin, oligomycin, ethidium bromide, and antimycin+oligomycin (Figure S3A). It’s notable that GPX4, along with GOT1, were among the most upregulated proteins in a previous proteomic profiling study in cells depleted of mtDNA (Bao et al., 2016) (Figure 4A), suggesting they may be induced by OXPHOS dysfunction. GPX4 is a selenoprotein (Maiorino et al., 1991; Ursini et al., 1985) that reduces lipid hydroperoxides to their corresponding alcohols to prevent accumulation of toxic byproducts such as malondialdehyde and 4-hydroxynonenal (Maiorino et al., 2018) which can lead to ferroptosis (Dixon et al., 2012; Stockwell et al., 2017; Yang et al., 2014). Moreover, therapy-resistant persister cancer cells are susceptible to loss of GPX4 function and this vulnerability has been proposed as a target for cancer therapies (Hangauer et al., 2017; Viswanathan et al., 2017).
Figure 4. Aggravation of Oligomycin’s Toxicity by Loss of GPX4.

A. Venn diagram showing the overlap between the 38 synthetic sick/lethal hits and the 100 most upregulated proteins in a previous proteomic profiling study in cells depleted of mtDNA (Bao et al., 2016).
B. Growth phenotypes of wild-type cells under a combination of oligomycin and GPX4 inhibition by 10 μM JKE-1674 for K562 in spent medium, HAP1 in fresh medium, and HeLa in fresh medium. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3).
C. Growth phenotypes of GPX4 KO cells in HAP1 under oligomycin treatment in fresh medium supplemented with ferroptosis inhibitors alpha-tocopherol (aTOC, 1 μM) or ferrostatin-1 (Fer-1, 1 μM), or pan-caspase inhibitor zVAD-fmk (1 μM). Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3).
D. Growth phenotypes of GPX4 KO cells in HAP1 under oligomycin with re-expression of, lGPX4, or lGPX4 supplemented with the catalytic site-specific GPX4 inhibitors JKE-1674 (10 μM) or ML 210 (10 μM) in fresh medium. Cell counts were performed 3 days post treatment (average +/− SEM, n = 2).
E. Immunoblots for GPX4 and loading control in wild-type K562, HeLa and HAP1 cells treated with antimycin or oligomycin for 3 days. A representative experiment is shown.
F. Mitochondrial GPX4 levels derived from a proteomics dataset mitochondrial proteome from the heart tissues of five conditional knockout mouse strains with OXPHOS dysfunction (Kuhl et al., 2017). The data shown represent the average +/− SEM (n≥3). In (B)-(D) and (F), *p<0.05, **p<0.01 or ***p<0.001 indicates two-tailed Student’s t-test p-value.
We confirmed the synthetic lethal interaction between loss of GPX4 activity and OXPHOS inhibition by oligomycin (where the screening effect was among the strongest), selecting three cell lines (K562, HAP1, and HeLa) capable of tolerating its loss (Figure 4B) (Meyers et al., 2017). In all three cell types tested, we observed a synthetic lethal interaction between oligomycin and JKE-1674, a new specific chemical inhibitor that acts by covalently binding the catalytic selenocysteine residue of GPX4 without eliminating its expression (Eaton et al., 2018) (Figure S3F) and with an efficacy similar to the commonly used GPX4 inhibitor ML 210 (Figure 4D). We observed that the strong synthetic lethal interaction in K562 requires the cells to be grown in spent medium, as fresh medium only leads to a mild growth defects (Figure S3B). We observed steady-state accumulation of lipid hydroperoxides in cells treated with both oligomycin and GPX4 inhibitor in spent medium (Figure S3C), consistent with the ferroptotic death phenotype. We confirmed an aggravating interaction between GPX4 KO HAP1 cells (Figure S3D) and oligomycin (Figures 4C, 4D). As expected, ferroptosis suppressors alpha-tocopherol and ferrostatin-1, but not the pan-caspase inhibitor zVAD-fmk, rescue the fitness defects of GPX4 KO cells under oligomycin treatment (Figure 4C).
We next sought to rescue this phenotype by re-expressing GPX4 in knockout cells. GPX4 encodes multiple protein isoforms with distinct subcellular localizations (Savaskan et al., 2007). The two major isoforms, short (sGPX4) and long (lGPX4), are distinguished by the first exon (E1a) which encodes the N-terminal sequences of both isoforms but is truncated in sGPX4 (Figure S3E). The 3’ UTR of both isoforms contain a canonical selenocysteine insertion sequence (SECIS) required for GPX4’s catalytic activity (Ingold et al., 2018). Using subcellular fractionation (Figure S3F) and microscopy (Figure S3G), we find that lGPX4 is exclusively localized to mitochondria, whereas the short sGPX4 is primarily localized in the cytosol. Of note, our subcellular fractionation result and a previous study in mice (Liang et al., 2009) indicate that sGPX4 can also be targeted to the mitochondria. Re-expression of the mitochondria-specific lGPX4 in GPX4 KO cells is sufficient to fully rescue the synthetic lethal interaction with oligomycin in a way that could be inhibited with JKE-1674 or ML 210 (Figure 4D). These experiments demonstrate that GPX4 activity within mitochondria is sufficient to rescue the synthetic lethal interaction, thus providing support for mitochondria being an important site for lipid hydroperoxide accumulation and ferroptosis under OXPHOS inhibition. These results also implicate an “intra-mitochondrial” synthetic lethal interaction between GPX4 loss and oligomycin toxicity.
Given that GPX4 loss is synthetic lethal with OXPHOS inhibition and previous proteomics analysis (Figure 4A), we asked whether GPX4 expression might rise in response to mitochondrial dysfunction as an adaptive response. Total GPX4 protein was up-regulated under oligomycin treatment in K562, HeLa, and HAP1 cells (Figure 4E). To evaluate whether up-regulation of GPX4 in response to OXPHOS dysfunction occurs in vivo, we re-analyzed the published mitochondrial proteomes from the heart tissues of five different genetic knockout models of mitochondrial disease (Kuhl et al., 2017) spanning defects in mtDNA replication (Twinkle), mtDNA maintenance (Tfam), mtDNA transcription (Polrmt), mt-mRNA stability and maturation (Lrpprc), and mitochondrial transcription termination (Mterf4). In all five models of mitochondrial disease, mitochondrial-localized GPX4 protein levels are increased (Figure 4F), providing strong evidence that mitochondrial GPX4 is induced in response to OXPHOS mitochondrial dysfunction in vivo. By showing that GPX4 is inducible in multiple cells types and tissues in the face of OXPHOS inhibition, and that its targeted expression within mitochondria can confer protection against OXPHOS inhibition, we propose GPX4 as an adaptive component for survival under OXPHOS dysfunction.
Genes whose Loss is Epistatically Buffered by Mitochondrial Inhibitors
Amongst the 154 genes involved in buffering interactions (i.e., ΔZDrug > 4.8), the majority (91) of gene knockouts show no growth advantage in the primary screen as compared with the wild-type cells under drug treatment, i.e., ZDrug ≤ 2.4 (Figure 1D). In this category (Figure S2C), the effect of gene loss in the presence of drug is eliminated or milder relative to what is observed in the presence of DMSO (and thus enrichment in drug vs. DMSO, which qualifies the gene for a buffering interaction). We describe these interactions as epistatic buffering, a term used in genetics to describe the non-additive effects of two deleterious gene disruptions. Not surprisingly, many of these epistatic buffering interactions are intra-mitochondrial (Figures S2B, S2C). We subsequently validated the buffering interaction between REXO2 loss and ethidium bromide in individually targeted CRISPR KO cells (Figure S4B), providing confirmation to a previous report that REXO2 loss can be tolerated by mammalian rho0 cells lacking mtDNA (Bruni et al., 2013). REXO2 encodes a mitochondrial protein that appears to be involved in mitochondrial nucleic acid degradation. Genes involved in mitochondrial RNA processing are generally buffered under ethidium bromide (Figure S4C). The processes downstream of mtDNA, such as mtRNA processing, are presumably no longer operative in the absence of mtDNA.
We identified a novel extra-mitochondrial epistatic buffering interaction between the loss of LARP1, which encodes a cytosolic protein, and oligomycin (Figure 5A). LARP1 loss also exhibits a sub-threshold interaction with ethidium bromide (Figure 5A). The buffering of LARP1 loss by mitochondrial inhibitors raises the hypothesis that LARP1 is involved in OXPHOS biology. Indeed, knocking out LARP1 gene in K562 leads to a partial reduction in oxygen consumption under basal and maximal respiration (Figure 5B). We also performed the glucose/galactose death assay as described in (Arroyo et al., 2016), and showed that LARP1 loss leads to increased cell death in galactose but not in glucose (Figure 5C). Both results suggest that LARP1 is required for intact OXPHOS capacity.
Figure 5. Loss of Cytosolic Protein LARP1 is Buffered under OXPHOS Inhibition.

A. Scatter plots of Z-scores highlighting knockout of LARP1 in oligomycin (top panel) or ethidium bromide (bottom panel) vs. DMSO. The gray dotted lines represent the cutoffs for interactions.
B. Oxygen consumption as assessed by the Seahorse Extracellular Flux (XFe96) Analyzer in K562 cells with sgRNAs directed against the genes indicated (average +/− SEM, n = 6).
C. Cell death as assessed by Annexin V staining and flow cytometry in K562 cells cultured in glucose or galactose containing medium with sgRNAs directed against the genes indicated (average +/− SEM, n = 3).
D. Heatmap of co-dependency based on hierarchical clustering of Pearson correlations between the mean log2-fold changes across sgRNAs for genes. Disjoint modules (demarcated with red triangles) were annotated by assigned GO term that was significantly enriched in that cluster (hypergeometric p ≤ 10−10). Black ticks represent genes that are listed in the MitoCarta2.0 database (Calvo et al., 2016). The red arrow denotes LARP1. E. Immunoblots for the specified mitochondrial proteins and LARP1 in K562 cells with sgRNAs directed against the genes indicated. A representative experiment is shown.
F. Real time PCR-based measurement of mtDNA relative to nuclear DNA (nDNA) in K562 cells with sgRNAs directed against the genes indicated (average +/− SEM, n = 3).
G. Taqman gene expression analysis of transcripts of OXPHOS components in K562 cells with sgRNAs directed against the genes indicated (average +/− SEM, n = 3). For (C) and (F), ns p>0.05 or ***<0.001 indicates two-tailed Student’s t-test p-value.
LARP1 is known to regulate cytosolic translation through binding to mRNA containing the 5’ terminal oligopyrimidine (TOP) motif, in response to mTOR signaling (Fonseca et al., 2015; Tcherkezian et al., 2014). While LARP1 homologs in S. cerevisiae (Kershaw et al., 2015) and Drosophila (Zhang et al., 2016) have been shown to regulate mitochondrial protein abundance, to our knowledge, involvement of LARP1 in the regulation of OXPHOS has not been previously reported in human or mammalian systems. To predict the specific function of LARP1 with human mitochondria, we considered co-dependency profiles (Figure 5D) across our chemical-genetic compendium to predict gene function (see Methods). Co-dependency profiling reveals LARP1 to be one of the few non-mitochondrial proteins within a tight cluster that is functionally related to mitochondrial translation (Figure S4A). Consistent with the hypothesis of LARP1 being involved in mitochondrial translation, the abundance of mtDNA-encoded MT-CO1 and MT-CO2 are reduced in LARP1 KO (Figure 5E). This reduction in MT-CO1 and MT-CO2 upon LARP1 loss without an apparent effect on mtDNA copy number (Figure 5F) or mtRNA level (Figure 5G) is consistent with a role in mitochondrial translation for LARP1. LARP1 loss also affects a subset of nuclear encoded mitochondrial proteins, notably complex I (NDUFB8) and complex II (SDHB) subunits (Figure 5E). The mechanism by which oligomycin or ethidium bromide, but not other mitochondrial inhibitors, buffers the loss of genes involved in mitochondrial translation including LARP1 (Figures S4A, S4D), requires further investigation.
Genes whose Loss Suppress the Toxic Effects of Mitochondrial Inhibitors
Amongst the collection of genes broadly characterized as buffering, we report 63 suppressors whose loss alleviates the toxic growth effects of mitochondrial inhibitors (Figure 2B). Our screening data predicts that knocking out these genes can genuinely reduce or reverse the proliferative defects of mitochondrial dysfunction. The losses of these genes either play an active role in compensating for or protecting against the underlying pathophysiology, or they prevent drug toxicity. This group of chemical-genetic interactions includes two recently identified genetic suppressors of mitochondrial dysfunction, ATPIF1 and vHL, while providing new information into the specificity of their suppressive actions. We find that loss of ATPIF1 suppresses the growth effects of antimycin+oligomycin, and ethidium bromide, but does not effectively suppress toxicity from piericidin, metformin, antimycin, oligomycin, or chloramphenicol (Figure 2B). This pattern is consistent with the notion that ATPIF1 is a strong genetic suppressor under drug treatments that compromises mitochondrial membrane potential (Chen et al., 2014). Co-treatment with antimycin+oligomycin collapses membrane potential acutely, whereas ethidium bromide depletes membrane potential more slowly due to chronic loss of both ETC and complex V. As ATPIF1 functions as an inhibitor of the ATPase activity of complex V, loss of ATPIF1 allows reversal of complex V to defend the membrane potential. This result points to the essential role of membrane potential for cell survival under severe mitochondrial dysfunction, consistent with classic results in S. cerevisiae (Veatch et al., 2009).
Loss of vHL, in contrast, suppresses antimycin, oligomycin and antimycin+oligomycin, but does not suppress other drugs including ethidium bromide. Of note, our screen has also identified TCEB2, which co-expressed with OXPHOS transcripts (Baughman et al., 2009) and its gene product physically interacts with vHL protein (Stebbins et al., 1999). Our screen has also revealed numerous genetic suppressors not previously reported – notably complex I genes (which we discuss later), and nuclear factors such as NAIF1, NR2F2, SIN3A and its corepressor complex component SUDS3, KLF16, C7orf26, and the PP2A regulator FAM122A (Figure 2B).
Suppression of Oligomycin’s Toxicity by Simultaneous Loss of Mitochondrial Complex I
Our screen has also identified numerous genetic suppressors not previously reported, but perhaps most striking, we find that many complex I subunits appear to score as genetic suppressors of oligomycin. Complex I represents the entry point to the electron transport chain and consists of ~45 subunits, 38 of which are encoded by the nuclear genome. Knockouts of a very large number of nuclear-encoded subunits and assembly factors are enriched under oligomycin (Figure 6A) and, to a lesser extent, antimycin+oligomycin (Figure S5A). Enrichment of complex I knockouts is weaker with antimycin treatment, and is not observed with other drug treatments (Figure S5A). We validated the alleviation of oligomycin’s toxicity by NDUFA9 loss (Figure 6B), and confirmed its drug specificity as only epistatic buffering effects were observed under piericidin or antimycin (Figure S5B). In addition, we found that the specific complex I inhibitor piericidin can alleviate oligomycin’s toxicity in multiple cell lines (Figure 6B). Metformin, which also targets complex I, can also alleviate oligomycin’s toxicity (Figure 6B).
While the loss of complex I can robustly alleviate oligomycin’s toxicity, the converse is not true as the loss of complex V components, with the possible exception of ATP5A1, is not alleviated or buffered by chemical inhibitors of complex I. The lack of reciprocity of interaction is likely due to the non-equivalence of pharmacologic inhibition of complex V and biallelic genetic loss of a component of complex V. Under oligomycin treatment, complex V is intact and OXPHOS is inhibited, typically leading to mitochondrial membrane hyperpolarization. On the other hand, biallelic genetic loss of a complex V subunit may lead to a different outcome since complex V can defend membrane potential through reversal of activity, as well as dissipate it. Moreover, the F1-ATPase of Complex V has been implicated in the maintenance of mtDNA (Contamine and Picard, 2000). As such, biallelic loss of a complex V component may affect the stability of other complexes of the electron transport chain or impact their expression due to defective mtDNA maintenance, which is not recapitulated by oligomycin action. Therefore, while the intra-OXPHOS suppression of complex V dysfunction by complex I loss has not been previously reported to our knowledge, it is restricted to specific modes of complex V inhibition modeled by oligomycin.
Suppression of Oligomycin’s Toxicity by Complex I Loss is not Simply Explained by Deficiency of Pyrimidines or Toxicity of Reactive Oxygen Species
We first sought to determine if an important resource required for proliferation is depleted under oligomycin but restored when complex I activity is simultaneously inhibited. One candidate is pyrimidines, as it has been shown that OXPHOS dysfunction leads to a stalling of dihydroorotate dehydrogenase (DHODH), a rate-limiting enzyme in de novo pyrimidine biosynthesis that requires oxidized coenzyme Q as a cofactor (Khutornenko et al., 2010). Though not well appreciated, complex I inhibition can actually up-regulate de novo pyrimidine biosynthesis (Shaham et al., 2010), owing to the fact that complex I inhibition makes a larger part of the oxidized Q pool available for other Q-linked enzymes including DHODH. Such a mechanism may be especially important when reoxidation of the coenzyme Q pool is limited or disabled with a blockade downstream (e.g. antimycin or oligomycin treatment). However, pyrimidine deficiency cannot explain the suppression by complex I inhibition as uridine was supplemented during screening and validation (Figures 6B, 6C). In addition, the interaction between complex I and complex V occurs to a similar extent when uridine is not supplemented and dialyzed serum is used (Figure 6C). In K562 cells, the uridine salvage pathway is operative as evidenced by the fact that loss of uridine-cytidine kinase UCK2 is synthetic lethal with antimycin (Figure 2A), a condition under which reoxidation of coenzyme Q is blocked and de novo pyrimidine synthesis is disabled, causing the cells to rely entirely on uridine salvage for pyrimidines. As such, pyrimidine levels are presumably supported by the salvage pathway, arguing against restoring de novo pyrimidine synthesis as the key mechanism of suppression.
We next sought to determine if oligomycin leads to the buildup of a toxin that can be cleared when complex I is simultaneously inhibited, the most obvious candidate being some form of ROS. Oligomycin treatment hyperpolarizes the mitochondrial membrane potential (as it prevents the proton gradient from being dissipated for ATP synthesis) and is a known driver of ROS formation, either by succinate-driven reverse electron transport (RET) (Chouchani et al., 2014) or via over-reduced states of the CoQ and NADH pools (Robb et al., 2018). We confirmed that oligomycin hyperpolarizes mitochondria in K562, and such hyperpolarization can be suppressed by simultaneous treatment with piericidin, or the mitochondria-specific uncoupler BAM15 (Kenwood et al., 2014) (Figure S5C). Simultaneous administration of an intermediate dose (1 μM) of uncoupler improves proliferation under oligomycin (Figure S5D). However, the level of oligomycin-induced mitochondrial superoxide is suppressed by BAM15 only when a high concentration (5 mM) of succinate is provided but not under normal culture condition (Figure S5E). Piericidin significantly increases mitochondrial superoxide under oligomycin (Figure S5E) in K562, which does not support complex I-RET being a significant source of mitochondrial superoxide. Furthermore, administration of antioxidants, including the specific complex I-RET inhibitor S1QEL (Brand et al., 2016), does not suppress oligomycin’s toxicity (Figure S5F). Hence, oligomycin’s toxicity, and the mechanism by which it is suppressed by complex I loss, cannot be simply explained by ROS toxicity.
Inhibiting Complex I Activity Re-Balances Redox Cofactors, Promotes Glycolysis and Reductive Carboxylation under Oligomycin Treatment
Next, we investigated the bioenergetic and metabolic consequences of complex I inhibition on oligomycin treated cells. As measured using a Seahorse XFe96 Extracellular Flux Analyzer (Agilent), inhibition of complex I with piericidin eliminates oligomycin-resistant respiration (Figure 6D), arguing against a suppression mechanism that involves reactivation of electron transport chain activity such as the alleviation enabled by uncouplers. When OXPHOS is dysfunctional, glycolysis is capable of compensating for ATP production as long as glucose is provided in the culture medium. As ATP availability is crucial for proliferation, we next asked if elevated glycolysis is part of the suppression mechanism by complex I loss. We measured glycolytic rates with a Seahorse assay termed GlycoPER (see Methods) and found it to be higher under piericidin+oligomycin than oligomycin alone in multiple cell lines (Figures 6E and S5G). To buttress the Seahorse results, we measured media lactate levels in K562 using targeted mass spectrometry, and observed an elevated lactate excretion under piericidin+oligomycin (Figure 6F). Furthermore, we found that pyruvate supplementation is essential for complex I inhibition to alleviate oligomycin’s toxicity (Figure S5H), as pyruvate withdrawal renders piericidin treatment detrimental to oligomycin treated cells. Under OXPHOS inhibition, pyruvate supplementation is required for NADH oxidation by the cytosolic lactate dehydrogenase to regenerate NAD+ for glycolysis (King and Attardi, 1989; Titov et al., 2016), consistent with glycolysis being instrumental for the alleviation.
To more broadly characterize the metabolic changes that arise from a combined piericidin+oligomycin treatment, we performed full-scan intracellular metabolomics using mass spectrometry in K562 cells. We identified 2-hydroxyglutarate (2-HG) to be the most up-regulated, statistically significant metabolite under piericidin+oligomycin when compared to oligomycin alone, followed by the non-essential amino acid proline which is not in the media used (Figure 6G). The elevation of 2-HG has been linked to reductive carboxylation of α-ketoglutarate (Mullen et al., 2014), presumably as a consequence of higher NADH/NAD+ ratio under mitochondrial dysfunction or hypoxia (Mullen et al., 2011; Wise et al., 2011). The whole cell NADH/NAD+ ratio is substantially elevated under piericidin+oligomycin (Figure S5I) when compared to oligomycin alone in our system, redox cofactor balance that favors reductive synthesis.
To confirm that reductive carboxylation is indeed promoted under combined piericidin+oligomycin treatment, we cultured K562 cells under DMSO, oligomycin, or piericidin+oligomycin with [U-13C]glutamine and monitored 13C incorporation in TCA cycle intermediates and other key metabolites over the course of 8 hours using mass spectrometry. Oxidative metabolism of uniformly labeled 13C-glutamine generates M+4 citrate, whereas reductive biosynthesis leads to M+5 citrate (Figure 6H). Glutamine-derived proline and 2-HG also contain five glutamine-derived 13C (M+5). Indeed, reductive synthesis of citrate from glutamine, as identified by the level of M+5 citrate, significantly increases under piericidin+oligomycin when compared to oligomycin alone (Figure 6I). While oligomycin reduces the overall citrate level, piericidin+oligomycin restores the citrate to a level comparable to the DMSO control. We observed an increased reliance on reductive synthesis of citrate under piericidin+oligomycin across multiple cell lines (Figure S5J). We also observed a significant increase in 2-HG under piericidin+oligomycin. The 2-HG is produced primarily from glutamine as indicated by the M+5 mass isotopomer (Figure 6I), providing additional evidence for an elevated flux in reductive carboxylation of α-ketoglutarate by one of the NAD(P)H-dependent dehydrogenases. Finally, the intracellular proline pool, whose level is restored under piericidin+oligomycin, contains a significant fraction derived from glutamine as evidenced by M+5 mass isotopomer, likely because proline can be derived from glutamate in an NADH and NADPH dependent pathway (Phang et al., 2015). An increase in proline from glutamine source implicates an intracellular redox state that favors reductive biosynthesis, consistent with the observed elevation of 2-HG and M+5 citrate.
Since reductive biosynthesis is largely driven by the reducing power of cytosolic NADPH, we sought to determine if the ability to suppress oligomycin by complex I inhibition is sensitive to the availability of cytosolic NADPH. First, we used G6PD KO, whose loss is synthetic lethal with complex I inhibition (Figure 3B), to reduce the formation of cytosolic NADPH, and observed a more severe proliferation defect under piericidin+oligomycin than oligomycin alone (Figure 6J). Next, we expressed the genetically encoded, water-forming NADPH oxidase TPNOX (Cracan et al., 2017) to promote consumption of cytosolic NADPH, and found that it blunted the suppression of oligomycin’s toxicity by complex I inhibition (Figure 6K). Both results support the involvement of reductive carboxylation in the suppression by complex I loss, as cytosolic NADPH is heavily used in reductive biosynthesis within cells (e.g. citrate, fatty acids, and proline). Collectively, these experiments demonstrate that in the setting of complex V inhibition, simultaneous loss of complex I boosts fitness via a mechanism that involves promoting glycolysis and reductive carboxylation through re-balancing redox cofactors.
DISCUSSION
We have utilized a CRISPR screening approach to discover new genetic modifiers of mitochondrial dysfunction. In total, we report chemical-genetic interactions involving 191 genes, of which 38 are classified as synthetic sick/lethal, 63 are suppressors, and 91 epistatically buffering (one gene is classified in two categories). Our compendium is high quality – our triplicate screens are tightly correlated, it recovers recently reported genetic suppressors (ATPIF1, vHL), genes involved in synthetic sick/lethal interactions (GOT1), and even in epistatic buffering (REXO2). We have been able to validate multiple novel interactors – namely G6PD, PFKP, GPX4, LARP1, and complex I. We have illustrated the utility of this compendium via four vignettes that explore some of the strongest suppressors and synthetic sick/lethal interactions identified.
More than half of the 101 genes whose loss are synthetic sick/lethal or suppressors (Figure 2) participate in only one chemical-genetic interaction, consistent with the notion that mitochondrial lesions each have very distinct patterns of genetic interactions. Our work provides unexpected genetic insights into the connectivity of OXPHOS both within mitochondria and with the rest of the cell. While 35% of the synthetic sick/lethal or candidate suppressor genes we report encode proteins resident outside of mitochondria (underscoring the coupling between processes within the organelle and the remainder of the cell), we were surprised to find that 65% of these genetic modifiers involve genes encoding mitochondrial proteins, far greater than expected by chance given that only 6% of all nuclear genes encode mitochondrial proteins. This observation reflects the fact that many pathways within the organelle locally sense and modify mitochondrial lesions. Moreover, the enrichment of intra-organelle genetic interactions relative to inter-organelle interactions has been observed in previously reported yeast genetic interactions screens (Costanzo et al., 2016; Hoppins et al., 2011). To the best of our knowledge, the extent of intra-organelle interactions in mammalian biology is underappreciated and has not been previously reported. In the coming years, as chemical-genetic and related approaches are applied to other organelles, it will be interesting to see if a similar pattern of “intra-organelle” versus “extra-organelle” interactions emerges in mammalian systems.
Extra-mitochondrial synthetic sick/lethal interactions are exemplified by genes involved in glycolysis or the pentose phosphate pathway. Genes associated with glycolysis are expected to be indispensable under OXPHOS inhibition, as glycolytic ATP production becomes essential in the face of OXPHOS dysfunction. The phosphofructokinase 1 (PFK-1), for which PFKP encodes one of its three isozymes, represents a key step that commits glucose to glycolysis, and is tightly regulated by multiple metabolites (Mor et al., 2011). PFKP has been shown to be highly up-regulated by Ras oncogene (Tanner et al., 2018) and stabilized by AKT in glioblastoma (Lee et al., 2017), and is implicated in promoting aerobic glycolysis. A highly regulated component of glycolysis, PFKP may play an especially important role in determining the utilization and fate of glucose when OXPHOS is inhibited. The pentose phosphate pathway gene G6PD is particularly important for NADPH homeostasis as it encodes the enzyme that generates the vast majority of cytosolic NADPH (Chen et al., 2019). Cytosolic NADPH production might become increasingly important in the face of OXPHOS dysfunction, as it is implicated in the reductive biosynthesis of citrate and fatty acids. Indeed, we observe a requirement for G6PD and cytosolic NADPH for the suppression of oligomycin’s toxicity by complex I loss. Furthermore, NADPH is used for the regeneration of glutathione, which plays important roles in ROS defense and redox homeostasis under OXPHOS dysfunction. Our work raises the very explicit hypothesis that individuals with G6PD deficiency – which can often be asymptomatic – will be at higher risk for manifesting mitochondrial diseases.
Our work suggests that the intra-mitochondrial synthetic sick/lethal hit GPX4 may be a part of a homeostatic feedback loop designed to guard against mitochondrial dysfunction. The up-regulation of GPX4 in response to OXPHOS dysfunction could be controlled by a nuclear transcriptional program, such as the ATF4-dependent integrated stress response (Bao et al., 2016). It is notable that GPX4 is also normally induced during the PGC-1α transcriptional program for mitochondrial biogenesis (Mootha et al., 2004), indicating that this is likely a part of a physiologic relationship whereby elevated OXPHOS activity is met with an increased need for GPX4. The interaction between OXPHOS dysfunction and loss of GPX4 is richly supported by in vivo reports of patients with mitochondrial disease, who can show elevated levels of toxic byproducts of lipid hydroperoxides such as malondialdehyde and 4-hydroxynonenal (Thompson Legault et al., 2015), and combined with the current screening results, raise the question of whether lipid peroxidation contributes to mitochondrial pathogenesis. OXPHOS dysfunction may lead to altered glutathione and NADPH metabolism, both of which play very important roles in the prevention of lipid peroxidation and GPX4 activity. Disrupting glutathione metabolism using erastin or buthionine sulfoximine, or by cysteine starvation, has been shown to induce ferroptotic cell death (Dixon et al., 2012; Gao et al., 2018). As previously discussed, our screens reveal that loss of G6PD exacerbates the growth defect caused by OXPHOS inhibition, raising the question of whether this interaction can be related to ferroptosis, as the pentose phosphate pathway is a major source of cellular NADPH for reductive synthesis of fatty acids and lipids. Lipid metabolism represents another important aspect for the regulation of ferroptosis (Doll et al., 2017; Zou et al., 2019). It was recently reported that inhibition of complexes I-IV of the electron transport chain suppresses ferroptosis induced by cysteine deprivation, specifically in cell lines that do not tolerate GPX4 loss (Gao et al., 2018). In contrast, our work suggests that the expression of GPX4 is induced in response to OXPHOS dysfunction, and that the inhibition of ATP synthase by oligomycin sensitizes cells to ferroptotic death when GPX4 is also inhibited. Future studies will be required to reconcile these related findings and how they operate in vivo in states such as cancer. Clearly, mitochondria play important roles in the regulation of ferroptosis.
We find that loss of the cytosolic protein LARP1 is itself toxic, but less toxic than expected in the setting of OXPHOS inhibition. Such a buffering interaction raises the hypothesis that LARP1 is functionally related to OXPHOS, which we experimentally confirm. Co-dependency profiles predict cytosolic LARP1 to regulate intra-mitochondrial protein translation, which we support with experimental evidence. While our finding represents the first report linking human LARP1 to mitochondrial function, previous studies have shown that LARP1 homologs in S. cerevisiae can affect the expression of many nuclear-encoded mitochondrial proteins (Kershaw et al., 2015). Likewise, a previous paper (Zhang et al., 2016) showed that the Drosophila homolog of LARP1 is recruited to the outer mitochondrial membrane and is involved in the synthesis of nuclear-encoded mitochondrial proteins. As both human LARP1 and mTOR signaling have been shown to regulate translation through the 5’ TOP motifs (Fonseca et al., 2015; Thoreen et al., 2012), it will be interesting to determine if TOP motifs are harbored at the 5’ UTR of any nuclear encoded genes that are important for mitochondrial translation, and whether mTOR is capable of regulating their expression.
One of the most surprising and striking intra-mitochondrial genetic suppressors we have identified is the alleviation of complex V dysfunction by simultaneous loss of complex I. Such an intra-mitochondrial interaction could not have been discovered by yeast genetic interaction mapping, as yeast does not have a complex I. We note that this type of intra-system suppression has previously been described in the context of antibiotic actions (Bollenbach et al., 2009; Yeh et al., 2009), but to our knowledge, has never before been reported in a mammalian system, and certainly not described in the context of mitochondrial physiology. A very large number of distinct genetic losses of complex I subunits suppress the toxicity of oligomycin (Figure 6A). We validate that NDUFA9 loss, piericidin or metformin can eliminate the toxic effect of oligomycin (Figures 6B, 6C).
The validation of complex I as a true suppressor of OXPHOS dysfunction may help to explain key observations from disease biology. Our work implies fitness benefits of complex I loss under certain circumstances, raising the question of whether it contributes to the proliferation of certain types of cancers such as renal oncocytoma and Hurthle cell thyroid carcinoma in which complex I loss is shown to be a driver event (Gopal et al., 2018a; Gopal et al., 2018b). It will be interesting to test if certain aspects of oligomycin’s effects, such as oligomycin-resistant respiration, mitochondrial hyperpolarization, or susceptibility to lipid peroxidation, are prominent in the cells of origin of these cancers such that loss of complex I is favorable. Moreover, our genetic interactions also support the notion that metformin targets complex I, and the unanticipated interaction between complex I and oligomycin raises a new mechanistic hypothesis about how metformin may alleviate aging (Barzilai et al., 2016). Specifically, the widely documented age associated decline in mitochondrial OXPHOS activity (Trounce et al., 1989) may have cellular pathologies that are actually be alleviated by inhibition of complex I via drugs such as metformin. Notably, it has been shown in C. elegans that a significant decline in ATP-linked oxygen consumption occurs over aging (Huang and Lin, 2018), implicating the loss of complex V activity as a signature for worm aging. Our work raises the specific hypothesis that metformin’s ability to prolong lifespan at the organismal level may be related to its ability to suppress complex V inhibition at the cellular level. The ability of complex I loss to improve fitness of OXPHOS limited cells could be related to the organismal loss of complex I subunits, which have occurred at least four times in eukaryotic evolution (Pagliarini et al., 2008).
Mechanistically, we show that loss of complex I activity increases glycolysis, promotes reductive carboxylation, and prevents mitochondrial hyperpolarization, all of which benefit cells under complex V inhibition. Our results are consistent with the observation that glycolysis and reductive carboxylation are mechanistically connected via NADH and NADPH redox cofactors (Brodsky et al., 2019; Gaude et al., 2018). Moreover, reductive synthesis of biomolecules such as citrate and proline may act as “sinks” for excessive reducing equivalents. Such re-balancing of redox networks may improve cell proliferation by optimizing electron flow while providing building blocks such as nucleotides, amino acids, and lipids. In E. coli, mutations that re-balance redox cofactors can result in metabolic flexibility and improved fitness (Long et al., 2018). Perhaps in mammalian systems, such “malleability” in metabolic networks is enabled by elimination of CI activity when CV is inhibited with oligomycin.
We anticipate that our inventory of chemical-genetic interactions can be useful in many ways for the biomedical research community. First, it helps to nominate new drug targets that are capable of specifically suppressing different types of mitochondrial dysfunction. An entire collection of such targets is valuable given the growing types of mitochondrial dysfunction that are linked to rare and age-associated, common human diseases. Perhaps counterintuitively, our work suggests that one way to suppress mitochondrial dysfunction is to actually inhibit a “second site” within mitochondria. Second, our inventory may help to nominate new drug combinations where monotherapy leads to a mitochondrial vulnerability that could also be targeted pharmacologically, as in certain cancers. Third, it will help to identify either inherited genetic factors or perhaps tissue-specific programs that may help to explain the penetrance and tissue specificity of mitochondrial disease. For example, our rich inventory of 101 genes now serve as candidate inherited genetic modifiers for mitochondrial disease. For example, our work raises the specific hypothesis that G6PD deficiency, which is found in hundreds of millions of individuals, may serve as a novel genetic modifier of mitochondrial disease. Finally, our work may have very basic implications for understanding mitochondrial evolution. Over millions of years of evolution, mitochondria have been effectively hard-wired within our cells – with nearly all of the original endosymbiont’s genes either completely lost or transferred to the nuclear genome, with brand new functionality encoded in the nuclear genome (Vafai and Mootha, 2012). The rich set of genetic interactions we’ve identified will help us to decipher the full regulatory and metabolic logic of this ancient compartmentalization.
STAR ★ METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vamsi K. Mootha (vamsi@hms.harvard.edu).
DATA AND CODE AVAILABILITY
All screening data generated during this study are provided as Supplementary Tables. Expression levels in K562 cells are based on RNAseq sample GSM854403 in GEO series GSE34740 (Slavoff et al., 2013). MATLAB and Python source codes are available at: https://github.com/yangli88
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
K562 (ATCC CCL-243), HeLa (ATCC CCL-2), 293T (ATCC CRL-3216), A375 (ATCC CRL-1619) and HT-29 (ATCC No. HTB-38) cells were obtained from ATCC. All experiments with wild-type cells or CRISPR-Cas9 mediated knockouts were performed under passage number 20 upon receipt from ATCC, and late passage cells were authenticated by STR profiling. HAP1 wild-type cells and gene-specific knockouts were obtained from Horizon Discovery and were authenticated by genomic PCR and sequencing.
METHOD DETAILS
Determination of Infection Conditions for CRISPR Pooled Screens
Optimal infection conditions were determined in order to achieve 30–50% infection efficiency, corresponding to a multiplicity of infection (MOI) of ~0.5–1. Spin-infections were performed in 12-well plate format with 3×106 cells each well. Optimal conditions were determined by infecting cells with different virus volumes (0, 100, 200, 400, 600, 800 μL) with a final concentration of 4 μg/mL polybrene in K562 cells. Cells were centrifuged for 2 hours at 930xg at 30 degrees Celsius. Approximately 24 hours after infection, cells were collected and 4×105 K562 cells from each infection were seeded in 2 wells of a 6-well plate, each with complete medium, one supplemented with the appropriate concentration of puromycin. Cells were counted 2 days post selection to determine the infection efficiency, comparing survival with and without puromycin selection. Volume of virus that yielded ~30–50% infection efficiency was used for screening.
Genome Scale CRISPR Screens with Brunello All-in-one Library
Brunello barcoded all-in-one library contains 77,441 sgRNA, which includes an average of 4 guides per gene and 1000 non–targeting control guides. Infection, selection and expansion were performed in three distinct replicates. Screening-scale infections were performed with the pre-determined volume of virus in the same 12-well format as the viral titration described above, and pooled 24 h post-centrifugation. Infections were performed with ~1.5×108 cells per replicate, in order to achieve a representation of at least 500 cells per sgRNA following puromycin selection (~4×107 surviving cells). Approximately 24 hours after infection, all wells within a replicate were pooled and were split into T225 flasks. 48 hours after infection, cells were selected with puromycin for 4–5 days to remove uninfected cells. After selection was complete, at least 4×107 of K562 cells were seeded in T225 flasks. Media with 10 mM Metformin, 10 nM Piericidin, 100 nM Antimycin, 10 nM Oligomycin, 10 nM Antimycin and 10 nM Oligomycin, 100 ng/mL Ethidium Bromide and 10 μg/mL Chloramphenicol was then added to the cells. Cells were passaged in fresh media containing drugs every 2–3 days. Cells were harvested 15 days after initiation of treatment.
For all screens, genomic DNA (gDNA) was isolated using Maxi (3×107−1×108 cells) kits according to the manufacturer’s protocol (Qiagen). PCR and sequencing were performed as previously described (Doench et al., 2016; Piccioni et al., 2018). Samples were sequenced on a HiSeq2000 (Illumina). Of note, two of our “day 15 samples”, one for DMSO and one for metformin, were lost during the PCR step, yielding a total of 22 (out of 24) successful 15 day screens (Table S2).
Gene-Specific CRISPR-Cas9 Knockouts
The two best sgRNAs from the Brunello-4 library were ordered as complementary oligonucleotides (Integrated DNA Technologies) and cloned into pLentiCRISPRv2. An sgRNA targeting EGFP was used as a negative control. Lentiviruses were produced according to Addgene’s protocol (Sanjana et al., 2014) and cells were selected with 2 μg/mL puromycin 24 hours post-infection. Puromycin was withdrawn 48 hours later and cells were maintained in for 10–20 addition days before analysis. Gene disruption efficiency was verified by protein immunoblotting. The sequences of the sgRNAs used are in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GPX4 | Abcam | Cat # ab125066 |
| Beta-Actin | Sigma | Cat # A5441 |
| ATP5B | Sigma | Cat # HPA001528 |
| TOMM20 | Santa Cruz | Cat # SC-11415 |
| MT-CO1 | Abcam | Cat # ab14705 |
| CYCS | BD Biosciences | Cat # 556432 |
| Human OXPHOS cocktail | Abcam | Cat # ab110411 |
| LARP1 | Abcam | Cat # ab86359 |
| FLAG M2 | Sigma | Cat # F1804 |
| PFKP | Sigma | Cat # HPA018257 |
| G6PD | Sigma | Cat # HPA000834 |
| HRP-linked anti-rabbit IgG | GE Healthcare | Cat # NA934 |
| HRP-linked anti-mouse IgG | GE Healthcare | Cat # NXA931 |
| IRDye 800CW Goat anti-Mouse IgG (H + L) | LI-COR Biosciences | Cat # 926–32210 |
| IRDye 800CW Goat anti-Rabbit IgG (H + L) | LI-COR Biosciences | Cat # 926–32211 |
| IRDye 680RD Goat anti-Mouse IgG (H + L) | LI-COR Biosciences | Cat # 926–68070 |
| IRDye 680RD Goat anti-Rabbit IgG (H + L) | LI-COR Biosciences | Cat # 926–68071 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| JKE-1674 | Eaton et al., 2018 | doi: 10.1101/376764 |
| DMEM, high glucose, pyruvate | GIBCO | Cat # 11995073 |
| Sodium Pyruvate | GIBCO | Cat # 11360070 |
| L-Glutamine | GIBCO | Cat # 25030081 |
| Fetal Bovine Serum | GIBCO | Cat # 26140079 |
| Fetal Bovine Serum, dialyzed | GIBCO | Cat # 26400044 |
| DMEM, no glucose, no glutamine, no phenol red | GIBCO | Cat # A1443001 |
| DMEM low Glucose, w/ L-Glutamine | US Biological | Cat # D9800 |
| Seahorse XF Base Medium with 5 mM HEPES | Agilent | Cat # 103575–100 |
| Phenol Red | Sigma | Cat # P3532 |
| D-(+)-Glucose solution | Sigma | Cat # G8644 |
| Galactose | Sigma | Cat # PHR1206 |
| Uridine | Sigma | Cat # U3750 |
| Penicillin-Streptomycin | GIBCO | Cat # 15140122 |
| Puromycin Dihydrochloride | GIBCO | Cat # 10131035 |
| G-418 solution | Sigma | Cat # 4727878001 |
| Cell-Tak | Corning | Cat # 354240 |
| Metformin / 1,1-Dimethylbiguanide HCl | Sigma | Cat # D150959 |
| Piericidin A | Enzo Life Sciences | Cat # ALX-380–235-M002 |
| Antimycin A | Sigma | Cat # A8674 |
| Oligomycin A | Sigma | Cat # 75351 |
| Ethidium bromide | Sigma | Cat # E1510 |
| Chloramphenicol-Water Soluble | Sigma | Cat # C3175 |
| Rotenone | Sigma | Cat # R8875 |
| 2-Deoxy-D-glucose | Sigma | Cat # D8375 |
| Carbonyl cyanide 3-chlorophenylhydrazone | Sigma | Cat # C2759 |
| BAM15 | Sigma | Cat # SML1760 |
| ML 210 | Sigma | Cat # SML0521 |
| (±)-α-Tocopherol | Sigma | Cat # T3251 |
| Ferrostatin-1 | Sigma | Cat # SML0583 |
| zVAD-fmk | Sigma | Cat # 219007 |
| N-Acetyl-L-cysteine | Sigma | Cat # A9165 |
| Sodium L-ascorbate | Sigma | Cat # A7631 |
| Dimethyl succinate | Combi-Blocks | Cat # QE-5973 |
| S1QEL1.1 | Sigma | Cat # SML1948 |
| MitoTEMPO | Sigma | Cat # SML0737 |
| Mn(III)TBAP | Cayman Chemical | Cat # 75850 |
| L-Glutathione reduced | Sigma | Cat # G4251 |
| L-Glutathione oxidized | Sigma | Cat # G4376 |
| Ammonium bicarbonate | Sigma | Cat # 09830 |
| Ammonium carbonate | Fluka | Cat # 74415 |
| Ammonium hydroxide | EMD Millipore | Cat # 533003 |
| LC-MS grade water | Fisher Scientific | Cat # W64 |
| NAD+ | Sigma | Cat # N0632 |
| NADH | Sigma | Cat # N8129 |
| 13C5 NAD+ | Toronto research chemicals | Cat # N407782 |
| d5-NADH | Toronto research chemicals | Cat # N201487 |
| Ammonium acetate | Sigma | Cat # 14267 |
| Acetonitrile | Fisher Scientific | Cat # A955 |
| XBridge BEH Amide column | Water | Cat # 186006091 |
| ZIC-philic column | Merck | Cat # 150460 |
| L-Glutamine 13C5, 99% | Cambridge Isotope Laboratories | Cat # CLM-1822-H-0.5 |
| TWEEN 20 | Sigma | Cat # P9416 |
| Odyssey Blocking Buffer | LI-COR Biosciences | Cat # 927–40000 |
| Dulbecco’s PBS | Sigma | Cat # D8537 |
| HBSS | Gibco | Cat # 14025076 |
| Critical Commercial Assays | ||
| Seahorse XFe96 FluxPaks | Agilent | Cat # 102416–100 |
| Seahorse XF Glycolytic Rate Assay Kit | Agilent | Cat # 103344–100 |
| Annexin V Alexa Fluor 647 conjugate | Invitrogen | Cat # A23204 |
| TMRM | Invitrogen | Cat # T668 |
| Hoechst 33342, Trihydrochloride, Trihydrate | Invitrogen | Cat # H3570 |
| BODIPY 581/591 C11 | Invitrogen | Cat # D3861 |
| MitoSOX Red | Invitrogen | Cat # M36008 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | Cat # 4369016 |
| TaqMan MT-ND1 | Thermo Fisher Scientific | Assay ID Hs02596873_s1 |
| TaqMan MT-ND2 | Thermo Fisher Scientific | Assay ID Hs02596874_g1 |
| TaqMan MT-CO1 | Thermo Fisher Scientific | Assay ID Hs02596864_g1 |
| TaqMan MT-CO2 | Thermo Fisher Scientific | Assay ID Hs02596865_g1 |
| TaqMan MT-CO3 | Thermo Fisher Scientific | Assay ID Hs02596866_g1 |
| TaqMan NDUFB8 | Thermo Fisher Scientific | Assay ID Hs00428204_m1 |
| TaqMan UQCRC2 | Thermo Fisher Scientific | Assay ID Hs00996395_m1 |
| TaqMan TBP | Thermo Fisher Scientific | Assay ID Hs00427620_m1 |
| Novex 4–20% Tris-Glycine Mini Gels | Thermo Fisher Scientific | Cat # XP04202BOX |
| Trans-Blot Turbo Midi Nitrocellulose Transfer Packs | BioRad | Cat # 1704159 |
| M-MLV Reverse Transcriptase | Promega | Cat # M1701 |
| Deposited Data | ||
| Expression levels in K562 based on RNAseq | Slavoff et al., 2013 | GEO GSE34740 |
| Experimental Models: Cell Lines | ||
| K562 | ATCC | CCL-243 |
| HeLa | ATCC | CCL-2 |
| 293T | ATCC | CRL-3216 |
| A375 | ATCC | CRL-1619 |
| HT-29 | ATCC | HTB-38 |
| HAP1 | Horizon Discovery | C631 |
| HAP1 GPX4 KO | Horizon Discovery | HZGHC005981c005 |
| HAP1 NDUFA9 KO | Horizon Discovery | HZGHC002487c004 |
| Oligonucleotides | ||
| GFP sgRNA control GGGCGAGGAGCTGTTCACCG | This paper | N/A |
| G6PD sgRNA 1 AGAGGTGCAGGCCAACAATG | This paper | N/A |
| G6PD sgRNA 2 TGCCCGTTCCCGCCTCACAG | This paper | N/A |
| PFKP sgRNA 1 GATGTGTGTCAAACTCTCGG | This paper | N/A |
| PFKP sgRNA 2 GCCGGATGATCAGATCCCAA | This paper | N/A |
| GPX4 sgRNA 1 AGAGATCAAAGAGTTCGCCG | This paper | N/A |
| GPX4 sgRNA 2 GAGCTGAGTGTAGTTTACTT | This paper | N/A |
| LARP1 sgRNA 1 TAGTGAATACTACTTCAGCG | This paper | N/A |
| LARP1 sgRNA 2 GCTGTTCCTAAACAGCGCAA | This paper | N/A |
| REXO2 sgRNA 1 CTTGCAGTCTGGCCTTACCA | This paper | N/A |
| REXO2 sgRNA 2 ATAATCAGGTTAGGACCCTG | This paper | N/A |
| Taqman mtDNA assay-ND2 Forward TGTTGGTTATACCCTTCCCGTACTA | Bao et al., 2016 | DOI: 10.7554/eLife.10575 |
| Taqman mtDNA assay-ND2 Reverse CCTGCAAAGATGGTAGAGTAGATGA | Bao et al., 2016 | DOI: 10.7554/eLife.10575 |
| Taqman mtDNA assay-ND2 Probe CCCTGGCCCAACCC | Bao et al., 2016 | DOI: 10.7554/eLife.10575 |
| Taqman nucDNA assay- AluYb8 Forward CTTGCAGTGAGCCGAGATT | Bao et al., 2016 | DOI: 10.7554/eLife.10575 |
| Taqman nucDNA assay- AluYb8 Reverse GAGACGGAGTCTCGCTCTGTC | Bao et al., 2016 | DOI: 10.7554/eLife.10575 |
| Taqman nucDNA assay- AluYb8 Probe ACTGCAGTCCGCAGTCCGGCCT | Bao et al., 2016 | DOI: 10.7554/eLife.10575 |
| Recombinant DNA | ||
| lentiCRISPR v2 | Addgene | Plasmid # 52961 |
| psPAX2 | Addgene | Plasmid # 12260 |
| pMD2.G | Addgene | Plasmid # 12259 |
| pWPI /Neo | Addgene | Plasmid # 35385 |
| pWPI/ lGPX4 / Neo | This paper | N/A |
| pWPI/ sGPX4 / Neo | This paper | N/A |
| pWPI/ TPNOX-FLAG / Neo | This paper | N/A |
| Software and Algorithms | ||
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Jupyter | Project Jupyter | https://jupyter.org |
| R | The R Foundation | http://www.R-project.org |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Seahorse Wave Desktop Software | Agilent | https://www.agilent.com/en/products/cell-analysis/cell-analysis-software/data-analysis/wave-desktop-2-6 |
| Compound Discoverer Software | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/compound-discoverer-software.html |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | BD | https://www.flowjo.com |
| MAGeCK | Li et al., 2014 | https://sourceforge.net/p/mageck/wiki/Home/ |
| Gene set enrichment analysis (GSEA) | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| MATLAB code for computing Z and ΔZ scores | This paper | https://github.com/yangli88 |
| Python code for computing p-values and FDRs | This paper | https://github.com/yangli88 |
Gene-Specific cDNA Rescue
SgRNA-resistant versions of sGPX4 and lGPX4 containing the 3’ SECIS sequence were in vitro synthesized (Integrated DNA Technologies) to encode silent mutations within the GPX4 sgRNA recognition sites. The cDNAs were cloned into pWPI-Neo (Visanji et al., 2011) and lentiviruses were produced and selected according to Addgene’s protocol. The sequences of sgRNA-resistant sGPX4 and lGPX4 are below, with lowercase letters denoting 3’ UTR. The original recognition sites for GPX4 sgRNA that were silently mutated were underscored.
sGPX4
ATGTGCGCGTCCCGGGACGACTGGCGCTGTGCGCGCTCCATGCACGAGTTTTCCGCCAAGGACATCGACGGGCACATGGTTAACCTGGACAAGTACCGGGGCTTCGTGTGCATCGTCACCAACGTGGCCTCCCAGTGAGGCAAGACCGAAGTGAATTATACCCAACTTGTCGACCTGCACGCCCGATACGCTGAGTGTGGTTTGCGGATACTGGCCTTCCCGTGTAACCAGTTCGGGAAGCAGGAGCCAGGGAGTAACGAAGAAATAAAGGAATTTGCAGCGGGCTACAACGTCAAATTCGATATGTTCAGCAAGATCTGCGTGAACGGGGACGACGCCCACCCGCTGTGGAAGTGGATGAAGATCCAACCCAAGGGCAAGGGCATCCTGGGAAATGCCATCAAGTGGAACTTCACCAAGTTCCTCATCGACAAGAACGGCTGCGTGGTGAAGCGCTACGGACCCATGGAGGAGCCCCTGGTGATAGAGAAGGACCTGCCCCACTATTTCTAGctccacaagtgtgtggccccgcccgagcccctgcccacgcccttggagccttccaccggcactcatgacggcctgcctgcaaacctgctggtggggcagacccgaaaatccagcgtgcaccccgccggaggaaggtcccatggcctgctgggcttggctcggcgcccccacccctggctaccttgtgggaataaacagacaaattagcctgctggataaaaaaa
lGPX4
ATGAGCCTCGGCCGCCTTTGCCGCCTACTGAAGCCGGCGCTGCTCTGTGGGGCTCTGGCCGCGCCTGGCCTGGCCGGGACCTCCTGCGCGTCCCGGGACGACTGGCGCTGTGCGCGCTCCATGCACGAGTTTTCCGCCAAGGACATCGACGGGCACATGGTTAACCTGGACAAGTACCGGGGCTTCGTGTGCATCGTCACCAACGTGGCCTCCCAGTGAGGCAAGACCGAAGTGAATTATACCCAACTTGTCGACCTGCACGCCCGATACGCTGAGTGTGGTTTGCGGATACTGGCCTTCCCGTGTAACCAGTTCGGGAAGCAGGAGCCAGGGAGTAACGAAGAAATAAAGGAATTTGCAGCGGGCTACAACGTCAAATTCGATATGTTCAGCAAGATCTGCGTGAACGGGGACGACGCCCACCCGCTGTGGAAGTGGATGAAGATCCAACCCAAGGGCAAGGGCATCCTGGGAAATGCCATCAAGTGGAACTTCACCAAGTTCCTCATCGACAAGAACGGCTGCGTGGTGAAGCGCTACGGACCCATGGAGGAGCCCCTGGTGATAGAGAAGGACCTGCCCCACTATTTCTAGctccacaagtgtgtggccccgcccgagcccctgcccacgcccttggagccttccaccggcactcatgacggcctgcctgcaaacctgctggtggggcagacccgaaaatccagcgtgcaccccgccggaggaaggtcccatggcctgctgggcttggctcggcgcccccacccctggctaccttgtgggaataaacagacaaattagcctgctggataaaaaaa
TPNOX Expression
A short peptide (SGGSGG) for vector control or cytosolic TPNOX (Cracan et al., 2017) was cloned into pWPI-Neo (Visanji et al., 2011). Lentiviruses were produced and according to Addgene’s protocol. The sequence of cytosolic TPNOX containing a C-terminal FLAG tag is below.
TPNOX
ATGAAGGTCACCGTGGTCGGATGCACCCATGCCGGCACCTTCGCCATCAAGCAAATCCTCGCTGAGCACCCTGACGCCGAGGTCACCGTCTACGAGAGGAACGATGTGATCTCCTTCCTGTCCTGTGGCATCGCCCTCTACCTGGGCGGAAAAGTGGCCGATCCCCAAGGCCTCTTCTACAGCTCCCCTGAAGAACTGCAGAAGCTGGGCGCTAATGTGCAGATGAACCACAACGTGCTGGCCATCGACCCTGACCAAAAGACCGTCACAGTCGAGGACCTCACCAATCACGCCCAGACCACCGAGTCCTACGACAAACTGGTGATGACCTCCGGAAGCTGGCCTATCGTGCCCAAAATCCCCGGCATCGACAGCGATAGGGTGAAGCTCTGCAAGAATTGGGCCCACGCCCAGGCTCTGATTGAGGACGCCAAGGAGGCCAAGAGGATCACCGTCATCGGCGCCGGATACATCGCCGCCGAACTGGCCGAGGCCTACTCCACAACAGGCCACGACGTCACCCTGATTGCCAGAAGCGCTAGGGTCATGAGGAAGTACTTCGATGCCGACTTCACCGACGTCATCGAACAGGACTACAGGGACCATGGCGTGCAACTCGCTCTGGGCGAGACAGTGGAGAGCTTCACCGACAGCGCCACCGGCCTCACAATCAAGACAGACAAGAACTCCTATGAGACCGACCTGGCCATCCTCTGCATTGGCTTTAGGCCCAACACAGACCTGCTGAAAGGCAAAGTGGACATGGCCCCTAACGGCGCCATCATTACCGACGACTACATGAGGTCCAGCAACCCTGATATTTTCGCTGCTGGCGACTCCGCCGCCGTCCATTACAACCCCACACACCAAAACGCCTACATTCCCCTCGCTACCAACGCCGTCAGGCAGGGAATCCTCGTCGGAAAGAACCTCGTCAAGCCCACAGTGAAGTACATGGGAACCCAGTCCAGCTCCGGACTGGCCCTCTATGACAGGACAATTGTCTCCACAGGCCTCACACTGGCCGCCGCCAAGCAACAAGGCCTCAATGCCGAGCAGGTCATCGTGGAGGACAACTATAGGCCCGAGTTCATGCCTTCCACCGAGCCCGTCCTCATGAGCCTGGTCTTCGACCCCGATACACACAGAATCCTGGGAGGCGCCCTGATGTCCAAATACGACGTGTCCCAGAGCGCTAATACCCTGTCCGTCTGCATCCAGAACGAGAACACCATCGATGACCTGGCCATGGTGGACATGCTGTTCCAGCCCAATTTCGACAGGCCCTTCAACTACCTGAACATTCTCGCCCAGGCTGCCCAAGCTAAAGTGGCCCAATCCGTCAACGCTGGAGGCAGCGATTACAAGGATGACGATGACAAG
Cell Death Assay
Cells were treated with OXPHOS inhibitors and grown in glucose (10 mM) or galactose (10 mM) media for 24 hours. Cell death was assessed by staining cells with Annexin V Alexa Fluor 647 conjugate (Invitrogen) according to the manufacturer’s protocol. Alexa Fluor fluorescence was analyzed by flow cytometry on a BD Accuri C6 (BD Biosciences).
Oxygen Consumption by the Seahorse XF Analyzer
1.5×105 K562 cells were plated on a Seahorse plate coated with Cell-Tak (Corning) in DMEM containing 5.5 mM glucose (D9800 US Biological) supplemented with 4 mM glutamine (Gibco). Oxygen consumption was recorded by a Seahorse XFe96 Analyzer (Agilent) using the mito stress test protocol, in which cells were sequentially perturbed by 2 μM oligomycin, 2 μM CCCP or 3 μM BAM15, and 0.5 μM piericidin + 0.5 μM antimycin.
Glycolytic Rate Assay by the Seahorse XF Analyzer
1.5×105 K562, 5×104 HeLa, or 5×104 HAP1 cells were plated on a Seahorse plate coated with Cell-Tak (Corning) in Seahorse XF Base Medium with 5 mM HEPES (Agilent) supplemented with 25 mM glucose (Sigma), 1 mM pyruvate (Gibco), and 4 mM glutamine (Gibco). Extracellular acidification rate was recorded by a Seahorse XFe96 Analyzer (Agilent) using the glycolytic rate assay protocol, in which cells were sequentially perturbed by 0.5 μM rotenone + 0.5 μM antimycin and 50 mM 2-Deoxy-D-glucose. The extracellular acidification rate (ECAR) was corrected for contribution from mitochondrial respiration using oxygen consumption data using the Seahorse Wave Desktop Software (Agilent), such that acidification is due primarily to lactate excretion. The corrected extracellular acidification rate, termed GlycoPER, was used as a proxy for glycolytic flux.
Polyacrylamide Gel Electrophoresis and Protein Immunoblotting
Cells were harvested, washed in PBS and lysed for 5min on ice in lysis buffer (50 mM Tris/HCl at pH 7.5, 150 mM NaCl, 1mM MgCl2, 1% NP-40, protease / phosphatase inhibitors). Lysates were clarified by centrifugation for 5 min at 2,000xg. Gel electrophoresis was performed using Novex Tris-Glycine 4–20% gels (Thermo Fisher Scientific). Semi-dry western blotting was performed using the Trans-Blot Turbo blotting system and PVDF membranes (BioRad). Immunoblotting was performed in 5% milk powder in PBS + 0.1% Tween-20 (Sigma) or in Odyssey Blocking Buffer (LI-COR Biosciences).
Quantification of mtDNA by Real-Time PCR
DNA was extracted from cells treated with OXPHOS inhibitors and analyzed by multiplex real-time quantitative PCR as described in (Bao et al., 2016). Custom-synthesized assays for the AluYb8 repeat element (nuclear DNA) and MT-ND2 (mitochondrial DNA) were obtained from Applied Biosystems.
Mitochondrial Membrane Potential Measurement
K562 Cells were treated with OXPHOS inhibitors and grown for 3 days. One hour prior to measurement, cells were concentrated to 106 cells/mL in phenol red-free media supplemented with OXPHOS inhibitors. TMRM (Invitrogen) was added to the cells at a final concentration of 10 nM. TMRM fluorescence was analyzed by confocal microscopy. In Figure S1E, cells were co-stained with Hoechst 33342 (Invitrogen) at a concentration of 0.5 μg/mL, and imaged under a spinning disk (Yokogawa) confocal microscope (Zeiss) equipped with a CCD camera (Hamamatsu). Image processing was performed using the ImageJ software (NIH). Maximum projection was taken across Z-stacks of 3 frames of view and the maximum intensity of each cell was recorded. In Figure S5C, at least 60 cells from 5 frames of view were analyzed for each treatment.
Growth Experiments
Follow-up for GPX4 was performed in K562, HAP1, and HeLa cells. Follow-up for NDUFA9 was performed in HAP1 cells. Follow-up for G6PD was performed in K562 and A375 cells. Follow-up for PFKP was performed in K562 and HT-29 cells. Follow-up for LARP1 and REXO2 was performed in K562 cells. Unless noted otherwise, cells were grown in DMEM (Gibco) with 25 mM glucose (Sigma), 4 mM L-glutamine (Gibco), 1 mM sodium pyruvate (Gibco), 10% fetal bovine serum (Gibco), 100U penicillin-100 μg/mL streptomycin (Gibco), and 50 μg/mL uridine (Sigma).
For follow-up of G6PD, PFKP, LARP1 and wild-type cells in K562, cells were seeded in triplicates at 2×105/mL (~12.5% of maximum cell density) in 2 mL media in 12-well plates. For follow-up of GPX4 and REXO2 in K562, cells were seeded in triplicates at 5×104/mL (~3% of maximum cell density) in 2 mL media in 12-well plates. Cell counts were performed 3 days after drug treatments. For follow-up in HAP1, HeLa and 293T, cells were seeded in triplicates at 5×104 per well (~10% confluency) in 2 mL media in 12-well plates. Counts were performed 3 days after plating. For follow-up in HT29 and A375, cells were seeded in triplicates at 1×105 per well (~20% confluency) in 2 mL media in 12-well plates. Counts were performed 3 days after plating. Population doubling was calculated as log2(final density/seeding density).
For spent media growth experiment in K562 (Figure 4B), cells were treated DMSO or oligomycin and grown for 3 days. Media were harvested and clarified by centrifugation and vacuum filtering. The spent media were supplemented with fresh OXPHOS inhibitors during the growth experiments. Cells were seeded in triplicates at 5×104/mL under spent media growth, as well as under fresh media growth in Figure S3B.
For cells infected with viruses carrying the pWPI lentivector, 500 μg/mL or 800 μg/mL of G-418 (Sigma) was used to select for stable lines in HAP1 and K562, respectively.
Lipid Peroxidation Measurement
Cells treated with various chemicals were harvested, washed with HBSS (Gibco), and resuspended in HBSS to 0.5 mL, and incubated with 2 μM BODIPY 581/591 C11 (Invitrogen) for 10 min at 37 °C. Cells were then washed twice in HBSS and analy zed by flow cytometry on a BD Accuri C6 (BD Biosciences). Quantification of BODIPY-C11 fluorescence was performed as described in (Dixon et al., 2012; Yang et al., 2014).
Mitochondrial Superoxide Measurement
Cells treated with various chemicals were harvested, washed with HBSS (Gibco), and resuspended in HBSS to 0.5 mL, and incubated with 5 μM MitoSOX Red (Invitrogen) for 10 min at 37 °C. Cells were then washed twice in HBSS and analy zed by flow cytometry on a BD Accuri C6 (BD Biosciences).
Isolation of Mitochondria
HAP1 cells were grown in 3–5 15 cm dishes until confluence. Cells were then harvested, washed in PBS, and washed once with 10 mL of MB buffer (210 mM mannitol, 70 mM sucrose, 10 mM HEPES-KOH at pH 7.4, 1mM EDTA, protease/phosphatase inhibitor). Cells were resuspended in 1 mL of MB buffer and transferred to 2 mL glass homogenizer (Kontes). Breaking of cells was achieved by pressing the potter (Kontes) up and down of the homogenizer about 35 times on ice. Treated cells were transferred to a 15 mL tube, with volume increased to 6 mL with MB buffer. The mixture was centrifuged at 2,000xg for 5 min and the pellet (nuclei and intact cells) was discarded. The supernatant, which contained the cytoplasm and mitochondria, was distributed to 1.5 mL Eppendorf tubes and centrifuged at 13,000xg for 10 min at 4 degree Celsius. The supernatant contained the cytosolic fraction whereas the small, solid brown pellets contained the mitochondria. The cytosolic fraction was saved. The mitochondrial pellets were washed by MB buffer once, resuspended in MB buffer, and saved. Western blotting was performed on the cytosolic and mitochondrial fractions as described in the “Polyacrylamide gel electrophoresis and protein immunoblotting” section.
Immunofluorescence Experiment
HAP1 cells were seeded in 35-mm glass bottom dishes (MatTek) and grown until ~70% confluent. Cells was fixed with 10% formalin in PBS at room temperature for 30 min, washed 3X in PBS, and blocked with PBS + 0.1% Triton X-100 + 1% BSA for 2 hours at room temperature. Cell were incubated with primary antibodies (1:250 anti-GPX4, 1:500 anti-CYCS) in PBS + 0.1% Triton X-100 + 1% BSA overnight at 4 degrees Celsius. Cells were washed 3X in PBS, and incubated with secondary antibodies (1:200) in PBS + 0.1% Triton X-100 + 1% BSA for 3 hours at room temperature. Cells were then washed 3X in PBS, stained with 1:2000 Hoechst 33342 (Invitrogen) for 10 min at room temperature, and imaged under a spinning disk (Yokogawa) confocal microscope (Zeiss) equipped with a CCD camera (Hamamatsu). Image processing was performed using the ImageJ software (NIH).
Gene Expression Assay by Real-Time PCR
Real-time quantitative PCR was performed using the TaqMan technology (Applied Biosystems). Briefly, total RNA was extracted with the RNeasy kit (Qiagen) and digested with DNAse-I (Qiagen). Reverse transcription was performed using MLV reverse transcriptase supplemented with RNasin and dNTP (Promega). Real-time qPCR was performed using the standard protocol for Taqman assay on a C1000 Touch thermal cycler (Bio-rad). All data were normalized to TBP. The Taqman probes used in this study are listed in the Key Resources Table.
Intracellular Metabolite Profiling using Liquid Chromatography-Mass Spectrometry (LCMS):
6×105 cells/well (K562) in 3 mL were seeded in 6-well plates in the presence of OXPHOS inhibitors. After 72 hours, equal number of cells were centrifuge at 600xg for 5 min at room temperature and the media were aspirated off. For intracellular metabolite profiling, cells were rinsed with 1 mL of PBS and metabolism was quenched by adding 1 mL of dry ice-cold 80% methanol. After incubation on ice for 20 min, the supernatant was collected after centrifugation of the sample at 21,000xg for 20 min at 4 °C, dried in a speed vacuum concentrator (Savant SPD1010, Thermo Fisher Scientific), and stored at −80°C until the analysis. Metabolite extracts were resuspended in 100 μL of 60/40 acetonitrile/water, vortexed, and sonicated in ice-cold water for 1 min. The extracts were then incubated on ice for 20 min and clarified by centrifugation at 21,000xg for 20 mins at 4°C. Nex t, 55 μL of supernatant was transferred to an autosampler vial and pooled QC sample was generated by combining around 20 μL of each sample.
Polar metabolite separation was done using Dionex Ultimate 3000 UHPLC system and ZIC-philic column (150×2.1 mm, 5 μm, Merck KGaA). Mobile phase A (MPA) was 20 mM ammonium carbonate in water, pH 9.6 (adjusted with ammonium hydroxide), and mobile phase B (MPB) was acetonitrile. The column was held at 27°C, injection volume 5 μL, and an autosampler temperature of 4°C. LC conditions at f low rate of 0.15 mL/min were: 0 min: 80% B, 0.5 min: 80% B, 20.5 min: 20% B, 21.3 min: 20% B, 21.5 min: 80% B with 7.5 min of column equilibration time. Mass spectrometry (MS) detection was performed using Q-Exactive plus Orbitrap mass spectrometer (Thermo Fisher Scientific) with an Ion Max source and HESI II probe operating in switch polarity mode. MS parameters were: sheath gas flow 30, aux gas flow 7, sweep gas flow 2, spray voltage 2.80 for negative & 3.80 for positive, capillary temperature 310°C, S-lens RF level 50 and aux gas heater temp 3 70°C. Data acquisition was done using Xcalibur 4.1 (Thermo Fisher Scientific) and performed in full scan mode with a range of 70–1000 m/z, resolution 70,000, AGC target 1×106 and maximum injection time of 80 ms. Data analysis was done using Compound Discoverer 3.0. Samples were injected in randomized order and pooled QC samples were injected regularly throughout the analytical batch. Metabolite annotation was done base on accurate mass (±5 ppm) and matching retention time (±0.3min)as well as MS/MS fragmentation pattern from the pooled QC sample against in-house retention time+MSMS library of reference chemical standards. Metabolites which have CV<20% in pooledQC, were used for the statistical analysis.
NADH/NAD+ Analysis using LCMS
After centrifugation and removal of media, samples were prepared as described in (Lu et al., 2018). In brief, cells were lysed with ice-cold 4/4/2 acetonitrile/methanol/water with 0.1M formic acid containing 13C5 NAD+ and d5-NADH internal standards, neutralized with 15% ammonium bicarbonate for 3 min, centrifuged at 21,000xg and 4°C, and injected 5 μL of supernatant on ZIC-philic column with above mentioned LC-MS parameters. External calibration curves of NAD+ and NADH were generated using 13C5 NAD+ and d5-NADH internal standards.
Stable Isotope Analysis of [U-13C]glutamine
For K562 cells, 6×105 cells/well in 3 mL were seeded in 6-well plates in the presence of OXPHOS inhibitors and after 48 hours, media in each plate were exchanged with isotope tracing media consisting of glutamine-free & uridine-free DMEM containing 25 mM glucose (Sigma), 1 mM sodium pyruvate (Gibco), 10% dialyzed FBS (Gibco), 100U penicillin-100 μg/mL streptomycin (Gibco), and supplemented with 2 mM [U-13C]Glutamine. After 8 hours of incubation, equal number of cells were centrifuged, media was aspirated off, and 1 mL of dry ice-cold 80% methanol was added to extract metabolite and incubated on ice for 20 min. Next, intracellular metabolite profiling was performed as described previously.
HAP1, HT29, and 293T cells were seeded in 6 well-plates at 106 cells per well and grown overnight. On the next day, media in each plate were exchanged as described previously with OXPHOS inhibitors. After 8 hours of incubation, media were aspirated off and tracer analysis was performed as described above. Natural abundance correction was performed using Stable Isotope labeling workflow within Compound Discoverer 3.0 (Thermo Fisher Scientific).
Media Lactate Analysis
After 3 days of cell growth, 30 μL of media were extracted with 120 μL of ice-cold acetonitrile, incubated on ice for 20 min, centrifuged at 21,000xg for 20 min at 4°C, and supernatant was transferred to an autosampler vial. Media metabolite separation was done using XBridge BEH amide (2.1×100mm, 2.5 μm, Waters Corporation) column on Dionex Ultimate 3000 UHPLC system. MPA was 5/95 acetonitrile/water, 20 mM ammonium acetate, pH 9.0 and MPB was acetonitrile. The column was held at 27°C, injecti on volume 10 μL, autosampler temperature 4°C. LC conditions at flow rate of 0.22 mL/min wer e : 0 min: 85% B, 0.5 min: 85% B, 9.00 min: 35% B, 11 min: 2% B, 12 min: 2% B, 13.5 min 85% B with 4.5 min of column equilibration time. Mass spectrometry (MS) detection was done performed using Q-Exactive plus Orbitrap mass spectrometer (Thermo Fisher Scientific) with an Ion Max source and HESI II probe operating in switch polarity mode. MS parameters were: sheath gas flow 50, aux gas flow 10, sweep gas flow 2, spray voltage 2.50 for negative & 3.80 for positive, capillary temperature 310°C, S-lens RF level 50 and aux gas heater temp 370 °C. Data a cquisition was done using Xcalibur 4.1 (Thermo Fisher Scientific) and performed in full scan mode with a range of 70–1000 m/z, resolution of 70,000, AGC target of 3×106, and maximum injection time of 80 ms. Data analysis was done as described previously.
QUANTIFICATION AND STATISTICAL ANALYSIS
Gene Scoring
Raw sgRNA read counts were normalized to reads per million and then log2 transformed using the following formula:
Log2 fold-change of each sgRNA was determined relative to the initial time point (day 0). The replicates were paired throughout the course of the screens.
For each gene and for each replicate, the mean log2 fold-change in the abundance of all 4 sgRNAs was calculated. Genes with low expression (log2 FPKM < 0) according to publicly available K562 RNA-seq dataset (sample GSM854403 in GEO series GSE34740) were removed. This step filtered out about 50% of all genes, leading to a gene set of 11,102 genes for further analysis. Log2 fold-changes were averaged by taking the mean across replicates. For each treatment, a null distribution was defined by the 3,726 genes with lowest expression (log2 FPKM = −7). We chose non-expressing genes instead of non-targeting guides for the null distributions since we observed a systematic, significant enrichment of non-targeting guides (Figure S1G) at the late time point. To score each gene within each treatment, its mean log2 fold-change across replicates was Z-score transformed, using the statistics of the null distribution defined as above. The normalized Z-score was computed as ΔZDrug = (ZDrug−ZDMSO)/√2, with the variance corrected by the √2 factor. Homoscedasticity was assumed as variances are approximately the same across the treatments (Figure S1G). Aggravating interactions (under a given drug treatment) were defined by ΔZDrug < −2.4, whereas buffering interactions were defined by ΔZDrug > 4.8. Among buffering interactions, suppressors additionally satisfied the condition ZDrug > 2.4, whereas the rest (ZDrug ≥ 2.4) were termed epistatic buffering. The two-sided p-values for the Z-scores and normalized Z-score were calculated assuming the irrelevant genes follow a normal distribution with the same mean and variance as null distribution defined by the genes with lowest expression. To control for family-wise error rates for the p-values, we computed false discovery rates (FDR) using the Benjamini-Hochberg (BH) approach (Benjamini, 1995)
To score each gene using MAGeCK (Li et al., 2014), normalized sgRNA read counts from the triplicates in each condition were used as input for MAGeCK v0.5.3 to obtain a p-value and FDR for gene enrichment or depletion relative to the reference samples (day 15 DMSO) MAGeCK was run with default parameters. The smaller value between positive and negative modes is shown in Table S3.
Pathway Analysis
Gene set enrichment analysis (GSEA) (Mootha et al., 2003) was performed on the list ranked by ΔZDrug using default parameters. A customized geneset database containing all KEGG Metabolism or Cellular Process pathways (92 total) with 15–500 genes was used for the GSEA. The KEGG Oxidative Phosphorylation and Complex I pathways were manually reviewed. All pathways with GSEA FDR < 0.05 were considered significant.
Hierarchical Clustering
Mean log2 fold-change across sgRNAs was computed for each gene each replicate and genes with low expression were filtered as described in the Gene scoring section. This gene set of 11,102 genes was further filtered such that only the top 10% genes with the largest variance across all samples were kept. The final gene set contains ~1,100 genes. The mean log2-fold changes across sgRNAs for genes were clustered with average linkage hierarchical clustering using Pearson correlation distance metrics. Hierarchical clustering was applied using SciPy 1.1.0. The MitoCarta2 human database was used for labeling known mitochondrial localized proteins (Calvo et al., 2016).
For gene function annotation, we downloaded GO terms for all genes from DAVID 6.8 (Huang da et al., 2009) for the GO_BP_DIRECT, GO_MF_DIRECT, and GO_CC_DIRECT annotations. For each node on the hierarchical clustering dendrogram, the hyper-geometric test p-value was calculated for all GO terms. Nodes were highlighted as key nodes if they contained at least one GO term with p < 10−10 and at least one term scoring higher than any other node. To reduce redundancy, a lower node was removed if the upper node contained more than 80% of its genes.
Supplementary Material
Figure S2. Additional Data Related to Figure 2 A. Scatter plots of Z-scores showing knockouts that are enriched or depleted in each pairwise drug vs. drug comparison. Axis label denotes the Z-score of the specified drug. Knockouts are scored by ΔZDrug-Drug = (ZDrug,Y-axis−ZDrug,X-axis)/√2. Enrichment (shown in red or magenta, ΔZDrug-Drug > 4.8) and depletion (shown in blue, ΔZDrug-Drug < −2.4) are used to define drug-specific buffering and synthetic sick/lethal interactions, respectively. Among buffering interactions, drug-specific suppressors (ZDrug, Y-axis > 2.4) are shown in red. Scatter plots are grouped based on the impact of drug treatment on proliferation (moderate or severe), according to the growth curves shown in Figure 1C. The gray dotted lines represent the cutoffs for drug-specific interactions.
B. Top categories of KEGG pathways among enriched knockouts by Gene Set Enrichment Analysis (GSEA). The 11,102 genes was ranked by ΔZDrug in descending order and GSEA was run in the positive mode. Pathways are ordered by the maximum of −log10 FDR across 7 drugs.
C. The 91 genes involved in epistatic buffering interactions that score in ≥ 1 drug (ΔZDrug > 4.8, and ZDrug ≤ 2.4). Genes are further divided into two panels. The left panel contains genes that have interactions with multiple drugs, and is ordered by the number of drugs under which interactions occur. The right panel contains genes with only drug-specific interactions, and genes are listed by the specific drug and ordered by the strength of interaction. Gene-drug pairs that do not score are colored white (−2.4 ≤ ΔZDrug ≤ 4.8).
Figure S3. Additional Data Related to Figure 4 A. Scatter plot of the fitness of a gene knockout in the specified drug (ZDrug) vs. its fitness in DMSO (ZDMSO). Gray dots denote all 11,102 genes in the analysis and the blue dot represents GPX4.
B. Growth phenotypes of cells under a combination of oligomycin and GPX4 inhibition by 10 μM JKE-1674 in K562 in fresh medium. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). **p<0.01 indicates two-tailed Student’s t-test p-value.
C. Lipid peroxidation of the K562 samples in Figure 4B as assessed by BODIPY581/591 C11 staining of cells. **p<0.01 indicates two-tailed Student’s t-test p-value.
D. Immunoblots for GPX4 and loading control HAP1 GPX4 KO cells.
E. Structures of mRNA variants encoding the short (sGPX) and long (lGPX) isoforms. The first exon (E1a) encodes the N-terminal sequences of both isoforms but is truncated in sGPX4. The 3’ UTR region contains a selenocysteine insertion sequence (SECIS).
F. Immunoblots for GPX4 and loading controls in isolated cytosolic and mitochondrial fractions from HAP1 GPX4 KO with re-expression of sGPX or lGPX4. Equal amount of total proteins was loaded into each lane. Right: immunoblot for GPX4 and loading control in HAP1 GPX4 KO cells with re-expression of lGPX4 supplemented with the catalytic site-specific GPX4 inhibitors JKE-1674 (10 μM) or ML 210 (10 μM).
G. Immunofluorescence for GPX4 in HAP1 GPX4 KO with re-expression of sGPX or lGPX4. Mitochondria were labeled with a cytochrome c antibody. Scale bar = 5 μm.
Figure S4. Additional Data Related to Figure 5 A. Clustergram of a gene module within “Mitochondrial translation” in Figure 5D. LARP1 is labeled with a red. Genes involved in mitochondrial translation by GO annotations are labeled blue. Genes currently listed in the MitoCarta2.0 (Calvo et al., 2016) database are boldfaced.
B. Validation of the buffering interaction between REXO2 loss and ethidium bromide in K562. Cell counts were performed 3 days post 10 ng/mL ethidium bromide treatment (average +/− SEM, n = 3). **<0.01 indicates two-tailed Student’s t-test p-value.
C. Scatter plot of Z-scores highlighting knockout of REXO2 in ethidium bromide vs. DMSO. Genes involved in mtRNA processing are highlighted magenta.
D. Scatter plots of Z-scores highlighting knockout of LARP1 in oligomycin (left panel) or ethidium bromide (right panel) vs. DMSO. Genes encoding for mitochondrial ribosomes or tRNA synthetases are highlighted purple, whereas genes encoding for cytosolic ribosomes or tRNA synthetases are highlighted dark gray.
Figure S5. Additional Data Related to Figure 6 A. Scatter plot of the fitness of a gene knockout in the specified drug (ZDrug) vs. fitness in DMSO (ZDMSO). Gray dots denote all 11,102 genes in the analysis and the red dots denote 50 nuclear genes that encode structural subunits and assembly factors of complex I.
B. Drug specificity in genetic suppression by NDUFA9 loss in HAP1 cells. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 2).
C. Membrane potential measurement by TMRM staining (red) and confocal microscopy in K562 cells after the specified treatment. Scale bar = 5 μm. Right: Quantification of TMRM in individual cells using the maximum intensity. ****<0.0001 indicates two-tailed Student’s t-test p-value.
D. Validation of BAM15 at 1 μM as suppressor of oligomycin in K562 and 293T cells. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). **<0.01 or ***<0.001 indicates two-tailed Student’s t-test p-value.
E. Mitochondrial superoxide measurement by mitoSOX staining and flow cytometry in K562 cells after the specified drug treatment. Dimethyl succinate was used at 5 mM and BAM15 was used at 1 μM. *<0.05 indicates two-tailed Student’s t-test p-value.
F. Growth phenotypes of cells under a combination of oligomycin and known antioxidants in K562: S1QEL (10 μM), NAC (2 mM), L-ascorbate (10 μM), a-Tocopherol (100 μM), mitoTEMPO (100 μM), Mn(III)TBAP (100 μM), low GSH/GSSG (10 μM/1 mM), and high GSH/GSSH (1 mM/10 μM). Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3).
G. Corrected extracellular acidification rate as assessed by the Seahorse Extracellular Flux (XFe96) Analyzer in HAP1 and HeLa after 3 days of drug treatment (average +/− SEM, n = 12).
H. Growth phenotypes of cells under a combination of piericidin and oligomycin in K562 in the absence of 1 mM pyruvate supplementation. Dialyzed serum was used. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). **p<0.01 indicates two-tailed Student’s t-test p-value.
I. Ratio of whole cell NADH to NAD+ levels in K562 using targeted mass spectrometry after 3 days of drug treatment (average +/− SEM, n = 3). *p<0.05 indicates two-tailed Student’s t-test p-value.
J. The 13C labeling pattern of citrate over the course of 8 hours upon supplementation with [U-13C]glutamine in K562, HAP1, 293T, and HT-29 (average +/− SEM, n = 3).
Table S2. Read count (normalized) matrix, related to Figure S1 and STAR Methods
Table S3. Log2 fold changes at the level of each sgRNA for each infection replicate (A,B,C), related to Figure S1 and STAR Methods
Figure S1. Additional Data Related to Figure 1 A. Cell death as assessed by Annexin V staining and flow cytometry in K562 cells cultured in glucose or galactose containing medium with the specified OXPHOS inhibitors.
B. Cell death as assessed by Annexin V staining and flow cytometry in K562 cells cultured in glucose or galactose containing medium with different doses of metformin.
C. Real time PCR-based measurement of mtDNA relative to nuclear DNA (nDNA) in K562 cells after 9 days of ethidium bromide treatment (average +/− SEM, n = 3 replicates from one experiment).
D. Immunoblots for the mtDNA encoded MT-CO1 and loading control in K562 cells after 9 days of ethidium bromide (EtBr) or chloramphenicol (CAP) treatment.
E. Membrane potential measurement by TMRM staining (red) and confocal microscopy in K562 cells after 3 days of antimycin+oligomycin treatment. Nuclei were stained with Hoechst 33342 (blue). Scale bar = 5 μm.
F. Principal component analysis (PC1: 89%, PC2: 3%) on guide abundance on day 15 across all biological replicates.
G. Histograms of average log2 fold changes for all genes (between day 15 samples and day 0 baseline). The average log2 fold change for each gene was calculated by taking the mean of its 4 sgRNAs. Biological triplicates were averaged by taking the mean. Shown are distributions for 3,726 non-expressed genes in K562 (dark blue), 1,000 non-targeting sgRNAs (maroon), and the core essential genes as defined in (Hart et al., 2015) (gray).
Table S4. Gene Set Enrichment Analysis (GSEA) for the screens, related to Figures 3A and S2B
Genome-wide CRISPR screens were performed with multiple mitochondrial inhibitors
Loss of G6PD, PFKP, or GPX4 is synthetic lethal with mitochondrial dysfunction
“Intra-mitochondrial” interactions are pervasive
Fitness defect from CV inhibition is alleviated by simultaneous loss of CI activity
A chemical-genetic map of the mitochondrion provides insights into the pathways that modulate its function
ACKNOWLEDGMENTS
We thank Dan Gottschling, David Botstein, Calvin Jan, Vasanthi Viswanathan, Sarah Calvo, and Jordan Wengrod for their valuable feedback on the manuscript; Jason Arroyo, Matthew Broadus, and Lev Sandler for technical assistance. This work was supported by funding from Calico Life Sciences LLC (S.B.V.) and NIH R35GM122455 (V.K.M.). VKM is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
V.K.M. is a paid advisor to Raze Therapeutics, Janssen Pharmaceuticals, and 5AM Ventures, S.B.V. is currently an employee of Sanofi Pharmaceuticals. T.-L.T., S.B.V., and V.K.M. are listed as inventors on a provisional patent application submitted by the Broad Institute on technology described in this paper.
SUPPLEMENTAL INFORMATION
Supplemental information includes 5 figures and 4 Supplemental Tables can be found with this article online.
REFERENCES
- Arroyo JD, Jourdain AA, Calvo SE, Ballarano CA, Doench JG, Root DE, and Mootha VK (2016). A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab 24, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Kantor HL, Katz LA, and Briggs RW (1986). Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science 232, 1121–1123. [DOI] [PubMed] [Google Scholar]
- Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, Vafai SB, Cox AG, Marutani E, Ichinose F, et al. (2016). Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N, Crandall JP, Kritchevsky SB, and Espeland MA (2016). Metformin as a Tool to Target Aging. Cell Metab 23, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Nilsson R, Gohil VM, Arlow DH, Gauhar Z, and Mootha VK (2009). A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet 5, e1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini YH,Y. (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B 57, 289–300. [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, and Sabatini DM (2015). An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollenbach T, Quan S, Chait R, and Kishony R (2009). Nonoptimal microbial response to antibiotics underlies suppressive drug interactions. Cell 139, 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Goncalves RL, Orr AL, Vargas L, Gerencser AA, Borch Jensen M, Wang YT, Melov S, Turk CN, Matzen JT, et al. (2016). Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab 24, 582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky AN, Odenwelder DC, and Harcum SW (2019). High extracellular lactate causes reductive carboxylation in breast tissue cell lines grown under normoxic conditions. PLoS One 14, e0213419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni F, Gramegna P, Oliveira JM, Lightowlers RN, and Chrzanowska-Lightowlers ZM (2013). REXO2 is an oligoribonuclease active in human mitochondria. PLoS One 8, e64670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, and Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellini MD, and Fiorelli G (2008). Glucose-6-phosphate dehydrogenase deficiency. Lancet 371, 64–74. [DOI] [PubMed] [Google Scholar]
- Chance B, and Williams GR (1955). Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem 217, 383–393. [PubMed] [Google Scholar]
- Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z, and Rabinowitz JD (2019). NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat Metab 1, 404–415. [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Birsoy K, Mihaylova MM, Snitkin H, Stasinski I, Yucel B, Bayraktar EC, Carette JE, Clish CB, Brummelkamp TR, et al. (2014). Inhibition of ATPIF1 ameliorates severe mitochondrial respiratory chain dysfunction in mammalian cells. Cell Rep 7, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contamine V, and Picard M (2000). Maintenance and integrity of the mitochondrial genome: a plethora of nuclear genes in the budding yeast. Microbiol Mol Biol Rev 64, 281–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. (2016). A global genetic interaction network maps a wiring diagram of cellular function. Science 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracan V, Titov DV, Shen H, Grabarek Z, and Mootha VK (2017). A genetically encoded tool for manipulation of NADP(+)/NADPH in living cells. Nat Chem Biol 13, 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hillig RC, Hilpmann A, Zimmermann K, Ryan MJ, Niehues M, Badock V, et al. (2018). Targeting a Therapy-Resistant Cancer Cell State Using Masked Electrophiles as GPX4 Inhibitors. bioRxiv. [Google Scholar]
- Fonseca BD, Zakaria C, Jia JJ, Graber TE, Svitkin Y, Tahmasebi S, Healy D, Hoang HD, Jensen JM, Diao IT, et al. (2015). La-related Protein 1 (LARP1) Represses Terminal Oligopyrimidine (TOP) mRNA Translation Downstream of mTOR Complex 1 (mTORC1). J Biol Chem 290, 15996–16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier AE, Thorburn DR, and Compton AG (2017). Mitochondrial energy generation disorders: genes, mechanisms and clues to pathology. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, and Jiang X (2018). Role of Mitochondria in Ferroptosis. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaude E, Schmidt C, Gammage PA, Dugourd A, Blacker T, Chew SP, Saez-Rodriguez J, O’Neill JS, Szabadkai G, Minczuk M, et al. (2018). NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol Cell 69, 581–593 e587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal RK, Calvo SE, Shih AR, Chaves FL, McGuone D, Mick E, Pierce KA, Li Y, Garofalo A, Van Allen EM, et al. (2018a). Early loss of mitochondrial complex I and rewiring of glutathione metabolism in renal oncocytoma. Proc Natl Acad Sci U S A 115, E6283–E6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal RK, Kubler K, Calvo SE, Polak P, Livitz D, Rosebrock D, Sadow PM, Campbell B, Donovan SE, Amin S, et al. (2018b). Widespread Chromosomal Losses and Mitochondrial DNA Alterations as Genetic Drivers in Hurthle Cell Carcinoma. Cancer Cell 34, 242–255 e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, et al. (2008). The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320, 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Collins SR, Cassidy-Stone A, Hummel E, Devay RM, Lackner LL, Westermann B, Schuldiner M, Weissman JS, and Nunnari J (2011). A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol 195, 323–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang SH, and Lin YW (2018). Bioenergetic Health Assessment of a Single Caenorhabditis elegans from Postembryonic Development to Aging Stages via Monitoring Changes in the Oxygen Consumption Rate within a Microfluidic Device. Sensors (Basel) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422 e421. [DOI] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, et al. (2016). Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasnos L, and Korona R (2007). Epistatic buffering of fitness loss in yeast double deletion strains. Nat Genet 39, 550–554. [DOI] [PubMed] [Google Scholar]
- Kenwood BM, Weaver JL, Bajwa A, Poon IK, Byrne FL, Murrow BA, Calderone JA, Huang L, Divakaruni AS, Tomsig JL, et al. (2014). Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol Metab 3, 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw CJ, Costello JL, Castelli LM, Talavera D, Rowe W, Sims PF, Ashe MP, Hubbard SJ, Pavitt GD, and Grant CM (2015). The yeast La related protein Slf1p is a key activator of translation during the oxidative stress response. PLoS Genet 11, e1004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, and Evstafieva AG (2010). Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Natl Acad Sci U S A 107, 12828–12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP, and Attardi G (1989). Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503. [DOI] [PubMed] [Google Scholar]
- Kuhl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A, Filipovska A, and Larsson NG (2017). Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Liu R, Li J, Zhang C, Wang Y, Cai Q, Qian X, Xia Y, Zheng Y, Piao Y, et al. (2017). Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat Commun 8, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Yoo SE, Na R, Walter CA, Richardson A, and Ran Q (2009). Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J Biol Chem 284, 30836–30844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long CP, Gonzalez JE, Feist AM, Palsson BO, and Antoniewicz MR (2018). Dissecting the genetic and metabolic mechanisms of adaptation to the knockout of a major metabolic enzyme in Escherichia coli. Proc Natl Acad Sci U S A 115, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Wang L, Chen L, Hui S, and Rabinowitz JD (2018). Extraction and Quantitation of Nicotinamide Adenine Dinucleotide Redox Cofactors. Antioxid Redox Signal 28, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, et al. (2014). Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiorino M, Chu FF, Ursini F, Davies KJ, Doroshow JH, and Esworthy RS (1991). Phospholipid hydroperoxide glutathione peroxidase is the 18-kDa selenoprotein expressed in human tumor cell lines. J Biol Chem 266, 7728–7732. [PubMed] [Google Scholar]
- Maiorino M, Conrad M, and Ursini F (2018). GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid Redox Signal 29, 61–74. [DOI] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, et al. (2004). Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A 101, 6570–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Mor I, Cheung EC, and Vousden KH (2011). Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harb Symp Quant Biol 76, 211–216. [DOI] [PubMed] [Google Scholar]
- Morais R, Gregoire M, Jeannotte L, and Gravel D (1980). Chick embryo cells rendered respiration-deficient by chloramphenicol and ethidium bromide are auxotrophic for pyrimidines. Biochem Biophys Res Commun 94, 71–77. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, et al. (2014). Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep 7, 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, and DeBerardinis RJ (2011). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MR, Doran E, and Halestrap AP (2000). Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348 Pt 3, 607–614. [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang JM, Liu W, Hancock CN, and Fischer JW (2015). Proline metabolism and cancer: emerging links to glutamine and collagen. Curr Opin Clin Nutr Metab Care 18, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccioni F, Younger ST, and Root DE (2018). Pooled Lentiviral-Delivery Genetic Screens. Curr Protoc Mol Biol 121, 32 31 31–32 31 21. [DOI] [PubMed] [Google Scholar]
- Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, James AM, and Murphy MP (2018). Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem 293, 9869–9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savaskan NE, Ufer C, Kuhn H, and Borchert A (2007). Molecular biology of glutathione peroxidase 4: from genomic structure to developmental expression and neural function. Biol Chem 388, 1007–1017. [DOI] [PubMed] [Google Scholar]
- Shaham O, Slate NG, Goldberger O, Xu Q, Ramanathan A, Souza AL, Clish CB, Sims KB, and Mootha VK (2010). A plasma signature of human mitochondrial disease revealed through metabolic profiling of spent media from cultured muscle cells. Proc Natl Acad Sci U S A 107, 1571–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, and Saghatelian A (2013). Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol 9, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins CE, Kaelin WG Jr., and Pavletich NP (1999). Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science 284, 455–461. [DOI] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner LB, Goglia AG, Wei MH, Sehgal T, Parsons LR, Park JO, White E, Toettcher JE, and Rabinowitz JD (2018). Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst 7, 49–62 e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherkezian J, Cargnello M, Romeo Y, Huttlin EL, Lavoie G, Gygi SP, and Roux PP (2014). Proteomic analysis of cap-dependent translation identifies LARP1 as a key regulator of 5’TOP mRNA translation. Genes Dev 28, 357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson Legault J, Strittmatter L, Tardif J, Sharma R, Tremblay-Vaillancourt V, Aubut C, Boucher G, Clish CB, Cyr D, Daneault C, et al. (2015). A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome. Cell Rep 13, 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, and Sabatini DM (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, and Mootha VK (2016). Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounce I, Byrne E, and Marzuki S (1989). Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet 1, 637–639. [DOI] [PubMed] [Google Scholar]
- Ursini F, Maiorino M, and Gregolin C (1985). The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta 839, 62–70. [DOI] [PubMed] [Google Scholar]
- Vafai SB, and Mootha VK (2012). Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383. [DOI] [PubMed] [Google Scholar]
- Veatch JR, McMurray MA, Nelson ZW, and Gottschling DE (2009). Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 137, 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visanji NP, Wislet-Gendebien S, Oschipok LW, Zhang G, Aubert I, Fraser PE, and Tandon A (2011). Effect of Ser-129 phosphorylation on interaction of alpha-synuclein with synaptic and cellular membranes. J Biol Chem 286, 35863–35873. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, and Lander ES (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, et al. (2014). Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 3, e02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, and Thompson CB (2011). Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A 108, 19611–19616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh PJ, Hegreness MJ, Aiden AP, and Kishony R (2009). Drug interactions and the evolution of antibiotic resistance. Nat Rev Microbiol 7, 460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, et al. (2007). AGEMAP: a gene expression database for aging in mice. PLoS Genet 3, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen Y, Gucek M, and Xu H (2016). The mitochondrial outer membrane protein MDI promotes local protein synthesis and mtDNA replication. EMBO J 35, 1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dancik V, et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 10, 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S2. Additional Data Related to Figure 2 A. Scatter plots of Z-scores showing knockouts that are enriched or depleted in each pairwise drug vs. drug comparison. Axis label denotes the Z-score of the specified drug. Knockouts are scored by ΔZDrug-Drug = (ZDrug,Y-axis−ZDrug,X-axis)/√2. Enrichment (shown in red or magenta, ΔZDrug-Drug > 4.8) and depletion (shown in blue, ΔZDrug-Drug < −2.4) are used to define drug-specific buffering and synthetic sick/lethal interactions, respectively. Among buffering interactions, drug-specific suppressors (ZDrug, Y-axis > 2.4) are shown in red. Scatter plots are grouped based on the impact of drug treatment on proliferation (moderate or severe), according to the growth curves shown in Figure 1C. The gray dotted lines represent the cutoffs for drug-specific interactions.
B. Top categories of KEGG pathways among enriched knockouts by Gene Set Enrichment Analysis (GSEA). The 11,102 genes was ranked by ΔZDrug in descending order and GSEA was run in the positive mode. Pathways are ordered by the maximum of −log10 FDR across 7 drugs.
C. The 91 genes involved in epistatic buffering interactions that score in ≥ 1 drug (ΔZDrug > 4.8, and ZDrug ≤ 2.4). Genes are further divided into two panels. The left panel contains genes that have interactions with multiple drugs, and is ordered by the number of drugs under which interactions occur. The right panel contains genes with only drug-specific interactions, and genes are listed by the specific drug and ordered by the strength of interaction. Gene-drug pairs that do not score are colored white (−2.4 ≤ ΔZDrug ≤ 4.8).
Figure S3. Additional Data Related to Figure 4 A. Scatter plot of the fitness of a gene knockout in the specified drug (ZDrug) vs. its fitness in DMSO (ZDMSO). Gray dots denote all 11,102 genes in the analysis and the blue dot represents GPX4.
B. Growth phenotypes of cells under a combination of oligomycin and GPX4 inhibition by 10 μM JKE-1674 in K562 in fresh medium. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). **p<0.01 indicates two-tailed Student’s t-test p-value.
C. Lipid peroxidation of the K562 samples in Figure 4B as assessed by BODIPY581/591 C11 staining of cells. **p<0.01 indicates two-tailed Student’s t-test p-value.
D. Immunoblots for GPX4 and loading control HAP1 GPX4 KO cells.
E. Structures of mRNA variants encoding the short (sGPX) and long (lGPX) isoforms. The first exon (E1a) encodes the N-terminal sequences of both isoforms but is truncated in sGPX4. The 3’ UTR region contains a selenocysteine insertion sequence (SECIS).
F. Immunoblots for GPX4 and loading controls in isolated cytosolic and mitochondrial fractions from HAP1 GPX4 KO with re-expression of sGPX or lGPX4. Equal amount of total proteins was loaded into each lane. Right: immunoblot for GPX4 and loading control in HAP1 GPX4 KO cells with re-expression of lGPX4 supplemented with the catalytic site-specific GPX4 inhibitors JKE-1674 (10 μM) or ML 210 (10 μM).
G. Immunofluorescence for GPX4 in HAP1 GPX4 KO with re-expression of sGPX or lGPX4. Mitochondria were labeled with a cytochrome c antibody. Scale bar = 5 μm.
Figure S4. Additional Data Related to Figure 5 A. Clustergram of a gene module within “Mitochondrial translation” in Figure 5D. LARP1 is labeled with a red. Genes involved in mitochondrial translation by GO annotations are labeled blue. Genes currently listed in the MitoCarta2.0 (Calvo et al., 2016) database are boldfaced.
B. Validation of the buffering interaction between REXO2 loss and ethidium bromide in K562. Cell counts were performed 3 days post 10 ng/mL ethidium bromide treatment (average +/− SEM, n = 3). **<0.01 indicates two-tailed Student’s t-test p-value.
C. Scatter plot of Z-scores highlighting knockout of REXO2 in ethidium bromide vs. DMSO. Genes involved in mtRNA processing are highlighted magenta.
D. Scatter plots of Z-scores highlighting knockout of LARP1 in oligomycin (left panel) or ethidium bromide (right panel) vs. DMSO. Genes encoding for mitochondrial ribosomes or tRNA synthetases are highlighted purple, whereas genes encoding for cytosolic ribosomes or tRNA synthetases are highlighted dark gray.
Figure S5. Additional Data Related to Figure 6 A. Scatter plot of the fitness of a gene knockout in the specified drug (ZDrug) vs. fitness in DMSO (ZDMSO). Gray dots denote all 11,102 genes in the analysis and the red dots denote 50 nuclear genes that encode structural subunits and assembly factors of complex I.
B. Drug specificity in genetic suppression by NDUFA9 loss in HAP1 cells. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 2).
C. Membrane potential measurement by TMRM staining (red) and confocal microscopy in K562 cells after the specified treatment. Scale bar = 5 μm. Right: Quantification of TMRM in individual cells using the maximum intensity. ****<0.0001 indicates two-tailed Student’s t-test p-value.
D. Validation of BAM15 at 1 μM as suppressor of oligomycin in K562 and 293T cells. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). **<0.01 or ***<0.001 indicates two-tailed Student’s t-test p-value.
E. Mitochondrial superoxide measurement by mitoSOX staining and flow cytometry in K562 cells after the specified drug treatment. Dimethyl succinate was used at 5 mM and BAM15 was used at 1 μM. *<0.05 indicates two-tailed Student’s t-test p-value.
F. Growth phenotypes of cells under a combination of oligomycin and known antioxidants in K562: S1QEL (10 μM), NAC (2 mM), L-ascorbate (10 μM), a-Tocopherol (100 μM), mitoTEMPO (100 μM), Mn(III)TBAP (100 μM), low GSH/GSSG (10 μM/1 mM), and high GSH/GSSH (1 mM/10 μM). Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3).
G. Corrected extracellular acidification rate as assessed by the Seahorse Extracellular Flux (XFe96) Analyzer in HAP1 and HeLa after 3 days of drug treatment (average +/− SEM, n = 12).
H. Growth phenotypes of cells under a combination of piericidin and oligomycin in K562 in the absence of 1 mM pyruvate supplementation. Dialyzed serum was used. Cell counts were performed 3 days post drug treatment (average +/− SEM, n = 3). **p<0.01 indicates two-tailed Student’s t-test p-value.
I. Ratio of whole cell NADH to NAD+ levels in K562 using targeted mass spectrometry after 3 days of drug treatment (average +/− SEM, n = 3). *p<0.05 indicates two-tailed Student’s t-test p-value.
J. The 13C labeling pattern of citrate over the course of 8 hours upon supplementation with [U-13C]glutamine in K562, HAP1, 293T, and HT-29 (average +/− SEM, n = 3).
Table S2. Read count (normalized) matrix, related to Figure S1 and STAR Methods
Table S3. Log2 fold changes at the level of each sgRNA for each infection replicate (A,B,C), related to Figure S1 and STAR Methods
Figure S1. Additional Data Related to Figure 1 A. Cell death as assessed by Annexin V staining and flow cytometry in K562 cells cultured in glucose or galactose containing medium with the specified OXPHOS inhibitors.
B. Cell death as assessed by Annexin V staining and flow cytometry in K562 cells cultured in glucose or galactose containing medium with different doses of metformin.
C. Real time PCR-based measurement of mtDNA relative to nuclear DNA (nDNA) in K562 cells after 9 days of ethidium bromide treatment (average +/− SEM, n = 3 replicates from one experiment).
D. Immunoblots for the mtDNA encoded MT-CO1 and loading control in K562 cells after 9 days of ethidium bromide (EtBr) or chloramphenicol (CAP) treatment.
E. Membrane potential measurement by TMRM staining (red) and confocal microscopy in K562 cells after 3 days of antimycin+oligomycin treatment. Nuclei were stained with Hoechst 33342 (blue). Scale bar = 5 μm.
F. Principal component analysis (PC1: 89%, PC2: 3%) on guide abundance on day 15 across all biological replicates.
G. Histograms of average log2 fold changes for all genes (between day 15 samples and day 0 baseline). The average log2 fold change for each gene was calculated by taking the mean of its 4 sgRNAs. Biological triplicates were averaged by taking the mean. Shown are distributions for 3,726 non-expressed genes in K562 (dark blue), 1,000 non-targeting sgRNAs (maroon), and the core essential genes as defined in (Hart et al., 2015) (gray).
Table S4. Gene Set Enrichment Analysis (GSEA) for the screens, related to Figures 3A and S2B
Data Availability Statement
All screening data generated during this study are provided as Supplementary Tables. Expression levels in K562 cells are based on RNAseq sample GSM854403 in GEO series GSE34740 (Slavoff et al., 2013). MATLAB and Python source codes are available at: https://github.com/yangli88