

Abstract
Wnt signaling has emerged in recent years as a major player in both nervous system development and adult synaptic plasticity. Of particular relevance to researchers studying learning and memory, Wnt signaling is critical for normal functioning of the hippocampus, a brain region that is essential for many types of memory formation and whose dysfunction is implicated in numerous neurodegenerative and psychiatric conditions. Impaired hippocampal Wnt signaling is implicated in several of these conditions, however, little is known about how Wnt signaling mediates hippocampal memory formation. This review will provide a general overview of Wnt signaling and discuss evidence demonstrating a key role for Wnt signaling in hippocampal memory formation in both normal and disease states. The regulation of Wnt signaling by ovarian sex steroid hormones will also be highlighted, given that the neuroprotection afforded by Wnt-hormone interactions may have significant implications for cognitive function in aging, neurodegenerative disease, and ischemic injury.
Keywords: hippocampus, β-catenin, GSK3β, Alzheimer’s disease, estradiol, progesterone
Within the past decade, Wnt signaling has emerged as a significant regulator of adult hippocampal plasticity and memory. Wnts have been studied outside of the nervous system for decades as key players in cardiac and bone diseases (Krishnan and others 2006; Marinou and others 2012; Pandey and Chandravati 2013), degenerative skeletal disorders (Church and Francis-West 2002), and cancers (Nusse and Varmus 1982; Nusse and Varmus 2012). In the nervous system, Wnts were initially studied in the context of neural development, where they are necessary for the development of brain regions including the hippocampus (Grove and Tole 1999; Lee and others 2000). Later studies determined that Wnts regulate synaptic plasticity in the adult hippocampus via synapse formation, dendritic morphogenesis, and long-term potentiation (LTP) (Chen and others 2006; Dickins and Salinas 2013; Rosso and Inestrosa 2013). Despite the well-known role of the hippocampus in learning and memory, it has only recently been established that Wnts contribute to hippocampal learning and memory, and that hippocampal Wnt signaling is dysregulated in neuropsychiatric and neurodegenerative diseases such as Alzheimer’s disease and Down syndrome (Caricasole and others 2004; Contestabile and others 2013). Nevertheless, the specific mechanisms through which Wnts contribute to memory formation remain poorly understood, as do the reasons why Wnt signaling goes awry in various disease states. Unlocking these mysteries may lead to novel therapeutics for a host of neurological and neurodegenerative disorders.
Wnts are evolutionarily conserved across many species and are believed to be at least 600 million years old (MacDonald and others 2009; Nusse and Varmus 2012). Found in a wide array of species from planaria to humans, Wnts play a key role in establishing polarity and determining axis development (Kusserow and others 2005; Petersen and Reddien 2009). The name “Wnt” is derived from the gene Wg (wingless) in Drosophila and int1 (integration-1) in rodents. The first Wnt gene, identified at the time as int1, was cloned from the mouse genome in 1982 and determined to be a proto-oncogene due to its role in regulating cell cycle progression and oncogenesis (Nusse and Varmus 1982). Interestingly, the Wnt gene was already being investigated under the name Wg in Drosophila for its role in regulating patterning phenotypes during embryogenesis (Nusslein-Volhard and Wieschaus 1980). In 1987, two independent mapping studies confirmed that Wg and int1 were the same gene (Baker 1987; Rijsewijk and others 1987). Therefore, Wg and int1 were combined in the early 1990s to form Wnt1 as it is known today (for review, see Nusse and Varmus 2012). A total of 19 Wnt genes have thus far been identified (Niehrs 2012).
Wnt proteins are secreted as lipid-modified glycoproteins of approximately 350 to 400 amino acids in length with a molecular weight of 40 kDa (Coudreuse and Korswagen 2007). All Wnts function as extracellular ligands (Coudreuse and Korswagen 2007; MacDonald and others 2009). Prior to their secretion, Wnts can be regulated in the endoplasmic reticulum by posttranslational modifications to determine the conditions under which a given Wnt will be secreted. The two major types of posttranslational Wnt modifications are lipidation/ acylation and N-glycosylation (Tang and others 2012). Although the details of Wnt sorting and secretion are still being elucidated and are discussed elsewhere (Bartscherer and Boutros 2008; Coudreuse and Korswagen 2007; Tang and others 2012), it appears that different posttranslational modifications affect the sorting, secretion, binding, and biological activity of each Wnt protein in unique ways. For example, N-glycosylation of one Wnt protein may enhance secretion of that Wnt, whereas N-glycosylation of another Wnt protein may decrease its expression. Adding to this complexity, Wnts can bind multiple Frizzled receptors, potentially eliciting different responses depending on the cellular environment.
Frizzled (Fzd) receptors are seven transmembrane domain proteins, similar to G-protein-coupled receptors, that are responsible for transducing the effects of Wnt proteins (Dijksterhuis and others 2014). There are currently 10 different known Fzd receptors (Fzd 1–10) to which the 19 Wnt ligands can bind. Fzd receptors facilitate Wnt signaling by interacting with numerous co-receptors including LRP5/6, RYK, ROR1/2 (Niehrs 2012). Regulation of Fzd receptors occurs through posttranslational modifications such as phosphorylation, ubiquitination, and deubiquitination, or through cleavage by metalloproteases (Dijksterhuis and others 2014). However, not all of these modifications have been demonstrated for all Fzd receptors in all species that express Wnts. The role of Fzd receptors and their co-receptors in adult nervous system function remains relatively unexplored and is likely to offer many new insights into the mechanisms underlying hippocampal function and dysfunction.
Wnt signaling occurs through both autocrine and paracrine mechanisms and can be classified as β-catenin-dependent or β-catenin-independent.β-Catenin-dependent Wnt signaling is referred to as “canonical,” and is the only Wnt pathway mediated by stabilization of β-catenin in the nucleus. β-Catenin-independent Wnt signaling is considered “non-canonical,” and includes both the JNK/Planar Cell Polarity (PCP) and Wnt-Calcium pathways. Each of the Wnt signaling pathways is discussed in turn below (also see Box 1). It should be noted that the same Wnt ligand can activate different Wnt signaling pathways in different cell types or environments. Thus, the various combinations of Wnt ligands and receptor/co-receptors present in a cell will ultimately determine which Wnt pathways are activated (for review, see Niehrs 2012). Additionally, the three Wnt signaling pathways can interact to some extent. For example, activation of the JNK/ PCP pathway inhibits the β-catenin-dependent signaling pathway (Sato and others 2010). The complicated interactions among Wnt ligands and Fzd receptors, as well as among Wnt signaling pathways themselves, may explain why alterations in Wnt signaling are linked with numerous neurological diseases (see “Wnt Signaling and Neurological Disease” below).
Box 1.
General overview of the three Wnt signaling pathways. A total of 19 Wnt ligands bind to 10 different Frizzled receptors. Activation of Wnt signaling occurs through one of three pathways: the canonical Wnt/β-catenin-dependent pathway or the non-canonical JNK/PCP or Wnt/Ca2+ pathways. Each Wnt pathway has general effects on cellular function (red), but also contributes in specific ways to regulating hippocampal structure and function (dark blue).
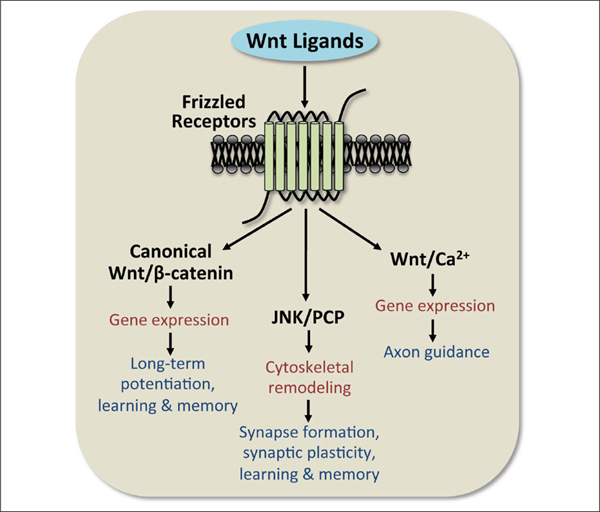
The Canonical Wnt/β-Catenin-Dependent Signaling Pathway
Canonical Wnt/β-catenin-dependent signaling is the most widely studied of the Wnt signaling pathways. The main biological effect of this pathway is to regulate the phosphorylation of β-catenin by GSK3β (glycogen synthase kinase-3β; Fig. 1A). Constitutively active GSK3β promotes degradation of β-catenin, which then prevents β-catenin from entering the nucleus and acting as a cofactor for TCF (T-cell-specific transcription factor)/LEF (lymphoid enhancer binding factor) transcription factors. The phosphorylation of β-catenin by GSK3β leads to recognition by β-Trcp (Liu and others 1999), an E3 ubiquitin ligase that targets β-catenin for proteosomal degradation (Stamos and Weis 2013) and prevents the regulation of downstream target genes. Upstream of GSK3β, canonical Wnt signaling is activated by binding of the Wnt ligand to a Fzd receptor, which then forms a complex with the co-receptor LRP5/6, and recruits Dishevelled (Dvl) to phosphorylate LRP (Bilic and others 2007; Niehrs 2012). The phosphorylation of LRP leads to the dephosphorylation of GSK3β, which prevents GSK3β from phosphorylating and degrading β-catenin. Thus, β-catenin is available to enter the nucleus and interact with TCF/LEF transcription factors to facilitate transcription of downstream target genes. However, β-catenin can also interact with cadherins to regulate transsynaptic plasticity and alter spine morphology (Murase and others 2002; Okuda and others 2007; Salinas and Price 2005; Vitureira and others 2012). This interaction is particularly important for the formation of new synapses in neural development or in response to synaptic events to facilitate synaptic plasticity. Therefore, β-catenin plays a significant role in nervous system function as both a regulator of gene expression and through interactions with cadherins.
Figure 1.
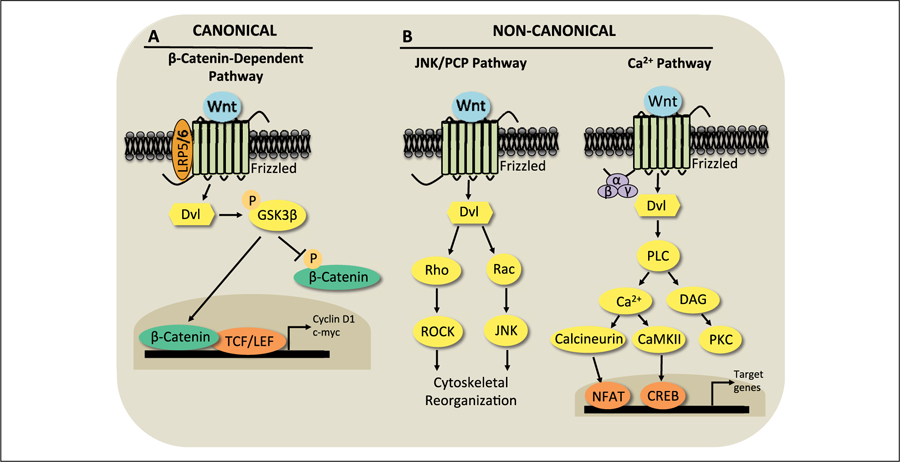
Wnt signaling is mediated by canonical and non-canonical pathways. (A) Canonical Wnt/β-catenin signaling is activated when a canonical Wnt ligand binds to a frizzled (Fzd) receptor and its co-receptor LRP 5/6. This leads Dishevelled (Dvl) to phosphorylate GSK3β on Serine 9, which inactivates GSK3β. This inactivation decreases phosphorylation of the transcriptional activator β-catenin and increases nuclear translocation of β-catenin, which then interacts with TCF/LEF transcriptional complexes to increase expression of downstream target genes such as Cyclin D1 and c-myc. (B) Wnt ligands binding to Fzd receptors can also activate the non-canonical JNK/planar cell polarity (PCP) or the Wnt/Ca2+ pathways. In the JNK/PCP pathway, activation of Dvl signals to Rho and Rac GTPases to activate ROCK and JNK, respectively, to regulate cytoskeletal reorganization. In the Wnt/Ca2+ pathway, G-protein mediated activation of phospholipase C increases intracellular Ca2+ and diacylglycerol (DAG). The rise in intracellular Ca2+ activates CaMKII and calcineurin to modulate transcriptional activity through regulation of CREB and NFAT, respectively. DAG activates the enzyme protein kinase C (PKC).
The JNK/PCP Pathway
The JNK/PCP pathway, classified as non-canonical Wnt signaling, is known for its role in regulating cytoskeletal dynamics. Similar to the β-catenin-dependent pathway, activation of the Fzd receptor typically forms a complex with a co-receptor such as RYK, MUSK, PTK7, Syndecan, Glypican, or ROR1/2, the latter of which is the primary vertebrate co-receptor that transduces the effects of the non-canonical ligand Wnt5a (Niehrs 2012). Activation of the Fzd receptor recruits Dvl to signal to the monomeric GTPases Rho and Rac (Fig. 1B). Rho stimulates ROCK, and Rac activates JNK to regulate activating transcription factor-2 (ATF-2) and downstream target genes that mediate cytoskeletal reorganization (Oliva and others 2013; Vivancos and others 2009) (Fig. 1B). Relevant to memory, the JNK/PCP pathway is an important pathway regulating the dendritic spine formation and stability that leads to synapse formation and hippocampal plasticity during development and adulthood (Luo 2000; Rex and others 2009; Rosso and others 2005; Tashiro and Yuste 2004).
The Wnt/Ca2+ Signaling Pathway
The other non-canonical Wnt signaling pathway, Wnt/ Ca2+, plays a significant role in axon guidance. This pathway is thought to elicit signaling through a rise in intracellular Ca2+ levels resulting from a G-protein-coupled signaling mechanism and activation of Dvl (Montcouquiol and others 2006; Slusarski and others 1997). The generation of diacylglycerol and inositol-(1,4,5)-triphosphate activates protein kinase C (PKC) and increases Ca2+ release from the endoplasmic reticulum, respectively. The rise in intracellular Ca2+ ultimately increases levels of calcineurin and Ca2+/calmodulin dependent protein kinase II (CaMKII), which regulate, respectively, the downstream transcription factors nuclear factor of activated T cells (NFAT) and cAMP response element binding protein (CREB) (Hogan and others 2003; Niehrs 2012; Oliva and others 2013; Rosso and Inestrosa 2013) (Fig. 1B). As such, the Wnt/Ca2+ pathway plays a significant role in mediating gene transcription (Hutchins and others 2011).
Wnt Signaling and Hippocampal Function
Some of the first clues that Wnt signaling was important for mammalian neural development came from studies demonstrating that Wnt signaling could regulate neural patterning, axonal remodeling, and synapse formation in the hippocampus and cerebellum (Galceran and others 2000; Grove and Tole 1999; Hall and others 2000; Lee and others 2000; Lucas and Salinas 1997). These findings laid the groundwork for subsequent research demonstrating the importance of Wnt signaling in adulthood for the functioning of brain regions such as the hippocampus. As will be discussed below, Wnt signaling is essential for the development and maintenance of hippocampal function. Therefore, understanding how Wnt signaling regulates hippocampal function may have important therapeutic implications for disorders in which the hippocampus plays a key role.
The involvement of Wnt signaling in the development of the various hippocampal subfields has been illustrated in several mutant mouse lines. For example, Wnt3a−/−mice do not develop a dentate gyrus or the CA subfields that are characteristic of a mature, functional hippocampus (Grove and Tole 1999; Lee and others 2000). Wnt3a regulates cell proliferation, and its loss substantially reduces the population of hippocampal progenitor cells necessary for the proliferation of the cells that comprise the mature hippocampus (Lee and others 2000). Interestingly, the surrounding neocortex and telencephalic choroid plexus epithelium are unaffected by genetic deletion of Wnt3a (Grove and Tole 1999), suggesting a specific role for Wnt3a in regulating the development of the hippocampus. Further evidence for a necessary role of Wnt signaling in hippocampal development comes from studies of mice that cannot express Lef1, a nuclear mediator of Wnt signaling (Galceran and others 2000). Specifically, Lef1 null mutants do not form a dentate gyrus, whereas mice carrying a mutant Lef1 that also interferes with Tcf gene functioning lack the entire hippocampus (Galceran and others 2000). These data illustrate that both Wnt ligands and downstream mediators of Wnt signaling, such as TCF/LEF genes, are critical for the formation of the hippocampus.
Within the normally developed hippocampus, Wnt signaling regulates synapse and circuit formation. At the presynaptic terminal, specific Wnts, such as Wnt3a and Wnt7a, increase the clustering of presynaptic proteins and promote synaptic vesicle release and recycling in cultured hippocampal neurons (Cerpa and others 2008; Varela-Nallar and others 2009). Wnt3a increases clustering of the presynaptic protein bassoon, whereas Wnt7a increases clustering of other presynaptic proteins including vGlut, synaptophysin, and synaptotagmin (Cerpa and others 2008; Ciani and others 2011; Varela-Nallar and others 2009). Pharmacological blockade of canonical Wnt/β-catenin signaling with the endogenous Wnt inhibitor Dickkopf-1 (Dkk-1) or the secreted frizzled-related protein-1 (sFRP-1) prevents the canonical Wnt ligand Wnt7a from increasing presynaptic protein clustering (Davis and others 2008), suggesting that such clustering may be mediated by β-catenin-dependent signaling (Xu and others 2014). Additionally, Wnt7a potently regulates synaptic vesicle recycling to modulate neurotransmitter release (Cerpa and others 2008). Interestingly, this effect is specific to Wnt7a, as synaptic vesicle recycling was only modestly affected by Wnt3a and was unaffected by Wnt1 or Wnt5a (Cerpa and others 2008). Recombinant Wnt7 has also been shown to increase the number of presynaptic active zones, suggesting that Wnt7 promotes the formation of new presynaptic terminals (Tabatadze and others 2014). The significance of Wnt7a in regulating hippocampal presynaptic plasticity is further demonstrated by evidence that Wnt7a/Dvl double mutants exhibit impaired neurotransmitter release, spine morphogenesis, and synaptic transmission in the hippocampus (Ahmad-Annuar and others 2006; Ciani and others 2011). Collectively, this evidence suggests that specific Wnts, particularly Wnt7a, play a critical role in regulating presynaptic function to facilitate hippocampal synapse formation.
Wnt ligands are also essential for postsynaptic assembly and synapse formation. For example, Wnt7a specifically increases the number and strength of excitatory, but not inhibitory, synapses in cultured hippocampal neurons by both increasing the formation and maturation of dendritic spines and promoting excitatory synaptic transmission (Ciani and others 2011). Wnt5a also facilitates a JNK-dependent increase in postsynaptic PSD95 clustering (Farias and others 2009). In contrast to the Wnt7a-induced formation of excitatory synapses (Ciani and others 2011), Wnt5a facilitates the formation of inhibitory synapses by increasing recycling of GABAA receptors and facilitating GABA receptor currents in a CaMKII-dependent manner (Cuitino and others 2010). As such, the differential roles of Wnt5a and Wnt7a in excitatory and inhibitory synaptic assembly highlights the specificity with which Wnts regulate synapse development in the hippocampus.
Within the hippocampus, the synthesis and release of Wnt ligands and Fzd receptors appear to be activity-dependent. Early studies in hippocampal neurons demonstrated that potassium-induced depolarization increased dendritic arborization in a manner that requires β-catenin and the release of unspecified Wnt proteins (Yu and Malenka 2003). In 2006, it was determined that Wnt3a is released in the hippocampus following tetanic stimulation in an NMDA-dependent manner (Chen and others 2006). It was proposed that NMDA receptors are activated via an increase in Ca2+ that facilitates an increase in Wnt3a release from the synapse, which then allows Wnts to bind to Fzd receptors and activate Wnt signaling (Chen and others 2006). In addition to Wnt release, NMDA receptors can also regulate Wnt synthesis. In particular, Ca2+-dependent activation of ERK through NMDA receptors leads to an increase in CREB at the Wnt2 promoter to facilitate the transcription of Wnt2 in hippocampal neurons (Wayman and others 2006). Interestingly, at least some Fzd receptors are regulated by synaptic activity as well. In response to high-frequency stimulation or potassium-mediated depolarization, Fzd5 is significantly increased at the cell surface (Sahores and others 2010). Because Fzd5 is believed to mediate the effects of Wnt7a (Sahores and others 2010), the activity-dependent increase in Fzd5 may be one mechanism through which Wnt7a facilitates spinogenesis and the formation of excitatory synapses (Ciani and others 2011).
Although much has been learned in the past decade about the effects of various Wnts on hippocampal morphology and plasticity, our understanding of how Wnts regulate hippocampal function remains rudimentary. Much of the work to date has been conducted in vitro or has been limited to an examination of hippocampal morphology and physiology. As such, the functional role of the Wnt signaling pathways in hippocampally-mediated behavior remains poorly understood. Such information is critically important, given evidence that Wnt dysfunction is characteristic of conditions such as Alzheimer’s disease (Caricasole and others 2004; Liu and others 2014; Purro and others 2012). To address this issue, a handful of studies have begun to pinpoint the role of Wnt signaling in hippocampal memory formation. This work will be described below.
Wnt Signaling and Hippocampal Memory
Learning-Induced Regulation of Wnts
Several groups have examined whether learning regulates Wnt protein expression in the hippocampus. The first study to address this issue demonstrated that levels of Wnt7 and Wnt5a, but not Wnt3, protein were significantly increased in male rats 7 days after spatial learning in a hippocampal-dependent Morris water maze task (Tabatadze and others 2012). This effect was persistent, as Wnt7 levels remained elevated for 30 days following spatial learning (Tabatadze and others 2012). The learning-induced changes in Wnt7 were observed in the granule cell layer, but not in CA3 (Tabatadze and others 2012), demonstrating subregion specificity of activity-induced changes. Moreover, potassium- or glutamate-induced transport of Wnt7 from cell bodies to neuronal processes was observed in primary hippocampal neurons, as was activity-induced Wnt7 release (Tabatadze and others 2012; Tabatadze and others 2014), suggesting that neural activity mobilizes Wnt7 to facilitate synaptic plasticity. On a more rapid timescale, an object training protocol that results in dorsal hippocampal-dependent memory consolidation produced a small, yet significant, increase in Wnt7a protein levels in male mice 5 minutes after training (Fortress and others 2013a) (Fig. 2A). This increase was transient, as levels did not differ from vehicle either 30 minutes or 4 hours after training. Also in male mice, contextual fear conditioning, a form of fear conditioning that requires the hippocampus, triggers a specific and significant increase in mRNA and protein levels of Wnt3a in the hippocampus 2 to 3 hours after training (Xu and others 2014). Wnt3a mRNA and protein levels returned to baseline 4 hours after training, suggesting a relatively brief learning-induced elevation of Wnt3a.
Figure 2.

Object training activates canonical Wnt/β-catenin-dependent signaling in the dorsal hippocampus of male mice. (A) Protein levels of Wnt7a were significantly increased relative to controls 5 minutes after training. (B) Phospho-GSK3β protein levels were significantly increased relative to controls 30 minutes after training. (C, D) At all time points following training, protein levels of β-catenin (C) and Cyclin D1 (D) were significantly increased relative to controls. Protein levels were normalized to β-actin. Each bar represents the mean ± SEM percent change from vehicle (*P ≤ 0.05 relative to controls). Insets show representative Western blots. Adapted with permission from Fortress and others (2013a).
Various forms of hippocampal learning also regulate canonical Wnt/β-catenin signaling proteins, as indicated by increases in phosphorylated GSK3β and total or active β-catenin within 30 minutes of object training in male mice (Fortress and others 2013a) (Fig. 2B and C), 2 hours of contextual fear conditioning in male mice (Xu and others 2014), or 12 hours of passive avoidance training in male rats (Conboy and others 2007). Object training also produced a rapid and sustained increase in protein levels of the TCF/LEF-induced gene Cyclin D1, which was elevated from 5 minutes to 4 hours after training (Fig. 2D). Rapid activation of the Wnt/Ca2+ pathway has also been observed within 15 minutes of contextual fear conditioning in male mice (Xu and others 2014). Together, these findings suggest that β-catenin-dependent and β-catenin-independent signaling can be rapidly activated in the hippocampus following learning.
Wnts as Essential Modulators of Hippocampal Memory
Although learning-induced changes suggest a possible role for Wnt signaling in hippocampal memory, they do not establish that such signaling is required for memory formation. The first study to demonstrate a necessary role for Wnt signaling in memory examined amygdala-dependent fear conditioning. This work found that infusion of Wnt1 or the endogenous Wnt/β-catenin antagonist Dkk-1 into the amygdala of male rats prior to training impaired fear memory consolidation without affecting learning (Maguschak and Ressler 2011). Because of the aforementioned data showing that learning increased Wnt7 levels in the hippocampus (Conboy and others 2007; Fortress and others 2013a; Tabatadze and others 2012), we hypothesized that Wnt signaling may also be necessary for hippocampal memory consolidation. Immediately after training in a hippocampal-dependent object task, we bilaterally infused vehicle or Dkk-1 into the dorsal hippocampus of male mice (Fig. 3A). Three different doses of Dkk-1 impaired object recognition memory consolidation 24 hours later (Fortress and others 2013a) (Fig. 3B), demonstrating that β-catenin-dependent Wnt signaling is necessary for object recognition memory consolidation. Similar findings have recently been reported for contextual fear memory in male mice. In this work, pretraining dorsal hippocampal infusions of Dkk-1 selectively impaired long-term contextual fear memory, suggesting that Wnt/β-catenin signaling is also critical for contextual fear memory consolidation (Xu and others 2014). In both studies (Fortress and others 2013a; Xu and others 2014), Dkk-1 inhibited canonical Wnt signaling by decreasing phosphorylated GSK3β and/or β-catenin (Fig. 3C and D). In our object recognition study, Dkk-1 also reduced protein levels of Wnt7a (Fig. 3E), TCF, LEF, and Cyclin D1 (Fig. 3F) in the dorsal hippocampus (Fortress and others 2013a). In contrast to Dkk-1 treatment, viral deletion of Dkk-1 in older mice enhances hippocampal neurogenesis and spatial working memory and reverses age-related memory consolidation impairments (Seib and others 2013). Together, these data provide persuasive evidence for a central role of Wnt/β-catenin signaling in hippocampal memory formation.
Figure 3.

Inhibition of canonical Wnt/β-catenin-dependent signaling in the dorsal hippocampus prevents object recognition memory consolidation and downstream cell signaling. (A) Behavioral paradigm used to test object recognition. Immediately after accumulating 30 seconds exploring two identical objects (training), mice received bilateral dorsal hippocampal infusions of vehicle or one of three doses of the canonical Wnt/β-catenin inhibitor Dkk-1. Twenty-four hours later, a time at which vehicle mice remember the familiar object, memory for the familiar object was tested by allowing mice to accumulate 30 seconds of time with a familiar and novel object. Because mice are drawn to novelty, they will explore the novel object more than chance (15 seconds) if they remember the familiar training objects. (B) Twenty-four hours after training, mice infused with vehicle, but not any dose of Dkk-1 (50 ng, 100 ng, 200 ng/hemisphere), spent significantly more time than chance (dashed line at 15 seconds) with the novel object (**P < 0.01 compared to chance). These data indicate that inhibition of Wnt signaling impaired memory consolidation. Bilateral dorsal hippocampal infusion of 50 ng Dkk-1 significantly decreased dorsal hippocampal Wnt7a (C) 5 minutes after infusion and increased GSK3β (D) protein levels 4 hours after infusion (*P ≤ 0.05 relative to controls). Similarly, bilateral dorsal hippocampal infusion of 50 ng of Dkk-1 significantly decreased β-Catenin (E) protein levels in the dorsal hippocampus 4 hours after infusion. Protein levels were normalized to β-actin. Each bar represents the mean ± SEM percent change from vehicle (*P ≤ 0.05 relative to vehicle-infused mice). Insets show representative Western blots. Adapted with permission from Fortress and others (2013a).
Another recent study demonstrates that Wnt3a can regulate contextual fear memory via both canonical Wnt/β-catenin signaling and Wnt/Ca2+ signaling. This study found that decreasing hippocampal Wnt3a expression with sequestering antibodies prevented acquisition and consolidation, but not expression, of contextual fear memory in male mice (Xu and others 2014). Interestingly, the data implicated Wnt/Ca2+ signaling in acquisition and Wnt/β-catenin in consolidation, indicating differential involvement of Wnt signaling pathways in learning and memory (Xu and others 2014). Accordingly, dorsal hippocampal infusion of exogenous Wnt3a activated both Wnt pathways and enhanced both fear acquisition and consolidation, suggesting that Wnt3a is both necessary and sufficient to regulate contextual fear memory.
The importance of Wnt3a and Wnt5a in facilitating other types of hippocampal memory was shown in another recent study using synthetic small molecules to selectively activate canonical and non-canonical Wnt signaling. Mice were chronically infused into hippocampal CA1 with WASP-1 (Wnt-activating small molecule), which triggers Wnt3a-induced canonical Wnt signaling, or with FOXY-5 (formylated Wnt5a-derived hexapeptide), which mimics Wnt5a-induced JNK/PCP signaling (Vargas and others 2014). Both WASP-1 and FOXY-5 enhanced synaptic plasticity, expression of hippocampal synaptic proteins, spatial memory, and object recognition memory (Vargas and others 2014), suggesting that both Wnt/β-catenin and JNK/PCP signaling regulate hippocampal spatial and object memory. Moreover, WASP-1 and FOXY-5 enhanced basal synaptic transmission and reversed impairments in both long-term potentiation and object recognition memory in a mouse model of Alzheimer’s disease (Vargas and others 2014), suggesting that activation of either Wnt pathway may reverse memory loss in neurodegenerative diseases such as Alzheimer’s.
Finally, recent work has suggested that dysfunctional Wnt signaling contributes to age-related hippocampal memory decline. In mice, Dkk-1 expression in the dentate gyrus increases with age, and genetic deletion of Dkk-1 in neural progenitor cells reversed age-related reductions in neurogenesis, TCF/LEF activity, dendritic complexity, and spatial working memory (Seib and others 2013). Other findings demonstrate that conditional deletion of the gene for Lrp6 in the mouse forebrain significantly decreases dendritic spine density in hippocampal CA1 and cortical pyramidal neurons, reduces hippocampal LTP, and impairs contextual fear conditioning in aged mice (Liu and others 2014). These data suggest that maintaining normal Wnt signaling may be essential for preventing age-related memory decline.
Collectively, several findings published in just the past 3 years demonstrate that multiple Wnt signaling pathways can regulate various forms of hippocampal memory in rodents. These results have significant implications for understanding the neural mechanisms underlying learning and memory, as the many Wnt ligands, Fzd receptors, and Wnt signaling pathways appear capable of modulating hippocampal function in myriad ways. Because hippocampal dysfunction is implicated in the etiology or symptomatology of conditions characterized by memory impairment, such as Alzheimer’s disease, Down syndrome, ischemia, stress, and aging (Box 2) (Busceti and others 2008; Cramer and Galdzicki 2012; Fjell and others 2014; Matrisciano and others 2011; Nikonenko and others 2009; Pandey and others 2014; Wingenfeld and Wolf 2014), better understanding how Wnt signaling regulates hippocampal memory may reveal new avenues for therapeutics to prevent or reverse memory loss in these conditions. Data linking Wnt signaling to neurological diseases will be discussed in the next section.
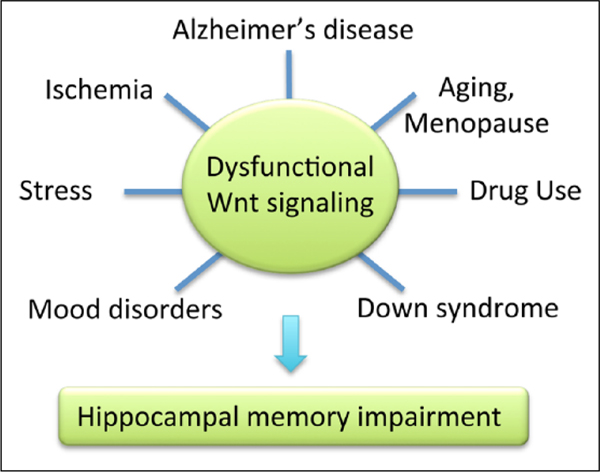
Box 2.
Dysfunctional Wnt signaling is associated with the etiology or symptomatology of multiple neurodegenerative and neuropsychiatric disorders, including Alzheimer’s disease, ischemia, drug use, Down syndrome, and mood disorders (depression, bipolar disorder). Aberrant Wnt signaling is also observed in aging and menopause, and after chronic stress. Each of these conditions is associated with hippocampal memory impairments, which may be the result of perturbations in Wnt signaling.
Wnt Signaling in Neurological Disease
A link between dysfunctional Wnt signaling and Alzheimer’s disease was established about a decade before the necessity of Wnt signaling for memory formation was demonstrated. Alzheimer’s disease is the most common form of dementia and is characterized by widespread deterioration in the brain, particularly in the hippocampus, medial temporal cortex, and prefrontal cortex. In addition to substantial neuron loss, the brains of Alzheimer’s patients are riddled with amyloid plaques, containing toxic beta-amyloid (Aβ) proteins, and neurofibrillary tangles, comprised of hyperphosphorylated tau filaments. Consistent with a beneficial role of Wnt signaling in memory, LRP6-mediated Wnt signaling is down-regulated and negatively correlated with neurotoxic Aβ40 and Aβ42 peptides in the temporal cortex of Alzheimer’s patients (Liu and others 2014). Levels of β-catenin are also reduced in the Alzheimer’s brain, and the destabilization of β-catenin by presenilin-1, mutations of which are associated with early-onset Alzheimer’s, increases Aβ-induced apoptosis (Zhang and others 1998). Interestingly, GSK3β actively hyperphosphorylates tau in neurons, as demonstrated by studies in which GSK3β inhibitors blocked Aβ-induced tau hyperphosphorylation (Caricasole and others 2004). The endogenous Wnt inhibitor Dkk-1 is also necessary for tau phosphorylation in Aβ-treated neurons, and expression of Dkk-1 is increased in Alzheimer’s brains relative to controls (Caricasole and others 2004; Liu and others 2014). In keeping with a role for Dkk-1 in tau phosphorylation, this increase occurs specifically in degenerating neurons and colocalizes with hyperphosphorylated tau, neurofibrillary tangles, and dystrophic neurites (Caricasole and others 2004). Dkk-1 is also required for the synaptic loss induced by Aβ in cultured rat hippocampal neurons (Purro and others 2012), suggesting that suppression of Wnt signaling by Dkk-1 increases the risk of Alzheimer’s pathology.
In contrast, activation of Wnt signaling can prevent the neurotoxic effects of Aβ. For example, activation of protein kinase C protected rat hippocampal neurons from Aβ-induced neurotoxicity by decreasing GSK3β activity and increasing β-catenin-induced gene expression (Garrido and others 2002). Similar effects were observed after treatment with Wnt3a (Garrido and others 2002). The benefits of Wnt3a were also shown in another study in which Wnt3a protected rat hippocampal neurons from Aβ-induced apoptosis by decreasing the phosphorylation of tau and GS3Kβ, increasing β-catenin, and enhancing cell survival (Alvarez and others 2004). Interestingly, the FDA-approved mood stabilizing agent lithium activates Wnt/β-catenin signaling by increasing GSK3β phosphorylation, and in vitro findings indicate that it also blocks the neurotoxic effects of Aβ in rat hippocampal neurons (Garrido and others 2002). In vivo, lithium reverses and prevents the neurodegenerative and memory-impairing effects of Aβ fibrils injected into the hippocampus of adult rats (De Ferrari and others 2003). Dietary lithium also reduces amyloid plaque formation and enhances hippocampal neurogenesis and hippocampal-dependent spatial memory in mice overexpressing amyloid precursor protein (TgCRND8) (Fiorentini and others 2010), supporting a key role for Wnt/β-catenin signaling in protecting hippocampal neurons from amyloid toxicity. Combined, the findings from Alzheimer’s patients and rodent models suggest that Wnt signaling is a key regulator of Alzheimer’s neuropathology and can protect against the cellular hallmarks of the disease.
Wnt signaling is also altered in numerous other conditions, including Down syndrome, Williams syndrome, ischemia, chronic stress, and drug use (Busceti and others 2008; Contestabile and others 2013; Mohn and others 2014; O’Brien and others 2004; Pandey and others 2014; Zhao and others 2005). For example, the Fzd9 receptor is mutated in Williams syndrome, and its deletion in mice causes hippocampal neuron loss and spatial memory deficits (Zhao and others 2005). In a mouse model of Down syndrome, lithium restores normal hippocampal neurogenesis and memory function by activating Wnt/β-catenin signaling (Contestabile and others 2013; Zhao and others 2005). Moreover, Dkk-1 is induced in the rodent hippocampus after cerebral ischemia (Cappuccio and others 2005), acute or chronic stress (Matrisciano and others 2011), or short-term treatment with ecstasy (MDMA) (Busceti and others 2008). The ischemia-induced increase in Dkk-1 was blocked by pretreatment with lithium or Dkk-1 antisense oligonucleotides (Cappuccio and others 2005). The ecstasy-induced increase in Dkk-1 was also accompanied by increased hippocampal tau hyperphosphorylation, reduced canonical Wnt/β-catenin signaling, and impaired spatial learning (Busceti and others 2008). Collectively, these studies suggest that impaired hippocampal Wnt signaling, particularly Wnt/β-catenin signaling, may contribute to the neuropathology and/or symptomatology of various neurological or neuropsychiatric conditions.
Wnt-Hormone Interactions and Hippocampal Function
Another way in which Wnts may mediate memory formation is by interacting with sex-steroid hormones, which are key modulators of hippocampal memory formation in their own right. Some neurodegenerative diseases (e.g., Alzheimer’s) and neuropsychiatric conditions (e.g., depression, anxiety disorders) are more prevalent in women than in men (Alzheimer’s Association 2011; Kessler and others 2005), potentially due to sex differences in circulating estrogens and progestins. Of particular relevance to hippocampal memory is the elevated risk of Alzheimer’s disease faced by menopausal women (Yaffe and others 1998; Zandi and others 2002). This risk is thought to be due to the precipitous drop in circulating estrogens and progestins that occurs at menopause (Sherwin and Henry 2008; Yaffe and others 2007). Given the importance of Wnt signaling to hippocampal memory and the ability of Wnt signaling to protect from Aβ neurotoxicity, interactions among estrogens, progestins, and Wnts may contribute to the etiology and/or symptomatology of Alzheimer’s. Understanding the extent to which these potential interactions influence hippocampal functioning could provide vital new insights into this disease. As such, Wnt-hormone interactions will be discussed in this section.
The hippocampus is exquisitely sensitive to sex-steroid hormones, such as 17β-estradiol (E2) and progesterone (P4). Both E2 and P4 facilitate hippocampal dendritic spinogenesis, synaptic transmission, synaptic plasticity, and neurogenesis (Foy and others 2010; Kato and others 2013). Moreover, these hormones regulate the same types of hippocampal memory modulated by Wnt signaling (Luine 2014; Tuscher and others 2014). Because numerous reviews have discussed the molecular mechanisms through which E2 and P4 mediate hippocampal memory formation (Brinton and others 2008; Fortress and Frick 2014; Frick 2009; Sellers and others 2014; Singh and others 2013; Tuscher and others 2014), this information will be reviewed only briefly here. Both E2 and P4 are synthesized from a cholesterol precursor in the gonads and de novo in brain areas including the hippocampus (Cui and others 2013; Kato and others 2013). E2 binds to intracellular estrogen receptors (ERs) ERα and ERβ located in the cytoplasm, or to membrane-associated receptors such as GPER or Gq-mER. P4 binds to the intracellular receptors PR-A and PR-B, and to the membrane-associated receptors PGRMC1/2 and mPRα–ε. Both E2 and P4 can induce either a classical genomic response, where receptor homo- or heterodimers act as transcription factors at the hormone response element on DNA, or a non-classical response that triggers rapid changes in cell-signaling mechanisms. For example, both E2 and P4 induce rapid changes in dorsal hippocampal ERK and mTOR signaling that are necessary for each hormone to enhance object recognition memory consolidation in female mice (Fig. 4) (Fernandez and others 2008; Fortress and others 2013b; Orr and others 2012). Furthermore, evidence from our laboratory suggests that E2 and P4 each rapidly regulate epigenetic processes, such as histone acetylation (Fig. 4), in the dorsal hippocampus to facilitate memory formation (Fortress and others 2014b; Heisler and others 2012; Zhao and others 2010; Zhao and others 2012).
Figure 4.
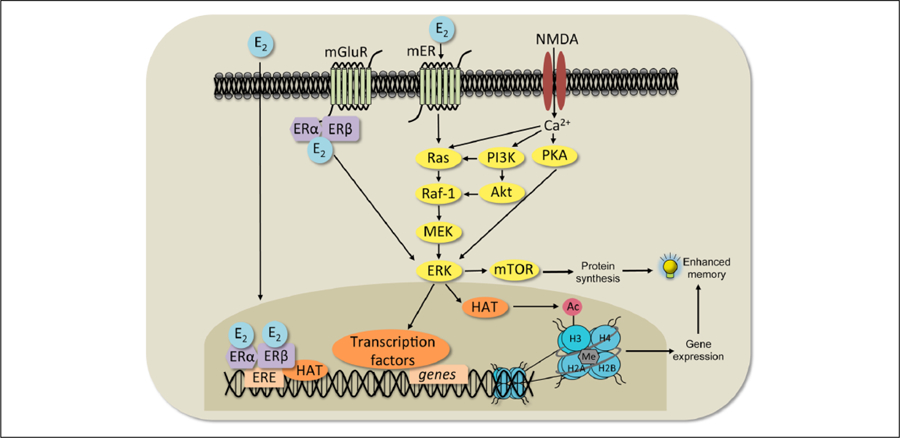
Classical and non-classical 17β-estradiol (E2) signaling mechanisms. In the classical response (left), E2 binds ERα and ERβ, which then translocate into the nucleus, bind to the estrogen response element (ERE) on DNA, and interact with co-regulatory proteins (including histone acetyltransferases, HAT) to influence transcription. In a non-classical response (center), E2 may affect cell signaling in several ways. It can bind to ERs that interact with metabotropic glutamate receptors (mGluRs) at the membrane and activate extracellular regulated kinase (ERK) signaling. E2 can also interact with NMDA receptors and membrane-bound ERs (mER) to activate the protein kinase A (PKA), phosphoinositol-3-kinase (PI3K), and mammalian target of rapamycin (mTOR) signaling pathways. mTOR signaling regulates the protein synthesis necessary for memory formation. Activation of ERK increases histone H3 acetylation (Ac). Both H3 acetylation and DNA methylation (Me) are necessary for E2 to enhance memory consolidation. Adapted with permission from Fortress and Frick (2014).
Estradiol/Wnt Interactions
Although no study has specifically tested the importance of Wnt-hormone interactions for hippocampal memory, the ability of E2 to regulate Wnt signaling in the hippocampus and cortex has been well established (Scott and Brann 2013). For example, E2 induces a long-term reduction in GSK3β activity and tau phosphorylation in primary hippocampal neurons (Cardona-Gomez and others 2004). In primary cortical neurons, E2 reduces GSK3β activity, increases the amount and stabilization of β-catenin, and increases TCF/LEF-induced gene transcription (Varea and others 2009). This gene transcription is blocked by an estrogen receptor antagonist and increased by estrogen receptor agonists (Varea and others 2009), suggesting that estrogen receptor activation influences Wnt/β-catenin-induced gene transcription. Indeed, in primary hippocampal cultures, β-catenin complexes with ERα and GSK3β, and E2 causes β-catenin to dissociate from this complex (Cardona-Gomez and others 2004), thereby allowing it to interact with other molecules. Moreover, reproductively senescent aged female rats exhibit higher hippocampal levels of Dkk-1 and β-catenin phosphorylation than young females, as well as a reduced ability of E2 to decrease Dkk-1 levels (Scott and others 2013), suggesting that long-term E2 deprivation may promote basal Wnt/β-catenin dysfunction in aging females.
Consistent with an increased risk of Alzheimer’s in postmenopausal females, E2 reduces degeneration associated with Aβ and tau hyperphosphorylation. In vitro, pretreatment of PC12 cells with E2 prevents an Aβ-induced activation of GSK3β and decrease in phosphorylation of the transcription factor CREB (Chen and others 2013), suggesting that regulation of GSK3β may be a key mechanism through which E2 protects against Aβ neurotoxicity. Similarly, E2 in PC12 and N2a cells rapidly phosphorylates GSK3β in an Akt-dependent manner, leading to a reduction in tau phosphorylation (Chen and others 2013; Shi and others 2008). In primary hippocampal neurons, the E2-induced phosphorylation of GSK3β decreases its intrinsic kinase activity, increases β-catenin stabilization, and increases the expression of Wnt7a and Wnt5a (Quintanilla and others 2005). The increased expression of Wnt7a and Wnt5a suggests that E2 may drive a positive Wnt feedback loop in which repression of GSK3β increases expression of Wnt ligands, thereby potentiating additional Wnt signaling. Such positive feedback could facilitate hippocampal function and protect hippocampal neurons from Aβ- and tau-induced neurodegeneration.
E2 is also neuroprotective in rodent models of ischemia. The negative consequences of ischemic injury, such as size of infarct, memory impairments, and hippocampal cell death, are more pronounced when E2 levels are low, such as after menopause (Koellhoffer and McCullough 2013; Zuo and others 2013). In ovariectomized rats, E2 protects hippocampal CA1 neurons from degeneration after global cerebral ischemia by suppressing Dkk-1 expression and tau hyperphosphorylation, and increasing β-catenin and Wnt3 levels (Scott and others 2013; Zhang and others 2008). Interestingly, the ability of E2 to promote Wnt/β-catenin signaling and protect against ischemic injury is lost 10 weeks after ovariectomy (Scott and others 2013), suggesting that long-term estrogen deprivation diminishes the neuroprotective effects of E2. Similar detrimental effects of long-term estrogen deprivation have been reported in studies showing that E2 loses its ability to enhance hippocampal LTP and memory formation in rats ovariectomized for several months prior to treatment (Daniel 2006; Smith and others 2010; Vedder and others 2014). These findings may help explain why postmenopausal women, who are estrogen deprived, are particularly susceptible to Alzheimer’s disease.
Progesterone/Wnt Interactions
Although most studies have focused on the role of E2 in regulating Wnt signaling, new evidence indicates that P4 robustly activates canonical Wnt/β-catenin signaling in the dorsal hippocampus. As described above, P4 requires ERK and mTOR signaling to facilitate object recognition memory consolidation in ovariectomized female mice (Orr and others 2012). Our laboratory recently reported that the P4-induced activation of ERK and mTOR is mediated by membrane-associated progesterone receptors (PRs) in the dorsal hippocampus (Fortress and others 2014a). In contrast, intracellular PRs activated Wnt/β-catenin signaling instead of ERK or mTOR signaling (Fortress and others 2014a) (Fig. 5). Specifically, infusion of the intracellular PR-A and PR-B agonist R5020 into the dorsal hippocampus increased protein levels of Wnt7a, β-catenin, and the β-catenin target gene c-myc in this brain region (Fig. 6A–C). The effects of R5020 were attenuated by the intracellular PR antagonist RU486 (Fig. 6A–C), supporting a role for activation of Wnt/β-catenin signaling by PR-A and PR-B (Fortress and others 2014a). Given that R5020 also enhanced object recognition memory consolidation, these data suggest that P4 may facilitate memory by activating Wnt/β-catenin signaling. Such an interaction is consistent with data from the uterus and mammary gland, where PR-A and PR-B require β-catenin-dependent signaling to promote the development of those organs (Boras-Granic and Hamel 2013; Robinson and others 2000; Satterfield and others 2008). Given that both P4 and β-catenin-dependent signaling contribute to hippocampal memory consolidation (Fortress and others 2013a; Xu and others 2014), our data suggest that P4-induced Wnt/β-catenin signaling may modulate hippocampal memory formation.
Figure 5.
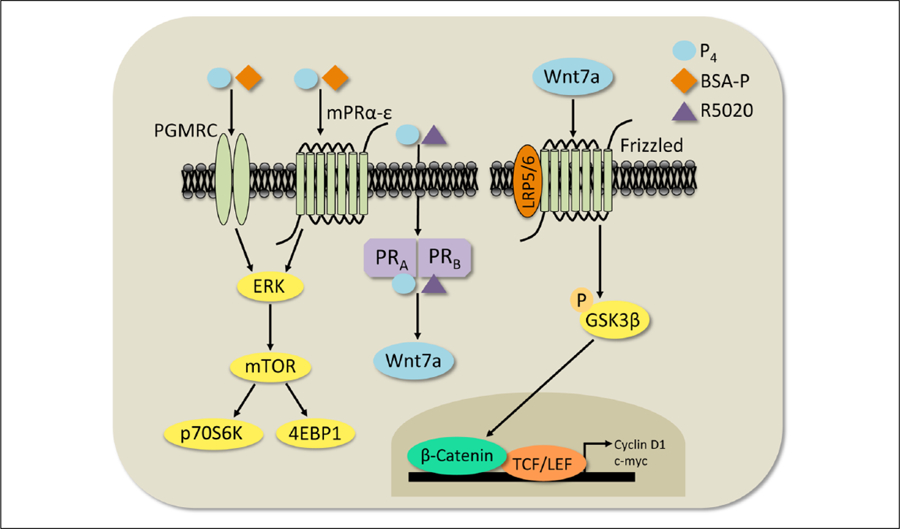
Diagram of hypothesized progesterone (P4)-mediated cell signaling mechanisms in the dorsal hippocampus. Our data suggest that P4 activates membrane or intracellular receptors to activate two different signaling cascades. P4, or the membrane-associated progesterone receptor (PR) agonist BSA-P, binds to PRs such as the PGMRCs or the mPRα–ε to trigger signaling through the ERK-dependent mTOR pathway (left). Alternatively, P4, or the intracellular agonist R5020, can bind to PR-A and PR-B to increase Wnt7a protein levels (center). The rapid rise in Wnt7a protein levels may then activate downstream canonical Wnt/β-catenin-dependent signaling (right).
Figure 6.

Progesterone (P4)-mediated activation of canonical Wnt/β-catenin-dependent signaling. Bilateral dorsal hippocampal infusion of P4 or the intracellular PR agonist R5020 significantly increased Wnt7a (A) and β-catenin (B) protein levels 5 minutes after infusion. Protein levels of Wnt7a and β-catenin were not affected by the membrane-associated PR agonist BSA-P, suggesting a role of intracellular PRs in activating Wnt signaling. Bilateral dorsal hippocampal infusion of P4 also significantly increased c-myc (C) protein levels 5 minutes after infusion. Protein levels were normalized to β-actin. Each bar represents the mean ± SEM percent change from vehicle (*P ≤ 0.05 relative to vehicle-infused mice). Insets show representative Western blots. Adapted with permission from Fortress and others (2014a).
Conclusions
As this review has illustrated, the multitude of ligands, receptors, and pathways that fall under the umbrella of “Wnt signaling” can have profound effects on the functioning of the hippocampus. Not only do Wnts direct hippocampal cytoarchitecture during embryogenesis, but they also regulate synapse formation and function in adulthood. As a result, hippocampal memory consolidation requires at least some forms of Wnt signaling. Wnts may be involved in the powerful modulatory effects of sex steroid hormones on hippocampal function, particularly in relation to age-related memory decline. The important role of Wnt signaling in protecting hippocampal neurons from Aβ- and ischemia-induced neurotoxicity, and regulating tau hyperphosphorylation, also positions Wnts as key players in neurodegenerative disease. Better understanding how the Wnt signaling pathways work and interact with other modulatory factors in the hippocampus should lead to important new insights into memory function and dysfunction in the coming decades.
Acknowledgments
We would like to thank Jennifer Tuscher for critical comments on this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the University of Wisconsin–Milwaukee, R01 DA038042, and an Ellison Medical Foundation/AFAR Postdoctoral Fellowship in Aging to AMF.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Ahmad-Annuar A, Ciani L, Simeonidis I, Herreros J, Fredj NB, Rosso SB, and others 2006. Signaling across the synapse: a role for Wnt and Dishevelled in presynaptic assembly and neurotransmitter release. J Cell Biol 174:127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez AR, Godoy JA, Mullendorff K, Olivares GH, Bronfman M, Inestrosa NC. 2004. Wnt-3a overcomes beta-amyloid toxicity in rat hippocampal neurons. Exp Cell Res 297:186–96. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s Association. 2011. 2011 Alzheimer’s disease facts and figures. Washington, DC: Alzheimer’s Association; p 31. [DOI] [PubMed] [Google Scholar]
- Baker NE. 1987. Molecular cloning of sequences from wingless, a segment polarity gene in Drosophila: the spatial distribution of a transcript in embryos. EMBO J 6:1765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartscherer K, Boutros M. 2008. Regulation of Wnt protein secretion and its role in gradient formation. EMBO Rep 9:977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, and others 2007. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316:1619–22. [DOI] [PubMed] [Google Scholar]
- Boras-Granic K, Hamel PA. 2013. Wnt-signalling in the embryonic mammary gland. J Mammary Gland Biol Neoplasia 18:155–63. [DOI] [PubMed] [Google Scholar]
- Brinton RD, Thompson RF, Foy MR, Baudry M, Wang J, Finch CE, and others 2008. Progesterone receptors: form and function in brain. Front Neuroendocrinol 29:313–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busceti CL, Biagioni F, Riozzi B, Battaglia G, Storto M, Cinque C, and others 2008. Enhanced tau phosphorylation in the hippocampus of mice treated with 3,4-methylenedioxymethamphetamine (“Ecstasy”). J Neurosci 28:3234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappuccio I, Calderone A, Busceti CL, Biagioni F, Pontarelli F, Bruno V, and others 2005. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J Neurosci 25:2647–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona-Gomez P, Perez M, Avila J, Garcia-Segura LM, Wandosell F. 2004. Estradiol inhibits GSK3 and regulates interaction of estrogen receptors, GSK3, and beta-catenin in the hippocampus. Mol Cell Neurosci 25:363–73. [DOI] [PubMed] [Google Scholar]
- Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, and others 2004. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J Neurosci 24:6021–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa W, Godoy JA, Alfaro I, Farias GG, Metcalfe MJ, Fuentealba R, and others 2008. Wnt-7a modulates the synaptic vesicle cycle and synaptic transmission in hippocampal neurons. J Biol Chem 283:5918–27. [DOI] [PubMed] [Google Scholar]
- Chen J, Park CS, Tang SJ. 2006. Activity-dependent synaptic Wnt release regulates hippocampal long term potentiation. J Biol Chem 281:11910–6. [DOI] [PubMed] [Google Scholar]
- Chen Y, Su Y, Run X, Sun Z, Wang T, Sun S, and others 2013. Pretreatment of PC12 cells with 17beta-estradiol prevents Abeta-induced down-regulation of CREB phosphorylation and prolongs inhibition of GSK-3beta. J Mol Neurosci 50:394–401. [DOI] [PubMed] [Google Scholar]
- Church VL, Francis-West P. 2002. Wnt signalling during limb development. Int J Dev Biol 46:927–36. [PubMed] [Google Scholar]
- Ciani L, Boyle KA, Dickins E, Sahores M, Anane D, Lopes DM, and others 2011. Wnt7a signaling promotes dendritic spine growth and synaptic strength through Ca(2)(+)/Calmodulin-dependent protein kinase II. Proc Natl Acad Sci U S A 108:10732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy L, Seymour CM, Monopoli MP, O’Sullivan NC, Murphy KJ, Regan CM. 2007. Notch signalling becomes transiently attenuated during long-term memory consolidation in adult Wistar rats. Neurobiol Learn Mem 88:342–51. [DOI] [PubMed] [Google Scholar]
- Contestabile A, Greco B, Ghezzi D, Tucci V, Benfenati F, Gasparini L. 2013. Lithium rescues synaptic plasticity and memory in Down syndrome mice. J Clin Invest 123: 348–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudreuse D, Korswagen HC. 2007. The making of Wnt: new insights into Wnt maturation, sorting and secretion. Development 134:3–12. [DOI] [PubMed] [Google Scholar]
- Cramer N, Galdzicki Z. 2012. From abnormal hippocampal synaptic plasticity in Down syndrome mouse models to cognitive disability in down syndrome. Neural Plast 2012:101542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Shen Y, Li R. 2013. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med 19:197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuitino L, Godoy JA, Farias GG, Couve A, Bonansco C, Fuenzalida M, and others 2010. Wnt-5a modulates recycling of functional GABAA receptors on hippocampal neurons. J Neurosci 30:8411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JM. 2006. Effects of oestrogen on cognition: what have we learned from basic research? J Neuroendocrinol 18:787–95. [DOI] [PubMed] [Google Scholar]
- Davis EK, Zou Y, Ghosh A. 2008. Wnts acting through canonical and noncanonical signaling pathways exert opposite effects on hippocampal synapse formation. Neural Dev 3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G, and others 2003. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry 8:195–208. [DOI] [PubMed] [Google Scholar]
- Dickins EM, Salinas PC. 2013. Wnts in action: from synapse formation to synaptic maintenance. Front Cell Neurosci 7:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijksterhuis JP, Petersen J, Schulte G. 2014. WNT/Frizzled signalling: receptor-ligand selectivity with focus on FZD-G protein signalling and its physiological relevance: IUPHAR Review 3. Br J Pharmacol 171:1195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias GG, Alfaro IE, Cerpa W, Grabowski CP, Godoy JA, Bonansco C, and others 2009. Wnt-5a/JNK signaling promotes the clustering of PSD-95 in hippocampal neurons. J Biol Chem 284:15857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez SM, Lewis MC, Pechenino AS, Harburger LL, Orr PT, Gresack JE, and others 2008. Estradiol-induced enhancement of object memory consolidation involves hippocampal ERK activation and membrane-bound estrogen receptors. J Neurosci 28:8660–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini A, Rosi MC, Grossi C, Luccarini I, Casamenti F. 2010. Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mutant mice. PloS One 5:e14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB. 2014. What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog Neurobiol 117:20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortress AM, Fan L, Orr PT, Zhao Z, Frick KM. 2013b. Estradiol-induced object recognition memory consolidation is dependent on activation of mTOR signaling in dorsal hippocampus. Learn Mem 20:147–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortress AM, Frick KM. 2014. Epigenetic regulation of estrogen-dependent memory. Front Neuroendocrinol 35: 530–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortress AM, Heisler JD, Frick KM. 2014a. The mTOR and canonical Wnt signaling pathways mediate the mnemonic effects of progesterone in the dorsal hippocampus. Hippocampus doi: 10.1002/hipo.22398. [DOI] [PubMed] [Google Scholar]
- Fortress AM, Kim J, Poole RL, Gould TJ, Frick KM. 2014b. 17β-estradiol regulates histone alterations associated with memory consolidation and increases Bdnf promoter acetylation in middle-aged female mice. Learn Mem 21:457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortress AM, Schram SL, Tuscher JJ, Frick KM. 2013a. Canonical Wnt signaling is necessary for object recognition memory consolidation. J Neurosci 33:12619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy MR, Baudry M, Akopian GK, Thompson RF. 2010. Regulation of hippocampal synaptic plasticity by estrogen and progesterone. Vitam Horm 82:219–39. [DOI] [PubMed] [Google Scholar]
- Frick KM. 2009. Estrogens and age-related memory decline in rodents: what have we learned and where do we go from here? Horm Behav 55:2–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galceran J, Miyashita-Lin EM, Devaney E, Rubenstein JL, Grosschedl R. 2000. Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development 127:469–82. [DOI] [PubMed] [Google Scholar]
- Garrido JL, Godoy JA, Alvarez A, Bronfman M, Inestrosa NC. 2002. Protein kinase C inhibits amyloid beta peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J 16:1982–4. [DOI] [PubMed] [Google Scholar]
- Grove EA, Tole S. 1999. Patterning events and specification signals in the developing hippocampus. Cereb Cortex 9:551–61. [DOI] [PubMed] [Google Scholar]
- Hall AC, Lucas FR, Salinas PC. 2000. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell 100:525–35. [DOI] [PubMed] [Google Scholar]
- Heisler JD, Boulware MI, Frick KM, Fortress AM. 2012. Time course of cell signaling alterations produced by dorsal hippocampal progesterone infusion. Soc Neurosci Abstr, Poster # 92.18.
- Hogan PG, Chen L, Nardone J, Rao A. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17:2205–32. [DOI] [PubMed] [Google Scholar]
- Hutchins BI, Li L, Kalil K. 2011. Wnt/calcium signaling mediates axon growth and guidance in the developing corpus callosum. Dev Neurobiol 71:269–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Hojo Y, Higo S, Komatsuzaki Y, Murakami G, Yoshino H, and others 2013. Female hippocampal estrogens have a significant correlation with cyclic fluctuation of hippocampal spines. Front Neural Circuits 7:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. 2005. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 62:617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koellhoffer EC, McCullough LD. 2013. The effects of estrogen in ischemic stroke. Transl Stroke Res 4:390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Bryant HU, Macdougald OA. 2006. Regulation of bone mass by Wnt signaling. J Clin Invest 116:1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusserow A, Pang K, Sturm C, Hrouda M, Lentfer J, Schmidt HA, and others 2005. Unexpected complexity of the Wnt gene family in a sea anemone. Nature 433:156–160. [DOI] [PubMed] [Google Scholar]
- Lee SM, Tole S, Grove E, McMahon AP. 2000. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development 127:457–67. [DOI] [PubMed] [Google Scholar]
- Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. 1999. beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc Natl Acad Sci U S A 96:6273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Tsai CW, Deak F, Rogers J, Penuliar M, Sung YM, and others 2014. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron 84:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas FR, Salinas PC. 1997. WNT-7a induces axonal remodeling and increases synapsin I levels in cerebellar neurons. Dev Biol 192:31–44. [DOI] [PubMed] [Google Scholar]
- Luine VN. 2014. Estradiol and cognitive function: past, present and future. Horm Behav 66:602–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. 2000. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci 1:173–80. [DOI] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. 2009. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17:9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguschak KA, Ressler KJ. 2011. Wnt signaling in amygdala-dependent learning and memory. J Neurosci 31: 13057–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinou K, Christodoulides C, Antoniades C, Koutsilieris M. 2012. Wnt signaling in cardiovascular physiology. Trends Endocrinol Metab 23:628–36. [DOI] [PubMed] [Google Scholar]
- Matrisciano F, Busceti CL, Bucci D, Orlando R, Caruso A, Molinaro G, and others 2011. Induction of the Wnt antagonist Dickkopf-1 is involved in stress-induced hippocampal damage. PloS One 6:e16447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn JL, Alexander J, Pirone A, Palka CD, Lee SY, Mebane L, and others 2014. Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol Psychiatry 19:1133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M, Crenshaw EB 3rd, Kelley MW. 2006. Noncanonical Wnt signaling and neural polarity. Annu Rev Neurosci 29:363–86. [DOI] [PubMed] [Google Scholar]
- Murase S, Mosser E, Schuman EM. 2002. Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 35:91–105. [DOI] [PubMed] [Google Scholar]
- Niehrs C. 2012. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol 13:767–79. [DOI] [PubMed] [Google Scholar]
- Nikonenko AG, Radenovic L, Andjus PR, Skibo GG. 2009. Structural features of ischemic damage in the hippocampus. Anat Rec (Hoboken) 292:1914–21. [DOI] [PubMed] [Google Scholar]
- Nusse R, Varmus HE. 1982. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 31:99–109. [DOI] [PubMed] [Google Scholar]
- Nusse R, Varmus H. 2012. Three decades of Wnts: a personal perspective on how a scientific field developed. EMBO J 31:2670–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E. 1980. Mutations affecting segment number and polarity in Drosophila. Nature 287:795–801. [DOI] [PubMed] [Google Scholar]
- O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, and others 2004. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci 24:6791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Yu LM, Cingolani LA, Kemler R, Goda Y. 2007. beta-Catenin regulates excitatory postsynaptic strength at hippocampal synapses. Proc Natl Acad Sci U S A 104:13479–84.17679699 [Google Scholar]
- Oliva CA, Vargas JY, Inestrosa NC. 2013. Wnts in adult brain: from synaptic plasticity to cognitive deficiencies. Front Cell Neurosci 7:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr PT, Rubin AJ, Fan L, Kent BA, Frick KM. 2012. The progesterone-induced enhancement of object recognition memory consolidation involves activation of the extracellular signal-regulated kinase (ERK) and mammalian target of rapamycin (mTOR) pathways in the dorsal hippocampus. Horm Behav 61:487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey GN, Rizavi HS, Tripathi M, Ren X. 2014. Region-specific dysregulation of glycogen synthase kinase-3beta and beta-catenin in the postmortem brains of subjects with bipolar disorder and schizophrenia. Bipolar Disord doi: 10.1111/bdi.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandravati Pandey S. 2013. Targeting Wnt-Frizzled signaling in cardiovascular diseases. Mol Biol Rep 40:6011–8. [DOI] [PubMed] [Google Scholar]
- Petersen CP, Reddien PW. 2009. Wnt signaling and the polarity of the primary body axis. Cell 139:1056–68. [DOI] [PubMed] [Google Scholar]
- Purro SA, Dickins EM, Salinas PC. 2012. The secreted Wnt antagonist Dickkopf-1 is required for amyloid beta-mediated synaptic loss. J Neurosci 32:3492–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Munoz FJ, Metcalfe MJ, Hitschfeld M, Olivares G, Godoy JA, and others 2005. Trolox and 17beta-estradiol protect against amyloid beta-peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. J Biol Chem 280:11615–25. [DOI] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, and others. 2009. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol 186:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijsewijk F, Schuermann M, Wagenaar E, Parren P, Weigel D, Nusse R. 1987. The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 50:649–57. [DOI] [PubMed] [Google Scholar]
- Robinson GW, Hennighausen L, Johnson PF. 2000. Side-branching in the mammary gland: the progesterone-Wnt connection. Genes Dev 14:889–94. [PubMed] [Google Scholar]
- Rosso SB, Inestrosa NC. 2013. WNT signaling in neuronal maturation and synaptogenesis. Front Cell Neurosci 7:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso SB, Sussman D, Wynshaw-Boris A, Salinas PC. 2005. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci 8:34–42. [DOI] [PubMed] [Google Scholar]
- Sahores M, Gibb A, Salinas PC. 2010. Frizzled-5, a receptor for the synaptic organizer Wnt7a, regulates activity-mediated synaptogenesis. Development 137:2215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas PC, Price SR. 2005. Cadherins and catenins in synapse development. Curr Opin Neurobiol 15:73–80. [DOI] [PubMed] [Google Scholar]
- Sato A, Yamamoto H, Sakane H, Koyama H, Kikuchi A. 2010. Wnt5a regulates distinct signalling pathways by binding to Frizzled2. EMBO J 29:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterfield MC, Song G, Hayashi K, Bazer FW, Spencer TE. 2008. Progesterone regulation of the endometrial WNT system in the ovine uterus. Reprod Fertil Dev 20: 935–46. [DOI] [PubMed] [Google Scholar]
- Scott EL, Brann DW. 2013. Estrogen regulation of Dkk1 and Wnt/beta-Catenin signaling in neurodegenerative disease. Brain Res 1514:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EL, Zhang QG, Han D, Desai BN, Brann DW. 2013. Long-term estrogen deprivation leads to elevation of Dickkopf-1 and dysregulation of Wnt/beta-Catenin signaling in hippocampal CA1 neurons. Steroids 78:624–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib DR, Corsini NS, Ellwanger K, Plaas C, Mateos A, Pitzer C, and others 2013. Loss of Dickkopf-1 restores neurogenesis in old age and counteracts cognitive decline. Cell Stem Cell 12:204–14. [DOI] [PubMed] [Google Scholar]
- Sellers K, Raval P, Srivastava DP. 2014. Molecular signature of rapid estrogen regulation of synaptic connectivity and cognition. Front Neuroendocrinol doi: 10.1016/j.yfrne.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Sherwin BB, Henry JF. 2008. Brain aging modulates the neuroprotective effects of estrogen on selective aspects of cognition in women: a critical review. Front Neuroendocrinol 29:88–113. [DOI] [PubMed] [Google Scholar]
- Shi HR, Zhu LQ, Wang SH, Liu XA, Tian Q, Zhang Q, and others 2008. 17beta-estradiol attenuates glycogen synthase kinase-3beta activation and tau hyperphosphorylation in Akt-independent manner. J Neural Transm 115: 879–888. [DOI] [PubMed] [Google Scholar]
- Singh M, Su C, Ng S. 2013. Non-genomic mechanisms of progesterone action in the brain. Front Neurosci 7:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarski DC, Corces VG, Moon RT. 1997. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature 390:410–3. [DOI] [PubMed] [Google Scholar]
- Smith CC, Vedder LC, Nelson AR, Bredemann TM, McMahon LL. 2010. Duration of estrogen deprivation, not chronological age, prevents estrogen’s ability to enhance hippocampal synaptic physiology. Proc Natl Acad Sci U S A 107:19543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamos JL, Weis WI. 2013. The β-catenin destruction complex. Cold Spring Harb Perspect Biol 5:a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabatadze N, McGonigal R, Neve RL, Routtenberg A. 2014. Activity-dependent Wnt 7 dendritic targeting in hippocampal neurons: plasticity- and tagging-related retrograde signaling mechanism? Hippocampus 24:455–65. [DOI] [PubMed] [Google Scholar]
- Tabatadze N, Tomas C, McGonigal R, Lin B, Schook A, Routtenberg A. 2012. Wnt transmembrane signaling and long-term spatial memory. Hippocampus 22:1228–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Wu Y, Belenkaya TY, Huang Q, Ray L, Qu J, and others 2012. Roles of N-glycosylation and lipidation in Wg secretion and signaling. Dev Biol 364:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. 2004. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci 26:429–40. [DOI] [PubMed] [Google Scholar]
- Tuscher JJ, Fortress AM, Kim J, Frick KM. 2014. Regulation of novel object recognition and object placement by ovarian sex steroid hormones. Behav Brain Res doi: 10.1016/j.bbr.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varea O, Garrido JJ, Dopazo A, Mendez P, Garcia-Segura LM, Wandosell F. 2009. Estradiol activates beta-catenin dependent transcription in neurons. PloS One 4:e5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela-Nallar L, Grabowski CP, Alfaro IE, Alvarez AR, Inestrosa NC. 2009. Role of the Wnt receptor Frizzled-1 in presynaptic differentiation and function. Neural Dev 4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JY, Fuenzalida M, Inestrosa NC. 2014. In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer’s disease model. J Neurosci 34:2191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedder LC, Bredemann TM, McMahon LL. 2014. Estradiol replacement extends the window of opportunity for hippocampal function. Neurobiol Aging 35:2183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitureira N, Letellier M, White IJ, Goda Y. 2012. Differential control of presynaptic efficacy by postsynaptic N-cadherin and beta-catenin. Nat Neurosci 15:81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivancos V, Chen P, Spassky N, Qian D, Dabdoub A, Kelley M, and others 2009. Wnt activity guides facial branchiomotor neuron migration, and involves the PCP pathway and JNK and ROCK kinases. Neural Dev 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, and others 2006. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron 50:897–909. [DOI] [PubMed] [Google Scholar]
- Wingenfeld K, Wolf OT. 2014. Stress, memory, and the hippocampus. Front Neurol Neurosci 34:109–20. [DOI] [PubMed] [Google Scholar]
- Xu N, Zhou WJ, Wang Y, Huang SH, Li X, Chen ZY. 2014. Hippocampal Wnt3a is necessary and sufficient for contextual fear memory acquisition and consolidation. Cereb Cortex doi: 10.1093/cercor/bhu121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, Barnes D, Lindquist K, Cauley J, Simonsick EM, Penninx B, and others 2007. Endogenous sex hormone levels and risk of cognitive decline in an older biracial cohort. Neurobiol Aging 28:171–8. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Sawaya G, Lieberburg I, Grady D. 1998. Estrogen therapy in postmenopausal women: Effects on cognitive function and dementia. JAMA 279:688–95. [DOI] [PubMed] [Google Scholar]
- Yu X, Malenka RC. 2003. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci 6:1169–77. [DOI] [PubMed] [Google Scholar]
- Zandi PP, Carlson MC, Plassman BL, Welsh-Bohmer KA, Mayer LS, Steffens DC, and others 2002. Hormone replacement therapy and incidence of Alzheimer disease in older women. JAMA 288:2123–29. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Wang R, Khan M, Mahesh V, Brann DW. 2008. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci 28:8430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, and others 1998. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 395:698–702. [DOI] [PubMed] [Google Scholar]
- Zhao C, Aviles C, Abel RA, Almli CR, McQuillen P, Pleasure SJ. 2005. Hippocampal and visuospatial learning defects in mice with a deletion of frizzled 9, a gene in the Williams syndrome deletion interval. Development 132:2917–27. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Fan L, Frick KM. 2010. Epigenetic alterations regulate the estradiol-induced enhancement of memory consolidation. Proc Natl Acad Sci U S A 107:5605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Fan L, Fortress AM, Boulware MI, Frick KM. 2012. Hippocampal histone acetylation regulates object recognition and the estradiol-induced enhancement of object recognition. J Neurosci 32:2344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo W, Zhang W, Chen NH. 2013. Sexual dimorphism in cerebral ischemia injury. Eur J Pharmacol 711:73–9. [DOI] [PubMed] [Google Scholar]