

Abstract
Acetylcholinesterase (AChE) is a pivotal enzyme in neurotransmission. Its inhibition leads to cholinergic crises and could ultimately result in death. A related enzyme butyrylcholinesterase (BChE) may act in the CNS as a co-regulator in terminating nerve impulses and is a natural plasma scavenger upon exposure to organophosphate (OP) nerve agents that irreversibly inhibit both enzymes. With the aim of improving reactivation of cholinesterases phosphylated by nerve agents sarin, VX, cyclosarin, and tabun, ten phenyltetrahydroisoquinoline (PIQ) aldoximes were synthesized by Huisgen 1,3 dipolar cycloaddition between alkyne- and azide-building blocks. The PIQ moiety may serve as a peripheral site anchor positioning the aldoxime moiety at the AChE active site. In terms of evaluated dissociation inhibition constants, the aldoximes could be characterized as high-affinity ligands. Nevertheless, high binding affinity of these oximes to AChE or its phosphylated conjugates did not assure rapid and selective AChE reactivation. Rather, potential reactivators of phosphylated BChE, with its enlarged acyl pocket, were identified, especially in case of cyclosarin, where the reactivation rates of the lead reactivator was 100- and 6-times that of 2-PAM and HI-6, respectively. Nevertheless, the return of the enzyme activity was affected by the nerve agent conjugated to catalytic serine, which highlights the lack of the universality of reactivators with respect to both the target enzyme and OP structure.
Keywords: organophosphate, antidote, nerve agent, acetylcholinesterase, butyrylcholinesterase, pralidoxime, docking, peripheral anionic site
1. Introduction
In the past decades, we have witnessed numerous cases of abuse of organophosphorus compounds (OPs) with nerve agents against civilians in war (Dolgin, 2013) or as means of terrorist attacks (Nakagawa et al., 2018; Vale et al., 2018) resulting in severe outcomes due to the irreversible inhibition of the pivotal enzyme acetylcholinesterase (AChE) and the related enzyme butyrylcholinesterase (BChE). The role of BChE is not essential, but it acts as a natural scavenger from tissue AChE inhibition by OPs and may serve as a CNS co-regulator of cholinergic neurotransmission (Raveh et al., 1993; Mesulam et al., 2002; Masson and Nachon, 2017). Currently used therapy in the event of OP poisoning, consisting of an oxime reactivator (2-PAM, obidoxime, HI-6), atropine and an anticonvulsant remains limiting due to variability in the oxime efficacy regarding different conjugated OPs (Gray, 1984; Dawson, 1994; Antonijević and Stojiljković 2007; Worek et al., 2016). Over the years, numerous approaches were employed in order to improve the reactivation efficacy which is primarily attributed to the nucleophilic displacement rate of OPs and apparent affinity of the reactivating oxime for the phosphylated cholinesterases (ChE). Therefore, efficacy varies with the bound OP and the structure of the oxime (Millard et al., 1999; Kovarik et al., 2004, 2006, 2019a; Ekström et al., 2006a, 2006b; Wille et al., 2010; Wong et al., 2000; Worek et al., 2002). Concepts for the development of new antidotes include miscellaneous ligands that form productive interactions with the ChE active centers and increases in enzyme binding affinity. Furthermore, a positive effect on reactivation efficiency was shown upon designing oximes that lack a permanent charge (Kalisiak et al., 2011; Sit et al., 2011, 2018; Radić et al. 2012; Kovarik et al., 2013; Rosenberg et al, 2017a, 2017b; Zorbaz et al., 2018a) or pro-drugs and amphipathic reactivators, like pro-2-PAM, that improve the delivery of the oxime to the central nervous system (Demar et al., 2010; Chambers et al., 2016).
The active site of ChE may be divided into two sub-sites: the catalytic site (CAS) at the base of the gorge, composed of a catalytic triad, oxyanion hole, acyl binding pocket, and choline binding site; and the peripheral anionic site (PAS) at the gorge rim (Taylor and Radić, 1994). The difference in amino acid residue composition of AChE and BChE active sites enables BChE to hydrolyze or bind larger substrates and ligands than AChE (Radić et al., 1993; Taylor et al., 1995; Saxena et al., 1999; Kovarik, 1999; Kovarik et al., 2003; Bosak et al., 2008). Encouraged by the hypothesis that the increase of the binding affinity of the ChE would enhance the reactivation efficacy, dual-binding mode reactivators containing ligand(s) that form interactions with both CAS and PAS (Ekström et al., 2009) or triple-binding mode compounds (Maraković et al., 2016) were designed and tested. Researchers also found design inspiration in agents used in the treatment of Alzheimer’s disease, like tacrine or donepezil, which interact with aromatic residues of AChE (Kryger et al., 1999; McHardy et al., 2014; Renou et al., 2016). An improved reactivation efficacy was shown for piperidine derivatives, tetrahydroacridine and tryptoline moieties containing reactivators, etc. (De Koning et al., 2011; Kliachyna et al., 2014; McHardy et al., 2014; Renou et al., 2014; Zorbaz et al., 2018a). Moreover, oximes comprising tetrahydroisoquinoline (TIQ) and phenyltetrahydroisoquinoline (PIQ) groups were presented as equal or more potent reactivators of VX- and tabun-inhibited AChE than standard oximes (Mercey et al., 2011, 2012; Wei et al., 2014, 2017; Zorbaz et al., 2018a). Further, Krasinski et al. (2005) reported micromolar to femtomolar affinity of the PIQ-comprising AChE inhibitors synthesized by in situ click chemistry. This and the fact that the triazole moiety was observed earlier to contribute synergistically to the total energy of binding of high-affinity AChE inhibitors (Manetsch et al., 2004) encouraged us to design triazole aldoximes linked to the PIQ moiety. In our recent study with triazole containing oximes, we reported a successful reactivation of tabun and other nerve agents inhibited AChE (Kovarik et al., 2019a, 2019b).
Herein, we report the synthesis of ten potential reactivators containing PIQ moiety using Huisgen 1,3 dipolar cycloaddition forming anti-triazoles. The family of reactivating oximes was tested as individual reactivators of human AChE and BChE inhibited by sarin, VX, cyclosarin, and tabun. We also estimated the AChE and BChE binding affinity for PIQ aldoximes in terms of the oxime-enzyme dissociation constants (Ki). Computational docking was used to visualize important interactions with the native or OP-conjugated enzyme and for the overall interpretation of reactivation kinetics.
2. Materials and methods
2.1. Chemistry
Reagents and solvents were purchased from Aldrich (St. Louis, MO, USA), Acros (Morris Plains, New Jersey, USA) or GFS (Columbus, Ohio, USA) and used without further purification. Alkynes and azides were prepared according to procedures from the literature (Krasiński et al., 2005; Kovarik et al., 2019a). 1H and 13C NMR spectra were recorded on a Varian INOVA-400 spectrometer in CDCl3 (7.26 ppm for 1H, 77.00 ppm 13C), d6-DMSO (2.50 ppm for 1H, 39.43 ppm for 13C) as standards.
Electrospray ionization mass spectrometry (ESI-MS) gave the following results: 1a: Solid (343 mg, 89%). ESI MS [M]+ for C32H39N6O3, calcd 555.3, found 555.4. 1b: Solid (363 mg, 92%). ESI MS [M]+ for C33H41N6O3, calcd 569.3, found 569.5. 1c: Solid (371 mg, 92%). ESI MS [M]+ for C34H43N6O3, calcd 583.3, found 583.5. 1d: Solid (370 mg, 96%). ESI MS [M]+ for C32H39N6O3, calcd 555.3, found 555.5. 1e: Solid (380 mg, 96%). ESI MS [M]+ for C33H41N6O3, calcd 569.3, found 569.4. 2a: Solid (380 mg, 96%). ESI MS [M]+ for C33H41N6O3, calcd 569.3, found 569.4. 2b: Solid (391 mg, 97%). ESI MS [M]+ for C34H43N6O3, calcd 583.3, found 583.5. 2c: Solid (406 mg, 99%). ESI MS [M]+ for C35H45N6O3, calcd 597.3, found 597.5. 2d: Solid (390 mg, 99%). ESI MS [M]+ for C33H41N6O3, calcd 569.3, found 569.5. 2e: Solid (398 mg, 99%). ESI MS [M]+ for C33H41N6O3, calcd 569.3, found 569.5.
2.2. Cholinesterase activity measurements
Recombinant human AChE, wild type, was prepared as described earlier (Cochran et al., 2011). Purified oligomeric BChE isolated from human plasma was kindly donated by Dr. David Lenz and the late Dr. Douglas Cerasoli, USAMRICD, Edgewood, MD.
For reactivation experiments, the enzyme was incubated with a ten-fold molar excess of nerve agents (NC Laboratory, Spiez, Switzerland) for an hour, fractionated on a Sephadex G-50 spin column (Roche Diagnostic GmbH, Mannheim, Germany), and then incubated with an oxime. Oxime stock solutions (100 or 10 mM) were made in DMSO and further dilutions were prepared in water. At specified time intervals, an aliquot was 100-fold diluted and upon addition of ATCh residual enzyme activity was measured. The same procedure was applied to the uninhibited enzyme and control activity was measured in the presence of an oxime at concentrations used for reactivation. Reactivation screening was done at a given oxime concentration (10 μM for AChE and 100 μM for BChE) and was monitored up to 24 h. No spontaneous reactivation of phosphylated cholinesterases was detected and enzyme was stable in 24 h. Detailed reactivation kinetics using a wider oxime concentration range of leads enabled the determination of reactivation constants: k2 (maximal first-order reactivation rate constant), Kox (apparent phosphylated enzyme-oxime dissociation constant) and kr (overall second-order reactivation rate constant). k2 and Kox were evaluated from the plot kobs vs oxime concentration where (Kovarik et al., 2004; Maček Hrvat et al., 2018). kr was the ratio of k2 and Kox.
Reversible inhibition of AChE and BChE was measured in the presence of aldoxime (0.004 - 20 μM). The enzyme-oxime dissociation constant (Ki) was evaluated from the Hunter-Downs equation and procedures described previously (Kovarik et al. 2006; Katalinić et al., 2017).
2.3. Computational molecular docking
A three-dimensional structure of mouse AChE (PDB code: 5EHN; Bourne et al., 2016) and human BChE (PDB code: 3DJY; Carletti et al., 2008) were used for docking. Aldoxime structures were modeled and minimized using the MMFF94 force field implemented in ChemBio3D Ultra 12.0 (PerkinElmer, Inc., Waltham, MA). Discovery Studio 2017 R2 with CDOCKER docking protocol using a CHARMm force field (BioVia Discovery Studio 2017R2 software, San Diego, USA) generated for each aldoxime 20 poses in the active site gorge of AChE or BChE (Maraković et al., 2016). Poses were scored and ranked according to the calculated CDOCKER energy from interactions between oxime and active site residues (Šinko, 2019).
3. Results
3.1. Synthesis
Aldoximes were synthesized using copper(I)-catalyzed azide-alkyne building block Huisgen’s [2+3] cycloaddition (CuAAC) to form a linking 1,4-triazole as shown on Scheme 1:
Scheme 1.
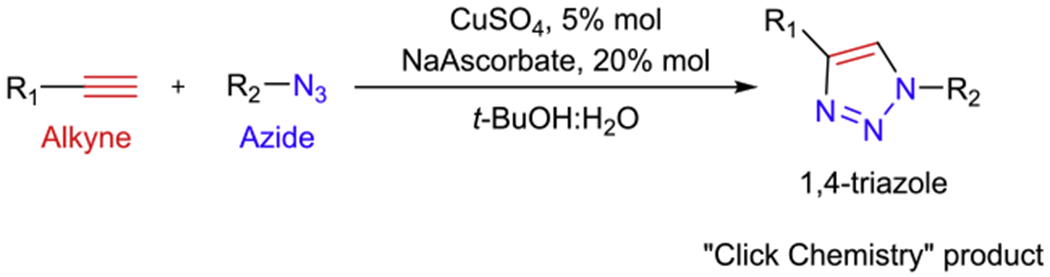
The copper(I)-catalyzed azide-alkyne cycloaddition employed to synthesize 1,4-triazole oximes.
Alkynes and azides used for the synthesis of these 10 compounds (Figure 1) were prepared as described previously (Krasinsky et al., 2005; Kovarik et al., 2019a). The general method for the synthesis of 1,4-triazole-oximes is described as follows. The alkyne (0.55 mmol) and the azide (0.55 mmol) were dissolved in the mixture of t-BuOH:H2O, 2:1 (3.3 mL), then sodium ascorbate (21.8 mg, 0.11 mmol, 20% mol) and CuSO4 (4.39 mg, 0.027 mmol, 5% mol) were added. The reaction was carried out for overnight at room temperature until LC/MS analysis showed no starting materials. Subsequently, the mixture was diluted with H2O (10 mL) and a solution of NaPF6 (185 mg, 1.1 mmol, 2 equiv.) in H2O (5 mL) was added. The mixture was cooled down to 0 °C and the hexafluorophosphate salt was separated, washed with H2O and dried on high-vacuum.
Figure 1.
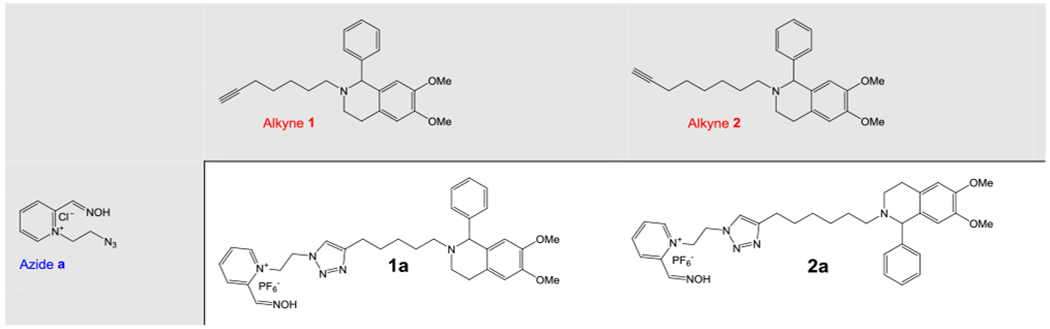
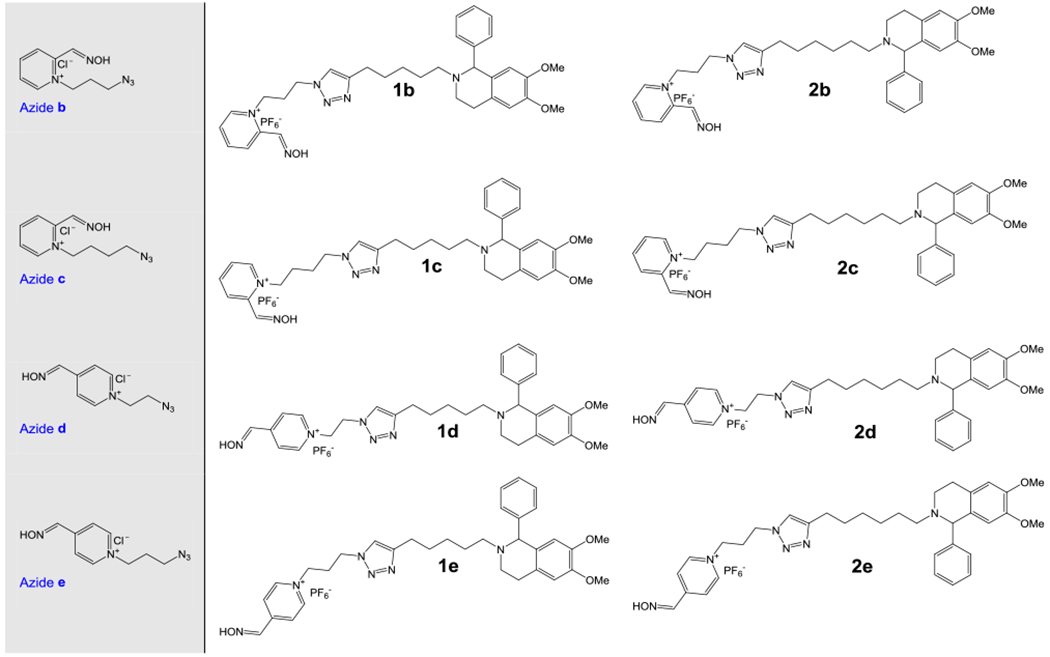
Chemical structures of alkyne (1-2) and azide (a-e) building blocks in copper(I)-catalyzed azide-alkyne building block [2+3] cycloaddition utilized for the synthesis of phenyltetrahydroisoquinoline aldoximes (1a-1e and 2a-2e).
3.2. Reversible inhibition of cholinesterases by aldoximes
The evaluated enzyme-oxime dissociation constants (Ki; Table 1) in nanomolar (for AChE) and sub-nanomolar to micromolar range (for BChE) classify the PIQ aldoximes as high-affinity binding inhibitors. Although both enzymes had the highest affinity (1/Ki) for aldoxime 2a, the affinity for AChE was about 12-times higher than that for BChE. Generally, most of the PIQ compounds could be considered as selective AChE inhibitors, since their inhibition of BChE was 5- (1a) to 955-fold (1b) less potent than for AChE. From a comparison of inhibition potencies and aldoxime structure no straightforward generalizations appear. The most potent AChE inhibitor (2a) differs from the less potent (1d) in length of linker having a 6- vs a 5-methylenes between the triazole ring and PIQ moiety, and ortho- vs para-positioned oxime group, respectively. Yet, aldoxime 1a, an ortho- analog of 1d, is among of the top three most potent AChE inhibitors. In the case of BChE, the Ki determined for the most potent BChE inhibitors (2a and 1a) and their analogs with a para-positioned oxime group (2d and 2a) differed 2- and 60-times, respectively. Interestingly, 1d and 2c had an almost identical Ki determined for BChE, even though they are structurally very different (shortest vs longest triazole containing linker and para- vs ortho-positioned oxime group, respectively). A similar situation as was found for oximes 1b and 1c.
Table 1.
Dissociation inhibition constants (Ki ± S.E.) of the phenyltetrahydroisoquinoline aldoximes for AChE and BChE determined at 25 °C.
| Oxime |
Ki (nM) |
BChE/AChE | |
|---|---|---|---|
| AChE | BChE | ||
| 1a | 15 ± 1 | 78 ± 7 | 5.2 |
| 1b | 8.9 ± 0.5 | 8500 ± 1300 | 955 |
| 1c | 67 ± 4 | 8600 ± 1700 | 128 |
| 1d | 130 ± 8 | 4900 ± 500 | 38 |
| 1e | 41 ± 4 | 10900 ± 1700 | 266 |
| 2a | 6.0 ± 0.7 | 71 ± 4 | 12 |
| 2b | 28 ± 3 | 5900 ± 500 | 211 |
| 2c | 32 ± 3 | 4800 ± 700 | 150 |
| 2d | 23 ± 2 | 147 ± 14 | 6.4 |
| 2e | 100 ± 9 | 7900 ± 1200 | 79 |
| HI-6a | 25000 ± 1400 | 215000 ± 11000 | 8.6 |
| 2-PAMa | 180000 ± 10000 | 390000 ± 48000 | 2.2 |
3.3. Oxime-assisted reactivation of cholinesterases inhibited by nerve agents
Ten phenyltetrahydroisoquinoline (PIQ) aldoximes (Figure 1) were screened for reactivation of sarin-, cyclosarin-, VX- and tabun-inhibited human AChE and BChE at a given oxime concentration, 10 μM for AChE and 100 μM for BChE, and the results were sorted in terms of the observed first-order reactivation rate (kobs) (Figure 2). If the maximal percentage of reactivation (Reactmax) reached in 24 h was less than 30%, kobs was not calculated. In case of AChE reactivation PIQ aldoximes were not more efficient than HI-6, although nine, five and two aldoximes reactivated more than 50% in case of cyclosarin, sarin, and VX inhibition, respectively. Tabun-inhibited AChE or BChE was resistant to reactivation by any of tested aldoximes (results not shown). Phosphylated BChE conjugates were more prone to be reactivated with PIQ aldoximes. All PIQ aldoximes reactivated more than 60% of cyclosarin- and sarin-inhibited BChE, and 40 - 50% of BChE activity in the case of VX inhibition (Figure 2). Moreover, several PIQ aldoximes were more potent reactivators of phosphylated BChE than HI-6 or 2-PAM, and for these detailed kinetic parameters were determined.
Figure 2.
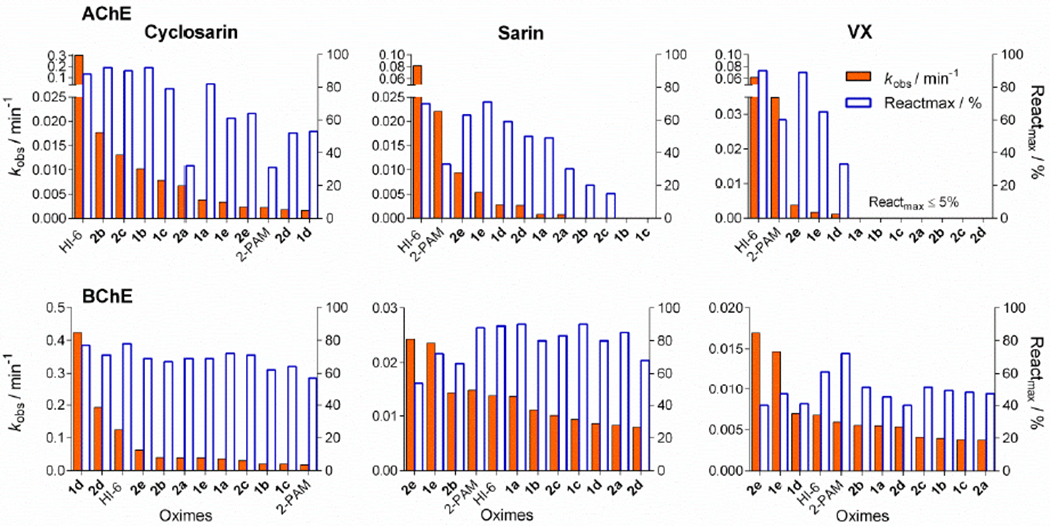
Reactivation screening of cyclosarin-, sarin- and VX- inhibited human AChE and BChE with 10μM (AChE) or 100 μM (BChE) phenyltetrahydroisoquinoline aldoximes. The observed reactivation rates (kobs) and maximal percentage of reactivation (Reactmax) within 24 h were evaluated and deviations were less than 10%. kobs was not evaluated when Reactmax was <30%. The results for tabun inhibition are not shown due to <5% reactivation within 24h.
Kinetic studies over a range of oxime concentrations were completed for the selected reactivators of cyclosarin- and sarin-inhibited BChE (Figure 3), and results were sorted in terms of kr as an overall measure of efficiency (Table 2). The cyclosarin-inhibited BChE activity was restored with selected aldoximes, 1d and 2d, up to 80% within a short time, 10 or 30 min, respectively. Both aldoximes have similar binding affinities (1/Kox) but the rate of dephosphylation (k2) was two-fold lower for 2d. It seems that the length of the triazole-containing linker affected the reactivation efficacy, because aldoximes 1d and 2d differ only by a five versus six methylene chain linked between the triazole ring and PIQ moiety, respectively (Figure 1). A 6- or a 100-fold higher kr with aldoxime 1d than with HI-6 or 2-PAM, respectively, seems to be due to a low binding affinity of cyclosarin-conjugated BChE for standard oximes (Table 2).
Figure 3.
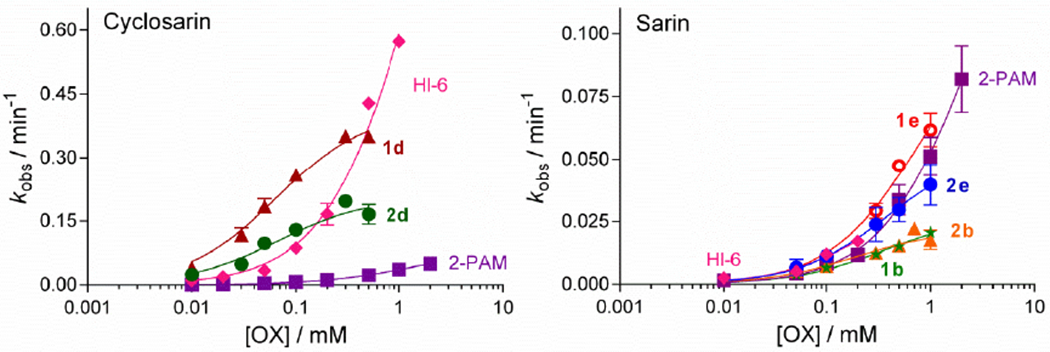
Reactivation kinetics of cyclosarin- and sarin-inhibited BChE by the selected aldoximes.
Table 2.
Reactivation parameters determined for selected aldoximes and human BChE inhibited by cyclosarin and sarin. The results are expressed as mean ± S.E. calculated from at least three experiments. Maximal percentage of reactivation (Reactmax) and the time (t) in which it was achieved were also determined.
| Inhibitor | Oxime | k2 / min−1 | Kox / μM | kr / M−1 min−1 | Reactmax / % | t / min |
|---|---|---|---|---|---|---|
| Cyclosarin | 1d | 0.40 ± 0.02 | 66 ± 8.0 | 6250 ± 55 | 80 | 10 |
| 2d | 0.21 ± 0.02 | 67 ± 16 | 3050 ± 520 | 70 | 30 | |
| HI-6 | 1.4 ± 0.27 | 1410 ± 390 | 1010 ± 95.0 | 90 | 30 | |
| 2-PAM | 0.08 ± 0.01 | 1200 ± 240.0 | 65 ± 10 | 80 | 60 | |
| Sarin | 1e | 0.11± 0.013 | 800 ± 170 | 140 ± 14 | 80 | 60 |
| 2e | 0.058 ± 0.014 | 410 ± 250 | 140 ± 50 | 60 | 60 | |
| 2b | 0.022 ± 0.0033 | 195 ± 89.0 | 110 ± 35 | 70 | 120 | |
| 1b | 0.030 ± 0.003 | 365 ± 84.0 | 70 ± 10 | 90 | 180 | |
| HI-6 | 0.041 ± 0.019 | 270 ± 200 | 150 ± 40 | 100 | 300 | |
| 2-PAM | 0.18 ± 0.05 | 2400 ± 995.0 | 75 ± 10 | 100 | 120 | |
In case of sarin, four selected aldoximes reactivated up to 90% of sarin-inhibited BChE within 3 hours. Although high percentage of reactivation was achieved with both standard oximes (Table 2), and 2-PAM had the highest potency for the nucleophilic displacement of sarin moiety, its overall reactivation potency was lower than that of tested oximes due to its low apparent binding affinity (high Kox). It is interesting to note that if one compares spacer length, shorter linker analogues (1b and 1e) showed faster reactivation rate but lower affinity than their counterparts (2b and 2e).
3.4. Molecular modelling of aldoxime complexes with AChE and phosphonylated BChE
The model of the complex between aldoxime 2a, the most potent AChE inhibitor, and mouse AChE is shown in Figure 4. It seems that the high affinity of native AChE for 2a is due to the multiple interactions of the aldoxime. The pyridinium ring of 2a was stabilized by Trp86 from the choline binding site and by His447, a residue of the catalytic triad, while the oxime group was close to the catalytic Ser203. H-bonding with Gly121 from the oxyanion hole is also a possibility. The PIQ of 2a, as expected, was located in the PAS, forming π-π interactions with Trp286, and H-bonds with Ser293 and Tyr341.
Figure 4.
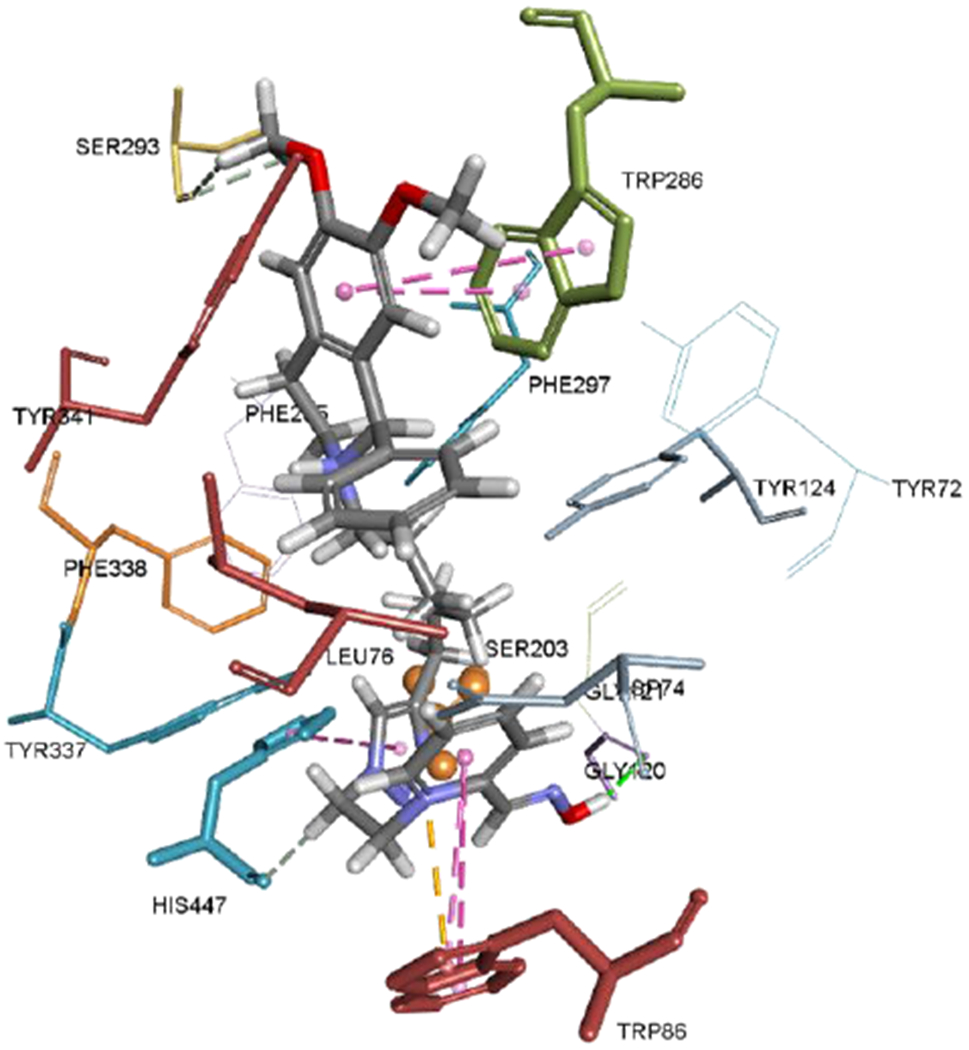
Positioning of the aldoxime 2a with a protonated nitrogen of phenyltetrahydroisoquinoline ring in the mouse AChE active site (PDB code: 5EHN; Bourne et al, 2016). The catalytic Ser203 is presented as an orange stick and ball. Hydrogen bonds are shown in green, electrostatic interactions in orange and π-π interactions in magenta.
Aldoxime 1d, a potent reactivator of cyclosarin-inhibited BChE, was modelled inside the model of BChE-cyclosarin conjugate using flexible docking allowing Leu286, Phe329, and Tyr332 to rotate freely. The minimised structure of the complex, presented in Figure 5 showed a favourable position of the oxime group for a nucleophilic attack with a distance of 4.6 Å from the phosphorus atom. The PIQ domain of 1d was stabilized outside the choline-binding site of BChE via multiple H-bonds involving Trp82, Asn83, Thr120, Pro285, and Asn289. The triazole ring of 1d was positioned close to Ala277. Due to a poorly defined PAS in BChE, lacking aromatic side chains, the conformation of 1d was so oriented to be stabilized by an intramolecular cation-π and π-π interaction between the pyridinium ring and PIQ moiety of 1d.
Figure 5.
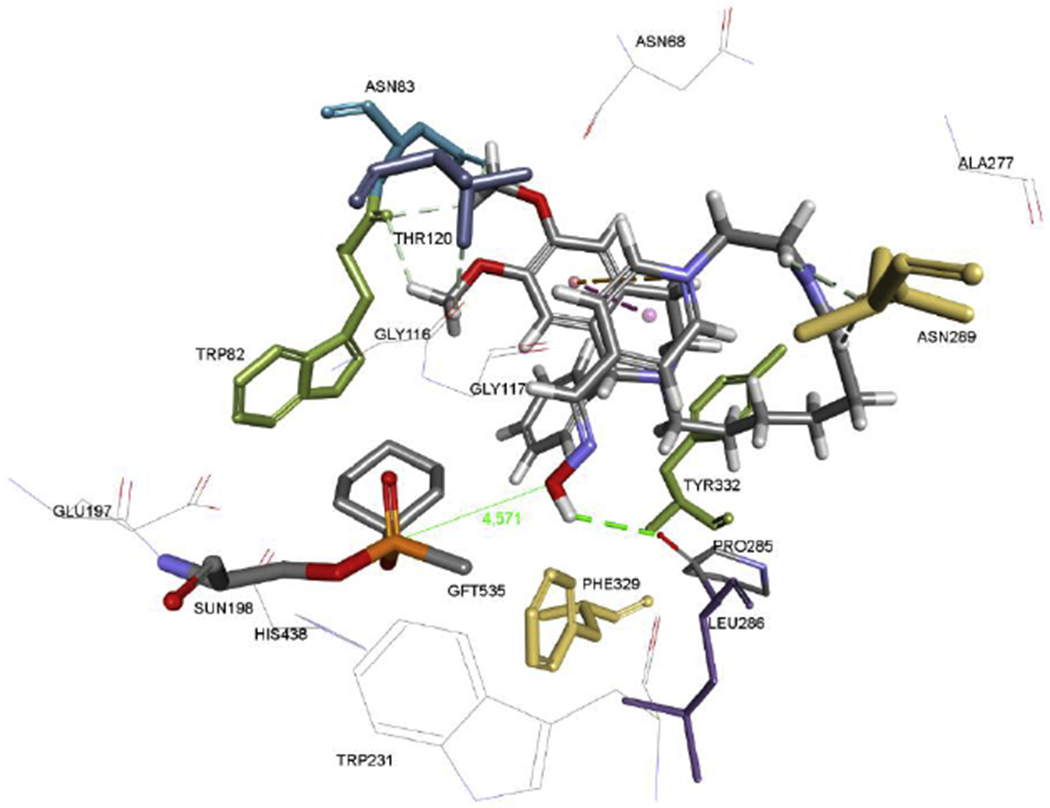
Minimized structure of aldoxime 1d in cyclosarin-phosphonylated BChE modelled from human BChE inhibited by tabun (PDB code: 3DJY; Carletti et al, 2008). Residues Leu286, Phe329 and Tyr332 were set to be flexible during docking. Hydrogen bonds are shown in green, π-π and alkyl-π interactions in orange.
4. Discussion
This study describes the design, synthesis and in vitro evaluation of the interactions of ten new aldoximes and, their potential as reactivators of cholinesterases inhibited by the nerve agents: sarin, cyclosarin, VX, and tabun. Oximes were synthesized using Huisgen 1,3 dipolar cycloaddition forming anti-triazoles. Alkyne building blocks comprising the PAS-binding ligand PIQ should anchor the compound in a way that the azide building block with a pyridinium ring and oxime group is directed towards phosphorate or phosphonate at the catalytic serine. The triazole ring formed by this cycloaddition should contribute to binding interactions within the active site gorge and additionally enhance the affinity of the enzyme for the compounds. This hypothesis was supported by the dissociation inhibition constants of AChE which were in nanomolar range (Table 1) classifying the tested aldoximes as its high-affinity ligands. Moreover, the molecular modeling study with aldoxime 2a confirmed the stabilization of its PIQ moiety in the PAS of AChE. Nevertheless, although we succeeded in improving the binding affinity of the native AChE for tested compounds, the efficacy of PIQ aldoximes as phosphylated AChE reactivators was not superior to HI-6. This may indicate that oximes are stabilized in an unfavorable position for efficient nucleophilic attack. It seems that triazole in linker affect the reactivation, since other PIQ aldoximes have been previously reported as equally or more potent reactivators than the standard oximes for AChE inhibited by various OPs (Mercey et al., 2011, 2012; Zorbaz et al., 2018a). This may indicate that the triazole linkage as a strong dipole confers a less favorable oxime orientation for nucleophilic attack, despite the high affinity of these compounds for unconjugated AChE.
Conformational changes and steric hindrance in the gorge induced by the OP conjugation (Eto, 1976; Millard et al., 1999; Ekström et al., 2006a, 2006b; Katalinić et al., 2018) consequently affect the possibility of the oxime to adopt the most favorable position for dephosphylation of catalytic serine. This was particularly noted in the case of tabun inhibition where there was no reactivation of either AChE or BChE with the tested aldoximes. Yet, in our recent paper (Kovarik et al., 2019a) we reported a potent reactivator (oxime 3A) of a phosphoramidated (tabun-AChE) conjugate, also synthesized by the Huisgen cycloaddition, but with a much shorter triazole containing linker between two pyridinium rings, when compared to PIQ aldoximes. On the other hand, although the active site of BChE allows more conformational freedom and flexibility of tested PIQ aldoximes than the active site of AChE, potential reactivators of cyclosarin- and sarin-, but not of VX- and tabun-inhibited BChE were identified emphasizing the complexity of reactivation as a consequence of the steric constraints of conjugated phosphorus moiety. It is interesting to note, that aldoxime 1d is more efficient reactivator of the cyclosarin-BChE conjugate than a PIQ containing aldoxime reported by Zorbaz et al. (2018a). This is probably due to its higher binding affinity and stabilization of the triazole ring in the cyclosarin-BChE conjugate, as shown by molecular modelling (Figure 5).
The determined dissociation inhibition constants of BChE were in micromolar or submicromolar range, and if compared to the nanomolar constants with these compounds for AChE, the tested aldoximes are preferably complexed by AChE. Furthermore, detailed kinetics of the reactivation of cyclosarin- and sarin-inhibited BChE showed that the affinity of the phosphonylated BChE conjugate for top reactivators decreased up to 400 times, when compared to native BChE (Table 1, Table 2). This is probably due to the conformational changes in the active site and steric hindrance with the binding of the OP which affects oxime embedding and orientation. Therefore, achieving a moderate apparent affinity optimizes reactivation and, excessively high affinity does not necessarily enhance reactivation rates. Multiple interactions of oxime with active site residues could stabilize the oxime in unproductive conformation, with the oximate directed away from the phosphorus conjugated to catalytic serine, therein not fulfilling the alignment for efficient dephosphylation. Alternatively, a spatially constrained active center gorge may restrict the angle of attack and the departure of the conjugated organophosphate from the gorge. Finally, the high affinities of these oxime reactivators for the native enzyme are likely to inhibit the residual active enzyme in an OP exposure interval. Accordingly, achieving maximal reactivation in vivo and minimal toxicity from the reactivator itself is best be achieved with a maximal reactivation rate and minimizing the affinity for the residual free enzyme.
To conclude, the efficacy of reactivator reveals an interplay between affinity for the residual free and conjugated enzymes, the optimal length of the linker (compound), position and orientation of the oximate with respect to the conjugated phosphorus. Moreover, one must consider the structure of the phosphylating agents since they also differentially restrict access to the gorge.
Highlights.
phenyltetrahydroisoquinoline moiety acts as peripheral anionic site ligand
potential reactivators of phosphylated BChE were identified
high binding affinity of AChE for oximes does not ensure efficient reactivation in situ oxime efficacy depends on the organophosphate conjugated to the cholinesterase
Acknowledgements
The authors thank Antonio Zandona for his help with several experiments and Dr. Christophe Curty for helpful assistance with purchasing organophosphates.
Funding: This work was supported by the National Institutes of Health Counteract Program [grant numbers U01 NS058046] and by the Croatian Science Foundation [grant number HrZZ-IP-2018-01-7683].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: The authors declare no conflict of interest.
References
- Antonijević B, Stojiljković MP, 2007. Unequal efficacy of pyridinium oximes in acute organophosphate poisoning. Clin. Med. Res 5(1), 71–82. doi: 10.3121/cmr.2007.701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosak A, Gazić I, Vinković V, Kovarik Z, 2008. Amino acid residues involved in stereoselective inhibition of cholinesterases with bambuterol. Arch. Biochem. Biophys 471(1), 72–76. [DOI] [PubMed] [Google Scholar]
- Bourne Y, Sharpless KB, Taylor P, Marchot P, 2016. Steric and dynamic parameters influencing in situ cycloadditions to form triazole inhibitors with crystalline acetylcholinesterase. J. Am. Chem. Soc 138(5), 1611–21. doi: 10.1021/jacs.5b11384 [DOI] [PubMed] [Google Scholar]
- Carletti E, Li H, Li B, Ekstrom F, Nicolet Y, Loiodice M, Gillon E, Froment MT, Lockridge O, Schopfer LM, Masson P, Nachon F, 2008. Aging of cholinesterases phosphylated by tabun proceeds through o-dealkylation. J. Am. Chem. Soc 130, 16011–16020. [DOI] [PubMed] [Google Scholar]
- Chambers JE, Chambers HW, Funck KE, Meek EC, Pringle RB, Ross MK, 2016. Efficacy of novel phenoxyalkyl pyridinium oximes as brain-penetrating reactivators of cholinesterase inhibited by surrogates of sarin and VX. Chem. Biol. Interact 259, 154–159. [DOI] [PubMed] [Google Scholar]
- Cochran R, Kalisiak J, Küçükkilinç T, Radić Z, Garcia E, Zhang L, Ho K-Y, Amitai G, Kovarik Z, Fokin VV, Sharpless KB, Taylor P, 2011. Oxime-assisted acetylcholinesterase catalytic scavengers of organophosphates that resist aging. J. Biol. Chem 286 (34), 29718–29724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson RM, 1994. Review of oximes available for the treatment of nerve agent poisoning. J. Appl. Toxicol 14, 317–331. [DOI] [PubMed] [Google Scholar]
- De Koning MC, Joosen MJ, Noort D, Van Zuylen A, Tromp MC, 2011. Peripheral site ligand-oxime conjugates: a novel concept towards reactivation of nerve agent inhibited human acetylcholinesterase. Bioorg. Med. Chem 19, 588–594. [DOI] [PubMed] [Google Scholar]
- Demar JC, Clarkson ED, Ratcliffe RH, 2010. Pro-2-PAM therapy for central and peripheral cholinesterases. Chem.-Biol. Interact 187, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin E, 2013. Syrian gas attack reinforces need for better anti-sarin drugs. Nat. Med 19, 1194–1195. [DOI] [PubMed] [Google Scholar]
- Ekström F, Akfur C, Tunemalm AK, Lundberg S, 2006a. Structural changes of phenylalanine 338 and histidine 447 revealed by the crystal structures of tabun-inhibited murine acetylcholinesterase. Biochemistry 45, 74–81. [DOI] [PubMed] [Google Scholar]
- Ekström F, Pang YP, Boman M, Artursson E, Akfur C, Börjegren S, 2006b. Crystal structures of acetylcholinesterase in complex with HI-6, Ortho-7 and obidoxime: structural basis for differences in the ability to reactivate tabun conjugates. Biochem. Pharmacol 72, 597–607. [DOI] [PubMed] [Google Scholar]
- Ekström F, Hörnberg A, Artursson E, Hammarström LG, Schneider G, Pang YP, 2009. Structure of HI-6-sarin-acetylcholinesterase determined by X-ray crystallography and molecular dynamics simulation: reactivator mechanism and design. PLoS One 4, e5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, 1976. Organic and biological chemistry, in: Zweig G (Ed), The Organophosphorus Pesticides CRC Press Inc., Cleveland, pp 142. [Google Scholar]
- Gray AP, 1984. Design and structure-activity relationships of antidote to organophosphorus anticholinesterase agents. Drug. Metab. Rev 15, 557–589. [DOI] [PubMed] [Google Scholar]
- Kalisiak J, Ralph EC, Zhang J, Cashman JR, 2011. Amidine-oximes: reactivators for organophosphate exposure. J. Med. Chem 54 (9), 3319–3330. [DOI] [PubMed] [Google Scholar]
- Katalinić M, Zandona A, Ramić A, Zorbaz T, Primožič I, Kovarik Z, 2017. New cinchona oximes evaluated as reactivators of acetylcholinesterase and butyrylcholinesterase inhibited by organophosphorus compounds. Molecules 22 (7), pii: E1234. doi: 10.3390/molecules22071234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katalinić M, Šinko G, Maček Hrvat N, Zorbaz T, Bosak A, Kovarik Z, 2018. Oxime-assisted reactivation of tabun-inhibited acetylcholinesterase analysed by active site mutations. Toxicology (406-407), 104–113. [DOI] [PubMed] [Google Scholar]
- Kliachyna M, Santoni G, Nussbaum V, Renou J, Sanson B, Colletier J-P, Arboléas M, Loiodice M, Weik M, Jean L, Renard P-Y, Nachon F, Baati R, 2014. Design, synthesis and biological evaluation of novel tetrahydroacridine pyridine- aldoxime and - amidoxime hybrids as efficient uncharged reactivators of nerve agent-inhibited human acetylcholinesterase. Eur. J. Med. Chem 78, 455–467. [DOI] [PubMed] [Google Scholar]
- Kovarik Z, 1999. Amino acid residues conferring specificity of cholinesterases. Period. Biol 101 (1), 7–15. [Google Scholar]
- Kovarik Z, Radić Z, Berman HA, Simeon-Rudolf V, Reiner E, Taylor P, 2003. Acetylcholinesterase active centre and gorge conformations analysed by combinatorial mutations and enantiomeric phosphonates. Biochem. J 373(1), 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik Z, Radic Z, Berman HA, Simeon-Rudolf V, Reiner E, Taylor P, 2004. Mutant cholinesterases possessing enhanced capacity for reactivation of their phosphonylated conjugates. Biochemistry 43, 3222–3229. [DOI] [PubMed] [Google Scholar]
- Kovarik Z, Ciban N, Radic Z, Simeon-Rudolf V, Taylor P, 2006. Active site mutant acetylcholinesterase interactions with 2-PAM, HI-6, and DDVP. Biochem. Biophys. Res. Commun 342, 973–978. [DOI] [PubMed] [Google Scholar]
- Kovarik Z, Maček N, Sit RK, Radić Z, Fokin VV, Sharpless KB, Taylor P, 2013. Centrally acting oximes in reactivation of tabun-phosphoramidated AChE. Chem.-Biol. Interact 203, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik Z, Kalisiak J, Maček Hrvat N, Katalinić M, Zorbaz T, Žunec S, Green C, Radić Z, Fokin VV, Sharpless KB, Taylor P, 2019a. Reversal of tabun toxicity enabled by a triazole-annulated oxime library - reactivators of acetylcholinesterase. Chem. Eur. J 25(16), 4100–4114. [DOI] [PubMed] [Google Scholar]
- Kovarik Z, Maček Hrvat N, Kalisiak J, Katalinić M, Sit RK, Zorbaz T, Radić Z, Fokin VV, Sharpless KB, Taylor P, 2019b. Counteracting tabun inhibition by reactivation by pyridinium aldoximes interacting with active center gorge mutations of acetylcholinesterase. Toxicol. Appl. Pharmacol 372, 40–46. [DOI] [PubMed] [Google Scholar]
- Krasiński A, Radić Z, Manetsch R, Raushel J, Taylor P, Sharpless KB, Kolb HC, 2005. In situ selection of lead compounds by click chemistry: target-guided optimization of acetylcholinesterase inhibitors. J. Am. Chem. Soc 127 (18), 6686–6692. [DOI] [PubMed] [Google Scholar]
- Kryger G, Silman I, Sussman JL, 1999. Structure of acetylcholinesterase complexed with E2020 (Aricept (R)): implications for the design of new anti-Alzheimer drugs. Structure 7(3), 297–307. [DOI] [PubMed] [Google Scholar]
- Maček- Hrvat N, Zorbaz T, Šinko G, Kovarik Z 2018. The estimation of oxime efficiency is affected by the experimental design of phosphylated acetylcholinesterase reactivation. Toxicol. Lett 293, 222–228. [DOI] [PubMed] [Google Scholar]
- Manetsch R, Krasiński A, Radić Z, Raushel J, Taylor P, Sharpless KB, & Kolb HC, 2004. In situ click chemistry: enzyme inhibitors made to their own specifications. J. Am. Chem. Soc 126(40), 12809–12818. [DOI] [PubMed] [Google Scholar]
- Maraković N, Knežević A, Vinković V, Kovarik Z, Šinko G, 2016. Design and synthesis of N-substituted-2-hydroxyiminoacetamides and interactions with cholinesterases. Chem.-Biol. Interact 259, 122–132. [DOI] [PubMed] [Google Scholar]
- Masson P, Nachon F, 2017. Cholinesterase reactivators and bioscavengers for pre- and post-exposure treatments of organophosphorus poisoning. J. Neurochem 142, 26–40. [DOI] [PubMed] [Google Scholar]
- McHardy SF, Bohmann JA, Corbett MR, Campos B, Tidwell MW, Thompson PM, Bemben CJ, Menchaca TA, Reeves TE, Cantrell WR Jr., Bauta WE, Lopez A, Maxwell DM, Brecht KM, Sweeney RE, McDonough J,2014. Design, synthesis, and characterization of novel, nonquaternary reactivators of GF-inhibited human acetylcholinesterase. Bioorg. Med. Chem. Lett 24, 1711–1714. [DOI] [PubMed] [Google Scholar]
- Mercey G, Verdelet T, Saint-André G, Gillon E, Wagner A, Baati R, Jean L, Nachon F, Renard P-Y, 2011. First efficient uncharged reactivators for the dephosphylation of poisoned human acetylcholinesterase. Chem. Comm 47 (18), 5295–5297. [DOI] [PubMed] [Google Scholar]
- Mercey G, Renou J, Verdelet T, Kliachyna M, Baati R, Gillon E, Arboléas M, Loiodice M, Nachon F, Jean L, Renard P-Y, 2012. Phenyltetrahydroisoquinoline-pyridinaldoxime conjugates as efficient uncharged reactivators for the dephosphylation of inhibited human acetylcholinesterase. J. Med. Chem 55 (23), 10791–10795. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O, 2002. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 110, 627–639. [DOI] [PubMed] [Google Scholar]
- Millard CB, Koellner G, Ordentlich A, Shafferman A, Silman I, Sussman JL, 1999. Reaction products of acetylcholinesterase and VX reveal a mobile histidine in the catalytic triad. J. Am. Chem. Soc 121, 9883–9884. [Google Scholar]
- Nakagawa T, Tu AT, 2018. Murders with VX: Aum Shinrikyo in Japan and the assassination of Kim Jong Nam in Malaysia. Forensic Toxicol 36, 542–544. [Google Scholar]
- Radić Z, Pickering NA, Vellom DC, Camp S, Taylor P, 1993. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 32, 12074–12084. [DOI] [PubMed] [Google Scholar]
- Radić Z, Sit RK, Kovarik Z, Berend S, Garcia E, Zhang L, Amitai G, Green C, Radić B, Fokin VV, Sharpless KB, and Taylor P (2012) Refinement of structural leads for centrally acting oxime reactivators of phosphylated cholinesterases. J Biol Chem 287: 11798–11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh L, Grunwald J, Marcus D, Papier Y, Cohen E, Ashani Y, 1993. Human butyrylcholinesterase as a general prophylactic antidote for nerve agent toxicity. In vitro and in vivo quantitative characterization. Biochem. Pharmacol 45 (12), 2465–2474. [DOI] [PubMed] [Google Scholar]
- Renou J, Loiodice M, Arboléas M, Baati R, Jean L, Nachon F, Renard P-Y, 2014. Tryptoline-3-hydroxypyridinaldoxime conjugates as efficient reactivators of phosphylated human acetyl and butyrylcholinesterases. Chem. Comm 50 (30), 3947–3950. [DOI] [PubMed] [Google Scholar]
- Renou J, Dias J, Mercey G, Verdelet T, Rousseau C, Gastellier A-J, Arboléas M, Touvrey-Loiodice M, Baati R, Jean L, Nachon F, Renard P-Y, 2016. Synthesis and in vitro evaluation of donepezil-based reactivators and analogues for nerve agent-inhibited human acetylcholinesterase. RSC Adv 6 (22), 17929–17940. [Google Scholar]
- Rosenberg YJ, Mao L, Jiang X, Lees J, Zhang L, Radić Z, and Taylor P (2017a) Post-exposure treatment with the oxime RS194B rapidly reverses early and advanced symptoms in macaques exposed to sarin vapor. Chem Biol Interact 274: 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg YJ, Wang J, Ooms T, Rajendran N, Mao L, Jiang X, Lees J, Urban L, Momper JD, Sepulveda Y, Shyong YJ, and Taylor P (2017b) Post-exposure treatment with the oxime RS194B rapidly reactivates and reverses advanced symptoms of lethal inhaled paraoxon in macaques. Toxicol Lett (in press) doi: 10.1016/j.toxlet.2017.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Redman AMG, Jiang X, Lockridge O, Doctor BP, 1999. Differences in active-site gorge dimensions of cholinesterase revealed by binding of inhibitors to human butyrylcholinesterase. Chem.-Biol. Interact 119–120, 61–69. [DOI] [PubMed] [Google Scholar]
- Simeon-Rudolf V, Kovarik Z, Radić Z, Reiner E, 1999. Reversible inhibition of acetylcholinesterase and butyrylcholinesterase by 4,4’-bipyridine and by a coumarin derivative. Chem.-Biol. Interact 119-120, 119–28. [DOI] [PubMed] [Google Scholar]
- Sit RK, Radić Z, Gerardi V, Zhang L, Garcia E, Katalinić M, Amitai G, Kovarik Z, Fokin VV, Sharpless KB, Taylor P, 2011. New structural scaffolds for centrally acting oxime reactivators of phosphylated cholinesterases. J. Biol. Chem 286, 19422–19430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit RK, Kovarik Z, Maček Hrvat N, Žunec S, Green C, Fokin VV, Sharpless KB, Radić Z, Taylor P, 2018. Pharmacology, pharmacokinetics, and tissue disposition of zwitterionic hydroxyiminoacetamido alkylamines as reactivating antidotes for organophosphate exposure. J. Pharmacol. Exp. Ther 367 (2), 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šinko G, 2019. Assessment of scoring functions and in silico parameters for AChE-ligand interactions as a tool for predicting inhibition potency. Chem.-Biol. Interact 308, 216–223. [DOI] [PubMed] [Google Scholar]
- Taylor P, Radić Z, 1994. The cholinesterases: from genes to proteins. Annu. Rev. Pharmacol. Toxicol 34, 281–320. [DOI] [PubMed] [Google Scholar]
- Taylor P, Radić Z, Hosea NA, Camp S, Marchot P, Berman HA, 1995. Structural bases for the specificity of cholinesterase catalysis and inhibition. Toxicol. Lett 82–83, 453–458. [DOI] [PubMed] [Google Scholar]
- Taylor P, Kovarik Z, Reiner E, Radić Z, 2006. Acetylcholinesterase: converting a vulnerable target to a template for antidotes and detection of inhibitor exposure. Toxicology 233(1-3), 70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale JA, Marrs TCOBE, Maynard RLCBE, 2018. Novichok: a murderous nerve agent attack in the UK. Clin. Toxicol 56(11), 1093–1097. [DOI] [PubMed] [Google Scholar]
- Wei Z, Liu Y-Q, Zhou X-B, Luo Y, Huang C-Q, Wang Y-A, Zheng Z-B, Li S, 2014. New efficient imidazolium aldoxime reactivators for nerve agent-inhibited acetylcholinesterase. Bioorganic Med. Chem. Lett 24 (24), 5743–5748. [DOI] [PubMed] [Google Scholar]
- Wei Z, Liu Y-Q, Wang S-Z, Yao L, Nie H-F, Wang Y-A, Liu X-Y, Zheng Z-B, Li S, 2017. Conjugates of salicylaldoximes and peripheral site ligands: Novel efficient nonquaternary reactivators for nerve agent-inhibited acetylcholinesterase. Bioorganic Med. Chem 25 (16), 4497–4505. [DOI] [PubMed] [Google Scholar]
- Wille T, Ekström F, Lee J-C, Pang Y-P, Thiermann H, Worek F, 2010. Kinetic analysis of interactions between alkylene-linked bis-pyridiniumaldoximes and human acetylcholinesterases inhibited by various organophosphorus compounds. Biochem. Pharmacol 80 (6), 941–946. [DOI] [PubMed] [Google Scholar]
- Wong L, Radić Z, Brüggemann RJM, Hosea N, Berman HA, Taylor P, 2000. Mechanism of oxime reactivation of acetylcholinesterase analyzed by chirality and mutagenesis. Biochemistry 39(19), 5750–5757. [DOI] [PubMed] [Google Scholar]
- Worek F, Reiter G, Eyer P, Szinicz L, 2002. Reactivation kinetics of acetylcholinesterase from different species inhibited by highly toxic organophosphates. Arch. Toxicol 76, 523–529. [DOI] [PubMed] [Google Scholar]
- Worek F, Thiermann H, Wille T, 2016. Oximes in organophosphate poisoning: 60 years of hope and despair. Chem.-Biol. Interact 259, 93–98. doi: 10.1016/j.cbi.2016.04.032 [DOI] [PubMed] [Google Scholar]
- Zorbaz T, Braïki A, Maraković N, Renou J, de la Mora E, Maček Hrvat N, Katalinić M, Silman I, Sussman JL, Mercey G, Gomez C, Mougeot R, Pérez B, Baati R, Nachon F, Weik M, Jean L, Kovarik Z, Renard P-Y, 2018a. Potent 3-Hydroxy-2-pyridine aldoxime reactivators of organophosphate-inhibited cholinesterases with predicted blood-brain barrier penetration. Chem. Eur. J 24 (38), 9675–9691. [DOI] [PubMed] [Google Scholar]
- Zorbaz T, Malinak D, Maraković N, Maček Hrvat N, Zandona A, Novotny M, Skarka A, Andrys R, Benkova M, Soukup O, Katalinić M, Kuca K, Kovarik Z, Musilek K, 2018b. Pyridinium oximes with ortho-positioned chlorine moiety exhibit improved physicochemical properties and efficient reactivation of human acetylcholinesterase inhibited by several nerve agents. J. Med. Chem 61 (23), 10753–10766. [DOI] [PubMed] [Google Scholar]
- Zorbaz T, Malinak D, Kuca K, Musilek K, Kovarik Z, 2019. Butyrylcholinesterase inhibited by nerve agents is efficiently reactivated with chlorinated pyridinium oximes. Chem.-Biol. Interact 307, 16–20. [DOI] [PubMed] [Google Scholar]