

ABSTRACT
Nascent transcription assays, such as global run-on sequencing (GRO-seq) and precision run-on sequencing (PRO-seq), have uncovered a myriad of unstable RNAs being actively produced from numerous sites genome-wide. These transcripts provide a more complete and immediate picture of the impact of regulatory events. Transcription factors recruit RNA polymerase II, effectively initiating the process of transcription; repressors inhibit polymerase recruitment. Efficiency of recruitment is dictated by sequence elements in and around the RNA polymerase loading zone. A combination of sequence elements and RNA binding proteins subsequently influence the ultimate stability of the resulting transcript. Some of these transcripts are capable of providing feedback on the process, influencing subsequent transcription. By monitoring RNA polymerase activity, nascent assays provide insights into every step of the regulated process of transcription.
KEWORDS: RNA polymerase II, enhancers, nascent RNA, gene expression, non-coding RNA function, eRNAs, transcription factors, chromatin structure
1. Introduction
Transcription varies from cell type to cell type, unlocking the information held within DNA. Understanding transcriptional regulation and the factors that control it is a grand challenge. Despite the many difficulties of deconstructing the biological rules of transcriptional regulation, the importance of this topic cannot be overstated, as most disease associated variation is noncoding and likely regulatory [1,2]. In fact, a startling 60–76.5% of disease associated single nucleotide polymorphisms (SNPs) are in enhancers, the major regulatory domains of DNA [1–4]. Enhancers are short regulatory regions densely bound by transcription factors [5–7].
Transcription factors (TFs) are the managers of the cellular factory, controlling everything from cellular identity to response to external stimuli [8]. Fundamentally, TFs are proteins that bind to specific DNA sequences and regulate the transcription apparatus. Many high throughput genomics assays have focused on evaluating DNA-protein interactions of TFs, e.g. binding. The workhorse of binding studies is chromatin immunoprecipitation (ChIP), which requires antibodies to the specific TF of interest [9–11]. ChIP experiments are useful in that they can allow researchers to obtain the genomic sequences bound by a particular TF in a specific cellular condition or state. Over the last two decades, ChIP studies have been the backbone of large scale genomics projects such as ENCODE and the Roadmap Epigenomics Project [12,13]. In general, ChIP studies revealed that most TFs display widespread binding across the genome; though many binding sites may be nonfunctional or spurious [14,15]. Despite the noise, ChIP data, along with other related assays, has been utilized to characterize the DNA recognition motif preferentially bound by a particular TF [9,11].
ChIP data informs on protein-DNA interactions but does not provide information on the regulatory function of the TF binding events. To understand the impact of binding on transcription, typically RNA-seq or another steady state RNA detection method is employed. This coupling of expression analysis with ChIP-seq, is preferably carried out in the presence of a TF perturbation, to identify bona fide functional TF binding sites. In some cases, a TF can be rapidly activated by a small molecule, compound, or specific condition. Alternatively a TF can be removed via knockdown or knockout. Unfortunately, it is inherently difficult to distinguish primary from secondary effects of the perturbation on the system if the perturbation itself is not abrupt. The detection of significant RNA changes immediately after perturbation requires either a large deviation in transcript steady state levels (which typically takes time to obtain) or excessive numbers of replicates (which is cost prohibitive) [16]. Thus the steady state approach is incapable of reliably detecting small changes at short time points.
In general, the major complication in using expression data is that expression studies are a poor readout on the transcriptional apparatus. RNA-seq, the typical approach to expression studies, is biased toward the detection of stable transcripts. Hence rapidly degraded RNAs are virtually undetectable. Most critically, steady state expression assays reflect not only transcription but also RNA processing, maturation and stability. So even when a change is detected in RNA-seq, it is unclear whether the change reflects alterations in transcription or transcript stability. Arguably a preferred approach would focus on deciphering the activity and behavior of the key transcriptional machinery, namely cellular RNA polymerases.
In eukaryotic cells, three RNA polymerases are responsible for all nuclear transcription: RNA polymerase I, II, and III. RNA polymerase I and III transcribe primarily ribosomes and structural RNAs, respectively. Critically important for cell growth and replication, these RNA polymerases have relatively well defined promoters [17]. In contrast, RNA polymerase II is the most versatile of the three polymerases, responsible for transcribing all protein coding mRNAs and many ncRNAs. Perhaps a consequence of this versatility, RNA polymerase II interacts with a wide range of transcription factors to regulate the precise locations and levels of its activity. Consequently, most regulators of transcription are known to alter RNA polymerase II activity. Therefore we focus on RNA polymerase II regulation in this review.
Nascent transcription assays provide a more concise readout on the immediate activity of cellular RNA polymerases, e.g. transcription. Generally nascent RNA refers to all transcripts pre-maturation (protocols reviewed in [18]). While many protocols provide insights into transcripts prior to complete maturation, here we specifically refer to nascent protocols that target newborn RNAs; those just coming into existence or those in the process of being made. Some nascent methods (PRO-seq and GRO-seq) label the RNAs with a marked nucleotide via nuclear run-on and then isolate the labeled RNAs via selective precipitation [19,20]. Other nascent methods select for newborn RNAs via precipitation of molecules that associate with nascent RNA such as polymerase or chromatin (mNET-seq, Chro-seq) [21–24]. In all cases, these methods are complementary to RNA polymerase ChIP but provide higher resolution and strand specific information. In contrast, other protocols are focused on recently synthesized RNA rather than newborn RNA. For example, SLAM-seq and Bru-Seq label live cells with marked nucleotides over extended time frames (hours) [25,26]. RNAs transcribed within those hours are subjected to processing events such as splicing and 3’ end cleavage. Additionally, RNAs with half lives shorter than hours are labeled and subsequently degraded. Here we focus on nascent protocols aimed specifically at newborn RNAs, as these approaches provide the most temporally immediate readout on the impact of regulators on RNA polymerase II.
We now know that nearly all RNA polymerase II loading and initiation sites produce one or more noncoding RNA transcripts, most of which are unstable [27]. These noncoding transcripts are called a variety of names, including long noncoding RNAs (lncRNA) [28], enhancer RNAs (eRNAs) [29], promoter upstream transcripts (PROMPTs) [30], upstream antisense RNAs (uaRNAs) [31], transcription start site–associated RNAs (TSSa-RNAs) [32] and short-lived non-coding transcripts (SLiTs) [33]. They are classified according to length, stability, and origin relative to protein-coding genes, but the boundaries between the classes are often far from distinct [34–37]. It has been argued that the unstable fraction may simply be a side effect of the transcription process, e.g. noise [38–40]. The term “noise” implies a certain degree of irrelevance; yet, the act of transcription itself can be critical to the regulation of the local genomic context (extensively reviewed in [41]). Furthermore, the mere presence of these transcripts, even when they are apparently nonfunctional and highly unstable, serve as markers of regulatory activity, and are hence informative about the regulation of RNA polymerase II.
2. Global characteristics of polymerase loading and transcription initiation
Decades of mechanistic studies (reviewed in [42]) have resulted in a well defined cycle of RNA polymerase II activity. Briefly, the first step in the process of transcription is recruitment of RNA polymerase II to the DNA. The pre-initiation complex positions RNA polymerase II at transcription start sites (TSS), adjacent to regulatory regions. This process of loading and initiation of RNA polymerase II was originally thought to occur proximal to promoters and be influenced by distal enhancers. Transcription factors modulate the efficiency of RNA polymerase II recruitment. After loading and formation of the pre-initiation complex (PIC), RNA polymerase II escapes the promoter, initiates RNA synthesis and subsequently pauses (reviewed in [32]). Pause release transitions RNA polymerase II into the elongation phase which gives rise to long pre-processed RNAs. Downstream of encountering a cleavage site, RNA polymerase II terminates transcription. A number of transcript processing steps are co-transcriptional and influence the final product: a mature transcript [43].
2.1. Transcription initiation of various RNAs involves the same RNA polymerase II machinery
Nascent transcription methods have widened the view of transcriptional regulation: hugely increasing the number of genomic loci where transcription initiation is known to occur. Moreover, the discovery of extensive genome-wide transcription led to the realization that transcription initiation occurs proximal to regulatory regions more generally. In K562 cells, an astonishing 72% of initiation sites are not promoter associated [44]. Initial inquiries into these nontraditional transcribed regions led to the exciting classification of new RNA subclasses such as PROMPTs and eRNAs [19,34,45,46]. While the function of these transcripts remains hotly debated, it is undeniable that they enrich our understanding of RNA polymerase activity and transcriptional regulation. The emerging picture is of a consistent mechanism underlying RNA polymerase loading and initiation, regardless of the location of initiation or the stability of the resulting transcripts [27].
Basic steps in early transcription such as PIC recruitment and formation, transcription initiation, capping, and promoter proximal pausing appear to be universal at all transcription start sites, regardless of the stability of the resulting transcript. ChIP studies on various components of the PIC as well as general TFs suggests a common RNA polymerase II machinery underlies bidirectional TSSs, including enhancer transcripts [27,47,48]. When the distance between the bidirectional initiation sites is sufficient, distinct Polymerase II, TBP and TFIIB peaks are observed, consistent with PIC formation occurring at two distinct TSS locations for each bidirectional pair [27,47]. Subsequent to PIC formation and transcription initiation, there is evidence that eRNAs/PROMPTs are capped similarly to mRNAs [34,49]. More surprisingly, given their relatively short final RNA length, recent studies suggest that in both Drosophila and mammalian cells, transcription of PROMPTs and eRNAs is regulated by pausing factors similarly to mRNA transcription [31,50]. Both mRNAs and eRNAs show the well documented proximal pausing of RNA Polymerase II at the region 20 to 70 base pairs downstream of each TSS [50]. These studies provide further evidence that early stages of transcription of eRNAs and PROMPTs is similar to that of mRNAs and stable noncoding RNAs.
Additionally, Mediator may have a consistent role at both stable and unstable transcripts. Mediator is a large multi-subunit protein complex well established to regulate many vital steps in the process of transcription (reviewed in [51]). The Mediator complex is believed to facilitate PIC assembly, regulate promoter escape of Pol II, and play a role in transcription activation at stimulus responsive genes [51]. Importantly, Mediator may provide a functional bridge to connect DNA bound TFs to the PIC and Pol II (reviewed in [51]). Immunoprecipitation assays have demonstrated that Mediator complex associates with upstream activated sequences (UAS’s) regardless of the transcription level of the associated gene [52]. Mediator has also been localized to and observed to function at long noncoding RNAs and super-enhancers [53–55]. Finally, loss of functional mediator has been demonstrated to decrease the association of RNA polymerase II at essentially all transcribed genes (reviewed in [56]).
2.2. Transcription initiation is predominantly bidirectional
The recent use of nascent transcription assays and various 5’ cap enrichment assays resulted in the discovery that mammalian transcription initiation is predominantly bidirectional, with two oppositely oriented distinct transcription start sites in close proximity [19,27,47,57–61] (Figure 1). Early recognition of bidirectional transcription at protein coding genes led to the unstable upstream transcript being dubbed either a uaRNA or PROMPT [31,47,48,62]. A large fraction of annotated protein coding genes, roughly 75%, have bidirectional transcription at their promoter [19,22,47]. Similarly, genomic regions that show evidence of transcriptional activity but do not produce stable transcripts in either direction, such as enhancer regions, were found to generally contain two opposing TSSs that produce two unstable transcripts termed enhancer RNAs or eRNAs [27,48,50,61,63]. Importantly, the distinction between enhancers and promoters has blurred with time [64,65], as they share common underlying sequence patterns, RNA polymerase activity, and chromatin accessibility (Figure 1) [27,57]. Genome wide, the distance between observed bidirectional TSS pairs is quite variable, averaging close to 175 base pairs but going up to a few hundred base pairs [47]. Henceforth we refer to the region between the two bidirectional TSSs as the RNA polymerase loading zone.
Figure 1.
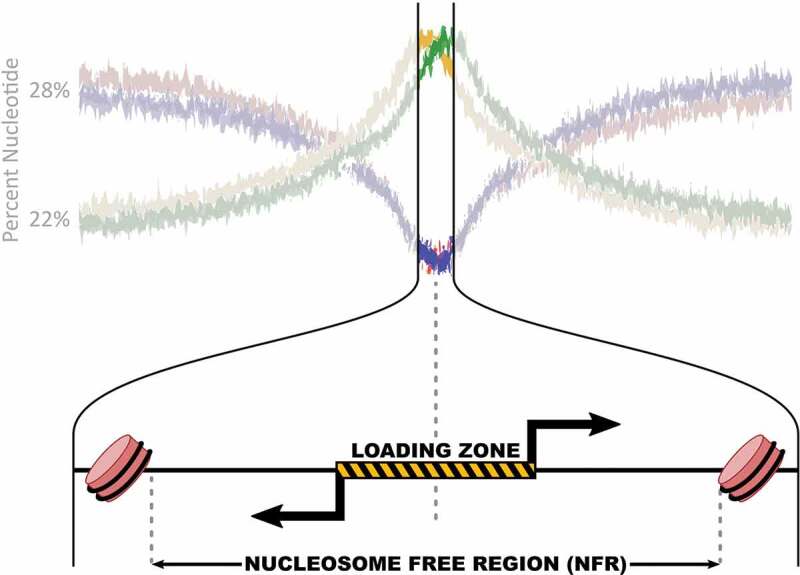
A typical RNA polymerase II loading and initiation region.
Sequence bias at RNA polymerase loading and initiation sites peaks at the epicenter of bidirectional transcription. Top: The percentage of A (red), T(blue), G(green) and C(yellow) nucleotides for all sites of RNA polymerase II loading and initiation genome wide (adapted from [44]). Bottom: Zoom in on epicenter of RNA polymerase loading and initiation, where typically two bidirectional transcription start sites (black arrows) originate from within a nucleosome free region. Rose barrels are nucleosomes.
Recently, single cell sequencing and single molecule imaging techniques have been utilized to refine our understanding of transcription initiation. These methods assay individual cells, in contrast to the population and time averaged data of typical nascent protocols. Within a cell, transcription occurs in regulated bursts of RNA production from individual loci [66–71]. The burst size, or the number of RNA transcripts produced by a single transcriptional burst, is likely influenced by core promoter elements and gene length of a given loci; whereas, the identify of regulating enhancer regions may primarily regulate burst frequency [70–72]. Interestingly, the initial polymerase loading event may bias the direction of subsequent transcriptional events co-occurring within the same transcriptional burst [73]. Exclusive transcription from one TSS within a bidirectional pair may be enforced by the biophysical properties, such as torsional strain and steric hindrance, at the loci induced by the initial transcription event [74]. Thus the bidirectional signal observed from nascent transcription assays reflects averaging across the cellular population rather than simultaneous activity at a single cell level.
2.3. Polymerase loading occurs at regions with a general nucleotide bias
Figure 1 shows the genome wide nucleotide composition surrounding all bidirectional sites of RNA polymerase loading and initiation [44]. Notably, some GC bias is present at nearly all sites of RNA polymerase II loading, regardless of the transcription level or stability of the resulting transcript. The GC bias of mRNA promoters is well studied and provides some insights into how nucleotide composition influences transcription [75,76]. Low GC content regions tend to favor closed chromatin [77,78]. In contrast, genes that are expressed globally across all tissues, such as housekeeping genes, have higher GC content at their promoters compared to tissue specific genes [78,79]. Tissue specific genes and enhancers have a somewhat lower GC bias [61,80,81]. One reason for the high GC content at TSSs is that transcription initiation coincides with CpG islands, stretches of sequence around 1000 bp that have a higher percentage of C and G bases and a low amount of methylation compared to the global genome [82].
2.4. Transcription initiation occurs in nucleosome free regions
DNA accessibility assays [83–85] (reviewed in [86]) have consistently demonstrated that regions of polymerase loading and initiation are contained within nucleosome free regions (NFRs) [47,57,61,80,83–87]. Well placed nucleosomes are found upstream and downstream of the loading zone at both enhancers and promoters, suggesting that the machinery involved in transcription may play a part in maintaining open chromatin at these regions [27,88,89]. Most TFs that recruit RNA polymerase have been shown to bind predominantly (94% in K562 [90]) to open chromatin regions [12]. It remains an important question as to the extent that transcription and transcription factors contribute to the establishment and maintenance of the nucleosome free region.
2.5. Transcription levels correlate with histone marks
Factors that influence transcription levels are likely to have a concomitant influence on the local chromatin marks. Transcription levels correlate with a variety of histone marks (Figure 2a). H3K4me1 tends to mark eRNAs, while H3K4me3 tends to mark promoters, providing a functional if imperfect bioinformatic tool for separating the two sets of transcribed genetic regions [80]. However, nascent transcription assays indicate that active histone modifications directly correlate with the transcriptional activity of each bidirectional region, regardless of whether the resulting transcripts are stable or not [27]. For example, H3K4 methylation status correlates with the overall RNA polymerase initiation levels, with increasing transcription leading to higher methylation status. However, studies on a histone demethylase complex (RACK7/KDM5C) suggest the relationship may not be that simple. Loss of RACK7-Histone Demethylase Complex resulted in increased transcription at enhancers and conversion from H3K4me1 to H3K4me3, suggesting RACK7 represses transcription [91]. Interestingly, RACK7 associated genes were unaffected. Therefore, either the RACK7 complex uniquely functions at enhancers or some mechanism drives RACK7 associated promoters to H3K4me3 status despite the presence of the demethylase.
Figure 2.
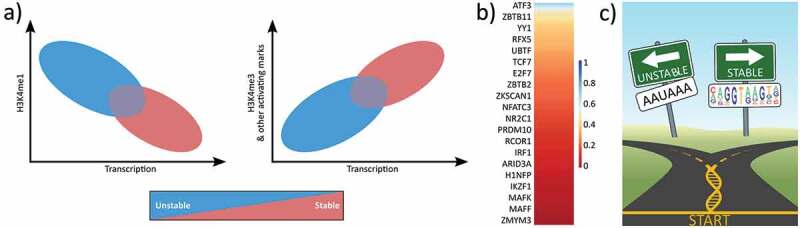
The differences between stable mRNAs and other unstable nascent transcripts.
(a) Cartoon showing the relationship between H3K4 methylation status (me1 and me3) and transcription (adapted from [27]). Stable transcripts include mRNAs and lncRNAs. (b) Most TFs preferentially bind non-promoter regions. Using ENCODE K562 ChIP and RefSeq annotations, the heatmap shows the fraction of ChIP sites that overlap promoters. 194 total TFs total but only every 10th row labeled. (c) Cartoon depicting early RNA processing decision. Encountering a splice site motif is correlated with message stability whereas encountering a cleavage motif is correlated with instability.
3. Genetically encoded signals regulate transcriptional outcomes
If RNA polymerase II initiation and loading is mechanistically consistent across the genome, then what regulatory processes influence the frequency of transcription and ultimately the stability of each transcript? A number of sequence features within the RNA polymerase loading zone, including recognition sites for a broad range of transcription factors, influence the efficiency of transcription initiation. The rich profile of transcription provided by nascent assays inherently informs on a broad class of regulatory proteins.
3.1. DNA sequences in transcription loading zone influence initiation frequency
Transcription factors are key orchestrators of rapid cellular responses to environmental cues, metabolic demands, and distinct developmental stages. TFs function by binding to DNA and altering the activity of cellular polymerases. Genome profiling of protein-DNA localization, e.g. binding, has led to tremendous insights into how TF binding specificity is achieved (reviewed in [9]). Recruitment of RNA polymerase, and thereby the quantity of transcription initiating from a given TSS, is driven by a wide variety of transcription factors, each with distinct binding profiles across the genome [12]. Transcription factors have distinct genome-wide binding profiles that can vary between cell types or conditions [90,92,93]. Some TFs binding predominantly at promoters whereas others are more enhancer specific (Figure 2(b)). Though the presence of a TF motif near a gene can be informative, not all TF binding events are functional [5,8,14,15,94–101]. Therefore, no aspect of TF binding provides information on the subsequent regulatory activity of the TF.
The width of the loading zone also influences the overall quantity of transcriptional initiation for each pair of initiation sites [47,102]. Sites of bidirectional transcription have larger loading zones that are more responsive to induction than unidirectional regions [47]. Larger loading zones may be more responsive simply because there is more genetic real estate for inducible TF binding [47]. Hence the length and identity of sequence elements embedded in the transcription loading zone quantitatively affect transcription initiation at that region [27,57,103].
3.2. DNA sequences in transcription loading zone influence strand bias of transcription
Decades of transcription research have defined the basic genetic requirements for efficient PIC recruitment at protein coding gene promoters, revealing the importance of core promoter sequences (reviewed in [104]). However, most of these studies have assumed that the majority of TSS regions are unidirectional and result in stable RNAs. The discovery of widespread bidirectional transcription led to questions about the extent that promoter elements influence the balance of transcription from each strand, as well as the quantity of total transcription from a TSS pair. Stable-unstable TSS pairs tend to reveal a large bias of increased transcription initiation from the stable transcript compared to the unstable RNA; whereas, unstable-unstable transcript pairs often reveal a more balanced ratio of initiation from both strands [27]. Genetic analysis of the effects of SNPs on bidirectional TSSs suggests that the sequences within and adjacent to the loading zone tune the ratio of divergent transcription events [102,105], e.g. the strand bias [78].
3.3. Genetic signals dictate RNA processing events that affect differential stability of transcripts
After polymerase initiation, the subsequent transcription and post-transcriptional processing of transcripts is quite diverse. The first major difference between stable processed transcripts and unstable enhancer associated transcripts is the frequency of pause release. In fact, one of the motivating goals of the development of nascent protocols was the study of pause release, a key regulatory step in transcription (reviewed in [106]). Though pausing appears to be regulated by pausing factors at both enhancer and promoter regions, average pause release appears to be faster at enhancer regions than at promoter regions [31,45,50]. Studies of pausing related factors NELF/PTefb imply that at some enhancers the eRNA may function in release of pausing factors from partner gene promoters [79].
Ultimately RNA processing signals lead to differential stability [107–109]. Upon pause release at mRNAs, RNA polymerase transitions to elongation and transcripts that encounter a 5’ splice site undergo splicing. Splicing conveys many benefits to the transcript including stability and nuclear export (reviewed in [110]). Upon splicing many cofactors associate with both polymerase and the RNA. For instance, a splice site signals snRNPs, SR proteins and nuclear export factors to bind to the RNA (reviewed in [110]). Downstream, cleavage of mRNAs is initiated by the low complexity A rich polyA signal (AAUAAA) or one of its alternatives and a polyA tail is added to the mRNAs further increasing the stability of the transcripts [111] . Meanwhile, the elongating polymerase proceeds past the canonical cleavage motif (reviewed in [112]). Subsequent termination of RNA polymerase II occurs several kb downstream of the cleavage site.
In contrast, unstable transcripts, such as eRNAs and PROMPTs, rarely encounter stabilizing factors. For instance few have splice sites (Figure 2(c)). Instead, there is evidence that synthesis of these unstable transcripts is often halted relatively close to the TSS. In the case of uaRNAs, transcripts show an enrichment of RNA cleavage signals near the transcription start site [107]. Importantly, when both a splice site and cleavage signal are present, the U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation [108,113]. Without splicing, the early occurrence of a cleavage site may target the transcript for premature cleavage and polyadenylation (PCPA), a signal for targeted nuclear degradation of the transcript [114]. Studies demonstrated some enhancer RNAs are polyadenylated while other eRNAs are not, implying there may be multiple mechanisms of termination [46,48,115]. Consistent with this idea, there is some evidence that the integrator complex may be essential for cleavage and processing of enhancer RNAs [53]. In any case, the termination of the transcript without splicing is likely sufficient to make the transcript both nuclear and unstable. Therefore, the major known genomic difference that distinguishes unstable RNA classes from stable RNAs is encoded by the initial 5’ RNA processing signals (reviewed in [29]).
Mammalian cells have distinct nuclear and cytoplasmic RNA degradation pathways. Therefore differential cellular localization also influences the stability of a transcript (reviewed in [116]). eRNAs and PROMPTs tend to be short and have limited half lives, reportedly in the range of 7.5 to 30 minutes; explaining why early RNA based detection methods initially failed to identify their existence [31,45]. In fact, the lack of stability complicates measurements of half life. Their rapid degradation involves the nuclear exosome targeting (NEXT) complex [109,117], as exosome depletion increases the levels of these short unstable transcripts [30,109,118].
4. Beyond genetics: using nascent assay analysis to dissect cell type or cellular condition specific deviations in transcriptional regulation
Each cell within an organism contains the same genome and therefore the same encoded regulatory sequences. Yet most multicellular organisms are built from a large variety of different cell types, each with a unique transcriptional program. Therefore, transcriptional regulation is inherently also context dependent – reliant on the subset of transcription factors and regulators present to not only define cellular state but also cellular responses to perturbations [119]. Nascent transcription provides a unique tool for understanding the context dependent nature of transcriptional regulation.
Transcription factor proteins can themselves be regulated at transcription, translation, post-translationally, or by cellular localization [120–124]. For example, the tumor suppressor transcription factor p53 modulates gene expression to control cell-cycle progression and apoptosis [125,126] (reviewed in [127]). MDM2 is the principal cellular antagonist of p53 via ubiquitination and subsequent degradation. Consequently, p53 is constitutively transcribed and translated yet its regulatory status is inherently dependent on the tightly regulated p53-MDM2 complex [128]. As each TF is regulated by a unique process, it is difficult to know which set of transcription factors are actually present and actively participating in transcriptional regulation, e.g. active, at any given time within a cell.
Binding of a TF can be readily measured, but it has long been observed that many TF binding sites do not appear to contribute to promoter activity at the nearby target gene [5,8,14,15,94–101]. The presence of apparently nonfunctional binding calls into question whether binding is, by itself, sufficient for altering RNA polymerase activity nearby [129,130]. However, the extent of apparently nonfunctional binding is difficult to estimate, as the regulatory impact of a binding event is typically accessed by changes in transcription at the associated protein-coding gene. Critically, most TF binding is not at promoters [131]. Therefore, establishing the regulatory impact of binding sites requires distal ChIP peaks to be assigned to the target gene they are thought to regulate. While target gene assignment is defined by the spatial organization of the genome, limits on the availability of high quality 3D data results in wide adoption of the simpler nearest gene approach, an assumption that is often incorrect [132,133].
4.1. Active transcription factor binding sites recruit RNA polymerase II
Intriguingly, nascent transcription studies have demonstrated a tight relationship between the activation of a transcription factor and increased transcription associated with the TF’s binding sites [115,134–137]. Activation of p53 via Nutlin-3a led to a concomitant production of eRNAs from a subset of p53 binding sites [135,138]. p53-dependent eRNAs are required for efficient transcriptional enhancement of corresponding target genes and induction of p53-dependent cell cycle arrest [138]. Similar results have been observed for activation of other TFs, including estrogen receptor [115], androgen receptor [134,139], PPARγ [140] and NFκB [136] (Figure 3(a)). In contrast, knocking out the repressive TF Rev-Erb led to recruitment of polymerase and subsequent eRNA production from the locations that had been bound by the repressive TF [141]. While the mechanistic details may vary, the overall pattern is consistent: activating TFs recruit polymerase proximal to the site of binding whereas repressors inhibit RNA polymerase recruitment (Figure 3(b)).
Figure 3.
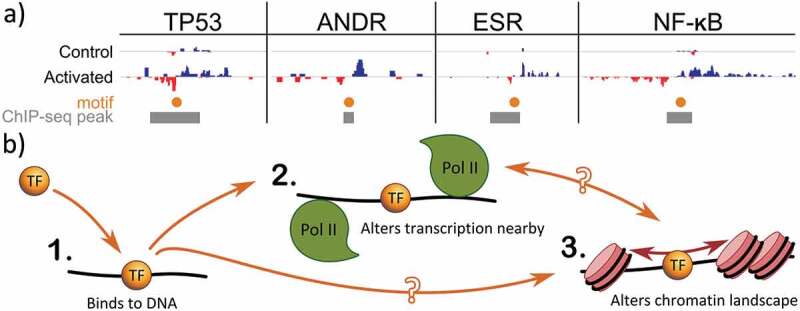
Active transcription factors alter local transcription and chromatin context.
(a) Activation of a TF leads to recruitment of RNA polymerase II and proximal bidirectional transcription. In four separate papers, activation of specific TFs results in concomitant increases in transcription levels of eRNAs (blue positive strand, red negative strand) associated with that TF’s binding events (gray boxes) and motifs (orange dots). Data from: TP53 [135], ANDR [139,159], ESR1 [115], NF-kB [136]; motifs from HOCOMOCO [160]. All data mapped to hg19 as described in [44]. (b) Distinct functions of transcription factors (in orange) include (1) binding to DNA, (2) recruitment of RNA polymerase II (green) and (3) altering the local chromatin landscape (rose). Recruitment of RNA polymerase II may contribute to alterations in chromatin or the TF may alter chromatin distinct from RNA polymerase II recruitment. In the case of a transcriptional repressor, the repressive transcription factor disrupts recruitment of RNA Polymerase II.
Thus the presence of transcripts immediately proximal to TF binding sites eliminates the need for assigning a binding site to a target gene, as transcription at the binding site itself can be utilized. This allows for a reevaluation of whether TF binding alone is sufficient for regulatory activity. Careful comparisons of ChIP data to nascent transcription assays indicate that most sites of RNA polymerase initiation overlap multiple different TF binding sites, in line with the fact that regulatory regions are dense with transcription factor binding motifs [104]. For any given TF, it is clear that RNA polymerase initiation associates with only a subset of bound sites – reinforcing that not all TF binding leads to alterations in RNA polymerase activity [142–145]. This observation led to the intriguing idea that those binding sites with transcription activity may, in fact, be the functional subset. Consistent with this idea, functional enhancers by CapStarr-seq are 5X more likely to have eRNAs associated [44,146]. Additionally, transcription of a gene is increased when the TF binding site nearby is also transcribed [44]. Therefore binding is a separate, but likely prerequisite, activity of a TF before regulatory activity.
It remains a mystery why some TF binding sites are able to recruit RNA polymerase whereas others do not. Many transcription factors work with cofactors, require a particular chromatin conformation, or prefer a certain DNA methylation status for functionality [9], hence the local context may strongly influence which sites are functional. It has been suggested that TF residency times influence functionality [99], though no comparison has yet been made between residency times and presence of a nascent transcript. Finally, ChIP is not without its own artifacts, including nonspecific antibodies and so called phantom peaks [15,100]. Additionally, it is also worth noting that transcription factors may have regulatory functions that are distinct from RNA polymerase recruitment, for example in altering local chromatin context (Figure 3(b)) [147,148]. Chromatin altering functions would not necessarily be reflected in alterations to RNA polymerase, yet may still be crucial to cellular function [149]. Moving ahead, additional studies are necessary to learn what features distinguish functional sites from the nonfunctional.
4.2. Profile of transcription initiation is predictive of transcription factor activity
Because active TFs recruit RNA polymerase nearby, the genome wide profile of bidirectional transcription (an indicator of RNA polymerase initiation) can be used to infer when a TF is participating in regulatory activity. Because transcription factor proteins can themselves be regulated at transcription, translation, post-translationally it is difficult to know which set of transcription factors are actually present and actively participating in transcriptional regulation in a cell.
Transcription factors actively participating in regulation will bind to their cognate motif and contribute to the recruitment (or suppression) of RNA polymerase at multiple loci across the genome. Furthermore, there is a dramatic co-localization of the motif with the RNA polymerase loading zone of the regulated loci [44]. This co-localization suggests that one can utilize nascent transcription to infer when a TF is actively participating in regulation. However, it isn’t as simple as looking at an individual locus. The density of motifs and ChIP binding profiles at any one locus precludes assigning responsibility for that transcript to any one factor (or even a small set). But when co-localization is considered genome wide, active TFs show dramatic co-occurrence of the motif with RNA polymerase loading zone, far more than is expected by chance [44,101]. Hence global TF activity can be inferred from a nascent transcription experiment with high confidence, even when TF responsibility at individual sites is difficult to ascertain.
When comparing across nascent transcription assay samples, it is possible to additionally identify changes in TF activity across conditions [44]. Strikingly, changes can be detected rapidly – within minutes of a perturbation [150] – time points early enough to assert that the observed transcription changes are the direct immediate result of the perturbation. Furthermore, analysis of nascent RNA at numerous time points has revealed both short term TF activity pulses and the temporal order in which TFs respond [44]. Thus analysis of nascent transcription data holds tremendous potential to unravel the regulatory network in response to both physiological perturbations and pharmaceuticals.
5. Some transcripts participate in regulation
Many noncoding RNAs, regardless of stability, are important to the transcription regulation process as an RNA. Years of work on lncRNAs indicates that many of these stable transcripts are regulatory [41,151,152]. Because of their short length and instability, there was a great deal of initial skepticism on whether enhancer RNAs have a functional role in regulation. However, the mere act of transcription has an important influence on the local chromatin architecture and TF activity profile in a region (reviewed in [41]). Evidence indicates that enhancer transcripts can also be important as RNAs, directly participating in transcriptional regulation through diverse mechanisms such as stabilizing transcription factor binding events or augmenting the activity of epigenetic modifiers (see Figure 4; also reviewed in [29]).
Figure 4.

Plethora of mechanisms where transcription at enhancers influences regulation.
Top: Instances where the resulting eRNA participates in transcriptional regulation. Enhancer RNAs display functionality through physical interactions with chromatin modulators in a sequence-dependent or independent manner. Most often the presence of eRNAs results in a bias toward gene upregulation. Studies have established direct eRNA effects on transcription regulation by interacting with proteins such as: transcription factors, chromatin writers/readers, 3D chromatin structural proteins, and pausing factors [153–155,157,158]. Bottom: Instances where the act of enhancer transcription impacts transcriptional regulation. Indirect ways in which enhancer transcription itself contributes to transcription regulation include: altering transcription factors, polymerase, or general transcription factor localization, participating in transcriptional interference, or affecting chromatin rearrangements (reviewed in [29,41]).
5.1. Specific protein-eRNA binding interactions can affect transcriptional events
Evidence supports the role of some enhancer RNAs in modulating transcription factor activities at regulatory regions. For example, under immune signaling activation BRD4 binds to enhancer elements primed with acetylated histones to stimulate eRNA transcription. Then bromodomains in BRD4 cooperate to interact with sequence specific eRNAs generated from BRD4 bound enhancers [153]. Additionally, in vitro binding assays demonstrate that BRD4 proteins associate more frequently with the acetylated histones H3K27 and H4K16 in the presence of eRNAs from BRD4/p53 positive enhancers. Similarly, the transcription factor Ying Yang 1 (YY1) interacts with eRNAs and, at least in in vitro assays, displays preferential affinity for distinct RNA sequences [154]. In this way, sequence specific eRNAs may reinforce the binding/stability of transcription factors at cis regulatory elements and promoters.
In other cases, the importance of the eRNA has been demonstrated, but the mechanism is not well understood. For example, repressive effect that NELF has upon RNA polymerase II elongation can be transiently silent under the presence of specific eRNAs [155]. Likewise, key eRNAs upstream of the Fos gene appear to be important for the appropriate transcription of Fos [156].
5.2. Some enhancer transcripts augment epigenetic modifiers
While most studies of the impact of these transcripts have focused on target genes in close proximity to the enhancer, recent evidence demonstrated the ability of eRNAs to act upon distal loci and even across chromosomes. For instance, during the onset of myogenesis, two particular enhancers upstream of the MyoD gene become activated to produce eRNAs. While the eRNA from the most proximal enhancer promotes higher occupancy of RNA polymerase II at the MyoD promoter, the more distal eRNA participates exclusively in the upregulation of downstream myogenic gene effectors [157]. Specifically, the distal enhancer, located on the mouse chromosome 7, generates a stable unidirectional transcript that specifically targets the Myogenin locus on chromosome 1. This regulatory noncoding transcript binds to multiple subunits of the cohesin complex and together they co-localize at the Myogenin gene to regulate its expression. This finding suggests a more elaborate functional role of a regulatory transcript, namely in the recruitment of chromatin organizers to establish transcriptionally active nuclear hot spots.
Interestingly, in some cases the sequence of the transcribed RNA may not matter. A recent study rigorously demonstrated that the histone acetyltransferase (HAT) CBP can bind RNA indiscriminately, e.g. independently of sequence characteristics. Importantly, a diverse set of eRNAs preferentially accumulate at the HAT domain and stimulate its catalytic activity resulting in hyperacetylation of lysines in histones, including H3K27 and H4K5 [158]. Notably, this eRNA-induced catalytic stimulation decreases at both low and high accumulation levels of eRNAs, suggesting the effect is tuned to respond to only moderate levels of enhancer RNAs.
6. Conclusion and discussion
By monitoring changes in RNA polymerase II activity, nascent transcription assays paint a more complete picture of regulation than one focused predominantly on protein-coding genes. Importantly, while this review is written from an RNA polymerase II centric viewpoint, some of the processes included may be incorrectly attributed to this polymerase. It remains to be seen to what extent RNA polymerase I and III are influenced by the regulators of RNA polymerase II.
RNA polymerase II steps through multiple stages during the transcription process and nascent transcription protocols have been informative on the regulators participating in every stage of the transcription process. The loading and initiation stage of RNA polymerase II is regulated by a large number of DNA binding transcription factors. Active transcription factor binding sites alter polymerase activity immediately proximal to the binding site. Therefore, evidence of RNA polymerase II loading and initiation, as measured by nascent transcription assays, provides a unique temporal and positional resolution on regulatory activity. Subsequent steps in RNA polymerase II activity: pause release, elongation and termination, depend on not only sequence features but also RNA binding proteins and other regulators. Ultimately this process gives rise to transcripts with a variety of lengths, stability, and post-transcriptional localization.
Transcript properties underly the definitions of the classes of noncoding RNAs (lncRNAs, PROMPTs, uaRNAs, eRNAs, SLITs). Yet transcription studies suggest these features exist on a continuum [58] and therefore there may be substantial overlap between the classes [41]. Intriguingly, some of these properties may even vary according to cell type or perturbation.
What these transcripts do remains a hot topic, but the answer is likely to be diverse (Figure 4), just as proteins have a diverse set of functions. The act of transcription is a critical contributor to the local regulatory context, interacting with and influencing both TFs and histones. Arguably the relationship between nucleosome free regions, histone marks, TF binding and RNA polymerase initiation is one of co-dependence, with each element both interacting and contributing to the local context. Given the vast numbers of transcripts produced in a eukaryotic cell, it is also likely that evolution has found ways to utilize some of the noncoding transcripts as regulators.
Funding Statement
This work was supported by the NIH under grant GM125871; NSF under grant DBI 1759949; and a Sie Postdoctoral Fellowship.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Corradin O, Scacheri PC.. Enhancer variants: evaluating functions in common disease. Genome Med. 2014;6(10):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mifsud B, Tavares-Cadete F, Young AN, et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet. 2015;47(6):598–606. [DOI] [PubMed] [Google Scholar]
- [4].Farh KK-H, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shlyueva D, Stampfel G, Stark A.. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15(4):272–286. [DOI] [PubMed] [Google Scholar]
- [6].Halfon MS. Studying transcriptional enhancers: the founder fallacy, validation creep, and other biases. Trends Genet. 2019;35(2):93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lewis MW, Li S, Franco HL. Transcriptional control by enhancers and enhancer RNAs. Transcription. 2019;1–16. DOI: 10.1080/21541264.2019.1695492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Spitz F, Furlong EEM. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13(9):613–626. [DOI] [PubMed] [Google Scholar]
- [9].Farnham PJ. Insights from genomic profiling of transcription factors. Nat Rev Genet. 2009;10(9):605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gade P, Kalvakolanu DV. Chromatin immunoprecipitation assay as a tool for analyzing transcription factor activity. Methods Mol Biol Clifton NJ. 2012;809:85–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lambert SA, Jolma A, Campitelli LF, et al. The human transcription factors. Cell. 2018;172(4):650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Birney E, Stamatoyannopoulos JA, Dutta A, et al.; ENCODE Project Consortium . Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bernstein BE, Stamatoyannopoulos JA, Costello JF, et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol. 2010;28(10):1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Spivakov M. Spurious transcription factor binding: non-functional or genetically redundant? BioEssays News Rev Mol Cell Dev Biol. 2014;36(8):798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Teytelman L, Thurtle DM, Rine J, et al. Highly expressed loci are vulnerable to misleading chip localization of multiple unrelated proteins. Proc Natl Acad Sci U S A. 2013;110(46):18602–18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hart SN, Therneau TM, Zhang Y,Poland GA, Kocher JP. Calculating sample size estimates for RNA sequencing data. J Comput Biol. 2013. Dec;20(12):970–978. doi: 10.1089/cmb.2012.0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schramm L, Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev. 2002;16(20):2593–2620. [DOI] [PubMed] [Google Scholar]
- [18].Wissink EM, Vihervaara A, Tippens ND, et al. Nascent RNA analyses: tracking transcription and its regulation. Nat Rev Genet. 2019;1–19. DOI: 10.1038/s41576-019-0159-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322(5909):1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kwak H, Fuda NJ, Core LJ, et al. Precise maps of rna polymerase reveal how promoters direct initiation and pausing. Science. 2013;339(6122):950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469(7330):368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mayer A, Di Iulio J, Maleri S, et al. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell. 2015;161(3):541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nojima T, Gomes T, Grosso ARF, et al. Mammalian NET-seq reveals genome-wide nascent transcription coupled to RNA processing. Cell. 2015;161(3):526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chu T, Rice EJ, Booth GT, et al. Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme. Nat Genet. 2018;50(11):1553–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Muhar M, Ebert A, Neumann T, et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science. 2018;360(6390):800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Paulsen MT, Veloso A, Prasad J, et al. Use of bru-seq and bruchase-seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 2014;67(1):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Core LJ, Martins AL, Danko CG, et al. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet. 2014;46(12):1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jarroux J, Morillon A, Pinskaya M. History, discovery, and classification of LncRNAs. Adv Exp Med Biol. 2017;1008:1–46. [DOI] [PubMed] [Google Scholar]
- [29].Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17(4):207–223. [DOI] [PubMed] [Google Scholar]
- [30].Lloret-Llinares M, Mapendano CK, Martlev LH, et al. Relationships between PROMPT and gene expression. RNA Biol. 2016;13(1):6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Flynn RA, Almada AE, Zamudio JR, et al. Antisense RNA polymerase II divergent transcripts are P-TEFb dependent and substrates for the RNA exosome. Proc Natl Acad Sci. 2011;108(26):10460–10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Seila AC, Calabrese JM, Levine SS, et al. divergent transcription from active promoters. Science. 2008;322(5909):1849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tani H. Short-Lived Non-Coding Transcripts (SLiTs): clues to regulatory long non-coding RNA. Drug Discov Ther. 2017;11(1):20–24. [DOI] [PubMed] [Google Scholar]
- [34].Preker P, Nielsen J, Kammler S, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322(5909):1851–1854. [DOI] [PubMed] [Google Scholar]
- [35].Espinosa JM. Revisiting LncRNAs: how do you know yours is not an ERNA? Mol Cell. 2016;62(1):1–2. [DOI] [PubMed] [Google Scholar]
- [36].Agirre X, Meydan C, Jiang Y, et al. Long non-coding RNAs discriminate the stages and gene regulatory states of human humoral immune response. Nat Commun. 2019;10(1):821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ørom UA, Shiekhattar R. Long noncoding RNAs usher in a new era in the biology of enhancers. Cell. 2013;154(6):1190–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ponjavic J, Ponting CP, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007;17(5):556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].van Bakel H, Nislow C, Blencowe BJ, et al. Most “Dark Matter” transcripts are associated with known genes. PLoS Biol. 2010;8(5):e1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lam MTY, Li W, Rosenfeld MG, et al. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci. 2014;39(4):170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kaikkonen MU, Adelman K. Emerging roles of non-coding RNA transcription. Trends Biochem Sci. 2018;43(9):654–667. [DOI] [PubMed] [Google Scholar]
- [42].Fuda NJ, Ardehali MB, Lis JT. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature. 2009;461(7261):186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bentley DL. coupling mrna processing with transcription in time and space. Nat Rev Genet. 2014;15(3):163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Azofeifa JG, Allen MA, Hendrix JR, et al. Enhancer RNA profiling predicts transcription factor activity. Genome Res. 2018;28(3):334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].De Santa F, Barozzi I, Mietton F, et al. A large fraction of extragenic RNA Pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim T-K, Hemberg M, Gray JM, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465(7295):182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Scruggs BS, Gilchrist DA, Nechaev S, et al. Upstream anti-sense promoters are hubs of transcription factor binding and active histone modifications. Mol Cell. 2015;58(6):1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Koch F, Fenouil R, Gut M, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol. 2011;18(8):956–963. [DOI] [PubMed] [Google Scholar]
- [49].Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Henriques T, Scruggs BS, Inouye MO, et al. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 2018;32(1):26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Allen BL, Taatjes DJ. The mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16(3):155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Grünberg S, Zentner GE. Genome-wide characterization of mediator recruitment, function, and regulation. Transcription. 2017;8(3):169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lai F, Orom UA, Cesaroni M, et al. Activating RNAs associate with mediator to enhance chromatin architecture and transcription. Nature. 2013;494(7438):497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pelish HE, Liau BB, Nitulescu II, et al. Mediator kinase inhibition further activates super-enhancer associated genes in AML. Nature. 2015;526(7572):273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Knoll ER, Zhu ZI, Sarkar D, et al. Role of the pre-initiation complex in mediator recruitment and dynamics. eLife. 2018;7. DOI: 10.7554/eLife.39633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Andersson R, Sandelin A, Danko CG. A unified architecture of transcriptional regulatory elements. Trends Genet. 2015;31(8):426–433. [DOI] [PubMed] [Google Scholar]
- [58].Mikhaylichenko O, Bondarenko V, Harnett D, et al. The degree of enhancer or promoter activity is reflected by the levels and directionality of ERNA transcription. Genes Dev. 2018;32(1):42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rhee HS, Pugh BF. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012;483(7389):295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Duttke SHC, Lacadie SA, Ibrahim MM, et al. Human promoters are intrinsically directional. Mol Cell. 2015;57(4):674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Andersson R, Gebhard C, Miguel-Escalada I, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Preker P, Almvig K, Christensen MS, et al. PROMoter upstream transcripts share characteristics with mrnas and are produced upstream of all three major types of mammalian promoters. Nucleic Acids Res. 2011;39(16):7179–7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Andersson R, Refsing Andersen P, Valen E, et al. Transcriptional directionality separate functionally distinct RNA species. Nat Commun. 2014;5:5336. [DOI] [PubMed] [Google Scholar]
- [64].Medina-Rivera A, Santiago-Algarra D, Puthier D, et al. Widespread enhancer activity from core promoters. Trends Biochem Sci. 2018;43(6):452–468. [DOI] [PubMed] [Google Scholar]
- [65].Kim T-K, Shiekhattar R. Architectural and functional commonalities between enhancers and promoters. Cell. 2015;162(5):948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yunger S, Rosenfeld L, Garini Y, et al. Single-allele analysis of transcription kinetics in living mammalian cells. Nat Methods. 2010;7(8):631–633. [DOI] [PubMed] [Google Scholar]
- [67].Suter DM, Molina N, Gatfield D, et al. Mammalian genes are transcribed with widely different bursting kinetics. Science. 2011;332(6028):472–474. [DOI] [PubMed] [Google Scholar]
- [68].Sanchez A, Golding I. Genetic determinants and cellular constraints in noisy gene expression. Science. 2013;342(6163):1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Arner E, Daub CO, Vitting-Seerup K, et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science. 2015;347(6225):1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Fukaya T, Lim B, Levine M. Enhancer control of transcriptional bursting. Cell. 2016;166(2):358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Larsson AJM, Johnsson P, Hagemann-Jensen M, et al. Genomic encoding of transcriptional burst kinetics. Nature. 2019;565(7738):251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Tunnacliffe E, Corrigan AM, Chubb JR. Promoter-mediated diversification of transcriptional bursting dynamics following gene duplication. Proc Natl Acad Sci. 2018;115(33):8364–8369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kouno T, Moody J, Kwon AT-J, et al. C1 CAGE detects transcription start sites and enhancer activity at single-cell resolution. Nat Commun. 2019;10(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Teves SS, Henikoff S. Transcription-generated torsional stress destabilizes nucleosomes. Nat Struct Mol Biol. 2014;21(1):88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhang L, Kasif S, Cantor CR, et al. GC/AT-content spikes as genomic punctuation marks. Proc Natl Acad Sci. 2004;101(48):16855–16860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cooper SJ, Trinklein ND, Anton ED, et al. Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res. 2006;16(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kudla G, Lipinski L, Caffin F, et al. High guanine and cytosine content increases MRNA levels in mammalian cells. PLoS Biol. 2006;4(6):e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Vinogradov AE. Dualism of gene GC content and CpG pattern in regard to expression in the human genome: magnitude versus breadth. Trends Genet. 2005;21(12):639–643. [DOI] [PubMed] [Google Scholar]
- [79].Vinogradov AE. Isochores and Tissue‐specificity. Nucleic Acids Res. 2003;31(17):5212–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–318. [DOI] [PubMed] [Google Scholar]
- [81].Visel A, Minovitsky S, Dubchak I, et al. VISTA enhancer browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35(Database issue):D88–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010;2010(2):pdb.prot5384-pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Henikoff JG, Belsky JA, Krassovsky K, et al. Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci. 2011;108(45):18318–18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Buenrostro JD, Giresi PG, Zaba LC, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zentner GE, Henikoff S. Surveying the epigenomic landscape, one base at a time. Genome Biol. 2012;13(10):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Keene MA, Corces V, Lowenhaupt K, et al. DNase I hypersensitive sites in drosophila chromatin occur at the 5’ ends of regions of transcription. Proc Natl Acad Sci. 1981;78(1):143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Jin J, Bai L, Johnson DS, et al. Synergistic action of RNA polymerases in overcoming the nucleosomal barrier. Nat Struct Mol Biol. 2010;17(6):745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Skene PJ, Hernandez AE, Groudine M, et al. The nucleosomal barrier to promoter escape by RNA polymerase II is overcome by the chromatin remodeler Chd1. eLife. 2014;3:e02042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wang J, Zhuang J, Iyer S, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22(9):1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Shen H, Xu W, Guo R, et al. Suppression of enhancer overactivation by a RACK7-histone demethylase complex. Cell. 2016;165(2):331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Håndstad T, Rye M, Močnik R, et al. Cell-type specificity of ChIP-predicted transcription factor binding sites. BMC Genomics. 2012;13:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Andrysik Z, Galbraith MD, Guarnieri AL, et al. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017;27(10):1645–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Wu M, Chan C. Learning transcriptional regulation on a genome scale: a theoretical analysis based on gene expression data. Brief Bioinform. 2012;13(2):150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Yang T-H, Wu W-S. Inferring functional transcription factor-gene binding pairs by integrating transcription factor binding data with transcription factor knockout data. BMC Syst Biol. 2013;7 Suppl 6:S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Whitfield TW, Wang J, Collins PJ, et al. Functional analysis of transcription factor binding sites in human promoters. Genome Biol. 2012;13(9):R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Cusanovich DA, Pavlovic B, Pritchard JK, et al. The functional consequences of variation in transcription factor binding. PLoS Genet. 2014;10(3):e1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Wu W-S, Lai F-J. Functional redundancy of transcription factors explains why most binding targets of a transcription factor are not affected when the transcription factor is knocked out. BMC Syst Biol. 2015;9(Suppl 6):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Lickwar CR, Mueller F, Hanlon SE, et al. Genome-wide protein-DNA binding dynamics suggest a molecular clutch for transcription factor function. Nature. 2012;484(7393):251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Jain D, Baldi S, Zabel A, et al. Active promoters give rise to false positive “Phantom Peaks” in ChIP-seq experiments. Nucleic Acids Res. 2015;43(14):6959–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wang Z, Chu T, Choate LA, et al. Identification of regulatory elements from nascent transcription using DREG. Genome Res. 2019;29(2):293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kristjánsdóttir K, Kwak Y, Tippens ND, et al. Population-scale study of ERNA transcription reveals bipartite functional enhancer architecture. bioRxiv. 2018;426908. DOI: 10.1101/426908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Rennie S, Dalby M, Lloret-Llinares M, et al. Transcription start site analysis reveals widespread divergent transcription in d. melanogaster and core promoter-encoded enhancer activities. Nucleic Acids Res. 2018;46(11):5455–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Butler JEF, Kadonaga JT. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev. 2002;16(20):2583–2592. [DOI] [PubMed] [Google Scholar]
- [105].Ibrahim MM, Karabacak A, Glahs A, et al. Determinants of promoter and enhancer transcription directionality in metazoans. Nat Commun. 2018;9(1):4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13(10):720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Almada AE, Wu X, Kriz AJ, et al. Promoter directionality is controlled by U1 SnRNP and polyadenylation signals. Nature. 2013;499(7458):360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Kaida D, Berg MG, Younis I, et al. U1 SnRNP protects Pre-MRNAs from premature cleavage and polyadenylation. Nature. 2010;468(7324):664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ntini E, Järvelin AI, Bornholdt J, et al. Polyadenylation site–induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol. 2013;20(8):923–928. [DOI] [PubMed] [Google Scholar]
- [110].Jo B-S, Choi SS. Introns: the functional benefits of introns in genomes. Genomics Inform. 2015;13(4):112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tian B, Manley JL. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 2013;38(6):312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Porrua O, Libri D. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat Rev Mol Cell Biol. 2015;16(3):190–202. [DOI] [PubMed] [Google Scholar]
- [113].Berg MG, Singh LN, Younis I, et al. U1 SnRNP determines MRNA length and regulates isoform expression. Cell. 2012;150(1):53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Schmid M, Jensen TH. Controlling nuclear RNA levels. Nat Rev Genet. 2018;19(8):518–529. [DOI] [PubMed] [Google Scholar]
- [115].Hah N, Murakami S, Nagari A, et al. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23(8):1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136(4):763–776. [DOI] [PubMed] [Google Scholar]
- [117].Andersen PR, Domanski M, Kristiansen MS, et al. The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat Struct Mol Biol. 2013;20(12):1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lubas M, Christensen MS, Kristiansen MS, et al. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell. 2011;43(4):624–637. [DOI] [PubMed] [Google Scholar]
- [119].Zhang Z, Lee J-H, Ruan H, et al. Transcriptional landscape and clinical utility of enhancer RNAs for ERNA-targeted therapy in cancer. Nat Commun. 2019;10(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Freiman RN, Tjian R. Regulating the regulators: lysine modifications make their mark. Cell. 2003;112(1):11–17. [DOI] [PubMed] [Google Scholar]
- [121].Saydam N, Georgiev O, Nakano MY, et al. Nucleo-cytoplasmic trafficking of metal-regulatory transcription factor 1 is regulated by diverse stress signals. J Biol Chem. 2001;276(27):25487–25495. [DOI] [PubMed] [Google Scholar]
- [122].Gill G. Post-translational modification by the small ubiquitin-related modifier SUMO has big effects on transcription factor activity. Curr Opin Genet Dev. 2003;13(2):108–113. [DOI] [PubMed] [Google Scholar]
- [123].Jackson SP. Regulating transcription factor activity by phosphorylation. Trends Cell Biol. 1992;2(4):104–108. [DOI] [PubMed] [Google Scholar]
- [124].Latchman DS. Eukaryotic transcription factors. Biochem J. 1990;270(2):281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Levine AJ. P53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331. [DOI] [PubMed] [Google Scholar]
- [126].Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression.. Cold Spring Harb Perspect Med. 2016;6(3):a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Hafner A, Bulyk ML, Jambhekar A, et al. The multiple mechanisms that regulate P53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20(4):199–210. [DOI] [PubMed] [Google Scholar]
- [128].Moll UM, Petrenko O. The MDM2-P53 Interaction. Mol Cancer Res MCR. 2003;1(14):1001–1008. [PubMed] [Google Scholar]
- [129].Slattery M, Zhou T, Yang L, et al. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci. 2014;39(9):381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Grossman SR, Zhang X, Wang L, et al. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc Natl Acad Sci. 2017;114(7):E1291–E1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].The ENCODE Project Consortium . An Integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Yao L, Berman BP, Farnham PJ. Demystifying the secret mission of enhancers: linking distal regulatory elements to target genes. Crit Rev Biochem Mol Biol. 2015;50(6):550–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Sanyal A, Lajoie BR, Jain G, et al. The long-range interaction landscape of gene promoters. Nature. 2012;489(7414):109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Wang D, Garcia-Bassets I, Benner C, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by ERNA. Nature. 2011;474(7351):390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Allen MA, Andrysik Z, Dengler VL, et al. Global analysis of P53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife. 2014;3:e02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Luo X, Chae M, Krishnakumar R, et al. Dynamic reorganization of the AC16 cardiomyocyte transcriptome in response to TNFα signaling revealed by integrated genomic analyses. BMC Genomics. 2014;15:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Yang F, Ma Q, Liu Z, et al. Glucocorticoid receptor: megaTransSwitching mediates the repression of an ERα-regulated transcriptional program. Mol Cell. 2017;66(3):321–331.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Melo CA, Drost J, Wijchers PJ, et al. ERNAs are required for P53-dependent enhancer activity and gene transcription. Mol Cell. 2013;49(3):524–535. [DOI] [PubMed] [Google Scholar]
- [139].Puc J, Kozbial P, Li W, et al. Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell. 2015;160(3):367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Step SE, Lim H-W, Marinis JM, et al. Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPARγ-driven enhancers. Genes Dev. 2014;28(9):1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Lam MTY, Cho H, Lesch HP, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498(7455):511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Vokes SA, Ji H, Wong WH, et al. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 2008;22(19):2651–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Ucar D, Beyer A, Parthasarathy S, et al. Predicting functionality of protein-DNA interactions by integrating diverse evidence. Bioinforma Oxf Engl. 2009;25(12):i137–i144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Krejcí A, Bernard F, Housden BE, et al. Direct response to notch activation: signaling crosstalk and incoherent logic. Sci Signal. 2009;2(55):ra1. [DOI] [PubMed] [Google Scholar]
- [145].MacIsaac KD, Lo KA, Gordon W, et al. A quantitative model of transcriptional regulation reveals the influence of binding location on expression. PLoS Comput Biol. 2010;6(4):e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Vanhille L, Griffon A, Maqbool MA, et al. High-throughput and quantitative assessment of enhancer activity in mammals by capstarr-seq. Nat Commun. 2015;6(1):6905. [DOI] [PubMed] [Google Scholar]
- [147].Mayran A, Drouin J. Pioneer transcription factors shape the epigenetic landscape. J Biol Chem. 2018;293(36):13795–13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell. 1998;92(3):307–313. [DOI] [PubMed] [Google Scholar]
- [149].Mivelaz M, Cao A-M, Kubik S, et al. Chromatin fiber invasion and nucleosome displacement by the Rap1 transcription factor. Mol Cell. 2019;S1097276519308032. DOI: 10.1016/j.molcel.2019.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Jonkers I, Kwak H, Lis JT. Genome-wide dynamics of pol ii elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife. 2014;3:e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15(6):423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Rahnamoun H, Lee J, Sun Z, et al. RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat Struct Mol Biol. 2018;25(8):687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Sigova AA, Abraham BJ, Ji X, et al. Transcription factor trapping by RNA in gene regulatory elements. Science. 2015;350(6263):978–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Schaukowitch K, Joo J-Y, Liu X, et al. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56(1):29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Carullo NVN, Simon RC, Salisbury AJ, et al. Enhancer RNAs are necessary and sufficient for activity-dependent neuronal gene transcription. bioRxiv. 2018;270967. DOI: 10.1101/270967 [DOI] [Google Scholar]
- [157].Tsai P-F, Dell’Orso S, Rodriguez J, et al. A muscle-specific enhancer RNA mediates cohesin recruitment and regulates transcription in trans. Mol Cell. 2018;71(1):129–141.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Bose DA, Donahue G, Reinberg D, et al. RNA binding to CBP stimulates histone acetylation and transcription. Cell. 2017;168(1–2):135–149.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Tan PY, Chang CW, Chng KR, et al. Integration of regulatory networks by NKX3-1 promotes androgen-dependent prostate cancer survival. Mol Cell Biol. 2012;32(2):399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Kulakovskiy IV, Vorontsov IE, Yevshin IS, et al. HOCOMOCO: towards a complete collection of transcription factor binding Models for human and mouse via large-scale chip-seq analysis. Nucleic Acids Res. 2018;46(D1):D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]