

ABSTRACT
Cardiometabolic affections greatly contribute to the global burden of disease. The susceptibility to obesity, cardiovascular disease, and type-2 diabetes, conditions that add to the cardiometabolic syndrome (CMS), was associated with the ancestral genetic composition and gut microbiota. Studies explicitly testing associations between genetic ancestry and gut microbes are growing. We here examined whether the host genetic ancestry was associated with gut microbiota composition, and distinguished the effects of genetic ancestry and non-genetic factors on human cardiometabolic health. We performed a cross-sectional study with 441 community-dwelling Colombian mestizos from five cities spanning the Andes, Pacific, and Caribbean coasts. We characterized the host genetic ancestry by genotyping 40 ancestry informative markers; characterized gut microbiota through 16S rRNA gene sequencing; assessed diet intake, physical activity, cigarette, and medicament consumption; and measured cardiometabolic outcomes that allowed calculating a CMS risk scale. On average, each individual of our cohort was 67 ± 6% European, 21 ± 5% Native American and 12 ± 5% African. Multivariable-adjusted generalized linear models showed that individuals with higher Native American and African ancestries had increased fasting insulin, body mass index and CMS risk, as assessed by the CMS risk scale. Furthermore, we identified 21 OTUs associated to the host genetic ancestry and 20 to cardiometabolic health. While we highlight novel associations between genetic ancestry and gut microbiota, we found that the effect of intestinal microbes was more likely to explain the variance in CMS risk scale than the contributions of European, Native American and African genetic backgrounds.
KEYWORDS: Ancestry informative markers, genetic admixture, mestizo, Colombia, Latin America, microbiome, obesity, metabolic syndrome
Introduction
Obesity, cardiovascular disease, and type 2 diabetes are notable contributors to the global burden of disease1 and add to the cardiometabolic syndrome (CMS).2 Seminal studies in monozygotic twins demonstrated that components of the CMS are heritable,3–5 but genome-wide association studies (GWAS) have failed to consistently uncover replicable variants across human populations, with notable exceptions.6,7 One possible explanation for this is that the identification of variants in candidate genes is highly dependent on the ethnic and geographic origin of the studied population.8 Differences in allele frequencies and linkage disequilibrium structure make difficult the extrapolation of results in human groups with different genetic backgrounds. Therefore, the ancestral genetic composition of the studied population becomes a key element in association studies.9
Additionally, the lack of replicability of many GWAS results across populations may be explained by the interactions between gene variants and non-genetic factors.10 The gut microbiota, that is, the set of microorganisms that naturally colonize the human intestine,11 is one of such factors. The gut microbiota has been shown to be central to CMS12–14 and to be shaped by human genetics.15,16 Despite the impact of recent discoveries on the relationship between gut microbes and human health, the degree to which associations found in one population can extend to another is still unclear. The geographic origin of human populations is one of the most important factors shaping the composition of this microbial community,17,18 yet it is unknown whether such pattern is explained by genetic or non-genetic factors correlated with geography and ancestry (e.g., diet, lifestyle). Studies explicitly testing associations between host genetic ancestry and gut microbiota are growing. Some suggested that broad ethnic differences could contribute to gut microbiota composition,19,20 while others found no association.21
In this study, we analyzed a cohort of Colombian adult mestizos whose genetic background is the product of extensive recent admixture.22 We fine-mapped the individual contributions of European, Native American and African genetic backgrounds using ancestry informative markers (AIMs), characterized gut microbiota through high-throughput 16S rRNA gene sequencing and measured numerous variables that informed about diet, lifestyle, and CMS risk. We aimed to determine whether the individual contribution of the three ethnicities mentioned above was associated with the composition of the gut microbiota, and gauge the effects of genetic ancestry and gut microbes on human cardiometabolic health.
Results
Ancestral genetic composition of the studied cohort
We performed a cross-sectional study in which we enrolled 441 adult Colombian mestizos in roughly similar proportions across five large cities spanning the Andes, the Caribbean and Pacific coasts (Bogota, Medellin, Cali, Barranquilla, and Bucaramanga); body mass index (BMI: lean, overweight, obese); sex (male, female); and age range (18–40 years, 41–62 years). We characterized the ancestral genetic composition in 440 of these participants using a panel of 40 AIMs that have been previously shown to discriminate among European, Native American and African populations23,24 (Table S1). One individual of our cohort could not be genotyped because we were not able to acquire DNA from blood. Overall, the 40 evaluated AIMs were in Hardy–Weinberg equilibrium (all p > .05 in exact Hardy–Weinberg tests).
On average, the ancestral genetic composition of each individual of our cohort was (mean ± SD) 0.674 ± 0.057 European (range: 0.469–0.788), 0.209 ± 0.048 Native American (0.089–0.397), and 0.117 ± 0.047 African (0.051–0.352) (Figure 1A). These proportions differed significantly among the cities where participants were enrolled (ANOVA for European: F4,431 = 2.84, p = .02; Native American: F4,431 = 7.46, p < .0001; African: F4,431 = 5.64, p = .0002): the European component was highest in Medellin (Northwestern Andes) and lowest in Barranquilla (Northern Caribbean); the Native American component highest in Bogota (Central Andes) and lowest in Medellin; and the African component highest in Barranquilla and lowest in Bogota (Figure 1B-D). In agreement with this, we found evidence of limited but significant genetic structure (mean Fst ± SE = 0.004 ± 0.001, 95% CI = 0.002–0.006). However, there was no evidence of isolation by distance, according to a Mantel test considering genetic (Fst/(1-Fst)) and (log-transformed) geographic distance matrices (r = −0.43, 95% CI = −0.80–0.14, two-tailed p = .44). Furthermore, we did not find significant differences in the ancestral genetic composition by other factors controlled by design (p > .10 in all ANOVAs for BMI, sex, and age range).
Figure 1.
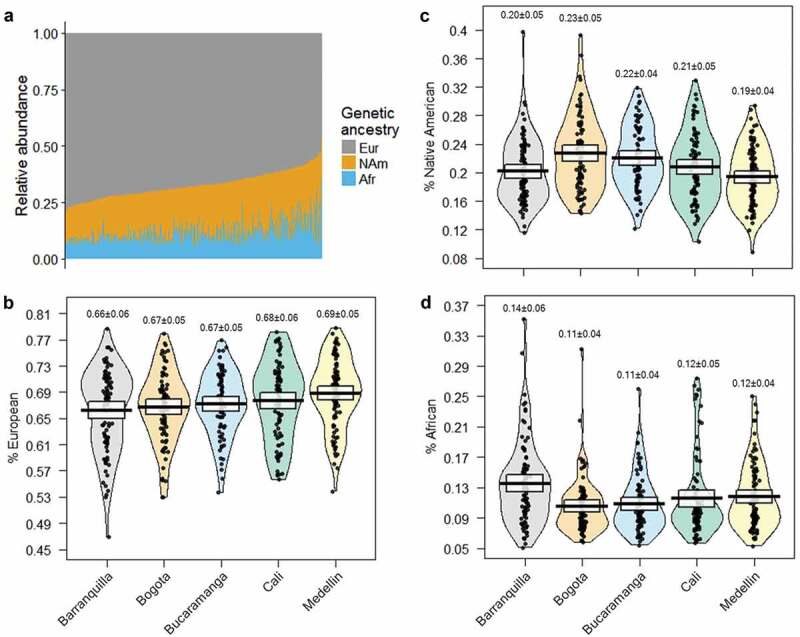
Contributions of European, Native American and African ancestries to the studied population. (A) Ancestral genetic composition across individuals (vertical bars). Data sorted by the European component. Eur = European; NAm = Native American; Afr = African. (B-D) Ancestral genetic composition along the five Colombian cities from which participants originated. The raw data, average, and 95% confidence intervals are shown in each plot. The mean and SD are given above each plot. Note the change in the scale among panels.
Next, we performed a robust principal component analysis (PCA) for compositional data based on the individual proportions of European, Native American and African, and found a gradient where the first component (PC1) distinguished Native American and African ancestries, whereas the second component (PC2) discerned between European and non-European ancestries (Figure 2A-C). In agreement with the above result, these two components differed among the cities from which participants originated (ANOVA for PC1: F4,431 = 7.45, p < .0001; PC2: F4,431 = 3.55, p = .007) but did not differ by BMI, sex or age range (p > .10 in all ANOVAs).
Figure 2.
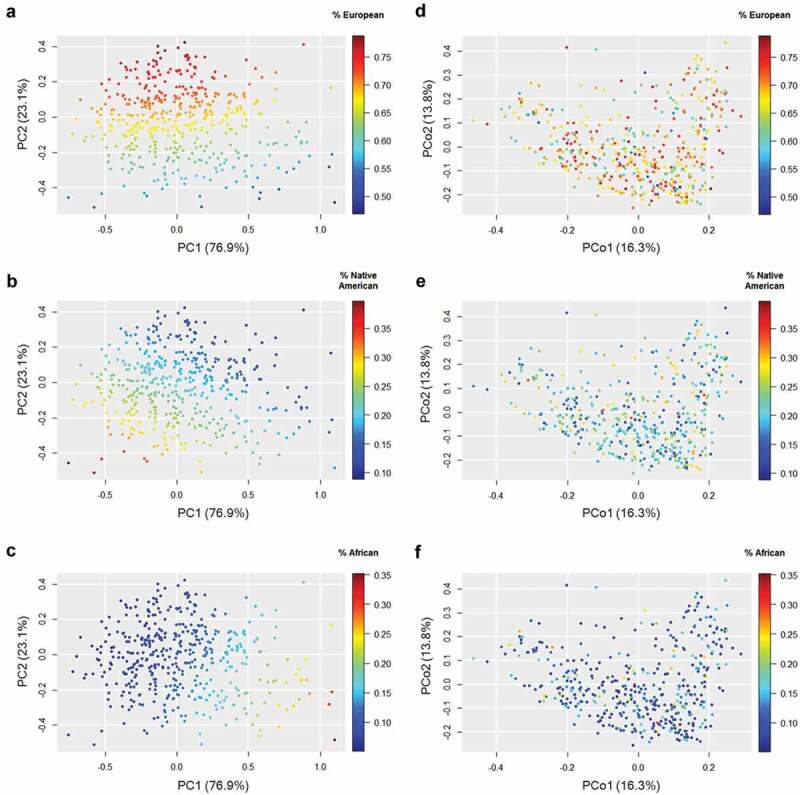
Ancestral genetic composition and gut microbiota composition in the studied population. Each set of panels shows the same cloud point colored by the contributions of each genetic ancestry. Robust principal components analysis (PCA) for compositional data based on the proportions of European (A), Native American (B) and African (C) ancestries. Principal coordinate analysis (PCoA) based on weighted UniFrac distances of the gut microbiota for European (D), Native American (E), and African (F) ancestries. The percentages on the axes represent the proportion of explained variation. Note the change in the scale among panels.
Associations between the host genetic ancestry and gut microbiota
Afterwards, we sought to examine whether the host genetic ancestry was associated with the composition of gut microbiota. We analyzed the complete microbial community through principal coordinates analysis (PCoA) using weighted UniFrac distances on rarefied sequence counts (3667 reads/sample) and found that the gut microbiota of Colombians formed a single point cloud of microbial abundances. Beta-diversity analyses indicated that differences in the composition of the microbial community were partly driven by the city of origin (PERMANOVA: R2 = 0.074, p = .001), BMI (R2 = 0.010, p = .001), and sex (R2 = 0.011, p = .001), but not by the age range (R2 = 0.003, p = .17).
We found limited evidence of a direct, unadjusted association between the host genetic ancestry and the complete microbial community. Procrustes analyses revealed no correlation between the weighted UniFrac distance matrix and the matrix of genetic ancestry (Procrustes correlation = 0.04, p = .98). There was no correlation either between the first two PCoA axes of microbiota composition and the PCA components of genetic ancestry (Procrustes correlation = 0.03, p = .92) (Figure 2D-F).
Since microbiota-ancestry associations could be masked by potential confounders and be restricted to specific groups of microbes, we fitted generalized linear models (GLMs) with negative binomial error distribution using rarefied OTU counts as dependent variables, and genetic PC1 and PC2 as explanatory variables. These models were adjusted for appropriate covariates, including the participants’ city of origin, sex, age range, diet intake (carbohydrate, protein, fat, and fiber), physical activity levels and CMS risk. The latter was assessed through a summary measure, the CMS risk scale, which totaled Z-scores of waist circumference, fasting insulin, triglycerides, diastolic blood pressure and high-sensitive C reactive protein (hs-CRP) (see Materials and Methods). These variables informed about general features of the CMS, namely abnormal body fat distribution, insulin resistance, atherogenic dyslipidemia, elevated blood pressure, and pro-inflammatory state, respectively.2 These GLMs indicated that the abundance of 21 OTUs was associated to the host genetic ancestry: 17 OTUs were associated to European, three to Native American and one to African (Figure 3; Table S2). These results were not affected by rarefaction depth since the patterns obtained with a deeper rarefaction (>10,000 reads/sample) were similar (not shown).
Figure 3.
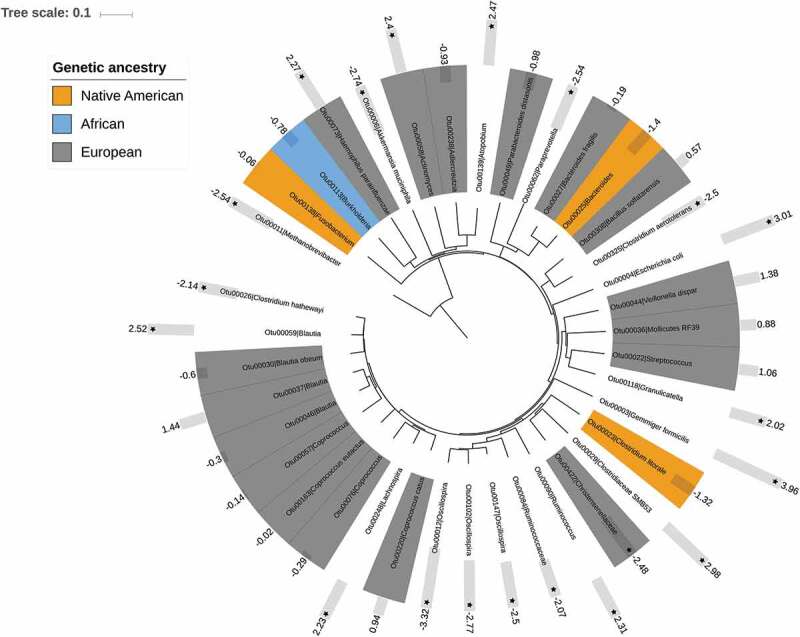
Neighbor-joining tree showing OTUs with significant associations with ancestry and cardiometabolic health.25 OTUs with a colored background were those associating with a particular ancestry (see legend). The bars denote CMS risk scale values (negative values = low risk, positive values = high risk) and their statistical significance (stars = p < .05 and q< 0.10).
CMS risk was better explained by gut microbiota composition than by the host genetic ancestry
We next examined whether gut microbes and the participants’ ancestral genetic composition each associated with variables related to cardiometabolic health, diet and lifestyle. We first divided the CMS risk scale by tertiles and found that individuals with higher cardiometabolic risk were more likely to be male, of older age, to have low levels of high-density lipoprotein (HDL) cholesterol, high levels of total cholesterol, low-density lipoprotein (LDL) cholesterol, very low-density lipoprotein (VLDL) cholesterol, and triglycerides, high levels of fasting glucose, glycated hemoglobin (HbA1c), fasting insulin, and insulin resistance (HOMA-IR), high levels of hs-CRP, high blood pressure, and adiposity (BMI, waist circumference and body fat), to regularly smoke and consume all kinds of medications, including anti-hypertensives and metformin, but not proton-pump inhibitors. In addition, they were more likely to suffer from coronary heart disease, as assessed by the Framingham score.26 While the levels of the CMS risk scale were not associated to the host genetic ancestry, diet intake or levels of physical activity, they were significantly associated to gut microbiota composition (i.e., PCoA axes) (Table 1).
Table 1.
Characteristics of the study population. Variables presented overall and according to tertiles of the CMS risk scale (low, intermediate, and high levels). Data presented as mean ± SE. P-values from ANOVA to the exception of sex, age range, smoking status, and medicament consumption (chi-squared tests).
| CMS risk scale |
|||||
|---|---|---|---|---|---|
| Overall | Tertile 1 (low) | Tertile 2 (intermediate) | Tertile 3 (high) | p-value | |
| n | 440 | 147 | 146 | 147 | |
| Sex (%) | <0.0001 | ||||
| Male | 0.48 | 0.33 | 0.46 | 0.64 | |
| Female | 0.52 | 0.67 | 0.54 | 0.36 | |
| Age range (%) | 0.05 | ||||
| 18–40 years | 0.47 | 0.55 | 0.41 | 0.45 | |
| 41–62 years | 0.53 | 0.45 | 0.59 | 0.55 | |
| Lipid profile | |||||
| HDL cholesterol (mg/dL) | 46 ± 1 | 52 ± 1 | 46 ± 1 | 40 ± 1 | <0.0001 |
| LDL cholesterol (mg/dL) | 115 ± 1 | 110 ± 3 | 120 ± 2 | 115 ± 3 | 0.02 |
| VLDL cholesterol (mg/dL) | 28.8 ± 1 | 17.7 ± 0.6 | 27.5 ± 1.0 | 40.5 ± 2.2 | <0.0001 |
| Total cholesterol (mg/dL) | 186 ± 2 | 178 ± 3 | 189 ± 3 | 190 ± 3 | 0.003 |
| Triglycerides (mg/dL) | 143 ± 5 | 87 ± 3 | 138 ± 5 | 203 ± 11 | <0.0001 |
| Glucose metabolism | |||||
| Fasting glucose (mmol/L) | 89 ± 1 | 82 ± 1 | 88 ± 1 | 96 ± 2 | <0.0001 |
| HbA1c (%) | 5.55 ± 0.03 | 5.37 ± 0.02 | 5.49 ± 0.05 | 5.77 ± 0.06 | <0.0001 |
| Fasting insulin (µU/ml) | 13.27 ± 0.41 | 8.04 ± 0.29 | 11.67 ± 0.39 | 19.62 ± 0.80 | <0.0001 |
| HOMA-IR | 3.12 ± 0.15 | 2.84 ± 0.33 | 2.97 ± 0.19 | 3.58 ± 0.22 | 0.0005 |
| Pro-inflammatory state | |||||
| hs-CRP (mg/L) | 3.15 ± 0.22 | 1.56 ± 0.11 | 2.63 ± 0.20 | 5.30 ± 0.58 | <0.0001 |
| Blood pressure | |||||
| Systolic (mm Hg) | 124 ± 1 | 112 ± 1 | 125 ± 1 | 136 ± 1 | <0.0001 |
| Diastolic (mm Hg) | 80 ± 1 | 71 ± 1 | 81 ± 1 | 88 ± 1 | <0.0001 |
| Body fat distribution | |||||
| BMI (kg/m2) | 27.9 ± 0.2 | 23.7 ± 0.2 | 28.2 ± 0.3 | 31.8 ± 0.4 | <0.0001 |
| Waist circumference (cm) | 92.8 ± 0.6 | 80.5 ± 0.6 | 93.3 ± 0.7 | 104.0 ± 0.9 | <0.0001 |
| Body fat (%) | 37.2 ± 0.3 | 33.9 ± 0.4 | 38.0 ± 0.4 | 39.6 ± 0.4 | <0.0001 |
| Cardiometabolic health | |||||
| CMS risk scale | 0.00 ± 0.16 | −3.83 ± 0.13 | 0.18 ± 0.07 | 3.57 ± 0.12 | <0.0001 |
| Framingham score | 0.52 ± 0.32 | −3.33 ± 0.53 | 1.38 ± 0.49 | 3.49 ± 0.48 | <0.0001 |
| Diet | |||||
| Calories (kcal/day) | 1931 ± 21 | 1944 ± 31 | 1921 ± 41 | 1922 ± 38 | 0.60 |
| Carbohydrates (g/day) | 266 ± 3 | 268 ± 5 | 265 ± 6 | 264 ± 5 | 0.69 |
| Protein (g/day) | 74 ± 1 | 74 ± 1 | 73 ± 1 | 74 ± 1 | 0.79 |
| Fat (g/day) | 63 ± 1 | 63 ± 1 | 62 ± 1 | 63 ± 1 | 0.54 |
| Fiber (g/day) | 17.7 ± 0.2 | 18.2 ± 0.4 | 17.5 ± 0.4 | 17.3 ± 0.4 | 0.21 |
| Lifestyle | |||||
| Physical activity (MET/min/week) | 5115 ± 264 | 5322 ± 434 | 5079 ± 412 | 5012 ± 528 | 0.18 |
| % Smoking (yes/no) | 0.13/0.87 | 0.09/0.91 | 0.12/0.88 | 0.18/0.82 | 0.08 |
| % Medicament consumption (yes/no) | 0.42/0.58 | 0.31/0.69 | 0.39/0.61 | 0.56/0.44 | <0.0001 |
| % Anti-hypertensives (yes/no) | 0.18/0.82 | 0.09/0.91 | 0.16/0.84 | 0.29/0.71 | <0.0001 |
| % Metformin (yes/no) | 0.03/0.97 | 0.00/1.00 | 0.03/0.97 | 0.06/0.94 | 0.01 |
| % Proton-pump inhibitors (yes/no) | 0.05/0.95 | 0.06/0.94 | 0.05/0.95 | 0.05/0.95 | 0.84 |
| Genetic ancestry | |||||
| European (%) | 67.37 ± 0.27 | 67.53 ± 0.43 | 68.02 ± 0.44 | 66.42 ± 0.52 | 0.10 |
| Native American (%) | 20.94 ± 0.23 | 20.66 ± 0.38 | 20.59 ± 0.38 | 21.63 ± 0.43 | 0.17 |
| African (%) | 11.69 ± 0.22 | 11.82 ± 0.38 | 11.39 ± 0.34 | 11.96 ± 0.44 | 0.71 |
| PC1 | 0.03 ± 0.02 | 0.04 ± 0.03 | 0.02 ± 0.03 | 0.02 ± 0.03 | 0.49 |
| PC2 | −0.02 ± 0.01 | −0.02 ± 0.01 | 0.0004 ± 0.02 | −0.05 ± 0.02 | 0.15 |
| Microbiota composition | |||||
| PCo1 | 0.00 ± 0.01 | 0.03 ± 0.01 | −0.005 ± 0.01 | −0.02 ± 0.01 | 0.006 |
| PCo2 | 0.00 ± 0.01 | −0.03 ± 0.01 | 0.006 ± 0.01 | 0.03 ± 0.01 | 0.001 |
We next fitted multivariable-adjusted GLMs using the aforementioned cardiometabolic factors as dependent variables, and ancestry PC1 and PC2, and gut microbiota PCo1 and PCo2 in the same models as explanatory variables. These models were adjusted by the participants’ city of origin, sex, age range, diet intake (carbohydrate, protein, fat, and fiber), levels of physical activity, smoking status and consumption medicaments of any kind. They indicated that both the host genetic ancestry and gut microbiota composition were significantly associated to cardiometabolic health. Individuals with higher non-European ancestries (i.e., Native American or African) had higher levels of fasting insulin, BMI and CMS risk scale, although the latter was borderline significant (p = .05, q = 0.06). In addition, gut microbiota was significantly associated with blood pressure, body fat distribution and cardiometabolic health (Table 2).
Table 2.
Associations between cardiometabolic health, host genetic ancestry, and gut microbiota composition. Scaled regression coefficients, p-values, and q-values are shown for each cardiometabolic factor.
| Genetic ancestry |
Gut microbiota composition |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PC1 |
PC2 |
PCo1 |
PCo2 |
|||||||||
| Scaled beta | P | q | Scaled beta | P | q | Scaled beta | P | q | Scaled beta | P | q | |
| Lipid profile | ||||||||||||
| HDL | 0.80 | 0.42 | 0.34 | −1.16 | 0.25 | 0.25 | 1.47 | 0.14 | 0.19 | −0.18 | 0.86 | 0.56 |
| LDL | 0.49 | 0.62 | 0.42 | −0.96 | 0.34 | 0.37 | 0.98 | 0.33 | 0.37 | 1.70 | 0.09 | 0.18 |
| VLDL | −0.94 | 0.35 | 0.26 | −1.20 | 0.23 | 0.22 | −0.43 | 0.66 | 0.31 | 0.50 | 0.62 | 0.31 |
| Total cholesterol | 0.10 | 0.92 | 0.53 | −1.78 | 0.08 | 0.12 | 1.35 | 0.18 | 0.19 | 1.47 | 0.14 | 0.19 |
| Triglycerides | −0.96 | 0.34 | 0.22 | −1.11 | 0.27 | 0.22 | −0.38 | 0.7 | 0.32 | 0.53 | 0.60 | 0.30 |
| Glucose metabolism | ||||||||||||
| Fasting glucose | −1.24 | 0.21 | 0.16 | −1.18 | 0.24 | 0.16 | −1.29 | 0.20 | 0.16 | −0.63 | 0.53 | 0.27 |
| HbA1c | 0.33 | 0.74 | 0.32 | −1.32 | 0.19 | 0.17 | −0.48 | 0.63 | 0.29 | −0.50 | 0.62 | 0.29 |
| Fasting insulin | −1.83 | 0.07 | 0.11 | −2.62 | 0.01 | 0.02 | −0.86 | 0.39 | 0.31 | 1.72 | 0.09 | 0.11 |
| HOMA-IR | −0.19 | 0.85 | 0.75 | 1.35 | 0.18 | 0.63 | −0.95 | 0.34 | 0.63 | 0.49 | 0.63 | 0.63 |
| Pro-inflammatory state | ||||||||||||
| hsCRP | 0.89 | 0.37 | 1.00 | 0.00 | 1.00 | 1.00 | −0.23 | 0.81 | 1.00 | 1.78 | 0.07 | 0.52 |
| Blood pressure | ||||||||||||
| Systolic | 0.04 | 0.97 | 0.99 | −0.36 | 0.72 | 0.92 | −2.81 | 0.005 | 0.02 | 3.67 | 0.0002 | 0.001 |
| Diastolic | 1.03 | 0.30 | 0.38 | −0.09 | 0.93 | 0.67 | −2.15 | 0.03 | 0.08 | 3.49 | 0.0005 | 0.002 |
| Body fat distribution | ||||||||||||
| BMI | −0.07 | 0.94 | 0.69 | −2.39 | 0.02 | 0.04 | −1.45 | 0.15 | 0.29 | 3.92 | <0.0001 | 0.0009 |
| Waist circumference | −0.70 | 0.48 | 0.27 | −1.62 | 0.11 | 0.13 | −1.34 | 0.18 | 0.18 | 3.51 | 0.0004 | 0.0009 |
| Body fat | −0.94 | 0.35 | 0.52 | −1.61 | 0.11 | 0.18 | −0.66 | 0.51 | 0.66 | 2.85 | 0.0004 | 0.01 |
| Cardiometabolic health | ||||||||||||
| CMS risk scale | −0.68 | 0.50 | 0.40 | −1.99 | 0.05 | 0.06 | −2.24 | 0.03 | 0.04 | 3.89 | <0.0001 | 0.0003 |
| Framingham score | 0.05 | 0.96 | 0.96 | 0.20 | 0.84 | 0.94 | 0.58 | 0.57 | 0.94 | 3.22 | 0.001 | 0.004 |
These GLMs allowed further examination of the contributions of the host genetic ancestry, gut microbiota composition, and their interaction to explain the variance in cardiometabolic factors. Based on likelihood-ratio tests and the Akaike information criterion (AIC), we found that the gut microbiota composition significantly explained more variance in the CMS risk scale, analyzed as a continuous variable, than genetic ancestry (model including genetic ancestry: partial R2 = 0.01; model including gut microbiota: partial R2 = 0.05; model including genetic ancestry × gut microbiota interaction: partial R2 = 0.006). Similar results were obtained for other cardiometabolic factors (Table S3).
Finally, multivariable-adjusted GLMs allowed identifying particular OTUs associated with cardiometabolic health outcomes. In this case, the CMS risk scale was set as the dependent variable and rarefied OTU counts as explanatory variables. These models were adjusted by host ancestry (PC1 and PC2), the participants’ city of origin, sex, age range, diet intake (carbohydrate, protein, fat, and fiber), levels of physical activity, smoking status, and medicament consumption. They indicated that the abundances of 10 OTUs were increased in individuals with higher CMS risk, including OTUs related to Gemmiger formicilis, Escherichia coli, Clostridiaceae SMB53, Blautia, Atopobium, and Haemophilus parainfluenzae, among others. In contrast, the abundance of other 10 OTUs related to Oscillospira, Akkermansia muciniphila, Paraprevotella, Methanobrevibacter, and Christensenellaceae, among others, was increased in individuals with low CMS risk (Figure 3; Table S2). Similar results were obtained with deeper rarefaction.
Discussion
The composition of gut microbiota and the host genetic background have been each associated to human cardiometabolic health.6,7,12–14 However, the evidence associating the microbial community and the host genetic ancestry remains sparse, despite latest efforts in different populations. A study performed in the USA including Caucasians, Asian-Pacific Islanders, Hispanics, and African Americans (N = 1673) found that 12 microbial genera and families varied by ethnicity.19 Another study in The Netherlands including Dutch, Ghanaians, Moroccans, Turks, African Surinamese, and South-Asian Surinamese (N = 2084) found that ethnicity contributed to explain the inter-individual dissimilarities in gut microbiota composition.20 In contrast, a study performed in Israel considering a variety of ancestries, including Ashkenazi, North African, Middle Eastern, Sephardi, Yemenite, and admixed (N = 1046) found that the gut microbiome was not significantly associated with genetic ancestry.21
In our Colombian cohort, we found that participants had an admixed genetic composition typical of Latin American mestizos, with predominance at the individual level of European ancestry, followed by Native American and African.22 Overall, the contribution of each ancestral component to the Colombian genetic makeup followed a previously described geographic pattern, where inhabitants of the inner, Andean regions (Bogota, Medellin, and Bucaramanga) had the highest European ancestry; those North and Northwest the lowest Native American ancestry (Medellin and Barranquilla); and those on the Caribbean and Pacific coasts (Barranquilla and Cali) the highest African ancestry.22,27 In this population, we highlight novel associations between gut microbiota composition and genetic ancestry, adding to the growing evidence that the host genetic background affects the composition of inner symbionts. Importantly, previous evidence on the relationship between gut microbiota and the host genetic ancestry has been based on self-perceived ethnicity. In this context, we are the first to fine-map the levels of admixture using genetic markers. Moreover, our robust statistical analyses allowed splitting apart the effect of the host genetic ancestry and confounding factors intimately related to it, such as diet, lifestyle, and geography (i.e., the cities where participants originated). Some of the microbes we found associated to ancestry have also been detected in populations with different genetic backgrounds. Christensenellaceae and Mollicutes RF39 were found increased in African Americans, Caucasians, and Hispanics in both the American Gut Project and the Human Microbiome Project.19 We found them associated to the European component in our mestizo population. Likewise, Coprococcus, Blautia, and Bacteroides contributed with ethnic-driven dissimilarities in a European cohort.20 We found Coprococcus and Blautia associated to the European ancestry, while one OTU of Bacteroides was associated to European and another OTU to Native American (Figure 3).
Concerning ancestry-health associations, multiethnic surveys demonstrated that the origin of human populations contributed to the genetic predisposition to CMS. We found that Colombians with higher Native American and African ancestries had higher fasting insulin levels, BMI and CMS risk, independent of potential non-genetic confounders, including sex, age, the participants’ city of origin, diet, and lifestyle. Studies in Mexican-Americans,28 US Native Americans,29 and Alaska Natives30 have shown a higher risk of type 2 diabetes in individuals of Amerindian ancestry. Likewise, Africans, African Americans, and genetically admixed individuals with high African ancestry have a higher risk of cardiometabolic disease.31–34
In addition to the evidence associating the host genetic ancestry and cardiometabolic health, we found that gut microbes were associated to CMS risk, as assessed by the CMS risk scale. We found that the microbiota composition was a better explanatory variable of the risk of cardiometabolic disease than the host genetic ancestry, and informed about abnormal body fat distribution, elevated blood pressure, and coronary heart disease risk. Further, we uncovered a list of 20 OTUs that were associated to CMS. This included microbes more abundant in patients with atherosclerotic disease, such as Escherichia coli and Atopobium;12 in type 2 diabetic patients, such as Clostridiaceae SMB53;35 and in unhealthy obese individuals, such as E. coli, Gemmiger formicilis, Clostridiaceae SMB53, and Haemophilus parainfluenzae.36,37 On the other hand, microbes such as Akkermansia muciniphila, Oscillospira, Methanobrevibacter, and Christensenellaceae were associated to healthy cardiometabolic states.15,37–40
Our study had several strengths, including a thorough sampling in various cities and an in-depth characterization of the studied cohort in terms of genetic ancestry (fine-mapped with genetic markers, in opposition to self-perceived ancestry), gut microbiota, cardiometabolic outcomes and non-genetic factors related to diet and lifestyle that allowed adjusting statistical models for potential confounding. However, we were limited by the relatively small sample size and by the fact that this was a cross-sectional study, so that we cannot distinguish cause and effect.
Collectively, our results indicate that two important features of human biology, the genome, and the microbiome, contribute to shaping the risk of cardiometabolic disease. Our study and others suggest that the gut microbiota is partly under the host genetic control,41–44 which might contribute to pervasive inter-population differences in the composition of this microbial community.17,18 However, our evidence indicated that gut microbiota could be a more important factor explaining the variance in CMS risk than genetic ancestry, suggesting routes to disease risk reduction via modulation of the microbial community.
Materials and methods
Study population
Between July and November 2014, we enrolled 441 mestizo adult men and women, living in the cities of Bogota, Medellin, Cali, Barranquilla, and Bucaramanga (Colombia, South America) (min-max distances between cities: 238–861 km). The national census indicates that these cities contribute about 30% of the Colombian population. Participants were enrolled in similar proportions according to the city of residence (19% Bogota, 22% Medellin, 20% Cali, 20% Barranquilla and 18% Bucaramanga), BMI (31% lean, 39% overweight and 30% obese), sex (48% male, 52% female), and age range (47% 18–40 years, and 53% 41–62 years). We excluded underweight participants (i.e., BMI <18.5 kg/m2), pregnant women, individuals who had consumed antibiotics or antiparasitics in the three months prior to enrollment, and individuals diagnosed with neurodegenerative diseases, current or recent cancer (<1 year), and gastrointestinal diseases (Crohn’s disease, ulcerative colitis, short bowel syndrome, diverticulosis or celiac disease).
The study followed the principles of the Declaration of Helsinki and had minimal risk according to the Colombian Ministry of Health (Resolution 8430 of 1993). Written informed consent was obtained from all the participants prior to the beginning of the study. The study was approved by the Bioethics Committee of SIU–University of Antioquia (act 14–24-588 dated May 28, 2014). A detailed description of the acquisition of these data can be found elsewhere.45
Genotyping of ancestry informative markers (AIMs)
The ancestral genetic composition of participants was assessed through a panel of 40 AIMs located on most chromosomes, chosen for having strong differences in allele frequency between European, Native American and African populations, and to be unlinked (Table S1). The selected AIMs have been previously used.27,46–48 Of these, 34 corresponded to insertion/deletion variants (InDel) and six to single nucleotide polymorphisms (SNP). Primers and PCR conditions followed specific protocols for each AIM. For InDels, genotypes were resolved with 1.5–2.0% agarose gel electrophoresis if the variant was >10 bp, otherwise with capillary electrophoresis in an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). SNPs were genotyped with PCR-RFLP and resolved with 2.5–3.0% agarose gel electrophoresis.
Analysis of the host genetic ancestry
The host genetic ancestry was analyzed as follows: genotypes for each AIM served to calculate the observed and expected allelic and genotypic frequencies, to test the Hardy–Weinberg equilibrium with an exact test,49 and to estimate the overall population structure (Fst) using the Weir and Cockerham estimator.50 The standard error and 95% confidence intervals of this estimator were calculated by jackknifing and bootstrapping over loci, respectively. Population-genetic analyses were performed with GenePop51 and FSTAT 2.9.3.52 Afterwards, we performed isolation by distance tests by correlating the genetic (Fst/(1-Fst)) and (log-transformed) geographic distance matrices using a Mantel test, as implemented in ecodist,53 with 10,000 permutations and 10,000 bootstrap iterations for calculating confidence intervals.
Next, a hidden Markov model approach was used to infer the individual genetic contributions of European, Native American and African ancestries using ADMIXMAP 3.7.54 This method models individual admixture using the genotypic information of all individuals and AIMs, the AIM’s physical position on the chromosome and the frequency of the largest allele in the parental populations. Allelic frequencies in the parental populations were previously reported for Europeans (Spain, Germany, England, Ireland), Native Americans (Maya, Pima, and Puebla) and Africans (Nigeria, Sierra Leone, Central African Republic, African-American, and Afro-Caribbean).55,56 The parameters used for running ADMIXMAP were: 40 loci, 440 diploid individuals, 250,000 iterations with a burn-in of 10,000 iterations, and a model of three populations.
The proportions of European, Native American, and African ancestries were compared across the five cities from which our participants originated, BMI (lean, overweight, obese), sex (male, female) and age range (18–40, 41–62 years) with ANOVA, after verifying homoscedasticity with the Fligner–Killeen test. Where necessary, data were transformed with natural logarithm for unbounded variables, or arcsine square root for proportions. We also performed a robust principal components analysis (PCA) for compositional data with the individual proportions of the three genetic ancestries using robCompositions.57 For this, the compositional dataset was transformed using the isometric log ratio, and a PCA was afterwards performed. The PC1 and PC2 components were compared across cities, BMI, sex and age range using ANOVA.
Characterization of the gut microbiota
Detailed laboratory and bioinformatic procedures can be found elsewhere.45 Briefly, each participant collected a fecal sample in a hermetically sealed, sterile receptacle provided by the research team. Samples were immediately refrigerated in household freezers and brought to a collection center within 12 h. Samples were stored on dry ice and sent to a central laboratory via next-day delivery. These procedures were standardized for all cities. Upon receipt, samples were kept at −80°C until DNA extraction. Total microbial DNA was extracted using the QIAamp DNA Stool Mini Kit (Qiagen; cat. No. 51504). Samples were randomized and the V4 region of the 16S rRNA gene was amplified with primers F515 and R806, sequenced with Illumina MiSeq v2, and processed as previously described.45
The gut microbiota was analyzed at the whole community level using principal coordinates analysis (PCoA) based on weighted UniFrac distances. These distances were computed on rarefied sequence counts (3667 reads/sample; mean number of reads/sample = 33,505; median = 28,572; range = 3667–102,660) with GuniFrac,58 and compared across cities, BMI, sex, and age range with permutational multivariate analysis of variance using distance matrices (PERMANOVA), as implemented in Vegan.59 Additional rarefaction was performed at 10,000 reads/sample. Microbiota analyses were performed at the OTU level. For this, we grouped sequences at 97% identity using the average neighbor algorithm60 and extracted OTUs that had median relative abundances ≥0.001% across all samples. The latter procedure guaranteed that the majority of sequences was analyzed (~83% of total reads) and minimized the impact of sequencing artifacts. OTUs were classified by consensus according to the Greengenes 13_8_99 taxonomy.61
CMS risk, diet, and lifestyle
We measured several variables that might interact with both gut microbiota and the host genetic ancestry. These included CMS risk factors (blood chemistry, blood pressure, and adiposity), diet intake (macronutrients and fiber) and lifestyle (physical activity, smoking status, medicament consumption). Detailed information about the measurement of these variables is presented elsewhere.36 Briefly, blood biochemical variables, including HDL, LDL, VLDL, total cholesterol, triglycerides, fasting glucose, HbA1c, fasting insulin, and hs-CRP, were measured using standard techniques routinely used in a clinical laboratory (Dinámica IPS, Medellin, Colombia). Blood insulin served to calculate the insulin resistance index using the homeostasis model assessment (HOMA-IR). The systolic and diastolic blood pressures were measured in mm Hg with a Rossmax AF701f digital tensiometer (Berneck, Switzerland). Adiposity was assessed through BMI (weight (kg)/height squared (m2)), waist circumference (cm) and percentage body fat (calculated with the thicknesses of four skinfolds: biceps, triceps, subscapular, and ileocrestal).
To assess the CMS risk, we constructed a summary scale, the CMS risk scale, by summing Z-scores of waist circumference, fasting insulin, triglycerides, diastolic blood pressure and hs-CRP (Z= [x-µ]/δ, where µ is the population mean and δ is the standard deviation of the population). Variables were log-transformed to adjust to a normal distribution before obtaining Z-scores. These variables were chosen because they informed about general features of CMS: abnormal body fat distribution, insulin resistance, atherogenic dyslipidemia, elevated blood pressure, and pro-inflammatory state, respectively.2 In addition, we calculated the Framingham coronary heart disease score using sex, age, diabetes status, smoking status, blood pressure, HDL and total cholesterol as predictor variables.26 Since the Framingham score did not consider individuals younger than 30 years, these were given the lowest age score (−1).
Daily intakes of macronutrients (g/day of carbohydrates, protein, and fat) and dietary fiber (g/day) were estimated with 24-h dietary recall interviews.62 Dietary recalls were randomly distributed in the different days of the week. Trained interviewers used validated forms, food models, geometric figures and full-size pictures to assess portion sizes and improve accuracy. Ten percent of the participants were interviewed a second time on a different day of the week, with a minimum of two days between consecutive evaluations, to estimate intra-individual variability. Dietary intake was obtained for each participant using the EVINDI 4.0 and PC-SIDE 1.0 software.
Levels of physical activity (number of metabolic equivalents per minute per week: MET/min/week) were assessed with the short form of the International Physical Activity Questionnaire.63 Smoking and medicament consumption were self-reported in specific questionnaires. For the latter, we considered all drugs taken by participants on a regular basis during the three months prior to enrollment, to the exception of over-the-counter vitamin and mineral supplements, phytotherapeutics and contraceptives. We discriminated drugs with potential effects on gut microbiota, such as anti-hypertensives, metformin, and proton-pump inhibitors. All measurements and questionnaires were performed by trained personnel.
Associations of the host genetic ancestry, gut microbiota, and CMS risk
The direct association between the host genetic ancestry and gut microbiota composition was assessed with Procrustes analyses.64 These were performed to examine, on one hand, the correlation between the weighted UniFrac distance matrix and the matrix of individual proportions of European, Native American and African. On the other hand, the correlation between the first two PCoA axes of weighted UniFrac distances (microbiota analysis) and the PCA components of the ancestry analysis. In both cases, microbiota matrices were set as targets and ancestry matrices as those to be rotated and scaled. Statistical significance was determined using 10,000 permutations.
To explore associations between the host genetic ancestry and gut microbiota composition, we fitted GLMs with negative binomial error distribution using rarefied sequence counts as dependent variable, ancestry PC1 and PC2 as explanatory variables, and the participants’ city of origin, sex, age range, diet intake (macronutrients and fiber), physical activity levels and the CMS risk scale, as covariates. Scaled regression coefficients were obtained and p-values were adjusted using the false discovery rate method using q-value.65
We next investigated associations of the host genetic ancestry and gut microbiota composition with cardiometabolic health. For this, we divided the CMS risk scale by tertiles (low, intermediate and high levels) and tested differences among them for each variable using ANOVA and chi-square tests. Where necessary, variables were transformed as described above.
Afterwards, we fitted GLMs to determine the effects of host genetic ancestry (PCA components) and gut microbiota (first two PCoA axes of weighted UniFrac) on the CMS risk scale. PCA components and PCoA axes were included in the same models. These models were adjusted by the participants’ city of origin, sex, age range, diet intake (macronutrients and fiber), levels of physical activity, smoking status and medicament consumption. GLMs served to examine the contributions of the host genetic ancestry, gut microbiota and their interaction in explaining CMS risk. For this, we constructed a basic model including the city of origin, sex, age, diet, and lifestyle. We then evaluated alternative models including the host genetic ancestry (PCA components), gut microbiota (first two PCoA axes of weighted UniFrac) and the host genetic ancestry × gut microbiota interaction. The first two alternative models were each compared against the basic model, the latter model was compared against the best preceding model. We obtained log-likelihoods of all models and evaluated their changes with likelihood ratio tests. The model selection was based on AIC. Models were fitted for the CMS risk scale, for individual components of CMS and for the Framingham coronary heart disease score.
Finally, to determine the associations between CMS risk scale and microbiota composition, we fitted GLMs in which the CMS risk scale was set as the dependent variable and rarefied OTU counts as explanatory variables. These models were adjusted by host ancestry (PC1 and PC2), the participants’ city of origin, sex, age range, diet intake (carbohydrate, protein, fat, and fiber), levels of physical activity, smoking status and medicament consumption. Scaled regression coefficients, p-values and q-values were obtained.
Funding Statement
This study was funded by Colciencias under grant 111565741349; Grupo Empresarial Nutresa, Universidad de Antioquia, Dinámica IPS, and EPS SURA. The funders of this work have not had any role in the study design; in the collection, analysis or interpretation of the data; in the writing of the report; and in the decision to submit the paper for publication.
Acknowledgments
We thank the participants who took part in the study. We are indebted to Roberto A. Jiménez and Luisa F. Mesa for their contributions during this project, and to GENMOL and Vidarium staff for their contributions during field and laboratory work. We are grateful to EPS SURA and Dinámica IPS for their support throughout the study, to the Centro de Computación Científica Apolo at Universidad EAFIT for hosting supercomputing resources (http://www.eafit.edu.co/apolo), and to the University of Michigan Medical School Host Microbiome Initiative for sequencing support. Some authors of this work collaborate through the Microbiome & Health Network.
Disclosure of potential conflicts of interest
We disclose that, while engaged in this project, JdlC-Z, EPV-M, and JSE were employed by a food company (Grupo Empresarial Nutresa). SJG-C, ELO-V, WR, and GB had nothing to disclose.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Institute for Health Metrics and Evaluation (IHME) . GBD Compare Data Visualization. 2016;
- 2.Alberti KGMM, Zimmet P, Shaw J.. Metabolic syndrome - A new world-wide definition. A consensus statement from the International Diabetes Federation. Diabet Med. 2006;23:469–480. doi: 10.1111/dme.2006.23.issue-5. [DOI] [PubMed] [Google Scholar]
- 3.Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256:51–54. doi: 10.1001/jama.1986.03380010055024. [DOI] [PubMed] [Google Scholar]
- 4.Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–1046. [DOI] [PubMed] [Google Scholar]
- 5.Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fall T, Ingelsson E. Genome-wide association studies of obesity and metabolic syndrome. Mol Cell Endocrinol. 2014;382:740–757. doi: 10.1016/j.mce.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 7.Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, Horikoshi M, Johnson AD, Ng MCY, Prokopenko I, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–244. doi: 10.1038/ng.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seldin MF, Pasaniuc B, Price AL. New approaches to disease mapping in admixed populations. Nat Rev Genet. 2011;12:523–528. doi: 10.1038/nrg3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pritchard JK, Rosenberg NA. Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet. 1999;65:220–228. doi: 10.1086/302449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Justice AE, Winkler TW, Feitosa MF, Graff M, Fisher VA, Young K, Barata L, Deng X, Czajkowski J, Hadley D, et al. Genome-wide meta-analysis of 241,258 adults accounting for smoking behaviour identifies novel loci for obesity traits. Nat Commun. 2017;8:14977. doi: 10.1038/ncomms14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The Human Microbiome Project Consortium, Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, Creasy HH, Earl AM, FitzGerald MG, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jie Z, Xia H, Zhong S-L, Feng Q, Li S, Liang S, Zhong H, Liu Z, Gao Y, Zhao H, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. 2017;8:845. doi: 10.1038/s41467-017-00900-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J, Bäckhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99–103. doi: 10.1038/nature12184. [DOI] [PubMed] [Google Scholar]
- 14.Walters WA, Xu Z, Knight R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 2014;588:4223–4233. doi: 10.1016/j.febslet.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodrich JK, Davenport ER, Clark AG, Ley RE. The relationship between the human genome and microbiome comes into view. Annu Rev Genet. 2017;51:413–433. doi: 10.1146/annurev-genet-110711-155532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mancabelli L, Milani C, Lugli GA, Turroni F, Ferrario C, van Sinderen D, Ventura M. Meta-analysis of the human gut microbiome from urbanized and pre-agricultural populations. Environ Microbiol. 2017;19:1379–1390. doi: 10.1111/1462-2920.13842. [DOI] [PubMed] [Google Scholar]
- 19.Brooks AW, Priya S, Blekhman R, Bordenstein SR. Gut microbiota diversity across ethnicities in the United States. PLoS Biol. 2018;16:e2006842. doi: 10.1371/journal.pbio.2006842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, Tremaroli V, Bakker GJ, Attaye I, Pinto-Sietsma SJ, et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat Med. 2018;24:1526–1531. doi: 10.1038/s41591-018-0160-1. [DOI] [PubMed] [Google Scholar]
- 21.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–215. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 22.Ruiz-Linares A, Adhikari K, Acuña-Alonzo V, Quinto-Sanchez M, Jaramillo C, Arias W, Fuentes M, Pizarro M, Everardo P, de Avila F, et al. Admixture in Latin America: geographic Structure, Phenotypic Diversity and Self-Perception of Ancestry Based on 7,342 Individuals. PLoS Genet. 2014;10:e1004572. doi: 10.1371/journal.pgen.1004541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonilla C, Shriver MD, Parra EJ, Jones A, Fernández JR. Ancestral proportions and their association with skin pigmentation and bone mineral density in Puerto Rican women from New York city. Hum Genet. 2004;115:57–68. doi: 10.1007/s00439-004-1125-7. [DOI] [PubMed] [Google Scholar]
- 24.Santos NPC, Ribeiro-Rodrigues EM, ÂKC R-D-S, Pereira R, Gusmão L, Amorim A, Guerreiro JF, Zago MA, Matte C, Hutz MH, et al. Assessing individual interethnic admixture and population substructure using a 48-insertion-deletion (INSEL) ancestry-informative marker (AIM) panel. Hum Mutat. 2010;31:184–190. doi: 10.1002/humu.21306. [DOI] [PubMed] [Google Scholar]
- 25.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.CIR.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 27.Cardona-Castro N, Cortés E, Beltrán C, Romero M, Badel-Mogollón JE, Bedoya G. Human Genetic Ancestral Composition Correlates with the Origin of Mycobacterium leprae Strains in a Leprosy Endemic Population. PLoS Negl Trop Dis. 2015;9(9):e0004045. doi: 10.1371/journal.pntd.0004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu HQ, Li Q, Lu Y, Hanis CL, Fisher-Hoch SP, Mccormick JB. Ancestral effect on HOMA-IR levels quantitated in an american population of Mexican origin. Diabetes Care. 2012;35:2591–2593. doi: 10.2337/dc12-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenlund KJ, Valdez R, Casper ML, Rith-Najarian S, Croft JB. Prevalence and correlates of the insulin resistance syndrome among Native Americans: the Inter-Tribal Heart Project. Diabetes Care. 1999;22:441–447. doi: 10.2337/diacare.22.3.441. [DOI] [PubMed] [Google Scholar]
- 30.Galloway JM. Cardiovascular health among American Indians and Alaska natives - Successes, challenges, and potentials. Am J Prev Med. 2005;29:11–17. doi: 10.1016/j.amepre.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Brancati FL, Kao WH, Folsom AR, Watson RL, Szklo M. Incident type 2 diabetes mellitus in African American and white adults: the Atherosclerosis Risk in Communities Study. JAMA. 2000;283:2253–2259. doi: 10.1001/jama.283.17.2253. [DOI] [PubMed] [Google Scholar]
- 32.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 33.Cheng CY, Reich D, Haiman CA, Tandon A, Patterson N, Elizabeth S, Akylbekova EL, Brancati FL, Coresh J, Boerwinkle E, et al. African ancestry and its correlation to type 2 diabetes in african americans: a genetic admixture analysis in three U.S. population cohorts. PLoS One. 2012;7:e32840. doi: 10.1371/journal.pone.0032840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chande AT, Rowell J, Rishishwar L, Conley AB, Norris ET, Valderrama-Aguirre A, Medina-Rivas MA, Jordan IK. Influence of genetic ancestry and socioeconomic status on type 2 diabetes in the diverse Colombian populations of Chocó and Antioquia. Sci Rep. 2017;7:17127. doi: 10.1038/s41598-017-17380-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Napolitano A, Miller S, Nicholls AW, Baker D, Van Horn S, Thomas E, Rajpal D, Spivak A, Brown JR, Nunez DJ. Novel gut-based pharmacology of metformin in patients with type 2 diabetes mellitus. PLoS One. 2014;9:e100778. doi: 10.1371/journal.pone.0100778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de la Cuesta-Zuluaga J, Corrales-Agudelo V, Carmona JA, Abad JM, Escobar JS. Body size phenotypes comprehensively assess cardiometabolic risk and refine the association between obesity and gut microbiota. Int J Obes. 2018;42:424–432. doi: 10.1038/ijo.2017.281. [DOI] [PubMed] [Google Scholar]
- 37.de la Cuesta-Zuluaga J, Mueller NT, Álvarez-Quintero R, Velásquez-Mejía EP, Sierra JA, Corrales-Agudelo V, Carmona JA, Abad JM, Escobar JS. Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometabolic disease risk factors. Nutrients [Internet]. 2019;11:51. Available from https://www.mdpi.com/2072-6643/11/1/51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Million M, Angelakis E, Maraninchi M, Henry M, Giorgi R, Valero R, Vialettes B, Raoult D. Correlation between body mass index and gut concentrations of Lactobacillus reuteri, Bifidobacterium animalis, Methanobrevibacter smithii and Escherichia coli. Int J Obes. 2013;37:1460–1466. doi: 10.1038/ijo.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konikoff T, Gophna U. Oscillospira: a central, enigmatic component of the human gut microbiota. Trends Microbiol. 2016;24:523–524. doi: 10.1016/j.tim.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 41.Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci U S A. 2010;107:18933–18938. doi: 10.1073/pnas.0910097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, Guttman DS, Griffiths A, Panaccione R, Otley A, et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet. 2016;48:1413–1417. doi: 10.1038/ng.3693. [DOI] [PubMed] [Google Scholar]
- 43.Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, Deelen P, Vatanen T, Schirmer M, Smeekens SP, et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48:1407–1412. doi: 10.1038/ng.3663. [DOI] [PubMed] [Google Scholar]
- 44.Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, Hov JR, Degenhardt F, Heinsen F-A, Rühlemann MC, Szymczak S, et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet. 2017;48:1396–1406. doi: 10.1038/ng.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de la Cuesta-Zuluaga J, Corrales-Agudelo V, Velásquez-Mejía EP, Carmona JA, Abad JM, Escobar JS. Gut microbiota is associated with obesity and cardiometabolic disease in a population in the midst of Westernization. Sci Rep. 2018;8:11356. doi: 10.1038/s41598-018-29687-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terreros MC, Alfonso-Sánchez MA, Novick GE, Luis JR, Lacau H, Lowery RK, Regueiro M, Herrera RJ. Insights on human evolution: an analysis of Alu insertion polymorphisms. J Hum Genet. 2009;54:603–611. doi: 10.1038/jhg.2009.86. [DOI] [PubMed] [Google Scholar]
- 47.Valencia DM, Naranjo CA, Parra MV, Caro MA, Valencia AV, Jaramillo CJ, Bedoya G. Association and interaction of AGT, AGTR1, ACE, ADRB2, DRD1, ADD1, ADD2, ATP2B1, TBXA2R and PTGS2 genes on the risk of hypertension in Antioquian population. Biomedica. 2013;33:598–614. [DOI] [PubMed] [Google Scholar]
- 48.Muñoz AM, Velásquez CM, Bedoya G. Cardio-metabolic parameters are associated with genetic admixture estimates in a pediatric population from Colombia. BMC Genet. 2016;17:93. doi: 10.1186/s12863-016-0402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louis EJ, Dempster ER. An exact test for Hardy-Weinberg and multiple alleles. Biometrics. 1987;43:805–811. doi: 10.2307/2531534. [DOI] [PubMed] [Google Scholar]
- 50.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution (N Y). 1984;38:1358–1370. [DOI] [PubMed] [Google Scholar]
- 51.Rousset F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 52.Goudet J. FSTAT (Version 1.2): A Computer Program to Calculate F-statistics. J Hered. 1995;86:485–486. doi: 10.1093/oxfordjournals.jhered.a111627. [DOI] [Google Scholar]
- 53.Goslee SC, Urban DL. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw. 2007;22:1–19. doi: 10.18637/jss.v022.i07. [DOI] [Google Scholar]
- 54.Hoggart CJ, Shriver MD, Kittles RA, Clayton DG, McKeigue PM. Design and analysis of admixture mapping studies. Am J Hum Genet. 2004;74:965–978. doi: 10.1086/382659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shriver MD, Parra EJ, Dios S, Bonilla C, Norton H, Jovel C, Pfaff C, Jones C, Massac A, Cameron N, et al. Skin pigmentation, biogeographical ancestry and admixture mapping. Hum Genet. 2003;112:387–399. [DOI] [PubMed] [Google Scholar]
- 56.Parra EJ, Marcini A, Akey J, Martinson J, Batzer MA, Cooper R, Forrester T, Allison DB, Deka R, Ferrell RE, et al. Estimating African American admixture proportions by use of population-specific alleles. Am J Hum Genet. 1998;63:1839–1851. doi: 10.1086/302148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Templ M, Hron K, Filzmoser P. robCompositions: an R-package for robust statistical analysis of compositional data. In: Pawlowsky-Glahn V, Buccianti A, editors. Compositional Data Analysis. Theory and Applications. Chichester (UK): John Wiley & Sons; 2011. p. 341–355. [Google Scholar]
- 58.Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, Wu GD, Collman RG, Bushman FD, Li H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics. 2012;28:2106–2113. doi: 10.1093/bioinformatics/bts342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. Vegan: community ecology package. R Packag. version 2. 3-1. 2015;264.
- 60.Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol. 2011;77:3219–3226. doi: 10.1128/AEM.02810-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. Isme J. 2012;6:608–610. doi: 10.1038/ismej.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson RK. Dietary intake–how do we measure what people are really eating? Obes Res. 2002;10:63S–68S. doi: 10.1038/oby.2002.123. [DOI] [PubMed] [Google Scholar]
- 63.Craig CL, Marshall AL, Sjostrom M, Bauman AE, Booth ML, Ainsworth BE, Pratt M, Ekelund U, Yngve A, Sallis JF, et al. International physical activity questionnaire: 12-country reliability and validity. Med Sci Sports Exerc. 2003;35:1381–1395. doi: 10.1249/01.MSS.0000078924.61453.FB. [DOI] [PubMed] [Google Scholar]
- 64.Peres-Neto PR, Jackson DA. How well do multivariate data sets match? The advantages of a procrustean superimposition approach over the Mantel test. Oecologia. 2001;129:169–178. doi: 10.1007/s004420100720. [DOI] [PubMed] [Google Scholar]
- 65.Storey JD, Bass AJ, Dabney A, Robinson D. Q-value estimation for false discovery rate control [Internet]. 2015; [cited 2018 Aug 17]. http://github.com/jdstorey/qvalue
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Storey JD, Bass AJ, Dabney A, Robinson D. Q-value estimation for false discovery rate control [Internet]. 2015; [cited 2018 Aug 17]. http://github.com/jdstorey/qvalue