

ABSTRACT
Despite continuous exposure to trillions of microbes, the intestinal immune system protects the mucosa by balancing barrier protection, tolerance, and immunity. As both sentinel and effector, the mucosal innate immune system plays a central role in coordinating these responses. By integrating signals from the intestinal microbiota, mononuclear phagocytes (MNPs) serve as a critical link in regulating effector functions of group 3 innate lymphoid cells (ILC3s). Our recent work identified the role for MNP production of the IBD-linked protein TNF-like ligand 1A (TL1A) in modulating microbial regulation of ILC3 barrier immunity. These findings highlight a broader role for ILC3s in local control of T cell immunity and their potential role in the pathogenesis and treatment of inflammatory disease.
KEYWORDS: Innate lymphoid cells, inflammatory bowel diseases, TL1A, OX40L
Introduction
Group 3 innate lymphoid cells (ILC3s) have emerged as a central regulator in barrier immunity .1,2 In response to inflammatory stimuli, including IL-1b and IL-23, ILC3s produce the cytokine IL-22 which promotes epithelial cell proliferation and healing (Figure 1) .3 Pleiotropic effects of IL-22 reinforce barrier immunity by inducing anti-microbial peptide production and maintaining microbial homeostasis. In addition, emerging data has identified a broader role for ILC3s in modulating mucosal immunity. The mechanisms guiding this response, including the impact of gut microbiota and IBD-associated genetic pathways, are an active area of research with important diagnostic and therapeutic potential.
Figure 1.
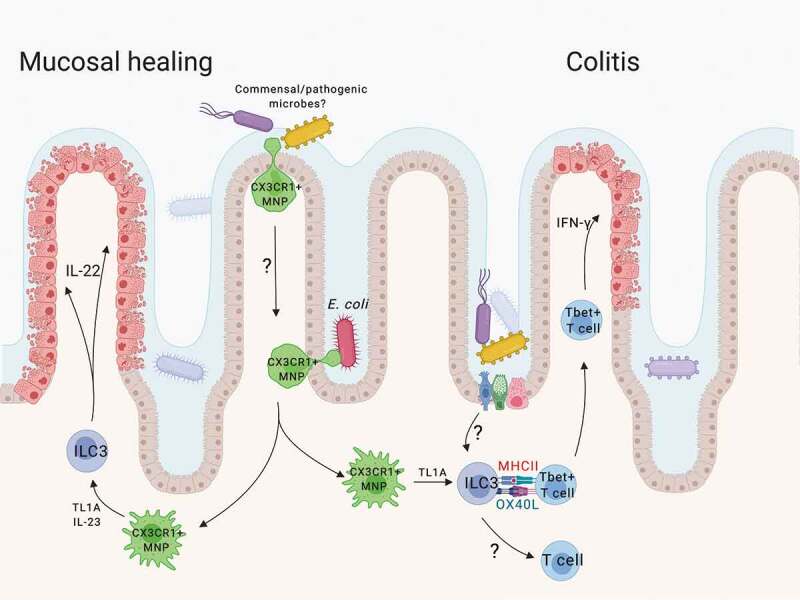
Microbial induction of TL1A regulates ILC3 barrier immunity. Mucosal-associated microbiota are sensed by intestinal CX3CR1+ MNPs and induce expression of cytokines and TL1A. TL1A enhances ILC3 production of effector cytokines, including IL-22, promoting mucosal healing associated with acute colitis. TL1A also induces MHCII+ ILC3 expression of the co-stimulatory molecule OX40L which can support mucosal inflammatory T cells. The broader impact of ILC3 expression of OX40L in regulating microbe-specific mucosal T cell immunity remains to be determined.
Intestinal mononuclear phagocytes (MNPs) act as sentinels for the intestinal immune system. CX3CR1+ MNPs can form transepithelial dendrites and sample the intestinal microbiota in an active process regulated by the microbiota .4 In the setting of dysbiosis, these largely tissue-resident MNPs can upregulate CCR7 and migrate to lymphatics carrying antigens from non-invasive luminal microbes .5 During inflammation, however, these MNPs expand and co-localize with ILC3s in the tissue .6–8 Following infection with Citrobacter rodentium, CX3CR1+ MNPs are required for mucosal protection via ILC3 production of IL-22 .7,8 Although genetic deletion models in mice revealed that CD103+ dendritic cells (DCs) were dispensable, 9 a coordinated response with conventional DCs may be required for protective ILC3 immunity .10 Thus, the MNP-ILC3 interface creates a functional unit linking microbial signals with ILC3 effector functions, but the mechanisms regulating their interactions are not clearly defined. In our recent paper, we defined a key role for MNP-derived TL1A in regulating ILC3 mucosal immunity. This addendum aims to contextualize the key mechanistic findings of this study in their contribution to inflammatory bowel disease (IBD) and mucosal immunology.
TL1A and innate lymphoid cells in IBD pathogenesis
To date, genome-wide association studies have linked over 200 genes with IBD, and many of these genes are implicated in immunoregulatory functions .11 One of the first and strongest IBD-linked polymorphisms discovered occurs in the Tumor Necrosis Factor Superfamily member 15 (TNFSF15) locus, which shares remarkable homology with TNFA .12 TNFSF15 variants span ethnic cohorts and are associated with more aggressive IBD. TNFSF15 encodes for the protein TNF-like ligand 1A (TL1A). Although the mechanism of how TL1A contributes to IBD pathogenesis may be pleiotropic, its function has been linked with intestinal immunomodulation. Previous reports have shown a role for TL1A overexpression in exacerbating T cell- and ILC2-dependent disease, as well as fibrosis ;13–15 however, recent data using mouse models with genetic deficiencies in TL1A or its monogamous receptor, called death receptor 3 (DR3), have revealed a potential protective role for TL1A during acute colitis .16 This duality could reflect multiple functions of TL1A under homeostatic or inflammatory conditions, which we reasoned could be explained by additional cellular effectors downstream of TL1A.
Indeed, our previous work, as well as others, identified TL1A as a potentiator of ILC3 cytokine production in vitro ;7,17 however, the role for TL1A in regulating ILC3 function in vivo was not known. To address this question, we generated a novel mouse model with an ILC3-specific deletion of DR3 .18 While deletion of DR3 did not impact intestinal ILC3 numbers or subsets during homeostasis, our results show that DR3-deficient ILC3s produce less IL-22 and are unable to reduce the severity of chemical or infectious colitis. This finding offers critical additional insight into the function of TL1A during acute colitis. Moreover, the protective role for TL1A in acute colitis offers a potential explanation for the strong evolutionary conservation of this genetic polymorphism which, in the context of either chronic inflammation or additional genetic risk, could negatively impact disease. Coupling of human tissue immunophenotyping with genotypic analysis will help offer critical insight into the impact of IBD-linked genetics on the contribution of ILC3 immunity.
MNP production of TL1A links IBD-associated gut microbes to ILC immunity
Previous work by our laboratory and others identified CX3CR1+ MNPs as critical regulators of ILC3 barrier immunity .7,8 However, the exact mechanisms of how CX3CR1+ MNPs regulate ILC3 function are not well defined. Our recent publication identified CX3CR1+ MNPs as the primary producers of intestinal TL1A in both mice and humans ;18 furthermore, IBD patients with active intestinal inflammation had higher CX3CR1+ MNP TL1A production than healthy controls, but the factors regulating this induction were unknown. While previous work identified bacteria and immune complexes as robust inducers of TL1A in vitro, 19 our in vivo mouse models identified gut microbes, and in particular adherent, mucosal-associated microbes, as critical for the induction of MNP-derived TL1A.
Using mice with a specific deletion of TL1A in CD11c+ CX3CR1+ MNPs, we demonstrated a critical requirement for MNP production of TL1A in regulating ILC3s in vivo. The mechanisms guiding MNP-ILC3 cellular interactions in the intestines require further study and there are several possibilities to consider. First, in contrast to the structure of the lymph nodes, which maximize antigen presentation to a variety of stochastically arranged T cell receptors, the mechanism by which MNPs and ILC3s interact is less clear. A recent study demonstrated that constitutive production of CXCL16 by MNPs in the small intestine functionally recruit CXCR6+ NKp46+ ILC3s to the lamina propria, enabling rapid and productive interactions. Likewise, TL1A signaling induces ILC3 production of GM-CSF, as well as other chemokines, which may be critical in promoting these MNP-ILC3 interactions. Second, microbial regulation may offer a mechanism for local organization of this response. Although mucosal adherence and MYD88 are required for MNP production of TL1A, it is not clear yet which types and/or strains of bacteria are most efficient in this process and whether this regulation will be different in the small intestine compared to the colon. A recent study revealed novel antigen access by CX3CR1+ MNPs in the colon through goblet cell-associated passages .20 The role for these and other points of regulated access to the mucosal-associated microbiota may impact these cellular interactions. Finally, the requirement for direct interaction between MNPs and ILC3s needs to be assessed. Membrane bound TL1A can be cleaved by the TNFα converting enzyme (TACE) to produce soluble TL1A. While recombinant soluble TL1A is sufficient to activate ILC3s in vitro, the role for TACE and direct interaction of MNPs and ILC3s in vivo needs to be assessed.
TL1A tunes IL-23 signaling in ILC3s
While TL1A stimulation alone can induce IL-22 production by ILC3s, our recent paper shows that TL1A synergizes with IL-23 stimulation to exponentially increase IL-22 production .18 In addition to TNFSF15, polymorphisms in IL23R are highly associated with IBD .21 The functional synergy in ILC3s may reflect the convergence of this genetic risk. Their respective signaling pathways in ILC3s appear to be unique, as TL1A activation of IL-22 requires p38-MAPK signaling and does not induce STAT3 phosphorylation directly. Reports in human macrophages shows that TL1A synergizes with NOD2 ligand MDP through non-canonical activation of IL-1b, and subsequently works in an autocrine fashion to enhance inflammatory cytokine production .22 In ILC3s, we showed that the synergy between TL1A and IL-23 is IL-1R- and MYD88-independent, but the mechanism by which TL1A tunes IL-23-induced transcription remains undefined. Furthermore, while some target gene expression is enhanced, TL1A independently activates or antagonizes the expression of others. Technical advances in sequencing technology to assess single-cell resolution and genomic accessibility may provide insight into the transcriptional mechanisms of this regulation. A mechanistic understanding of the synergy between these two highly linked IBD pathways will help identify novel therapeutic strategies.
ILC3s as regulators of mucosal T cell immunity
Data continue to emerge illustrating a role for ILC3s in regulating local T cell immunity directly and indirectly via cytokine production. Indirectly, ILC3-derived IL-22 induces epithelial cells to secrete the acute phase reactant SAA, which promotes IL-17 production by lamina propria T cells .23 In addition, GM-CSF production by ILC3s recruits local macrophages and supports polarization of regulatory T cells .24 Directly, MNP derived IL-1b induces ILC3 production of IL-2 in the small intestine, which can support regulatory T cells .25 During IBD, ILC3 production of IL-2 is reduced, resulting in the diminished regulatory effect of ILC3s.
In addition to cytokine production, ILC3 expression of MHCII can shape antigen-specific T cell responses in the intestine .26 Although splenic ILC3s can express conventional B7 co-stimulatory molecules, intestinal ILC3s do not .27 While this lack of ‘signal 2ʹ may limit T cell responses during homeostasis, 26 our recent study showed that TL1A induced during colitis drives expression of the co-stimulatory molecule OX40L on intestinal MHCII+ ILC3s. Expanding on previous work which showed that lymphoid tissue inducer cell expression of OX40L was critical for maintaining T cell memory in secondary lymphoid tissue, 28 our recent work shows that both TL1A and OX40L ligand are required for ILC3s to support inflammatory colonic T cell responses in a transfer T cell colitis model (Figure 1) .18
This work collectively highlights two key questions that will need to be addressed in understanding the impact of ILC3 regulation of mucosal T cell immunity to gut microbes. First, does local regulation of ILC3s enable tissue-specific regulation of T cell effectors? These mucosal mechanisms described above may have less of an impact on systemic infection or vaccine immunity and subsequently may provide a portal for selective targeting of intestinal immunity. Second, how broadly do ILC3s regulate microbe-specific mucosal T cell immunity? OX40L, for example, is also required for the effective generation of regulatory T cells to suppress transfer T cell colitis29 and emerging data suggest that OX40L expression by ILC3s may provide this signal for regulatory T cells .30 Selective genetic deletion models coupled with antigen-specific T cell reagents are needed to define the potential contribution of ILC3s in this local immune regulation.
Conclusions
Our recent data identify the importance of MNP-production of TL1A in linking gut microbes to ILC3 immunity. With stereotypical changes noted in the microbiome of patients with IBD, it will be important to understand the microbial characteristics that contribute to TL1A-dependent immunity – both the protective acute response and the inflammatory chronic response. In addition, the synergy between TL1A and IL-23 highlights the importance of the MNP-ILC3 interface in IBD. Therapies designed to target this interface may enable selective mucosal regulation. Finally, the role for ILC3s and ILC3 expression of OX40L in regulating the inflammatory response in IBD still needs to be determined. Pharmacologic targeting of the MNP-ILC3 axis coupled with diagnostic genetic and microbiome stratification has the potential to guide therapeutic strategies for IBD.
Funding Statement
This work was supported in part by grants from the Crohn’s and Colitis Foundation (R.S.L), NIH 1R03DK111852 and 1R01 DK114252 (R.S.L), NIH Medical Scientist Training Program grant T32GM07739 (J.G.C.), and the Charina Foundation (R.S.L.); Crohn’s and Colitis Foundation of America [Senior Research Award]; National Institute of Diabetes and Digestive and Kidney Diseases [1R03DK111852-01A1]; National Institute of Diabetes and Digestive and Kidney Diseases [R01 DK114252]; Charina Endowment Fund.
Acknowledgments
We would like to thank the members of the Longman lab for their thoughtful feedback and scientific contribution to this work. We would like to acknowledge our colleagues Stephan Targan, David Shih, and David Withers for providing critical feedback and key resources. We would like to thank Ellen Scherl, the clinical research staff at the Jill Roberts Center for IBD, and the patients that contributed to our research.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Sonnenberg GF, Artis D.. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med. 2019;129(7):2640–2650. doi: 10.1172/JCI124617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castellanos JG, Longman RS. The balance of power: innate lymphoid cells in tissue inflammation and repair. J Clin Invest. 2019;130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. 2015;528(7583):560–564. doi: 10.1038/nature16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307(5707):254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 5.Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature. 2013;494:116–120. doi: 10.1038/nature11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, Mack M, Shpigel N, Boneca IG, Murphy KM, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 2012;37(6):1076–1090. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 7.Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, Swaminath A, Bonneau R, Scherl EJ, Littman DR. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. 2014;211(8):1571–1583. doi: 10.1084/jem.20140678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Satoh-Takayama N, Serafini N, Verrier T, Rekiki A, Renauld JC, Frankel G, Di Santo JP. The chemokine receptor CXCR6 controls the functional topography of interleukin-22 producing intestinal innate lymphoid cells. Immunity. 2014;41(5):776–788. doi: 10.1016/j.immuni.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Welty NE, Staley C, Ghilardi N, Sadowsky MJ, Igyarto BZ, Kaplan DH. Intestinal lamina propria dendritic cells maintain T cell homeostasis but do not affect commensalism. J Exp Med. 2013;210(10):2011–2024. doi: 10.1084/jem.20130728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, Wu X, Thomas SR, Lee W-L, Turkoz M, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. 2013;14(9):937–948. doi: 10.1038/ni.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L, Rice DL, Gutierrez-Achury J, Ji S-G, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49(2):256–261. doi: 10.1038/ng.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siakavellas SI, Bamias G. Tumor necrosis factor-like cytokine TL1A and its receptors DR3 and DcR3: important new factors in mucosal homeostasis and inflammation. Inflamm Bowel Dis. 2015;21(10):2441–2452. doi: 10.1097/MIB.0000000000000492. [DOI] [PubMed] [Google Scholar]
- 13.Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, Sciumè G, Richard AC, Hayes ET, Gomez-Rodriguez J, et al. The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol. 2014;7(4):958–968. doi: 10.1038/mi.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shih DQ, Zheng L, Zhang X, Zhang H, Kanazawa Y, Ichikawa R, Wallace KL, Chen J, Pothoulakis C, Koon HW, et al. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol. 2014;7(6):1492–1503. doi: 10.1038/mi.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett R, Zhang X, Koon HW, Vu M, Chang JY, Yeager N, Nguyen MA, Michelsen KS, Berel D, Pothoulakis C, et al. Constitutive TL1A expression under colitogenic conditions modulates the severity and location of gut mucosal inflammation and induces fibrostenosis. Am J Pathol. 2012;180(2):636–649. doi: 10.1016/j.ajpath.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Butto LF, Buela KA, Jia LG, Lam M, Ward JD, Pizarro TT, Cominelli F. Death receptor 3 signaling controls the balance between regulatory and effector lymphocytes in SAMP1/YitFc mice with Crohn’s disease-like ileitis. Front Immunol. 2018;9:362. doi: 10.3389/fimmu.2018.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahn YO, Weeres MA, Neulen ML, Choi J, Kang SH, Heo DS, Bergerson R, Blazar BR, Miller JS, Verneris MR. Human group3 innate lymphoid cells express DR3 and respond to TL1A with enhanced IL-22 production and IL-2-dependent proliferation. Eur J Immunol. 2015;45(8):2335–2342. doi: 10.1002/eji.201445213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castellanos JG, Woo V, Viladomiu M, Putzel G, Lima S, Diehl GE, Marderstein AR, Gandara J, Perez AR, Withers DR, et al. Microbiota-induced TNF-like ligand 1A drives group 3 innate lymphoid cell-mediated barrier protection and intestinal T cell activation during colitis. Immunity. 2018. e5;49(6):1077–1089. doi: 10.1016/j.immuni.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shih DQ, Kwan LY, Chavez V, Cohavy O, Gonsky R, Chang EY, Chang C, Elson CO, Targan SR. Microbial induction of inflammatory bowel disease associated gene TL1A (TNFSF15) in antigen presenting cells. Eur J Immunol. 2009;39(11):3239–3250. doi: 10.1002/eji.200839087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knoop KA, McDonald KG, Kulkarni DH, Newberry RD. Antibiotics promote inflammation through the translocation of native commensal colonic bacteria. Gut. 2016;65(7):1100–1109. doi: 10.1136/gutjnl-2014-309059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314(5804):1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hedl M, Abraham C. A TNFSF15 disease-risk polymorphism increases pattern-recognition receptor-induced signaling through caspase-8-induced IL-1. Proc Natl Acad Sci U S A. 2014;111(37):13451–13456. doi: 10.1073/pnas.1404178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, Lee J-Y, Ziel J, Miraldi E, Domingos A, et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell. 2016;164(1–2):324. doi: 10.1016/j.cell.2015.12.047. [DOI] [PubMed] [Google Scholar]
- 24.Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, Merad M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343:1249288. doi: 10.1126/science.1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, Putzel GG, Kabata H, Kelsen JR, Baldassano RN, et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature. 2019;568:405–409. doi: 10.1038/s41586-019-1082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, Withers DR, Hugues S, Farrar MA, Reith W, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science. 2015;348(6238):1031–1035. doi: 10.1126/science.aaa4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, Pieters J, Tacchini-Cottier F, Rolink A, Acha-Orbea H, et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc Natl Acad Sci U S A. 2014;111(35):12835–12840. doi: 10.1073/pnas.1406908111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Withers DR, Gaspal FM, Mackley EC, Marriott CL, Ross EA, Desanti GE, Roberts NA, White AJ, Flores-Langarica A, McConnell FM, et al. Cutting edge: lymphoid tissue inducer cells maintain memory CD4 T cells within secondary lymphoid tissue. J Immunol. 2012;189(5):2094–2098. doi: 10.4049/jimmunol.1201639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griseri T, Asquith M, Thompson C, Powrie F. OX40 is required for regulatory T cell-mediated control of colitis. J Exp Med. 2010;207(4):699–709. doi: 10.1084/jem.20091618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng T, Suo C, Chang J, Yang R, Li J, Cai T, Qiu J. ILC3-derived OX40L is essential for homeostasis of intestinal tregs in immunodeficient mice. Cell Mol Immunol. 2019. doi: 10.1038/s41423-019-0200-x. [DOI] [PMC free article] [PubMed] [Google Scholar]