

ABSTRACT
The gut microbial community greatly changes in early life, influencing infant health and subsequent host physiology, notably through its collective metabolism, including host–microbiota interplay of bile acid (BA) metabolism. However, little is known regarding how the development of the intestinal microbial community is associated with maturation of intestinal BA metabolism. To address this, we monitored the succession of gut bacterial community and its association with fecal BA profile in the first 3 y of ten healthy Japanese infants. The BA profiles were classified into four types, defined by high content of conjugated primary BA (Con type), unconjugated primary BA (chenodeoxycholic acid and cholic acid) (Pri type), ursodeoxycholic acid (Urs type), and deoxycholic and lithocholic acid (Sec type). Most subjects begun with Con type or Pri type profiles during lactation and eventually transited to Sec type through Urs type after the start of solid food intake. Con type and Pri type were associated with Enterobacteriaceae-dominant microbiota corresponding to the neonatal type or Bifidobacterium-dominant microbiota corresponding to lactation type, respectively. Urs type subjects were strongly associated with Ruminococcus gnavus colonization, mostly occurring between Pri type and Sec type. Sec type was associated with adult-type complex microbiota dominated by a variety of Firmicutes and Bacteroidetes species. Addressing the link of the common developmental passage of intestinal BA metabolism with infant’s health and subsequent host physiology requires further study.
KEYWORDS: Infant gut microbiota, bile acid metabolism, Bifidobacterium, Ruminococcus gnavus, Bile salt hydrolase
Introduction
The symbiotic relationship between host and gut microbes begins at birth. Although various environmental factors, such as the mode of delivery, milk feeding, antibiotic use, prebiotic and probiotic treatment, and the timing of solid food intake, influence colonization by certain species, there is a common trend in the development of intestinal microbiota in the beginning of life.1 After birth, the gut microbiota of neonatal is transiently dominated by Enterobacteriaceae and Staphylococcus.2 During lactation, the intestinal microbiota is stabilized by the domination of Bifidobacterium, solely utilizing human milk oligosaccharides or nonhuman oligosaccharide supplied as alternatives in formula milk.2,3 After the start of solid food intake, a variety of anaerobic species replace Bifidobacterium, and adult-type microbiota, dominated by the phyla Bacteroidetes and Firmicutes, is eventually established.1,4–6 The developing microbiota has a great impact on infant health and subsequent host physiology, notably through the development of the digestive tract, immune system, and metabolic homeostasis.6,7 Therefore, failure in gut microbiota development may lead to diseases such as food allergies,8,9 asthma,10 atopic dermatitis,11 and childhood obesity.12,13
Bile acids (BAs) constitute the primary component of bile. BAs are secreted into the small intestine as conjugated forms and aid the absorption of dietary lipids. While most BAs are reabsorbed in the ileum and returned to the liver, unabsorbed BAs enter into the large intestine and are, therein, deconjugated and metabolized into secondary BAs by the microbial community.14 Deconjugation is catalyzed by bile salt hydrolase (BSH) widely present among intestinal bacteria.15 Two types of enzymatic conversions are involved in the generation of secondary BAs: 7α-dehydroxylase converts cholic acid (CA) to deoxycholic acid (DCA) and chenodeoxycholic acid (CDCA) to lithocholic acid (LCA), and 7-epimerase converts CDCA to ursodeoxycholic acid (UDCA).16
In addition to their role in digestion, BAs have some hormonal functions, as ligands for a nuclear farnesoid X receptor (FXR) and a cell membrane G-protein-coupled receptor (TGR-5), which initiate a variety of signaling cascades relevant to the regulation of lipid, sugar, and energy metabolism, as well as BA synthesis feedback control.17 Furthermore, BAs are known to regulate intestinal cell proliferation by modulating signaling via the epidermal growth factor receptor (EGFR) in addition to FXR, which is critical for mucosal inflammation and barrier function.18 Due to differences in the BA molecules in the interaction mode with the receptors, BA composition, largely defined by gut microbiota, is suggested to be important for these hormonal actions.19,20 On the other hand, it is known that a high concentration of BAs, notably secondary BAs, is cytotoxic and carcinogenic, which promote colon or liver cancer.16,21 It is also known that BAs are bactericidal and provide selective pressure for colonization of bile-sensitive bacteria.22,23
Taken together, there is a complex cross talk between the intestinal microbial community and the host via BAs, which may determine host health and gut microbiota. Specifically, in infancy when the gut microbiota and host physiological functions are underdeveloped, the BA-mediated cross talk must have a significant impact on host health in the present and future life. However, little is known about the occurrence of intestinal BAs in association with the development of gut microbiota in infancy. To address this notion, here, we carefully monitored the successions of bacterial compositions and BA profiles in the first 3 y of ten healthy Japanese infants.
Results
Development of the fecal bacterial community in the first 3 y of life
To investigate the dynamics of the fecal bacterial community in 3 y after birth, we conducted the high-throughput sequencing of the 16S rRNA gene amplicons obtained from ten infants containing both vaginally delivered and cesarean delivered infants and both breast-fed and formula-fed infants (see details in Supplementary Table 1). We obtained stool samples from the infants at 1, 3, 6, 12, 24, and 36 months after birth (six stool samples per subject). A total of 3,222,630 sequencing reads from 60 samples (average 53,710 ± 26,650 reads per sample; good coverage >0.988 for all samples) were obtained and used to determine the taxonomic composition of each sample.
Figure 1(a) shows changes in the relative abundance of five dominant families in ten subjects during the first 3 y of life. The Bifidobacteriaceae-dominant microbiota was observed in all infants at 6 months, while Enterobacteriaceae appeared as the predominant family instead of Bifidobacteriaceae in some infants in earlier period. After 6 months, the Bifidobacteriaceae-dominant microbiota was replaced by the adult-type microbiota dominated by Lachnospiraceae and Bacteroidaceae.
Figure 1.
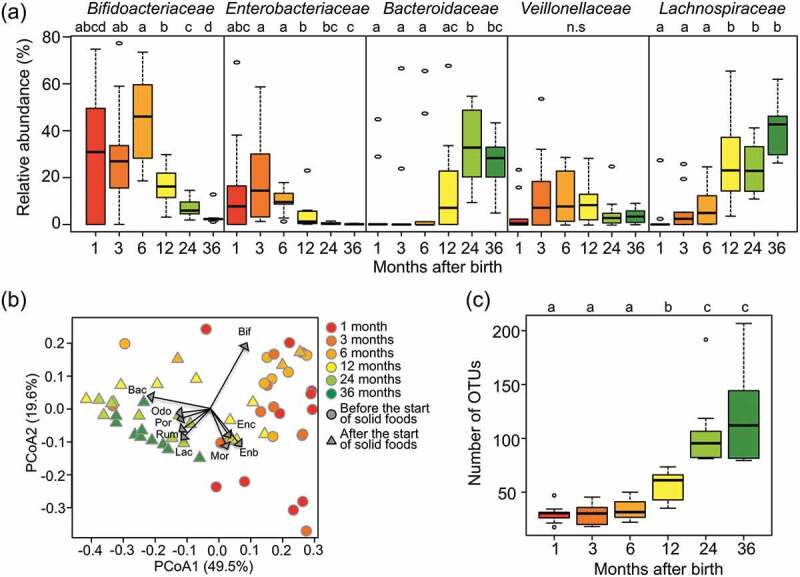
Fecal microbiota compositions over the first 3 y of life.
(a) Succession of top five bacteria families in the first 3 y. Box plots show the distribution of the relative abundance of Bifidobacteriaceae, Enterobacteriaceae, Bacteroidaceae, Veillonellaceae, and Lachnospiraceae among the ten subjects at the indicated age. Different letters (a–d) indicate significant differences between ages (p < 0.05, Wilcoxon rank-sum test with Benjamini–Hochberg correction). n.s: non-significantly. (b) Principal Coordinate Analysis (PCoA) plots based on weighted Unifrac distances, calculated using the OTU compositions and phylogeny. Circle plots showed samples before the start of solid food intake and triangle plots showed samples after that. Regression of sample distribution in the PCoA plot to relative abundance of each bacteria family was calculated using the Envfit R program, and loading of bacteria families whose R2 was higher than 0.01 were plotted by dashed arrow vector. Differences in community structures between before and after the start of solid foods were analyzed statistically using the ‘adonis’ function with 999 permutations in the QIIME pipeline. Bif: Bifidobacteriaceae, Enc: Enterococcaceae, Enb: Enterobacteriaceae, Mor: Moraxellaceae, Lac: Lachnospiraceae, Rum: Ruminococcaceae, Por: Porphyromonadaceae, Odo: Odoribacteraceae, Bac: Bacteroidaceae. (c) Alpha-diversity of fecal microbiota in the first 3 y of life. Distribution box plots showing the number of OTUs detected in each subject are displayed at each sampling point. Different letters (a–c) indicate significant differences between ages (p < 0.05, Wilcoxon rank-sum test with Benjamini–Hochberg correction).
The beta-diversity of the microbiota among the samples was profiled by principal coordinate analysis (PCoA) according to the pairwise weighted UniFrac distance between samples and displayed by a biplot with the vector of bacteria loading at the family level (Figure 1(b)). The PCoA plot clearly classified the microbiota before and after the start of solid food intake (p < 0.001, adonis). Samples collected before the start of solid food intake were highly loaded with Bifidobacteriaceae and/or Enterobacteriaceae. On the other hand, samples collected after the start of solid food intake were highly loaded with Lachnospiraceae, Ruminococcaceae, and/or Bacteroidaceae.
Figure 1(c) shows that the alpha-diversity of the fecal microbiota of infants in 3 y of life was evaluated according to the number of operational taxonomic units (OTUs). The number of OTUs was kept low during the first 6 months, then increased until 2 y of age, and stabilized eventually.
Development of the intestinal BA metabolism in the first 3 y of life
We measured the concentrations of 15 major BAs in fecal samples of ten subjects in the first 3 y (Supplementary Table 2). We classified these BAs into the groups of conjugated BA, primary BA (CA+CDCA), secondary BA (DCA+LCA), and UDCA, and the time course of concentrations of each group was graphed in addition to that of total BA concentration in Figure 2(a). The secondary BA group suddenly appeared and became dominant at 2 y old, while the other groups and total BA, notably UDCA, show a tendency to peak at 12 months after the birth. To focus on the change in bacterial BA metabolism in the developmental microbiota in infancy, we calculated the ratio of deconjugation, epimerization, and dehydroxylation to total BA in each sample by using the individual data of each fecal BA concentration, and the time course for the first 3 y was graphed for each individual (Figure 2(b)). In seven subjects, BAs were mostly deconjugated from the beginning, while the deconjugation was delayed in the other three subjects. Epimerization of BA tended to become highly active at 12–24 months and decrease thereafter. Dehydroxylation appeared after 24–36 months of birth in all subjects. Taken together, it was suggested that gut microbiota of infants develops with a gain of functions involved in BA metabolism, namely deconjugation, epimerization, and dehydroxylation.
Figure 2.
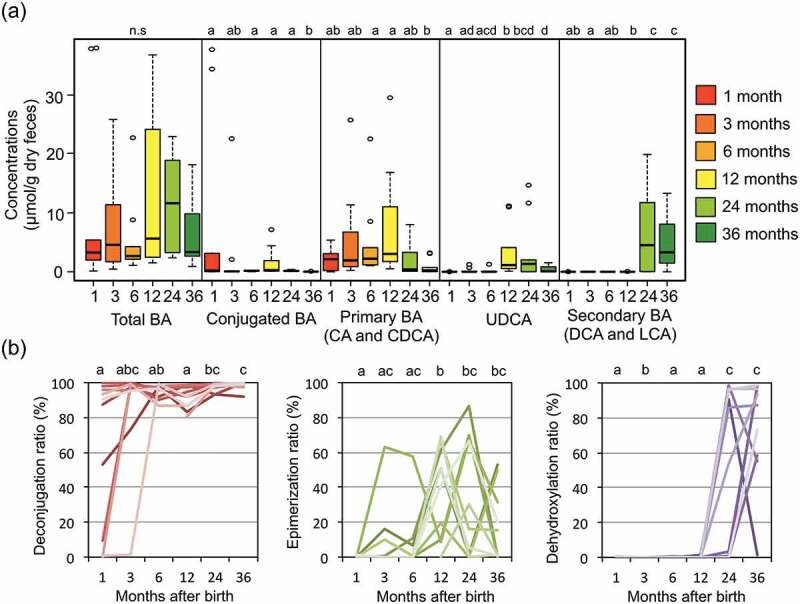
Succession of bile acid composition over the first 3 y of life.
(a) Box plot showing the distribution of the concentrations of each BA group among the ten subjects at the indicated age. Wilcoxon rank-sum test with Benjamini–Hochberg correction was performed to examine the statistical difference among the ages and the significant differences (p < 0.05) are indicated by different alphabets (a–d). n.s: nonsignificantly. (b) Individual plot showing the change of deconjugation ratio (CA+CDCA+DCA+LCA+UDCA to total BA), epimerization ratio (UDCA to CDCA+LCA+UDCA+T-CDCA+T-LCA+T-UDCA+G-CDCA+G-LCA+GUDCA), and dehydroxylation ratio (DCA+LCA to total BA). Wilcoxon rank-sum test with Benjamini–Hochberg correction was performed to examine the statistical difference among the ages, and the significant differences (p < 0.05) are indicated by different alphabets (a–c).
Subsequently, a cluster analysis was performed using the whole individual BA data. As a result, four main clusters were obtained, each associated with the developmental stage for BA metabolism and termed Con, Pri, Urs, and Sec type, respectively (Figure 3(a)). Con type, clustered by three 1-month samples and one 3-month sample, was characterized by a high abundance of primary conjugated BAs such as TCDCA and TCA (Supplementary Table 2). The Pri type, mainly clustered by samples before 12 months, was dominated by primary BAs, particularly CDCA. Urs type, mainly clustered by 12–24-month samples, was characterized by a high ratio of UDCA followed by CDCA. Sec type, mainly clustered by 24 and 36-month samples, was dominated by DCA and LCA. The total abundance of BAs in each type is shown in Figure 3(b). Con type showed higher total BA concentration than the other three types (p = 0.032, Wilcoxon rank-sum test). The alpha-diversity, represented by the number of OTUs in Figure 3(c), increased stepwise from Con to Sec type with the significant difference among BA types. Figure 3(d) shows a change in BA type in the first 3 y of ten tested infants. All of these infants mostly followed the common sequence from Pri to Sec via Urs, while three subjects began with the Con type and two subjects skipped Urs type. It is interesting that one of the two subjects without Urs type delayed in the start of solid food intake until 10 months, which might cause the absence of Urs type. The trend from Con type to Sec type was similarly observed both male and female groups, although the number of female subjects was limited (Supplementary Table 3).
Figure 3.
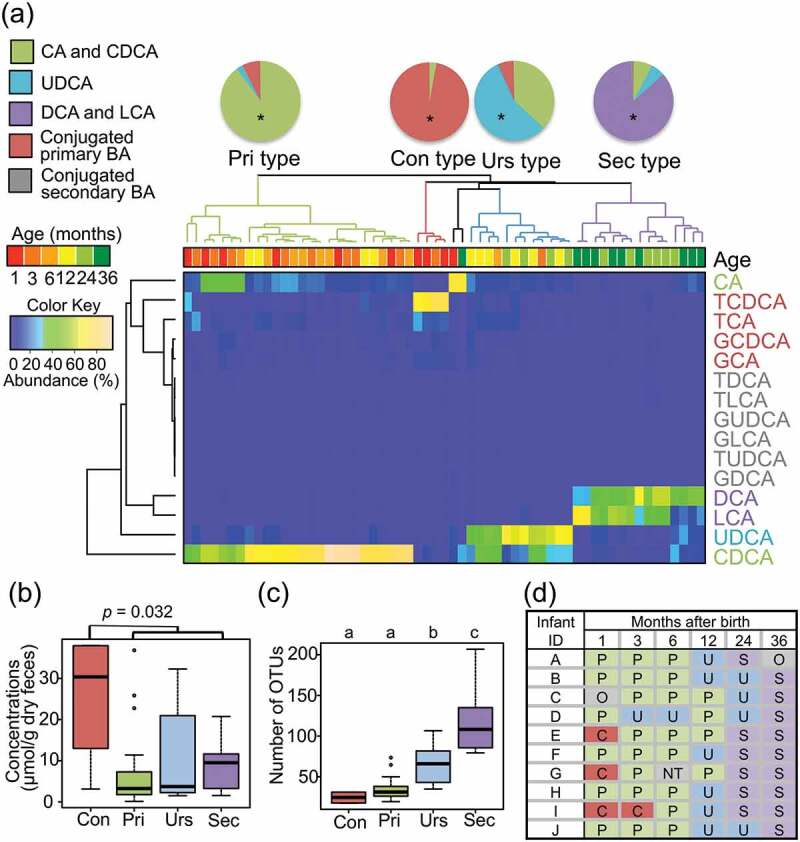
Characterization of BA types in the first 3 y of life.
(a) Clustering of samples using their BA composition. Age colored by sampling time and clusters colored by BA type are shown in the dendrogram. The relative abundance of each BA molecule was displayed in the heat map according to the above color key. These clusters were characterized by a high abundance of deconjugated primary BAs mainly by CDCA; conjugated primary BA mainly by T-CDCA, epimerized BA (UDCA), and dehydrated BAs (DCA and LCA); and termed Pri type, Con type, Urs type, and Sec type, respectively. The pie charts show the BA composition for each BA type. Asterisks in the pie charts indicate statistically significant differences among BA type by Wilcoxon rank-sum test with Benjamini–Hochberg correction (p < 0.05). (b) The total concentration of BAs in feces. Box plots show the distribution of total BA concentration in each BA type group. A non-parametric Mann–Whitney U-test was used to test the significance of the difference in the total BA concentration between Con type and other type. (c) The number of OTUs in samples in each BA type group. Distribution box plots showing the number of OTUs detected are displayed at each BA type. Different alphabets (a–c) above each BA type group data represent the statistically significant difference among the BA type (p < 0.05 in Wilcoxon rank-sum test with Benjamini–Hochberg correction). (d) Succession of BA types in the first 3 y of life of ten infants. C: Con type; P: Pri type; U: Urs type; S: Sec type; O: Other; NA: not available due to the lack of sample.
The correlation between gut microbiota and BA type
To correlate BA type with fecal microbiota, a linear discriminant analysis effect size (LEfSe) analysis was performed using whole bacteria composition data. Subsequently, bacterial taxa showing significant LDA score in LEfSe analysis were subjected to a correlation analysis of their abundance to the concentration of associated BA type. As shown in Figure 4(a,b), Con type infants were highly colonized by unclassified Enterobacteriaceae, while Pri type correlated with Bifidobacterium including Bifidobacterium breve and Bifidobacterium longum as dominant species. Actually, dominant colonization of Bifidobacterium appeared in almost of all Pri type subjects, and the relative abundance of Bifidobacterium significantly correlates with the concentration of primary BA in the first 36 months (-log10p = 9.58, r = 0.72, in Pearson correlation analysis, Supplementary Table 4), suggesting that Bifidobacterium largely contributes to the deconjugation of BA in infant's intestine. Urs type significantly correlated with Ruminococcus gnavus and unclassified Rikenellaceae. Actually, dominant colonization of R. gnavus was observed and the relative abundance of R. gnavus significantly correlates with the UDCA abundance in the first 3 y (-log10p = 2.02, r = 0.34, in Pearson correlation analysis, Supplementary Table 4), suggesting that R. gnavus largely contributes to the production of UDCA in infant’s intestine. Sec type correlated with a wide range of taxonomic groups belonging to the order Clostridiales, such as Roseburia, Ruminococcus, Faecalibacterium, and Eubacterium, as well as two specific Bacteroidetes species, namely Bacteroides stercoris and Parabacteroides distasonis.
Figure 4.
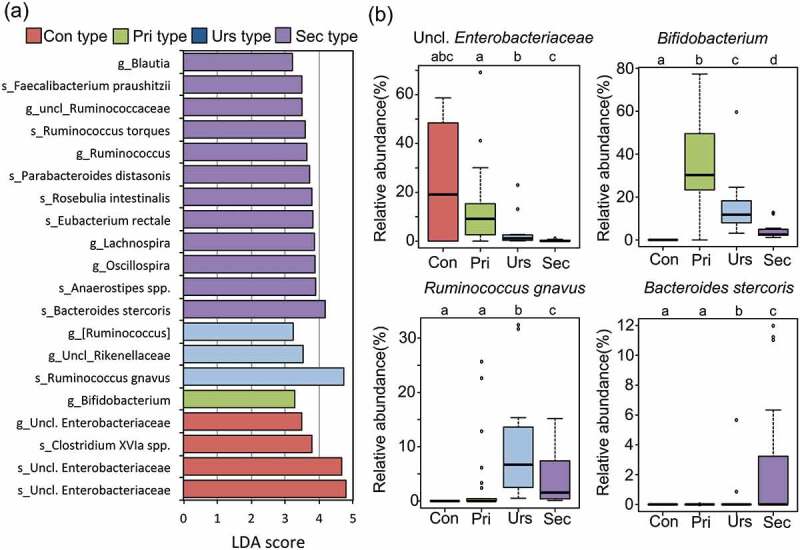
Bacterial taxa differently populated among BA type groups.
(a) LDA score of taxonomic group differently populated among BA type groups. Linear discriminant analysis effect size (LEfSe) analysis identified the bacterial taxa positively associated with each BA type. Operational taxonomic units (OTUs) whose average abundance, among all samples, was higher than 0.1% were subjected to LDA. LDA scores were analyzed using the one-against-all strategy. Taxonomic groups showing LDA scores >2.0. g: genus; s: OTU. The species name represents a closely related species to each OTU with an assignment score higher than the cutoff value of 0.8. (b) Box plots showing the distribution of the relative abundance of key taxa in each BA group. Different letters (a–d) indicate significant differences in the relative abundance of each bacterial taxonomy among BA types (p < 0.05, Wilcoxon rank-sum test with Benjamini–Hochberg correction).
Discussion
To investigate the associations between the developing gut microbiota and intestinal BA metabolism in infants, we conducted this longitudinal study with ten healthy Japanese subjects. As a result, we found that intestinal BA metabolism develops via four stages defined by the high abundance of conjugated primary BAs (Con type), unconjugated primary BAs (Pri type), UDCA (Urs type), and secondary BAs of DCA and LCA (Sec type), each associated with neonatal-type, lactation-type, weaning-type, and adult-type microbiota, respectively.1
In the first 6 months of life, Pri type or Con type was observed. Con type, which was associated with microbiota lacking in Bifidobacterium, was found in three out of the ten subjects. These subjects were highly colonized by Enterobacteriaceae, the pattern of which has been found by Matsuki et al.2 However, taking into account the fact that Enterobacteriaceae-dominant microbiota appears during a neonatal period before Bifidobacteriaceae-dominant microbiota is established with considerable individual variation in the duration of the transition period, it is suspected that the other subjects also carried Con type before the first sampling. Total BA concentration was significantly high in Con type, suggesting that the Con type microbiota with less variety of species including Enterobacteriaceae but not Bifidobacterium is uniquely established under the low expression of BA transporter in the neonatal period or that Con type induces a high level of BA synthesis.24,25 There is a report to support the latter possibility, which has demonstrated that gnotobiotic mice monocolonized by BSH-deleted Bacteroides increase total BA concentration in the cecum and feces compared to that colonized by wild-type Bacteroides and indicated that deconjugated BAs controls the BA synthesis through the activation of FXR signaling.25 This suggests the importance of the transition from Con to Pri type for the development of BA homeostasis in infancy.
Pri type was observed during the lactation period of all tested subjects, in association with the domination of Bifidobacterium. This coincides with the fact that Bifidobacterium is a largest source of BSH in the human intestine,26 while Enterobacteriaceae lacks BSH.15,27 Deconjugated BAs, which are known to be more hydrophobic and more bactericidal than the conjugated form,28 appear to produce a more selective environment for colonizing microbes. It can be said that Bifidobacterium is an initial key player in the developing microbiota in infant's intestine.
It is noticeable that Urs type preceded Sec type in most cases. Two subjects did not show Sec type in these 3 y, one of which delayed in the start of solid food intake until 10 months. The Urs type was uniquely associated with R. gnavus. Indeed, R. gnavus has been reported to be an UDCA producer with 7α-epimerization activity by 7α- and 7β-hydroxysteroid dehydrogenase,29 notably at the start of solid foods.30 The beneficial pharmaceutical aspect of UDCA is well known and commonly administrated for the treatment or prevention of various diseases or symptom associated with disorder of BA metabolism,31 while the antagonistic mode of action of UDCA via FXR, resulting in the activation of BA synthesis, has been demonstrated.32,33 Meanwhile, it has been also reported that R. gnavus colonized the gut microbiota of most children by 2 y after birth with mucin-degrading and utilizing capability.34–36 Blanton et al. reported that Malawi’s undernourished infants at 6- and 18-month old had fewer bacterial species and less R. gnavus and Clostridium symbiosum than healthy infants.5 Furthermore, it was demonstrated that the feeding of two bacterial species, R. gnavus and C. symbiosum, ameliorated growth and metabolic abnormalities which had occurred in mice transplanted with the undernourished donor stool.5 Taken together with our results, UDCA-producing R. gnavus appears to act as a second key player in promoting BA secretion,29,31 enabling children to efficiently absorb lipids and fat-soluble vitamins, and eventually aids healthy growth of children.37
At 3 y of age, DCA and LCA became major components of BA in feces, and all subjects except one became Sec type. The Sec type was associated with colonization by a wide range of taxonomic groups belonging to Bacteroidetes and Firmicutes. Eventually, stable adult-type microbiota was established with high alpha-diversity and Sec type BA metabolism.
Presently, BAs are regarded as important hormones beyond digestive surfactants, which are involved in a wide range of physiological functions, including glucose and lipid metabolism, energy homeostasis, and the modulation of immune response.19,20 However, the mode of interaction with the receptors varies among the BA molecules. For example, it is known that CDCA functions as a potent FXR agonist, resulting in the reduction of BA and cholesterol biosynthesis, whereas UDCA had negligible activities and even functions in an antagonistic way, resulting in increased cholesterol synthesis and triglyceride level.38–41 It is also known that conjugated BAs show a unique agonistic activity against S1PR2 and muscarinic receptors, while microbiota-produced secondary BAs strongly activate TGR-5.42,43 Therefore, it is suspected that the BA types successively appeared in growing infants have distinctive impacts on host physiology at each developmental stage.
In conclusion, this study using ten healthy Japanese newborns has shown that gut microbiota is developed stepwise through the definite pattern in association with the maturation of BA metabolism. Its link with the development of host physiology is rather interesting but yet to be studied. Further, it should also address how the growth condition and living environment affect the microbiota-associated BA metabolism. Large-scale studies including disease patients are warranted to address abnormal BA profiles linked with certain diseases or impaired growth in children.
Material and methods
Subject recruitment and sample collection
The study protocol was approved by the Ethics Committees of the Faculty of Agriculture, Kyushu University (No. 445–03), and the Faculty of Medicine, Kyushu University (No. 27–140 and 30–265). Informed written consent was obtained from the parents of all participants prior to enrollment. The exclusion criteria were gestation of 34 weeks or less, congenital anomalies, and chronic disease. Ten subjects, who were born in Kyushu island between May 2014 and August 2014, were enrolled in this study. Infant stool samples were collected at 1, 3, and 6 months and 1, 2, and 3 y of age by their parents. The mean (SD) collection months were as follows: 1.1 (0.16), 3.3 (0.54), 6.0 (0.15), 12.2 (0.54), 24.3 (0.88), and 36.4 (1.08) (see detail in Supplementary Table 1). Follow-up contents, mode of delivery, milk feeding, the start of solid food intake, and use of antibiotics were conducted in following 3 y after birth. Characteristics of subjects in this study are shown in Supplementary Table 1. Fresh feces were collected by their parents in two sterile tubes (size 76 × 20 mm), each with or without 2 mL of RNAlater (Ambion, Inc.). Collected samples were kept under chilled condition until transport to the laboratory. Then, samples in RNAlater were stored at 4°C until further processing for DNA extraction. Samples without RNAlater were stored at −80°C until further processing for BA extraction. For BA extraction, fecal samples were freeze-dried to remove all moisture and their subsequent dry weight measured. The freeze-dried feces were kept at −80°C.
DNA extraction
The microbial genomic DNA was extracted using the bead-beating method as previously described.2 Twenty milligrams of each fecal sample were washed by phosphate buffered salts solution twice. After removing PBS, fecal samples were suspended in 300 μL of extraction buffer (100 mM Tris-HCl, pH 9.0, 40 mM EDTA, and 1% SDS; final concentrations), 300 mg of 0.1-mm-diameter glass beads, and 500 μL of Tris-EDTA buffer-saturated phenol in a 2 mL screw-cap tube. Microbial cells were lysed by mechanical disruption using FastPrep FP 120 (BIO 101) at a power level of 5.0 for 30 s. The mixtures were centrifuged at 15,000 g for 5 min, and the upper layers were subjected to phenol/chloroform/isoamyl alcohol extraction, followed by isopropanol and ethanol precipitation. Finally, the dried DNA samples were suspended in 0.2 mL TE buffer and stored at −30°C until sequencing by Illumina Miseq.
16S rRNA gene sequencing
The variable region, V1-V2, of the 16S rRNA gene, was amplified from the bacterial genomic DNA using the universal primer Tru 27F (5ʹ-CGCTCTTCCGATCTCTGAGRGTTTGATYMTGGCTCAG-3ʹ) and Tru 354R (5ʹ-TGCTCTTCCGATCTGACCTGCCTCCCGTAGGAGT-3ʹ) and TaKaRa Ex Taq HS (Takara Bio).2,44 The amplified products were then used as templates in the second PCR to further amplify using barcode-tagged primers. Both PCRs were performed as described previously.8 Amplicons from the second PCR were purified using FastGene Gel/PCR Extraction Kit (Nippon Genetics Co., Ltd.), and the DNA concentrations were measured using PicoGreen® ds DNA Assay Kit (Life Technologies) in a Tecan M200 (excitation at 480 nm and emission at 520 nm). A pool of equal amounts (200 ng) of amplicons from each sample was purified by electrophoresis in 2% (w/v) agarose gel, followed by extraction from the gel using FastGene Gel/PCR Extraction Kit. Then, the amplicon mixture was subjected to paired-end sequencing by Illumina MiSeq (Illumina Inc.).
Gut microbiota analysis based on 16S rRNA gene sequence
The obtained sequences were processed using the Uparse pipeline in Usearch version 9.2 (http://drive5.com/useach/manual/upp_ill_pe.html).45 First, the pair of sequence reads were merged using fastq_mergepairs script with mismatched windows up to 30 bases. After quality filtering, dereplication, discarding of singletons, and length trimming, the merged sequences were clustered into 733 OTUs, each representing greater than 97% identity, using the UPARSE-OTU algorithm. The taxonomy of each OTU was assigned at phylum to genus level using the Greengenes reference sequence database (gg_13_8) and the UCLUST algorithm (assign_taxonomy.py) in QIIME version 19.1 (http://qiime.org/).46 The taxonomic composition of each fecal sample was determined based on the OTU table using the QIIME summarize_taxa_througy_plots.py command. To identify a closely related species, the representative sequence of each OTU was subjected to SINTAX algorithm47 with RDP training setv16 and cut-off 0.8.
Alpha-diversity analysis
The alpha-diversity for each sample was calculated using three standard indices (observed species – the number of detected OTUs, Phylogenetic diversity (PD) whole tree, and the Shannon–Wiener index) and the OTU table rarified to 5,000 sequences per sample, with 10 random interactions using QIIME alpha_rarefaction.py command.
Beta-diversity analysis
Beta-diversity was computed with the OTU table using UniFrac. Weighted UniFrac distances were used as inputs for PCoA based on the OTU table, purified to 5,000 sequences per sample using QIIME beta_diversity_through_plots.py command. Differences in community structures between before and after the start of solid foods were analyzed statistically based on the weighted Unifrac metric using the ‘adonis’ function with 999 permutations in the QIIME pipeline.
Extraction and quantification of BAs
Extraction of BAs was performed according to the method described by Hagio et al.48 Freeze-dried feces (100 mg) were ground thoroughly. Ethanol (1 mL) was added to the ground samples to extract BAs. Nor deoxycholic acid (NDCA) (20 nM, final concentration) was added as an internal standard for each sample. After homogenization, samples were heated at 60°C for 30 min on a heating block. After cooling to room temperature, the samples were heated at 100°C on the heating block for 3 min and centrifuged at 1,600 g for 10 min at 15°C. The supernatants were collected. Ethanol (1 mL) was added to the precipitates and mixed vigorously by vortexing for 1 min. The samples were centrifuged at 11,200 g for 1 min at 15°C, and the supernatants were collected. This extraction was repeated one more time. Pooled extracts from each sample were evaporated, and then, 1 mL of methanol was added to the dried extracts. The methanol extracts were purified using an HLB cartridge (Waters), which is a co-polymeric resin which provides both lipophilic and hydrophilic retention characteristics, according to the manufacturer’s instructions.
The BAs were quantified by liquid chromatography with triple quadrupole mass spectrometry (LCMS-8050, Shimazu). The autosampler was kept at 15°C during successive measurements. The sample (2 μL) was injected into a TSKgel ODS-100S column (5 μm, 2.0 mm × 150 mm; Tosoh Corporation) at 40°C and eluted by a gradient consisting of two solvents. Solvent A was acetonitrile/water (20:80) containing 10 mM ammonium acetate. Solvent B was acetonitrile/water (80:20) containing 10 mM ammonium acetate. The elution gradient over 56 min at a flow rate of 0.2 mL/min was as follows: 0–20 min (5–35% B), 20–25 min (35–80% B), 25–40 min (80% B), 40–41 min (80–5% B), and 41–56 min (5% B). The mass spectrometry was analyzed at a negative electrospray ionization mode, in which selective ion monitoring (SIM) was conducted at Q1 for quantitation of deprotonated ion of each BAs, while multiple reaction monitoring (MRM) was conducted through Q1 to Q3 to confirm the BA molecular species of SIM peaks. Optimization of the MRM transitions of the BA and the scheduled MRM operation parameter is described by Muto et al.49 Concentration of each BA was calculated based on the SIM peak area and calibration curve (R2 > 0.96) prepared using different concentrations of standard BAs and was then adjusted with the spiked NDCA. Underdetected limitation had 0.2 nmol/g dry feces.
Linear discriminant analysis effect size (LEfSe)
LEfSe was calculated by using the online galaxy version (http://huttenhower.sph.harvard.edu/galaxy/root, version 1.0.0)50. Any genus with a max abundance higher than 0.1%, among all samples, was subjected to the linear discriminant analysis (LDA). LDA was analyzed using the one-against-all strategy and genera with scores higher than 2.0 were selected.
Statistical analysis
Statistical analyses were performed using the R software, version 3.2.2 (http://www.r-project.org/) and Excel 2011 (Microsoft). For comparing bacterial abundance, alpha-diversity, and BA concentration and relative abundance, the Pairwise Wilcoxon rank-sum test with Benjamini–Hochberg adjustment was used. Clustering of BA types was performed based on the BA composition of each sample, in which hierarchical clustering was calculated based on complete linkage of Euclidean distances matrix by the heatmap.2 function in the R gplots package. Correlations between the relative abundance of bacteria and BA across samples were calculated with Pearson’s correlation coefficient in R version 3.2.2.
Accession number of 16S rRNA gene sequences
Raw sequence data were deposited in the DNA Data Bank of Japan (DDBJ) sequence read archive (DRA007726) under BioProject no PRJDB5316, which contains links and access to stool sampling data under BioSample SAMD00154533 to SAMD0015492.
Funding Statement
This study was supported in part by JSPS KAKENHI Grant Numbers JP25304006, JP 15H04480, and JP 17H04620, and Kyushu University Grant Numbers JA3BE17602.
Abbreviation
- BA
bile acid
- BSH
bile salt hydrolase
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- DCA
deoxycholic acid
- FXR
farnesoid X receptor
- LCA
lithocholic acid
- OTU
operational taxonomic unit
- PCoA
principal coordinate analysis
- TGR-5
G-protein-coupled bile acid receptor
- UDCA
ursodeoxycholic acid
Acknowledgments
We thank all families who provided fecal samples. We also thank the staff at Kyushu University Hospital for their cooperation in this study. We appreciate the technical assistance for the operation of MiSeq and LCMS-8050 from the Center for Advanced Instrumental and Educational Supports, Faculty of Agriculture, Kyushu University.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Tanaka M, Nakayama J.. Development of the gut microbiota in infancy and its impact on health in later life. Allergol Int. 2017;66:515–522. doi: 10.1016/j.alit.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 2.Matsuki T, Yahagi K, Mori H, Matsumoto H, Hara T, Tajima S, Ogawa E, Kodama H, Yamamoto K, Yamada T, et al. A key genetic factor for fucosyllactose utilization affects infant gut microbiota development. Nat Commun. 2016;7:11939. doi: 10.1038/ncomms11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitsuoka T. Development of functional foods. Biosci Microbiota Food Heal. 2014;33:117–128. doi: 10.12938/bmfh.33.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vallès Y, Artacho A, Pascual-García A, Ferrús ML, Gosalbes MJ, Abellán JJ, Francino MP. Microbial succession in the gut: directional trends of taxonomic and functional change in a birth cohort of spanish infants. PLoS Genet. 2014:10. doi: 10.1371/journal.pgen.1004406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanton LV, Charbonneau MR, Salih T, Barratt MJ, Venkatesh S, Ilkaveya O, Subramanian S, Manary MJ, Trehan I, Jorgensen JM, et al. Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children. Science. 2016;351(6275). doi: 10.1126/science.aad3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subramanian S, Blanton LV, Frese SA, Charbonneau M, Mills DA, Gordon JI. Cultivating healthy growth and nutrition through the gut microbiota. Cell. 2015;161:36–48. doi: 10.1016/j.cell.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen QN, Himes JE, Martinez DR, Permar SR, Hogan DA. The impact of the gut microbiota on humoral immunity to pathogens and vaccination in early infancy. PLoS Pathog. 2016;12(12):e1005997. doi: 10.1371/journal.ppat.1005997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka M, Korenori Y, Washio M, Kobayashi T, Momoda R, Kiyohara C, Kuroda A, Saito Y, Sonomoto K, Nakayama J. Signatures in the gut microbiota of Japanese infants who developed food allergies in early childhood. FEMS Microbiol Ecol. 2017;93:1–11. doi: 10.1093/femsec/fix099. [DOI] [PubMed] [Google Scholar]
- 9.Azad MB, Konya T, Guttman DS, Field CJ, Sears MR, HayGlass KT, Mandhane PJ, Turvey SE, Subbarao P, Becker AB, et al. Infant gut microbiota and food sensitization: associations in the first year of life. Clin Exp Allergy Clin Exp Allergy. 2015;45(3):632–643. doi: 10.1111/cea.12487. [DOI] [PubMed] [Google Scholar]
- 10.Arrieta M, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, Kuzeljevic B, Gold MJ, Britton HM, Lefebvre DL, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med. 2015;7. doi: 10.1126/scitranslmed.aab2271. [DOI] [PubMed] [Google Scholar]
- 11.Ismail IH, Boyle RJ, Licciardi PV, Oppedisano F, Lahtinen S, Robins-Browne RM, Tang MLK. Early gut colonization by Bifidobacterium breve and B. catenulatum differentially modulates eczema risk in children at high risk of developing allergic disease. Pediatr Allergy Immunol. 2016;27:838–846. doi: 10.1111/pai.12646. [DOI] [PubMed] [Google Scholar]
- 12.Riva A, Borgo F, Lassandro C, Verduci E, Morace G, Borghi E, Berry D. Pediatric obesity is associated with an altered gut microbiota and discordant shifts in Firmicutes populations. Environ Microbiol. 2017;19:95–105. doi: 10.1111/1462-2920.13463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanislawski MA, Dabelea D, Wagner BD, Iszatt N, Dahl C, Sontag MK, Knight R, Lozupone CA, Eggesbø M, Pettigrew MM. Gut microbiota in the first 2 years of life and the association with body mass index at age 12 in a Norwegian birth cohort. MBio. 2018;9:1–14. doi: 10.1128/mBio.01751-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wahlström A, Sayin SI, Marschall HU, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA. 2008;105:13580–13585. doi: 10.1073/pnas.0804437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jason MR, Dae JK, Phillip BH, Jasmohan SB. Bile acids and the gut microbiome. Curr Opin Gastoenterol. 2014;30:332–338. doi: 10.1097/MOG.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E, Thomas G, Barbu V, Humbert L, Despras G, et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut. 2013;62:531–539. doi: 10.1136/gutjnl-2012-302578. [DOI] [PubMed] [Google Scholar]
- 19.Vítek L, Haluzík M. The role of bile acids in metabolic regulation. J Endocrinol. 2016;228:R85–96. doi: 10.1530/JOE-15-0469. [DOI] [PubMed] [Google Scholar]
- 20.Schaap FG, Trauner M, Jansen PLM. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11:55–67. doi: 10.1038/nrgastro.2013.151. [DOI] [PubMed] [Google Scholar]
- 21.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 22.Begley M, Gahan CGM, Hill C. The interaction between bacteria and bile. F FEMS Microbiol Rev. 2005;29:625–651. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe M, Fukiya S, Yokota A. Comprehensive evaluation of the bactericidal activities of free bile acids in the large intestine of humans and rodents. J Lipid Res. 2017;58:1143–1152. doi: 10.1194/jlr.M075143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui JY, Aleksunes LM, Tanaka Y, Fu ZD, Guo Y, Guo GL, Lu H, Zhong X, Klaassen CD. Bile acids via FXR initiate the expression of major transporters involved in the enterohepatic circulation of bile acids in newborn mice. Am J Physiol Liver Physiol. 2012;302:G979–96. doi: 10.1152/ajpgi.00370.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao L, Seaton SC, Ndousse-fetter S, Adhikari AA, Dibenedetto N, Mina AI, Banks AS, Bry L, Devlin AS. A selective gut bacterial bile salt hydrolase alters host metabolism. eLife. 2018;7:e37182. doi: 10.7554/eLife.37182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim G, Lee BH. Biochemical and molecular insights into bile salt hydrolase in the gastrointestinal microflora – a review. Asian-Aust J Anim Sci. 2005;18:1505–1512. doi: 10.5713/ajas.2005.1505. [DOI] [Google Scholar]
- 27.Imamura T, Sakanomot N, Tamaki M, Hirano S. Transformation of bile acids by members of the Enterobacteriaceae. Nihon Saikingaku Zasshi. 1979;34:513–520. doi: 10.3412/jsb.34.513. [DOI] [PubMed] [Google Scholar]
- 28.Sannasiddappa TH, Lund PA, Clarke SR. In vitro antibacterial activity of unconjugated and conjugated bile salts on Staphylococcus aureus. Front Microbiol. 2017;8:1–11. doi: 10.3389/fmicb.2017.01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J, Arai H, Nakamura Y, Fukiya S, Wada M, Yokota A. Contribution of the 7β-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J Lipid Res. 2013;54:3062–3069. doi: 10.1194/jlr.M039834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Devlin AS, Fischbach MA. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol. 2015;11:685–690. doi: 10.1038/nchembio.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ðanić M, Stanimirov B, Pavlović N, Goločorbin-Kon S, Al-Salami H, Stankov K, Mikov M. Pharmacological applications of bile acids and their derivatives in the treatment of metabolic syndrome. Front Pharmacol. 2018;9:1–20. doi: 10.3389/fphar.2018.01382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gioiello A, Cerra B, Mostarda S, Guercini C, Pellicciari R, Macchiarulo A. Bile acid derivatives as ligands of the farnesoid X receptor: molecular determinants for bile acid binding and receptor modulation. Curr Top Med Chem. 2014;14:2159–2174. doi: 10.2174/1568026614666141112100208. [DOI] [PubMed] [Google Scholar]
- 33.Peet DJ, Janowski BA, Mangelsdorf DJ, Dev OG, Forman BM, Willy PJ, Dev G, Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids : natural ligands for an orphan nuclear receptor. Science. 1998;284:1365. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 34.Sagheddu V, Patrone V, Miragoli F, Puglisi E, Morelli L. Infant early gut colonization by lachnospiraceae: high frequency of Ruminococcus gnavus. Front Pediatr. 2016;4:57. doi: 10.3389/fped.2016.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crost EH, Tailford LE, Le Gall G, Fons M, Henrissat B, Juge N, de Crécy-Lagard V. Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS One. 2013;8:e76341. doi: 10.1371/journal.pone.0076341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 37.Kotb MA, Street A, Manial E. Molecular mechanisms of ursodeoxycholic acid toxicity & side effects : ursodeoxycholic acid freezes regeneration & induces hibernation mode. Int J Mol Sci. 2012;13(7):8882–8914. doi: 10.3390/ijms13078882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/seicnce.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/S1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 40.Mueller M, Thorell A, Claudel T, Jha P, Koefeler H, Lackner C, Hoesel B, Fauler G, Stojakovic T, Einarsson C, et al. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol. 2015;62:1398–1404. doi: 10.1016/j.jhep.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez FJ, Jiang C, Bisson WH, Patterson AD. Inhibition of farnesoid X receptor signaling shows beneficial effects in human obesity. J Hepatol. 2015;62:1234–1236. doi: 10.1016/j.jhep.2015.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuipers F, Bloks VW, Groen AK. Beyond intestinal soap – bile acids in metabolic control. Nat Rev Endocrinol. 2014;10:488. doi: 10.1038/nrendo.2014.60. [DOI] [PubMed] [Google Scholar]
- 43.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes gary. Science. 2011;334:105–109. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim S-W, Suda W, Kim S, Oshima K, Fukuda S, Ohno H, Morita H, Hattori M. Center. Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 2013;20:241–253. doi: 10.1093/dnares/dst006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 46.Justin K, Jesse S, William AW, Antonio G, Caporaso JG, Rob K. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr Protoc Bioinforma. 2011;10:1–7. doi: 10.1002/0471250953.bi1007s36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edgar R. SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv. 2016:74161. doi: 10.1101/074161. [DOI] [Google Scholar]
- 48.Hagio M, Matsumoto M, Fukushima M, Hara H, Ishizuka S. Improved analysis of bile acids in tissues and intestinal contents of rats using LC/ESI-MS. J Lipid Res. 2009;50:173–180. doi: 10.1194/jlr.D800041-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Muto A, Takei H, Unno A, Murai T, Kurosawa T, Ogawa S, Iida T, Ikegawa S, Mori J, Ohtake A, et al. Detection of Δ4-3-oxo-steroid 5β-reductase deficiency by LC-ESI-MS/MS measurement of urinary bile acids. J Chromatogr B Anal Technol Biomed Life Sci. 2012;900:24–31. doi: 10.1016/j.jchromb.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 50.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.