

ABSTRACT
Members of the MYC family of proto-oncogenes are the most commonly deregulated genes in all human cancers. MYC proteins drive an increase in cellular proliferation and facilitate multiple aspects of tumor initiation and progression, thereby controlling all hallmarks of cancer. MYC's ability to drive metabolic reprogramming of tumor cells leading to biomass accumulation and cellular proliferation is the most studied function of these oncogenes. MYC also regulates tumor progression and is often implicated in resistance to chemotherapy and in metastasis. While most oncogenic functions of MYC are attributed to its role as a transcription factor, more recently, new roles of MYC as a pro-survival factor in the cytoplasm suggest a previously unappreciated diversity in MYC's roles in cancer progression. This review will focus on the role of MYC in invasion and will discuss the canonical functions of MYC in Epithelial to Mesenchymal Transition and the cytoplasmic functions of MYC-nick in collective migration
KEYWORDS: EMT, MYC, MYC-nick, metastasis, migration
The MYC family of transcription factors
MYC genes are evolutionarily conserved, appearing first in primitive pro-metazoans such as the choanoflagellate Monosiga brevicollis.1 While lower organisms have one myc gene, mammalian cells harbor three independent genes that encode for the proteins: MYC (ubiquitously expressed), MYCN (neuronal variant) and MYCL (lung variant). Due to their critical role in regulating cellular biology, MYC proteins are essential for embryonic development and for adult self-renewing tissues.2 Knocking out MYC and MYCN, but not MYCL, causes early embryonic lethality in mice.3 Aberrant activation of any MYC family member by DNA amplification, transcriptional upregulation, or protein stabilization leads to elevated MYC levels and contributes to tumorigenesis.3,4,5,6 This deregulation of MYC renders cells insensitive to environmental cues causing an “egotistic” reactivation of proliferative programs, thereby contributing to cancer.7,8 Because MYC can promote almost every aspect of cellular transformation, it has captured the attention of cancer biologists, virologists, biochemists, and geneticists for more than three decades.9,10,11
MYC proteins are basic helix-loop-helix leucine zipper (bHLH-LZ) nuclear transcription factors12,13 (Fig. 1). The bHLH-LZ domain of MYC is responsible for heterodimerization with MAX and binding to DNA.14 MYC:MAX complexes bind to regulatory regions present in promoters of target genes named E-boxes.14,15,16 MYC:MAX can also regulate transcription by binding to non-E-box sites located in promoters or enhancers.17,18,19 More recently, MYC was proposed to prevent pausing of RNA polymerase II and, hence, cause widespread acceleration in the transcription of already active genes.20,21
Figure 1.
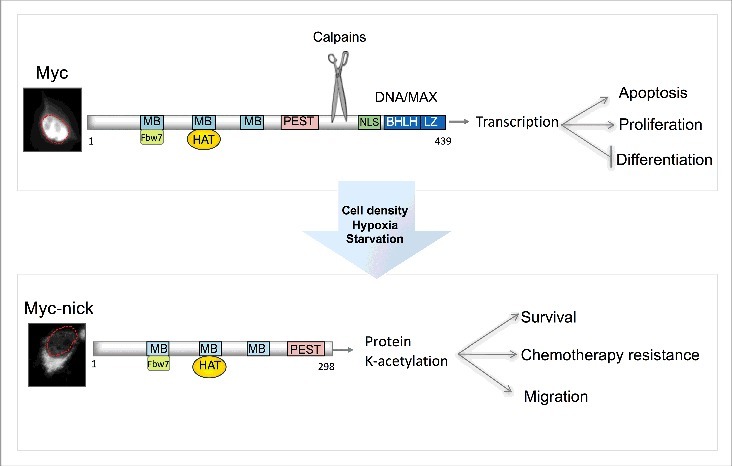
Schematic representation of MYC and MYC-nick. MYC is predominantly localized in the nucleus (outlined in red) where it drives the expression of genes that promote cell proliferation. MYC-nick is localized in the cytoplasm and promotes cell survival and migration. In conditions of stress (such as high cell density, hypoxia and nutrient starvation) calpain-mediated cleavage of MYC produces MYC-nick. (MB) MYC Box I-IV. BHLH-DNA binding and heterodimerization domain. (HAT) Histone Acetyl Transferases such as GCN5. PEST (proline, glutamic acid, serine and threonine-rich domain). (NLS) nuclear localization sequence.
In addition to the bHLH-LZ and nuclear localization sequences near the C-terminus, MYC also has conserved domains named MYC Boxes (I-IV) in the N-terminal part of the protein.22 The first of these domains, MBI, comprises a “degron” that targets MYC for ubiquitin-mediated proteasomal degradation.23,24 GSK3β promotes this degradation by phosphorylating residues in MBI, making MYC a substrate for the SCFFbw7 E3 ligase.25 Inactivation of SCFFbw7, inactivation of GSKβ, or mutations in MBI (e.g. T58A) that prevent GSK3β-mediated phosphorylation avert MYC degradation and elevate MYC levels in cancer cells.25,26 The second of the MYC boxes, MBII, plays a key role in the regulating MYC's transcriptional activity by recruiting histone acetyltransferases (HAT) such as GCN5, TIP60, and TIP48 to sites of transcription.27,28,29
MYC's transcriptional signature contains genes involved in Epithelial to Mesenchymal Transition
MYC-regulated transcription activates a signature of genes that promote uptake, synthesis, and recycling of macromolecules including fatty acids, amino acids, and nucleotides. Thereby, MYC leads to metabolic reprogramming, which is responsible for augmented cell proliferation and tumor initiation.7,30-32 Among the transcriptional targets induced by MYC are also genes that modulate the interaction of tumor cells with their microenvironment.33,34 For example, MYC directly induces the expression of the innate immune regulator CD47 (cluster of differentiation 47) and the adaptive immune checkpoint PD-L1 (programmed death-ligand 1), which are critical for evasion of antitumor immune response.35 Moreover, by increasing the expression of interleukin 1β and VEGF, and by suppressing thrombospondin-1 (TSP-1), MYC activates angiogenesis, thereby facilitating the delivery of nutrients to tumor cells and allowing for escape of cancer cells into the blood stream.36,37
A direct requirement for MYC in metastasis was established using several animal models including mouse models of lung and prostate cancer.38,39 Deregulated MYC activity was shown to synergize with other oncogenic pathways to promote cell migration and metastasis. For example, a cooperation between deregulated MYC and Wnt signaling results in profound morphological changes in mammary epithelial cells that enables anchorage-independent growth and invasiveness.40 MYC was also found to cooperate with c-RAF to robustly drive lung cancer metastasis.38,41 The synergistic activity of MYC and the RAS family of GTPases in cancer cells has been the target of extensive investigation.42 RAS activates a signaling cascade that leads to MYC protein stabilization in cancer cells.41,43 MYC, in turn, is required for the maintenance, progression, and metastasis of RAS-driven tumors such as lung cancer.44 MYC was also shown to induce the transcription of RhoA, which cooperates with MYC in cellular transformation.45 More recent studies demonstrated that MYC can reduce the assembly of RhoA-mediated stress fibers and focal adhesions, thereby preventing excessive adhesion to substrate and facilitating directional migration. Thus indicating that MYC likely augments the oncogenic functions of RhoA by reducing prolonged substrate adhesion.46
Several reports propose that the increase in metastatic behavior of MYC-expressing tumor cells is derived from MYC's ability to induce cell motility by transcriptionally downregulating the expression of E-cadherin.47 Downregulation of E-cadherin is a hallmark of Epithelial to Mesenchymal Transition (EMT).48 EMT is often characterized by a downregulation in the expression of adhesion molecules and epithelial markers, accompanied by an upregulation of mesenchymal markers including vimentin and N-cadherin.48 The reduction in E-cadherin leads to destabilization of adherens junction and loss of epithelial phenotype.48 MYC and MYCN were shown to induce the expression of the microRNA mir-9, which targets E-cadherin mRNA.49 Downregulation of E-cadherin by mir-9 leads to loss of epithelial phenotype and results in increased cell motility and invasion.49 In agreement with a role in cancer progression, mir-9 levels are directly associated with poor prognosis in breast cancer patients.49 MYC was also found to upregulate the expression of the master regulator of EMT, SNAIL.50 SNAIL is a transcriptional repressor that prevents the expression of E-cadherin causing EMT during embryonic development and during metastasis.51,52
Cytoplasmic functions of MYC-nick
Although originally identified as a transcriptional regulator, recent studies have revealed a previously unappreciated role for MYC in the cytoplasm as a pro-survival factor.54 The cytoplasmic functions of MYC are executed by a truncated MYC variant named MYC-nick, which is produced by a proteolytic cleavage of MYC.52 In addition to being degraded by the proteasome, MYC and MYCN are also targeted by calcium-dependent cysteine proteases from the calpain family.53 Calpain-mediated cleavage of MYC degrades the C-terminus, which is essential for DNA binding and transcription, while preserving the N-terminus intact. The 298 N-terminal fragment gives rise to MYC-nick, a transcriptionally inactive cytoplasmic form of MYC (Fig. 1). The cleavage sites for calpains on MYC are conserved from choanoflagelates to humans (Conacci-Sorrell, unpublished results) implying a conserved mechanism to regulate MYC levels and activity.
Calpain-mediated cleavage can generate protein fragments that have functions that are different than those of the parent protein.54 Consistent with this possibility, we found that MYC-nick is an active cytosolic factor capable of driving cell survival and cell migration in a process that appears to depend on protein acetylation.53,55 In the cytoplasm, MYC-nick forms a complex with acetyltransferases such as GCN5 and promotes acetylation of cytoplasmic proteins, including ATG3 and α-tubulin, which are both required for cell survival.53, 55 (Fig. 1).
The cleavage of MYC by calpains and the presence of functional MYC-nick is required for cell survival during the process of muscle differentiation.52 Preventing MYC cleavage using pharmacological inhibitors or mutating the calpain cleavage site on MYC prevents the differentiation of myoblasts into myotubules and causes apoptosis.53 Similarly, regulated increase in calpain activity was shown to be necessary for terminal differentiation of multiple cell types including muscle cells.56 Importantly, mutations in the muscle-specific calpain 3, which impair its proteolytic activity cause limb girdle muscular dystrophy 2A (LGMD2A).57 While normal calpain activity is necessary for embryonic development, hyperactivation of calpains in adult tissues is associated with increased tumorigenesis and invasiveness.58 Our work, described below, proposes that this rise in invasive behavior of cells harboring hyperactivation of calpains is, at least in part, triggered by the production of MYC-nick, which facilitates cell survival and migration.
MYC-nick promotes cell survival and collective migration
While full-length MYC is the most studied MYC variant in cancer progression, we recently demonstrated that MYC-nick plays a critical role in cancer cell survival, resistance to chemotherapeutic agents, and migration.53,55,59 MYC is constitutively converted into MYC-nick in the cytoplasm of most normal and cancer cells. However, in cells exposed to environmental stress such as high cell density, hypoxia, and nutrient deprivation MYC-nick is the predominant form of MYC. MYC-nick is abundant in the cytoplasm of tumor tissues such as colon, breast, prostate, rhabdomyosarcoma, neuroblastoma, medullobastoma, lymphoma, and others,55 indicating an active role for MYC-nick in these cells. Moreover, MYC-nick preserves the MBI domain and oncogenic conditions that inhibit GSK3β, downregulate SCFFbw7, or prevent MYC phosphorylation (e.g., the T58A mutation) stabilize MYC-nick in tumor cells.53
Ectopic expression of MYC-nick in colon cancer cell lines promotes an increase in survival of cells when subjected to nutritional stress such as deprivation of growth factors, glucose and glutamine.55 Furthermore, MYC-nick attenuates apoptosis of cancer cells grown in the presence of chemotherapeutic agents such as etoposide, cisplatin, oxaliplatin, and imatinib in a process that requires autophagy.55
Concomitant with increased survival, MYC-nick also promotes cell migration; however, this induced migration does not involve EMT. MYC-nick expression does not induce mesenchymal morphology or alter the levels of the known EMT drivers such as N-cadherin, SNAIL, SLUG, TWIST, and vimentin.55 MYC-nick-expressing migratory cells travel directionally in small clusters of epithelial-shaped cells that detach from spheroids of tumor cells cultured in 3D (Fig. 2). The migratory properties induced by MYC-nick in colon cancer cells grown in 3D cultures reasembles collective migration. Collective migration is characterized by a cluster of cells creating an invasive front where leader cells modify the extracellular matrix generating a path for follower cells.60 Collective invasion is emerging as a newly appreciated route for metastases in human cancers including breast cancer.61,62
Figure 2.
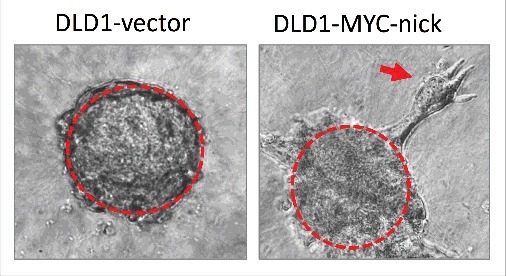
Spheroids of DLD1 cells expressing empty vector (left) or MYC-nick (right) were cultured in 3D media containing 50% collagen for 4 days. Note that MYC-nick expressing cells undergo collective migration in collagen. The red arrow points to a group of migratory cells disconnecting from the “parent” spheroid.
In vivo studies of prostate and breast cancer have shown that MYC is relocalized from the nucleus to the cytoplasm (indicative of MYC-nick) during metastasis.39,63 Furthermore, MYCN localization was shown to shift from the nucleus to the cytoplasm in differentiating migratory neurons of the neural crest, retinal ganglion cells, spinal ganglia,64 and Purkinje cells.64,65 This relocalization of MYC to the cytoplasm is suggestive of MYC-nick and indicates that MYC-nick also regulates cell migration during embryonic development.
MYC-nick induces fascin expression
How does MYC-nick promote cell migration? Our observations suggest that MYC-nick drives dramatic changes in cellular morphology and promotes motility. MYC-nick expression modifies the actin cytoskeleton by inducing the formation of sharp and long-lived filopodia at the leading edge of migrating cells (Fig. 2).55 Filopodia are membrane protrusions involved in cell-cell interactions, nutrient sensing, and migration.66 Often filopodia-like structures are considered an indication of the metastatic potential of cancer cells.67
The actin bundling protein fascin is critical for stabilizing actin filaments in filopodia and invadopodia and thereby drives metastasis in solid tumors.68,69 While fascin expression is elevated in metastatic cells, its levels are very low in normal adult tissues. Moreover, fascin1-deficient mice are viable and fertile.70 Fascin is dispensable for normal adult tissues and is necessary for cancer cell migration, thus targeting fascin could be a promising strategy to treat metastatic cancer.
Fascin mRNA and protein are dramatically upregulated in MYC-nick-expressing colon cancer cells. Hence silencing fascin in these cells diminishes cell migration measured in 2D and 3D cultures.71 While fascin stimulates the migration of colon cancer cells, it is not sufficient to promote filopodia formation, indicating that MYC-nick regulates additional components of the actin cytoskeleton which are also required to generate filopodia. Moreover, these results suggest that fascin can promote cell migration without necessarily modulating filopodia and that filopodia may play an important role in sensing and directing migration rather than propelling cultured cells forward. Likely filopodia and invadopodia are more important for metastasis in vivo where migratory cells encounter greater environmental resistance.
MYC-nick promotes sustained activation of Cdc42
In addition to upregulating fascin levels, MYC-nick also drives activation of the cytoskeletal effector protein Cdc42. The Rho family of small GTPases, which includes Rho, Rac and Cdc42, has been shown to regulate multiple aspects of cellular biology including actin dynamics and cellular migration. Rho GTPases cycle between active (GTP bound) and inactive (GDP bound) states by binding and hydrolyzing GTP. Rho family GTPases control actin organization to form different cellular structures: Rho promotes the organization of actin filaments into stress fibers, Rac functions in lamellipodia formation, while Cdc42 is responsible for regulating filopodia formation.72 Cdc42 is necessary for embryonic development and its aberrant expression in a variety of tumor types is correlated with poor prognosis.73 Activated Cdc42 mediates a signaling cascade leading to the regulation of over twenty downstream effectors, including protein kinases (such as PAK1), lipid kinases (such as PI3K), scaffolding proteins, and cytoskeletal interacting factors.74,75 These targets, in turn, promote changes in cell polarity, adhesion, migration, actin cytoskeleton remodeling, and membrane trafficking. Therefore, by activating Cdc42, MYC-nick could regulate a broad range of biological processes that lead to an increase in cancer cell fitness.
We found that MYC-nick activates Cdc42 in colon cancer cells such as DLD1 and HCT116 and treating MYC-nick-expressing cells with inhibitors of Cdc42 completely abolishes filopodia formation.55 Surprisingly, MYC-nick promotes a sustained activation of Cdc42 that can persist for 24 hours after stimulation with growth factors, such as EGF. As with fascin knock down, Cdc42 inhibition does not affect the viability of MYC-nick expressing cells, further supporting the notion that MYC-nick-stimulated survival and migration are independent events.55
Previous studies show that suppression of Cdc42 blocks cell proliferation and cell cycle progression of Ras-transformed cells, but not MYC transformed cells.76 These results may suggest that MYC-driven cancers are dependent on Cdc42 for metastasis in a process that is mediated by MYC-nick. Whereas Cdc42 is sufficient to induce filopodia in colon cancer cells, synergy between Cdc42 and fascin is necessary to promote maximal migration of colon cancer cells expressing deregulated MYC-nick. In agreement with the premise that MYC-nick induces fascin and Cdc42 activity to drive migratory behavior, we found that MYC-nick, Cdc42, and fascin are dramatically elevated at the invasive front of human colon cancer tumors. This indicates that these proteins may indeed coordinate cell migration and thereby activate cancer metastasis in vivo.55
MYC-nick-induced cell migration is mediated by its binding to acetyltransferases
The specific molecular mechanisms by which MYC-nick promotes fascin mRNA induction and the activation of Cdc42 remain to be determined. A likely possibility is that these events are mediated by the ability of MYC-nick to promote acetylation of cytoplasmic proteins when complexed with acetyltransferases. The MBII region (amino acids 106–143) located within the N-terminal segment of MYC and MYC-nick constitutes a binding site for recruitment of acetyltransferases. Deleting MBII has no effect on MYC-nick expression levels and localization, but reduces its ability to promote cell survival, chemoresistance, and migration. In agreement, deletion of MBII on MYC-nick also attenuates fascin transcription and blocks Cdc42 activation. Thus, binding to acetyltransferases appears to play a crucial role in mediating MYC-nick's biological functions.53,55
Among non-histone acetylation targets, fascin was previously found to be acetylated in the cytoplasm.77 Indeed, we found fascin to be highly acetylated in colon cancer cells. Whereas acetylation of fascin was independent of MYC-nick, fascin expression levels were directly proportional to the presence of active MYC-nick. Because MYC-nick does not have a DNA binding domain and cannot directly regulate transcription, the mechanism by which MYC-nick regulates fascin expression is likely indirect. One possibility is that transcriptional regulators, which are among the proteins acetylated in a MYC-nick-dependent manner, activate the fascin promoter in cancer cells.
Similarly, the acetylation events that mediate Cdc42 activation by MYC-nick remain to be defined, including unexplored acetylated lysines on Cdc42 and its regulatory proteins. The role of protein acetylation in regulating the Ras superfamily of GTPases is beginning to be explored. For example, acetylation of Ran GTPases regulates nearly all aspects of Ran functions including nucleotide exchange.78 Rho, Rac and Cdc42 can also be acetylated and these modifications are also likely to play an important role in the regulation of these GTPases.78 Moreover, alterations within microtubules driven by MYC-nick-induced acetylation of α-tubulin may result in changes in the actin cytoskeleton.53 Crosstalk between microtubules and the actin cytoskeleton is known to influence migration, cell polarity, and division.79,80 Furthermore, microtubules can target filopodia and regulate their movement and density.81 While Cdc42 and fascin were previously found to influence each other's levels and activity,72 MYC-nick-driven Cdc42 activation and fascin expression in colon cancer cells are not codependent or coregulated.
Conclusion
In summary, MYC can drive not only cell transformation and cancer initiation, but also migration and metastasis (Fig. 3). MYC expression drives migration by two mechanisms: 1. full-length MYC causes repression of E-cadherin and induces EMT, 2. MYC-nick drives activation of Cdc42 and expression of fascin, which promote filopodia formation and collective invasion. Thus blocking transcriptional functions of MYC with MYC:MAX inhibitors, may not be sufficient to treat metastatic tumors, as this approach would not block MYC-nick driven chemo-resistance and metastasis. Therefore, identifying and characterizing molecular pathways regulated by MYC-nick may lead to the delineation of critical effectors of MYC-driven tumorigenesis and provide novel possibilities to treat metastatic cancer.
Figure 3.
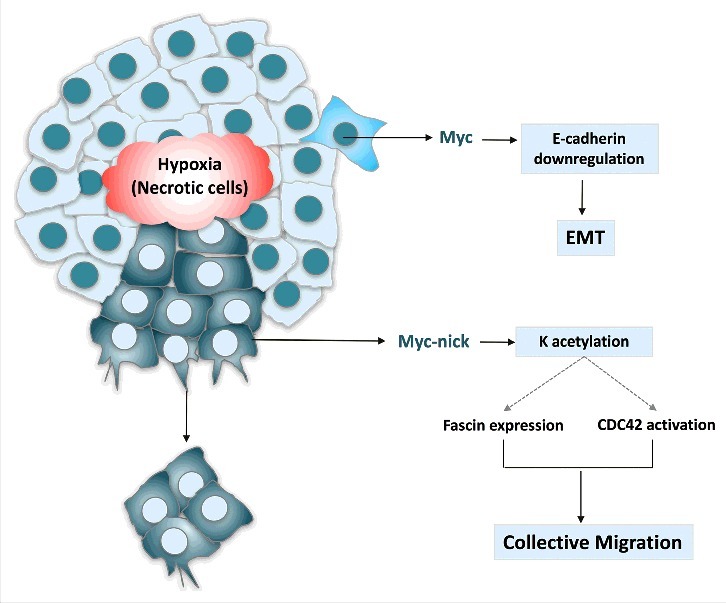
MYC activates the transcription of genes that promote proliferation and represses the expression of E-cadherin leading to EMT. MYC-nick, however is highly expressed in the cytoplasm of migratory cancer cells. It drives the expression of fascin and the activation of CDC42. Both fascin induction and CDC42 activation require the MBII domain on MYC-nick, which recruits HATs and promote acetylation of cytoplasmic proteins. (K = lysine).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Dr. Sandra L. Schmid, Dr. Peter Michaely, Dr. William Michael Henne, Dr. Marcel Mettlen, Dr. Erik Welf, and Roshni Ray for valuable input in editing the manuscript.
Funding
This work was funded by the Cancer Prevention and Research Institute of Texas (CPRIT) 50C0579903 and Welch Foundation 52C1049001. Maralice Conacci Sorrell is the Virginia Murchison Linthicum Scholar in Medical Research.
References
- 1.Young SL, Diolaiti D, Conacci-Sorrell M, Ruiz-Trillo I, Eisenman RN, King N. Premetazoan ancestry of the Myc-Max network. Mol Biol Evol. 2011;28:2961-2971. doi: 10.1093/molbev/msr132. PMID:21571926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hurlin PJ. Control of vertebrate development by MYC. Cold Spring Harb Perspect Med. 2013;3:a014332. doi: 10.1101/cshperspect.a014332. PMID:24003246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatton KS, Mahon K, Chin L, Chiu FC, Lee HW, Peng D, Morgenbesser SD, Horner J, DePinho RA. Expression and activity of L-Myc in normal mouse development. Mol Cell Biol. 1996;16:1794-1804. doi: 10.1128/MCB.16.4.1794. PMID:8657155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121-1124. doi: 10.1126/science.6719137. PMID:6719137 [DOI] [PubMed] [Google Scholar]
- 5.Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, McBride OW, Bertness V, Hollis GF, Minna JD. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature. 1985;318:69-73. doi: 10.1038/318069a0. PMID:2997622 [DOI] [PubMed] [Google Scholar]
- 6.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al.. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899-905. doi: 10.1038/nature08822. PMID:20164920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci U S A. 2012;109:8983-8988. doi: 10.1073/pnas.1203244109. PMID:22615405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsieh AL, Walton ZE, Altman BJ, Stine ZE, Dang CV. MYC and metabolism on the path to cancer. Semin Cell Dev Biol. 2015;43:11-21. doi: 10.1016/j.semcdb.2015.08.003. PMID:26277543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dang CV. MYC on the path to cancer. Cell. 2012;149:22-35. doi: 10.1016/j.cell.2012.03.003. PMID:22464321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duesberg PH, Bister K, Vogt PK. The RNA of avian acute leukemia virus MC29. Proc Natl Acad Sci U S A. 1977;74:4320-4324. doi: 10.1073/pnas.74.10.4320. PMID:200913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu SS, Lai MM, Vogt PK. Genome of avian myelocytomatosis virus MC29: analysis by heteroduplex mapping. Proc Natl Acad Sci U S A. 1979;76:1265-1268. doi: 10.1073/pnas.76.3.1265. PMID:86989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheiness D, Fanshier L, Bishop JM. Identification of nucleotide sequences which may encode the oncogenic capacity of avian retrovirus MC29. J Virol. 1978;28:600-610. PMID:214581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H. Sequence-specific DNA binding by the c-Myc protein. Science. 1990;250:1149-1151. doi: 10.1126/science.2251503. PMID:2251503 [DOI] [PubMed] [Google Scholar]
- 14.Prendergast GC, Ziff EB. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science. 1991;251:186-189doi: 10.1126/science.1987636. PMID:1987636 [DOI] [PubMed] [Google Scholar]
- 15.Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211-1217. doi: 10.1126/science.2006410. PMID:2006410 [DOI] [PubMed] [Google Scholar]
- 16.Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, Land H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature. 1992;359:423-426. doi: 10.1038/359423a0. PMID:1406955 [DOI] [PubMed] [Google Scholar]
- 17.Eilers M, Eisenman RN. Myc's broad reach. Genes Dev. 2008;22:2755-2766. doi: 10.1101/gad.1712408. PMID:18923074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orian A, van Steensel B, Delrow J, Bussemaker HJ, Li L, Sawado T, Williams E, Loo LW, Cowley SM, Yost C, et al.. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17:1101-1114. doi: 10.1101/gad.1066903. PMID:12695332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115-1129. doi: 10.1101/gad.1067003. PMID:12695333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al.. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68-79. doi: 10.1016/j.cell.2012.08.033. PMID:23021216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432-445. doi: 10.1016/j.cell.2010.03.030. PMID:20434984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976-990. doi: 10.1038/nrc2231. PMID:19029958 [DOI] [PubMed] [Google Scholar]
- 23.Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18:717-726. doi: 10.1093/emboj/18.3.717. PMID:9927431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flinn EM, Busch CM, Wright AP. myc boxes, which are conserved in myc family proteins, are signals for protein degradation via the proteasome. Mol Cell Biol. 1998;18:5961-5969. doi: 10.1128/MCB.18.10.5961. PMID:9742113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol. 2004;14:1852-1857. doi: 10.1016/j.cub.2004.09.083. PMID:15498494 [DOI] [PubMed] [Google Scholar]
- 26.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085-9090. doi: 10.1073/pnas.0402770101. PMID:15150404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20:556-562. doi: 10.1128/MCB.20.2.556-562.2000. PMID:10611234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003;4:575-580. doi: 10.1038/sj.embor.embor861. PMID:12776177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bellosta P, Hulf T, Balla Diop S, Usseglio F, Pradel J, Aragnol D, Gallant P. Myc interacts genetically with Tip48/Reptin and Tip49/Pontin to control growth and proliferation during Drosophila development. Proc Natl Acad Sci U S A. 2005;102:11799-11804. doi: 10.1073/pnas.0408945102. PMID:16087886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anso E, Mullen AR, Felsher DW, Matés JM, Deberardinis RJ, Chandel NS. Metabolic changes in cancer cells upon suppression of MYC. Cancer Metab. 2013;1:7. doi: 10.1186/2049-3002-1-7. PMID:24280108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. 2013;3:a014217. doi: 10.1101/cshperspect.a014217. PMID:23906881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-1033. doi: 10.1126/science.1160809. PMID:19460998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shchors K, Shchors E, Rostker F, Lawlor ER, Brown-Swigart L, Evan GI. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006;20:2527-2538. doi: 10.1101/gad.1455706. PMID:16980582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, et al.. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060-1065. doi: 10.1038/ng1855. PMID:16878133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, et al.. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227-231. doi: 10.1126/science.aac9935. PMID:26966191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16:2530-2543. doi: 10.1101/gad.1024602. PMID:12368264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brandvold KA, Neiman P, Ruddell A. Angiogenesis is an early event in the generation of myc-induced lymphomas. Oncogene. 2000;19:2780-2785doi: 10.1038/sj.onc.1203589. PMID:10851079 [DOI] [PubMed] [Google Scholar]
- 38.Rapp UR, Korn C, Ceteci F, Karreman C, Luetkenhaus K, Serafin V, Zanucco E, Castro I, Potapenko T. MYC is a metastasis gene for non-small-cell lung cancer. PLoS One. 2009;4:e6029. doi: 10.1371/journal.pone.0006029. PMID:19551151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho H, Herzka T, Zheng W, Qi J, Wilkinson JE, Bradner JE, Robinson BD, Castillo-Martin M, Cordon-Cardo C, Trotman LC. RapidCaP, a novel GEM model for metastatic prostate cancer analysis and therapy, reveals myc as a driver of Pten-mutant metastasis. Cancer Discov. 2014;4:318-333. doi: 10.1158/2159-8290.CD-13-0346. PMID:24444712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cowling VH, D'Cruz CM, Chodosh LA, Cole MD. c-Myc transforms human mammary epithelial cells through repression of the Wnt inhibitors DKK1 and SFRP1. Mol Cell Biol. 2007;27:5135-5146. doi: 10.1128/MCB.02282-06. PMID:17485441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerkhoff E, Houben R, Löffler S, Troppmair J, Lee JE, Rapp UR. Regulation of c-myc expression by Ras/Raf signalling. Oncogene. 1998;16:211-216. doi: 10.1038/sj.onc.1201520. PMID:9464539 [DOI] [PubMed] [Google Scholar]
- 42.Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10:57-67. doi: 10.4161/cc.10.1.14449. PMID:21200143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3:169-179. doi: 10.1016/S1097-2765(00)80308-1. PMID:10078200 [DOI] [PubMed] [Google Scholar]
- 44.Soucek L, Whitfield JR, Sodir NM, Massó-Vallés D, Serrano E, Karnezis AN, Swigart LB, Evan GI. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504-513. doi: 10.1101/gad.205542.112. PMID:23475959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan CH, Lee SW, Li CF, Wang J, Yang WL, Wu CY, Wu J, Nakayama K, Kang HY, Huang HY, et al.. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat Cell Biol. 2010;12:457-467. doi: 10.1038/ncb2047. PMID:20383141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sauzeau V, Berenjeno IM, Citterio C, Bustelo XR. A transcriptional cross-talk between RhoA and c-Myc inhibits the RhoA/Rock-dependent cytoskeleton. Oncogene. 2010;29:3781-3792. doi: 10.1038/onc.2010.134. PMID:20453885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cowling VH, Cole MD. E-cadherin repression contributes to c-Myc-induced epithelial cell transformation. Oncogene. 2007;26:3582-3586. doi: 10.1038/sj.onc.1210132. PMID:17146437 [DOI] [PubMed] [Google Scholar]
- 48.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178-196. doi: 10.1038/nrm3758. PMID:24556840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, et al.. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247-256. PMID:20173740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith AP, Verrecchia A, Fagà G, Doni M, Perna D, Martinato F, Guccione E, Amati B. A positive role for Myc in TGFbeta-induced Snail transcription and epithelial-to-mesenchymal transition. Oncogene. 2009;28:422-430. doi: 10.1038/onc.2008.395. PMID:18978814 [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Shi J, Chai K, Ying X, Zhou BP. The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets. 2013;13:963-972. doi: 10.2174/15680096113136660102. PMID:24168186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415-428. doi: 10.1038/nrc2131. PMID:17508028 [DOI] [PubMed] [Google Scholar]
- 53.Conacci-Sorrell M, Ngouenet C, Eisenman RN. Myc-nick: a cytoplasmic cleavage product of Myc that promotes alpha-tubulin acetylation and cell differentiation. Cell. 2010;142:480-493. doi: 10.1016/j.cell.2010.06.037. PMID:20691906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorimachi H, Hata S, Ono Y. Expanding members and roles of the calpain superfamily and their genetically modified animals. Exp Anim. 2010;59:549-566. doi: 10.1538/expanim.59.549. PMID:21030783 [DOI] [PubMed] [Google Scholar]
- 55.Conacci-Sorrell M, Ngouenet C, Anderson S, Brabletz T, Eisenman RN. Stress-induced cleavage of Myc promotes cancer cell survival. Genes Dev. 2014;28:689-707. doi: 10.1101/gad.231894.113. PMID:24696454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dedieu S, Poussard S, Mazères G, Grise F, Dargelos E, Cottin P, Brustis JJ. Myoblast migration is regulated by calpain through its involvement in cell attachment and cytoskeletal organization. Exp Cell Res. 2004;292:187-200. doi: 10.1016/j.yexcr.2003.08.014. PMID:14720518 [DOI] [PubMed] [Google Scholar]
- 57.Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C, Pasturaud P, Roudaut C, et al.. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27-40. doi: 10.1016/0092-8674(95)90368-2. PMID:7720071 [DOI] [PubMed] [Google Scholar]
- 58.Moretti D, Del Bello B, Allavena G, Maellaro E. Calpains and cancer: friends or enemies? Arch Biochem Biophys. 2014;564:26-36. doi: 10.1016/j.abb.2014.09.018. PMID:25305531 [DOI] [PubMed] [Google Scholar]
- 59.Conacci-Sorrell M, Eisenman RN. Post-translational control of Myc function during differentiation. Cell Cycle. 2011;10:604-610. doi: 10.4161/cc.10.4.14794. PMID:21293188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campbell K, Casanova J. A role for E-cadherin in ensuring cohesive migration of a heterogeneous population of non-epithelial cells. Nat Commun. 2015;6:7998. doi: 10.1038/ncomms8998. PMID:26272476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639-1651. doi: 10.1016/j.cell.2013.11.029. PMID:24332913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445-457. doi: 10.1038/nrm2720. PMID:19546857 [DOI] [PubMed] [Google Scholar]
- 63.Pietilainen T, Lipponen P, Aaltomaa S, Eskelinen M, Kosma VM, Syrjänen K. Expression of c-myc proteins in breast cancer as related to established prognostic factors and survival. Anticancer Res. 1995;15:959-964. PMID:7645986 [PubMed] [Google Scholar]
- 64.Wakamatsu Y, Watanabe Y, Shimono A, Kondoh H. Transition of localization of the N-Myc protein from nucleus to cytoplasm in differentiating neurons. Neuron. 1993;10:1-9. doi: 10.1016/0896-6273(93)90236-K. PMID:8427698 [DOI] [PubMed] [Google Scholar]
- 65.Okano HJ, Park WY, Corradi JP, Darnell RB. The cytoplasmic Purkinje onconeural antigen cdr2 down-regulates c-Myc function: implications for neuronal and tumor cell survival. Genes Dev. 1999;13:2087-2097. doi: 10.1101/gad.13.16.2087. PMID:10465786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aratyn YS, Schaus TE, Taylor EW, Borisy GG. Intrinsic dynamic behavior of fascin in filopodia. Mol Biol Cell. 2007;18:3928-3940. doi: 10.1091/mbc.E07-04-0346. PMID:17671164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shibue T, Brooks MW, Inan MF, Reinhardt F, Weinberg RA. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov. 2012;2:706-721. doi: 10.1158/2159-8290.CD-11-0239. PMID:22609699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ozerhan IH, Ersoz N, Onguru O, Ozturk M, Kurt B, Cetiner S. Fascin expression in colorectal carcinomas. Clinics (Sao Paulo). 2010;65:157-164. doi: 10.1590/S1807-59322010000200007. PMID:20186299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Machesky LM, Li A. Fascin: Invasive filopodia promoting metastasis. Commun Integr Biol. 2010;3:263-270. doi: 10.4161/cib.3.3.11556. PMID:20714410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamakita Y, Matsumura F, Yamashiro S. Fascin1 is dispensable for mouse development but is favorable for neonatal survival. Cell Motil Cytoskeleton. 2009;66:524-534. doi: 10.1002/cm.20356. PMID:19343791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anderson S, Poudel KR, Roh-Johnson M, Brabletz T, Yu M, Borenstein-Auerbach N, Grady WN, Bai J, Moens CB, Eisenman RN, et al.. MYC-nick promotes cell migration by inducing fascin expression and Cdc42 activation. Proc Natl Acad Sci U S A. 2016;113:E5481-5490. doi: 10.1073/pnas.1610994113. PMID:27566402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53-62. doi: 10.1016/0092-8674(95)90370-4. PMID:7536630 [DOI] [PubMed] [Google Scholar]
- 73.Arias-Romero LE, Chernoff J. Targeting Cdc42 in cancer. Expert Opin Ther Targets. 2013;17:1263-1273. doi: 10.1517/14728222.2013.828037. PMID:23957315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cotteret S, Chernoff J. The evolutionary history of effectors downstream of Cdc42 and Rac. Genome Biol. 2002;3:REVIEWS0002. doi: 10.1186/gb-2002-3-2-reviews0002. PMID:11864373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang HW, Shin MG, Lee S, Kim JR, Park WS, Cho KH, Meyer T, Heo WD. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol Cell. 2012;47:281-290. doi: 10.1016/j.molcel.2012.05.007. PMID:22683270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stengel KR, Zheng Y. Essential role of Cdc42 in Ras-induced transformation revealed by gene targeting. PLoS One. 2012;7:e37317. doi: 10.1371/journal.pone.0037317. PMID:22719838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834-840. doi: 10.1126/science.1175371. PMID:19608861 [DOI] [PubMed] [Google Scholar]
- 78.Knyphausen P, Kuhlmann N, de Boor S, Lammers M. Lysine-acetylation as a fundamental regulator of Ran function: Implications for signaling of proteins of the Ras-superfamily. Small GTPases. 2015;6:189-195. doi: 10.1080/21541248.2015.1103399. PMID:26507377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goode BL, Drubin DG, Barnes G. Functional cooperation between the microtubule and actin cytoskeletons. Curr Opin Cell Biol. 2000;12:63-71. doi: 10.1016/S0955-0674(99)00058-7. PMID:10679357 [DOI] [PubMed] [Google Scholar]
- 80.Suzuki M, Kabir SR, Siddique MS, Nazia US, Miyazaki T, Kodama T. Myosin-induced volume increase of the hyper-mobile water surrounding actin filaments. Biochem Biophys Res Commun. 2004;322:340-346. doi: 10.1016/j.bbrc.2004.07.111. PMID:15313212 [DOI] [PubMed] [Google Scholar]
- 81.Schober JM, Komarova YA, Chaga OY, Akhmanova A, Borisy GG. Microtubule-targeting-dependent reorganization of filopodia. J Cell Sci. 2007;120:1235-1244. doi: 10.1242/jcs.003913. PMID:17356063 [DOI] [PubMed] [Google Scholar]