

Two enzymes that have recently been identified from the hyperthermophilic archaeon A. pernix as components of the archaeal mevalonate pathway do not require ATP for their reactions. This pathway, therefore, might consume less energy than other mevalonate pathways to produce precursors for isoprenoids. Thus, the pathway might be applicable to metabolic engineering and production of valuable isoprenoids that have application as pharmaceuticals. The archaeal mevalonate pathway was successfully reconstructed in E. coli cells by introducing several genes from the methanogenic or hyperthermophilic archaeon, which demonstrated that the pathway requires the same components even in distantly related archaeal species and can function in bacterial cells.
KEYWORDS: archaea, isoprenoid, lycopene, methanogen, mevalonate pathway
ABSTRACT
The mevalonate pathway is a well-known metabolic route that provides biosynthetic precursors for myriad isoprenoids. An unexpected variety of the pathway has been discovered from recent studies on microorganisms, mainly on archaea. The most recently discovered example, called the “archaeal” mevalonate pathway, is a modified version of the canonical eukaryotic mevalonate pathway and was elucidated in our previous study using the hyperthermophilic archaeon Aeropyrum pernix. This pathway comprises four known enzymes that can produce mevalonate 5-phosphate from acetyl coenzyme A, two recently discovered enzymes designated phosphomevalonate dehydratase and anhydromevalonate phosphate decarboxylase, and two more known enzymes, i.e., isopentenyl phosphate kinase and isopentenyl pyrophosphate:dimethylallyl pyrophosphate isomerase. To show its wide distribution in archaea and to confirm if its enzyme configuration is identical among species, the putative genes of a lower portion of the pathway—from mevalonate to isopentenyl pyrophosphate—were isolated from the methanogenic archaeon Methanosarcina mazei, which is taxonomically distant from A. pernix, and were introduced into an engineered Escherichia coli strain that produces lycopene, a red carotenoid pigment. Lycopene production, as a measure of isoprenoid productivity, was enhanced when the cells were grown semianaerobically with the supplementation of mevalonolactone, which demonstrates that the archaeal pathway can function in bacterial cells to convert mevalonate into isopentenyl pyrophosphate. Gene deletion and complementation analysis using the carotenogenic E. coli strain suggests that both phosphomevalonate dehydratase and anhydromevalonate phosphate decarboxylase from M. mazei are required for the enhancement of lycopene production.
IMPORTANCE Two enzymes that have recently been identified from the hyperthermophilic archaeon A. pernix as components of the archaeal mevalonate pathway do not require ATP for their reactions. This pathway, therefore, might consume less energy than other mevalonate pathways to produce precursors for isoprenoids. Thus, the pathway might be applicable to metabolic engineering and production of valuable isoprenoids that have application as pharmaceuticals. The archaeal mevalonate pathway was successfully reconstructed in E. coli cells by introducing several genes from the methanogenic or hyperthermophilic archaeon, which demonstrated that the pathway requires the same components even in distantly related archaeal species and can function in bacterial cells.
INTRODUCTION
The mevalonate (MVA) pathway is a well-known metabolic route that supplies isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMAPP) as biosynthetic precursors for isoprenoids, which are the largest group of natural compounds (1, 2). The pathway was discovered in the late 1950s through studies with eukaryotes such as yeast and had been considered the sole metabolic pathway that could synthesize IPP and DMAPP until the methylerythritol phosphate (MEP) pathway was isolated from a wide range of bacteria. Recent studies, however, have revealed an unexpected variety in the MVA pathway (3–9). Organisms that include most archaea and some Chloroflexi bacteria have been shown to possess partially modified pathways, which proceed through a few intermediates that do not exist in the canonical eukaryotic MVA pathway. To date, three different modified pathways have been discovered: the haloarchaea-type MVA pathway (4, 7), the Thermoplasma-type MVA pathway (3, 8, 9), and the archaeal MVA pathway (6). The archaeal MVA pathway, which was named because of its probable conservation in most archaea, was recently discovered in a study by our group using a hyperthermophilic archaeon, Aeropyrum pernix (Fig. 1). We proposed that two previously undiscovered enzymes, phosphomevalonate dehydratase (PMDh) and anhydromevalonate phosphate decarboxylase (AMPD), were involved in the pathway. Along with known isopentenyl phosphate kinase (IPK), PMDh and AMPD function in A. pernix cells to convert mevalonate 5-phosphate (MVA5P) into IPP via the specific intermediates trans-anhydromevalonate 5-phosphate (tAHMP) and isopentenyl phosphate. In contrast, in the eukaryotic MVA pathway, the conversion from MVA5P into IPP is catalyzed by phosphomevalonate kinase and diphosphomevalonate decarboxylase via mevalonate 5-pyrophosphate. These two pathways have a common upstream portion where the conversion of three molecules of acetyl coenzyme A (acetyl-CoA) into MVA5P is catalyzed by acetoacetyl-CoA thiolase, 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthase, HMG-CoA reductase, and mevalonate kinase (MVK). Also, the interconversion between IPP and DMAPP is commonly catalyzed by isopentenyl pyrophosphate:dimethylallyl pyrophosphate isomerase (IDI), although two unrelated types of the enzyme have now been discovered (1). The homologous genes encoding PMDh and AMPD are conserved in a wide range of archaea, such as thermophiles and methanogens, suggesting that most archaea utilize the archaeal MVA pathway. It remains unclear, however, if the same set of enzymes is utilized in pathways from species distant from A. pernix.
FIG 1.
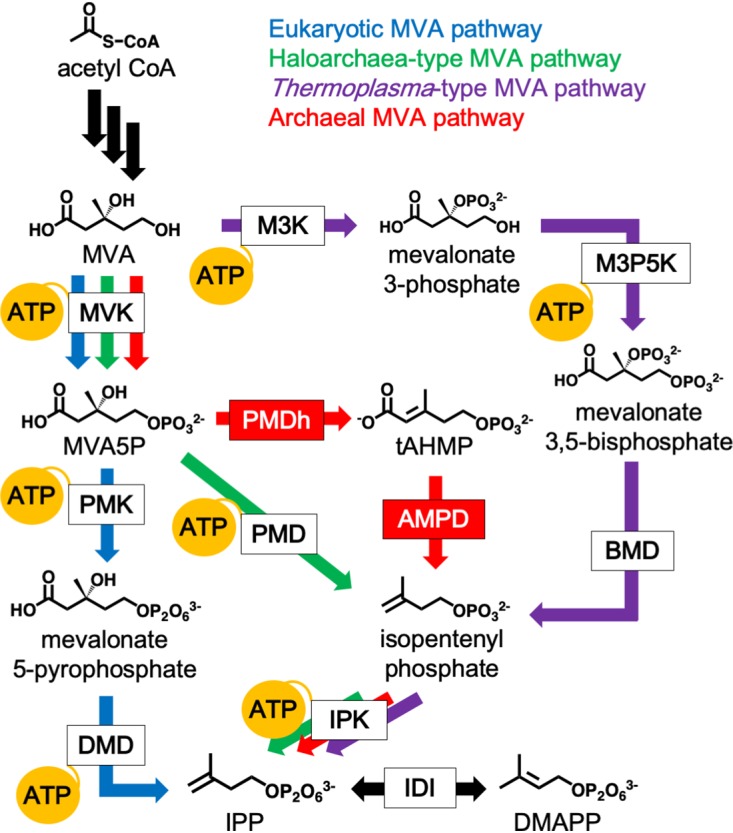
The variety of MVA pathways and their ATP consumption. The canonical eukaryotic MVA pathway, which is utilized by almost all eukaryotes, some bacteria, and archaea of the order Sulfolobales, is shown by blue arrows. The haloarchaea-type MVA pathway, found in all archaea of the class Halobacteria and in some bacteria of the phylum Chloroflexi, is shown by green arrows. The Thermoplasma-type MVA pathway, utilized in archaea of the order Thermoplasmatales, is shown by purple arrows. The archaeal MVA pathway, probably conserved in a wide range of archaea, is shown by red arrows. Black arrows indicate common reactions. The reactions that consume ATP are indicated by yellow balloons. The abbreviated names of enzymes are boxed, and those in red boxes, i.e., PMDh and AMPD, were discovered in a recent study (6). BMD, bisphosphomevalonate decarboxylase; DMD, diphosphomevalonate decarboxylase; M3K, mevalonate 3-kinase; M3P5K, mevalonate 3-phosphate 5-kinase; PMD, phosphomevalonate decarboxylase; PMK, phosphomevalonate kinase.
Metabolic engineering of microorganisms with either an exogenous or an endogenous MVA pathway is often adopted to increase the productivity of valuable isoprenoids as pharmaceuticals and industrial raw materials (10, 11). For example, introduction of the MVA pathway in Escherichia coli, which intrinsically possesses the MEP pathway, leads to much higher isoprenoid production (12). The MVA pathway used for this purpose was mainly the eukaryotic version, although some studies have tested the abilities of haloarchaea-type and Thermoplasma-type pathways to enhance isoprenoid biosynthesis (13, 14). To convert one molecule of MVA into IPP, the recently discovered archaeal MVA pathway consumes two molecules of ATP, which is required by both MVK and IPK. This ratio of ATP consumption to IPP formation is lower than those of the other MVA pathways, which require three ATP molecules for the same conversion process. Therefore, the archaeal MVA pathway is considered an “energy-saving” example, which might be advantageous when the pathway is utilized for metabolic engineering to enhance isoprenoid productivity.
In the present study, the effect that the archaeal MVA pathway exerts on isoprenoid production was studied by introducing ortholog genes from a methanogenic archaeon, Methanosarcina mazei, into E. coli cells. M. mazei was chosen because it is taxonomically distant from A. pernix and because it is a mesophile, whose enzymes are expected to show a high level of activity at the growth temperature of E. coli. The utility of the recently discovered enzymes PMDh and AMPD was also confirmed via gene deletion and complementation analysis. The results of this study corroborate the configuration of the archaeal MVA pathway reported in the previous study (6) and suggest its applicability for metabolic engineering.
RESULTS
Reconstruction of the archaeal MVA pathway in E. coli cells.
We set out to construct plasmids for the reconstruction of the archaeal MVA pathway in E. coli. To simplify the plasmid construction, we decided to exclude the genes for the upper half of the pathway up to the formation of MVA, which are the genes of acetoacetyl-CoA thiolase, HMG-CoA synthase, and HMG-CoA reductase. As mevalonolactone (MVL) is spontaneously incorporated into E. coli cells and then hydrolyzed to give MVA, we added it to a growth medium to achieve the biosynthesis of IPP/DMAPP through the lower half of the pathway. In this approach, the contribution of the exogenous MVA pathway for isoprenoid production could be easily discriminated from that of the endogenous methylerythritol phosphate (MEP) pathway by removing MVL from the medium.
We separated the enzymes of the lower pathway into well-studied and poorly studied groups. The plasmid harboring the genes encoding MVK, IPK, and IDI, which have been studied in detail (1, 2), was constructed first. The genes of M. mazei MVK, E. coli IDI, and M. mazei IPK were successively inserted into the same vector to construct the plasmid pBAD-mMP3. We utilized the bacterial IDI gene because its expression was known to significantly enhance isoprenoid biosynthesis (15, 16) and also because we found it difficult to realize the recombinant expression of the M. mazei enzyme. Then we constructed the plasmids that encode the relatively uncharacterized enzymes, which included the large and small subunits of PMDh (PMDh-L and PMDh-S, respectively), AMPD, and prenylated flavin mononucleotide (FMN) synthase (PFS). PFS is the enzyme that catalyzes the formation of prenylated FMN (PF), which is the cofactor likely required for the activity of AMPD. The feasibility of the recombinant expression of the putative ortholog genes from M. mazei was unclear, and, therefore, we used the corresponding genes from A. pernix as a positive control, because their recombinant expression in E. coli had already been reported (6). The amino acid sequence identities between the A. pernix enzymes and corresponding M. mazei homologs are 36.8% for PMDh-L, 40.4% for PMDh-S, 39.7% for AMPD, and 36.3% for PFS. We constructed two plasmids, pBAD-mUA4 and pBAD-aUA4, which harbor the four genes from M. mazei and A. pernix, respectively. Finally, the artificial operons, including the putative or verified genes of AMPD, PFS, PMDh-L, and PMDh-S, were amplified from pBAD-mUA4 and pBAD-aUA4 and then inserted into pBAD-mMP3 to construct pBAD-mMP7 and pBAD-aMP7, respectively (Fig. 2A).
FIG 2.

The schemes of plasmid construction and lycopene production in E. coli cells. (A) The construction of plasmids for the expression of the archaeal MVA pathway. The DNA fragments containing AMPD, PFS, PMDh-L, and PMDh-S amplified from pBAD-mUA4, pBAD-msUA4, pBAD-aUA4, and pBAD-asUA4 were inserted into pBAD-mMP3 digested with EcoRI by in vivo homologous recombination to construct pBAD-mMP7, pBAD-msMP7, pBAD-aMP7, and pBAD-asMP7, respectively. (B) The pathway of lycopene production in E. coli cells via the archaeal MVA pathway. MVL is spontaneously incorporated into E. coli cells and then hydrolyzed to give MVA. Lycopene is biosynthesized from IPP and DMAPP by the actions of endogenous E. coli farnesyl pyrophosphate synthase and exogenous carotenogenic enzymes from Pantoea ananatis, i.e., geranylgeranyl pyrophosphate synthase (CrtE), phytoene synthase (CrtB), and phytoene desaturase (CrtI).
To investigate the isoprenoid productivity of E. coli, we exploited an engineered system for the production of lycopene, which is a red-colored carotenoid pigment biosynthesized from IPP and DMAPP and therefore used as a measure of isoprenoid biosynthesis (Fig. 2B). E. coli cells harboring the plasmid pACYC-IBE (15), which contains three carotenogenic genes sufficient for the biosynthesis of lycopene in E. coli, were transformed with pBAD-mMP7, pBAD-aMP7, or an empty vector, pBAD18, as a negative control to construct the mMP7, aMP7, and NC strains, respectively. After aerobic cultivation of each strain at 30°C for 24 h in a rich medium with or without MVL, the cells were harvested. Lycopene was extracted with acetone to be quantified by its specific UV absorption (Fig. 3). As shown in Fig. 3A, the cells of the NC strain showed a deep red color in either the absence or presence of MVL, suggesting that the endogenous MEP pathway is effective under these conditions. In contrast, lycopene production in the mMP7 and aMP7 strains, which was standardized by dividing it by the optical density at 600 nm (OD600) of the cell culture, was much lower than that in the NC strain even when MVL was added to the medium, due to unclear reasons. One possibility is that high-level expression of the recombinant proteins might deprive the cells of energy for isoprenoid production. When fed MVL, the mMP7 strain did not largely increase the lycopene production over growth in the absence of MVL, while lycopene production was decreased in the aMP7 strain.
FIG 3.
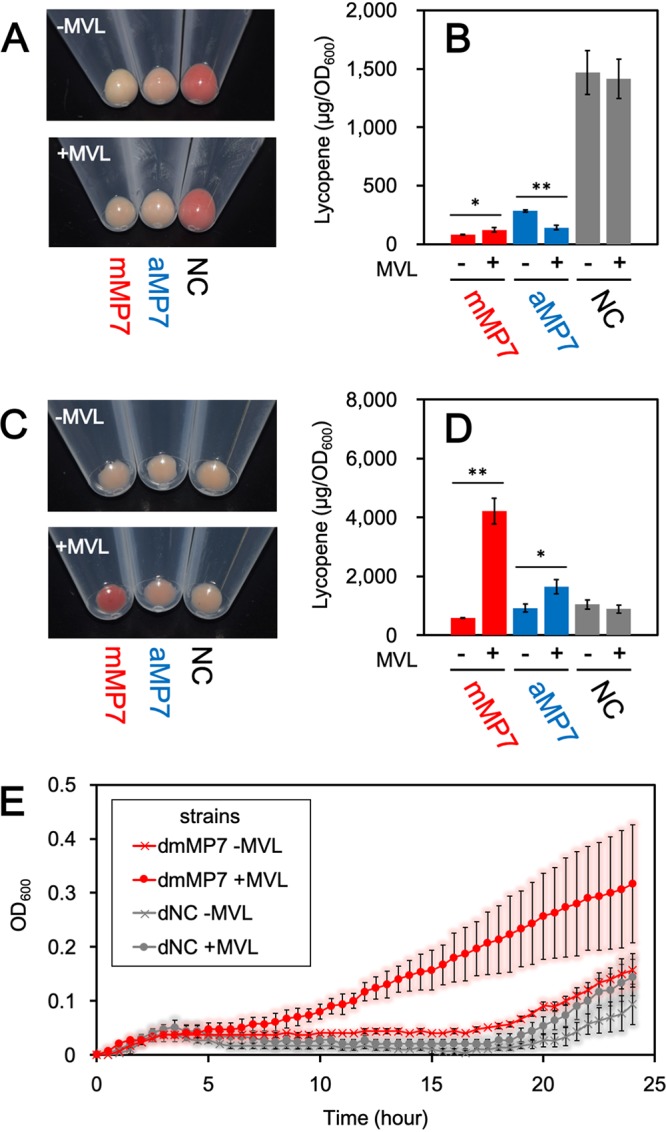
Lycopene production in E. coli strains harboring the archaeal MVA pathway. (A) The cell pellet of each strain cultivated aerobically. (B) The amount of lycopene extracted from each strain cultivated aerobically in the medium supplemented with or without 5.0 mg/ml MVL. Cultivation and lycopene extraction were performed in triplicate. Student’s t test values: **, P < 0.01; *, P < 0.03. (C) The cell pellet of each strain cultivated semianaerobically. (D) The amount of lycopene extracted from each strain cultivated semianaerobically in the medium supplemented with or without 5.0 mg/ml MVL. (E) The growth curves of dmMP7 and dNC strains with or without supplementation of MVL. Each strain was cultivated semianaerobically in a medium containing 100 μM fosmidomycin. Cultivation and measurement of OD600 were performed in triplicate.
Therefore, we tried to improve the efficiency of the archaeal MVA pathway, at least compared with that of the endogenous MEP pathway. In a previous study we reported the sensitivity of PMDh to oxygen (6), which led us to expect a reduction in the efficiency of the pathway during aerobic growth. Therefore, the cells were cultivated semianaerobically by putting paraffin wax on the medium and growing at 30°C for 24 h. Under these growth conditions, the lycopene production of the NC strain was significantly reduced, which served our purpose of determining the feasibility of the exogenous archaeal MVA pathway (Fig. 3C and D). Although the mMP7 strain produced a smaller amount of lycopene than the NC strain did in the absence of MVL, its lycopene productivity increased 7.3-fold when MVL was supplemented and became higher than that of the NC strain. When MVL was not supplemented, the lycopene production of the aMP7 strain was comparable to that of the NC strain, but it increased 1.8-fold with the addition of MVL to the medium. These results suggest that the archaeal enzymes expressed in E. coli cells form a metabolic route to synthesize the precursors for isoprenoid biosynthesis.
There still remains a possibility, however, that the introduction of exogenous genes and the supplement of MVL enhance the activity of the endogenous MEP pathway by some unknown method. To directly prove that the reconstructed lower half of the archaeal MVA pathway actually functions in E. coli cells, the effect of the MEP pathway must be removed. Because the complete inhibition of the MEP pathway is lethal to E. coli, we performed growth-complementation assays using fosmidomycin, a well-known inhibitor of 1-deoxy-d-xylulose 5-phosphate reductoisomerase, which is the second enzyme in the MEP pathway. Although some E. coli strains were reported to be insensitive to fosmidomycin, we utilized the DH1 strain, which was sensitive to that inhibitor (Fig. S1 in the supplemental material) (17). The strain was transformed with either pBAD-mMP7 or pBAD18 to construct dmMP7 and dNC strains, respectively. The growth of each strain in an LB medium containing fosmidomycin was monitored by measuring OD600 (Fig. 3E). The growth of the dNC strain was significantly inhibited until ∼20 h after the start of cultivation, even when MVL was added to the culture medium, indicating that fosmidomycin effectively inhibited the growth of the E. coli DH1 strain and that the supplement of MVL did not affect its growth. In the case of the dmMP7 strain, cell growth was similarly inhibited by fosmidomycin when MVL was not supplemented. In contrast, the growth of the dmMP7 strain was markedly recovered by the addition of MVL, which clearly shows that the reconstructed archaeal MVA pathway can function to synthesize IPP and DMAPP from MVA in E. coli cells.
PFS genes from different origins and their effect on lycopene production.
AMPD belongs to the UbiD-type decarboxylase family, which requires the recently discovered coenzyme PF (18–20). The formation of prenylated FMN (PF) is catalyzed by the flavin prenyltransferase PFS (21). Although E. coli is known to possess endogenous PFS for the biosynthesis of ubiquinone, we added a PFS gene from archaea for the construction of the plasmids pBAD-mMP7 and pBAD-aMP7 because our study on A. pernix AMPD showed that the enzyme was mostly expressed in E. coli as an apo-form that lacked PF (unpublished data). Interestingly, most of the characterized PFSs to date are known to utilize dimethylallyl phosphate as a substrate for the prenylation of FMN, whereas PFS from Saccharomyces cerevisiae, called PAD1, is known to use DMAPP instead (20–22). DMAPP is a ubiquitous biosynthetic precursor of isoprenoids. Because dimethylallyl phosphate is considered to be supplied in E. coli cells by the hydrolysis of DMAPP, we expected that S. cerevisiae PFS might work better in E. coli cells by comparison with archaeal orthologs. In expectation of improvement in the isoprenoid productivity through the archaeal MVA pathway, we replaced the archaeal PFS genes in pBAD-mMP7 and pBAD-aMP7 with the PFS gene from S. cerevisiae to construct the plasmids pBAD-msMP7 and pBAD-asMP7, respectively. E. coli harboring pACYC-IBE was transformed with pBAD-msMP7 or pBAD-asMP7 to develop the strains msMP7 and asMP7, respectively. After semianaerobic cultivation for 24 h at 30°C, the lycopene production of each strain was measured. Contrary to our expectations, however, lycopene production in the asMP7 and msMP7 strains was not increased by the addition of MVL (Fig. 4). This result can be explained either by the failure of the expression of S. cerevisiae PFS in these systems or by the hypothesis that PFS from the same archaeal species is preferable for the activation of AMPD, but it suggests at least that endogenous E. coli PFS is insufficient for the activation of recombinantly expressed AMPD.
FIG 4.

Replacement of archaeal PFS genes with the yeast ortholog. (A) The cell pellet of each strain cultivated semianaerobically. (B) The amount of lycopene extracted from each strain cultivated semianaerobically in the medium supplemented with or without 5.0 mg/ml MVL. Cultivation and lycopene extraction were performed in triplicate. Student’s t test values: **, P < 0.01; *, P < 0.03.
Gene deletion and complementation studies of the archaeal MVA pathway.
The lycopene productivity of E. coli harboring either pBAD-aMP7 or pBAD-mMP7 was improved by supplementation with MVL, which suggests that the archaeal MVA pathway was successfully reconstructed in E. coli cells, but it did not definitively show that all the introduced archaeal genes were responsible for the pathway. Although the reconstruction of the canonical MVA pathway in E. coli cells has been studied intensively, there remains the possibility that either of the poorly studied reactions catalyzed by the recently discovered enzymes PMDh and AMPD might be catalyzed nonenzymatically or by endogenous enzymes in E. coli cells. To exclude this possibility, we performed deletion and complementation of the genes of PMDh and AMPD using the above-mentioned system that utilizes lycopene production in E. coli. First, we constructed the plasmid pBAD-mMPdelAMPD by inserting the artificial operon containing the genes of PFS, PMDh-L, and PMDh-S from M. mazei into pBAD-mMP3. The constructed plasmid corresponds to the AMPD-deleted version of pBAD-mMP7. To complement the gene deficiency, the gene encoding M. mazei AMPD was inserted into pBAD-mMPdelAMPD, although at a position downstream of the MVK gene, and the complemented plasmid was named pBAD-mMPcomAMPD. In a similar manner, an artificial operon containing the genes of AMPD and PFS was inserted into pBAD-mMP3 to construct pBAD-mMPdelPMDh, which corresponded to the PMDh-deleted version of pBAD-mMP7. The genes encoding M. mazei PMDh-L and PMDh-S were inserted into pBAD-mMPdelPMDh at a location downstream of the MVK gene to construct a complemented plasmid, pBAD-mMPcomPMDh. The plasmid pBAD-mMPdelAMPD, pBAD-mMPcomAMPD, pBAD-mMPdelPMDh, or pBAD-mMPcomPMDh was introduced into E. coli harboring pACYC-IBE to construct E. coli strains delAMPD, comAMPD, delPMDh, and comPMDh, respectively. Fig. 5 shows the results from the measurement of lycopene produced in each strain, which was semianaerobically cultivated for 24 h at 30°C. Lycopene productivity of the E. coli strains delAMPD and delPMDh was not enhanced by the addition of MVL to the medium. In contrast, the complemented strains recovered the MVL-evoked increase in lycopene production. The addition of MVL to the comAMPD and comPMDh strains resulted in lycopene productivity that was 7.7- and 11-fold higher, respectively, than that in strains cultivated without MVL. These results suggest that both AMPD and PMDh are strictly required for the reconstruction of the archaeal MVA pathway in E. coli.
FIG 5.

Gene deletion and complementation studies. (A) The cell pellet of each strain cultivated semianaerobically. (B) The amount of lycopene extracted from each strain cultivated semianaerobically in the medium supplemented with or without 5.0 mg/ml MVL. Cultivation and lycopene extraction were performed in triplicate. Student’s t test value: **, P < 0.01.
DISCUSSION
Reconstruction of the archaeal MVA pathway (albeit the lower half of it) successfully increased the isoprenoid productivity of E. coli in the presence of MVL. The effect appeared stronger when a pathway was constructed using M. mazei enzymes rather than A. pernix enzymes, likely because the enzymes from the mesophilic species act more efficiently than those from the hyperthermophilic species in bacterial cells grown at 30°C. Importantly, these results suggest a set of enzymes from M. mazei that are required for the archaeal MVA pathway, which agrees with the results found in our previous study of the pathway from A. pernix (6). M. mazei is classified in the phylum Euryarchaeota, which is taxonomically distant from A. pernix in the phylum Crenarchaeota. Nevertheless, both M. mazei and A. pernix likely possess the enzymes MVK, PMDh, AMPD, and IPK, which are the same set used for the conversion of MVA into IPP, along with PFS that is used in the formation of PF and is required for the activation of AMPD. The putative orthologs of the enzymes, along with those of highly conserved acetoacetyl-CoA thiolase, HMG-CoA synthase, and HMG-CoA reductase for the upper half of the pathway and of IDI for the biosynthesis of DMAPP, are encoded in the genomes of most archaea, which therefore are considered to possess the archaeal MVA pathway.
The archaeal MVA pathway consumes fewer ATP molecules than the other MVA pathways and would therefore be expected to offer advantages for the bio-production of isoprenoids, particularly in cases where the host cells cannot obtain sufficient ATP because of poor respiration. Essentially, this pathway could be suitable for anaerobes. This idea is supported by the fact that almost all anaerobic archaea seem to possess the archaeal MVA pathway whereas some, although not all, aerobic archaea such as haloarchaea, Sulfolobus spp., and Thermoplasma spp. utilize other MVA pathways (3, 6–9, 23). Indeed, enhancement of lycopene production in E. coli through the activation of the archaeal MVA pathway, which was achieved by feeding MVL to the host, was significant only when E. coli cells were grown semianaerobically. This high efficiency of the pathway under anaerobic growth conditions could be the result of its “energy-saving” nature, while it is also conceivable that the archaeal MVA pathway might be inhibited under oxidative growth conditions owing to, for example, the sensitivity of PMDh to oxidation. If that is the case, the archaeal MVA pathway might be promising if applied to bio-production on a large scale where a sufficient supply of oxygen is problematic. This could be assessed by improving the current experimental system such that MVA would be biosynthesized in E. coli cells by introducing the upper half of the MVA pathway and more effective genes could be selected from various archaeal species, allowing the optimization of expression levels.
MATERIALS AND METHODS
General procedures.
Restriction enzyme digestions, transformations, and other standard molecular biological techniques were carried out as described by Sambrook et al. (24).
Cultivation of microorganisms.
M. mazei Gö1 and A. pernix were provided by the RIKEN BRC through the Natural Bio-Resource Project of the MEXT, Japan. M. mazei was cultured in a DSMZ120 Methanosarcina medium at 30°C. A. pernix was cultured in a JXT medium supplemented with 4 mM Na2S2O3 · 5H2O at 85°C. S. cerevisiae was cultured in yeast extract-peptone-dextrose (YPD) medium at 30°C.
Plasmid construction for the expression of archaeal MVA pathway genes.
All plasmids used in this study are listed in Table 1. PCRs were performed using KOD plus DNA polymerase (TOYOBO, Japan), and the primers are shown in Table 2. The genomic DNA of M. mazei, A. pernix, and S. cerevisiae was extracted from cells using a DNA extraction kit, Geno Plus Mini (Viogene, USA).
TABLE 1.
Plasmids and E. coli strains used in this study
| Plasmid or strain | Relevant characteristics | Reference or source |
|---|---|---|
| Plasmids | ||
| pBAD18 | AmpR, pBR322, PARA | 27 |
| pBAD-MVK | AmpR, pBR322, PARA, MM_1762 | This study |
| pBAD-mMP2 | AmpR, pBR322, PARA, JW2857, MM_1762 | This study |
| pBAD-mMP3 | AmpR, pBR322, PARA, MM_1763, JW2857, MM_1762 | This study |
| pBAD-mUA1 | AmpR, pBR322, PARA, MM_1524 | This study |
| pBAD-mUA2 | AmpR, pBR322, PARA, MM_1525, MM_1524 | This study |
| pBAD-mUA3 | AmpR, pBR322, PARA, MM_1871, MM_1525, MM_1524 | This study |
| pBAD-msUA3 | AmpR, pBR322, PARA, YDR538W, MM_1525, MM_1524 | This study |
| pBAD-mUA4 | AmpR, pBR322, PARA, MM_1526, MM_1871, MM_1525, MM_1524 | This study |
| pBAD-msUA4 | AmpR, pBR322, PARA, MM_1526, YDR538W, MM_1525, MM_1524 | This study |
| pBAD-mMP7 | AmpR, pBR322, PARA, MM_1526, MM_1871, MM_1525, MM_1524, MM_1763, JW2857, MM_1762 | This study |
| pBAD-msMP7 | AmpR, pBR322, PARA, MM_1526, YDR538W, MM_1525, MM_1524, MM_1763, JW2857, MM_1762 | This study |
| pBAD-aUA1 | AmpR, pBR322, PARA, APE_2089 | This study |
| pBAD-aUA2 | AmpR, pBR322, PARA, APE_2087.1, APE_2089 | This study |
| pBAD-aUA3 | AmpR, pBR322, PARA, APE_1647, APE_2087.1, APE_2089 | This study |
| pBAD-asUA3 | AmpR, pBR322, PARA, YDR538W, APE_2087.1, APE_2089 | This study |
| pBAD-aUA4 | AmpR, pBR322, PARA, APE_2078, APE_1647, APE_2087.1, APE_2089 | This study |
| pBAD-asUA4 | AmpR, pBR322, PARA, APE_2078, YDE538W, APE_2087.1, APE_2089 | This study |
| pBAD-aMP7 | AmpR, pBR322, PARA, APE_2078, APE_1647, APE_2087.1, APE_2089, MM_1763, JW2857, MM_1762 | This study |
| pBAD-asMP7 | AmpR, pBR322, PARA, APE_2078, YDE538W, APE_2087.1, APE_2089, MM_1763, JW2857, MM_1762 | This study |
| pBAD-mMPdelAMPD | AmpR, pBR322, PARA, MM_1871, MM_1525, MM_1524, MM_1763, JW2857, MM_1762 | This study |
| pBAD-mMPcomAMPD | AmpR, pBR322, PARA, MM_1871, MM_1525, MM_1524, MM_1763, JW2857, MM_1762, MM_1526 | This study |
| pBAD-mMPdelPMDh | AmpR, pBR322, PARA, MM_1526, MM_1871, MM_1763, JW2857, MM_1762 | This study |
| pBAD-mMPcomPMDh | AmpR, pBR322, PARA, MM_1526, MM_1871, MM_1763, JW2857, MM_1762, MM_1525, MM_1524 | This study |
| pACYC-IBE | TetR, p15A, CrtE, CrtI, CrtB | 15 |
| E. coli strain | ||
| TOP10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 Δ lacX74 recA1 araD139 Δ(araleu)7697 galU galK rpsL (StrR) endA1 nupG | Life Technologies, USA |
| ME9783 (SN1071) | F− thr-1 leuB6 thi-1 lacY1 galK2 ara-4 xyl-5 mtl-1 proA2 his-60 argE3 rpsL31 tsx-33 supE44 recB21 recC22 sbcA23 Δ(hsdR::frt) | NBRP-E. coli (NIG, Japan) (26) |
| DH1 | F− recA1 endA1 gyrA96 thi-1 hsdR17(r-K, m-K) supE44 relA1 λ- | NBRP-E. coli (NIG, Japan) |
| NC | TOP10 with pBAD18 and pACYC-IBE | This study |
| mMP7 | TOP10 with pBAD-mMP7 and pACYC-IBE | This study |
| msMP7 | TOP10 with pBAD-msMP7 and pACYC-IBE | This study |
| aMP7 | TOP10 with pBAD-aMP7 and pACYC-IBE | This study |
| asMP7 | TOP10 with pBAD-asMP7 and pACYC-IBE | This study |
| delAMPD | TOP10 with pBAD-mMPdelAMPD and pACYC-IBE | This study |
| comAMPD | TOP10 with pBAD-mMPcomAMPD and pACYC-IBE | This study |
| delPMDh | TOP10 with pBAD-mMPdelPMDh and pACYC-IBE | This study |
| comPMDh | TOP10 with pBAD-mMPcomPMDh and pACYC-IBE | This study |
| dNC | DH1 with pBAD18 | This study |
| dmMP7 | DH1 with pBAD-mMP7 | This study |
TABLE 2.
Primers used in this study
| Gene | Primer sequence | Constructed plasmid |
|---|---|---|
| MM_1762 | forward: 5′-AGCTGGGTACCAAGAGATATAATATGGTTTCATGTTCTGCGCC-3′ | pBAD-MVK |
| reverse: 5′-GATGCTCTAGATTAATCGACCTTCAACCCCTGTTC-3′ | ||
| JW2857 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAGAGGAATTATAATGATAATGCAAACGGAACAC-3′ | pBAD-mMP2 |
| reverse: 5′-CTTGGTACCGAGCTCGAATTATTATTTAAGCTGGGTAAATGCAG-3′ | ||
| MM_1763 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAGGAGAAATATAATGAATGCTTCAAACGAACC-3′ | pBAD-mMP3 |
| reverse: 5′-CATTATAATTCCTCTGAATTATTATACCCTTTTATCCGGACTG-3′ | ||
| MM_1524 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAAGAGGAATATTATGATTCCCATTAAATTCAAAGG-3′ | pBAD-mUA1 |
| reverse: 5′-CCGGGTACCGAGCTCGAATTATTATTTTTTTGCCTGCCCTTC-3′ | ||
| MM_1525 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAAGGAGAATTATATGTATCTAACAAAAGAAGAAGAACAAATC-3′ | pBAD-mUA2 |
| reverse: 5′-CATAATATTCCTCTTGAATTATTAATGGGAATCACCTGCC-3′ | ||
| MM_1871 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAGGAGGAATAATATGGAAATAATTGTAGGCATCAG-3′ | pBAD-mUA3 |
| reverse: 5′-CATATAATTCTCCTTGAATTATTATCCGTTGTTCTCCAAATC-3′ | ||
| YDR538W | forward: 5′-TTTTTTTGGGCTAGCGAATTCAGGAGGAATAATATGCTCCTATTTCCAAGAAGAAC-3′ | pBAD-msUA3 |
| reverse: 5′-CATATAATTCTCCTTGAATTATTACTTGCTTTTTATTCCTTCCC-3′ | pBAD-asUA3 | |
| MM_1526 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAAGAAGATTATTATGACTTATAGAGATTTTATAGGCCG-3′ | pBAD-mUA4 |
| reverse: 5′-CATATTATTCCTCCTGAATTATTATTCTTCCGGAATTATTGC-3′ | pBAD-msUA4 | |
| MM_1526–MM_1524 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAAGAAGATTATTATGACTTATAGAGATTTTATAGGCCG-3′ | pBAD-mMP7 |
| reverse: 5′-CATTATATTTCTCCTGAATTATTATTTTTTTGCCTGCCCTTC-3′ | pBAD-msMP7 | |
| APE_2089 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAAGAGGAATATTATGGAGGTCCTCAGGTACAG-3′ | pBAD-aUA1 |
| reverse: 5′-CCGGGTACCGAGCTCGAATTATTAGGGCTGGCCAGG-3′ | ||
| APE_2087.1 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAAGGAGAATTATATGTACCTGACGAGGGAGG-3′ | pBAD-aUA2 |
| reverse: 5′-CATAATATTCCTCTTGAATTTTACGACACCAGCTTTGGC-3′ | ||
| APE_1647 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAGGAGGAATAATATGTCCTGCAGACCTAGCTCGG-3′ | pBAD-aUA3 |
| reverse: 5′-CATATAATTCTCCTTGAATTATTAATCCCCCTCCTCGG-3′ | ||
| APE_2078 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAAGAAGATTATTATGGAGGATGCGAGTCTAAAG-3′ | pBAD-aUA4 |
| reverse: 5′-CATATTATTCCTCCTGAATTATTAGTCTCCCGGCTGGTG-3′ | pBAD-asUA4 | |
| APE_2078–APE_2089 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAAGAAGATTATTATGGAGGATGCGAGTCTAAAG-3′ | pBAD-aMP7 |
| reverse: 5′-CATTATATTTCTCCTGAATTATTAGGGCTGGCCAGG-3′ | pBAD-asMP7 | |
| MM_1871/MM_1525/MM_1524 | forward: 5′-TTTTTTTGGGCTAGCGAATTCAGGAGGAATAATATGGAAATAATTGTAGGCATCAG-3′ | pBAD-mMPdelAMPD |
| reverse: 5′-CATTATATTTCTCCTGAATTATTATTTTTTTGCCTGCCCTTC-3′ | ||
| MM_1526 | forward: 5′-GGAACAGGGGTTGAAGGTCGATTAATCTAGCAAGAAGATTATTATGACTTATAGAGATTTTATAGG-3′ | pBAD-mMPcomAMPD |
| reverse: 5′-CAAGCTTGCATGCCTGCAGGTCGACTCTAGATTATTCTTCCGGAATTATTGC-3′ | ||
| MM_1526/MM_1871 | forward: 5′-TCCATACCCGTTTTTTTGGGCTAGCGAATTCAAGAAGATTATTATGACTTATAGAGATTTTATAGGCCG-3′ | pBAD-mMPdelPMDh |
| reverse: 5′-TTGAAGCATTCATTATATTTCTCCTGAATTATTATCCGTTGTTCTCCAAATC-3′ | ||
| MM_1525/MM_1524 | forward: 5′-GGAACAGGGGTTGAAGGTCGATTAATCTAGCAAGGAGAATTATATGTATCTAACAAAAGAAGAAG-3′ | pBAD-mMPcomPMDh |
| reverse: 5′-CAAGCTTGCATGCCTGCAGGTCGACTCTAGATTATTTTTTTGCCTGCCCTTC-3′ |
The MM_1762 gene, encoding MVK, was amplified using the genomic DNA of M. mazei as a template. The amplified gene was digested by restriction enzymes KpnI and XhoI, and was then ligated into pBAD18, where it was digested by the same restriction enzymes to construct pBAD-MVK. The JW2857 gene, encoding IDI from E. coli, was amplified using the plasmid pJBEI-2999 (25) as a template. The amplified gene and EcoRI-digested pBAD-MVK were introduced into the E. coli ME9783 (also called SN1071) strain (NBRP [National BioResource Project]-E. coli, NIG, Japan), which enabled in vivo E. coli cloning (iVEC) based on homologous recombination (26). Then, a plasmid in which JW2857 was inserted was extracted from an E. coli clone and was designated pBAD-mMP2. In a similar manner using the E. coli ME9783 strain, the MM_1763 gene, encoding putative IPK, was amplified and inserted into EcoRI-digested pBAD-mMP2 to construct pBAD-mMP3.
MM_1524, encoding putative PMDh-S from M. mazei, was amplified and inserted into EcoRI-digested pBAD18 using the E. coli ME9783 strain to construct pBAD-mUA1. MM_1525 encoding putative PMDh-L from M. mazei was amplified and inserted into EcoRI-digested pBAD-mUA1 to construct pBAD-mUA2. MM_1871, encoding putative PFS from M. mazei, was amplified and inserted into EcoRI-digested pBAD-mUA2 to construct pBAD-mUA3. MM_1526, encoding putative AMPD from M. mazei, was amplified and inserted into EcoRI-digested pBAD-mUA3 to construct pBAD-mUA4. A DNA fragment containing MM_1526, MM_1871, MM_1525, and MM_1524 was amplified from pBAD-mUA4 and then inserted into EcoRI-digested pBAD-mMP3 to construct pBAD-mMP7.
To construct pBAD-msMP7, in which MM_1871 in pBAD-mMP7 was replaced by the S. cerevisiae PFS gene YDR538W, the amplified YDR538W was inserted into EcoRI-digested pBAD-mUA2 to construct pBAD-msUA3. MM_1526 was then inserted into EcoRI-digested pBAD-msUA3 to construct pBAD-msUA4. A DNA fragment containing MM_1526, YDR538W, MM_1525, and MM_1524 was amplified from pBAD-msUA4 and then inserted into EcoRI-digested pBAD-mMP3 to construct pBAD-msMP7.
APE_2089, encoding A. pernix PMDh-S, was amplified and inserted into EcoRI-digested pBAD18 to construct pBAD-aUA1. APE_2087.1, encoding A. pernix PMDh-L, was amplified and inserted into EcoRI-digested pBAD-aUA1 to construct pBAD-aUA2. APE_1647, encoding A. pernix PFS, was amplified and inserted into EcoRI-digested pBAD-aUA2 to construct pBAD-aUA3. APE_2078, encoding A. pernix AMPD, was amplified and inserted into EcoRI-digested pBAD-aUA3 to construct pBAD-aUA4. A DNA fragment containing APE_2078, APE_1647, APE_2087.1, and APE_2089 was amplified from pBAD-aUA4 and then inserted into EcoRI-digested pBAD-mMP3 to construct pBAD-aMP7.
To replace the APE_1647 in pBAD-aMP7 with YDR538W, encoding S. cerevisiae PFS, YDR538W was amplified and inserted into EcoRI-digested pBAD-aUA2 to construct pBAD-asUA3. APE_2078 was then inserted into EcoRI-digested pBAD-asUA3 to construct pBAD-asUA4. A DNA fragment containing APE_2078, YDR528W, APE_2087.1, and APE_2089 was amplified from pBAD-asUA4 and then inserted into EcoRI-digested pBAD-mMP3 to construct pBAD-asMP7.
Plasmid construction for gene deletion and complementation studies.
To construct the AMPD-deleted version of pBAD-mMP7, named pBAD-mMPdelAMPD, a DNA fragment containing MM_1871, MM_1525, and MM_1524 was amplified using pBAD-mUA3 as a template and then inserted into EcoRI-digested pBAD-mMP3 using the E. coli ME9783 strain to construct pBAD-mMPdelAMPD. To complement the deletion of AMPD, MM_1526 was amplified and inserted into XbaI-digested pBAD-mMPdelAMPD to construct pBAD-mMPcomAMPD.
To construct a PMDh-deleted version of pBAD-mMP7, named pBAD-mMPdelPMDh, a DNA fragment containing MM_1526 and MM_1871 was amplified using pBAD-mUA4 as a template and then inserted into EcoRI-digested pBAD-mMP3 to construct pBAD-mMPdelPMDh. To complement the deletion of PMDh, a DNA fragment containing MM_1525 and MM_1524 was amplified from pBAD-mUA4. The DNA fragment was inserted into XbaI-digested pBAD-mMPdelPMDh to construct pBAD-mMPcomPMDh.
Lycopene production in E. coli.
The E. coli TOP10 strain was transformed with both pACYC-IBE (15), which contains carotenogenic genes crtI, crtB, and crtE from Pantoea ananatis, and each of the plasmids constructed for the expression of the genes of the archaeal MVA pathway (shown in Table 1). All strains are listed in Table 1. Each strain was precultured at 37°C in 5 ml of LB medium supplemented with 100 mg/liter ampicillin and 20 mg/liter tetracycline. After their OD600 reached 0.5, these cultures were diluted to an OD600 of 0.1 in 5 ml LB medium containing 100 mg/liter ampicillin, 20 mg/liter tetracycline, 0.02% l-arabinose, and 5.0 mg/ml MVL. As a negative control, each strain was inoculated in the same medium without MVL. Then each strain was aerobically cultivated at 30°C for 24 h by shaking at 160 rpm. For semianaerobic cultivation, 3 ml of paraffin wax was put on the same medium, and each strain was cultivated at 30°C for 24 h, with shaking at 160 rpm to avoid cell aggregation and precipitation. After cultivation, the cells were harvested and disrupted by sonication in 500 μl of acetone. The homogenate was centrifuged at 22,000 × g for 5 min, and the supernatant was recovered. The precipitate was resuspended with 500 μl of acetone and centrifuged again. The supernatant was recovered and mixed with the first extract. The amounts of lycopene in the extracts were determined from its absorption at 475 nm using UV-2460 (Shimadzu, Japan) and the extinction coefficient ε = 185,000 M−1 cm−1.
Inhibition of the MEP pathway and complementation test.
The E. coli DH1 strain (NBRP-E. coli, NIG, Japan) was transformed with pBAD-mMP7 or pBAD18 to construct the dmMP7 and dNC strains, respectively. Each strain was precultured at 37°C in 5 ml of LB medium supplemented with 100 mg/liter ampicillin. Fifty microliters of the preculture, whose OD600 was adjusted to 0.5, was inoculated into 5 ml of LB medium, containing 100 mg/liter ampicillin, 0.2% l-arabinose, 5.0 mg/ml MVL, and 100 μM fosmidomycin (Sigma-Aldrich, USA). To cultivate in semianaerobic conditions, 3 ml of paraffin wax was put on the culture medium. Each strain was cultivated at 30°C by shaking at 160 rpm for 24 h. The OD600 of the cell culture was measured every 30 min with an ODBox-C (Taitec, Japan).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by JSPS KAKENHI grants number 17H05437, 18K19170, and 19H04651 (to H.H.); by grants-in-aid from Novozymes Japan, the Institute for Fermentation, Osaka, the Noda Institute for Scientific Research, and the Nagase Scientific Technology Foundation (to H.H.); and, by JSPS KAKENHI grant number 19J21282 (to R.Y.).
We are grateful to Susumu Asakawa of Nagoya University for his help in the cultivation of M. mazei.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Kuzuyama T, Hemmi H, Takahashi S. 2010. Mevalonate pathway in bacteria and archaea, p 493–516. In Mander L, Liu H-W (ed), Comprehensive natural products II. Chemistry and biology, vol 1 Elsevier, Oxford, United Kingdom. [Google Scholar]
- 2.Miziorko HM. 2011. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys 505:131–143. doi: 10.1016/j.abb.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azami Y, Hattori A, Nishimura H, Kawaide H, Yoshimura T, Hemmi H. 2014. (R)-mevalonate 3-phosphate is an intermediate of the mevalonate pathway in Thermoplasma acidophilum. J Biol Chem 289:15957–15967. doi: 10.1074/jbc.M114.562686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dellas N, Thomas ST, Manning G, Noel JP. 2013. Discovery of a metabolic alternative to the classical mevalonate pathway. Elife 2:e00672. doi: 10.7554/eLife.00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grochowski LL, Xu H, White RH. 2006. Methanocaldococcus jannaschii uses a modified mevalonate pathway for biosynthesis of isopentenyl diphosphate. J Bacteriol 188:3192–3198. doi: 10.1128/JB.188.9.3192-3198.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayakawa H, Motoyama K, Sobue F, Ito T, Kawaide H, Yoshimura T, Hemmi H. 2018. Modified mevalonate pathway of the archaeon Aeropyrum pernix proceeds via trans-anhydromevalonate 5-phosphate. Proc Natl Acad Sci U S A 115:10034–10039. doi: 10.1073/pnas.1809154115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.VanNice JC, Skaff DA, Keightley A, Addo J, Wyckoff GJ, Miziorko HM. 2014. Identification in Haloferax volcanii of phosphomevalonate decarboxylase and isopentenyl phosphate kinase as catalysts of the terminal enzymatic reactions in an archaeal alternate mevalonate pathway. J Bacteriol 196:1055–1063. doi: 10.1128/JB.01230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vinokur JM, Cummins MC, Korman TP, Bowie JU. 2016. An adaptation to life in acid through a novel mevalonate pathway. Sci Rep 6:39737. doi: 10.1038/srep39737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinokur JM, Korman TP, Cao Z, Bowie JU. 2014. Evidence of a novel mevalonate pathway in archaea. Biochemistry 53:4161–4168. doi: 10.1021/bi500566q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phulara SC, Chaturvedi P, Gupta P. 2016. Isoprenoid-based biofuels: homologous expression and heterologous expression in prokaryotes. Appl Environ Microbiol 82:5730–5740. doi: 10.1128/AEM.01192-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirby J, Keasling JD. 2008. Metabolic engineering of microorganisms for isoprenoid production. Nat Prod Rep 25:656–661. doi: 10.1039/b802939c. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, Zada B, Wei G, Kim SW. 2017. Metabolic engineering and synthetic biology approaches driving isoprenoid production in Escherichia coli. Bioresour Technol 241:430–438. doi: 10.1016/j.biortech.2017.05.168. [DOI] [PubMed] [Google Scholar]
- 13.Kang A, George KW, Wang G, Baidoo E, Keasling JD, Lee TS. 2016. Isopentenyl diphosphate (IPP)-bypass mevalonate pathways for isopentenol production. Metab Eng 34:25–35. doi: 10.1016/j.ymben.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Motoyama K, Sobue F, Kawaide H, Yoshimura T, Hemmi H. 2019. Conversion of mevalonate 3-kinase into 5-phosphomevalonate 3-kinase by single amino acid mutations. Appl Environ Microbiol 85:e00256-19. doi: 10.1128/AEM.00256-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hemmi H, Ohnuma S, Nagaoka K, Nishino T. 1998. Identification of genes affecting lycopene formation in Escherichia coli transformed with carotenoid biosynthetic genes: candidates for early genes in isoprenoid biosynthesis. J Biochem 123:1088–1096. doi: 10.1093/oxfordjournals.jbchem.a022047. [DOI] [PubMed] [Google Scholar]
- 16.Kajiwara S, Fraser PD, Kondo K, Misawa N. 1997. Expression of an exogenous isopentenyl diphosphate isomerase gene enhances isoprenoid biosynthesis in Escherichia coli. Biochem J 324:421–426. doi: 10.1042/bj3240421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang A, Meadows CW, Canu N, Keasling JD, Lee TS. 2017. High-throughput enzyme screening platform for the IPP-bypass mevalonate pathway for isopentenol production. Metab Eng 41:125–134. doi: 10.1016/j.ymben.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 18.Marshall SA, Payne KAP, Leys D. 2017. The UbiX-UbiD system: the biosynthesis and use of prenylated flavin (prFMN). Arch Biochem Biophys 632:209–221. doi: 10.1016/j.abb.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 19.Payne KA, White MD, Fisher K, Khara B, Bailey SS, Parker D, Rattray NJ, Trivedi DK, Goodacre R, Beveridge R, Barran P, Rigby SE, Scrutton NS, Hay S, Leys D. 2015. New cofactor supports alpha,beta-unsaturated acid decarboxylation via 1,3-dipolar cycloaddition. Nature 522:497–501. doi: 10.1038/nature14560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White MD, Payne KA, Fisher K, Marshall SA, Parker D, Rattray NJ, Trivedi DK, Goodacre R, Rigby SE, Scrutton NS, Hay S, Leys D. 2015. UbiX is a flavin prenyltransferase required for bacterial ubiquinone biosynthesis. Nature 522:502–506. doi: 10.1038/nature14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang PH, Khusnutdinova AN, Luo F, Xiao J, Nemr K, Flick R, Brown G, Mahadevan R, Edwards EA, Yakunin AF. 2018. Biosynthesis and activity of prenylated FMN cofactors. Cell Chem Biol 25:560–570. doi: 10.1016/j.chembiol.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Arunrattanamook N, Marsh ENG. 2018. Kinetic characterization of prenyl-flavin synthase from Saccharomyces cerevisiae. Biochemistry 57:696–700. doi: 10.1021/acs.biochem.7b01131. [DOI] [PubMed] [Google Scholar]
- 23.Nishimura H, Azami Y, Miyagawa M, Hashimoto C, Yoshimura T, Hemmi H. 2013. Biochemical evidence supporting the presence of the classical mevalonate pathway in the thermoacidophilic archaeon Sulfolobus solfataricus. J Biochem 153:415–420. doi: 10.1093/jb/mvt006. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 25.Peralta-Yahya PP, Ouellet M, Chan R, Mukhopadhyay A, Keasling JD, Lee TS. 2011. Identification and microbial production of a terpene-based advanced biofuel. Nat Commun 2:483. doi: 10.1038/ncomms1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nozaki S, Niki H. 2019. Exonuclease III (XthA) enforces in vivo DNA cloning of Escherichia coli to create cohesive ends. J Bacteriol 201:e00660-18. doi: 10.1128/JB.00660-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.