

Chronic nonhealing diabetic foot ulcers (DFUs) are a serious complication of diabetes and are further exacerbated by bacterial colonization. The microbial burden in the wound of each individual displays diverse morphological and physiological characteristics with unique patterns of host-pathogen interactions, antibiotic resistance, and virulence. Treatment involves empirical decisions until definitive results on the causative wound pathogens and their antibiotic susceptibility profiles are available. Hence, there is a need for rapid and accurate detection of these polymicrobial communities for effective wound management. Deciphering microbial communities will aid clinicians to tailor their treatment specifically to the microbes prevalent in the DFU at the time of assessment. This may reduce DFUs associated morbidity and mortality while impeding the rise of multidrug-resistant microbes.
KEYWORDS: 16S metagenomics, antibiotic resistance, diabetic foot ulcer, wound microbiome
ABSTRACT
Diabetic foot ulcer (DFU) is a major complication of diabetes with high morbidity and mortality rates. The pathogenesis of DFUs is governed by a complex milieu of environmental and host factors. The empirical treatment is initially based on wound severity since culturing and profiling the antibiotic sensitivity of wound-associated microbes is time-consuming. Hence, a thorough and rapid analysis of the microbial landscape is a major requirement toward devising evidence-based interventions. Toward this, 122 wound (100 diabetic and 22 nondiabetic) samples were sampled for their bacterial community structure using both culture-based and next-generation 16S rRNA-based metagenomics approach. Both the approaches showed that the Gram-negative microbes were more abundant in the wound microbiome. The core microbiome consisted of bacterial genera, including Alcaligenes, Pseudomonas, Burkholderia, and Corynebacterium in decreasing order of average relative abundance. Despite the heterogenous nature and extensive sharing of microbes, an inherent community structure was apparent, as revealed by a cluster analysis based on Euclidean distances. Facultative anaerobes (26.5%) were predominant in Wagner grade 5, while strict anaerobes were abundant in Wagner grade 1 (26%). A nonmetric dimensional scaling analysis could not clearly discriminate samples based on HbA1c levels. Sequencing approach revealed the presence of major culturable species even in samples with no bacterial growth in culture-based approach. Our study indicates that (i) the composition of core microbial community varies with wound severity, (ii) polymicrobial species distribution is individual specific, and (iii) antibiotic susceptibility varies with individuals. Our study suggests the need to evolve better-personalized care for better wound management therapies.
IMPORTANCE Chronic nonhealing diabetic foot ulcers (DFUs) are a serious complication of diabetes and are further exacerbated by bacterial colonization. The microbial burden in the wound of each individual displays diverse morphological and physiological characteristics with unique patterns of host-pathogen interactions, antibiotic resistance, and virulence. Treatment involves empirical decisions until definitive results on the causative wound pathogens and their antibiotic susceptibility profiles are available. Hence, there is a need for rapid and accurate detection of these polymicrobial communities for effective wound management. Deciphering microbial communities will aid clinicians to tailor their treatment specifically to the microbes prevalent in the DFU at the time of assessment. This may reduce DFUs associated morbidity and mortality while impeding the rise of multidrug-resistant microbes.
INTRODUCTION
Chronic nonhealing foot ulcers are a major complication in diabetic individuals, contributing to significantly higher morbidity and mortality (1). Infected foot ulcers are the leading cause of diabetes related lower limb amputations and are a significant predictor of mortality (2). Globally, it is estimated that lower limb amputation is 10 to 20 times more common in people with diabetes than among nondiabetics (3). In India, approximately 45,000 diabetic patients undergo amputation every year; infected neuropathic foot contributes to 75% of these cases (4). Current practice of management of infections is based on expert assessment, which relies on phenotyping of the wound based on clinical features. This involves administration of broad-spectrum antibiotics for preliminary control of the wound until evidence-based data are available from time-consuming culture-based tests (5). However, there is considerable controversy on the usefulness of qualitative microbiology for infection diagnosis (6) since only less than 1% of known microbial species are successfully cultured (7).
Prevalent genera isolated by culture-based methods from diabetic foot ulcers (DFUs) include Staphylococcus, Streptococcus, Pseudomonas, Enterococcus, Bacteroides, and Prevotella (8, 9). Studies indicate that the high incidence of multidrug-resistant (MDR) organisms in DFUs is due to the increased and inappropriate use of broad-spectrum antibiotics. Addressing this issue requires reliable, rapid, and effective tools that provide an accurate assessment of the DFU microbiota. The increasing accessibility and affordability of molecular methods provides an efficient culture-independent alternative to probe the wound microbiome (10). Hence, we have analyzed the wound microbiota associated with different clinical phenotypes of infected wound ulcers using 16S rRNA gene based next-generation sequencing (NGS).
RESULTS
Microbial data based on culture.
All wound samples (diabetic wounds [DW], n = 100; nondiabetic wounds [NDW], n = 22) were prospectively enrolled for culture-based assays and 16S rRNA metagenomics. Patient demographic, clinical metadata, and culture-based assessment data for the microbial profiles are summarized (Table 1). A total of 158 bacterial strains were obtained from 122 ulcer samples (130 strains from 100 DW; 28 strains from 22 NDW) using culture-based techniques. Gram-negative bacilli were predominant members of the wound microbiome in both DW and NDW cohorts (Table 2). The maximum resistance among Staphylococcus aureus strains (n = 35) was against ciprofloxacin (60.7%). All enterococci isolates (n = 10) were sensitive to ampicillin and tetracycline, while higher resistance was seen toward amoxicillin-clavulanic acid (62.5%) and ciprofloxacin (57.1%) (Table 3).
TABLE 1.
Demographic and microbiological data of foot ulcer samples processed for sequencinga
| Characteristics | DW | NDW |
|---|---|---|
| No. of samples | 100 | 22 |
| No. of males | 88 | 19 |
| No. of females | 12 | 3 |
| Median age (yr) ± SD | 62 ± 10.26 | 57 ± 14.30 |
| No. of patients | ||
| Age, <60 yr | 44 | 14 |
| Age, ≥60 yr | 56 | 8 |
| Range | ||
| Neutrophils (%) | 45–87 | 38–94 |
| Eosinophils (%) | 0.2–21 | 0.3–21 |
| Random glucose (mg dl–1) | 71–534 | 84–155 |
| Glucose postprandial levels (mg dl–1) | 46–456 | 84–153 |
| Glucose fasting levels (mg dl–1) | 42–427 | 85–93 |
| HbA1c (%) | 5.3–16.3 | 4.88–6.1 |
| Monomicrobial (%) | 47 | 23 |
| Polymicrobial (%) | 31 | 45 |
| No growth in culture (%) | 22 | 33 |
| No. (%) | ||
| Gram-positive strains | 44 (34) | 5 (18) |
| Gram-negative strains | 86 (66) | 23 (82) |
| Total no. of strains obtained (culture based) | 130 | 28 |
DW, diabetic wounds; NDW, nondiabetic wounds; HbA1c, glycated hemoglobin level.
TABLE 2.
Wound microbiological data based on culture-based assaysa
| Bacterial species | No. (%) of: |
|
|---|---|---|
| DW | NDW | |
| Gram-negative strains | 86 (66) | 23 (82) |
| Acinetobacter spp. | 11 (8.4) | 2 (7.1) |
| Citrobacter spp. | 7 (5.3) | |
| Escherichia coli | 4 (3) | |
| Enterobacter spp. | 4 (3) | 1 (3.5) |
| Klebsiella pneumoniae | 5 (3.8) | 1 (3.5) |
| Proteus spp. | 9 (6.9) | 1 (3.5) |
| Pseudomonas aeruginosa | 25 (19.2) | 4 (14.2) |
| Unidentified | 21 (16.2) | 14 (50.0) |
| Gram-positive strains | 44 (34) | 5 (17.8) |
| Enterococci | 9 (6.9) | 1 (3.5) |
| MRCONS | 7 (5.3) | |
| MRSA | 10 (7.6) | 3 (10.7) |
| MSCONS | 6 (4.6) | 1 (3.5) |
| MSSA | 7 (5.3) | |
| CONS | 1 (0.76) | |
| Streptococci | 3 (2.3) | |
| Unidentified | 1 (0.76) | |
| Total | 130 | 28 |
DW, Diabetic wounds; NDW, nondiabetic wounds; CONS, coagulase-negative staphylococci; MRCONS, methicillin-resistant coagulase negative staphylococci; MSCONS, methicillin-sensitive coagulase-negative staphylococci; MRSA, methicillin-resistant S. aureus; MSSA, methicillin-sensitive S. aureus.
TABLE 3.
Antibiotic resistance in bacterial strains isolated from wound samples
| Antibiotic(s) | Antibiotic resistance (%) for: |
|||||||
|---|---|---|---|---|---|---|---|---|
| Gram-negative bacteria |
Gram-positive bacteria |
|||||||
| Acinetobacter spp. (n = 13) | Citrobacter spp. (n = 7) | Enterobacter spp. (n = 5) | Proteus spp. (n = 10) | P. aeruginosa (n = 29) | Enterococci (n = 10) | S. aureus (n = 35) | Streptococci (n = 3) | |
| Amikacin | 18.2 | 28.6 | 25.0 | 33.3 | 16.7 | 9.7 | 0.0 | |
| Amoxicillin-clavulanic acid | 80.0 | 62.5 | 29.2 | 100.0 | ||||
| Ampicillin | 40.0 | 0.0 | 50.0 | |||||
| Aztreonam | 36.4 | 28.6 | 50.0 | 66.7 | 24.0 | |||
| Cefazoline | 0.0 | 66.7 | ||||||
| Cefepime | 30.0 | 0.0 | 25.0 | 50.0 | 13.0 | |||
| Ceftazidime | 50.0 | 0.0 | 60.0 | 50.0 | 5.9 | |||
| Ceftriaxone | 33.3 | 28.6 | 25.0 | 50.0 | 15.0 | |||
| Chloramphenicol | 28.6 | 34.5 | 0.0 | |||||
| Ciprofloxacin | 66.7 | 16.7 | 20.0 | 40.0 | 26.1 | 57.1 | 60.7 | 100.0 |
| Erythromycin | 16.7 | 38.7 | 0.0 | |||||
| Gentamicin | 30.0 | 16.7 | 50.0 | 20.0 | 25.0 | 25.0 | ||
| Linezolid | 37.5 | 38.7 | 0.0 | |||||
| Rifampin | 28.6 | 31.8 | 50.0 | |||||
| Teicoplanin | 28.6 | 14.3 | 0.0 | |||||
| Tetracycline | 0.0 | 4.6 | 0.0 | |||||
| Vancomycin | 25.0 | 16.7 | 50.0 | |||||
Microbial patterns determined by NGS.
Following sequence analysis and quality control, a total of 15,306,481 reads were taxonomically categorized to the genus or species level. Rarefaction analysis showed that the sequencing depth was enough to ascertain for the overall species richness across all samples. The major phyla identified in the DW cohort and NDW cohort were Proteobacteria (71.3%, 77.4%), Firmicutes (14.7%, 10.2%), Actinobacteria (7.9%, 4.7%), and Bacteroidetes (5.7%, 6.5%), respectively (Fig. 1A). A total of 276 and 154 bacterial genera was obtained from DW and NDW cohorts, respectively (Table 4), with Alcaligenes and Pseudomonas consistently present across all cohorts. With a cutoff of >1% relative abundance (RA) in at least one sample in a given cohort, 37 genera were common among the two cohorts. Of the total 86 genera, 43 genera were unique to DW samples, while 6 were unique to NDW cohort (Fig. 1B). The DW samples were further subdivided into five Wagner grades (WG1 to WG5) based on wound severity. Among the different Wagner grades, WG3 and WG2 harbored the maximum number of genera, while WG3 and WG4 had higher number of unique genera (Table 4). With a cutoff of >1% RA in at least one sample in a given wound grade, 15 genera were found common among these five grades, while 5, 5, 12, 9, and 2 genera were unique to WG1 to WG5 samples, respectively (Fig. 1C). The percentage of facultative anaerobes was the highest in severe wounds (26.5%); however, strict anaerobes (26%) were more prevalent in WG1 (Fig. 1D). Wound samples from females were consistently enriched in Burkholderia across DW and NDW cohorts. Proteus was found almost exclusively in male samples (9.97% versus 0.15% in DW, 21.5% versus 0% in NDW) (Fig. 1E).
FIG 1.

Microbial diversity and distribution across different cohorts. (A) Microbial diversity across the two cohorts based on phylum. Vertical bars represent relative abundances for different microbial phyla from binned OTUs in each sample. Only phyla with a relative abundance of >1% in more than one sample have been plotted. (B) Number of OTUs based on bacterial genera shared across the two cohorts; diabetic wounds (DW) and nondiabetic wounds (NDW). (C) Bacterial OTUs shared across the five different wound grades (WG1 to WG5) based on clinical severity of the wound. Genera with an abundance of >1% in at least one sample were considered. (D) Distribution of OTUs based on all genera across the five WGs stratified by oxygen requirement. (E) Average relative abundance (ARA) of bacteria in wound microbiome of diabetic and nondiabetic ulcer samples based on gender (only genera with a >1% average relative abundance in >50% of the samples in the respective DW and NDW cohorts were plotted).
TABLE 4.
Occurrence of bacterial genera across different Wagner grades of diabetic foot ulcers (n = 96)a
| Wound grade | Total no. of samples | Total no. of reads | Avg no. of reads/sample | Total no. of genera | No. of unique genera | No. (range) of bacterial genera (avg) |
|---|---|---|---|---|---|---|
| WG1 | 9 | 1,053,970 | 117,107 | 113 | 17 | 15–42 (28.44) |
| WG2 | 25 | 3,745,016 | 149,800 | 175 | 23 | 7–74 (30.4) |
| WG3 | 29 | 4,555,290 | 157,078 | 186 | 28 | 5–62 (27.56) |
| WG4 | 23 | 4,046,690 | 175,943 | 167 | 26 | 6–95 (28.34) |
| WG5 | 10 | 1,234,243 | 123,424 | 105 | 6 | 12–24 (25.1) |
| DW | 100 | 15,306,481 | 153,064 | 276 | 107 | 5–95 (27.82) |
| NDW | 22 | 3,836,024 | 174,365 | 154 | 11 | 3–52 (28.68) |
The column heading “No. of unique genera” refers to presence only in the respective wound grade, i.e., a complete absence in other wound grades. “DW” includes four samples whose wound grades were not determined. DW, diabetic wounds; NDW, nondiabetic wounds.
Core diabetic wound microbiome.
In the DW cohort, 40 of 276 operational taxonomic units (OTUs) had an average RA (ARA) of >1% and occurred in more than five samples. A plot of ARA against occurrence for these OTUs could be divided into four quadrants (Q1 to Q4). Q1 consisted of OTUs with high occurrence and ARA, including Alcaligenes and Pseudomonas, while Q2 harbored OTUs showing high occurrence but low ARA (Achromobacter, Bacillus, Burkholderia, Corynebacterium, Staphylococcus, Acinetobacter, Methylobacterium, and Streptococcus) and formed the core diabetic wound microbiome (Fig. 2A). Analysis of NDW cohort yielded a similar core microbiome with an exception of Methylobacterium (Fig. 2B). Core microbiome members displayed variability in their relative abundance across wounds of various severities (Fig. 3).
FIG 2.
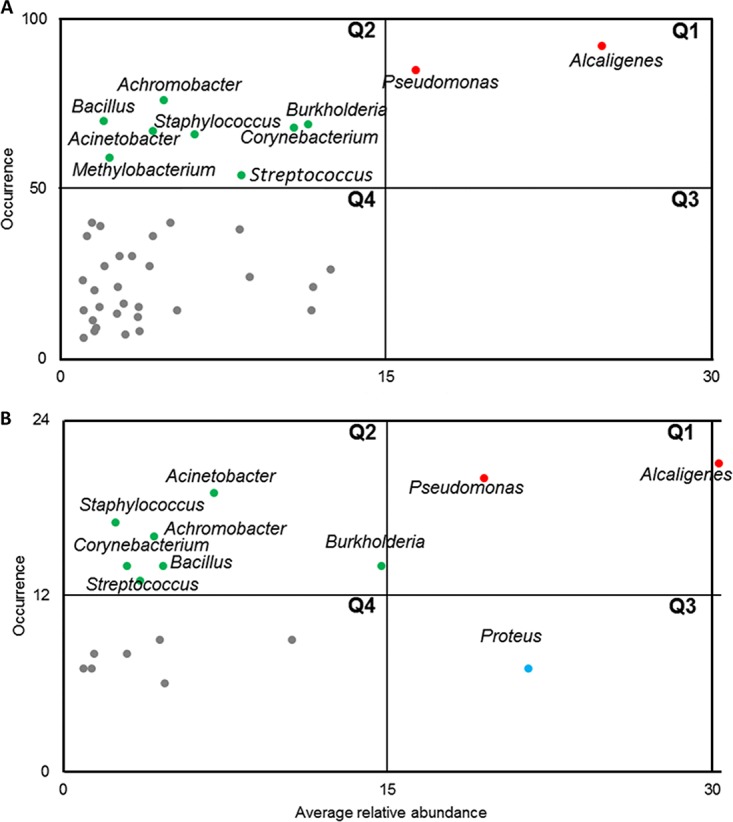
Core microbiome of DW and NDW cohorts. (A) Dominant microbes among diabetic wound samples. (B) Dominant microbes among nondiabetic wound samples. The average relative abundance of each bacterial OTU (at genus level) was plotted against number of samples in which the OTU was present. Only genera with >1% ARA and present in >5 samples were plotted. The scatter plot was divided into four quadrants at the midpoints of the maximum and minimum value of each axis. Q1 represents genera with a high average relative abundance and high occurrence, Q2 represents genera with a low average relative abundance and high occurrence, Q3 represents genera with a high average relative abundance but low occurrence, and Q4 represents genera with a low average relative abundance and low occurrence.
FIG 3.

Representation of the core microbiome members of each Wagner grade as a circular chord diagram (A) and bar plots (B) comparing the average relative abundance of the bacterial genera across each Wagner grade with significant differences (two-way ANOVA with Tukey post hoc test; P < 0.05 [*], P < 0.01 [**], and P < 0.001 [***]). Bacterial OTUs at the genus level with an average relative abundance of >1% in >50% of the total samples per Wagner grade were included for plotting the graph, and all other genera were included as “others.” The top right quadrant in the outer track represents Wagner grades; bacterial genera are represented in the inner track, and the scale is shown in the respective tracks. The scales for bacterial genera represent their contribution in each Wagner grade while the scales in Wagner grade represent the contribution by each genus toward the overall abundance. The width of the connecting chords indicates the abundance, while the shade represents the Wagner grade.
Microbial community structures within the DW cohort.
Alpha diversity metrics (Margalef’s index, Simpson index, and Hill’s index) revealed that samples from WG5 were relatively more diverse with respect to bacterial assemblage compared to other wound grades (Fig. 4A). A two-dimensional nonmetric dimensional scaling (NMDS) plot for all OTUs of the DW cohort showed no clear clustering based on Wagner grade (Fig. 4B) indicating high levels of heterogeneity. We performed a cluster analysis by partitioning around medoids (PAM) with genera (>1% average relative abundance in >5 samples) belonging to Q1 and Q2 of the DW cohort to determine whether there were any underlying clusters. The most natural clustering was observed with three Euclidean clusters EUC1, EUC2 and EUC3 (n = 16, 59, and 25, respectively) that were selected for further analysis (Fig. 5A). These clusters differed significantly in OTU diversity but not in richness or evenness. EUC2 harbored the greatest diversity compared to EUC1 and EUC3 (P = 0.0027 and 0.0014, respectively) (Fig. 5B). Clusters varied significantly (P < 0.0001, two-way analysis of variance [ANOVA]) with respect to the abundance of Alcaligenes, Pseudomonas, Corynebacterium, and Burkholderia (Fig. 5C).
FIG 4.
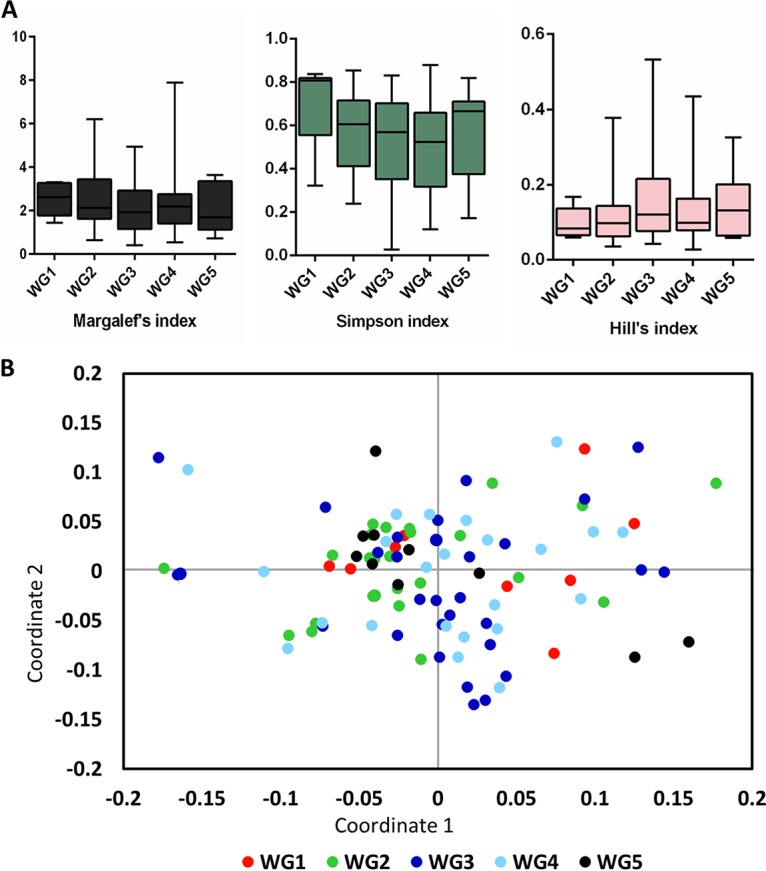
Variation of the diabetic wound microbiome based on Wagner grade. (A) Alpha diversity metrics were calculated for diabetic wound samples stratified by Wagner grade (WG1 to WG5). (B) NMDS with normalized data were performed for the samples, and the results are stratified based on Wagner grade.
FIG 5.

Clustering of DW based on Euclidean distances of normalized abundances of OTUs. (A) Euclidean distances of 100 DW samples were subjected to clustering by partitioning around medoids (PAM). Each point represents one diabetic wound sample. The choice of centroids/clusters (k) was decided based on average silhouette score. The first two principal components, explaining 56.9% of the point variability, with clustering of k = 3 medoids is shown. The three Euclidean clusters (EUCs) and samples within these clusters are marked with different colors and symbols, respectively. The silhouette score for the above clusters was 0.34. (B) Alpha diversity metrics calculated for diabetic wound samples stratified by their representative Euclidean clusters. (C) Average relative abundance values for the four genera that varied significantly (P < 0.0001, two-way ANOVA) across these clusters are plotted.
HbA1c levels and DW microbiome.
Reports suggest that glucose levels can modulate microbiota and determine the host response (11). Hence, we categorized a subset of DW samples (n = 50) into those with “poor glucose control” (HbA1c ≥7.5%) and “good glucose control” (HbA1c <7.5%) (12) and studied their microbial community structure. Poor glucose control samples had a higher percentage of actinobacteria (9.6% versus 1.7%) (Fig. 6A), although alpha diversity metrics were similar between these two cohorts (Fig. 6B). An NMDS analysis could not clearly discriminate between good control and poor control samples (Fig. 6C).
FIG 6.
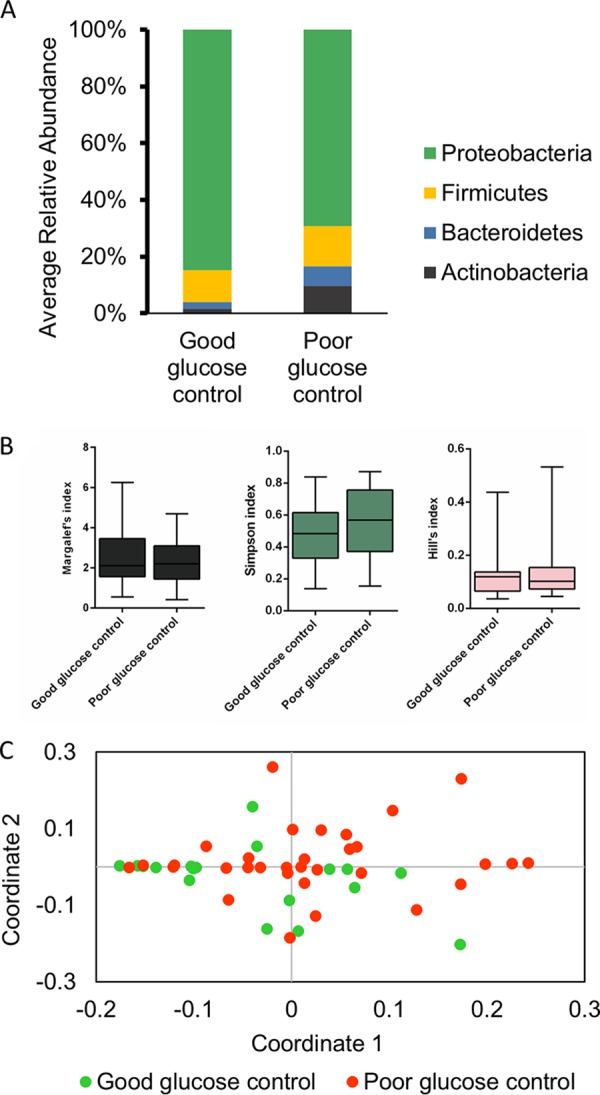
Microbial distribution in diabetic wound samples based on HbA1c values. Bacterial OTUs were taxonomically classified to the phylum level. Samples were classified into good (<7.5% HbA1c) and poor (≥7.5% HbA1c) glucose control. (A) Average relative abundance of each OTU categorized by its phylum. (B) Alpha diversity indices for the two cohorts. (C) NMDS results with normalized data for samples and stratified based on glucose control.
Microbiome in culture-negative DW samples.
While 22/100 DW samples failed to show any microbial growth in culture, NGS revealed presence of several culturable OTUs such as P. aeruginosa (11/22), S. aureus (7/22), and Acinetobacter baumannii (7/22) in culture-negative samples. To explore the microbiome of the culture negative samples, a principal-component analysis (PCA) was performed to compare culture-negative and culture-positive DW samples. Majority of the culture-negative samples formed a tight group with Alcaligenes as the strongest variable responsible for the cluster (Fig. 7).
FIG 7.

Microbiome diversity in culture negative DW samples based on NGS data. A principal-component analysis (PCA) graph developed from normalized data was constructed using NGS-based identification, and culture-negative samples (based on culture results, n = 22) were identified. A PCA biplot identified Alcaligenes as the major contributor to the clustering.
DISCUSSION
Infected foot ulcers remain a major complication of diabetes and treatment is often guided by empirical assessment based on clinical presentation. Despite gain in knowledge of the application of NGS technologies in the study of bacterial infections, there is a paucity in their translation to routine practice in clinical microbiology (13). Our culture-based methods identified S. aureus and P. aeruginosa as the frequently reported wound pathogens as has been reported earlier (11). However, our NGS-based study identified a diverse core microbiome of Alcaligenes, Pseudomonas, Burkholderia, Corynebacterium, Streptococcus, Staphylococcus, Achromobacter, Acinetobacter, Methylobacterium, and Bacillus consistent across all diabetic wounds. Certain trends were observed, such as an increase in facultative anaerobes with increasing wound severity and preferential presence of genera such as Proteus and Burkholderia based on gender. This suggests that, although there is a “core wound microbiome” prevalent among the samples, our study opens up an opportunity to use NGS for personalized medicine as we could observe quite a bit of variance in selected microbial genera across the samples. Knowledge of the wound pathogens needs to be complemented with their resistance patterns for it to be clinically relevant. Toward this, we carried out resistance profiles via MIC tests and found that isolates had higher resistance toward all antibiotics tested. The presence of a polymicrobial consortium with differing microbial resistance patterns in wound microenvironment could play a significant role in delaying wound healing process.
While studies based on 16S metagenomics have profiled chronic wounds from neuropathic or ischemic diabetic foot ulcers (14, 15), there are very few studies that offer direct comparison of culture-based approach with sequence-based techniques. Culture-dependent techniques remain the most widely available diagnostic tool for infection diagnosis (14). However, their effectiveness in capturing the entire repertoire of microbes and especially strict anaerobes is highly debated (16). Bacteria are rarely found as monomicrobial entities and, as the wound progresses, the presence of necrotic tissue as well as a poor supply of oxygen and significant biofilm production favors the growth of facultative or obligate anaerobes (5, 17). In our NGS study, a steady increase in facultative anaerobes was observed as the wound progressed, with the exception of WG5. The most frequently identified anaerobes were Prevotella, Finegoldia, Propionibacterium, and Clostridium among several others, as observed earlier (18). The large proportion of obligate anaerobes in WG1 is alarming and suggests that synergistic interactions between anaerobes and aerobes might also contribute to wound chronicity. The lack of anaerobes in severe wounds in our study could be due to the sampling technique employed. Though the Levine technique for sampling of microbes is considered as a superior technique for fresh and viable wounds, a majority of the samples collected in this study included eschar and necrotic areas especially in higher wound grades. Therefore, we chose a Z-stroke method covering the entire breadth of the wound. However, both the Z-stroke and Levine techniques are surface sampling techniques that have limited potential in capturing anaerobes found in deeper areas of the wound. These are best captured by invasive sampling techniques such as tissue biopsy or needle aspiration (19).
Culture-based techniques vastly underrepresent the diversity of wound microbiomes (5, 14). In our study, of the 100 DW samples, 22% of the samples were culture negative. However, NGS analysis of these samples revealed a vast majority of microbes, including isolates of Pseudomonas and Staphylococcus. Comparison of culture-negative samples with culture-positive samples showed tight clustering of culture-negative samples with Alcaligenes as the major genus responsible for clustering. Alcaligenes faecalis is an aerobic Gram-negative nosocomial pathogen (20, 21) that is generally regarded as a colonizer rather than a causative pathogen (22). However, recent research has explored its role in wound exacerbation (23). We hypothesize that the presence of Alcaligenes might interfere with the growth of other readily culturable microbes, providing the illusion of culture-negative results. This highlights the need to study microbial interactions with respect to microbial profiling of wounds to understand the dynamics of polymicrobial composition on wound microbiome.
Wound samples from females were consistently enriched in Burkholderia spp. across the cohorts; however, Burkholderia arboris and B. cenocepacia were found only in wound samples from males. Burkholderia spp. are reported to be animal and plant pathogens and implicated in cystic fibrosis and chronic granulomatous disease with the major site of infection being skin (24). Mukhopadhyay et al. (25) reported that Burkholderia primarily affects people involved in agricultural activities. Although, in our study, the majority of samples were collected from individuals with agricultural background, this still does not account for the preferential selection with respect to gender. We also observed a higher occurrence of Proteus in the male cohort, and further studies on this aspect might provide valuable clues on preferential microbial colonization in wound environment.
Despite these trends, no clear clusters based on wound severity were observed. We found a core microbiome consisting of genera such as Alcaligenes, Pseudomonas, Burkholderia, and Achromobacter whose composition differed among wounds of varying severity. Though several studies have indicated Pseudomonas as a major DFU pathogen, very few have highlighted the presence of Alcaligenes in wound microbiome. Alcaligenes is a Gram-negative nosocomial pathogen generally regarded as a colonizer rather than a causative pathogen. However, its consistent colonization of the wounds is a cause for major concern. Another such nosocomial pathogen is Achromobacter, shown to be a reservoir of beta-lactamase resistance genes and implicated in infections such as osteomyelitis, pneumonia, and peritonitis but rarely reported in DFU. Among the major genera, we observed several commonly reported wound pathogens, including Bacillus, Corynebacterium, Staphylococcus, Acinetobacter, and Streptococcus (26, 27). In addition, we found rarely reported pathogens such as Burkholderia and Methylobacterium. Burkholderia is a nutritionally diverse nonfermenting Gram-negative aerobe. B. cepacia and B. cenocepacia are highly transmissible, inherently resistant, and associated with high morbidity and mortality (28). Methylobacterium is a slow-growing aerobic microbe reported as a nosocomial pathogen (29). Quite disconcertingly, three of the common microbes across the wounds were nosocomial pathogens that are often clinically disregarded as colonizers. This necessitates the need for proper infection control strategies and studies to assess how these pathogens can contribute to wound exacerbation. In the absence of trends based on wound severity, we tested whether any correlation could be found with HbA1C levels and the DFU microbiome. We did not find any significant correlation unlike previous reports (14, 30), and this could be because of the heterogeneity of our samples with mixed phenotypes of wounds of varying severity.
Several studies exist on the dysbiosis of microbes in different disease states. However, our results show that there is an extensive sharing of wound microbes between different cohorts, diabetic or nondiabetic in nature. Hence, while our description of a core diabetic wound microbiome provides an attractive target for establishing a first-line point-of-care device to identify the pathogens involved; a longitudinal follow-up will provide critical insights on the microbial factors that contribute to pathogenicity and thereby, wound chronicity. Future studies may require adoption of multiple culture techniques to identify large number of culturable bacteria and invasive sampling to characterize anaerobes that are found deep inside the wounds (19). In addition, aspects such as expression of genes involved in virulence, biofilm formation, and antibiotic resistance, along with microbial interactions in the wound milieu, require better understanding (31, 32).
MATERIALS AND METHODS
Study design.
A prospective hospital-based study was approved by the Institutional Ethical Committee. Diabetic subjects above 18 years of age with the presence of a foot ulcer etiologically related to diabetes were included (DW, diabetic wounded) in the study. Samples from nondiabetic subjects with wounds (NDW) were also included. Wound grading was based on Wagner-Meggitt system (33). Exclusion criteria included subjects who (i) received antibiotics 1 week prior to sample collection, (ii) had wounds not related to diabetes or its complications, or (iii) had specific infections or neoplasms.
All subjects were recruited from the Department of Surgery at Kasturba Hospital, Manipal, India, with informed consent. Samples were collected from patients admitted to Kasturba Medical Hospital in Manipal from March 2016 to September 2017. Two swabs were collected for each wound by the Z technique (18). Briefly, cotton swabs were rolled in a broad Z stroke or zig-zag fashion across the breadth of the wound, which included eschar and necrotic areas. At least two swab samples were collected per subject from the lower-extremity area (including wounds on the toe, heel, plantar, medial, lateral, and dorsal aspects of the foot) for microbiological and NGS-based analysis, transported in a sterile polypropylene tubes (HiMedia, India), and processed within 2 h. Clinical metadata, including the levels of HbA1c (%) and random, fasting, and postprandial blood glucose levels (mg dl−1), were noted.
Microbiological processing.
One of the wound swabs was processed for aerobic bacteria by streaking on MacConkey agar (HiMedia) (34). Individual colonies were processed by biochemical and microbiological techniques (35). These included the triple sugar iron test, the mannitol motility test, the urease test, the citrate test, the indole test, the hydrogen sulfide test, and the methyl red test for the identification of Gram-negative bacteria and the coagulase test, the bile esculin agar test, and the catalase test for the identification of Gram-positive bacteria. Catalase-negative, coagulase-positive bacteria were further tested by using the Kirby-Bauer disk diffusion test with cefoxitin antibiotic. The MIC was determined according to CLSI regulations (36) using HiComb MIC strips (HiMedia).
Molecular processing.
DNA was extracted from swab specimens according to the standard phenol-chloroform (37)-based extraction method, and the DNA concentration was checked by using a Qubit 2.0 fluorometer (Invitrogen Life Technologies). Genome sequencing libraries were prepared by amplifying 50 ng of bacterial DNA with two primer sets targeting the V2, V3, V4, V67, V8, and V9 hypervariable regions of 16S rRNA (38). The amplification reactions were combined and cleaned using an Agencourt AMPure XP bead-based PCR purification system (Beckman Coulter). Purified amplicon pools (100 ng) were end repaired and barcoded using an Ion Plus Fragment Library kit and Ion Xpress Barcode Adapters, respectively. The library quality and quantity of the desired fragment length (∼250 bp) was assessed with the DNA high-sensitivity kit in the 2100 Bioanalyzer (Agilent Technologies). Each library was diluted to obtain a DNA concentration of 26 pM. Equal volumes of all libraries were combined and processed with Ion One Touch 2 and Ion One Touch ES systems, and sequencing was performed using an Ion Personal Genome Machine with a 318 v2 chip (Thermo Fisher Scientific).
Data analysis.
After sequencing, base calling and demultiplexing of the sequencing runs were performed by Torrent Suite v5.0 (Thermo Fisher Scientific) using the default parameters. Taxonomic classification was made using QIIME (Quantitative Insights Into Microbial Ecology) software based on curated MicroSEQ 16S Reference library v2013.1 and curated Greengenes v13.5 in IonReporter software (v5.2) at 97% similarity. Phylum abundance graphs and alpha diversity indices were calculated using vegan R packages (http://www.flutterbys.com.au, https://cran.r-project.org), while beta diversity and alpha rarefaction values were calculated using PAST 3.0 (https://folk.uio.no). The relative abundance (RA) of OTUs in each sample was calculated as: RA = (number of reads for a particular OTU/total number of reads) × 100. The average RA (ARA) for each OTU was calculated as the mean RA across all samples. Circular chord diagrams representing core microbiome members in each Wagner grade were visualized using online table viewer functionality of CIRCOS (http://mkweb.bcgsc.ca/tableviewer/). Differences in bacterial richness, diversity, and evenness values were calculated with Kruskal-Wallis test with a P value of <0.05 considered significant. For beta diversity, two-dimensional NMDS ordination was performed to study the dissimilarity in bacterial composition based on Euclidean distances with relative abundance data (39). Genera were classified into aerobe, anaerobe, or facultative anaerobe using BacDive database (40). Cluster analysis was performed by PAM based on data from the Euclidean distance matrix of core microbiome members. The validity of clustering and number of centroids was determined using the average silhouette score (14). Levels of significance were set at 0.05 and analyzed by two-way ANOVA and a post hoc Tukey test.
Data availability.
Sequencing data for all metagenomes (obtained from wound samples of diabetic and nondiabetic subjects) analyzed in this study is deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA596613.
ACKNOWLEDGMENTS
This study was supported by funding from TIFAC-CORE in Pharmacogenomics, DST-FIST (SR/FST/LSI-515/2011(C) 20/03/2013), Indo-German Science and Technology Centre and Manipal Academy of Higher Education, Manipal, India. The funders of the study had no role in in the study design, data collection, data analysis, data interpretation, or writing of the manuscript.
We thank Ganesh Acharya, Keerthi Kulal, Ramya Kurmoli, and Neethi Satish for their assistance in sample processing.
REFERENCES
- 1.Pereira S, Moura J, Carvalho E, Empadinhas N. 2017. Microbiota of chronic diabetic wounds: ecology, impact, and potential for innovative treatment strategies. Front Microbiol 8:1791. doi: 10.3389/fmicb.2017.01791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brennan MB, Hess TM, Bartle B, Cooper JM, Kang J, Huang ES, Smith M, Sohn MW, Crnich C. 2017. Diabetic foot ulcer severity predicts mortality among veterans with type 2 diabetes. J Diabetes Complications 31:556–561. doi: 10.1016/j.jdiacomp.2016.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ministry of Health and Family Welfare. 2016. The diabetic foot: prevention and management in India. Department of Health and Family Welfare, New Delhi, India: http://qi.nhsrcindia.org/sites/default/files/The%20Diabetic%20foot_0.pdf. [Google Scholar]
- 4.International Diabetes Federation. 2017. IDF diabetes atlas, 8th ed International Diabetes Federation, Brussels, Belgium. [Google Scholar]
- 5.Smith K, Collier A, Townsend E, O’Donnell L, Bal A, Butcher J, Mackay W, Ramage G, Williams C. 2016. One step closer to understanding the role of bacteria in diabetic foot ulcers: characterising the microbiome of ulcers. BMC Microbiol 16:1–12. doi: 10.1186/s12866-016-0665-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murali TS, Kavitha S, Spoorthi J, Bhat DV, Prasad AS, Upton Z, Ramachandra L, Acharya RV, Satyamoorthy K. 2014. Characteristics of microbial drug resistance and its correlates in chronic diabetic foot ulcer infections. J Med Microbiol 63:1377–1385. doi: 10.1099/jmm.0.076034-0. [DOI] [PubMed] [Google Scholar]
- 7.Grice EA, Snitkin ES, Yockey LJ, Bermudez DM, NISC Comparative Sequencing Program, Liechty KW, Segre JA. 2010. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc Natl Acad Sci U S A 107:14799–14804. doi: 10.1073/pnas.1004204107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowler PG, Duerden BI, Armstrong DG. 2001. Wound microbiology and associated approaches to wound management. Clin Microbiol Rev 14:244–269. doi: 10.1128/CMR.14.2.244-269.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papudeshi B, Haggerty J, Doane M, Morris M, Walsh K, Beattie D, Pande D, Zaeri P, Silva G, Thompson F, Edwards R, Dinsdale E. 2017. Optimizing and evaluating the reconstruction of Metagenome-assembled microbial genomes. BMC Genomics 18:1–13. doi: 10.1186/s12864-017-4294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franzosa E, Hsu T, Sirota-Madi A, Shafquat A, Abu-Ali G, Morgan X, Huttenhower C. 2015. Sequencing and beyond: integrating molecular ‘omics’ for microbial community profiling. Nat Rev Microbiol 13:360–372. doi: 10.1038/nrmicro3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grice EA, Segre JA. 2011. The skin microbiome. Nat Rev Microbiol 9:244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suh S, Joung JY, Jin SM, Kim MY, Bae JC, Park HD, Lee MS, Lee MK, Kim JH. 2014. Strong correlation between glycaemic variability and total glucose exposure in type 2 diabetes is limited to subjects with satisfactory glycaemic control. Diabetes Metab 40:272–277. doi: 10.1016/j.diabet.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Besser J, Carleton HA, Gerner-Smidt P, Lindsey RL, Trees E. 2018. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin Microbiol Infect 24:335–341. doi: 10.1016/j.cmi.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardner SE, Hillis SL, Heilmann K, Segre JA, Grice EA. 2013. The neuropathic diabetic foot ulcer microbiome is associated with clinical factors. Diabetes 62:923–930. doi: 10.2337/db12-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolcott RD, Hanson JD, Rees EJ, Koenig LD, Phillips CD, Wolcott RA, Cox SB, White JS. 2016. Jan-Feb. Analysis of the chronic wound microbiota of 2,963 patients by 16S rDNA pyrosequencing. Wound Repair Regen 24:163–174. doi: 10.1111/wrr.12370. [DOI] [PubMed] [Google Scholar]
- 16.Loesche M, Gardner SE, Kalan L, Horwinski J, Zheng Q, Hodkinson BP, Tyldsley AS, Franciscus CL, Hillis SL, Mehta S, Margolis DJ, Grice EA. 2017. Temporal stability in chronic wound microbiota is associated with poor healing. J Invest Dermatol 137:237–244. doi: 10.1016/j.jid.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suryaletha K, John J, Radhakrishnan M, George S, Thomas S. 2018. Metataxonomic approach to decipher the polymicrobial burden in diabetic foot ulcer and its biofilm mode of infection. Int Wound J 15:473–481. doi: 10.1111/iwj.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith ME, Robinowitz N, Chaulk P, Johnson K. 2014. Comparison of chronic wound culture techniques: swab versus curetted tissue for microbial recovery. Br J Community Nurs 19:S22–S26. doi: 10.12968/bjcn.2014.19.Sup9.S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charles PGP, Uçkay I, Kressmann B, Emonet S, Lipsky BA. 2015. The role of anaerobes in diabetic foot infections. Anaerobe 34:8–13. doi: 10.1016/j.anaerobe.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 20.Wisplinghoff H. 2017. Pseudomonas spp., Acinetobacter spp., and miscellaneous Gram-negative bacilli, p 1579–1599 In Cohen J, Powderly WG, Opal SM (ed), Infectious diseases. Elsevier Health Sciences, Philadelphia, PA. [Google Scholar]
- 21.Amoureux L, Bador J, Verrier T, Mjahed H, De Curraize C, Neuwirth C. 2016. Achromobacter xylosoxidans is the predominant Achromobacter species isolated from diverse non-respiratory samples. Epidemiol Infect 144:3527–3530. doi: 10.1017/S0950268816001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alibi S, Ferjani A, Boukadida J, Cano ME, Fernández-Martínez M, Martínez-Martínez L, Navas J. 2017. Occurrence of Corynebacterium striatum as an emerging antibiotic-resistant nosocomial pathogen in a Tunisian hospital. Sci Rep 7:9704. doi: 10.1038/s41598-017-10081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalan L, Meisel J, Loesche M, Horwinski J, Soaita I, Chen X, Uberoi A, Gardner S, Grice E. 2019. Strain- and species-level variation in the microbiome of diabetic wounds is associated with clinical outcomes and therapeutic efficacy. Cell Host Microbe 25:641–655. doi: 10.1016/j.chom.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burns JL. 2018. Burkholderia cepacia complex and other Burkholderia species, 871–873 In Long SS, Prober CG, Fischer M (ed), Principles and practice of pediatric infectious diseases. Elsevier, Philadelphia, PA. [Google Scholar]
- 25.Mukhopadhyay C, Vandana K, Chaitanya T, Shaw T, Bhat HV, Chakrabarty S, Paul B, Mallya S, Murali TS, Satyamoorthy K. 2015. Genome sequence of a Burkholderia pseudomallei clinical isolate from a patient with community-acquired pneumonia and septicemia. Genome Announc 3:e00915-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malone M, Johani K, Jensen S, Gosbell I, Dickson H, Hu H, Vickery K. 2017. Next-generation DNA sequencing of tissues from infected diabetic foot ulcers. EBioMedicine 21:142–149. doi: 10.1016/j.ebiom.2017.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Citron DM, Goldstein EJC, Merriam CV, Lipsky BA, Abramson MA. 2007. Bacteriology of moderate-to-severe diabetic foot infections and in vitro activity of antimicrobial agents. J Clin Microbiol 45:2819–2828. doi: 10.1128/JCM.00551-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.France MW, Dodd ME, Govan JR, Doherty CJ, Webb AK, Jones AM. 2008. The changing epidemiology of Burkholderia species infection at an adult cystic fibrosis centre. J Cyst Fibros 7:368–372. doi: 10.1016/j.jcf.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Kovaleva J, Degener JE, Van Der Mei HC. 2014. Methylobacterium and its role in health care-associated infection. J Clin Microbiol 52:1317–1321. doi: 10.1128/JCM.03561-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grice EA, Segre JA. 2012. Interaction of the microbiome with the innate immune response in chronic wounds. Adv Exp Med Biol 946:55–68. doi: 10.1007/978-1-4614-0106-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalan L, Loesche M, Hodkinson BP, Heilmann K, Ruthel G, Gardner SE, Grice EA. 2016. Redefining the chronic-wound microbiome: fungal communities are prevalent, dynamic, and associated with delayed healing. mBio 7:e01058-16. doi: 10.1128/mBio.01058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalan L, Grice EA. 2018. Fungi in the wound microbiome. Adv Wound Care (New Rochelle) 7:247–255. doi: 10.1089/wound.2017.0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie X, Bao Y, Ni L, Liu D, Niu S, Lin H, Li H, Duan C, Yan L, Huang S, Luo Z. 2017. Bacterial profile and antibiotic resistance in patients with diabetic foot ulcer in Guangzhou, southern China: focus on the differences among different Wagner’s grades, IDSA/IWGDF grades, and ulcer types. Int J Endocrinol 2017:8694903–8694912. doi: 10.1155/2017/8694903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekhar MS, Unnikrishnan MK, Rodrigues GS, Vyas N, Mukhopadhyay C. 2018. Antimicrobial susceptibility pattern of aerobes in diabetic foot ulcers in a South-Indian tertiary care hospital. Foot (Edinb) 37:95–100. doi: 10.1016/j.foot.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Forbes BA, Sahm DF, Weissfeld AS. 2007. Bailey and Scott’s diagnostic microbiology, 12th ed Mosby/Elsevier, St. Louis, MO. [Google Scholar]
- 36.Clinical and Laboratory Standards Institute. 2018. Performance standards for antimicrobial susceptibility testing, 28th ed Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 37.Green MR, Sambrook J. 2012. Molecular cloning: a laboratory manual, 4th ed Cold Spring Harbor Press, Cold Spring Harbor, NY. [Google Scholar]
- 38.Barb J, Oler A, Kim H, Chalmers N, Wallen G, Cashion A, Munson P, Ames N. 2016. Development of an analysis pipeline characterizing multiple hypervariable regions of 16S rRNA using mock samples. PLoS One 11:e0148047-18. doi: 10.1371/journal.pone.0148047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramette A, Tiedje JM. 2007. Biogeography: an emerging cornerstone for understanding prokaryotic diversity, ecology, and evolution. Microb Ecol 53:197–207. doi: 10.1007/s00248-005-5010-2. [DOI] [PubMed] [Google Scholar]
- 40.Söhngen C, Podstawka A, Bunk B, Gleim D, Vetcininova A, Reimer LC, Ebeling C, Pendarovski C, Overmann J. 2016. BacDive: the bacterial diversity metadatabase. Nucleic Acids Res 44:D581–D585. doi: 10.1093/nar/gkv983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequencing data for all metagenomes (obtained from wound samples of diabetic and nondiabetic subjects) analyzed in this study is deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA596613.