

Current agricultural and livestock practices contribute to fecal contamination in the environment and the spread of food- and waterborne disease and antibiotic resistance genes (ARGs). Traditionally, the level of pollution and risk to public health are assessed by culture-based tests for the intestinal bacterium Escherichia coli. However, the accuracy of these traditional methods (e.g., low accuracy in quantification, and false-positive signal when PCR based) and their suitability for sediments remain unclear. We collected sediments for a time series metagenomics study from one of the most highly productive agricultural regions in the United States in order to assess how agricultural runoff affects the native microbial communities and if the presence of Shiga toxin-producing Escherichia coli (STEC) in sediment samples can be detected directly by sequencing. Our study provided important information on the potential for using metagenomics as a tool for assessment of public health risk in natural environments.
KEYWORDS: antibiotic resistance, metagenomics, microbial ecology, microbial source tracking, sediment microbial communities
ABSTRACT
Little is known about the public health risks associated with natural creek sediments that are affected by runoff and fecal pollution from agricultural and livestock practices. For instance, the persistence of foodborne pathogens such as Shiga toxin-producing Escherichia coli (STEC) originating from these practices remains poorly quantified. Towards closing these knowledge gaps, the water-sediment interface of two creeks in the Salinas River Valley of California was sampled over a 9-month period using metagenomics and traditional culture-based tests for STEC. Our results revealed that these sediment communities are extremely diverse and have functional and taxonomic diversity comparable to that observed in soils. With our sequencing effort (∼4 Gbp per library), we were unable to detect any pathogenic E. coli in the metagenomes of 11 samples that had tested positive using culture-based methods, apparently due to relatively low abundance. Furthermore, there were no significant differences in the abundance of human- or cow-specific gut microbiome sequences in the downstream impacted sites compared to that in upstream more pristine (control) sites, indicating natural dilution of anthropogenic inputs. Notably, the high number of metagenomic reads carrying antibiotic resistance genes (ARGs) found in all samples was significantly higher than ARG reads in other available freshwater and soil metagenomes, suggesting that these communities may be natural reservoirs of ARGs. The work presented here should serve as a guide for sampling volumes, amount of sequencing to apply, and what bioinformatics analyses to perform when using metagenomics for public health risk studies of environmental samples such as sediments.
IMPORTANCE Current agricultural and livestock practices contribute to fecal contamination in the environment and the spread of food- and waterborne disease and antibiotic resistance genes (ARGs). Traditionally, the level of pollution and risk to public health are assessed by culture-based tests for the intestinal bacterium Escherichia coli. However, the accuracy of these traditional methods (e.g., low accuracy in quantification, and false-positive signal when PCR based) and their suitability for sediments remain unclear. We collected sediments for a time series metagenomics study from one of the most highly productive agricultural regions in the United States in order to assess how agricultural runoff affects the native microbial communities and if the presence of Shiga toxin-producing Escherichia coli (STEC) in sediment samples can be detected directly by sequencing. Our study provided important information on the potential for using metagenomics as a tool for assessment of public health risk in natural environments.
INTRODUCTION
Nearly half of the major produce-associated Escherichia coli O157:H7 outbreaks in the United States between 1995 and 2006 have been traced to spinach or lettuce grown in the Salinas Valley of California (1). Fecal contamination of produce can be caused by exposure to contaminated irrigation or flood water, deposition of feces by wildlife or livestock, or field application of manure as fertilizer (2, 3). From a public health perspective, more information is needed on the risk of exposure to animal fecal contamination, as recent studies suggest that exposure to water impacted by cow feces may present public health risks that are similar or equal to those from human fecal contamination. For example, cattle are a reservoir of the major foodborne pathogen Shiga toxin-producing E. coli (STEC) (4, 5). Environmental contamination by animal feces from farms is an emerging public health issue not only as a source of pathogens but also as a source of antibiotic resistance genes (ARGs) (6). Antibiotics are regularly administered to livestock at prophylactic concentrations to prevent infection, and food animal production is responsible for a significant proportion of total antibiotic use (7). Such practices are known to contribute to the prevalence of ARGs in the environment (8–10), which can spread rapidly to other microbes via horizontal gene transfer, including to human pathogens of clinical importance (11, 12). Surprisingly, there is very little regulation of antibiotic use in the U.S. livestock industry, even though these operations can be major contributors to fecal pollution and the spread of ARGs in the environment (13, 14).
Our previous culture- and PCR-based surveys of the Salinas watershed, and particularly, Gabilan and Towne Creeks (here called GABOSR and TOWOSR, respectively), indicated persistent presence of STEC in water and sediments (15, 16) and a potentially significant public health risk. Continued prevalence of STEC in both GABOSR and TOWOSR sites is hypothesized to be linked to the presence of cattle upstream. For instance, in several cases, STEC strains isolated from cattle fecal samples were identical to those found in water and sediment based on multilocus variable-number tandem-repeat analysis (MLVA) typing. Indeed, the prevalence of STEC was strongly correlated with runoff due to rainfall (1, 16). However, hydrologic modeling and surveys indicated that pathogen levels in streams were due not only to overland flow but also to contributions from sediment (17, 18). These observations were further supported by several examples of identical MLVA types isolated from both water and sediment at the same location or downstream during periods of drought (1, 15). Furthermore, the levels of pathogen in the water column and sediment are difficult to measure and are generally underestimated when using culture-based tests due to the predominance of biofilms and viable-but-not-culturable (VBNC) bacteria (19). Determining accurate pathogen levels is also problematic when using culture-independent quantitative PCR (qPCR) tests, because these tests may detect small fragments of highly degraded DNA long after the living microbe and pathogens have been inactivated (20). Furthermore, PCR methods do not give the complete picture of total functional and/or taxonomic shifts occurring in the sampled microbial communities. Therefore, metagenomic characterization of the creek sediments should provide independent quantitative insights into the effect of agricultural practices on the surrounding environment.
River and creek sediments are among the most diverse communities sequenced to date and are largely undersampled (21, 22). Moreover, the sediments studied to date are exclusively from highly and/or historically polluted environments with various industrial or sewage inputs; thus, each sediment is characterized by its unique properties in terms of flow dynamics, chemical environment, climatic conditions, and anthropogenic inputs (22–28). Accordingly, previous studies on the effect of anthropogenic inputs on sediments in lotic (free-flowing) aquatic systems have yielded mixed results on how surrounding land use practices impact sediment communities or were not directly relevant. Furthermore, in order to properly quantify the effect of anthropogenic antibiotic inputs, appropriate controls (e.g., pristine sampling sites) are needed to determine baseline levels of ARGs and other genes (13, 29).
In this study, we examined the effect of agricultural runoff on microbial communities from creek sediments in the Salinas watershed and whether community structure correlated with precipitation or culture-based detection of STEC. We sampled upstream sites with reduced human and cattle presence as a baseline for comparison with the abundance of anthropogenic signals (i.e., human and cow gut microbiome and ARGs) observed in the downstream sites that receive inputs from cattle ranches and produce farms. By combining culture-based STEC data with metagenome-based ARGs and animal host microbiome signals, we assessed the effect of cattle ranching runoff on the creek sediments at multiple independent levels, providing for more robust conclusions and interpretations. Furthermore, we compared the data from these sites to other publicly available sediment, soil, and river water metagenomes from both highly pristine and polluted environments in order to validate our results and assess anthropogenic pollution levels relative to those from other similar habitats.
RESULTS
Description of sampling sites.
Six sites from three creeks in the Salinas River Valley in California were included in this study. Two of the sites (collectively referred to as the “downstream” samples/sites) are impacted by cattle ranching but vary in the level of agricultural activities in the directly surrounding area. Cattle have direct access to creeks at both locations, and no effort is made to exclude them. At GABOSR, the cattle have access 2.38 km upstream from the sampling location, and cattle access for TOWOSR is 0.68 km upstream. The creeks are isolated at the sampling locations but converge further downstream before emptying into the Salinas River. Gabilan (GABOSR) is directly downstream of organic strawberry produce fields that use both green and poultry manure fertilizer and has cattle ranching upstream of the strawberry farm. The second site, Towne Creek (TOWOSR), is roughly 2 km north of GABOSR but does not have any abutting agricultural fields directly upstream and only receives input from cattle ranches. Ten samples from each of the two downstream sites, GABOSR and TOWOSR, collected over a 9-month period from September 2013 through June 2014 were selected for metagenome sequencing based on precipitation levels and detection of pathogenic E. coli via enrichment culture (Table 1). An additional seven samples from four upstream sites (collectively referred to as the “upstream” samples/sites) were included to serve as upstream controls for metagenomic comparison (Table 1 and Fig. 1). The samples from these locations included three samples collected ∼10 km upstream from Gabilan (GABOSR control) in March 2016 (GC1 to -3), two samples collected ∼3 km upstream from Towne Creek (TOWOSR control) in April 2017 (TC1 and TC2), and finally, one sample from each of two sites on the west side of the Salinas River (West Salinas), ∼60 km and 110 km southeast from the downstream sites collected in May 2017 (WS1 and WS2, respectively). The latter two samples are not upstream of GABOSR or TOWOSR but were included because they are more pristine sites with no known history of cattle impact, as opposed to the GC and TC samples, which may have had minimal inputs from previous cattle grazing.
TABLE 1.
Culture-based detection of STEC and precipitation data reported in inches
| Sample IDa | Date collected (mo/day/yr) | STECb | No. of copies stx2/μg DNAc | Precipitation (in)d
|
|
|---|---|---|---|---|---|
| Day 1 | 5-day sum | ||||
| GABOSR | |||||
| G130904 | 9/4/13 | − | 8.1 | 0 | 0 |
| G140116 | 1/16/14 | + | 8 | 0 | 0.01 |
| G140128 | 1/28/14 | + | 0 | 0 | 0 |
| G140210 | 2/10/14 | + | 4.4 | 0.01 | 1.1 |
| G140224 | 2/24/14 | + | 1.8 | 0 | 0 |
| G140301 | 3/1/14 | + | 1.5 | 0.33 | 2.01 |
| G140319 | 3/19/14 | − | 0 | 0 | 0.01 |
| G140402 | 4/2/14 | + | 1.4 | 0.03 | 1.04 |
| G140415 | 4/15/14 | − | 0 | 0 | 0 |
| G140611 | 6/11/14 | − | 2.4 | 0 | 0 |
| TOWOSR | |||||
| T130904 | 9/4/13 | − | 14.2 | 0 | 0 |
| T130918 | 9/18/13 | + | 15.3 | 0 | 0 |
| T131023 | 10/23/13 | − | 0 | 0 | 0 |
| T131230 | 12/30/13 | + | 3.9 | 0 | 0 |
| T140116 | 1/16/14 | − | 0 | 0 | 0.01 |
| T140128 | 1/28/14 | + | 0 | 0 | 0 |
| T140210 | 2/10/14 | − | 1.7 | 0.01 | 1.1 |
| T140224 | 2/24/14 | − | 1.5 | 0 | 0 |
| T140319 | 3/19/14 | − | 0 | 0 | 0.01 |
| T140611 | 6/11/14 | − | 0 | 0 | 0 |
| Upstream GABOSR control | |||||
| GC1 | 3/9/16 | − | 0 | 0 | 2.84 |
| GC2 | 3/9/16 | + | 0 | 0 | 2.84 |
| GC3 | 3/9/16 | − | 0 | 0 | 2.84 |
| Upstream TOWOSR control | |||||
| TC1 | 4/19/17 | + | 0 | 0 | 0.45 |
| TC2 | 4/19/17 | − | 0 | 0 | 0.45 |
| West Salinas | |||||
| WS1 | 5/4/17 | − | 0 | 0 | 0 |
| WS2 | 5/4/17 | − | 0 | 0 | 0 |
ID, identifier; GABOSR, Gabilan at Old Stage Road; TOWOSR, Towne Creek at Old Stage Road.
Samples in which STEC was detected by PCR of enrichment cultures are listed as either positive (+) or negative (−).
Copy number of the Shiga toxin gene (stx2) was determined via ddPCR.
Precipitation levels (in inches) for the day of sample collection (day 1) and the sum of precipitation levels for five days prior to the sampling day (5-day sum) were obtained from the California Irrigation Management Information System database (http://ipm.ucanr.edu/WEATHER/) for North Salinas weather station (the closest monitoring station to the downstream sites).
FIG 1.

Location of sampling sites in the Salinas Valley, CA, and sampling scheme for time series metagenomics. Sampling site for Gabilan (GABOSR in red) and Towne Creek (TOWOSR in yellow). The upstream controls for Gabilan (GC) and Towne Creek (TC) are also indicated by the same colors. The red line shows the flow of the creek from GC to GABOSR, the yellow line shows the flow from TC to TOWOSR, and the blue line shows the confluence of the two creeks before flowing into the Salinas River. Orange pins mark the West Salinas sites (WS1 and WS2) included as less agriculturally impacted controls. Orange lines show the flow of these creeks from the sampling point to the Salinas River, except for WS2, whose confluence point with the Salinas River is 70 kilometers upstream from where WS1 creek intersects and is not shown in the map. The North Salinas weather station (NS; green star) is approximately 11 km southeast of GABOSR and was the closest weather monitoring station to all samples shown in the subset map. Global position system (GPS) coordinates for all sampling locations are provided in Table S1 in the supplemental material. (Inset) Location of the Salinas Valley in the state of California (map courtesy of the U.S. Geological Survey). (Map data are from Google, SIO, NOAA, U.S. Navy, NGA, GEBCO, CSUMB, SFML, CA OPC, Landsat/Copernicus, MBARI.)
Description of metagenomes and sequence coverage of microbial community.
A total of 27 metagenomic samples, ranging in size from 8.7 to 20.1 million reads (2.5 to 5 Gbp) after trimming, were recovered from the six locations (see Table S2 in the supplemental material). For all samples, less than 28% of the total community (average, 18.6%) was covered by our sequencing efforts as determined by Nonpareil analysis (see Fig. S1). Consequently, the assembly of the metagenomes was limiting (e.g., the N50 values were poor, as shown in Table S2), consistent with our previous analyses of soil and sediment communities (30) and those of a few other metagenomic studies of river sediments. Thus, an unassembled short-read-based strategy was used for all subsequent analyses (paired-end nonoverlapping reads with an average length of 132 to 145 bp per data set), unless noted otherwise. A total of 7.2 × 108 protein sequences were predicted from the short reads, with an average of 2.7 × 107 sequences per sample. The number of protein sequences that could be annotated to the Swiss-Prot database in each sample ranged between 10% and 16% (average, 14.5%) of the total sequences.
OTU characterization and alpha diversity assessment.
A total of 466,421 reads containing fragments of the 16S or 18S rRNA gene were detected in all 27 metagenomes with an average of 601 (± 55) reads per million reads. All data sets were dominated by bacteria, with only 0.6% and 3.0% of the total rRNA reads, on average, having archaeal or eukaryotic origin, respectively. Closed-reference operational taxonomic unit (OTU) picking at 97% nucleotide identity threshold resulted in a total of 25,764 OTUs from 349,886 reads for all 27 samples and an average of 4,465 OTUs per sample. Since the coverage was similar for all data sets, the numbers of OTUs shared between all samples were compared without any further normalization. Only 138 OTUs (0.5%) were shared among all 27 samples, while 9,500 (36.9%) of the OTUs were present in only one sample. The OTU rarefaction plot showed that diversity was not saturated (see Fig. S2A), which agreed with the low number of shared OTUs and the Nonpareil estimates on the shotgun data reported above (Fig. S1).
Alpha diversity observed in the California samples was compared to alpha diversities in three publicly available river sediment metagenomes from Montana that had similar land use inputs (i.e., agricultural or small towns) and were the most appropriate data for comparison among lotic sediment metagenomes currently available (21). Species richness and diversity in Montana samples were significantly lower than in California samples (P = 2.3 × 10−4 and 0.006, respectively) (Fig. S2). Within California sites, diversity and evenness were similar; however, average species richness in GABOSR was significantly lower than in TOWOSR and the upstream samples (P = 0.034 and 4.1 × 10−4, respectively).
Taxonomic composition and functional diversity of water-sediment microbial communities.
OTUs were analyzed further to characterize the taxonomic profile of the communities sampled. Proteobacteria and Bacteroidetes were the most abundant phyla across most samples. However, some of the upstream samples had a higher abundance of Actinobacteria (see Fig. S3A). Class-level taxonomic distributions were consistent over time for GABOSR samples and revealed the high abundance of Betaproteobacteria (>19% to 24% of total sequences). TOWOSR samples varied more over time; five samples (T130918, T131230, T140128, T140210, and T140611) had a higher abundance of Deltaproteobacteria and Bacteroidia, and one sample (T140116) had a higher abundance of Cyanobacteria. The upstream samples also showed a similar community composition to and had higher relative abundance of Alphaproteobacteria (11% to 17%) than the downstream samples (Fig. S3B). These results were consistent with the TrEMBL taxonomic classification of protein-coding metagenomic reads, which were dominated by Bacteria (∼95.2% per sample) (see Fig. S4).
Microbial community structure and dynamics in Salinas River valley creeks.
Location was the strongest factor affecting clustering patterns observed in principal-component analysis (PCA) ordinations of all distance matrices analyzed (see Fig. S5). ADONIS analysis in the R package vegan (using location as a categorical variable) yielded a P value of <0.001 and R2 values of 0.44, 0.67, 0.41, and 0.56 for MASH, functional gene, OTU Bray-Curtis (16S-BC), and OTU weighted UniFrac (16S-WUF), respectively. This result was confirmed by correlation analysis of the nonmetric multidimensional scaling (NMDS) ordinations to all metadata variables using the envfit function in vegan. After using Bonferroni’s correction for multiple comparisons, location had the strongest correlation to all ordinations (MASH: P = 0.001, R2 = 0.879; functional gene: P = 0.001, R2 = 0.845; 16S-BC: P = 0.001, R2 = 0.0.787; 16S-WUF: P = 0.001, R2 = 0.726) and was the only significant variable for MASH (Fig. 2) and 16S rRNA gene-based measures of beta-diversity (see Fig. S6B and C) among those parameters evaluated. The functional gene ordination was also correlated, albeit weakly, to total 5-day precipitation (P = 0.028, R2 = 0.359) (Fig. S6A). To control for spatial variance, a more rigorous distance-based redundancy analysis (db-RDA [31]) was used on constrained NMDS ordinations, which allows the influence of a matrix of conditioning variables (i.e., location) to be “removed” prior to analysis. No significant associations (P > 0.05) were found in the functional gene and OTU Bray-Curtis ordinations; however, the MASH and OTU weighted UniFrac distances were significantly associated with sampling time (analysis of variance [ANOVA]: F = 1.274, P = 0.031; F = 2.174, P = 0.04, respectively).
FIG 2.
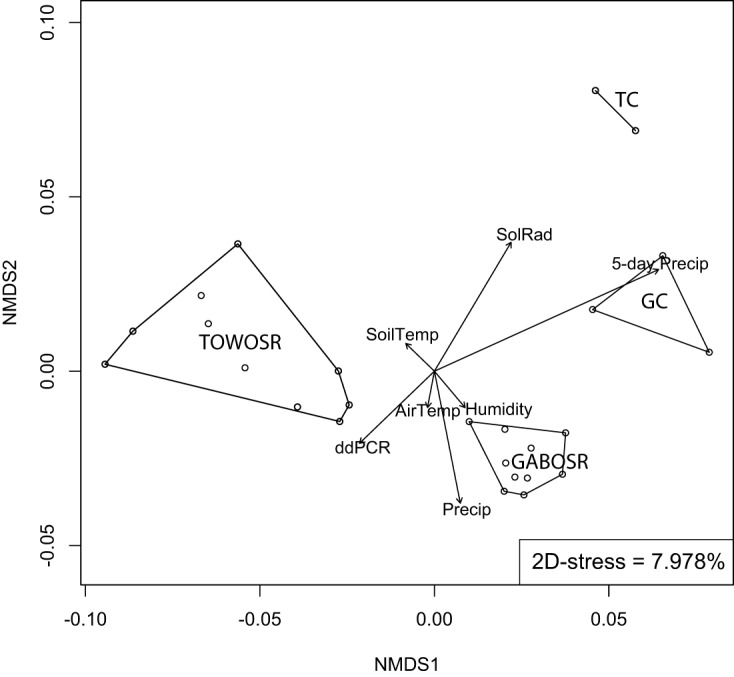
Effect of environmental parameters on microbial community structure. The graph shows nonmetric multidimensional scaling (NMDS) of the sequenced communities based on whole-community MASH distances. Each dot represents a metagenome sample, and metagenomes from the same location are connected by lines. Location (i.e., the polygons or lines) was the only variable that significantly correlated with the ordination. Arrowed vectors indicate correlations with other variables; however, none of these reached statistical significance (using the envfit function in the R package vegan). SoilTemp, AirTemp, SolRad, Precip, 5-day Precip, and ddPCR represent soil temperature, air temperature, solar radiation, day-of precipitation, 5-day precipitation, and digital droplet PCR counts for STEC, respectively.
Detection of E. coli by culture but not metagenomes.
The abundance of reads annotated as E. coli in the metagenomes based on BLASTN (nucleotide level) search against an STEC reference genome was low for all samples (∼0.002% of total reads). Samples with the highest relative abundance of metagenomic reads matching to E. coli were negative for all culture-based tests (Table 2), which indicated spurious in silico results (e.g., reads from non-E. coli genomes matching to conserved genes such as the rRNA operon). In addition, when using imGLAD (32), a tool developed by our team to deal with spurious matches, to predict the probability that E. coli was present in the metagenome, all samples yielded a P value of 1 (i.e., 0 probability of presence), which suggested that any E. coli populations (including STEC) were below the imGLAD estimated limit of detection for the metagenomic data sets in hand (i.e., 3% coverage of the E. coli genome at a minimum of 0.12× sequencing depth). The absolute abundance of the STEC based on droplet digital PCR (ddPCR) was also low (on the order of ∼1 in 108 cells, assuming average molecular weight of a base pair of DNA is 660 g/mol, 5 Mb genome size, and 1 copy stx/genome) or absent in all samples, which supports our bioinformatic-derived conclusions that E. coli was probably too low in abundance to be detected by our metagenomic sequencing effort (Table 2).
TABLE 2.
Culture-based versus in silico E. coli detection
| Location | Sample ID | Detection method |
|||
|---|---|---|---|---|---|
| Culture baseda
|
In silicob
|
||||
| EcO157 | STEC | No. reads matching | % relative abundance | ||
| GABOSR | G130904 | − | − | 1,652 | 0.0062 |
| G140116 | − | + | 576 | 0.0019 | |
| G140128 | − | + | 406 | 0.0020 | |
| G140210 | − | + | 936 | 0.0032 | |
| G140224 | − | + | 644 | 0.0024 | |
| G140301 | − | + | 886 | 0.0026 | |
| G140319 | − | − | 866 | 0.0028 | |
| G140402 | − | + | 1,112 | 0.0030 | |
| G140415 | − | − | 711 | 0.0022 | |
| G140611 | − | − | 1,050 | 0.0029 | |
| TOWOSR | T130904 | − | − | 516 | 0.0022 |
| T130918 | − | + | 255 | 0.0011 | |
| T131023 | − | − | 606 | 0.0023 | |
| T131230 | − | + | 379 | 0.0013 | |
| T140116 | − | − | 505 | 0.0019 | |
| T140128 | − | + | 367 | 0.0016 | |
| T140210 | − | − | 607 | 0.0035 | |
| T140224 | + | − | 780 | 0.0026 | |
| T140319 | − | − | 459 | 0.0016 | |
| T140611 | − | − | 1,495 | 0.0037 | |
| Upstream GABOSR | GC1 | − | − | 411 | 0.0016 |
| GC2 | − | + | 419 | 0.0017 | |
| GC3 | − | − | 423 | 0.0015 | |
| Upstream TOWOSR | TC1 | − | + | 478 | 0.0016 |
| TC2 | − | − | 626 | 0.0017 | |
| West Salinas | WS1 | − | − | 958 | 0.0032 |
| WS2 | − | − | 601 | 0.0022 | |
Culture-based methods to detect E. coli in resuspended sediment/water samples included an enrichment culturing step followed by Shiga toxin (stx) PCR procedures to detect specific virulence genes and genotypes as described in Materials and Methods. Detection of E. coli O157 (EcO157) was determined using enzyme-linked immunosorbent assay (ELISA) serotyping and a sample was positive for STEC if PCR and/or ELISA data yielded a positive result.
In silico methods included a blastn search of metagenomic reads against an STEC reference genome with a 95% identity and 97% read coverage cutoff for a read match, which was then normalized by dividing by the total number of reads per metagenome. The two samples with highest relative abundance of reads matching the STEC reference genome are in boldface font.
Differentially abundant functions and taxa between locations.
Of the 1,105 SEED subsystems (pathways) and 1,806 taxonomic groups identified, 911 and 408 were significantly differentially abundant (DA) with adjusted P values (Padj) of <0.05 for subsystems and taxa, respectively. Using pairwise comparisons between GABOSR, TOWOSR, and the 7 upstream samples, 184 SEED subsystems had a Log2 fold change (L2FC) of >1, while 273 taxa had an L2FC of >2, which were grouped into 36 and 35 broader functional and taxonomic categories, respectively (as described in Data Sets S1 and S2 in the supplemental material). The magnitudes of the L2FC differences were somewhat low overall, with average L2FCs of 1.82 and 3.71 for DA functional genes and taxa, respectively. Still, this analysis revealed several notable trends that were consistent between the functional SEED and taxonomy results (Fig. 3 and S7). More specifically, iron acquisition genes appeared more abundant in the upstream samples, particularly in the samples collected upstream of TOWOSR (TC1 and TC2). Plant-associated and photosynthesis genes were more abundant in the more pristine samples (WS1 and WS2). Consistently, members of the Alphaproteobacteria (e.g., Rhizobiales) (see Data Set S2) were more abundant upstream. Additional taxa that were more abundant in the upstream sites included those that are typically associated with soil and aquatic habitats (e.g., Gemmatimonadetes and Armatimonadetes), which indicated that these sites may indeed receive less anthropogenic inputs.
FIG 3.

Functional profiles of creek sediment microbial communities. The heat map shows SEED subsystems that were differentially abundant between locations (TOWOSR, GABOSR, and the upstream controls) with Padj of <0.05. Color scale indicates the abundance relative to the average of all samples (increasing from blue to red). Letters T or G and date in the column names represent the sample site (TOWOSR or GABOSR) and collection date, respectively. TC, GC, and WS represent the upstream TOWOSR control, GABOSR control, and West Salinas, respectively.
Sample T140116 was enriched for both cyanobacteria based on OTU analysis (Fig. S7) and photosynthesis genes (Fig. 3). TOWOSR appeared to be significantly more abundant in genes for anaerobic processes such as anoxygenic photosynthesis and methanogenesis, along with genes related to archaeal DNA, RNA, and protein metabolism (all organisms known to carry out methanogenesis are Archaea). Consistently, the two TOWOSR samples (T140128 and T140210) which were most abundant in archaeal and methanogenesis genes were also the most abundant in Archaea and methanotrophs from the order Methylococcales relative to the other sites. Other genes associated with anaerobic metabolisms, such as anoxygenic photosynthesis and sulfur metabolism genes (see Fig. 5), were congruent with taxonomic results that showed anoxygenic photosynthetic phyla Chlorobi (green sulfur bacteria) and Chloroflexi (green nonsulfur) and the family Chromatiaceae as well as known sulfur-metabolizing and anaerobic groups (e.g., Thiobacillus and Clostridia) to be more prevalent in the TOWOSR samples (Fig. S7). Additionally, the TOWOSR samples, in general, were more abundant in the Firmicutes and Bacteroidetes, which include gut-associated in addition to environmental members. Sample T140210 from TOWOSR was particularly enriched in specific enteric taxa, i.e., Endomicrobia and Fibrobacteres, which are rumen bacteria associated with cellulose degradation.
FIG 5.

Abundances of selected antibiotic resistance and production genes in the Salinas Valley metagenomes. (Left) Abundance (expressed as genome equivalents) of tetM, oxyT, and KSα genes for the 27 sites included in this study. (Right) Linear regression of tetM versus oxyT or KSα gene abundances. TC1 was an outlier for tetM abundance and was removed from this analysis.
Collectively, these results indicated that our annotation and grouping methods were robust, e.g., archaeal taxa identified as more abundant in TOWOSR samples were consistent with an increased frequency of archaeal functional genes such as methanogenesis in these samples. These results also suggested that TOWOSR samples might be more anaerobic, which could potentially indicate an effect of runoff and eutrophication as a result of human activity at this location. It could also be that this is the result of natural factors that we did not test here, and so we tried to look at specific DNA signals for anthropogenic pollution such as human and cow gut microbiome signal (see below). Also, Actinobacteria (i.e., common soil microbes and antibiotic producers) were all significantly more abundant in the upstream sites, which provides further evidence in support of this system being a natural source of ARGs (see below).
Quantifying anthropogenic and agricultural inputs.
(i) ARGs are more abundant in California samples than in other similar environments. The abundance of ARGs in each data set was determined by blastp search against the Comprehensive Antibiotic Resistance Database (CARD [33]). The most abundant ARGs detected are shown in Fig. S8 and Table S3 in the supplemental material. A comparison of selected metagenomic data sets that included metagenomes from agricultural sediments from Montana (MT) and soils from Illinois (Urbana [Urb] and Havana [Hav]), more pristine/remote samples from the Kalamas River (Kal) and Alaskan permafrost (AK), and a highly polluted sample from the Ganges River (Agra) was performed in order to benchmark the level of anthropogenic signal observed in the Salinas Valley against that in other environments. The abundance of ARGs in the California samples was significantly greater than in the other environmental metagenomes included here (Kruskal-Wallis χ2 =19.44, P = 0.0002) (Fig. 4A).
FIG 4.

Abundances of ARG, human gut (HG), and cow gut sequences in the Salinas Valley metagenomes compared to those in other environmental metagenomes. The box-and-whisker plots show the interquartile ranges for the abundances, with open dots indicating samples that exceeded 1.5× the interquartile range. The “upstream” metagenomes represent the seven more pristine control samples, i.e., three samples collected upstream from GABOSR, two collected upstream from TOWOSR, and two sites on the west side of the Salinas River that were farthest upstream from the rest of the sites (for more details, see main text and Fig. 1). The other environmental metagenomes (other env) included 3 river sediments, 2 agricultural soils, 1 permafrost soil, and 2 river water samples from the Kalamas and Ganges Rivers.
(ii) Abundance of genes associated with antibiotics used in cattle. To better assess the impact (if any) of ARGs related to cattle ranching, we built ROCker models, an approach for finding metagenomic reads containing a target gene of interest that is more accurate than simple homology searches (34), targeting tetracycline resistance (tetM) and production (oxyT) genes, since tetracyclines are among the most common antibiotics used in livestock (35). We also built a model targeting genes encoding the ketosynthase alpha subunit (KSα), which are involved in the synthesis of many antibiotics, including tetracyclines (36). The antibiotic production genes were quantified in order to the test the hypothesis that if (the high abundance of) ARGs naturally occur (as opposed to being human induced) then their abundance should correlate with that of the antibiotic production genes. To exclude the effect of potentially confounding variables, only the California samples were used for linear regression analysis of the abundances of antibiotic production and resistance genes, and gene abundance was expressed as genome equivalents (GE), or the fraction of total genomes containing the target gene of interest assuming the gene is single copy, as is usually the case for bacterial genes. In cases where the genes are in multiple copies, the GE will likely be >1 and would indicate genes per cell and not the fraction of genomes per total genomes. However, we did not observe cases of GE values >1, which indicated that our assumption was generally robust. ROCker analysis showed an abnormally high abundance of tetM in sample TC1 (Fig. 5, left), which was thus considered an outlier and excluded from the linear regression analysis. The high abundance in TC1 was presumably attributed to the fact that tetM has the widest host range of all tetracycline resistance (tet) genes due to its association with highly mobile conjugative transposons that behave similarly to plasmids and have several antirestriction systems (37, 38). oxyT abundance did not significantly correlate to tetM abundance (r2 = 0.031); however, KSα showed a moderate correlation to tetM (r2 = 0.280) (Fig. 5, right).
(iii) Abundance of cow and human gut microbiomes. The abundance of cow or human gut reads in the California Creek and reference metagenomes from other environments was determined by BLASTN search against a custom cow gut database and the Integrated Gene Catalog (IGC) of human gut microbiome genes (39), respectively. The IGC is referred to as the Human Gut Database (HG) here for clarity. The signal from the Ganges River (Agra) sample greatly exceeded that from all other samples in both the absolute number (Table 3) and relative abundance expressed as genome equivalents (GE), i.e., the fraction of total genomes containing human gut genes assuming a single copy of each gene per genome (33.5 GE; 8 to 100× more abundant than all other samples) (Fig. 4B). There was a significant difference between the HG abundance averages observed in California metagenomes and the 8 metagenomes from 5 other habitats evaluated here (Kruskal-Wallis P = 0.015). However, after correcting for multiple comparisons, none of the groups were significantly different (Wilcoxon rank sum P > 0.1). Within California samples, there was no significant difference overall between abundances observed in the downstream samples and the average abundances of the upstream control samples (Kruskal-Wallis P = 0.169).
TABLE 3.
Number of unique reference genes detected from CARD and human and cow gut databases
| Samples with unique genes detected | No. of samples | No. of genes from: |
||
|---|---|---|---|---|
| CARDa | Human gut database | Cow gut database | ||
| All samples | 35 | 1,776 | 167,481 | 15,497 |
| TOWOSR | 10 | 693 | 1,192 | 1,704 |
| GABOSR | 10 | 983 | 1,356 | 124 |
| Upstream controls | 7 | 760 | 1,135 | 136 |
| MT sediments | 3 | 441 | 1,522 | 116 |
| Agricultural soils | 2 | 722 | 9,877 | 270 |
| AK permafrost | 1 | 642 | 245 | 50 |
| Kalamas River | 1 | 475 | 3,952 | 554 |
| Ganges River (Agra) | 1 | 827 | 137,409 | 5,900 |
| Total reference genes in database | 2,820 | 9,879,896 | 459,176 | |
CARD, Comprehensive Antibiotic Resistance Database.
The abundance of different cow gut genes had a similar trend to the human gut data (Table 3). However, two samples from TOWOSR (T140210 and T140611) showed an elevated signal for cow sequences (Fig. 4C). Despite these two samples from TOWOSR with a higher level of cow gut signal, the average gene abundances were similar for California samples overall, and no significant difference was detected between the means compared to those from the other environmental metagenomes and the seven upstream control samples (Kruskal-Wallis P = 0.090) (Fig. 4C).
DISCUSSION
Analyses of planktonic microbial communities in rivers over time and land use have shown that these communities vary by average genome size, location, amount of sunlight, and nutrient concentrations (40) as well as by sampling time, more so than space (41). However, the results presented here suggested that community composition of Salinas Valley creek sediments is structured primarily by spatial separation, and the local weather parameters tested here did not have a significant effect (Fig. 2). More detailed in situ metadata than those obtained here, such as nutrient concentrations (e.g., organic carbon and biological oxygen demand), are needed in order to discern the processes that are driving community diversity and structure within each Salinas Valley site. For example, anaerobic taxa and processes related to methane and sulfur metabolism and anoxygenic photosynthesis were significantly more abundant in TOWOSR (Fig. 3 and supplemental material), which could indicate higher influence from agricultural runoff, lower permeability of the corresponding sediments by oxygen, or some other environmental factor that was not reflected by the local weather parameters measured here. It should be mentioned, however, that we did not observe any significant differences in the type of sediment sampled (e.g., percentage of fine sand) between the different sampling sites. Hence, the lower oxygen permeability appears to be a less plausible explanation for the functional differences observed than higher eutrophication (or another reason).
We compared abundances of metagenomic reads annotated as ARG and human or cow gut microbiome in order to assess levels of anthropogenic impacts on Salinas Valley creek sediment communities. No significant difference was detected between the downstream samples and the upstream controls for any of the three anthropogenic indicators (Fig. 4), which suggested that the land use practices surrounding the creeks do not have a major or lasting impact on the natural community and the inputs are likely diluted or attenuate faster than the intervals sampled here. To gain further quantitative insights, we then benchmarked abundances observed in the creek sediments from this study against those in metagenomes from other environments. These included agricultural sediments and soils, permafrost, and river water from both pristine and polluted habitats. GABOSR, TOWOSR, and the upstream samples all had significantly higher ARG abundances than the average of the other environments tested here (Fig. 4A). This high background level of reads annotated as ARGs suggested that the Salinas Valley creek sediments are a natural reservoir for these genes. Furthermore, genes encoding resistance to synthetic antibiotics such as florfenicol (fexA and floR) and ciprofloxacin (qnrS), one of the most widely used antibiotics in humans worldwide, were absent or detected in very low abundance (less than 10 reads matching) in our data sets. Spurious matches to conserved gene regions can occur when analyzing short reads like the ones here, but the signal was not large enough to warrant further investigation using precise and targeted methods (e.g., ROCker). Overall, the absence of genes encoding resistance to more recently introduced synthetic antibiotics provides further evidence that the ARG signal observed in the Salinas Valley is likely autochthonous in origin. Future studies could involve deeper sequencing (higher community coverage) in order to recover long contigs and thus determine the genomic background of the ARGs and if they are associated with mobile elements or plasmids for improved public health risk assessment. Still, our results highlight the importance of having a baseline or “pristine” sample to discern anthropogenic from naturally occurring ARGs and have important implications for monitoring the spread of ARGs in the environment. For instance, without the upstream control samples, this study could have (speciously) concluded that GABOSR and TOWOSR are elevated in ARGs as a result of cattle ranching. However, the similar abundances found in the upstream samples indicated that the signal detected downstream could be inherent to this environment and that a more targeted analysis of specific ARGs was required to determine if the effect of cattle could be detected.
Tetracycline resistance genes have been shown to increase and correlate with anthropogenic inputs along a river estuary system (42), suggesting that they can be useful indicators of anthropogenic pollution. However, tetracycline resistance genes are also found in other pristine or natural environments (29, 43–45) and therefore can also be considered part of the autochthonous gene pool in some habitats. Here, we tested the hypothesis that if tetracycline resistance genes are naturally occurring, the production enzymes for tetracycline should also follow similar abundance patterns, as antibiotic resistance and biosynthesis genes are often on the same operon to ensure antibiotic-producing species are resistant to the product they synthesize (46). Thus, we expected to see a correlation between abundances of the tetracycline resistance gene tetM and its associated production genes (oxyT and KSα) if this system is not under heavy selection pressure of human-introduced antibiotics. The abundance of tetM in the Salinas Valley creek sediments was not correlated to that of oxyT and only moderately correlated to KSα (Fig. 5). oxyT had very low abundance (fewer than 8 reads matching per sample), which suggested that the lack of correlation to tetM could be due to database limitations. That is, only a few reference oxyT genes are publicly available (13 sequences), and these likely do not capture the total diversity of this gene found in the environment. KSα, on the other hand, represents a broad class of synthesis genes for many different antibiotics, with many more sequences in the reference databases; thus, a better estimate of antibiotic production potential was obtained based on these genes. Overall, these findings further supported that this ecosystem is a natural reservoir for ARGs, and the presence of tetracycline resistance is not likely to be solely caused by inputs from the cattle ranches. However, future investigations could involve additional antibiotic production gene references for more robust conclusions.
Compared to the other pristine or rural environmental metagenomes such as for agricultural sediments and soils, permafrost, and river water, the abundances of reads annotated as human gut in the California sediments were not significantly different overall. However, the Ganges River (Agra) sample, collected from one of the most densely populated and highly polluted areas surrounding the river (Agra, Uttar Pradesh, India), was 1 to 2 orders of magnitude more abundant for human gut (open circle in Fig. 4B) than the rest of the samples used in our study. Thus, a high human gut signal was expected for the Ganges River, consistent with previous results (47), and served as a reference to assess relative levels of human fecal contamination. The rest of the samples included in our comparisons were from rural/agricultural or more remote areas, with lower population density, and consistently had lower signals of human fecal contamination than the Agra sample. Therefore, the low abundances of human gut sequences observed in Salinas Valley were consistent with the lower levels of human activity/density input than in more human- and animal-populated sites, such as the Ganges River used for comparison here, and indicated that our annotation and filtering methods were robust. Collectively, these results showed that metagenomics of river/creek sediments provide a reliable means for assessing the magnitude of the human presence/activity, consistent with recent studies of other riverine ecosystems (41, 47).
Contrary to the results for human gut, the abundances of cow gut signal in the California samples were not consistent with our expectations. The TOWOSR and GABOSR sites are directly downstream of large cattle ranch operations, and identical pathogen recovery from water and upstream cattle indicated the cattle ranches were the source of fecal contamination (1). As such, we expected to see a higher level of cow signal in the downstream metagenome samples, yet the abundance was not significantly different from the other environments or the upstream controls (Fig. 4B and C). Notably, two of the samples from TOWOSR (T140210 and T140611) showed elevated signal for cow that was similar to the abundance observed in the highly polluted Ganges River reference metagenome (Fig. 4C). These samples (especially T140210) had a higher abundance of the rumen enteric and cellulose degrading taxa (Endomicrobia and Fibrobacteres) (Fig. S7), which supports the conclusion that these samples contained runoff from cattle; however, the signal might be patchy or muted in the sediment and require more frequent sampling and/or larger sampling volumes than those used here to detect these signals.
Additionally, we were unable to detect any E. coli populations in any of the metagenomes, including samples that were positive for STEC via enrichment culture, indicating that it is not an abundant member of the sediment community (Table 2). This was consistent with imGLAD estimates that the sequencing effort applied to our metagenomes imposed a limit of detection for E. coli, and ddPCR results that showed abundance of STEC was low or absent in all samples. Overall, these results suggested that using shotgun metagenomics may not be sensitive (or economical) enough as a monitoring tool to detect a relatively low abundance microorganism in lotic sediments at the level of sequencing effort applied here, which was insufficient partly because of the extremely high community diversity (Fig. S1). More than the 2.5 to 5 Gbp/sample sequencing effort applied in this study would have been required to detect ∼10 E. coli cells in a sample according to our estimates, which is not economical based on current standards and costs. More specifically, obtaining the imGLAD minimum threshold of 0.12× coverage for an STEC genome (5 Mbp) in our metagenome libraries (average, 4 Gbp) would require 0.6 Mbp of STEC reads, or 0.015% of the total metagenome, which translates to a relatively large number of cells in situ. For example, assuming 108 total cells/g of sediment, it would require ∼104 STEC cells/g of sediment to robustly detect in the metagenomes (or 100 times more sequencing for detecting ∼10 cells/g). Thus, the limit of detection of metagenomics, as applied here, was not low enough and should be combined with methods that offer lower detection limits and more precise counts (such as ddPCR).
Rivers are highly dynamic ecosystems and therefore subject to higher random variation and sampling artifacts that likely affect the dilution of the exogenous (human) input. Furthermore, our samples represent relatively small volumes of sediment (∼10 g), and the resulting metagenomic data sets did not saturate the sequence diversity in the DNA extracted from these samples (Fig. S1), which might introduce further experimental noise and stochasticity. Despite these technical limitations, our data consistently showed little evidence that agricultural or cattle ranching activities have a significant effect on the creek sediment microbial communities. The underlying reason for these results remains speculative but could include sediment absorption or dilution by the creek waters and should be the subject of future research in order to better understand the impact of these activities on the environment. Additionally, the functional and taxonomic diversity observed between our samples could not be attributed to the environmental and weather variables measured, especially for the TOWOSR samples that showed extensive sample heterogeneity (diversity). These results suggested that shorter intervals between sampling as well as more detailed in situ geochemical data will be needed to elucidate the fine scale processes driving the community composition within each location. Although the continued presence of STEC in Salinas watershed sediments is a public health risk, we did not find evidence that runoff from human activities has a substantial effect on the sediment microbial community compared to that at more pristine sites. An imperative objective for public health is to assess how and where current agricultural practices impact the environment in order to determine best practices. Our study also provided important information on using metagenomics as a tool for public health risk studies of river water and sediment habitats, including what sampling volumes and frequencies to use, what amount of sequencing to apply, and what bioinformatics analyses to perform on the resulting data for future public health risk studies of river water and sediment habitats. Finally, the ROCker models developed here for tetracycline resistance and production genes should be useful for robustly examining the prevalence of these genes in other samples and habitats.
MATERIALS AND METHODS
Sample collection and enrichment method for STEC.
Sediment samples were collected from watersheds at public-access locations (see Table S1 in the supplemental material). Weather information was downloaded from the California Irrigation Management Information System database (http://ipm.ucanr.edu/WEATHER/) for the day of and 5 days prior to the sampling day from the closest monitoring station to the downstream sites (Table 1). Approximately 250 ml of sediment was collected by dragging an open sterile bottle attached to a 7.62-m telescoping pole along the bottom of the stream in the upstream direction and in such a way that the majority of the sample was undisturbed sediment. Nevertheless, some mixing with the water column occurred. Sediment at GABOSR contained more sand than TOWOSR or any of the control locations. Nevertheless, even at GABOSR, the collection was selectively silt (with fine sand occupying less than 10% by volume). As such, an effort was made to collect comparable samples at different locations. Additionally, only the top 1 to 2 cm of sediment was collected. All samples were transported on ice and processed within 24 h. Sediment was resuspended in the lab just prior to sampling to ensure a uniform subsample. DNA from 10 g of the resuspended sediment/water mix was purified for sediment DNA using a MoBio PowerSoil DNA extraction kit according to the manufacturer’s protocol. A separate 100 ml of the sample was used for enrichment and isolation of STEC as previously described (15).
PCR-based quantification method for STEC.
Droplet digital PCR (ddPCR; Bio-Rad) was performed on sediment DNA according to the method described by Cooley et al. (19). Each 20-μl reaction mixture contained 10 μl Bio-Rad Supermix for Probes, 2 μl primer (0.3 μM final concentration) and probe (0.2 μM), up to 1 μg DNA, 1.2 μl MgCl2 (1.5 mM), and 0.2 μl HindIII (0.2 U/μl). Primer and probe sequences were as previously published for STEC (19). Droplets were created with Droplet Generation Oil for Probes in the QX-200 droplet generator (Bio-Rad) and amplified for 5 min at 95°C, 45 cycles at 95°C for 30 s and 60°C for 90 s, and then 5 min at 72°C, and 5 min at 98°C. Droplets were processed with the QX-200 droplet reader and template levels were predicted by QuantaSoft software version 1.7.4 (Bio-Rad).
DNA sequencing and bioinformatics sequence analysis.
(i) Metagenomic sequencing and community coverage estimates. Shotgun metagenomic sequencing libraries were prepared using the Illumina Nextera XT library prep kit and a HiSEQ 2500 instrument as described previously (48). Short reads were passed through quality filtering and trimming as described previously (49). In short, sequences were trimmed with a PHRED score cutoff of 20 and minimum length of 50 bp. Only paired reads with both sisters longer than 50 bp after trimming were used for further analysis. Average community coverage and diversity were estimated using Nonpareil 3.0 (30) with kmer kernel and default parameters. Sequences were assembled with IDBA (50) using kmer values ranging from 20 to 80.
(ii) Taxonomic analysis of rRNA gene sequences. Metagenomic reads containing short subunit (SSU) rRNA genes were extracted with Parallel-Meta v.2.4.1 using default parameters (51). Closed reference OTU picking at 97% nucleotide identity with taxonomic assignment against the Greengenes database (52) was performed using MacQiime v.1.9.1 (53) with the reverse strand matching parameter enabled and the uclust clustering algorithm (54). Alpha diversity was calculated as the true diversity of order one (equivalent to the exponential of the Shannon index) and corrected for unobserved species using the Chao-Shen correction (55) as implemented in the R package entropy (56). Richness was estimated using the Chao1 index (57), and evenness was calculated from the estimated values of diversity divided by richness. Significant differences in taxonomic diversity, evenness, and richness were assessed using two-sided t tests. Multiple rarefactions were performed on OTU tables as implemented in MacQiime v.1.9.1 (rarifying up to the minimum number of counts per sample: option -e 5,596).
(iii) Determination of the total community bacterial fraction. To determine whether bacterial gene abundances needed to be corrected for relative bacterial fraction in the total metagenome libraries, the relative abundance of Bacteria, Archaea, and Eukarya was estimated in each data set by searching a subset (∼1 × 105 reads per sample) of randomly selected protein-coding reads against the TrEMBL database (58) (downloaded May 2018) using DIAMOND blastx v.0.9.22.123 (59) with the “–more sensitive” option and E value cutoff of 1 × 10−5. The TrEMBL identifiers (IDs) for best hit matches were summarized at the domain level using custom scripts and the metadata files available at ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/taxonomic_divisions/. No significant difference in the relative abundance of Bacteria was found between the different samples; thus, no correction for bacterial fraction was applied to gene abundance calculations.
(iv) Functional and ARG annotation of metagenomic sequences. Protein prediction was performed using FragGeneScan adopting the Illumina 0.5% error model (60). Resulting amino acid sequences were searched against the Swiss-Prot database (downloaded June 2017) (58) and Comprehensive Antibiotic Resistance Database (CARD; downloaded May 2017) (26) using blastp (61) for functional annotation. Best matches to the Swiss-Prot database with >80% query coverage, >40% identity, and >35-amino-acid alignment length were kept for further analyses. A more stringent cutoff was used for best matches to the CARD (>40% identity over >90% of the read length) to minimize false-positive matches.
(v) Detection of cow and human gut microbiome-associated sequences. Searches for cow gut-associated sequences were performed using our own collection of cow fecal metagenomes from six individual cows collected in Georgia, USA. DNA extracted from cow fecal material underwent the same library preparation, DNA sequencing, and quality trimming and processing as described above. Predicted genes (as nucleotides) from all six individual cows were pooled and dereplicated at 95% identity using the CD-HIT algorithm (options: -n 10, -d 0 [62]) resulting in 459,176 nonredundant cow gut metagenome “database” sequences. Human gut-associated sequences were assessed based on comparisons of short reads against the Integrated Gene Catalog (IGC) of human gut microbiome genes (39), here referred to as Human Gut Database (HG) for clarity. The abundance of cow and human gut signals in the short-read metagenomes was determined based on the number of reads from each data set matching these reference sequences using blastn v2.2.29 with a filtering cutoff of >95% identity and >90% query length coverage. Due to undersampling of the total community diversity at our sequencing depth, these more comprehensive whole-gut-microbiome databases were preferred over a specific suite of biomarkers for anthropogenic pollution, which are less likely to be detected in the metagenomes by chance than in the whole cow or human gut microbiome.
(vi) Abundance of specific antibiotic resistance and production genes using ROCker. Dynamic filtering cutoff models targeting a tetracycline resistance gene (tetM) and two antibiotic production genes (oxyT and KSα) were designed with ROCker v1.3.1, as previously described (34). Reference sequences for model building were manually selected from public databases, and models were built for 150-bp reads and default parameters. The reference sequences and ROCker models are available at http://enve-omics.ce.gatech.edu/rocker/models. Short reads were searched against the reference sequences used to build the model with blastx. The ROCker models were used to filter matches, which were subsequently divided by the median reference gene length in order to calculate sequencing coverage and were then normalized for genome equivalents as described below. Correlation between abundances of antibiotic production and resistance genes was determined using linear regression.
(vii) Quantification of genome equivalents. Average genome size and genome sequencing depth (i.e., the average sequencing depth of single-copy genes) were determined for each sample using MicrobeCensus v1.0.6 with default parameters (63). The sequencing depth of reference genes with a given annotation was estimated for each data set (in reads/base pairs) and then divided by the corresponding average genome sequencing depth and summed to give the total GEs per sample.
(viii) MASH and multivariate analysis. MASH v1.0.2 (64) was used to assess overall whole-community similarity among metagenomes in a reference database-independent approach (option -s 100000). Functional gene and 16S rRNA gene-based OTU count matrices were median normalized using the R package DESeq2 (v.1.16.1 [65]). Pairwise Bray-Curtis and weighted UniFrac (16S only) dissimilarity indexes of the normalized counts were used for principal-component analysis (PCA) and nonmetric multidimensional scaling (NMDS) analysis in order to assess whole-community gene functional and taxonomic (16S rRNA gene OTUs) similarity. The significance of metadata parameters on the NMDS ordinations was determined using the ecodist and envfit functions of the R package vegan v2.4.4 (indices included location, sampling time, ddPCR counts for STEC, same day precipitation, 5-day precipitation, solar radiation, air temperature, soil temperature, and humidity). The two west Salinas samples (WS1 and WS2) were excluded from this analysis in order to minimize confounding variation of temporal and spatial differences. To control for spatial variance, a more rigorous distance-based redundancy analysis (db-RDA) (31) was used to investigate the correlation to metadata using the capscale function in the R package vegan (including same indices as described above, but with condition [location] constraint on ordinations).
(ix) In silico detection of E. coli in sample metagenomes. The presence of any E. coli in the metagenomes was determined using a blastn search of short reads against an STEC reference genome (GenBank accession no. NC_002695) that had been filtered to remove nondiagnostic (i.e., highly conserved among phyla) regions with MyTaxa (66). Only matches with nucleotide identity >95% and alignment length >97% were used to calculate relative abundance of E. coli in the metagenomes. This level of sequence diversity (nucleotide identity >95%) encompasses well the diversity within the E. coli-Shigella sp. group; thus, any E. coli populations present in the metagenomes at high enough abundance would be detected at this filtering cutoff. The best hit output from blastn was also analyzed with imGLAD (32), a tool that can estimate the probability of presence and limit of detection of a reference/target genome in a metagenome.
(x) Determination of differentially abundant taxa and gene functions. Functional annotations of the recovered protein sequences were summarized into several hierarchical ranks, including metabolic pathways and individual protein families based on the SEED classification system (67). The 16S rRNA gene OTUs were placed into taxonomic groups based on the lowest rank of taxonomic classification (genus, family, etc.) shared by 90% or more of the sequences within the OTU using MacQiime v.1.9.1 (53). DA functional annotation terms (subsystems) or OTUs were identified in samples grouped by location (e.g., pairwise comparison of all 10 TOWOSR versus all 10 GABOSR and versus all 7 upstream “pristine control” sites) using the negative binomial test and false-discovery rate (Padj < 0.05) as implemented in DESeq2 v1.16.1 (65). Subsystems with a Log2 fold change (L2FC) of >1 or taxa with L2FC of >2 were manually grouped into broader categories based on known functional or taxonomic similarities, respectively (Fig. 3 and S7), which were then normalized by library size (per million read library). A larger L2FC cutoff was used for taxa to account for the larger data set size and allow for inspection of the taxa contributing most to differential abundance between the locations. The taxonomic assignment of these DA taxa was confirmed against the SILVA database (downloaded October 2018 [68]). Each subsystem or taxonomic category was then divided by its average sequencing depth across all samples to provide unbiased counts for presentation purposes.
(xi) Comparison of putative anthropogenic signals observed in California sediments to metagenomes from other environments. Publicly available metagenomes from other studies were used to compare abundances of reads annotated as ARG, HG, and cow gut with the results obtained for the California sediment data sets reported here. These metagenomes included three Montana River sediments (MT) (21), two temperate agricultural soils from Illinois (Hav and Urb) (69), an Alaskan tundra soil (AK) (70), one sample from the Ganges River near Agra, Uttar Pradesh (47), and one from the Kalamas River in Greece (Kal) (41). Short-read metagenomes for MT samples were downloaded from MG-RAST (71) (MG-RAST IDs 4481974.3, 4481983.3, and 4481956.3). The remaining data sets were obtained from the NCBI short read archive (SRA) database (Hav, ERR1939174; Urb, ERR1939274; AK, ERR1035437; Agra, SRR6337690; Kal, SRR3098772). Reads from these metagenomes were comparable to the ones from this study (100- to 150-bp paired-end Illumina sequencing) and underwent the same trimming, annotation (against the CARD, HG, and cow gut databases only), and gene count normalization protocol as described above. The Kruskal-Wallis test in R was performed to determine significantly different mean abundances between groups. Alpha diversity and taxonomic comparisons were performed (for MT data sets only) based on metagenomic reads containing fragments of the 16S rRNA gene, which were identified as described above.
Data availability.
Short reads for both the cow gut and CA sediment metagenomes have been deposited in the SRA database (submission IDs PRJNA545149 and PRJNA545542, respectively).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the USDA (award 2030-42000-050-10), the US National Science Foundation (award numbers 1511825 and 1831582 to K.T.K.), and the US National Science Foundation Graduate Research Fellowship under grant number DGE-1650044. The funding agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Cooley M, Carychao D, Crawford-Miksza L, Jay MT, Myers C, Rose C, Keys C, Farrar J, Mandrell RE. 2007. Incidence and tracking of Escherichia coli O157:H7 in a major produce production region in California. PLoS One 2:e1159. doi: 10.1371/journal.pone.0001159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mantha S, Anderson A, Acharya SP, Harwood VJ, Weidhaas J. 2017. Transport and attenuation of Salmonella enterica, fecal indicator bacteria and a poultry litter marker gene are correlated in soil columns. Sci Total Environ 598:204–212. doi: 10.1016/j.scitotenv.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 3.Jay MT, Cooley M, Carychao D, Wiscomb GW, Sweitzer RA, Crawford-Miksza L, Farrar JA, Lau DK, O'Connell J, Millington A, Asmundson RV, Atwill ER, Mandrell RE. 2007. Escherichia coli O157:H7 in feral swine near spinach fields and cattle, central California coast. Emerg Infect Dis 13:1908–1911. doi: 10.3201/eid1312.070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soller JA, Schoen ME, Bartrand T, Ravenscroft JE, Ashbolt NJ. 2010. Estimated human health risks from exposure to recreational waters impacted by human and non-human sources of faecal contamination. Water Res 44:4674–4691. doi: 10.1016/j.watres.2010.06.049. [DOI] [PubMed] [Google Scholar]
- 5.Probert WS, Miller GM, Ledin KE. 2017. Contaminated stream water as source for Escherichia coli O157 illness in children. Emerg Infect Dis 23:1216–1218. doi: 10.3201/eid2307.170226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.WHO. 2014. Antimicrobial resistance: an emerging water, sanitation and hygiene issue. WHO, Geneva, Switzerland. [Google Scholar]
- 7.Landers TF, Cohen B, Wittum TE, Larson EL. 2012. A review of antibiotic use in food animals: perspective, policy, and potential. Public Health Rep 127:4–22. doi: 10.1177/003335491212700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jechalke S, Kopmann C, Rosendahl I, Groeneweg J, Weichelt V, Krögerrecklenfort E, Brandes N, Nordwig M, Ding G-C, Siemens J, Heuer H, Smalla K. 2013. Increased abundance and transferability of resistance genes after field application of manure from sulfadiazine-treated pigs. Appl Environ Microbiol 79:1704–1711. doi: 10.1128/AEM.03172-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Y-G, Johnson TA, Su J-Q, Qiao M, Guo G-X, Stedtfeld RD, Hashsham SA, Tiedje JM. 2013. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc Natl Acad Sci U S A 110:3435–3440. doi: 10.1073/pnas.1222743110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karkman A, Pärnänen K, Larsson D. 2018. Fecal pollution explains antibiotic resistance gene abundances in anthropogenically impacted environments. Nat Commun 10:80. doi: 10.1038/s41467-018-07992-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh TR, Weeks J, Livermore DM, Toleman MA. 2011. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect Dis 11:355–362. doi: 10.1016/S1473-3099(11)70059-7. [DOI] [PubMed] [Google Scholar]
- 12.Maal-Bared R, Bartlett KH, Bowie WR, Hall ER. 2013. Phenotypic antibiotic resistance of Escherichia coli and E. coli O157 isolated from water, sediment and biofilms in an agricultural watershed in British Columbia. Sci Total Environ 443:315–323. doi: 10.1016/j.scitotenv.2012.10.106. [DOI] [PubMed] [Google Scholar]
- 13.Durso LM, Cook KL. 2014. Impacts of antibiotic use in agriculture: what are the benefits and risks? Curr Opin Microbiol 19:37–44. doi: 10.1016/j.mib.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 14.Berendonk TU, Manaia CM, Merlin C, Fatta-Kassinos D, Cytryn E, Walsh F, Bürgmann H, Sørum H, Norström M, Pons M-N, Kreuzinger N, Huovinen P, Stefani S, Schwartz T, Kisand V, Baquero F, Martinez JL. 2015. Tackling antibiotic resistance: the environmental framework. Nat Rev Microbiol 13:310–317. doi: 10.1038/nrmicro3439. [DOI] [PubMed] [Google Scholar]
- 15.Cooley MB, Jay-Russell M, Atwill ER, Carychao D, Nguyen K, Quiñones B, Patel R, Walker S, Swimley M, Pierre-Jerome E, Gordus AG, Mandrell RE. 2013. Development of a robust method for isolation of Shiga toxin-positive Escherichia coli (STEC) from fecal, plant, soil and water samples from a leafy greens production region in California. PLoS One 8:e65716. doi: 10.1371/journal.pone.0065716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooley MB, Quiñones B, Oryang D, Mandrell RE, Gorski L. 2014. Prevalence of Shiga toxin producing Escherichia coli, Salmonella enterica, and Listeria monocytogenes at public access watershed sites in a California Central Coast agricultural region. Front Cell Infect Microbiol 4:30. doi: 10.3389/fcimb.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dorner SM, Anderson WB, Slawson RM, Kouwen N, Huck PM. 2006. Hydrologic modeling of pathogen fate and transport. Environ Sci Technol 40:4746–4753. doi: 10.1021/es060426z. [DOI] [PubMed] [Google Scholar]
- 18.Petit F, Clermont O, Delannoy S, Servais P, Gourmelon M, Fach P, Oberlé K, Fournier M, Denamur E, Berthe T. 2017. Change in the structure of Escherichia coli population and the pattern of virulence genes along a rural aquatic continuum. Front Microbiol 8:609. doi: 10.3389/fmicb.2017.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooley MB, Carychao D, Gorski L. 2018. Optimized co-extraction and quantification of DNA from enteric pathogens in surface water samples near produce fields in California. Front Microbiol 9:448. doi: 10.3389/fmicb.2018.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bae SW, Wuertz S. 2009. Discrimination of viable and dead fecal bacteroidales bacteria by quantitative PCR with propidium monoazide. Appl Environ Microbiol 75:2940–2944. doi: 10.1128/AEM.01333-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibbons SM, Jones E, Bearquiver A, Blackwolf F, Roundstone W, Scott N, Hooker J, Madsen R, Coleman ML, Gilbert JA. 2014. Human and environmental impacts on river sediment microbial communities. PLoS One 9:e97435. doi: 10.1371/journal.pone.0097435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abia ALK, Alisoltani A, Keshri J, Ubomba-Jaswa E. 2018. Metagenomic analysis of the bacterial communities and their functional profiles in water and sediments of the Apies River, South Africa, as a function of land use. Sci Total Environ 616–617:326–334. doi: 10.1016/j.scitotenv.2017.10.322. [DOI] [PubMed] [Google Scholar]
- 23.Bowen JL, Ward BB, Morrison HG, Hobbie JE, Valiela I, Deegan LA, Sogin ML. 2011. Microbial community composition in sediments resists perturbation by nutrient enrichment. ISME J 5:1540–1548. doi: 10.1038/ismej.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu M, Zhang Q, Xia C, Zhong Y, Sun G, Guo J, Yuan T, Zhou J, He Z. 2014. Elevated nitrate enriches microbial functional genes for potential bioremediation of complexly contaminated sediments. ISME J 8:1932–1944. doi: 10.1038/ismej.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costa PS, Reis MP, Ávila MP, Leite LR, de Araújo FMG, Salim ACM, Oliveira G, Barbosa F, Chartone-Souza E, Nascimento A. 2015. Metagenome of a microbial community inhabiting a metal-rich tropical stream sediment. PLoS One 10:e0119465. doi: 10.1371/journal.pone.0119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graves CJ, Makrides EJ, Schmidt VT, Giblin AE, Cardon ZG, Rand DM. 2016. Functional responses of salt marsh microbial communities to long-term nutrient enrichment. Appl Environ Microbiol 82:2862–2871. doi: 10.1128/AEM.03990-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Negi V, Lal R. 2017. Metagenomic analysis of a complex community present in pond sediment. J Genomics 5:36–47. doi: 10.7150/jgen.16685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huber DH, Ugwuanyi IR, Malkaram SA, Montenegro-Garcia NA, Lhilhi Noundou V, Chavarria-Palma JE. 2018. Metagenome sequences of sediment from a recovering industrialized Appalachian River in West Virginia. Genome Announc 6:e00350-18. doi: 10.1128/genomeA.00350-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D'Costa VM, King CE, Kalan L, Morar M, Sung WWL, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R, Golding GB, Poinar HN, Wright GD. 2011. Antibiotic resistance is ancient. Nature 477:457–461. doi: 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-R LM, Konstantinidis KT. 2014. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 30:629–635. doi: 10.1093/bioinformatics/btt584. [DOI] [PubMed] [Google Scholar]
- 31.Legendre P, Anderson MJ. 1999. Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monogr 69:1–24. doi: 10.1890/0012-9615(1999)069[0001:DBRATM]2.0.CO;2. [DOI] [Google Scholar]
- 32.Castro JC, Rodriguez-R LM, Harvey WT, Weigand MR, Hatt JK, Carter MQ, Konstantinidis KT. 2018. imGLAD: accurate detection and quantification of target organisms in metagenomes. PeerJ 6:e5882. doi: 10.7717/peerj.5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, Bhullar K, Canova MJ, De Pascale G, Ejim L, Kalan L, King AM, Koteva K, Morar M, Mulvey MR, O’Brien JS, Pawlowski AC, Piddock LJV, Spanogiannopoulos P, Sutherland AD, Tang I, Taylor PL, Thaker M, Wang W, Yan M, Yu T, Wright GD. 2013. The Comprehensive Antibiotic Resistance Database. Antimicrob Agents Chemother 57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orellana LH, Rodriguez-R LM, Konstantinidis KT. 2017. ROCker: accurate detection and quantification of target genes in short-read metagenomic data sets by modeling sliding-window bitscores. Nucleic Acids Res 45:e14. doi: 10.1093/nar/gkw900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.US-FDA. 2015. Antimicrobials sold or distributed for use in food-producing animals. Food and Drug Administration, Department of Health and Human Services, Washington, DC. [Google Scholar]
- 36.Morlon H, O’Connor TK, Bryant JA, Charkoudian LK, Docherty KM, Jones E, Kembel SW, Green JL, Bohannan B. 2015. The biogeography of putative microbial antibiotic production. PLoS One 10:e0130659. doi: 10.1371/journal.pone.0130659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salyers AA, Shoemaker NB, Stevens AM, Li LY. 1995. Conjugative transposons: an unusual and diverse set of integrated gene transfer elements. Microbiol Rev 59:579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts MC. 2005. Update on acquired tetracycline resistance genes. FEMS Microbiol Lett 245:195–203. doi: 10.1016/j.femsle.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, Juncker AS, Manichanh C, Chen B, Zhang W, Levenez F, Wang J, Xu X, Xiao L, Liang S, Zhang D, Zhang Z, Chen W, Zhao H, Al-Aama JY, Edris S, Yang H, Wang J, Hansen T, Nielsen HB, Brunak S, Kristiansen K, Guarner F, Pedersen O, Doré J, Ehrlich SD, Bork P, Wang J, MetaHIT Consortium. 2014. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 40.Van Rossum T, Peabody MA, Uyaguari-Diaz MI, Cronin KI, Chan M, Slobodan JR, Nesbitt MJ, Suttle CA, Hsiao WWL, Tang PKC, Prystajecky NA, Brinkman F. 2015. Year-long metagenomic study of river microbiomes across land use and water quality. Front Microbiol 6:1405. doi: 10.3389/fmicb.2015.01405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meziti A, Tsementzi D, Ar Kormas K, Karayanni H, Konstantinidis KT. 2016. Anthropogenic effects on bacterial diversity and function along a river-to-estuary gradient in Northwest Greece revealed by metagenomics: diversity patterns along a river-to-estuary gradient. Environ Microbiol 18:4640–4652. doi: 10.1111/1462-2920.13303. [DOI] [PubMed] [Google Scholar]
- 42.Chen B, Liang X, Huang X, Zhang T, Li X. 2013. Differentiating anthropogenic impacts on ARGs in the Pearl River Estuary by using suitable gene indicators. Water Res 47:2811–2820. doi: 10.1016/j.watres.2013.02.042. [DOI] [PubMed] [Google Scholar]
- 43.Allen HK, Moe LA, Rodbumrer J, Gaarder A, Handelsman J. 2009. Functional metagenomics reveals diverse beta-lactamases in a remote Alaskan soil. ISME J 3:243–251. doi: 10.1038/ismej.2008.86. [DOI] [PubMed] [Google Scholar]
- 44.Cytryn E. 2013. The soil resistome: the anthropogenic, the native, and the unknown. Soil Biol Biochem 63:18–23. doi: 10.1016/j.soilbio.2013.03.017. [DOI] [Google Scholar]
- 45.Yang J, Wang C, Shu C, Liu L, Geng J, Hu S, Feng J. 2013. Marine sediment bacteria harbor antibiotic resistance genes highly similar to those found in human pathogens. Microb Ecol 65:975–981. doi: 10.1007/s00248-013-0187-2. [DOI] [PubMed] [Google Scholar]
- 46.Martín MF, Liras P. 1989. Organization and expression of genes involved in the biosynthesis of antibiotics and other secondary metabolites. Annu Rev Microbiol 43:173–206. doi: 10.1146/annurev.mi.43.100189.001133. [DOI] [PubMed] [Google Scholar]
- 47.Zhang S-Y, Tsementzi D, Hatt JK, Bivins A, Khelurkar N, Brown J, Tripathi SN, Konstantinidis KT. 2019. Intensive allochthonous inputs along the Ganges River and their effect on microbial community composition and dynamics. Environ Microbiol 21:182–196. doi: 10.1111/1462-2920.14439. [DOI] [PubMed] [Google Scholar]
- 48.Johnston ER, Kim M, Hatt JK, Phillips JR, Yao Q, Song Y, Hazen TC, Mayes MA, Konstantinidis KT. 2019. Phosphate addition increases tropical forest soil respiration primarily by deconstraining microbial population growth. Soil Biol Biochem 130:43–54. doi: 10.1016/j.soilbio.2018.11.026. [DOI] [Google Scholar]
- 49.Rodriguez-R LM, Overholt WA, Hagan C, Huettel M, Kostka JE, Konstantinidis KT. 2015. Microbial community successional patterns in beach sands impacted by the Deepwater Horizon oil spill. ISME J 9:1928–1940. doi: 10.1038/ismej.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng Y, Leung HCM, Yiu SM, Chin F. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 51.Su X, Pan W, Song B, Xu J, Ning K. 2014. Parallel-META 2.0: enhanced metagenomic data analysis with functional annotation, high performance computing and advanced visualization. PLoS One 9:e89323. doi: 10.1371/journal.pone.0089323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 55.Chao A, Shen T-J. 2003. Nonparametric estimation of Shannon’s index of diversity when there are unseen species in sample. Environ Ecol Stat 10:429–443. doi: 10.1023/A:1026096204727. [DOI] [Google Scholar]
- 56.Hausser J, Strimmer K. 2009. Entropy Inference and the James-Stein estimator, with application to nonlinear gene association networks 16. J Mach Learn Res 10:1469–1484. [Google Scholar]
- 57.Chao A. 1984. Nonparametric estimation of the number of classes in a population. Scand J Stat 11:265–270. [Google Scholar]
- 58.UniProt Consortium. 2017. UniProt: the universal protein knowledgebase. Nucleic Acids Res 45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 60.Rho M, Tang H, Ye Y. 2010. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res 38:e191. doi: 10.1093/nar/gkq747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu L, Niu B, Zhu Z, Wu S, Li W. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nayfach S, Pollard KS. 2015. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol 16:51. doi: 10.1186/s13059-015-0611-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. 2016. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17:132. doi: 10.1186/s13059-016-0997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luo C, Rodriguez-R LM, Konstantinidis KT. 2014. MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res 42:e73. doi: 10.1093/nar/gku169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang H-Y, Cohoon M, de Crécy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Rückert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glöckner FO. 2014. The SILVA and “all-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res 42:D643–D648. doi: 10.1093/nar/gkt1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Orellana LH, Chee-Sanford JC, Sanford RA, Löffler FE, Konstantinidis KT. 2018. Year-round shotgun metagenomes reveal stable microbial communities in agricultural soils and novel ammonia oxidizers responding to fertilization. Appl Environ Microbiol 84:e01646-17. doi: 10.1128/AEM.01646-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnston ER, Rodriguez-R LM, Luo C, Yuan MM, Wu L, He Z, Schuur EAG, Luo Y, Tiedje JM, Zhou J, Konstantinidis KT. 2016. Metagenomics reveals pervasive bacterial populations and reduced community diversity across the Alaska tundra ecosystem. Front Microbiol 7:579. doi: 10.3389/fmicb.2016.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keegan KP, Glass EM, Meyer F. 2016. MG-RAST, a metagenomics service for analysis of microbial community structure and function. Methods Mol Biol 1399:207–233. doi: 10.1007/978-1-4939-3369-3_13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Short reads for both the cow gut and CA sediment metagenomes have been deposited in the SRA database (submission IDs PRJNA545149 and PRJNA545542, respectively).