

Abstract
Ubiquitin-conjugating E2 enzymes are central to the ubiquitination cascade and have been implicated in cancer and other diseases. Despite strong interest in developing specific E2 inhibitors, the shallow and exposed active site has proven recalcitrant to targeting with reversible small-molecule inhibitors. Here, we used phage display to generate highly potent and selective ubiquitin variants (UbVs) that target the E2 backside, which is located opposite to the active site. A UbV targeting Ube2D1 did not affect charging but greatly attenuated chain elongation. Likewise, a UbV targeting the E2 variant Ube2V1 did not interfere with the charging of its partner E2 enzyme but inhibited formation of diubiquitin. In contrast, a UbV that bound to the backside of Ube2G1 impeded the generation of thioester-linked ubiquitin to the active site cysteine of Ube2G1 by the E1 enzyme. Crystal structures of UbVs in complex with three E2 proteins revealed distinctive molecular interactions in each case, but they also highlighted a common backside pocket that the UbVs used for enhanced affinity and specificity. These findings validate the E2 backside as a target for inhibition and provide structural insights to aid inhibitor design and screening efforts.
Keywords: Ubiquitin, E2 enzymes, Phage display, Protein engineering, Inhibitors
Introduction
The ubiquitin proteasome system (UPS) regulates the stability and function of thousands of proteins in essentially all cellular processes, and aberrant function of the UPS is associated with many diseases, including cancer, neurodegeneration, and infectious disease [1–3]. Consequently, there is great interest in developing inhibitors of UPS components, both to aid basic research and to provide therapeutic leads [4–6]. Indeed, general proteasome inhibitors have proven effective for cancer therapy and have raised interest in the development of inhibitors that target specific components of the UPS to afford more focused targeting of particular signaling networks [7,8].
Ubiquitin (Ub), the central player in the UPS, is added to protein substrates by a sequential cascade of Ub-activating (E1), Ub-conjugating (E2), and Ub-ligating (E3) enzymes [9]. Residing at the center of the cascade, E2 enzymes largely determine Ub chain topology and are responsible for recruiting E3 ligases and their substrates, and thus, they may represent more specific targets for therapeutic intervention [10,11]. However, direct targeting of the E2 catalytic site with small molecules has proven difficult, presumably because of the shallow and exposed nature of the site, and only a few compounds that attach covalently to the nucleophilic cysteine have been reported [12–14].
Recent discovery of CC0651, a reversible small-molecule inhibitor of E2 Ube2R1, has shown that inhibition can be achieved by other means, as structural studies revealed an allosteric mode of inhibition with CC0651 binding away from the active site [15,16]. Indeed, despite a small and simple structural fold, E2 enzymes engage in numerous protein-protein interactions through distinct regions of the enzyme [10,17], raising the possibility of additional modes of inhibition.
In some E2 enzymes, a surface opposite to the catalytic site—the “backside”—mediates noncovalent interactions with Ub, small Ub-like modifier (SUMO), or E3 ligases [10]. In the Ube2D family, backside binding of Ub is required for chain elongation [18]. Similarly, members of the Ube2V family of E2 variants, which lack a catalytic cysteine but work with E2 enzymes to facilitate chain elongation, depend on Ub binding to the backside [19]. Ub has also been shown to bind to the backside of Ube2G2, but the functional consequences are not known [20]. However, the activity of Ube2G2 is enhanced by backside interactions with E3 ligases [21]. In other examples, Ube2E3 [22,23] and Ube2B [24] family members are inhibited or activated, respectively, by backside interactions with Ub, and Ube2I activity is enhanced by interactions with SUMO [25]. Thus, it is clear that many E2 families use backside interactions to enhance, inhibit, or modulate catalytic activity, and thus, targeting of this region could provide opportunities to modulate biological activity for therapeutic benefit.
In all known cases, the backside interactions with Ub are weak affinity [10,18,26], but structures of Ube2V2 [19] and three Ube2D [18,27,28] family members in complex with Ub show a large interaction surface. Thus, we targeted E2 backsides with Ub variants (UbVs) engineered to bind with high affinity and specificity, as we have done previously with deubiquitinating enzymes [29], CRL E3 ligases [30], and HECT E3 ligases [31]. Here, we show that phage-displayed UbV libraries can yield tight and specific binders for E2 backsides. UbVs targeting various E2 proteins affect function by different means, impeding charging in one case and attenuating chain elongation in another. Structures of three distinct E2-UbV complexes reveal the molecular basis for potency and selectivity and highlight interactions with a common backside pocket. Our results validate the backside as a target for the development of selective inhibitors of E2 activity.
Results
Analysis of E2-Ub backside interactions and UbV library design
The Protein Data Bank (PDB) contains structures of three E2 enzymes (Ube2D1, D2, and D3) and one E2 variant (Ube2V2) with Ub bound to their backsides. Superposition of these structures showed that Ub binds to each protein in a similar way (average pairwise root-mean-square deviation [RMSD] = 1.70 Å) (Fig. 1A), and thus, we focused on the structure of the Ub-Ube2D1 complex for further analysis.
Fig. 1. Structural rationale and library design for UbVs targeting E2 backsides.

(A) Superposition of Ub.wt in complex with Ube2D1 (blue, PDB: 3PTF), Ube2D2 (red, PDB: 4V3L), Ube2D3 (yellow, PDB: 2FUH), or Ube2V2 (green, PDB: 1ZGU). The superposition was performed with complete coordinates for the complexes, but for clarity, only Ube2D1 is shown (gray) with the active site cysteine shown as a red sphere. (B) Sequence alignment of the residues in the Ub-binding backsides of the human Ube2D, Ube2V, Ube2G, Ube2A/B, and Ube2E family members. Residues that are identical or similar across families are shaded orange or yellow, respectively. Residues that are identical within the Ube2D, Ube2V, Ube2G, Ube2A/B, or Ube2E families are shaded blue, green, gray, pink, or purple, respectively. The numbering for Ube2D1 is shown above the sequences. The Ub-binding positions were defined as those residues at the interface in the majority of structures. (C) The Ub-binding backside of Ube2D1. Ube2D1 is shown as a gray tube with backside residues that are conserved or unconserved across the Ub-binding families shown as red or gray spheres, respectively. (D) The designs of libraries UbV-E2 and UbV-USP mapped onto the structure of Ub (PDB: 1UBQ). The Ub main chain is shown as a gray tube. Diversified positions are shown as spheres colored as follows: region 1, magenta; region 2, cyan; region 3, red. Gray spheres indicate residues that were not diversified in library UbV-E2 but were diversified in library UbV-USP.
We defined the backside of Ube2D1 as 21 residues that make contact with Ub, and we aligned these residues in the sequences of the human E2 family. Among the 13 members of the five E2 families known to interact with Ub at the backside surface (Fig. 1B), we noted high conservation of small residues at positions 22 (typically Ser) and 24 (typically Gly). In contrast, among the 20 E2 enzymes not known to interact with Ub at the backside surface, these positions were not conserved and were typically occupied by larger residues (Supplementary Fig. S1). Notably, other residues within the backside were not conserved across families but were conserved within families. For example, conservation was observed at all 19 positions in the Ube2V family, 16 of 19 positions in the Ube2D family, and 10 of 19 positions in the Ube2G family. These results suggest that the different families use different molecular interactions to mediate recognition of Ub, but nonetheless, they share a common motif of small residues at positions 22 and 24. Positions 22 and 24 are adjacent to each other in the center of the backside (Fig. 1C), suggesting that small residues at these positions may be required to enable Ub to dock without steric hindrance. Indeed, the requirement for Ser at position 22 has been noted previously, and it has been shown that an Arg substitution abrogates Ub binding [18].
On the Ub side of the interface, we defined the E2-binding backside as 21 residues that make contact with Ube2D1. These residues overlap extensively with the previously defined ubiquitin-specific protease (USP)-binding site [29]. Nevertheless, there are significant differences in that the E2-binding site does not include eight residues in the USP-binding site (Fig. 1D). We constructed a phage-displayed library of 3 × 1010 unique UbVs (library UbV-E2) in which the positions within the E2-binding site were diversified with a soft randomization strategy that favored the wild-type (wt) sequence but allowed for diversity across the entire site, as described previously for library UbV-USP, which targeted the USP-binding site [29].
Selection and characterization of UbV binders for E2 proteins
We used libraries UbV-E2 and UbV-USP to perform binding selections for Ube2D1, Ube2V1, Ube2G1, and Ube2G2 and identified 22, 56, 9, and 18 unique binding clones, respectively (Supplementary Table S1). These unique UbV-phage clones were analyzed further using phage enzyme-linked immunosorbent assays (ELISAs) for binding to various other E2 proteins to isolate the most specific binders for each E2 protein and using competitive phage ELISAs to estimate affinities. This screening process yielded highly selective UbVs (Fig. 2A) that were purified and assessed for binding to a panel of 33 E2 proteins (Fig. 2B). The UbVs selected for binding to Ube2D1, Ube2G1, or Ube2G2 were absolutely selective, whereas the most selective Ube2V1 binder (UbV.V1.1) also recognized UbeV2. The Ube2V1 screen also yielded a variant (UbV.V1.2) that exhibited binding across the Ube2V, D, and G families. We verified that each UbV did not bind to a variant of its cognate E2 protein bearing a Ser to Arg substitution at the position corresponding to position 22 of Ube2D1, which has been shown to abrogate binding of Ub to the backside of Ube2D1 [18].
Fig. 2. Selective and tight binding of UbVs to E2 proteins.

(A) Sequence alignment of Ub.wt and UbVs selected for binding to Ube2D1 (D1.1), Ube2V1 (V1.1 and V1.2), Ube2G1 (G1.1), or Ube2G2 (G2.1). The alignment shows only those positions that were diversified in the UbV libraries, and positions that were conserved as the wt sequence are indicated by dashes. (B) The binding specificities of UbVs (y-axis) are shown across the human E2 family (x-axis), as assessed using ELISAs. FLAG-tagged UbV proteins were added to immobilized E2 proteins, and bound UbV proteins were detected by the addition of anti-FLAG-HRP and colorimetric development of TMB peroxidase substrate. The normalized mean value of absorbance at 450 nm is shaded in a purple gradient (white = 0 and purple = 1). “S22R” refers to E2 variants containing an Arg residue in place of Ser22 (Ube2D1 numbering). (C) IC50 values, determined using competition ELISA, for solution-phase E2 proteins inhibiting binding of UbVs to immobilized E2 proteins. Subsaturating concentrations of the indicated UbV proteins were incubated with serial dilutions of E2 proteins as indicated and binding to immobilized cognate E2 protein was detected. The IC50 value was defined as the concentration of solution-phase E2 protein that inhibited 50% of the binding of the UbV protein to the immobilized E2 protein.
To further assess specificity and affinity, the purified UbVs were analyzed using competitive ELISAs (Fig. 2C). UbV.D1.1 bound tightly to Ube2D1 but not to the other members of the Ube2D family. UbV.V1.1 bound with essentially equal affinity to Ube2V1 and V2 but did not bind to Ube2D1 or G1. UbV.G1.1 and UbV.G2.1 bound tightly to Ube2G1 or G2 but did not bind to Ube2G2 or G1, respectively, and neither UbV bound to Ube2D1 or V1. These results show that we were able to isolate highly selective and potent UbVs that likely bind to the backsides of E2 proteins.
Structural analysis of UbVs in complex with E2 proteins
We solved the structures of UbV.D1.1, UbV.V1.1, and UbV.G1.1 in complex with their cognate E2 proteins at 2.10 Å, 2.55 Å, and 2.35 Å resolution, respectively (Table 1, Supplementary Fig. 2). The structure of Ube2V1-UbV.V1.1 was solved in complex with its E2 partner Ube2N/Ubc13. Backbone superpositions showed that the Ube2D1-UbV.D1.1(Fig. 3A) and Ube2V1-UbV.V1.1 (Fig. 3B) complex structures were essentially identical to the structures of each E2 protein in complex with Ub.wt(RMSD = 0.70 or 1.34 Å, respectively). The structure of Ube2G1 in complex with Ub.wt is not known, but the superposition of the apo Ube2G1 structure with the Ube2G1-UbV.G1.1 complex structure (Fig. 3C) showed virtually no differences in the two Ube2G1 molecules (RMSD = 0.66 Å). We also superposed the three structures of UbVs in complex with E2 proteins (Fig. 3D), which demonstrate that the UbVs dock on a similar region of the backside, although there was some variability in the backbone superpositions (mean pairwise RMSD = 1.60 Å). Taken together, these results show that the UbVs bind to the backsides of their cognate E2 proteins in a manner that is very similar to that observed for Ub.wt.
Table 1.
Data collection and refinement statistics.
| Ube2D1 + Ubv.D1.1 | Ube2V1 + Ubv.V1.1 + Ube2N | Ube2G1 + Ubv.G1.1 | |
|---|---|---|---|
| Data collection | |||
| Wavelength | 0.9792 | 0.9792 | 0.9795 |
| Space group | P212121 | P32 | P3221 |
| Cell dimensions a, b, c (Å) | 51.12, 52.56, 96.75 | 91.44, 91.44, 92.45 | 66.54, 66.54, 225.1 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 120 | 90, 90, 120 |
| Resolution, Å | 50–2.10 | 40–2.55 | 46–2.35 |
| Rmerge, % | 10.4 (88.1) | 6.5 (95.0) | 6.0 (36.7) |
| CC1/2 | 0.999 (0.610) | 0.993(0.572) | 0.967 (0.913) |
| I/σ | 30.2 (1.0) | 26.7 (1.1) | 23.3 (2.8) |
| Completeness, % | 99.9 (100) | 99.8 (99.9) | 99.6 (98.9) |
| Reflections total/unique | 112423/15572 | 142899/28109 | 168582/24851 |
| Redundancy | 7.2 (6.7) | 5.1 (4.9) | 6.8 (6.2) |
| Refinement | |||
| Rwork/Rfree | 0.214/0.231 | 0.271/0.287 | 0.206/0.257 |
| No. of atoms | |||
| Protein | 3402 | 9966 | 7428 |
| Water | 24 | 0 | 45 |
| B-factors, Å2 | |||
| Protein | 51.8 | 104.4 | 70.2 |
| water | 39.8 | – | 61.5 |
| R.M.S deviations | |||
| Bond lengths, Å | 0.002 | 0.004 | 0.002 |
| Bond angles, ° | 0.60 | 0.63 | 0.58 |
| Ramachandran plot | |||
| Most favored regions (%) | 99.1 | 98.1 | 99.1 |
| Allowed regions (%) | 0.9 | 1.9 | 0.9 |
| Disallowed regions (%) | 0.0 | 0.0 | 0.0 |
| MolProbity Score | 1.09 | 1.13 | 0.84 |
| PDB ID code | 6D4P | 6D6I | 6D68 |
Values in parentheses are for the highest resolution shell.
Fig. 3. Superpositions of structures of UbVs and Ub.wt in complex with E2 proteins.

Ub.wt and its associated E2 protein are colored gray. The UbV and its associated E2 protein are colored as follows: UbV.D1.1 (orange) and Ube2D1 (blue), UbV.V1.1 (yellow) and Ube2V1 (green), UbV.G1.1 (magenta) and Ube2G1 (cyan). Active site cysteines in E2 enzymes are shown as red spheres. (A) Superposition of Ube2D1-UbV.D1.1 complex with Ube2D1-Ub.wt complex (PDB: 3PTF). (B) Superposition Ube2V1-UbV.V1.1 complex with Ube2V2-Ub.wt complex (PDB: 1ZGU). (C) Superposition of Ube2G1-UbV.G1.1 complex with apo Ube2G1 structure (PDB: 2AWF). (D) Superposition of Ube2D1-UbV.D1.1, Ube2V1-UbV.V1.1 and Ube2G1-UbV.G1.1 complexes.
Comparing binding interfaces, UbVs and their cognate E2 proteins interacted through similar surfaces, with UbV.D1.1, UbV.V1.1, and UbV.G1.1 using 790 Å2, 657 Å2, and 707 Å2 of surface area, respectively, to interact with 751 Å2, 586 Å2, and 680 Å2 of surface area on Ube2D1, V1, and G1, respectively. However, the UbVs differed in the contacts made by the residues that were substituted relative to Ub.wt and thus, used different molecular mechanisms to achieve enhanced affinity. In UbV.D1.1, 6 of 10 substitutions made contact with Ube2D1 and formed a cluster that constituted essentially half of the total interface (Fig. 4A, left panel). In contrast, a cluster of only three of eight substitutions in UbV.V1.1 made contact with Ube2V1 (Fig. 4A, center panel). Eight of 10 substitutions in UbV.G1.1 made contact with Ube2G1, and these were dispersed across the surface (Fig. 4A, right panel).
Fig. 4. Interactions at the UbV-E2 interfaces.
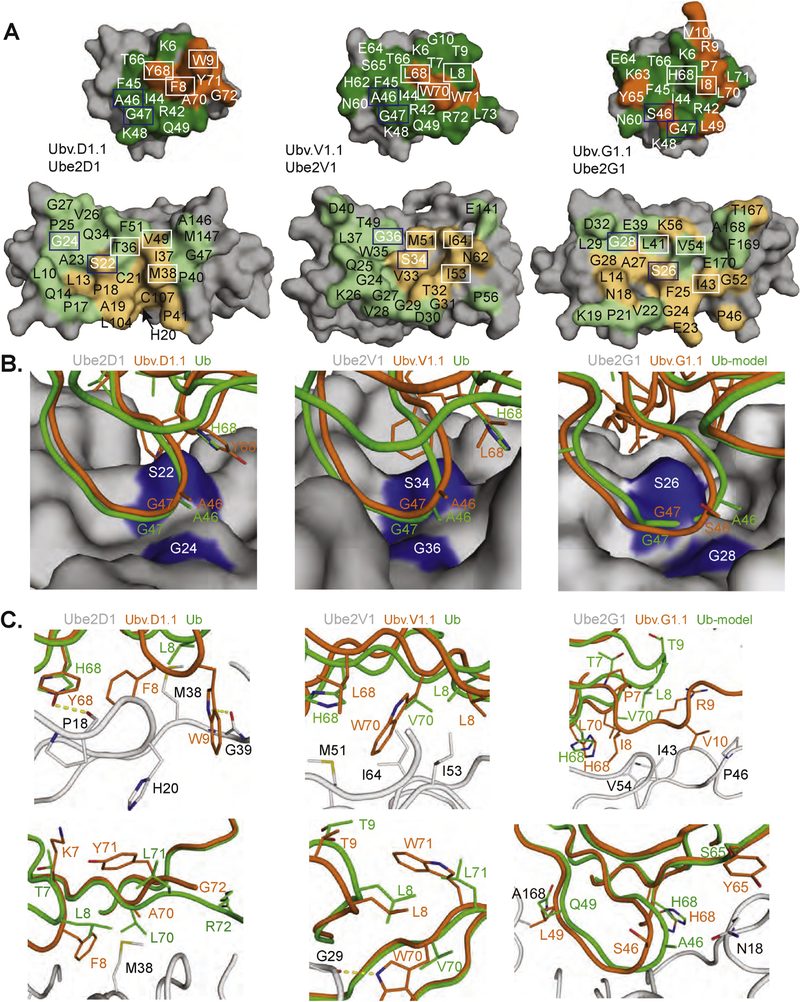
(A) Contact surfaces at the interface between UbV.D1.1 and Ube2D1 (left), UbV.V1.1 and Ube2V1 (center), and UbV.G1.1 and Ube2G1 (right). The complex is shown in an open book view with the UbV at the top and the E2 protein at the bottom. The proteins are shown as molecular surfaces with noncontact residues colored gray. Substituted or wt contact residues on the UbV are colored orange or green, respectively. Residues on the E2 that contact substituted or wt residues on the UbV are colored light orange or light green, respectively. UbV and E2 residues forming the hydrophobic pocket and adjacent contact surface are boxed in white or blue and are shown as sticks in panels B and C. (B,C) Interactions between the E2 backside pocket and UbV side chains are shown for Ube2D1and UbV.D1.1 (left), Ube2V1 and UbV.V1.1 (center), and Ube2G1 and UbV.G1.1 (right). The E2 protein from each UbV-E2 complex is shown as a surface in (B) or as a gray ribbon with indicated side chains in (C). The interacting side chains and associated backbones of the UbV, or Ub.wt for comparison, are colored orange or green, respectively. In the case of Ube2G1, a structure in complex with Ub.wt is not available, and thus, the complex was modeled by superposition of Ub.wt with UbV.G1.1 in the UbV.G1.1-Ube2G1 structure.
Molecular basis for enhanced affinity and specificity of UbVs
Despite the significant differences in the location and nature of UbV substitutions, the complex structures revealed similar “backside pockets” on the E2 proteins, which the UbVs used for both conserved and new interactions relative to Ub.wt. Interactions that were conserved across the three UbVs and with Ub.wt mainly involved small residues at positions 46 and 47, which interacted with small residues at positions 22* and 24* of the E2 proteins (Fig. 4B, E2 residues are marked with asterisks throughout). Adjacent to these conserved interactions, each E2 protein contains a hydrophobic patch that interacted with the cognate UbV through residues that are substituted relative to Ub.wt and often differ among UbVs (Fig. 4C).
The structures revealed the importance of the Ser22*/Gly24* pair that is conserved among the E2 proteins and is adjacent to the hydrophobic patch that accommodates the hydrophobic substitutions in the UbVs (Fig. 4A). In the structures of Ub.wt in complex with Ube2D1 or Ube2V2, Ser22* and Gly24* on the E2 protein are in contact with Ala46 and Gly47 on Ub.wt, and these small residues form a tightly packed cluster. Aside from a conservative Ser substitution at position 46 in UbV.G1.1 (Fig. 2A), these residues are conserved as the Ub.wt sequence among all three UbVs. The structures of the UbV-E2 complexes show very similar and close packing of these small residues across the interfaces (Fig. 4B), indicating that small residues at these positions are crucial for Ub.wt and UbV binding to the E2 backside and explaining why E2 backsides with large side chains at positions 22 and/or 24 are not known to bind Ub.wt.
In UbV.D1.1, four of the six substituted contact residues (Phe8, Trp9, Tyr68, and Tyr71) form an aromatic cluster that expands the hydrophobic patch formed by the wt residues Ile44, Phe45, and Ala46 (Fig. 4A, left panel). UbV.D1.1 buries more surface area compared with Ub.wt in complex with Ube2D1 (789 Å2 versus 659 Å2), and this is essentially all contributed by Phe8 and Trp9. While the backbone around Tyr68 and Tyr71 does not change appreciably relative to that of Ub.wt, the β1-β2 loop region including Phe8 and Trp9 moves closer to Ube2D1. Consequently, Phe8 and Trp9 are buried in hydrophobic cavities that are not occupied in the Ub.wtUbe2D1 complex. These new hydrophobic interactions are augmented by enhanced hydrophobic interactions mediated by Tyr68 in place of His68 and two backbone carbonyl H-bonds to Pro18* and Gly39* of Ube2D1 mediated by Trp9 and Tyr71 (Fig. 4C, left upper panel). Among the other substitutions in UbV.D1.1 relative to Ub.wt, the smaller Ala70 in place of Val70 alleviates a clash with Met38*, while the side chains of Lys7 and Tyr71 face away from the interface. Disorder of the remaining UbV.D1.1 tail residues in the crystal structure suggests that Gly72 and Ala75 substitutions do not contribute to the interaction with Ube2D1 (Fig. 4C, left bottom panel).
Despite the hydrophobic nature of most contacts, UbV.D1.1 is specific for Ube2D1, as it does not bind appreciably to any other E2 protein using competitive ELISA, including the close relatives Ube2D2, D3, and D4 (Fig. 2B and C). This E2 selectivity may be influenced by a difference at position 20, which is occupied by His in Ube2D1 or Gln in the other Ube2D family members. In the UbV.D1.1-Ube2D1 complex, the His20* side chain points away from Trp9, whereas superposition of the Ube2D2 structure shows that the Gln20* side chain would point towards Trp9 and cause a steric clash (Supplementary Fig. 3). We were unable to find any reported Ube2D2–4 structures in which Gln20* adapted a conformation that would permit UbvD1.1 to bind in the manner permitted by His20* in Ube2D1 (Supplementary Fig. S3, panel B). Two distinct side chain rotamers observed for Gln20* in the structures of apo-Ube2D2 (PDB entry: 3TGD) and the Ube2D2~Ub/RNF38/UbB complexes (PDB entries: 4V3K and 4V3L) would both clash with Trp9 of a superposed Ubv.D1.1. The importance of this difference for the selectivity of UbV.D1.1 was further supported by the finding that a His to Gln substitution at position 20* in Ube2D1 abrogated binding to UbV.D1.1 (Fig. 2C). Together, these results highlight the importance of a backside hydrophobic patch on Ube2D1 that accommodates the aromatic substitutions Phe8, Trp9, and Tyr68 in UbV.D1.1 to confer high affinity and specificity to this interaction.
UbV.V1.1 contains three substitutions that contact Ube2V1, and these are all hydrophobic residues (Leu68, Trp70, and Trp71, Fig. 4A, center panel). Leu68 and Trp70 interact with a hydrophobic patch on Ube2V1 in a manner analogous to Tyr68 and Phe8 of UbV.D1.1, respectively, suggesting that these residues are important for the enhanced affinity of UbV.V1.1. Residues Leu8 and Trp70 of Ube2V1 make contacts with the hydrophobic pocket around Met51*, Ile53*, and Ile64* on Ube2V1, which are analogous to those formed by Phe8 of UbV.D1.1 with Ube2D1 (Fig. 4C, center upper panel). Trp71 does not make significant contact with Ube2V1, suggesting that it plays a minor role in binding (Fig. 4C, center lower panel). The importance of Leu68 and Trp70 relative to Trp71 is supported by the fact that, among 56 unique UbVs selected for binding to Ube2V1, Leu68 and Trp70 are observed in 64% and 82% of the sequences, respectively, whereas Trp71 is only observed in 5% of the sequences (Supplementary Table S1), suggesting a strong selective pressure for Leu68 and Trp70 but not for Trp71. Moreover, UbV.V1.2, which binds broadly to the Ube2V, D, and G families (Fig. 2B), contains only three substitutions and two of these are Leu68 and Trp70 (Fig. 2A), suggesting that these two substitutions enhance binding to the backsides of many E2 proteins. Thus, it appears that UbVs selected for binding to Ube2V1 make use of enhanced interactions with a backside pocket, which is similar to the backside pocket on Ube2D1 that is engaged by UbVD1.1, although the specific details of the molecular interactions differ.
The analysis of UbV.G1.1 was complicated by a β1-strand exchange that caused dimerization in the crystal (Supplementary Fig. 4). The UbV.G1.1 dimer is unlikely to be an artifact, because similar strand-exchanged dimers have been observed in at least two other UbV structures reported recently [32,33]. The strand-exchanged region in UbV.G1.1 contains four consecutive substitutions relative to Ub.wt and dramatically alters the β1-β2 loop conformation. Nonetheless, Ube2G1 also possesses a backside hydrophobic patch similar to that of Ube2D1, but the residues in UbV.G1.1 that interact with this patch differ from those in UbV.D1.1 that interact with Ube2D1. The regions occupied by Tyr68 and Phe8 in UbV.D1.1 are occupied by the same positions of UbV.G1.1, but the residue types are different (His68 and Ile8). In Ube2G1, the region that is analogous to the pocket in Ube2D1 that is occupied by Trp9 from UbV.D1.1 is occupied by a smaller Val10 substitution in UbV.G1.1. This surface cavity in Ube2G1 is quite large and could likely accommodate an even larger residue to further optimize this interface (Fig. 4C, right panel). Two other substitutions in this region, Pro7 and Arg9, do not make significant contributions to the interface with Ube2G1. Finally, Ser46 is a small side chain oriented away from the Ser22*/ Gly24* E2 surface, whereas Leu49 and Tyr65 pack favorably against Ala168* or Gln19* of Ube2G1, respectively. Thus, UbV.G1.1 also interacts with a backside pocket on Ube2G1 that is analogous to the backside pocket on Ube2D1 that interacts with UbVD1.1.
Taken together, the three UbV-E2 complex structures show how E2 backsides can accommodate tight and specific binding of UbVs. Our analysis highlights the Ser22*/Gly24* pair in E2 proteins, which necessitates conservation of small residues at positions 46 and 47 of Ub moieties to enable close packing at the interface. Adjacent to this conserved cluster of small residues, each E2 protein contains a hydrophobic patch that is not optimally occupied by Ub.wt, and hydrophobic substitutions in each UbV establish favorable contacts with this region to enhance affinity and specificity.
Effects of UbVs on E2 function
We performed enzymatic assays to assess the functional effects of UbVs binding to the backsides of E2 proteins. Ube2D1 contains an active-site cysteine and acts as an autonomous E2 ligase to assemble poly-Ub chains on substrates [18]. Formation of Ube2D1~Ub was unaffected by the presence of Ubv.D1.1 or Ub.wt lacking its two C-terminal glycines (Ub.1–74), which were both non-functional for thioester bond formation but were capable of backside interactions with Ube2D1 (Fig. 5A).
Fig. 5. Effects of UbVs on E2 activity.

(A) Time course of Ube2D1~Ub formation in the presence of UbV.D1.1 or Ub.1-74 (left panel) with quantitation of Ube2D1~Ub relative to a 10 min reaction with Ube2D1 alone (right panel). Ube2D1 charging reactions containing 0.20 μM E1, 5 μM Ube2D1, 25 μM Ub-FL, 2 mM ATP, and 5 mM MgCl2 were incubated with or without 25 μM UbV.D1.1 or Ub.1–74. Quantification of Ube2D1~Ub was performed on fluorescent-scanned images processed using ImageQuant TL (V8.1.0.0) (B) Processive Ub chain formation by Ube2D1 in the presence of RBX1 or TRIM25 RING domain. Ubiquitination reactions containing 0.20 μM E1, 5 mM Ube2D1, 20 μM RING, and 12.5 μM Ub-Fluor were incubated with or without 50 μM UbV.D1.1 for 30 min. (C) Time course of processive Ub chain formation by Ube2D1 or Ube2D2 in the presence of Rbx1 and UbV.D1.1. Reactions containing 0.20 μM E1, 5 μM Ube2D1, 5 μM E3 RING, and 25 μM Ub-Fluor were incubated with or without 25 μM UbV.D1.1 for the indicated time. (D) Effects of UbV.V1.1 on di-Ub formation by Ube2N and Ube2V2. Reaction mixtures containing 0.25 μM E1, 5 μM Ube2N, 5 μM Ube2V2 or Ube2V1, 20 μM Ub, 2 mM ATP, and 5 mM MgCl2 were incubated with or without 20 μM UbV.V1.1. Formation of Ube2N~Ub and di-Ub was visualized using SDS-PAGE and Coomassie blue staining.
RING-mediated Ub chain formation by Ube2D1 is enhanced by Ub backside interactions through an allosteric mechanism that increases E2-RING affinity [28]. Self-assembly of multiple Ube2D1~Ub complexes mediated through backside Ub interactions also promoted Ub chain formation by Ube2D13 [34]. In the presence of UbV.D1.1, processive Ub chain formation by Ube2D1 was reduced in reactions containing Rbx1 or TRIM25 RING domains (Fig. 5B). In contrast, Ube2D2 did not detectably interact with Ubv.D1.1 (Fig. 2C), which therefore did not attenuate Ub chain formation by Ube2D2 (Fig. 5C), suggesting that the effect on Ube2D1 was not due to nonspecific termination of Ub chain formation. The differences in these results suggest that Ubv.D1.1 backside binding may impact both RING-dependent allosteric effects and Ube2D1~Ub self-assembly that contribute to processive chain formation by Ube2D1.
Ube2V1 and Ube2V2 lack active-site cysteines, but they work with the E2 enzyme Ube2N to facilitate Ub chain formation by binding and positioning Lys63 of the acceptor Ub for isopeptide bond formation with the charged Ub on Ube2N [35,36]. In the presence of Ube2V2, UbV.V1.1 did not inhibit charging of Ube2N, but it prevented di-Ub formation in a manner similar to the Ube2V2-S34R mutation that disrupts Ub binding to the backside (Fig. 5D). Formation of a Ub-UbV.V1.1 dimer (analogous to di-Ub) was not detected, presumably because UbV.V1.1 contains a Trp substitution in place of the wt Lys at position 63 and, thus, is not an isopeptide acceptor. Modeling the Ube2N/Ube2V1/UbV.V1.1 complex on a Ube2N-RING/Ube2V1 complex containing a donor Ub on Ube2N (PDB: 5AIT) showed a distance of 11 Å between the C-terminus of the donor Ub and an acceptor Lys63 modeled in place of Trp63 of UbV.V1.1 (Supplementary Fig. 5). Thus, UbVs that bind to the backsides of Ube2D1 or Ube2V1/2 do not inhibit charging of the E2 active-site cysteine but do attenuate formation of Ub chains, which is the expected result, as the backsides of these proteins are known to facilitate chain formation through binding of Ub.wt [18,35,36].
UbV.G1.1 interacted with the backside of Ube2G1 and reduced the levels of Ube2G1~Ub conjugate in charging reactions (Fig. 6A). This effect required binding of UbV.G1.1 to the backside of Ube2G1, as a Ube2G1-S22R mutant protein compromised for backside binding was efficiently charged with Ub by E1 enzyme in the presence of UbV.G1.1. Higher contrast of the fluorescent Ub scan (upper region of right panel) detected E1~Ub, providing further support that UbV.G1.1 does not directly inhibit E1 function. Furthermore, comparison of the rate of Ube2G1~Ub formation in the presence UbV.G1.1 showed that charging of Ube2G1-S26R saturated within 30 min while the level of Ube2G1~Ub did not reach 50% over a similar time course (Fig. 6B).
Fig. 6. UbV.G1.1 inhibits Ub charging of Ube2G1.

(A) Effect of UbV.G1.1 on Ube2G1~Ub formation. Charging reactions containing 0.20 μM E1, 5 μM Ube2G1 or Ube2G1-S26R, 25 μM Ub-Fluor, 2 mM ATP, and 5 mM MgCl2 were incubated with indicated amounts of UbV.G1.1 for 30 min. Coomassie blue staining (left panel) and fluorescent scanning (center panel) of the gel.are shown. The level of Ube2G1~Ub and Ube2G1-S26R~Ub was quantified (right panel) relative to a 30 min reaction in the absence of UbV.G1.1. (B) Time course of Ube2G1~Ub formation in the presence of Ubv.G1.1. Ube2G1 and Ube2G1-S26R charging reactions were performed with 62.5 μM UbV.G1.1 for the indicated times and analyzed as described earlier. (C,D) Discharge of (C) Ube2G1 or (D) Ube2G1-S26R in the presence of Ubv.G1.1. Ube2G1 or Ube2G1-S26R was charged with Ub-Fluor for 30 min; the discharge reaction was initiated by addition of 1 unit apyrase, 40 mM lysine, 25 mM EDTA, and 50 μM UbV.G1.1. After the indicated time, the reaction was stopped with SDS sample loading buffer. Gels were scanned at 480 nm on a Typhoon fluorescence scanner (GE LifeSciences); bands were quantified using ImageQuant TL (GE LifeSciences) and visualized using Coomassie blue staining
Inhibition of Ube2G1 charging by UbV.G1.1 could occur through steric blockage of E1 engagement of the Ube2G1/UbV.G1.1 complex or allosteric effects on the E2 active site that promote discharge of Ube2G1~Ub. To examine these possibilities, we monitored the loss of Ub from precharged Ube2G1~Ub and Ube2G1-S26R~Ub in the presence or absence of UbV.G1.1 (Fig. 6C and D). After charging for 30 min, the amount of Ube2G1~Ub was monitored after addition of apyrase, lysine, and EDTA to stop further E1 function. No significant differences were detected between the rates of Ub discharge of Ube2G1 or Ube2G1-S26R in the presence or absence of UbV.G1.1. Thus, backside binding of UbV.G1.1 to preformed Ube2G1~Ub does not detectably alter Ub discharge.
Together, these results suggest that binding of UbV.G1.1 to the backside of Ube2G1 may prevent E1-mediated Ub charging through a steric mechanism on the E2-UbV complex. Our crystal structure revealed that UbV.G1.1 forms a strand-exchanged UbV dimer with each protamer bound to a distinct Ube2G1 subunit to form a 2:2 complex (Supplementary Fig. 5). To determine if a Ube2G1-UbV.G1.1 higher order complex could pose a steric clash that competes with an E1 interaction, the structure of an E1-E2 complex of Ubc1 and Ubc15 (PDB: 5KNL) [37] was superposed with our E2-UbV complex (Supplementary Fig. 6). Alignment of the E2 domains allowed examination of potential clashes between the backside-bound UbV.G1.1 on Ube2G1 and the Ub-fold domain (Ufd) of the Saccharomyces pombe E1 protein (Supplementary Fig. 6A). For comparison, we also superposed the Ube2D1/Ub and Ube2D1/UbV.D.1.1 complexes and observed minor clashes with backside-bound Ubv.D1.1 or Ub.wt, which could be overcome by alternate rotamer modeling of side chains (Supplementary Fig. 6B and C). Overlay of each complex showed that the rotation of UbV.G1.1 on the backside of Ube2G1 relative to the position of UbV.D1.1 or Ub on the backside of Ube2D1 could account for a potential E1 steric clash that prevents charging of the Ube2G1-UbV.G1.1 complex (Supplementary Fig. 6D). This analysis is consistent with our findings that UbV.G1.1 reduced charging of Ube2G1, whereas UbV.D1.1 did not affect charging of Ube2D1.
Discussion
Using phage-displayed libraries, we generated UbVs that target E2 protein backsides with high affinity and specificity. In the case of Ube2D1 and Ube2V1, the UbVs did not affect charging at the active site but did attenuate chain formation. In contrast, a UbV that targeted the backside of Ube2G1 inhibited charging of Ub despite the long distance between the backside and the active site. Blockage of E2 charging by a steric mechanism was demonstrated previously for a Ub-fold protein (MUB) that binds the backside of the Ubc8 E2 enzyme and prevents Ub charging through steric clash with the E1 enzyme [38]. E3 ligases that occupy the backsides of E2 proteins (e.g., Ube2G2-gp78; PDB: 3H8K) [21] may also inhibit E1-mediated charging. Thus, it is clear that the backside of a significant subset of E2 proteins can be targeted to inhibit or attenuate catalytic function.
Despite the differences among the various UbVs, structural analysis reveals a common docking mode for UbVs bound to E2 proteins, which is very similar to that observed for Ub.wt docking with Ube2V1 and Ube2D family members. Detailed comparison of the molecular interactions at the interfaces of UbV-E2 and Ub.wt-E2 complexes showed that enhanced affinity is largely achieved through hydrophobic interactions between substituted residues in the UbVs and a common backside pocket in the E2 proteins. However, despite the hydrophobic nature of these contacts, the interactions are highly specific as each UbV is able to distinguish its cognate E2 protein among other E2 proteins. The discovery of a common hydrophobic pocket on the backside of a subset of the E2 family raises the possibility that small molecules could be designed to modulate E2 activity for therapeutic benefit. In this regard, the UbV proteins will be useful intracellular probes to assess biological effects of E2-backside blockade. Moreover, they may prove useful for enabling inhibitor discovery through displacement screens, and the UbV-E2 complex structures may inform inhibitor design.
Materials and Methods
Selection of E2-binding UbVs
Phage-displayed library UbV-E2 was constructed as described previously for library UbV-USP except that UbV-coding sequence terminated at position 76 instead of the originally longer constructs including two additional residues to eliminate the possibility of E1-mediated conjugation [29,39]. His-tagged and biotinylated E2 proteins were expressed and purified from Escherichia coli BL21(DE3), as described [40]. Phage pools representing the libraries were cycled through rounds of binding selections with biotinylated E2 protein immobilized on Nunc Maxisorp 96-well plate (Fisher Scientific, Nepean, ON, Canada) coated with neutravidin (ThermoFisher Scientific, Rockford, IL, USA). After the fifth round, phage from individual clones were assayed for binding to the target using phage ELISA as described [41], and 50 to 100 positive clones were subjected to DNA sequence analysis.
ELISAs for assessing specificity and affinity
UbVs with N-terminal His and FLAG tags were expressed and purified from E. coli BL21(DE3) as described [29]. For specificity assessment, ELISAs were performed as described [29] and binding of UbV to biotinylated E2 protein immobilized on neutravidin-coated plates was detected with an anti-FLAG antibody (Sigma-Aldrich, F1804, 1:5000). For determination of IC50 values, competition ELISAs were performed as described [42] with protein immobilization and detection similar to the specificity ELISA. Briefly, a constant subsaturating concentration of UbV protein was incubated with serial dilutions of each E2 protein tested and binding to immobilized cognate E2 protein was detected. The IC50 value was defined as the concentration of solution-phase E2 protein that inhibited 50% of the binding of UbV protein to immobilized E2 protein.
Protein purification and structure determination
His-tagged E2 and Ubv proteins for phage display were expressed in E. coli and purified using Ni-NTA chromatography using standard techniques, as described [16,29]. Expression constructs for Ube2G18−160, Ube2G21−165, Ube2V15−147, and Ube2N3−152 in the pET28-LIC vector were obtained from the Structural Genomics Consortium Toronto. Ube2D11−147, Rbx136−108, TRIM251−82, and Ub.1–74 were cloned into pProEx HTb. Mutation of Ube2D1-S22R, Ube2D1-H20Q, Ube2V2-S34R, Ube2G1-S26R, and Ube2G2-V25S was generated and confirmed using sequence analysis. All proteins were expressed in E. coli BL21(DE3) Codon+ cells (Agilent Technologies) induced with 0.25 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) for 14–18 h at 18C. Harvested cells were resuspended in 20 mM HEPES pH 7.5, 400 mM NaCl, 5 mM β-mercaptoethanol, lysed by passage through a cell homogenizer (Avestin Inc.) and spun at 30,000g. The supernatant was applied to a HiTrap nickel-chelating HP column (GE Healthcare) equilibrated in lysis buffer with 5 mM imidazole. Protein eluted with buffer containing 300 mM imidazole was incubated overnight with tobacco etch virus (TEV) or thrombin protease. Cleaved protein was dialyzed in HiTrap loading buffer and flowed over a subtractive HiTrap nickel-chelating column, then concentrated for injection onto a 24- or 120-mL Superdex S75 column (GE Healthcare) equilibrated in 20 mM HEPES pH 7.5, 100 mM NaCl, and 0.5 mM dithiothreitol (DTT). Fractions containing purified protein of interest were concentrated to 10–35 mg/mL and stored at −80C. Fluorescent Ub was prepared as described [16].
Crystals of the Ube2D1-UbV.D1.1 complex were grown at 20C in sitting drops containing 0.2 μL of 350 μM protein with 0.2 μL well solution containing 24% PEG2000-MME, 0.2 M trimethylamine N-oxide, Tris-HCl, pH 8.0, and cryoprotected with 20% glycerol. Diffraction data were collected at 100K on beamline NE-CAT 24-ID-E (APS, Chicago, Il). Molecular replacement was performed using Phaser [43] with Ube2D1 and Ub (PDB: 3PTF) as separate search objects [27]. Crystals of the Ube2V1-UbV.V1.1-Ube2N1 complex were grown at 20°C in hanging drops containing 1 μL of each protein at 500 μM with 1.0 μL of 0.2 M LiSO4, 25% PEG3350, 0.1 M Tris-HCl, pH 8.5, and cryoprotected with 20% glycerol. Molecular replacement was performed using Ube2N, Ube2V2, and Ub (PDB: 5AIT) as separate search objects [27]. Crystals of the Ube2G1-UbV.G1.1 complex were grown at 20°C in hanging drops containing 1 μL of protein with 1 μL of 25% PEG1500, 0.1 M SPG buffer (succinic acid, sodium dihydrogen phosphate, glycine in a 2:7:7 M ratio) pH 8.0, and cryoprotected with 20% glycerol. Molecular replacement was performed with Ube2G1 (PDB: 2AWF) and Ub (PDB: 1FXT) as separate search objects [44,45]. Model building and refinement were performed using Coot [46] and Phenix [47]. Ramachandran and other statistics are in Table 1. All protein structure figures were generated using PyMOL (www.pymol.org). Software used in this project was curated using SBGrid [48].
E2 charging and Ub linkage assays
Reactions were performed in 20 mM HEPES pH 7.5, 100 mM NaCl, 5 mM MgCl2, 250 nM UBE1 enzyme (Boston Biochem, catalog number E304), 5 μM E2 enzyme, 25–50 mM Ub-Fluor (4:1 ratio of unlabeled and fluorescently labeled Ub) [16], with or without the indicated concentration of UbV or Ub.1–74. The reaction was initiated by adding 2 mM ATP, incubated for the indicated time at 30°C, and stopped with nonreducing SDS sample loading buffer. To differentiate thioester-linked intermediates from isopeptide-linked intermediates, 10 μL of the reaction was treated with 250 μM DTT and heated at 90°C for 5 min before SDS-PAGE. Gels were scanned at 480 nm on a Typhoon fluorescence scanner (GE LifeSciences), and bands were quantified using ImageQuant TL (GE LifeSciences) and visualized using Coomassie blue staining. Line graphs were plotted with nonlinear regression curve fits and generated using Prism (V 5.0a).
Processive ubiquitination assays
Reactions were performed in 20 mM HEPES pH 7.5, 100 mM NaCl, 5 mM MgCl2, 250 nM UBE1 enzyme, 5 μM E2 enzyme, 5 μM Rbx1 or TRIM25 with 12.5–25 μM Ub or Ub-Fluor, with or without 25 μM or the indicated amount of UbV. The reaction was initiated by adding 2 mM ATP, incubated for 30 min or the indicated time at 30°C, and stopped with SDS sample loading buffer. Gels were scanned at 480 nm on a Typhoon fluorescence scanner (GE LifeSciences), and bands were quantified using ImageQuant TL (GE LifeSciences) and visualized using Coomassie blue staining. Line graphs were plotted with nonlinear regression curve fits and generated using Prism (V 5.0a).
Ub discharge assay
Ube2G1 and Ube2G1-S26R were charged for 30 min at 30°C in a reaction consisting of 25 μM total Ub-Fluor, 0.5 μM UBE1 enzyme, 12.5 μM Ube2G1, and 1 mM ATP. The discharge reactions were initiated by addition of 1 unit apyrase, 40 mM lysine, 25 mM EDTA, and 50 μM UbV.G1.1 for the indicated time and stopped with SDS sample loading buffer. Gels were scanned at 480 nm on a Typhoon fluorescence scanner (GE LifeSciences), and bands were quantified using ImageQuant TL (GE LifeSciences) and visualized using Coomassie blue staining. Line graphs were plotted with nonlinear regression curve fits and generated using Prism (V 5.0a).
Accession Numbers
The coordinates and the structure factors have been deposited in the Worldwide Protein Data Bank (wwPDB) with accession numbers 6D4P, 6D6I, and 6D68.
Supplementary Material
Acknowledgments
This work was supported by the Canadian Institutes of Health Research, as follows: foundation grant (FDN143277) to F. S., operating grant (MOP-136956) to S. S. S., and Ontario Trillium Scholarship to P. G. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on 24-ID-C beamline is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Abbreviations used:
- Ub.wt
ubiquitin wild type
- Ub
ubiquitin
- UbV
ubiquitin variant
- UPS
ubiquitin proteasome system
- E1
Ub-activating enzyme
- E2
Ub-conjugating enzyme
- E3
Ub-ligating enzyme
- SUMO
small Ub-like modifier
- DTT
dithiothreitol
- PDB
protein data bank
- wwPDB
Worldwide Protein Data Bank
- ELISA
enzyme-linked immunosorbent assay
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2019.09.024.
References
- [1].Hoeller D, Dikic I, Targeting the ubiquitin system in cancer therapy, Nature 458 (2009) 438–444. [DOI] [PubMed] [Google Scholar]
- [2].Ding F, Xiao H, Wang M, Xie X, Hu F, The role of the ubiquitin-proteasome pathway in cancer development and treatment, Front. Biosci. (Landmark Ed.) 19 (2014) 886–895. [DOI] [PubMed] [Google Scholar]
- [3].Huang X, Dixit VM, Drugging the undruggables: exploring the ubiquitin system for drug development, Cell Res. 26 (2016) 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Komander D, Rape M, The ubiquitin code, Annu. Rev. Biochem. 81 (2012) 203–229. [DOI] [PubMed] [Google Scholar]
- [5].Komander D, The emerging complexity of protein ubiquitination, Biochem. Soc. Trans. 37 (2009) 937–953. [DOI] [PubMed] [Google Scholar]
- [6].Sheridan C, Drug makers target ubiquitin proteasome pathway anew, Nat. Biotechnol. 33 (2015) 1115–1117. [DOI] [PubMed] [Google Scholar]
- [7].Jagannath S, Durie B, Wolf J, Camacho E, Irwin D, Lutzky J, et al. , First-line therapy with bortezomib (formerly PS-341) in patients with multiple myeloma (MM), J. Clin. Oncol. 22 (2004) 6551. [Google Scholar]
- [8].Cao B, Li J, Mao X, Dissecting bortezomib: development, application, adverse effects and future direction, Curr. Pharm. Des. 19 (2013) 3190–3200. [DOI] [PubMed] [Google Scholar]
- [9].Hershko A, Ciechanover A, The ubiquitin system, Annu. Rev. Biochem. 67 (1998) 425–479. [DOI] [PubMed] [Google Scholar]
- [10].Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS, E2 enzymes: more than just middle men, Cell Res. 26 (2016) 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ye Y, Rape M, Building ubiquitin chains: E2 enzymes at work, Nat. Rev. Mol. Cell Biol. 10 (2009) 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Strickson S, Campbell DG, Emmerich CH, Knebel A, Plater L, Ritorto MS, et al. , The anti-inflammatory drug BAY 11–7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system, Biochem. J. 451 (2013) 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sanders MA, Brahemi G, Nangia-Makker P, Balan V, Morelli M, Kothayer H, et al. , Novel inhibitors of Rad6 ubiquitin conjugating enzyme: design, synthesis, identification, and functional characterization, Mol. Cancer Ther. 12 (2013) 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pulvino M, Liang Y, Oleksyn D, DeRan M, Van Pelt E, Shapiro J, et al. , Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A, Blood 120 (2012) 1668–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ceccarelli DF, Tang X, Pelletier B, Orlicky S, Xie W, Plantevin V, et al. , An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme, Cell 145 (2011) 1075–1087. [DOI] [PubMed] [Google Scholar]
- [16].Huang H, Ceccarelli DF, Orlicky S, St-Cyr DJ, Ziemba A, Garg P, et al. , E2 enzyme inhibition by stabilization of a low-affinity interface with ubiquitin, Nat. Chem. Biol. 10 (2014) 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van Wijk SJ, Timmers HT, The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins, FASEB J. 24 (2010) 981–993. [DOI] [PubMed] [Google Scholar]
- [18].Brzovic PS, Lissounov A, Christensen DE, Hoyt DW, Klevit RE, A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination, Mol. Cell 21 (2006) 873–880. [DOI] [PubMed] [Google Scholar]
- [19].Lewis MJ, Saltibus LF, Hau DD, Xiao W, Spyracopoulos L, Structural basis for non-covalent interaction between ubiquitin and the ubiquitin conjugating enzyme variant human MMS2, J. Biomol. NMR 34 (2006) 89–100. [DOI] [PubMed] [Google Scholar]
- [20].Bocik WE, Sircar A, Gray JJ, Tolman JR, Mechanism of polyubiquitin chain recognition by the human ubiquitin conjugating enzyme Ube2g2, J. Biol. Chem. 286 (2011) 3981–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Das R, Mariano J, Tsai YC, Kalathur RC, Kostova Z, Li J, et al. , Allosteric activation of E2-RING finger mediated ubiquitylation by a structurally-defined specific E2 binding region of gp78, Mol. Cell 34 (2009) 674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nguyen L, Plafker KS, Starnes A, Cook M, Klevit RE, Plafker SM, The ubiquitin-conjugating enzyme, UbcM2, is restricted to monoubiquitylation by a two-fold mechanism that involves backside residues of E2 and Lys48 of ubiquitin, Biochemistry 53 (2014) 4004–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schumacher FR, Wilson G, Day CL, The N-terminal extension of UBE2E ubiquitin-conjugating enzymes limits chain assembly, J. Mol. Biol. 425 (2013) 4099–4111. [DOI] [PubMed] [Google Scholar]
- [24].Miura T, Klaus W, Gsell B, Miyamoto C, Senn H, Characterization of the binding interface between ubiquitin and class I human ubiquitin-conjugating enzyme 2b by multidimensional heteronuclear NMR spectroscopy in solution, J. Mol. Biol. 290 (1999) 213–228. [DOI] [PubMed] [Google Scholar]
- [25].Knipscheer P, van Dijk WJ, Olsen JV, Mann M, Sixma TK, Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation, Embo J. 26 (2007) 2797–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Choi YS, Lee YJ, Lee SY, Shi L, Ha JH, Cheong HK, et al. , Differential ubiquitin binding by the acidic loops of Ube2g1 and Ube2r1 enzymes distinguishes their Lys-48-ubiquitylation activities, J. Biol. Chem. 290 (2015) 2251–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bosanac I, Phu L, Pan B, Zilberleyb I, Maurer B, Dixit VM, et al. , Modulation of K11-linkage formation by variable loop residues within UbcH5A, J. Mol. Biol. 408 (2011) 420–431. [DOI] [PubMed] [Google Scholar]
- [28].Buetow L, Gabrielsen M, Anthony NG, Dou H, Patel A, Aitkenhead H, et al. , Activation of a primed RING E3-E2-ubiquitin complex by non-covalent ubiquitin, Mol. Cell 58 (2015) 297–310. [DOI] [PubMed] [Google Scholar]
- [29].Ernst A, Avvakumov G, Tong J, Fan Y, Zhao Y, Alberts P, et al. , A strategy for modulation of enzymes in the ubiquitin system, Science 339 (2013) 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gorelik M, Orlicky S, Sartori MA, Tang X, Marcon E, Kurinov I, et al. , Inhibition of SCF ubiquitin ligases by engineered ubiquitin variants that target the Cul1 binding site on the Skp1-F-box interface, Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 3527–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang W, Wu KP, Sartori MA, Kamadurai HB, Ordureau A, Jiang C, et al. , System-wide modulation of HECT E3 ligases with selective ubiquitin variant probes, Mol. Cell 62 (2016) 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Manczyk N, Veggiani G, Gish GD, Yates BP, Ernst A, Sidhu SS, et al. , Dimerization of a ubiquitin variant leads to high affinity interactions with a ubiquitin interacting motif, Protein Sci. 28 (2019) 848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gabrielsen M, Buetow L, Kowalczyk D, Zhang W, Sidhu SS, Huang DT, Identification and characterization of mutations in ubiquitin required for non-covalent dimer formation, Structure 27 (9) (2019. September 3) 1452–1459.e4, 10.1016/j.str.2019.06.008 PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Brzovic PS, Klevit RE , Ubiquitin transfer from the E2 perspective: why is UbcH5 so promiscuous? Cell Cycle 5 (2006) 2867–2873. [DOI] [PubMed] [Google Scholar]
- [35].Hau DD, Lewis MJ, Saltibus LF, Pastushok L, Xiao W, Spyracopoulos L, Structure and interactions of the ubiquitin-conjugating enzyme variant human Uev1a: implications for enzymatic synthesis of polyubiquitin chains, Biochemistry 45 (2006) 9866–9877. [DOI] [PubMed] [Google Scholar]
- [36].VanDemark AP, Hofmann RM, Tsui C, Pickart CM, Wolberger C, Molecular insights into polyubiquitin chain assembly: crystal structure of the Mms2/Ubc13 heterodimer, Cell 105 (2001) 711–720. [DOI] [PubMed] [Google Scholar]
- [37].Lv Z, Rickman KA, Yuan L, Williams K, Selvam SP, Woosley AN, et al. , S. Pombe Uba1-Ubc15 structure reveals a novel regulatory mechanism of ubiquitin E2 activity, Mol. Cell 65 (2017) 699–714.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lu X, Malley KR, Brenner CC, Koroleva O, Korolev S, Downes BP, A MUB E2 structure reveals E1 selectivity between cognate ubiquitin E2s in eukaryotes, Nat. Commun. 7 (2016) 12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sidhu SS, Lowman HB, Cunningham BC, Wells JA, Phage display for selection of novel binding peptides, Methods Enzymol. 328 (2000) 333–363. [DOI] [PubMed] [Google Scholar]
- [40].Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, et al. , VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface, Structure 6 (1998) 1153–1167. [DOI] [PubMed] [Google Scholar]
- [41].Tonikian R, Zhang Y, Boone C, Sidhu SS, Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries, Nat. Protoc. 2 (2007) 1368–1386. [DOI] [PubMed] [Google Scholar]
- [42].Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME, Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay, J. Immunol. Methods 77 (1985) 305–319. [DOI] [PubMed] [Google Scholar]
- [43].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software J. Appl. Crystallogr. 40 (2007) 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sheng Y, Hong JH, Doherty R, Srikumar T, Shloush J, Avvakumov GV, et al. , A human ubiquitin conjugating enzyme (E2)-HECT E3 ligase structure-function screen, Mol. Cell Proteomics 11 (2012) 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hamilton KS, Ellison MJ, Barber KR, Williams RS, Huzil JT, McKenna S, et al. , Structure of a conjugating enzyme-ubiquitin thiolester intermediate reveals a novel role for the ubiquitin tail, Structure 9 (2001) 897–904. [DOI] [PubMed] [Google Scholar]
- [46].Emsley P, Cowtan K, Coot: model-building tools for molecular graphics, Acta Crystallogr. D Biol. Crystallogr. 60 (2004) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [47].Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr. D Biol. Crystallogr. 66 (2010) 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, et al. , Collaboration gets the most out of software, Elife 2 (2013) e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.