

Abstract
Light-sensitive proteins can be used to perturb signaling networks in living cells and animals with high spatiotemporal resolution. We recently engineered a protein heterodimer that dissociates when irradiated with blue light and demonstrated that by fusing each half of the dimer to termini of a protein that it is possible to selectively block binding surfaces on the protein when in the dark. Upon activation with light, the dimer dissociates and exposes the binding surface, allowing the protein to bind its partner. Critical to the success of this system, called Z-lock, is that the linkers connecting the dimer components to the termini are engineered so that the dimer forms over the appropriate binding surface. Here, we develop and test a protocol in the Rosetta molecular modeling program for designing linkers for Z-lock. We show that the protocol can predict the most effective linker sets for three different light-sensitive switches, including a newly designed switch that binds the Rho-family GTPase Cdc42 upon stimulation with blue light. This protocol represents a generalized computational approach to placing a wide variety of proteins under optogenetic control with Z-lock.
Keywords: Optogenetics, Protein design, LOV2 domain, Rosetta, Z-lock
Graphical Abstract
Introduction
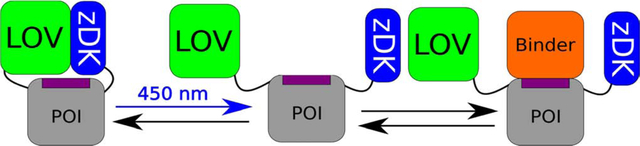
Light-responsive proteins offer spatiotemporal control of protein activity inaccessible to drug-based approaches[1–5], allowing biological processes to be rapidly and reversibly perturbed, in seconds to minutes, in specific locations in a cell or tissue. This has been widely used to interrogate the function of neurons[6,7], and certain of these tools, particularly channelrhodopsins[8], have been more broadly applied to controlling protein function with light. Engineered light-activatable proteins have been used to localize proteins to specific cellular components[9–12], oligomerize them[3,13], or control their activity[14,15], as with the photoactivatable Rac1 (PA-Rac) system[16]. In PA-Rac, the effector binding surface of the GTPase Rac1 domain is sterically occluded from binding effectors by the second light sensitive Light Oxygen Voltage (LOV2) domain from phototropin 1 of Avena sativa, which is fused to the N-terminus of Rac1. When irradiated by 450 nm light, the Jα helix at the C-terminus of the LOV2 domain rapidly becomes disordered as per normal LOV2 kinetics (< 1 sec at 30 C), [17] [freeing the LOV2 domain from the Rac1 surface and exposing the effector binding site. The process is reversible with the switch returning to the closed state in the dark with a half-time of approximately 60 seconds at 23°C [18]. If control on different time scales is needed, there exist mutations to the LOV2 domain that shorten or lengthen the half-life in the lit state[16,19,20]. A crystal structure of PA-Rac revealed that the LOV2 domain was able to block effector binding because specific interactions between the surface of the LOV2 domain and the surface of Rac1 helped to pin Rac1 against the LOV2 domain in the dark[16]. To control the activity of the GTPase Cdc42 with the same approach, it was necessary to redesign the surface of Cdc42 so that similar favorable interactions could be made with the LOV2 domain.
Recently, we developed a general system, called Z-lock, for photocaging proteins with the LOV2 domain that does not require the formation of a specific interface with the protein that is being caged [21]. Z-lock makes use of a small protein domain, zDK, which we previously engineered to interact with the LOV2 domain in the dark but not in the light. With Z-lock, the LOV2 domain is fused to one terminus of the protein that is being caged and zDK is fused to the other terminus. If the linkers connecting the two domains to the termini of the protein are an appropriate length, zDK interacts with the LOV2 domain in the dark and forms a complex over the surface of the protein. In the light, zDK releases from the LOV2 domain, exposing the surface of the protein to its binding partners. If the linkers are too short, the LOV2 domain and zDK are unable to interact in the dark and there is little caging. If the linkers are longer than needed, it is more likely that the zDK/LOV2 complex will form in a location that does not sterically occlude the target binding surface. Here, we describe a protocol we have developed in the molecular modeling program Rosetta to aid in the design of linkers for Z-lock. In addition to revealing which linker lengths are most appropriate for caging, the Rosetta protocol determines if the protein termini are appropriately positioned to place the zDK/LOV2 complex over the target binding surface. We first benchmark the computational protocol with data we previously obtained when caging the proteins cofilin and αTAT with Z-lock, and then use it to design a photosensitive binder of the GTPase Cdc42.
Results
In the Z-lock design protocol, a model is first constructed with zDK and LOV2 fused to the termini of the protein of interest (POI) with the linkers adopting random conformations and zDK not binding with the LOV2 domain. In order to build this model, it is necessary to have a high-resolution structure or high confidence homology model of the POI. There are two variants of zDK, zDK1 and zDK2, that can be used with the Z-lock system. With zDK1 the N-terminus of the LOV2 domain must be fused to the C-terminus of the POI as the LOV2 domain C-terminus is buried at the interface in the zDK1/LOV2 complex. With zDK2, the LOV2 domain and zDK2 can be fused to either terminus of the POI.
The second stage of the protocol is to perform structure prediction to determine if the linkers are long enough to allow zDK to interact with the LOV2 domain and to determine if the zDK/LOV2 complex is likely to block the surface of interest on the POI. Structure prediction is performed with Monte Carlo sampling with the Rosetta low resolution (centroid) energy function combined with distance constraints to direct the formation of a native-like complex between zDK and LOV. Each Monte Carlo move is either a random perturbation to backbone torsion angles in the two linkers or a fragment insertion, the latter being used primarily to speed the backbone sampling by sampling many torsions simultaneously in a structurally sound way. During a fragment insertion, the backbone torsion angles for three consecutive residues in the linker are replaced with torsion angles values pulled from 3-residue pieces of naturally occurring proteins that have similar sequences to the linker that is being designed. Following the Monte Carlo sampling, gradient-based minimization of the torsion angles in the linker is used to fall into a local minimum. The conformations of the individual domains (zDK, LOV2 and the POI) are held constant during the simulation. Many independent trajectories are performed, each starting from different random linker conformations, to map out the range of linker structures that allow for the zDK/LOV2 complex to form. If the linkers are too short for zDK to reach LOV, then none or very few of the trajectories will end up with zDK docked against LOV, even though distance constraints are being used to drive formation of the complex.
To predict if Z-lock is likely to block binding of the POI to its binding partner, we have developed two simulation strategies. The first approach is employed in cases where a high-resolution structure (or homology model) is available of the complex that will be inhibited with Z-lock. In this case, two sets of structure prediction simulations of the zDK/LOV2 linkers are performed: one set with the POI bound to its binding partner and one set with the binding partner absent. If the zDK/LOV2 interaction forms in the absence of the binding partner, it is a desirable result as it indicates that the linkers are long enough for the two proteins to interact. However, if the zDK/LOV2 interaction also forms in the presence of the binding partner (illustrated in the “too long” scenario of fig 1, panel B) it is an undesirable result as it suggests that the two binding events (zDK binding LOV2 and the POI binding to its partner) are not mutually exclusive. Therefore, to identify linkers favorable for caging we scan for linker lengths that allow zDK and LOV2 to interact in the absence of the binding partner, but disfavor interaction when the POI is bound to its binding partner (Fig 1, panel D).
Figure 1.

Inhibiting protein binding interactions with the light sensitive protein pair, LOV-zDK, and linker design. (a) By physically coupling the light-sensitive binding between zDK and the LOV2 domain to a protein of interest via flexible linkers, the binding of specific interactors can be reversibly mediated with light. (b) The behavior of a given switch design is a function of the length of the linkers used. Linkers that are too short to permit the formation of the optogenetic interface will fail to cage the protein of interest in either state, as LOV and zDK will never interact; if the length of the linkers is excessive, the formation of the LOV-zDK interface will not prevent the binding of the binding partner to the protein of interest. In the optimal case, the formation of the LOV-zDK complex is mutually exclusive with the accessibility of the binding site. (c) The fraction of models from a large number of docking trajectories that successfully form the optogenetic zDK-LOV complex can then be compared in the presence and absence of the binder. If the linkers are too short, no complex will be seen in either the presence or absence of the binder. If they are too long, a high proportion of complexes will form in both cases. (d) The fraction forming in the presence of the binder can then be subtracted from that forming in the absence of the binder; the linker set with the biggest difference between the two states is predicted to be optimal.
Our second simulation strategy applies in cases where a structure is not available of the POI bound to its partner, but there is information about which residues on the surface of the POI are important for binding to its partner. In this case, structure prediction is just performed with the POI fused to zDK and LOV2 and the resulting models are interrogated to determine if the zDK/LOV2 complex forms adjacent to the residues on the POI that are important for binding to its partner.
To search for effective Z-lock linkers, structure prediction simulations are performed with a panel of linker lengths going from short to long. As we are not trying to enforce specific conformations upon the linkers, we have computationally and experimentally tested linkers consisting only of serines and glycines. Because of the varied shapes of the POI, zDK and LOV, it can also be helpful to test asymmetric linkers lengths, i.e. where the zDK-POI and the POI-LOV2 linkers are not the same length as each other. Many structure prediction trajectories are performed with each set of linker lengths and the linker lengths that promote formation of the zDK/LOV2 complex over the desired surface of the POI are good candidates for experimental studies.
To test our computational protocol, we performed simulations with three systems that have been caged with Z-lock: the binding of cofilin to actin, the binding of α-tubulin acetyltransferase (αTAT) to tubulin, and the binding of a CBD domain to the GTPase Cdc42. We recently reported the experimental characterization of the cofilin and αTAT switches [21], while the caged Cdc42 binder is a new switch that we experimentally characterize here.
Caging cofilin.
The first optogenetic tool that we examined with our modeling protocol used Z-lock to prevent non-muscle cofilin from binding to actin, which causes depolymerization and severing of actin filaments as part of cytoskeletal remodeling[22,23]. In vivo, this serves to convert old actin filaments to monomeric actin[24] for re-integration into new filaments, maintaining G-actin levels to enable cell motility[24,25]. The actin/cofilin complex has an interface area of 1162 A2 as determined by CoCoMaps[26], with the primary structural component on the cofilin side of the interface being a single alpha helix running from residues 263 to 278 and the beta hairpins flanking it. A line drawn between the terminal residues of cofilin would pass through one end of the helix forming the majority of the interface, suggesting that the actin interface may be caged by constraining the optogenetic components to the region directly between the termini.
In the development of Z-lock cofilin three sets of linkers with varying lengths were experimentally tested: zDK2-GSGGG-Cofilin-GSG-LOV2 (linker set 1X), zDK2-GSGGG-Cofilin-GGSGG-LOV2 (2X) and zDK2-GSGGGCofilin-GGSGGSGG-LOV2 (3X). Binding to actin in the lit state and dark state was initially characterized using mutations to the LOV2 domain that mimic the photo-activated state by inducing the Jα helix to unfold (A532E I536E)[27] and mimic the dark state by disrupting signal transduction from the flavin to the Jα helix (C450A) and stabilizing the folded state of the Jα helix (L514K, G528A, L531E, N539E) [28][12]. Dark state mimics were also kept primarily in the dark as it has been shown that signals can still transduced in some cases with the C450A mutation[29]. Binding to actin was measured using co-sedimentation experiments as part of the cofilin sensor design process [21]. With the shortest linker (1X), 51% of the Z-lock molecules co-sedimented with actin in the dark state, while 31% and 15% co-sedimented with 2X and 3X respectively. These results indicated that the shorter linkers were not providing strong caging in the dark, possibly because the linkers were not long enough to allow the zDK/LOV2 interaction to readily form. The 3X form of the switch was carried forward for additional experimental studies and was shown to provide light-responsive control of cofilin activity in living cells.
To determine the capacity for our modeling protocol to predict the effect of different length linkers on the caging of Z-lock cofilin, we ran 10,000 independent simulations of the zDK2-cofilin-LOV2 construct in the presence and absence of actin and tabulated what fraction of the simulations produced models with zDK2 bound to LOV2. We refer to this as the closed state. To be considered closed, a model needed to satisfy an energy filter to exclude non-physical models and a constraint filter to verify that zDK2 and LOV2 adopted relative positions consistent with the zDK2-LOV2 co-crystal structure 5DJT; models that passed the first check but failed the second were considered open. The fraction of low-scoring conformations that were closed was compared for the simulations in the absence of actin and for the simulations in the presence of actin. Ideal linkers should allow the zDK2/LOV2 interaction to form (i.e. close) in the absence of actin, but not allow the interaction to form in the presence of actin. An absence of closed models in the presence of actin indicates that the formation of the zDK2/LOV2 interaction and the binding of actin are in competition with each other, a necessary condition for regulating actin binding. To further explore the effects of longer and shorter linker lengths than were experimentally tested, we also computationally tested a single glycine as the cofilin-LOV2 linker (G), as well as GGSx4(4X) and GGSx5(5X), consisting of the 3X linker set with one and two additional SGG repeats on the cofilin-LOV2 linker respectively.
In the simulations, the 3X linker set showed the largest decrease in interface closure caused by the presence of actin [Figure 2]. The 2X linker also demonstrated some caging capacity, while very little caging was predicted for 1X and 0X. The simulations indicate that the 1X and G the linkers are too short to allow the zDK2/LOV2 interaction to form, even in the absence of actin. These predictions are consistent with the co-sedimentation experiments that showed that the 1X linker set binds strongly to actin even in the dark state, and that 3X is more strongly caged in the dark than 2X [21]. The molecular modeling also predicts that 3X will more effectively block actin binding than switches with longer linkers, 4X and 5X. In the case of 4X and 5X, the linkers are long enough that it becomes possible to simultaneously form a complex with actin and form the zDK2/LOV2 complex.
Figure 2.

Figure 2. Modeling linkers for Z-lock cofilin. Various linkers were modeled in the presence and absence of actin to establish which linkers should most effectively regulate binding to cofilin. (a) The optimal-length linker set “3X” positioned the zDK(blue)/LOV(green) interface over cofilin (grey) such that zDK sterically clashed with actin (orange.) (b) The shorter linker set “G” did not permit the formation of a zDK/LOV2 interface even in the monomer state (the pictured model is with the constraint term downweighted to 0 to emulate the predicted behavior of this construct in cells). (c) The longer linker sets permitted the simultaneous formation of an actin-cofilin interface and a zDK/LOV2 interface. (d) The fraction of modeling trajectories that resulted in a well-formed interface between zDK and LOV2 was calculated for simulations with and without actin and the difference of these two fractions was plotted to establish the linker length most likely to inhibit cofilin binding in the dark. The linker length predicted to be most effective (3X) is in good agreement with experimental results that indicated that the 3X showed less binding to actin in the dark than the 2X and 1X linker sets. As expected, the dark-state efficacy of the linkers decreased sharply with decreasing length, a trend that continued with the short linker (blue, “G”), and the longer linkers (4X and 5X) exhibited a more gradual decline in predicted efficacy.
Caging αTAT.
Alpha-tubulin acetyltransferase (αTAT) preferentially acetylates microtubules over free tubilin and promotes microtubile degradation[30]. To create Z-lock αTAT, zDK1 was fused to the N-terminus of the catalytic domain of αTAT and the LOV2 domain was fused to the C-terminus. A fixed length linker with 5 gly-ser repeats was placed between zDK1 and αTAT and three different linkers were experimentally tested for connecting the C-terminus of αTAT to LOV: a 4 residue linker with 2 gly-ser (GS2) repeats, a 6 residue linker with 3 gly-ser (GS3) repeats, and an 8 residue linker with 4 gly-ser (GS4) repeats. The Z-lock variants were tested by probing levels of microtubule acetylation in 293TLinXE cells in the dark versus light. All three switches showed some light-dependent activity, with the GS3 linker showing the largest fold change in acetylation between the dark and lit state. The GS2 linker demonstrated higher background activity in the dark than the GS3 linker. To determine if our modeling protocol could predict this behavior we ran simulations with the GS2, GS3 and GS4 linkers as well as a single GS and a 5GS linker. The GS and GS5 linkers were simulated to more completely map out what is occuring when the linkers are shortened or lengthened.
Unlike with cofilin, there is no crystal structure of αTAT bound to its substrate. However, there is a structure of the αTAT catalytic domain[31] and mutational studies have identified a set of surface residues located in a large groove that are critical for interacting with microtubules. To computationally test which Z-lock constructs would be most effective at regulating acetylation we ran 10000 independent structure prediction trajectories for each linker variant and examined what fraction of the trajectories resulted in models consistent with blocking binding to tubulin. To be consistent with blocking tubulin binding the model needed to have a favorable energy (i.e. few steric clashes), have zDK1 and LOV2 appropriately docked against each other, and be adjacent to the tubulin binding site on αTAT. We observed clear differences between the linkers in the simulations. The GS and GS2 linkers rarely blocked binding because the linkers were not long enough to allow the zDK1/LOV2 interface to form. This result is consistent with the increased background activity observed experimentally for GS2 in the dark. The GS3 and GS4 linkers most consistently caged the interface, and the GS5 linker resulted in weaker caging because the zDK1/LOV2 complex more often formed in locations that were not adjacent to the tubulin binding site on αTAT. Overall, the results were in agreement with the trends observed experimentally.
When we originally engineered and tested Z-lock αTAT, we hypothesized that Z-lock may also regulate the binding of the cofactor, acetyl CoA, to the enzyme. In the Rosetta simulations we did not observe zDK1/LOV2 associating over the acetyl CoA binding site, and we did not observe any strain on the protein that would allosterically regulate acetyl CoA binding. These results suggest that perturbation of cofactor binding is not important to the mechanism of the switch.
Caging CBD.
To test our linker design protocol on a case without prior experimental data, an inhibitor was constructed around the CDC42-binding CBD domain of WASP, which plays a role in establishing cell polarity in complex with CDC42 and Par3[32] as well as chromosomal protein composition[33] and more general regulation of cell polarity[34,35] and has previously been placed under optogenetic control [36]. To optogenetically control the affinity of the CBD domain to CDC42 with LOV2, zDK2 was added to its N terminus and LOV2 to its C terminus, with glycine-serine dimers placed between the zDK2 and CBD domains. We computationally predicted the effects of adding zero to five glycine-serine dimers. The CBD-based switch is different than the cofilin and αTAT switches in that the CBD domain is partially disordered when it is not bound to CDC42. Cofilin and αTAT are well folded in the presence and absence of their binding partners. The intrinsic flexibility of the CBD domain provides an alternative mechanism for controlling its activity with Z-lock. When bound to CDC42, the N-terminal region of the CBD domain forms a long beta strand that partners with a beta strand on the surface of CDC42. When the CBD domain is not bound to CDC42, this beta strand is intrinsically disordered. We hypothesized that Z-lock could block binding to CDC42 by not only forming a steric block, but also by constraining the flexible N-terminal region of the CBD domain in a distorted, non-binding conformation. To allow for this mechanism of control in our simulations, we allowed the N-terminal beta strand of the CBD domain to adopt alternative conformations in our simulations of the switch not bound to CDC42.
As with cofilin, two types of simulations were performed: simulations with Z-lock Wasp CBD bound to its binding partner, CDC42, and simulations of the unbound state. In the simulations all of the switch variants could form the zDK2/LOV2 complex in the absence of CDC42 and the construct with no additional linker residues (GS0) was least susceptible to forming it in the complex state. The GS0 construct was expressed in LinXe cells as either a lit state or dark state variant (as described for the cofilin switches) and pulldown experiments were used to measure binding to CDC42 [Figure 4]. When zDK2/LOV2 was expressed at lower levels, over a 5-fold reduction in binding to CDC42 was observed with the dark state variant compared to the lit state variant.
Figure 4.

Computational design and experimental testing of a light-sensitive binder for Cdc42. zDK2(blue) was fused to the N-terminus of the Cdc42 binding motif from Wasp (Wasp CBD, grey) and the LOV2 domain was fused to the C-terminus of Wasp CBD. Simulations were performed in the presence and absence of Cdc42 (orange). (A) In the absence of Cdc42, the n-terminal beta strand of the CBD domain (grey) was allowed to adopt alternative conformations which allowed the the zDK2/LOV2 interaction to form with short linkers. The model shown (GS0) is a direct fusion with no linker residues. (B) With the GS0 linker set, zDK2 is not able to reach the LOV2 domain when the CDB is bound to Cdc42, indicating that Cdc42 binding and zDK2/LOV2 formation will be in competition and that this should be an effective switch. (C) Simulations with longer linkers (GS1-GS4) indicates that caging will be diminished as the linkers are lengthened. (D/E) Experimental analysis of the affinity of dark state and lit state variants of zDK2-GS0-CBD-LOV2 for Cdc42 as determined by pulldown experiments. In panel D the relative amounts of the switch that bound to Cdc42 are quantified for different expression levels. The top portion of panel E shows western blots used to detect the amount of protein in the pulldown and the bottom portion of panel E shows western blots used to detect the total amount of protein in the cell lysate. Lower binding is evident with the dark state variant (top left of panel E).
Discussion
A challenge in optimizing the linkers for the Z-lock system is the ambiguous nature of negative experimental results. Linkers that are too short do not permit zDK and the LOV2 domain to interact and so the switch is permanently “on”. Linkers that are too long allow the zDK/LOV2 interaction to form in locations that do not block binding to the protein of interest and therefore the switch is also permanently “on”. Here, we have developed and benchmarked a molecular modeling protocol that can distinguish between the two scenarios and indicates which linker lengths should provide maximal caging with Z-lock. Although it was not the case for the three systems studied here, the modeling should also provide an indication of whether a protein is amenable to caging with Z-lock. If no combination of linker lengths favor the zDK/LOV2 complex forming over the protein binding surface of interest, than that protein is not a good candidate for caging with Z-lock.
Materials and Methods
To validate a given set of linkers for their suitability in caging a given interface, it is necessary to construct a model of those domains in both the presence and absence of the binding partner of interest, then perturb the linker backbone torsions to create an ensemble of models representing possible relative orientations of the optical components to each other and the protein of interest; the entire ensemble can then be analyzed to estimate the relative ease of formation of the optogenetic interface and fraction of formed interfaces that also cage the desired interface. The first step, that of forming the model itself, can be done manually by aligning the components to each other using standard molecular modeling software, but was done automatically via domain assembly software created for the Rosetta macromolecular modeling suite that automatically generates linker atomic coordinates for a provided sequence, links them to the provided domains via idealized peptide bonds, and outputs the relevant kinematic inheritance tree for downstream linker perturbation. An example of this component file is provided as supplementary figure S4.
The RosettaScripts executable was then used to perturb the backbone torsional dihedrals of the linker residues in centroid mode according to the RosettaScript listed as supplementary figure S1. As the force field terms required to favor the formation of an interface only function at ranges that are small in comparison to the entire sampleable space, the score function was biased towards favorable interfaces by including constraints to favor distances between the alpha carbons of the LOV-zDK interface residues seen in the relevant LOV-zDK crystal structure. A sample constraint file of this type is included in supplementary figure S3, and an example fold tree is provided as supplementary figure S2. Flags files and command lines for running both scripts are supplementary figures S6–S9. Decoy generation itself consisted of a combination of fragment insertion into the linkers and random backbone torsion perturbation under the control of a simulated annealing Monte Carlo function. This step is followed by gradient-based minimization under the same constraints to optimize the linker torsions. As these constraints also artificially force the optogenetic components closer than they would be randomly in the absence of an interface, it is necessary to differentiate formed interfaces from unformed interfaces through a low numerical cutoff on the score term representing the total deviation of the interface alpha carbons from their ideal positions. Typically, 2000 such models are independently generated to assay a single set of linkers, 1000 in the monomer state and 1000 in the complex state; if the complex state is unknown, 2000 trajectories of the monomer state are used instead. In many cases, the set of 2000 trajectories was then repeated 10 times to establish error bars. The fraction of models that successfully formed interfaces (as determined by the constraint term being below a numerical cutoff, usually 5) was then compared in the case of the sensor alone relative to the sensor with the binding partner of interest positioned over the interface. If the selected linkers are too short, a large fraction of models will fail to form interfaces in both the monomer and complex cases as the protein of interest separates the optogenetic components too widely; if they are too long, the interface will form away from the interface of interest, creating a “leaky” sensor in which the closed state is not mutually exclusive with binding. This state is detectable as a high proportion of models with formed interface in both the monomer and complex states. The linker residues of optimal length will display a high proportion of formed interfaces in the monomer state but not in the complex state.
For the cofilin sensor, the complex state used was that of the cofilin-actin complex in the co-crystal structure (pdb id: 5YU8). For the CBD sensor, structures of CDC42 (2ODB) and WASP CBD (1CEE) were aligned according to the complex in the structure 1NF3. For the ATAT sensor, the structure of ATAT alone (4GS4) was used. The LOV2 and zdk structures were extracted from pdb deposition 5EFW. The LOV2 domain is the second light sensitive Light Oxygen Voltage domain from phototropin 1 of Avena sativa.
Plasmid construction
LOV2 dark mutant (C450A, L514K, G528A, L531E, and N538E) and lit mutant (I510E and I539E) were used for Z-lock-WASP dark and lit, respectively. These cDNA fused with zDK1-WASP CBD (Cdc42 binding domain) was cloned into pTriEx-mVenus vector. pEF-BOS-GST and pGEX-Cdc42 Q61L were provided by Kozo Kaibuchi (Nagoya University, Japan). Human Cdc42 Q61L cDNA was excised from pGEX plasmid and cloned into pEF-BOS-GST to express GST tagged Cdc42 Q61L in mammalian cells.
Pulldown assay
LinXe cells were cultured in DMEM supplemented with 10% fetal bovine serum (Gemini Bio-Product, CA, USA) and GlutaMax (Thermofisher Scientific, MA, USA). For pull down assay, the cells were seeded in 6-well plate one day before the transfection. The plasmids were transfected by Fugene 6 transfection reagent (Promega, WI, USA) according to the manufacture’s instruction. The following amount of the plasmid was used for transfection. pTriEx-mVenus-Z-lock-WASP lit, 400 to 1600 ng; pTriEx-mVenus-Z-lock-WASP dark, 50 to 200 ng; pTriEx-mVenus-CBD and mutant CBD, 150 ng; pEF-BOS-GST and pEF-BOS-GST-Cdc42 Q61L, 500 ng. pBabePuro was used to equalize the total amount of the plasmid. 2.1 mg plasmids in total was mixed with 10.5 ml of Fugene 6 in Opti-MEM (Thermofisher Scientific). After 24 hours transfection, the cells were lysed with the lysis buffer (25 mM Tris-HCl [pH 7.5], 1% NP-40, 10 mM MgCl2, 0.1% sodium dodecyl sulfate, 100 mM NaCl) supplemented with the protease inhibitor cocktail (Sigma, MA) by rocking the plate for 30 minutes at 4°C. The lysates were centrifuged by 12,000g for 5 minutes at 4°C to remove debris. The supernatant was incubated with glutathione agarose beads (Thermofisher Scientific) at 4°C for 1 hour. After washing the beads three times with the lysis buffer, the bound proteins were dissolved in SDS-PAGE sample buffer. Immunoblot was used to detect the bound proteins. The following antibodies were used; anti-GFP antibody (JF-8; Takara, Japan), anti-GST antibody (91G1; Cell Signaling Technology, MA, USA), DyLight 800-conjugated anti-rabbit IgG antibody (Cell Signaling Technology), and Dylight 680-conjugated anti-mouse IgG antibody (Cell Signaling Technology). The fluorescence was visualized by ChemiDoc (BioRad, CA).
Supplementary Material
Figure 3.

Modeling linkers for z-Lock αTAT. In lieu of a structure of the αTAT/tubulin complex, interface caging was determined by counting contacts between residues of zDK/LOV and surface residues of αTAT that are known to be critical for acetyltransferase activity (red spheres). (A) Models were considered to be caging if at least 30 α-carbons of either LOV2 or zDK were within 20 Ã of any critical residue, (B) while an interface was considered open if the zDK/LOV2 interaction could not form or (C) if the zDK/LOV2 complex was not adjacent to the tubulin binding site. While the fraction of caged models did not differ between the GS3 and GS4 linker sets, with dynamic ranges of 2 and 1.87 respectively, the GS2 linker set, with a dynamic range of 1.33, did show significantly fewer caging interfaces. GS3 was shown to be the most effective linker set experimentally [21].
Highlights.
Optogenetics provides spatiotemporal control of signaling in living systems.
The photosensitive Z-lock system regulates activity by occluding binding sites.
Rosetta can be used to design linkers that maximize caging with Z-lock.
The Z-lock design protocol can be used to photocage a wide variety of proteins.
Acknowledgements
We thank Neha Pankow and Bei Liu for helpful discussions regarding the Z-lock system. This work was supported by the NIH grants R35GM131923(BK) and R35GM122596 (KH).
Abbreviations
- LOV2
Light Oxygen Voltage 2
- PA-Rac
Photoactivatable Rac1
- POI
Protein of Interest
- αTAT
α-tubulin acetyltransferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kodani S, Soya S, Sakurai T. Optogenetic Manipulation of Neural Circuits During Monitoring Sleep/wakefulness States in Mice. J Vis Exp. 2019. doi: 10.3791/58613 [DOI] [PubMed] [Google Scholar]
- 2.Camporeze B, Manica BA, Bonafé GA, Ferreira JJC, Diniz AL, de Oliveira CTP, et al. Optogenetics: the new molecular approach to control functions of neural cells in epilepsy, depression and tumors of the central nervous system. Am J Cancer Res. 2018;8: 1900–1918. [PMC free article] [PubMed] [Google Scholar]
- 3.Govorunova EG, Sineshchekov ОА, Spudich JL. Three Families of Channelrhodopsins and Their Use in Optogenetics (review). Neurosci Behav Physiol. 2019;49: 163–168. [Google Scholar]
- 4.O’Banion CP, Lawrence DS. Optogenetics: A Primer for Chemists. Chembiochem. 2018;19: 1201–1216. [DOI] [PubMed] [Google Scholar]
- 5.Repina NA, Rosenbloom A, Mukherjee A, Schaffer DV, Kane RS. At Light Speed: Advances in Optogenetic Systems for Regulating Cell Signaling and Behavior. Annu Rev Chem Biomol Eng. 2017;8: 13–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang WJ, Chang WP, Shyu BC. Suppression of cortical seizures by optic stimulation of the reticular thalamus in PV-mhChR2-YFP BAC transgenic mice. Mol Brain. 2017;10: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Assaf F, Schiller Y. The antiepileptic and ictogenic effects of optogenetic neurostimulation of PV-expressing interneurons. J Neurophysiol. 2016;116: 1694–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegemann P, Nagel G. From channelrhodopsins to optogenetics. EMBO Molecular Medicine. 2013. pp. 173–176. doi: 10.1002/emmm.201202387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fenno LE, Deisseroth K. Optogenetic Tools for Control of Neural Activity. Neuromethods. 2014. pp. 73–86. doi: 10.1007/978-1-62703-785-3_5 [DOI] [Google Scholar]
- 10.Wehler P, Niopek D, Eils R, Di Ventura B. Optogenetic Control of Nuclear Protein Import in Living Cells Using Light-Inducible Nuclear Localization Signals (LINuS). Current Protocols in Chemical Biology. 2016. pp. 131–145. doi: 10.1002/cpch.4 [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Hahn KM. LOVTRAP: A Versatile Method to Control Protein Function with Light. Curr Protoc Cell Biol. 2016;73: 21.10.1–21.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Vilela M, Winkler A, Tarnawski M, Schlichting I, Yumerefendi H, et al. LOVTRAP: an optogenetic system for photoinduced protein dissociation. Nat Methods. 2016;13: 755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmerman SP, Hallett RA, Bourke AM, Bear JE, Kennedy MJ, Kuhlman B. Tuning the Binding Affinities and Reversion Kinetics of a Light Inducible Dimer Allows Control of Transmembrane Protein Localization. Biochemistry. 2016;55: 5264–5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jäschke A Genetically encoded RNA photoswitches as tools for the control of gene expression. FEBS Letters. 2012. pp. 2106–2111. doi: 10.1016/j.febslet.2012.05.040 [DOI] [PubMed] [Google Scholar]
- 15.Liu Q, Tucker CL. Engineering genetically-encoded tools for optogenetic control of protein activity. Curr Opin Chem Biol. 2017;40: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461: 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terazima M Studies of photo-induced protein reactions by spectrally silent reaction dynamics detection methods: applications to the photoreaction of the LOV2 domain of phototropin from Arabidopsis thaliana. Biochim Biophys Acta. 2011;1814: 1093–1105. [DOI] [PubMed] [Google Scholar]
- 18.Pudasaini A, El-Arab KK, Zoltowski BD. LOV-based optogenetic devices: light-driven modules to impart photoregulated control of cellular signaling. Front Mol Biosci. 2015;2: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yazawa M, Sadaghiani AM, Hsueh B, Dolmetsch RE. Induction of protein-protein interactions in live cells using light. Nat Biotechnol. 2009;27: 941–945. [DOI] [PubMed] [Google Scholar]
- 20.Möglich A, Ayers RA, Moffat K. Design and signaling mechanism of light-regulated histidine kinases. J Mol Biol. 2009;385: 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stone OJ, Pankow N, Liu B, Sharma VP, Eddy RJ, Wang H, et al. Optogenetic control of cofilin and αTAT in living cells using Z-lock. Nat Chem Biol. 2019;15: 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlier MF, Ressad F, Pantaloni D. Control of actin dynamics in cell motility. Role of ADF/cofilin. J Biol Chem. 1999;274: 33827–33830. [DOI] [PubMed] [Google Scholar]
- 23.Condeelis J How is actin polymerization nucleated in vivo? Trends Cell Biol. 2001;11: 288–293. [DOI] [PubMed] [Google Scholar]
- 24.Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401: 613–616. [DOI] [PubMed] [Google Scholar]
- 25.Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145: 1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vangone A, Spinelli R, Scarano V, Cavallo L, Oliva R. COCOMAPS: a web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics. 2011;27: 2915–2916. [DOI] [PubMed] [Google Scholar]
- 27.Harper SM, Christie JM, Gardner KH. Disruption of the LOV-Jalpha helix interaction activates phototropin kinase activity. Biochemistry. 2004;43: 16184–16192. [DOI] [PubMed] [Google Scholar]
- 28.Richter G, Weber S, Römisch W, Bacher A, Fischer M, Eisenreich W. Photochemically Induced Dynamic Nuclear Polarization in a C450A Mutant of the LOV2 Domain of theAvenasativaBlue-Light Receptor Phototropin. Journal of the American Chemical Society. 2005. pp. 17245–17252. doi: 10.1021/ja053785n [DOI] [PubMed] [Google Scholar]
- 29.Yee EF, Diensthuber RP, Vaidya AT, Borbat PP, Engelhard C, Freed JH, et al. Signal transduction in light–oxygen–voltage receptors lacking the adduct-forming cysteine residue. Nature Communications. 2015. doi: 10.1038/ncomms10079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalebic N, Martinez C, Perlas E, Hublitz P, Bilbao-Cortes D, Fiedorczuk K, et al. Tubulin acetyltransferase αTAT1 destabilizes microtubules independently of its acetylation activity. Mol Cell Biol. 2013;33: 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedmann DR, Fan J, Marmorstein R. Structure of the alpha-tubulin acetyltransferase, alpha-TAT1. 2012. doi: 10.2210/pdb4gs4/pdb [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marques E, Klefström J. Par6 family proteins in cancer. Oncoscience. 2015;2: 894–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dormoy V, Tormanen K, Sutterlin C. Par6 is at the mother centriole and controls centrosomal protein composition through a Par6 -dependent pathway. Journal of Cell Science. 2013. pp. 860–870. doi: 10.1242/jcs.121186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozdamar B Regulation of the Polarity Protein Par6 by TGF Receptors Controls Epithelial Cell Plasticity. Science. 2005. pp. 1603–1609. doi: 10.1126/science.1105718 [DOI] [PubMed] [Google Scholar]
- 35.Thiery JP, Huang R. Linking Epithelial-Mesenchymal Transition to the Well-Known Polarity Protein Par6. Developmental Cell. 2005. pp. 456–458. doi: 10.1016/j.devcel.2005.03.002 [DOI] [PubMed] [Google Scholar]
- 36.Leung DW, Otomo C, Chory J, Rosen MK. Genetically encoded photoswitching of actin assembly through the Cdc42-WASP-Arp2/3 complex pathway. Proc Natl Acad Sci U S A. 2008;105: 12797–12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.