

Abstract
Objectives:
Despite safe handling guidelines published by several groups, health care worker exposure to hazardous drugs continues to occur due to suboptimal engineering controls and low use of protective equipment. Simple, multi-target and specific analytical methods are needed so that acute exposures to these drugs in the workplace can be assessed rapidly. Our aim was to develop an analytical method for simultaneous detection and quantification of widely used cancer drugs to rule out accidental acute chemotherapy exposures in health care workers.
Methods:
We examined the feasibility of alternate high-performance liquid chromatographic-tandem mass spectrometry methods to simultaneously detect eighteen chemotherapy analytes in plasma and urine. The linear concentration ranges tested during assay development were 0.1–50 ng/mL. After development of a multi-analyte assay protocol, plasma samples (n = 743) from a multi-center cluster-randomized clinical trial (n = 12 sites) of an hazardous drug educational intervention were assayed. Confirmatory assays were performed based on the individual acute-spill case-histories.
Results:
An innovative HPLC-multiple reaction monitoring-information dependent acquisition-enhanced production ion (MRM-IDA-EPI) analytical method was developed to simultaneously detect: cytarabine, gemcitabine, dacarbazine, methotrexate, topotecan, mitomycin, pemetrexed, irinotecan, doxorubicin, vincristine, vinblastine, ifosamide, cyclophosphamide, vinorelbine, bendamustine, etoposide, docetaxel, and paclitaxel. The retention times ranged from 4 min to 13 min for the analytical run. The limit of detection (MRM-IDA-EPI) and limit of quantitation (MRM) was 0.25 ng/mL and 0.1 ng/mL, respectively for most analytes. No detectable plasma concentrations were measured at baseline, post-intervention and in cases of documented acute spills. Use of a secondary tandem mass spectrometry approach was able to successfully rule out false positive results.
Conclusions:
Development of a sensitive high-throughput multi-analyte cancer chemotherapy assay is feasible using an MRM-IDA-EPI method. This method can be used to rapidly rule out systemic exposure to accidental acute chemotherapy spills in health care workers.
Keywords: Antineoplastic drugs, HPLC-MS/MS, occupational exposure, biological monitoring
Introduction
Cancer chemotherapy agents are hazardous drugs (HDs) that pose significant occupational health risks to clinical personnel from the point of procurement to administration. In 2004, the National Institute for Occupational Safety and Health (NIOSH) issued an alert that summarized these health risks, such as skin rashes and adverse reproductive outcomes (including infertility, spontaneous abortions, and congenital malformations), and risk for leukemia and other cancers posed by HD exposures.1 An estimated eight million health care workers are potentially exposed to HDs in the United States each year.2 This risk potential has culminated in numerous recommendations from societies and regulatory bodies, which include the centralization of the preparation of cytotoxic drugs in dedicated areas with safety hoods and using personal protective equipment (PPE).3–7
Recently, the United States Pharmacopeia (USP) issued an enforceable standard (USP<800>) for handling HDs in health care settings that will impact the storage, transportation, preparation and administration of these agents. This general chapter will be combined with USP<795> and USP<797> general chapters to form a more comprehensive compounding standard by 1 December 2019.7 Currently, USP standards are recognized in several provisions of the federal Food, Drug, and Cosmetic Act (FDCA) and regulatory policies of several state boards of pharmacy. California is the first state to require full compliance with USP<800>. As a consequence, enforcement of these standards by the US Food and Drug Administration, Center for Medicare & Medicaid Services and The Joint Commission are expected. This single-source standard serves to protect pharmacists, technicians, nurses, physicians, physician assistants, home health care workers, veterinarians, veterinary technicians and any other health care workers who access facilities where HDs are prepared and administered. This standard does not currently recommend biological monitoring with the exception that it may be helpful as a follow-up to an acute HD spill.
Over 18 million cancer chemotherapy doses are administered annually in the US alone, which places oncology nurses at an exceptional risk for acute HD spill exposure.8 Statewide surveys have revealed that one out of six ambulatory care nurses reported skin or eye exposure to chemotherapy in the past year.9 Several studies have developed analytical methods demonstrating health care worker biological exposure to cancer chemotherapy during preparation and administration of the agent.10–29 We previously documented measurable pemetrexed, docetaxel, and cisplatin plasma concentrations in a sample of nurses and pharmacists who consented to participating in a six-month prospective study of acute HD spills. We subsequently performed a multicenter cluster randomized controlled trial to compare an educational module on HD handling to the same educational module plus survey feedback and biological data from participants.30 Biological data included plasma sampling at baseline and post-intervention assessment, as well as with the occurrence of any reported drug spill during the study to assay for presence of the HD. Given the breadth of potential HD spills, we focused on assay development of the 20 most widely used cancer chemotherapy agents in ambulatory oncology settings. Herein, we report on the development of an innovative multi-analyte assay and the results of biomonitoring as a follow-up to acute spills of cancer chemotherapy drugs. The presented analytical approach supports the provision of feedback to health care workers who experience an accidental spill of widely used chemotherapy agents.
Methods
Materials and reagents
Dacarbazine (DACA) was purchased from Tokyo Chemical Industry Co. (Portland, OR, USA). Paclitaxel (TAX) and docetaxel (DOCE) were purchased from Fisher Scientific (Pittsburgh, PA, USA). Ifosfamide (IF), cyclophosphamide (CP), bendamustine (BEN), irinotecan (IRI), topotecan (TOP), etoposide (ETOP), vincristine (VCR), vinblastine (VBL), vinorelbine (VIN), methotrexate (MTX), pemetrexed (PTR), gemcitabine (GCA), fludarabine (FLD), doxorubicin(DOXO) and mitomycin (MIT) were purchased from Sigma Chemical Co. (St Louis, MO, USA). Standard purity was ≥98% for all analytes. Irinotecan-d10 (IS1), doce-taxel-d9 (IS2), bendamustine-d6 (IS3), topotecan-d6 (IS4) were used as internal standards (IS). All ISs were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Acetonitrile, methanol and formic acid were HPLC-MS-grade and were obtained from Fisher Scientific (Pittsburgh, PA, USA). Deionized water was obtained from a Milli-Q Plus system (Millipore, Bedford, MA, USA). Control human plasma samples from pooled donors were purchased from Valley Biomedical (Winchester, VA, USA).
Preparation of analyte standard solutions
Individual stock solutions of the standards and internal standards, all at 1.0 mg/mL, were prepared by dissolving appropriate amounts of the analyte in dimethyl sulfoxide (DMSO). This solvent was selected due to nonpolar analytes such as paclitaxel and in order to maintain potency of the stock solutions. These solutions were found to be stable for at least six months when stored in the dark and refrigerated (2–8°C) conditions. Composite working standard solutions of all analytes were prepared by combining the above solutions and diluting with methanol:acetonitrile (1:1, v:v) to a final concentration of 20 μg/mL. The internal standard working solutions were prepared with methanol: acetonitrile (1:1) and 0.1% formic acid. For the method limits evaluation (limits of detection, selectivity, method validation), composite calibration working solutions were prepared by diluting the above solutions with blank plasma to obtain analyte concentrations suitable for the experiments. When unused, stock solutions were stored at −80°C, while working solutions of standards and internal standards at −20°C. The working standard mixtures were freshly prepared before use.
Sample extraction procedure
An aliquot of 80 μL plasma samples were mixed with 120 μL of internal standard solution, then shaken for 10 min and centrifuged at 4000 r/min for 10 min at 4°C. The supernatant was transferred to a 96-well plate and 30 μL was injected for LC-MS analysis.
Liquid chromatography and mass spectrometry
High-performance liquid HPLC–MS/MS analyses were performed in a system consisting of a Shimadzu Nexera XR ultra high-performance liquid chromatograph system (Shimadzu Scientific Instruments, Columbia, MD, USA), connected in series to a 5500 QTRAP hybrid triple quadrupole linear ion trap mass spectrometer equipped with a TurboIonSpray source (Sciex, Redwood City, CA, USA), operated in the positive electrospray ionization mode (ESI). The analytical instrumentation was controlled by Analyst 1.6.2 (Sciex).
HPLC separation of the analytes was performed with a 150 mm × 4.6 mm Xbridge C18 column (3.5 μm particle size) (Waters Corporation, 34 Maple Street Milford, MA, USA) and a mobile phase consisting of ACN and water, both acidified with 0.1% formic acid, at a flow rate of 0.7 mL/min. The initial conditions (2% ACN) were maintained for 2 min, then the following gradient elution scheme was used: 2–5% A in 1 min, 5–20% A in 1 min, 20–70% A in 7 min, and then to 95% in the following 0.1 min, which was held for a further 2.9 min; finally, the column was equilibrated for 3 min to the initial conditions. This gradient provided a chromatographic analysis time of 17 min.
MS/MS analyses were performed in the scheduled MRM and MRM-IDA-EPI mode. A total of 22 transitions in positive mode were monitored with an MRM pause time of 3 ms for the 18 analytes and 4 ISs. This pause time refers to the brief period of time for the mass spectrometer to reset between transients. The Scheduled MRM™ algorithm was used with an MRM detection window of 60 s and a target scan time of 0.7 s in Analyst®1.6 Software. For increased confidence in compound identification, information dependent acquisition (IDA) criteria were employed in order to automatically trigger the acquisition of EPI scans for any compounds that were detected by the MRM scans. EPI spectra at a scan speed of 10,000 Da/s were acquired using a dynamic fill time for optimal MS/MS quality, and generated using standardized collision energy (CE) of 40 V with collision energy spread (CES) of 15 V to ensure a characteristic MS/MS pattern independently of the compound’s fragmentation efficiency. All source and instrument parameters for the monitored analytes were tuned by infusing each single standard solution at a concentration of 0.5 μg/mL by a syringe pump (flow rate 10 μL/min). All the source parameters were checked (and revised as necessary) in flow injection analysis with the same chromatographic conditions (flow and solvent composition). Nitrogen was used as curtain, nebulizer, drying and collision gas (15, 50, 50 and “medium,” respectively, manufacturer’s units); drying gas temperature was set at 450°C. The most important MS parameters for MRM and EPI acquisition of the 18 target compounds are summarized in Table 2. The limits of detection (LOD) and quantitation (LOQ) were defined as the concentrations yielding signal intensity 3 and 10 times the background value, respectively. The LOQ is often either equivalent to LOD or a much higher concentration. However, in this particular study the assessment of LOQ and LOD were based on two different methods. The LOD was based on MRM-IDA-EPI scan, while the LLOQ was based on MRM scan. Because the LOD is based on an IDA process, the sensitivity is lower than that of the conventional MRM scan.
Table 2.
Lower limit of detection (LLOD) with MRM-IDA-EPI scan and lower limit of quantitation (LLOQ) with MRM scan, and calibration profile of 18 chemotherapy agents in plasma.
| Analyte | LLODa (ng/mL) | LLOQa (ng/mL) | Slope | Intercept | R2 | Linear range (ng/mL) |
|---|---|---|---|---|---|---|
| 1. Gemcitabine (GCA) | 1.00 | 0.50 | 0.00927 | 7.8445E-04 | 0.99523 | 0.50–50 |
| 2. Dacarbazine (DACA) | 0.50 | 0.50 | 0.01128 | 0.00263 | 0.99767 | 0.50–50 |
| 3. Fludarabine (FLD) | 2.50 | 0.10 | 0.03272 | 0.00122 | 0.99266 | 0.10–50 |
| 4. Methotrexate (MTX) | 0.25 | 0.10 | 0.28728 | 0.01918 | 0.99535 | 0.10–50 |
| 5. Topotecan (TOP) | 0.10 | 0.10 | 0.03797 | 1.3332E-05 | 0.99619 | 0.10–50 |
| 6. Mitomycin (MIT) | 0.25 | 0.10 | 0.05579 | 7.9954E-05 | 0.99853 | 0.10–50 |
| 7. Pemetrexed (PTR) | 0.25 | 0.10 | 0.03728 | −1.9923E-04 | 0.99745 | 0.10–50 |
| 8. Irinotecan (IRI) | 0.50 | 0.10 | 0.02392 | 3.8156E-04 | 0.99327 | 0.10–50 |
| 9. Vincristine (VCR) | 0.25 | 0.10 | 0.03453 | −9.0884E-05 | 0.99642 | 0.10–50 |
| 10. Doxorubicin (DOXO) | 0.25 | 0.10 | 0.01980 | 0.00249 | 0.99705 | 0.10–50 |
| 11. Vinblastine (VBL) | 0.50 | 0.10 | 0.04683 | −5.6620E-04 | 0.99543 | 0.10–50 |
| 12. Vinorelbine (VIN) | 1.00 | 0.10 | 0.06812 | −0.00168 | 0.99748 | 0.10–50 |
| 13. Bendamustine (BEN) | 0.10 | 0.10 | 0.19672 | 3.7972E-04 | 0.99671 | 0.10–50 |
| 14. Ifosfamide (IF) | 0.10 | 0.10 | 0.12948 | −3.8529E-04 | 0.99693 | 0.10–50 |
| 15. Cyclophosphamide (CP) | 0.25 | 0.10 | 0.08087 | 0.00123 | 0.99613 | 0.10–50 |
| 16. Etoposide (ETOP) | 0.25 | 0.10 | 0.04023 | 1.7948E-04 | 0.99783 | 0.10–50 |
| 17. Docetaxel (DOCE) | 0.5 | 1.00 | 4.15799E-04 | 2.37008E-04 | 0.99673 | 1.00–50 |
| 18. Paclitaxel (TAX) | 0.5 | 1.00 | 0.00209 | 0.00195 | 0.99665 | 1.00–50 |
The LLOD and LLOQ were derived using two different methods (see Table 2 caption). LLOQ is often equivalent or a higher concentration than LLOD; however, LLOD relied on IDA while LLOQ did not and so was less sensitive in this scenario.
Clinical trial plasma sample handling and processing
A four-year cluster randomized controlled trial known as the Drug Exposure Feedback and Education for Nurses’ Safety (DEFENS) study was conducted at 12 cancer centers in the US. Specific information regarding the clinical trial design, conceptual framework, study measures, spill reporting and plasma sampling has been published previously.30 In brief, participants provided blood for plasma sampling at baseline, after the educational assessment and if they experienced a chemotherapy drug spill. Sampling was performed 2 h after the spill with collection of 5 mL of blood in heparinized tubes. For cases with documented spill events, a second blood sample was collected 24 h after the first one. Plasma was harvested after centrifugation of the blood at 1000–2000 × g at 4°C. Plasma samples were stored frozen at −20°C or lower and shipped the next day on dry ice from the study site to the University of Michigan, Pharmacokinetic Core laboratory. Samples were analyzed in batches during the study period. Details regarding the spill event were completed using a brief report mechanism submitted by participants through a secure website. This report detailed the specific agent associated with the spill, time and date of occurrence, estimated quantity of spill, body areas exposed to the HD and estimated duration of exposure.
Results
This study focused on 20 HDs that are the most commonly administered chemotherapy agents in ambulatory oncology settings with chemical properties suitable for analysis. The developed LC-MS/MS method allowed the simultaneous detection of 18 of them on the positive ionization mode. Unfortunately, the method could not be extended to two agents, fludarabine and fluorouracil because they had stronger responses in the negative ionization mode while all other agents were best ionized in the positive mode. In order to exclude a cross contamination of the internal standard (IS), deuterated compounds were chosen: toptecan-d6, irinotecan-d10, bendamustine-d6 and docetaxel-d9 were used as IS for those ADs with similar retention time (RT). Details of the optimization approach are provided below.
Optimization of HPLC-MS/MS multi-target screening method
The HPLC method was optimized to enable best separation of 18 ADs in 13 min (17 min including the column reconditioning). The unresolved drugs presented different scan events in MRM mode. A typical chromatogram, obtained from the analysis of a calibration sample with 50 ng/mL of each analyte, 10 ng/mL for IS1 and IS4 and 100 ng/mL for IS2 and IS3 is shown in Figure 1. The ESI–MS/MS conditions were optimized for each analyte after infusion of individual standard solutions (500 ng/mL). The positive ESI mode provided higher signal intensity for the 18 HDs selected. The positive/negative ion-switching method had been tested for monitoring those 18 HDs together with fludarabine and fluorouracil in negative ESI mode, but the other analytes’ signal intensity dropped dramatically (data not shown). As a consequence, fludarabine and fluorouracil were excluded in favor of maintaining higher sensitivity to other agents in the panel (Table 1).
Figure 1.
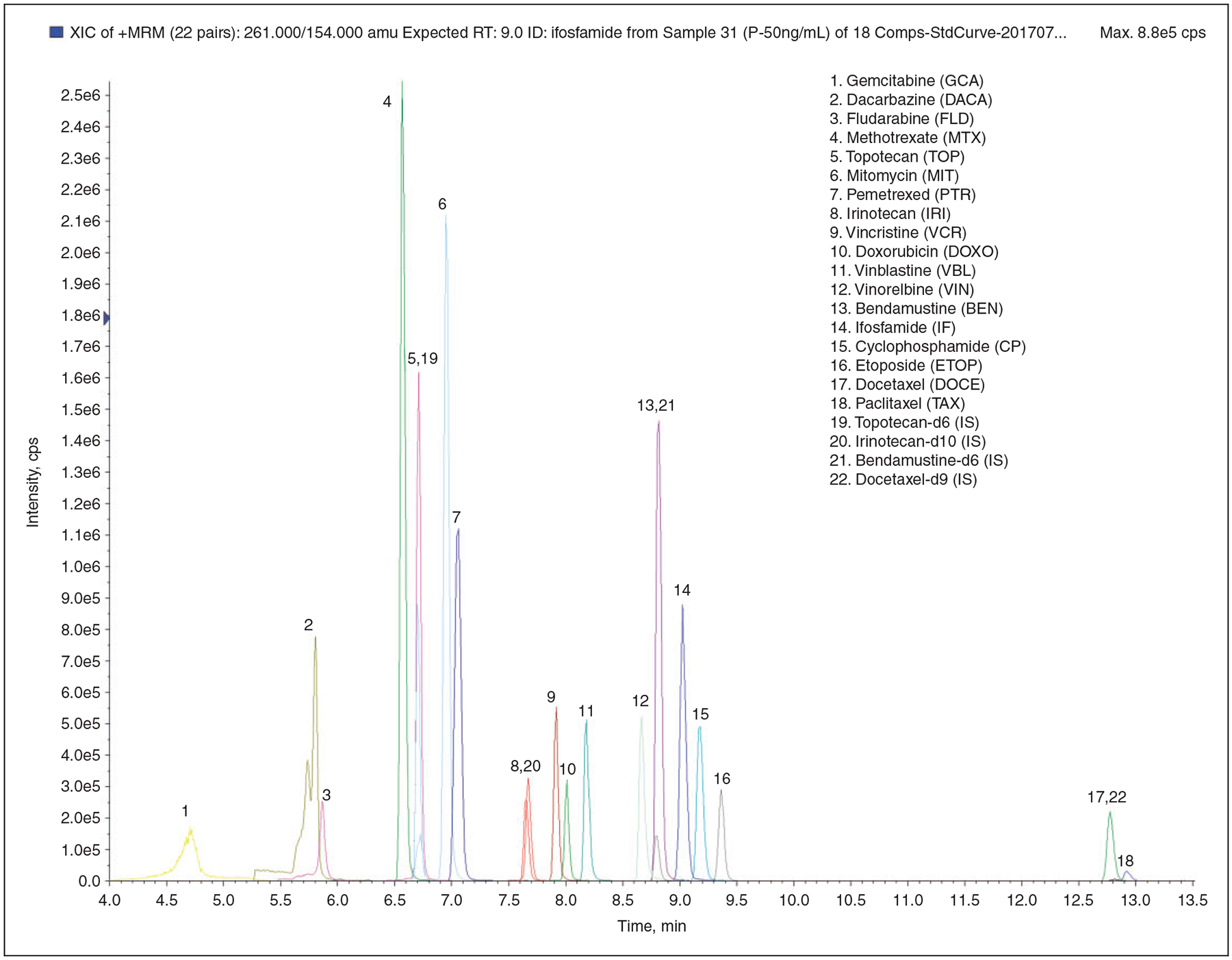
Overlaid MRM chromatograms of 18 chemotherapy drugs at 50 ng/mL in plasma.
Table 1.
Key LC-MS/MS parameters for multiple reaction monitoring (MRM) and enhanced product ion (EPI) acquisition of the 18 target compounds.
| Analyte | Retention time (min) | MRM transitions (m/z) | Declustering potential (V) | Collision energy (V) | Collision exit potential (V) | MS/MS fragments (m/z) |
|---|---|---|---|---|---|---|
| Gemcitabine (GCA) | 4.71 | 264/112 | 60 | 23 | 10 | 264.1, 246.0, 198.0, 194.9, 151.0, 112.1 |
| Dacarbazine (DACA) | 5.80 | 183/166 | 60 | 23 | 10 | 183.1, 166.0, 138.1, 123.0 |
| Fludarabine (FLD) | 5.87 | 366/154 | 50 | 22 | 10 | 366.0, 154.1, 134.0 |
| Methotrexate (MTX) | 6.57 | 455/308 | 60 | 30 | 10 | 455.2, 326.0, 308.2, 175.1, 134.0 |
| Topotecan (TOP) | 6.71 | 422/377 | 60 | 32 | 10 | 422.2, 377.0, 349.1, 333.2, 320.0, 305.2 |
| Mitomycin (MIT) | 6.95 | 335/242 | 60 | 19 | 10 | 335.2, 274.1, 242.2, 227.0, 214.1, 199.2 |
| Pemetrexed (PTR) | 7.05 | 428/281 | 60 | 28 | 10 | 428.3, 382.1, 299.1, 281.1, 253.3, 163.2, 119.0 |
| Irinotecan (IRI) | 7.67 | 587/502 | 60 | 43 | 10 | 587.3, 543.4, 502.3, 458.3, 414.4, 331.2, 303.1, 195.2, 167.0, 124.1 |
| Vincristine (VCR) | 7.92 | 413/353 | 60 | 23 | 10 | 413.3, 392.2, 383.0, 362.3, 353.3 |
| Doxorubicin (DOXO) | 8.01 | 544/397 | 60 | 19 | 10 | 544.0, 397.2, 379.1, 361.0, 346.0, 333.0, 321.0 |
| Vinblastine (VBL) | 8.18 | 406/271 | 100 | 25 | 10 | 406.3, 397.1, 376.1, 346.4, 271.9 |
| Vinorelbine (VIN) | 8.66 | 390/122 | 60 | 19 | 10 | 658.2, 626.2, 598.3, 566.0, 360.5, 122.0 |
| Bendamustine (BEN) | 8.81 | 358/304 | 60 | 40 | 10 | 358.1, 340.1, 322.2, 304.2, 242.2, 228.4 |
| Ifosfamide (IF) | 9.03 | 261/154 | 60 | 32 | 10 | 261.0, 233.0, 182.2, 154.0, 120.1 |
| Cyclophosphamide (CP) | 9.17 | 261/140 | 60 | 35 | 10 | 261.1, 233.2, 142.0, 140.0, 106.2, 104.0 |
| Etoposide (ETOP) | 9.36 | 589/229 | 60 | 23 | 10 | 589.3, 383.3, 299.2, 247.1, 229.1, 217.1, 199.1, 185.2 |
| Docetaxel (DOCE) | 12.8 | 808/226 | 20 | 20 | 10 | 808.4, 591.2, 531.2, 527.2, 509.2, 345.3, 327.2, 309.1, 281.1, 226.2, 181.9 |
| Paclitaxel (TAX) | 12.9 | 854/286 | 60 | 20 | 10 | 854.4, 569.2, 509.2, 447.2, 286.3 |
| Topotecan-d6 (IS) | 6.70 | 428/377 | 100 | 25 | 10 | - |
| Irinotecan-d10 (IS) | 7.65 | 597/133 | 100 | 47 | 10 | - |
| Bendamustine-d6 (IS) | 8.79 | 364.1/232 | 100 | 50 | 10 | - |
| Docetaxel-d9 (IS) | 12.8 | 839a/549 | 60 | 34 | 10 | - |
Sodium adduct was monitored.
The goals of HPLC-MS/MS optimization method were to obtain the most sensitivity by increasing the dwell time, while diminishing the total scan time. Thus, the so-called Scheduled MRM™ algorithm was adopted. The Scheduled MRM™ algorithm used knowledge of the retention time (RT) of each analyte so that each MRM transition was only monitored using a short time window. At any one point in time, the numbers of concurrent MRM transitions are significantly reduced, resulting in much higher duty cycles for each analyte. The software calculated the maximum dwell times for the co-eluting compounds while still maintaining the desired cycle time for the best signal-to noise ratio, accuracy and reproducibility by maintaining the same, or even improving the number of points across the peak. As a result, Scheduled MRM™ allowed the monitoring of many more MRM transitions in a single acquisition without compromising data quality.
Despite the high selectivity of MRM detection, we identified a risk of false positive findings due to interfering matrix signals. As a consequence a second MRM was monitored per analyte and the ratio of quantifier to qualifier transition was calculated for each unknown sample and compared to the MRM ratio of standards for identification. Previous reports suggest that relying only on MRM ratios for identification can result in a significant number of false positive results for compound identification, especially if the targeted analytes have a low fragmentation efficiency (many low intensity product ions).31–33 The sensitivity can be dramatically decreased if both quantifier and qualifier transitions are applied in the same run. Thus, only quantifier transitions were applied in the present study. But for improved accuracy, identification can be performed using full scan MS/MS experiments and comparison of the unknown with a standard spectrum. Therefore, dependent MS/MS spectra were acquired in the EPI mode of the QTRAP® 5500 system after being triggered from a Scheduled MRM™ IDA survey scan. The rapidly collected high-quality MS/MS data was used to increase the confidence of detection. Once any HD residue is detected and confirmed by the MS/MS data, the MRM survey scan data can be used for quantification.
Linearity and sensitivity
The developed method was found to be linear over the studied concentration range for all compounds (correlation coefficient 0.99266 to 0.99853). The calculated LODs by MRM-IDA-EPI scan and LOQs by MRM scan ranged from 0.10 to 10.0 ng/mL and from 0.10 to1.00 ng/mL in plasma, from 1.00 to 5.00 ng/mL and from 0.10 to 1.00 ng/mL in urine respectively, as shown in Table 2.
Clinical trial plasma results
A total of 743 plasma samples were assayed from 378 unique participants from 12 centers over the sampling period. There were 132 kits deployed to assess spills reported during this period and 64 participants were sampled on the day of spill or within 24 h post-exposure. The compounds by frequency of exposure were paclitaxel (32.6%), doxorubicin (13.5%), etoposide (13.5%), gemcitabine (11.5%), bendamustine (11.5%), docetaxel (9.6%), irinotecan (5.7%), and cyclophosphamide (2.0%). All plasma sample measurements were below the lower limit of detection as outlined in Table 2. Plasma samples considered to be positive were ruled out as false positive and excluded by further comparison of the MS/MS data against the analytical standards.
Discussion
Cancer chemotherapy regimens that were traditionally administered in the inpatient setting are increasingly being administered in the ambulatory and home settings. This transition reduces costs to patients and the health care organization while improving patient satisfaction with the care that is received. In the US, approximately 23 million adult patient visits occur annually for chemotherapy, of which 84% are delivered primarily by nurses in the ambulatory setting. We previously conducted a statewide survey of oncology nurses to examine the likelihood of self-reported accidental exposure to cancer chemotherapy. We specifically sought to understand the influence of practice environment, nursing workload, and safety standards on this exposure risk. Skin or eye exposures to cancer chemotherapy were reported in 16.9% of nurses surveyed and the likelihood of exposure was related to staffing and resources as well as chemotherapy dose verification by another nurse. These findings highlighted the need for a system to provide definitive feedback to nurses about the systemic exposure risk associated with acute chemotherapy spills. The issuance of the USP<800> standard implies that a larger number of institutions will have to come under compliance with health safety protections for a large pool of health care workers who handle HDs.
Most health care systems are unlikely to have the resources or sample numbers necessary to establish their own analytical assays to provide definitive risk assessment when an acute spill occurs. Most studies that have reported systemic exposures to acute spills have relied on analytical methods that monitored one or a handful of analytes at the same time.12,14,16,17,22,24,28 However, health care workers within a setting can be exposed to a multitude of HDs in a given day. Centralized analytical laboratories are also unlikely to have the throughput and technical staff to manage sample assay requests for these acute spill events for individual analyte assessments. As a consequence, development of a multi-analyte platform represents a necessary cost-effective solution to address this challenge of sample assay for a large health care network or statewide initiative centered on definitive risk assessment. Current analytical methods that have been developed to monitor multiple HDs require tedious sample preparation that hinders feasibility of time-sensitive biological monitoring.17,23,29 We demonstrated the ability to rapidly and reliably measure 18 widely used cancer chemotherapy drugs in plasma using a simplified sample processing method and state-of-the-art mass spectrometry analytical techniques.
This analytical approach was used to screen and quantify cancer chemotherapy in a large cohort of nurses at baseline and in participants exposed to a spill. Fortunately, no cancer chemotherapy concentrations were detectable in plasma implying: (1) a low likelihood of cutaneous absorption and/or (2) cutaneous absorption with limited distribution into systemic circulation. In addition, most participating sites employed engineering controls, including biological safety cabinets for compounding and closed system transfer devices during drug preparation and administration. Our previously published trial results suggest that personal protective use was suboptimal at baseline and did not improve with study interventions.30 This unchanged low use of PPE implies that either a low potential for cutaneous absorption or cutaneous absorption with limited systemic distribution most likely accounted for undetectable plasma concentrations of these cancer chemotherapy agents. However, these findings are limited to acute spill based assessment of parent compounds that may not reflect chronic cancer chemotherapy exposures detectable through surrogate analytes of exposure such as metabolites.
Conclusions
Definitive risk assessment through biological sampling is a relevant consideration for health care workers’ who handle HDs such as chemotherapy. Since workers within a health care facility are likely to be exposed to multiple drugs, cost- and time-saving procedures for simultaneous analysis of different compounds are required. In the current study, the combination of HPLC and scheduled MRM-IDA-EPI mass spectrometric method permitted analysis of 18 HDs in a single chromatographic run. The present method focuses not only on the quantification but also on the confident detection of the trace HD residue in plasma samples, to eliminate false positive results. Since there are no guidelines for biologic exposure or acceptable intake and exposure limits set for HDs,25,34 tedious, cost- and time-consuming LC-MS methods for quantification are not justified. The HDs monitoring methodology described here is simple, cost- and time-saving. Moreover, the collected MS/MS data enable detection of HDs more reliably, which can inform a feasible approach to definitive risk assessment of HDs.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This paper was supported by grant number 1 R01 OH 010582, funded by the National Institute for Occupational Safety and Health, Centers for Disease Control and Prevention. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention or the Department of Health and Human Services.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Burroughs GE CT, McDiarmid MA, Mead KR, et al. Preventing occupational exposures to antineoplastic and other hazardous drugs in health care settings. National Institute for Occupational Safety and Health 2004; NIOSH publication number 2004–165. [Google Scholar]
- 2.Polovich M. Safe handling of hazardous drugs. Online J Issues Nurs 2004; 9: 6. [PubMed] [Google Scholar]
- 3.American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health Syst Pharm 2006; 63: 1172–1193. [Google Scholar]
- 4.Neuss MN, Polovich M, McNiff K, et al. 2013 updated American Society of Clinical Oncology/Oncology Nursing Society chemotherapy administration safety standards including standards for the safe administration and management of oral chemotherapy. J Oncol Pract 2013; 9: 5 s–13 s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polovich M. Safe handling of hazardous drugs. 2nd ed. Pittsburgh: Oncology Nursing Society, 2011. [Google Scholar]
- 6.Polovich M, Olsen MM and LeFebvre KB. Chemotherapy and biotherapy guidelines and recommendations for practice, 4th ed. Pittsburgh: Oncology Nursing Society, 2014. [Google Scholar]
- 7.Walton AL, Eisenberg S and Friese CR. Hazardous drugs: legislative and regulatory efforts to improve safe handling. Clin J Oncol Nurs 2017; 21: 254–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boiano JM, Steege AL and Sweeney MH. Adherence to safe handling guidelines by health care workers who administer antineoplastic drugs. J Occup Environ Hyg 2014; 11: 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boiano JM, Steege AL and Sweeney MH. Adherence to precautionary guidelines for compounding antineoplastic drugs: a survey of nurses and pharmacy practitioners. J Occup Environ Hyg 2015; 12: 588–602. [DOI] [PubMed] [Google Scholar]
- 10.Turci R, Sottani C, Spagnoli G, et al. Biological and environmental monitoring of hospital personnel exposed to antineoplastic agents: a review of analytical methods. J Chromatogr B Analyt Technol Biomed Life Sci 2003; 789: 169–209. [DOI] [PubMed] [Google Scholar]
- 11.Sottani C, Tranfo G, Bettinelli M, et al. Trace determination of anthracyclines in urine: a new high-performance liquid chromatography/tandem mass spectrometry method for assessing exposure of hospital personnel. Rapid Commun Mass Spectrom 2004; 18: 2426–2436. [DOI] [PubMed] [Google Scholar]
- 12.Sottani C, Tranfo G, Faranda P, et al. Highly sensitive high-performance liquid chromatography/selective reaction monitoring mass spectrometry method for the determination of cyclophosphamide and ifosfamide in urine of health care workers exposed to antineoplastic agents. Rapid Commun Mass Spectrom 2005; 19: 2794–800. [DOI] [PubMed] [Google Scholar]
- 13.Sottani C, Rinaldi P, Leoni E, et al. Simultaneous determination of cyclophosphamide, ifosfamide, doxorubicin, epirubicin and daunorubicin in human urine using high-performance liquid chromatography/electrospray ionization tandem mass spectrometry: bioanalytical method validation. Rapid Commun Mass Spectrom 2008; 22: 2645–2659. [DOI] [PubMed] [Google Scholar]
- 14.Pieri M, Castiglia L, Basilicata P, et al. Biological monitoring of nurses exposed to doxorubicin and epirubicin by a validated liquid chromatography/fluorescence detection method. Ann Occup Hyg 2010; 54: 368–376. [DOI] [PubMed] [Google Scholar]
- 15.Nussbaumer S, Fleury-Souverain S, Antinori P, et al. Simultaneous quantification of ten cytotoxic drugs by a validated LC-ESI-MS/MS method. Anal Bioanal Chem 2010; 398: 3033–3042. [DOI] [PubMed] [Google Scholar]
- 16.Ndaw S, Denis F, Marsan P, et al. Biological monitoring of occupational exposure to 5-fluorouracil: urinary alpha-fluoro-beta-alanine assay by high performance liquid chromatography tandem mass spectrometry in health care personnel. J Chromatogr B Analyt Technol Biomed Life Sci 2010; 878: 2630–2634. [DOI] [PubMed] [Google Scholar]
- 17.Fabrizi G, Fioretti M, Rocca LM, et al. DESI-MS2: a rapid and innovative method for trace analysis of six cytostatic drugs in health care setting. Anal Bioanal Chem 2012; 403: 973–983. [DOI] [PubMed] [Google Scholar]
- 18.Nussbaumer S, Geiser L, Sadeghipour F, et al. Wipe sampling procedure coupled to LC-MS/MS analysis for the simultaneous determination of 10 cytotoxic drugs on different surfaces. Anal Bioanal Chem 2012; 402: 2499–2509. [DOI] [PubMed] [Google Scholar]
- 19.Pretty JR, Connor TH, Spasojevic I, et al. Sampling and mass spectrometric analytical methods for five antineoplastic drugs in the healthcare environment. J Oncol Pharm Pract 2012; 18: 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vyas N, Yiannakis D, Turner A, et al. Occupational exposure to anti-cancer drugs: a review of effects of new technology. J Oncol Pharm Pract 2014; 20: 278–287. [DOI] [PubMed] [Google Scholar]
- 21.B’Hymer C, Connor T, Stinson D, et al. Validation of an HPLC-MS/MS and wipe procedure for mitomycin C contamination. J Chromatogr Sci 2015; 53: 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teschke CY, Hon K, Shen H, et al. Antineoplastic drug contamination in the urine of Canadian healthcare workers. Int Arch Occup Environ Health 2015; 88: 933–941. [DOI] [PubMed] [Google Scholar]
- 23.Fabrizi G, Fioretti M and Mainero Rocca L. Dispersive solid phase extraction procedure coupled to UPLC-ESI-MS/MS analysis for the simultaneous determination of thirteen cytotoxic drugs in human urine. Biomed Chromatogr 2016; 30: 1297–1308. [DOI] [PubMed] [Google Scholar]
- 24.Canal-Raffin M, Khennoufa K, Martinez B, et al. Highly sensitive LC-MS/MS methods for urinary biological monitoring of occupational exposure to cyclophosphamide, ifosfamide, and methotrexate antineoplastic drugs and routine application. J Chromatogr B Analyt Technol Biomed Life Sci 2016; pii: S1570–0232(16)31088–1. [DOI] [PubMed] [Google Scholar]
- 25.Kibby T. A review of surface wipe sampling compared to biologic monitoring for occupational exposure to antineoplastic drugs. J Occup Environ Hyg 2017; 14: 159–174. [DOI] [PubMed] [Google Scholar]
- 26.Guichard N, Guillarme D, Bonnabry P, et al. Antineoplastic drugs and their analysis: a state of the art review. Analyst 2017; 142: 2273–2321. [DOI] [PubMed] [Google Scholar]
- 27.Mathias PI, Connor TH and B’Hymer C. A review of high performance liquid chromatographic-mass spectrometric urinary methods for anticancer drug exposure of health care workers. J Chromatogr B Analyt Technol Biomed Life Sci 2017; 1060: 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hedmer M, Tinnerberg H, Axmon A, et al. Environmental and biological monitoring of antineoplastic drugs in four workplaces in a Swedish hospital. Int Arch Occup Environ Health 2008; 81: 899–911. [DOI] [PubMed] [Google Scholar]
- 29.Zhou JY, Gao SH, Zhang F, et al. Liquid chromatography-tandem mass spectrometry method for simultaneous determination of seven commonly used anticancer drugs in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2012; 906: 1–8. [DOI] [PubMed] [Google Scholar]
- 30.Friese CR, Yang J, Mendelsohn-Victor K, et al. Randomized controlled trial of an intervention to improve nurses’ hazardous drug handling. Oncol Nurs Forum 2019; 46: 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schurmann A, Dvorak V, Cruzer C, et al. False-positive liquid chromatography/tandem mass spectrometric confirmation of sebuthylazine residues using the identification points system according to EU directive 2002/657/EC due to a biogenic insecticide in tarragon. Rapid Commun Mass Spectrom 2009; 23: 1196–1200. [DOI] [PubMed] [Google Scholar]
- 32.Bueno MJ, Aguera A, Gomez MJ, et al. Application of liquid chromatography/quadrupole-linear Ion trap mass spectrometry and time-of-flight mass spectrometry to the determination of pharmaceuticals and related contaminants in wastewater. Anal Chem 2007; 79: 9372–9384. [DOI] [PubMed] [Google Scholar]
- 33.Gros M, Petrovic M and Barcelo D. Tracing pharmaceutical residues of different therapeutic classes in environmental waters by using liquid chromatography/quadrupole-linear ion trap mass spectrometry and automated library searching. Anal Chem 2009; 81: 898–912. [DOI] [PubMed] [Google Scholar]
- 34.Easty AC, Coakley N, Cheng R, et al. Safe handling of cytotoxics: guideline recommendations. Curr Oncol 2015; 22: e27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]